Embed Size (px)
Citation preview

a-Cardiac myosin heavy chain (MYH6) mutationsaffecting myofibril formation are associated withcongenital heart defects
Javier T. Granados-Riveron1, Tushar K. Ghosh1, Mark Pope1, Frances Bu’Lock2,
Christopher Thornborough2, Jacqueline Eason3, Edwin P. Kirk4,5,6, Diane Fatkin4,7,8,9,
Michael P. Feneley4,7,8,9, Richard P. Harvey4,7,8, John A.L. Armour1 and J. David Brook1,∗
1Institute of Genetics, School of Biology, Queen’s Medical Centre, University of Nottingham, Nottingham, UK,2Department of Paediatric Cardiology, Glenfield Hospital, Leicester, UK, 3Clinical Genetics Service, City Hospital,
Nottingham, UK, 4Victor Chang Cardiac Research Institute, Darlinghurst, New South Wales, Australia, 5Department of
Medical Genetics, Sydney Children’s Hospital, Randwick, New South Wales, Australia, 6Faculty of Medicine, School of
Women’s and Children’s Health, University of New South Wales, New South Wales, Australia, 7Faculty of Science and8Faculty of Medicine, University of New South Wales, Australia and 9Department of Cardiology, St Vincent’s Hospital,
Darlinghurst, New South Wales, Australia
Received May 27, 2010; Revised and Accepted July 20, 2010
Congenital heart defects (CHD) are collectively the most common form of congenital malformation. Studies ofhuman cases and animal models have revealed that mutations in several genes are responsible for both familialand sporadic forms of CHD. We have previously shown that a mutation in MYH6 can cause an autosomal domi-nant form of atrial septal defect (ASD), whereas others have identified mutations of the same gene in patientswith hypertrophic and dilated cardiomyopathy. In the present study, we report a mutation analysis of MYH6 inpatients with a wide spectrum of sporadic CHD. The mutation analysis of MYH6 was performed in DNA samplesfrom 470 cases of isolated CHD using denaturing high-performance liquid chromatography and sequenceanalysis to detect point mutations and small deletions or insertions, and multiplex amplifiable probe hybridiz-ation to detect partial or complete copy number variations. One non-sense mutation, one splicing site mutationand seven non-synonymous coding mutations were identified. Transfection of plasmids encoding mutant andnon-mutant green fluorescent protein-MYH6 fusion proteins in mouse myoblasts revealed that the mutationsA230P and A1366D significantly disrupt myofibril formation, whereas the H252Q mutation significantlyenhances myofibril assembly in comparison with the non-mutant protein. Our data indicate that functionalvariants of MYH6 are associated with cardiac malformations in addition to ASD and provide a novel potentialmechanism. Such phenotypic heterogeneity has been observed in other genes mutated in CHD.
INTRODUCTION
The heart is the first functional organ to form in mammals andits development is controlled by a complex network of factorsand processes that we are only starting to understand. Disrup-tions to heart development cause congenital heart disease(CHD), the most common birth defect in humans. It hasbeen estimated that the incidence of moderate and severe
forms of CHD occurs in 6 out of every 1000 live births. Ifbicuspid aortic valve and trivial ventricular septal defects aretaken into account, the incidence rises to 19/1000 and 75/1000, respectively (1).
A genetic component of CHD aetiology has been suggestedby numerous epidemiological studies (2). Approximately aquarter of CHD cases occur as part of a complex withdefects in other organs in the form of a sporadic malformative
∗To whom correspondence should be addressed at: Institute of Genetics, Queen’s Medical Centre, University of Nottingham, Nottingham NG7 2UH,UK. Tel: +44 1158230345; Fax: +44 1158230313; Email: [email protected]
# The Author 2010. Published by Oxford University Press. All rights reserved.For Permissions, please email: [email protected]
Human Molecular Genetics, 2010, Vol. 19, No. 20 4007–4016doi:10.1093/hmg/ddq315Advance Access published on July 23, 2010
at Periodicals Departm
ent , Hallw
ard Library, U
niversity of Nottingham
on Novem
ber 29, 2011http://hm
g.oxfordjournals.org/D
ownloaded from

association, Mendelian syndrome or chromosomal abnormal-ity. The remaining three-quarters of cases occur as isolateddefects, most of them as sporadic events but a small proportionas familial cases, showing Mendelian patterns of inheritance(3–5). We have previously shown that a missense substitutionwithin the IQ domain of the a-cardiac myosin heavy chain(MYH6), expressed in the human atria, causes an autosomaldominant form of secundum atrial septal defect (ASD) withincomplete penetrance (6). However, others have foundmutations of MYH6 in patients with hypertrophic (HCM)and dilated cardiomyopathy [DCM (7–9)]. Studies in animalmodels have shown that deficiency during development ofthe atrial myosin in zebrafish, chicken and Xenopus tropicalisresults in malformed hearts (10–12). In order to evaluatethe role of mutations of MYH6 in sporadic CHD, we per-formed a denaturing high-performance liquid chromato-graphy (dHPLC) scan and sequence analysis to detect pointmutations, small deletions and insertions, and multiplexamplifiable probe hybridization (MAPH) to detect partial orcomplete copy number variations (deletions or duplications)of the coding and regulatory segments of the gene in acohort of DNA samples from 470 CHD patients. We testedthe ability of several of the MYH6 mutant myosin moleculesto form myofibrils in differentiated myoblasts.
RESULTS
Mutations of MYH6 are associated with otherCHDs in addition to ASD
We have conducted a mutational scan of the coding regions ofthe MYH6 gene in a cohort of 470 CHD patients (see Sup-plementary Material, Table S1). We have identified a stop-codon mutation, a splice acceptor site mutation as well asseven missense mutations. All nine mutations were absent in480 ethnically matched control subjects (960 chromosomes)and are described in what follows in the order that theyoccur in the MYH6 gene and are summarized in Table 1.The position of the following mutations is given with refer-ence to sequence accession NM_002471.2 for the MYH6cDNA and NP_002462.2 for the protein.
Four mutations were found within the segment of the MYH6gene encoding the motor domain. A 755G.C transversion(A230P, Fig. 1A) was detected in exon 8 in a 32-year-oldfemale patient diagnosed in infancy with tricuspid atresia, hypo-plastic right ventricle, large secundum ASD and valvular andsupravalvular pulmonary stenosis. A Fontan operation was per-formed at 12 years of age. The mutation was transmitted by thefather, who has left ventricular hypertrophy. An unaffectedsister also carries the variant. The alanine residue found in thenon-mutant protein is conserved in every muscle myosinsequence available and is predicted to disrupt the helicalconfiguration of the segment contiguous to the Switch-1 loopN-terminus. In exon 9, an 829C.G transversion (H252Q,Fig. 1B) was identified in a 16-year-old patient with transpositionof the great arteries. The mother, with persistence of the foramenovale, and the unaffected grandmother carry the mutation. The252 residue is located within a conserved segment and is pre-dicted to prevent the formation of a salt-bridge normallypresent between Strands 6 and 7 of the b-sheet forming part of
the transducer region. A non-sense mutation within exon 14,1568G.T (E501Stop, Fig. 1C), was discovered in a 7-year-oldmale patient with tricuspid atresia, restrictive ventricular septaldefect and hypoplastic right ventricle. The mutation is presentin the mother and maternal grandfather, both of whom are unaf-fected. Two unaffected siblings were found not to be carriers.This non-sense mutation is predicted to encode a truncatedpeptide of 500 residues containing most of the segments requiredfor nucleotide and actin binding but lacking most of the lower50 kDa domain and the entire 20 kDa domain as well as theneck and rod regions. In exon 18, a 2165G.A (V700M,Fig. 1D) mutation was found in a female patient with a largepatent foramen ovale. The mutation was absent in two clinicallynormal sons. This protein mutation is predicted to hinder themovements of the SH1 helix at different stages of the myosincycle.
Within the segment of the MYH6 gene encoding the myosinrod, five mutations were detected. In exon 26, a 3413C.Atransversion (R1116S, Fig. 1E) was found in a 69-year-oldfemale patient with an ASD. No samples were availablefrom other members of the family. The substitution occursat the ‘g’ heptad position of the second heptad unit of the11th 28-residue repeat. Within exon 29, a 4164C.A transver-sion (A1366D, Fig. 1F) was detected in a 2-year-old femalepatient with aortic valve stenosis. The variant was found inthe father who has septal dyskinesia and minor subaorticridge, in a male sibling with subaortic ridge and patentforamen ovale, in a second male sibling with patent foramenovale and in an unaffected dyzygotic twin sister (Fig. 2A).
Table 1. Phenotype of MYH6 mutations found in CHD
Mutation Subject Phenotype
Non-sense mutationE501Stop Proband TA
Mother UnaffectedGrandfather Unaffected
Splice site mutationIVS37-2A.G Proband ASD
Mother UnaffectedMissense mutations
A230P Proband TAFather LVHSister Unaffected
H252Q Proband TGAMother PFOGrandmother Unaffected
V700M Proband PFOR1116S Proband ASDA1366D Proband AS
Father SDK, SARSibling SAR, PFOSibling PFOSibling Unaffected
A1443D Proband ASDR1865Q Proband ASD
Mother DIVCSibling VSDSibling Unaffected
TA, tricuspid atresia; PTA, persistent truncus arteriosus; ASD, atrial septaldefect; LVH, left ventricular hypertrophy; TGA, transposition of great arteries;PFO, persistence of foramen ovale; AS, aortic stenosis; SDK, septal dyskinesis;SAR, subaortic ridge; DIVC, dilated inferior vena cava.
4008 Human Molecular Genetics, 2010, Vol. 19, No. 20
at Periodicals Departm
ent , Hallw
ard Library, U
niversity of Nottingham
on Novem
ber 29, 2011http://hm
g.oxfordjournals.org/D
ownloaded from
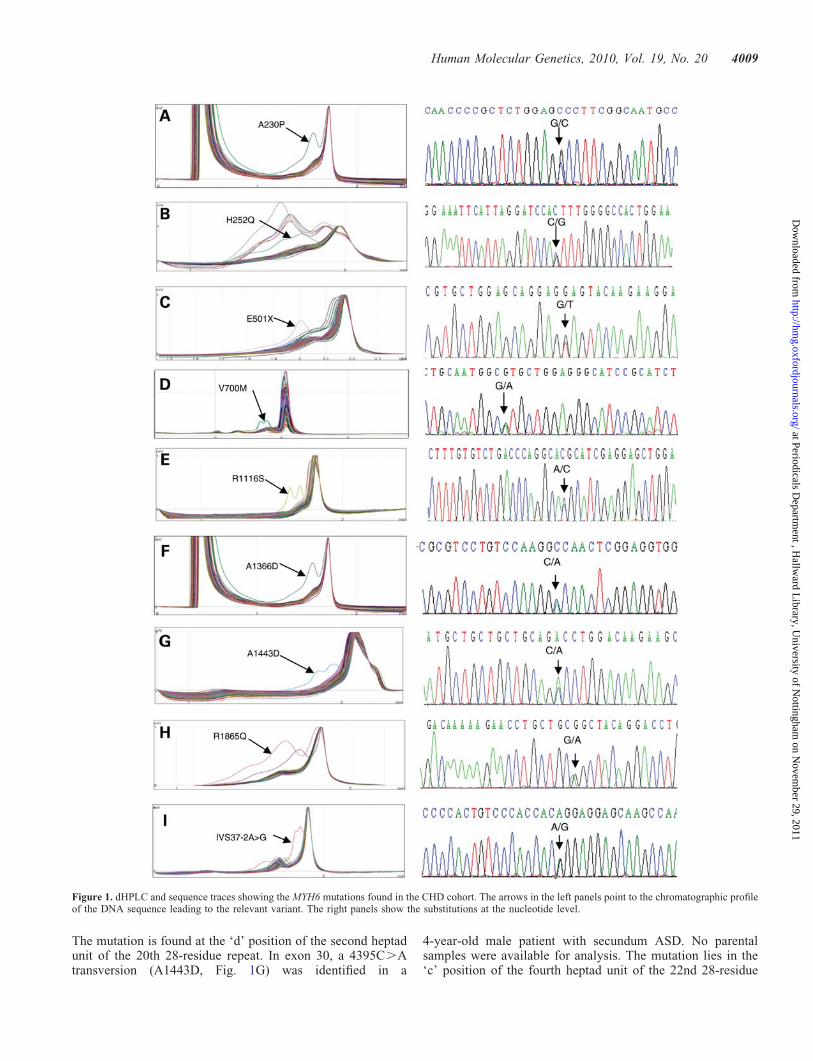
The mutation is found at the ‘d’ position of the second heptadunit of the 20th 28-residue repeat. In exon 30, a 4395C.Atransversion (A1443D, Fig. 1G) was identified in a
4-year-old male patient with secundum ASD. No parentalsamples were available for analysis. The mutation lies in the‘c’ position of the fourth heptad unit of the 22nd 28-residue
Figure 1. dHPLC and sequence traces showing the MYH6 mutations found in the CHD cohort. The arrows in the left panels point to the chromatographic profileof the DNA sequence leading to the relevant variant. The right panels show the substitutions at the nucleotide level.
Human Molecular Genetics, 2010, Vol. 19, No. 20 4009
at Periodicals Departm
ent , Hallw
ard Library, U
niversity of Nottingham
on Novem
ber 29, 2011http://hm
g.oxfordjournals.org/D
ownloaded from
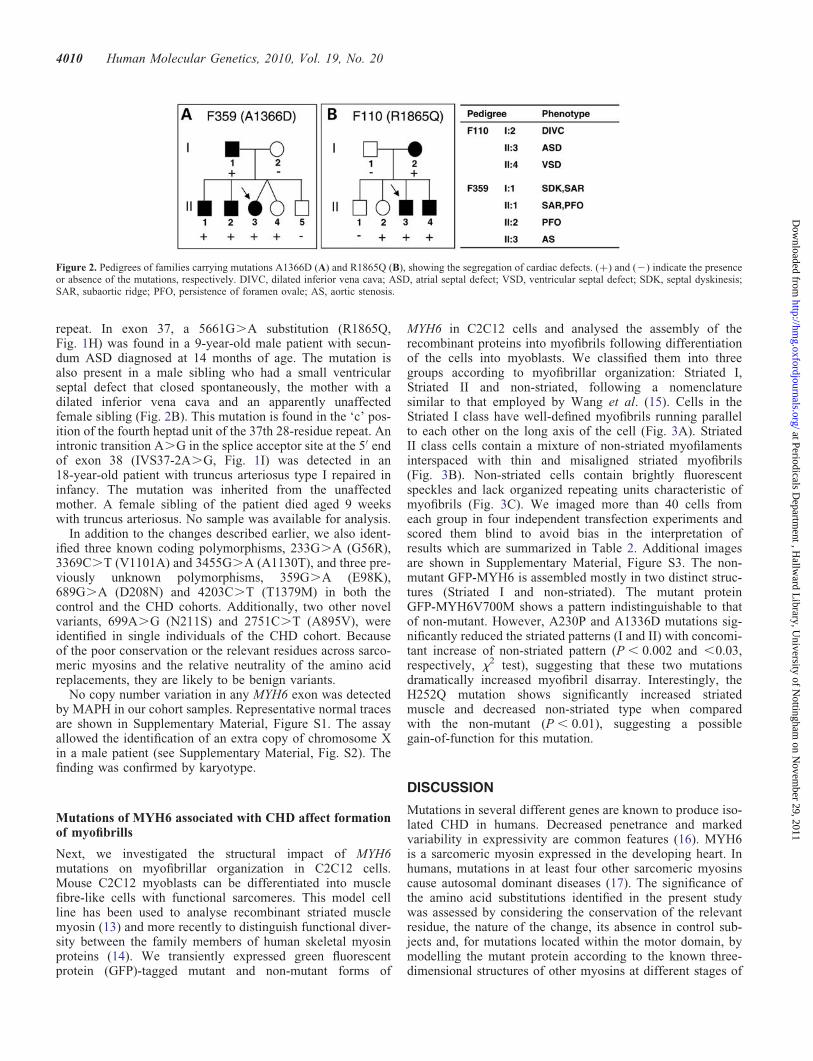
repeat. In exon 37, a 5661G.A substitution (R1865Q,Fig. 1H) was found in a 9-year-old male patient with secun-dum ASD diagnosed at 14 months of age. The mutation isalso present in a male sibling who had a small ventricularseptal defect that closed spontaneously, the mother with adilated inferior vena cava and an apparently unaffectedfemale sibling (Fig. 2B). This mutation is found in the ‘c’ pos-ition of the fourth heptad unit of the 37th 28-residue repeat. Anintronic transition A.G in the splice acceptor site at the 5′ endof exon 38 (IVS37-2A.G, Fig. 1I) was detected in an18-year-old patient with truncus arteriosus type I repaired ininfancy. The mutation was inherited from the unaffectedmother. A female sibling of the patient died aged 9 weekswith truncus arteriosus. No sample was available for analysis.
In addition to the changes described earlier, we also ident-ified three known coding polymorphisms, 233G.A (G56R),3369C.T (V1101A) and 3455G.A (A1130T), and three pre-viously unknown polymorphisms, 359G.A (E98K),689G.A (D208N) and 4203C.T (T1379M) in both thecontrol and the CHD cohorts. Additionally, two other novelvariants, 699A.G (N211S) and 2751C.T (A895V), wereidentified in single individuals of the CHD cohort. Becauseof the poor conservation or the relevant residues across sarco-meric myosins and the relative neutrality of the amino acidreplacements, they are likely to be benign variants.
No copy number variation in any MYH6 exon was detectedby MAPH in our cohort samples. Representative normal tracesare shown in Supplementary Material, Figure S1. The assayallowed the identification of an extra copy of chromosome Xin a male patient (see Supplementary Material, Fig. S2). Thefinding was confirmed by karyotype.
Mutations of MYH6 associated with CHD affect formationof myofibrills
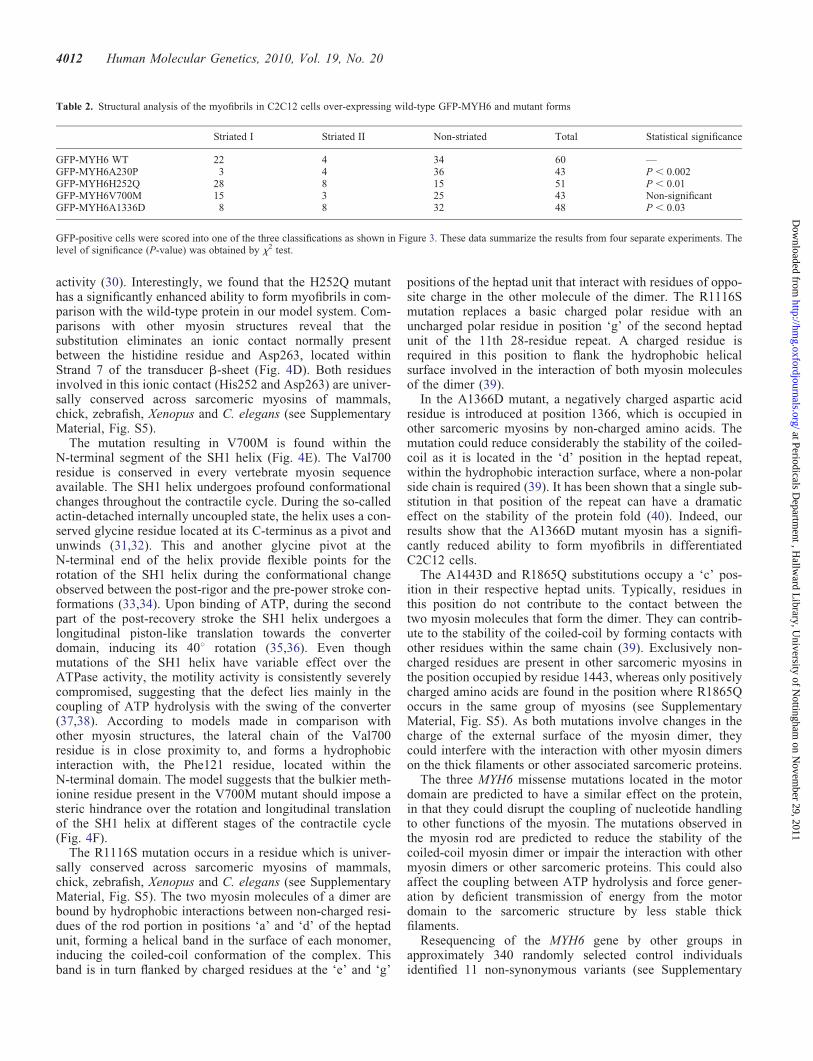
Next, we investigated the structural impact of MYH6mutations on myofibrillar organization in C2C12 cells.Mouse C2C12 myoblasts can be differentiated into musclefibre-like cells with functional sarcomeres. This model cellline has been used to analyse recombinant striated musclemyosin (13) and more recently to distinguish functional diver-sity between the family members of human skeletal myosinproteins (14). We transiently expressed green fluorescentprotein (GFP)-tagged mutant and non-mutant forms of
MYH6 in C2C12 cells and analysed the assembly of therecombinant proteins into myofibrils following differentiationof the cells into myoblasts. We classified them into threegroups according to myofibrillar organization: Striated I,Striated II and non-striated, following a nomenclaturesimilar to that employed by Wang et al. (15). Cells in theStriated I class have well-defined myofibrils running parallelto each other on the long axis of the cell (Fig. 3A). StriatedII class cells contain a mixture of non-striated myofilamentsinterspaced with thin and misaligned striated myofibrils(Fig. 3B). Non-striated cells contain brightly fluorescentspeckles and lack organized repeating units characteristic ofmyofibrils (Fig. 3C). We imaged more than 40 cells fromeach group in four independent transfection experiments andscored them blind to avoid bias in the interpretation ofresults which are summarized in Table 2. Additional imagesare shown in Supplementary Material, Figure S3. The non-mutant GFP-MYH6 is assembled mostly in two distinct struc-tures (Striated I and non-striated). The mutant proteinGFP-MYH6V700M shows a pattern indistinguishable to thatof non-mutant. However, A230P and A1336D mutations sig-nificantly reduced the striated patterns (I and II) with concomi-tant increase of non-striated pattern (P , 0.002 and ,0.03,respectively, x2 test), suggesting that these two mutationsdramatically increased myofibril disarray. Interestingly, theH252Q mutation shows significantly increased striatedmuscle and decreased non-striated type when comparedwith the non-mutant (P , 0.01), suggesting a possiblegain-of-function for this mutation.
DISCUSSION
Mutations in several different genes are known to produce iso-lated CHD in humans. Decreased penetrance and markedvariability in expressivity are common features (16). MYH6is a sarcomeric myosin expressed in the developing heart. Inhumans, mutations in at least four other sarcomeric myosinscause autosomal dominant diseases (17). The significance ofthe amino acid substitutions identified in the present studywas assessed by considering the conservation of the relevantresidue, the nature of the change, its absence in control sub-jects and, for mutations located within the motor domain, bymodelling the mutant protein according to the known three-dimensional structures of other myosins at different stages of
Figure 2. Pedigrees of families carrying mutations A1366D (A) and R1865Q (B), showing the segregation of cardiac defects. (+) and (2) indicate the presenceor absence of the mutations, respectively. DIVC, dilated inferior vena cava; ASD, atrial septal defect; VSD, ventricular septal defect; SDK, septal dyskinesis;SAR, subaortic ridge; PFO, persistence of foramen ovale; AS, aortic stenosis.
4010 Human Molecular Genetics, 2010, Vol. 19, No. 20
at Periodicals Departm
ent , Hallw
ard Library, U
niversity of Nottingham
on Novem
ber 29, 2011http://hm
g.oxfordjournals.org/D
ownloaded from

the contractile cycle (see Materials and Methods). Addition-ally, rod mutations were evaluated by considering the compat-ibility of the replacing residue with its position within therelevant heptad repeat.
The variant E501Stop encodes a 500 amino acid residue-truncated myosin peptide retaining most of the actin andnucleotide-binding segments but lacking the C-terminal partof the head, the neck and tail regions. Typically, transcriptswith premature stop codons located .50 bp 5′ of the last
exon–exon boundary are degraded through the non-sense-mediated decay (NMD) pathway, preventing the productionof the mutant protein (NMD-sensitive mutations). However,those with non-sense mutations located 3′ to this point arenot degraded (NMD-insensitive mutations), allowing thesynthesis of a truncated product (18). In general, NMD-sensitive mutations induce recessive phenotypes, whereasNMD-insensitive mutations can cause dominant disease, astruncated proteins compete with the full-length products forbinding to interacting partners and substrates, thus exerting adominant-negative effect (19). E501Stop is predicted to bean NMD-sensitive mutation. It has been noted, however, thatin humans, even in the presence of identical non-sensemutations, the efficiency of NMD shows individual (20,21)as well as inter-tissue (22,23) variation, modifying the severityand progression of disease. The incomplete penetranceobserved in the family carrying the E501Stop mutationcould be explained by decreased efficiency of NMD in theproband, where the truncated MYH6 would compete withthe non-mutant peptide for actin and/or adenosine triphosphate(ATP) binding.
The A.G transition in the splice acceptor site of intron 37is likely to induce skipping of exon 38 which encodes asegment of the assembly-competent domain (24).
The A230P mutation occurs within a segment of eightresidues which is completely conserved across sarcomericmyosins of mammals, chick, zebrafish, Xenopus and Caenor-habditis elegans (see Supplementary Material, Fig. S5), andis located at the C-terminal segment of the HG helix, whichis directly connected to the Switch-1 loop (Fig. 4A). Thisregion forms hydrogen-bond-mediated contacts with the phos-phate moiety of ATP and is responsible for the kinetic coup-ling between nucleotide binding and the variation of theaffinity for actin during the ATPase cycle (25,26). Thismutation is likely to alter the position of critical Switch-1nucleotide-binding residues by disrupting the helicalsegment adjacent to it. Based on predictions from crystalstructure comparisons (see Materials and Methods), the substi-tution abolishes a backbone/backbone hydrogen bond betweenthe alanine at position 230 and another alanine residue at pos-ition 227, introducing a hydrophobic lateral chain in the other-wise hydrophilic inner surface of the helix. Moreover, thissubstitution renders the segment incompatible with a tighthelical structure, as persistence of this configuration wouldinduce a steric clash between the mutant proline residue andAla227 (Fig. 4B). A comprehensive genetic analysis inC. elegans sarcomeric myosins has demonstrated thatmutations diminishing ATPase activity severely affect thickfilament assembly (27). Our results show that the A230Pmutation significantly disrupts assembly of the myosin mol-ecules into higher-order structures.
The H252Q variant is found in the C-terminal end of Strand6 of the b-sheet forming part of the transducer region(Fig. 4C), the configuration of which changes from fullytwisted at the rigor-like state to untwisted at the pre-powerstroke state (28). The distortion of the b-sheet contributes tothe coupling of the 50 kDa cleft closure, upon actin binding,to the decrease of nucleotide affinity (29). It has been shownthat mutations in the myosin transducer can have differenteffects. One such mutation, Mhc5, confers enhanced ATPase
Figure 3. Structural organization of wild-type GFP-MYH6 and mutants in themyofibrils of differentiated C2C12 cells. Based on the structural organization,they are grouped into three classes. (A) Striated I, cells have well-definedmyofibrils running parallel to each other along the long axis of the cells.(B) Striated II, cells contain a mixture of non-striated myofibrils interspacedwith thin and misaligned striated myofibrils. (C) Non-striated, cells containbrightly fluorescent speckles and lack organized repeating units characteristicsof myofibrils. The inset are enlargements of respective regions showing themyofibril structure.
Human Molecular Genetics, 2010, Vol. 19, No. 20 4011
at Periodicals Departm
ent , Hallw
ard Library, U
niversity of Nottingham
on Novem
ber 29, 2011http://hm
g.oxfordjournals.org/D
ownloaded from
activity (30). Interestingly, we found that the H252Q mutanthas a significantly enhanced ability to form myofibrils in com-parison with the wild-type protein in our model system. Com-parisons with other myosin structures reveal that thesubstitution eliminates an ionic contact normally presentbetween the histidine residue and Asp263, located withinStrand 7 of the transducer b-sheet (Fig. 4D). Both residuesinvolved in this ionic contact (His252 and Asp263) are univer-sally conserved across sarcomeric myosins of mammals,chick, zebrafish, Xenopus and C. elegans (see SupplementaryMaterial, Fig. S5).
The mutation resulting in V700M is found within theN-terminal segment of the SH1 helix (Fig. 4E). The Val700residue is conserved in every vertebrate myosin sequenceavailable. The SH1 helix undergoes profound conformationalchanges throughout the contractile cycle. During the so-calledactin-detached internally uncoupled state, the helix uses a con-served glycine residue located at its C-terminus as a pivot andunwinds (31,32). This and another glycine pivot at theN-terminal end of the helix provide flexible points for therotation of the SH1 helix during the conformational changeobserved between the post-rigor and the pre-power stroke con-formations (33,34). Upon binding of ATP, during the secondpart of the post-recovery stroke the SH1 helix undergoes alongitudinal piston-like translation towards the converterdomain, inducing its 408 rotation (35,36). Even thoughmutations of the SH1 helix have variable effect over theATPase activity, the motility activity is consistently severelycompromised, suggesting that the defect lies mainly in thecoupling of ATP hydrolysis with the swing of the converter(37,38). According to models made in comparison withother myosin structures, the lateral chain of the Val700residue is in close proximity to, and forms a hydrophobicinteraction with, the Phe121 residue, located within theN-terminal domain. The model suggests that the bulkier meth-ionine residue present in the V700M mutant should impose asteric hindrance over the rotation and longitudinal translationof the SH1 helix at different stages of the contractile cycle(Fig. 4F).
The R1116S mutation occurs in a residue which is univer-sally conserved across sarcomeric myosins of mammals,chick, zebrafish, Xenopus and C. elegans (see SupplementaryMaterial, Fig. S5). The two myosin molecules of a dimer arebound by hydrophobic interactions between non-charged resi-dues of the rod portion in positions ‘a’ and ‘d’ of the heptadunit, forming a helical band in the surface of each monomer,inducing the coiled-coil conformation of the complex. Thisband is in turn flanked by charged residues at the ‘e’ and ‘g’
positions of the heptad unit that interact with residues of oppo-site charge in the other molecule of the dimer. The R1116Smutation replaces a basic charged polar residue with anuncharged polar residue in position ‘g’ of the second heptadunit of the 11th 28-residue repeat. A charged residue isrequired in this position to flank the hydrophobic helicalsurface involved in the interaction of both myosin moleculesof the dimer (39).
In the A1366D mutant, a negatively charged aspartic acidresidue is introduced at position 1366, which is occupied inother sarcomeric myosins by non-charged amino acids. Themutation could reduce considerably the stability of the coiled-coil as it is located in the ‘d’ position in the heptad repeat,within the hydrophobic interaction surface, where a non-polarside chain is required (39). It has been shown that a single sub-stitution in that position of the repeat can have a dramaticeffect on the stability of the protein fold (40). Indeed, ourresults show that the A1366D mutant myosin has a signifi-cantly reduced ability to form myofibrils in differentiatedC2C12 cells.
The A1443D and R1865Q substitutions occupy a ‘c’ pos-ition in their respective heptad units. Typically, residues inthis position do not contribute to the contact between thetwo myosin molecules that form the dimer. They can contrib-ute to the stability of the coiled-coil by forming contacts withother residues within the same chain (39). Exclusively non-charged residues are present in other sarcomeric myosins inthe position occupied by residue 1443, whereas only positivelycharged amino acids are found in the position where R1865Qoccurs in the same group of myosins (see SupplementaryMaterial, Fig. S5). As both mutations involve changes in thecharge of the external surface of the myosin dimer, theycould interfere with the interaction with other myosin dimerson the thick filaments or other associated sarcomeric proteins.
The three MYH6 missense mutations located in the motordomain are predicted to have a similar effect on the protein,in that they could disrupt the coupling of nucleotide handlingto other functions of the myosin. The mutations observed inthe myosin rod are predicted to reduce the stability of thecoiled-coil myosin dimer or impair the interaction with othermyosin dimers or other sarcomeric proteins. This could alsoaffect the coupling between ATP hydrolysis and force gener-ation by deficient transmission of energy from the motordomain to the sarcomeric structure by less stable thickfilaments.
Resequencing of the MYH6 gene by other groups inapproximately 340 randomly selected control individualsidentified 11 non-synonymous variants (see Supplementary
Table 2. Structural analysis of the myofibrils in C2C12 cells over-expressing wild-type GFP-MYH6 and mutant forms
Striated I Striated II Non-striated Total Statistical significance
GFP-MYH6 WT 22 4 34 60 —GFP-MYH6A230P 3 4 36 43 P , 0.002GFP-MYH6H252Q 28 8 15 51 P , 0.01GFP-MYH6V700M 15 3 25 43 Non-significantGFP-MYH6A1336D 8 8 32 48 P , 0.03
GFP-positive cells were scored into one of the three classifications as shown in Figure 3. These data summarize the results from four separate experiments. Thelevel of significance (P-value) was obtained by x2 test.
4012 Human Molecular Genetics, 2010, Vol. 19, No. 20
at Periodicals Departm
ent , Hallw
ard Library, U
niversity of Nottingham
on Novem
ber 29, 2011http://hm
g.oxfordjournals.org/D
ownloaded from
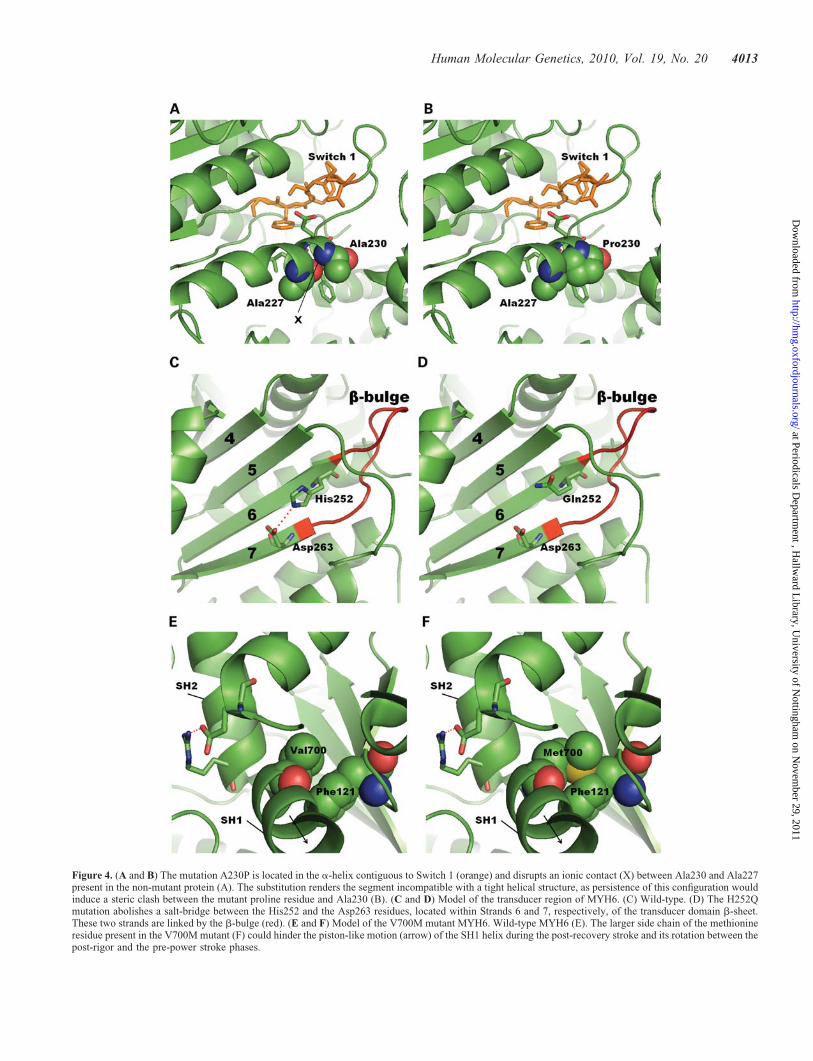
Figure 4. (A and B) The mutation A230P is located in the a-helix contiguous to Switch 1 (orange) and disrupts an ionic contact (X) between Ala230 and Ala227present in the non-mutant protein (A). The substitution renders the segment incompatible with a tight helical structure, as persistence of this configuration wouldinduce a steric clash between the mutant proline residue and Ala230 (B). (C and D) Model of the transducer region of MYH6. (C) Wild-type. (D) The H252Qmutation abolishes a salt-bridge between the His252 and the Asp263 residues, located within Strands 6 and 7, respectively, of the transducer domain b-sheet.These two strands are linked by the b-bulge (red). (E and F) Model of the V700M mutant MYH6. Wild-type MYH6 (E). The larger side chain of the methionineresidue present in the V700M mutant (F) could hinder the piston-like motion (arrow) of the SH1 helix during the post-recovery stroke and its rotation between thepost-rigor and the pre-power stroke phases.
Human Molecular Genetics, 2010, Vol. 19, No. 20 4013
at Periodicals Departm
ent , Hallw
ard Library, U
niversity of Nottingham
on Novem
ber 29, 2011http://hm
g.oxfordjournals.org/D
ownloaded from

Material, Text S1), only two of which show significant conser-vation across sarcomeric myosins of several mammals, chick,zebrafish, Xenopus and C. elegans. In contrast, all of the sevennon-synonymous changes we report show conservation withrespect to amino acid replacement (see SupplementaryMaterial, Fig. S5).
As in many other cases of mutations of genes associatedwith CHD (16), incomplete penetrance and phenotypic varia-bility are a common finding in the families in the presentstudy. Also, previous analyses have reported mutations ofMYH6 in cases of HCM and DCM (7–9). Conversely, variantsof the ACTC1 and MYH7 genes, both of which are mutated inHCM and DCM, have been described in patients with isolated(41) or associated with ventricular non-compaction CHD(42,43). Thus, it would appear that the effects of somemutations in cardiac muscle sarcomeric proteins manifestduring development, causing CHD, whereas others cause car-diomyopathy during postnatal life. This may be explained byrecent data suggesting that early myocardial contractility andintracardiac haemodynamic forces (44) are major factors con-trolling heart development, in particular, chamber (11) andendocardial cushion formation (45). Thus, we suggest thatother genes mutated in cardiomyopathy should be alsoassessed as candidates for CHD, both Mendelian and sporadic.
MATERIALS AND METHODS
Denaturing high-performance liquid chromatography
Peripheral blood samples from all participants were collectedafter informed consent and study approval by the local ethicscommittees. Genomic DNA was extracted from blood usingthe QIAmp DNA blood Midi or Maxi Kit (Qiagen, Hilden,Germany). Anonymous human control DNA was obtainedfrom the European Collection of Cell Culture (Salisbury,UK). Mutational analysis by dHPLC was performed asdescribed previously (46). Briefly, in order to cover the 39exons of the MYH6 gene, 35 polymerase chain reaction(PCR) amplicons were designed. In general, each ampliconspanned the complete length of an individual exon, plusshort segments of flanking intronic sequence to either side,to allow the detection of mutations of splicing regulatoryelements in those locations. In situations where two relativelysmall exons were close to each other, a single amplicon wasdesigned to cover them both. Large exons were covered bytwo overlapping amplicons (see Supplementary Material,Table S2). PCR reactions were carried out using patientDNA samples prepared from blood leucocytes according tostandard protocols. After DNA amplification, a final hybridiz-ation step was performed, starting at 958C and reducing thetemperature by 1.58C per minute to 258C in order to favourthe formation of heteroduplexes. The sequence of eachindividual amplicon was analysed using the Navigator soft-ware (Transgenomic, Omaha, NE, USA) to determine themelting profile of each DNA fragment and select theoptimal dHPLC temperatures. PCR products were processedusing the dHPLC WAVE System (Transgenomic). Ampliconsfrom samples showing a dHPLC trace suggestive ofvariation were sequenced using standard protocols. Potentially
deleterious variants were screened by dHPLC in 480 ethni-cally matched control subjects.
Multiplex amplifiable probe hybridization
MAPH was performed as described previously (47). Briefly,49 MHY6 and 6 control PCR amplicons of different and uni-formly spaced lengths (see Supplementary Material,Fig. S6), which could be resolved as discrete bands by electro-phoresis, were designed to span single or pairs of exons withless than two potential cross-hybridization targets. For exonswith more than two potential cross-hybridization targets,amplicons spanning segments of the flanking introns wereused instead (see Supplementary Material, Table S2 andFig. S4). PCR products of the amplicons were obtained andcloned in the EcoRV site of the pZero2 vector (Invitrogen,Carlsbad, CA, USA). In order to produce probes, the plasmidswere used as templates for PCR amplifications with vector-specific primers PZA and PZB (47). The probes were purifiedand pooled to produce the final probe mix. One microgram ofgenomic DNA from each patient was fixed to a 4 × 4 mmnylon filter membrane (Osmonics, Westborough, MA, USA)and hybridized with the probe mix. Filters were washed toremove the excess probe and boiled to recover specificallybound probe. The recovered probe solution was used as tem-plate for a PCR amplification reaction with FAM-labelledPZA primer. FAM-labelled PCR products were analysed byelectrophoresis in an ABI PRISM 3100 Genetic Analyzer(Applied Biosystems, Foster City, CA, USA) (SupplementaryMaterial, Fig. S1). The areas of the peaks from the tracesobtained from control and test samples were compared toestablish copy number differences.
Structural interpretation of MYH6 mutations
The interpretation of the structural consequences of specificMYH6 mutations was performed as described previously forMYH7 variants (48). Mutation models were made by compari-son of MYH6 protein with the structures of other myosins(Protein Data Bank ID: 2MYS, post-rigor state; 1Q5G,nucleotide-free state; 1B7T, internally uncoupled state).Images were prepared using the PyMOL Molecular GraphicsSystem software v0.99 (DeLano Scientific, Palo Alto, CA,USA).
Construction of plasmids
Full-length mouse Myh6 cDNA was PCR-amplified fromIMAGE clone 14406689 (GenBank Accession numberBC132667.1) and cloned into the pEGFP-C1 vector (Clontech,Mountain View, CA, USA) at SalI site to generatepEGFP-MHY6 WT. The resulting plasmid encodes a fusionprotein GFP-MYH6 WT (GFP fused to the N-terminus ofmyosin heavy chain). Mutant constructs pEGFP-MYH6A230P, pEGFP-MYH6 H252Q, pEGFP-MYH6 V700M andpEGFP-MYH6 H1336D were generated from the wild-typeconstruct by PCR using a site-directed mutagenesis kit(Stratagene, La Jolla, CA, USA). All the clones weresequence-verified. The plasmid DNAs were prepared for celltransfection assays using Endofree Maxi Kit (Qiagen).
4014 Human Molecular Genetics, 2010, Vol. 19, No. 20
at Periodicals Departm
ent , Hallw
ard Library, U
niversity of Nottingham
on Novem
ber 29, 2011http://hm
g.oxfordjournals.org/D
ownloaded from

Cell transfection and imaging
Mouse C2C12 myoblasts were cultured in Dulbecco’s modi-fied Eagle’s medium (DMEM) supplemented with 10% fetalbovine serum (FBS; Invitrogen). For cell transfection assays,1 × 106 cells were transfected with 18.0 mg of plasmid DNAusing a Nucleofector Kit V (Lonza, Basel, Switzerland).Transfected cells were grown on glass cover slips in six-wellplates in DMEM containing 10% FBS for 24 h and then trans-ferred to differentiating media (DMEM containing 1% horseserum) to induce differentiation. The cells were culturedfurther for 7 days in differentiation media. On Day 8, cellson coverslips were fixed in 4% paraformaldehyde for15 min, washed extensively with phosphate-buffered salineand finally mounted on glass slides. Laser scanning confocalmicroscopy was performed on LSM 510 META (Carl Zeiss,Oberkochen, Germany) using a 63x 1.4 oil objective.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
ACKNOWLEDGEMENTS
The authors are grateful to the patients and families participat-ing in the study.
Conflict of Interest statement. None declared.
FUNDING
This work was supported by the British Heart Foundation(Grant RG/07/010). J.T.G.-R. held a scholarship from theMexican Council of Science and Technology (CONACYT).
REFERENCES
1. Hoffman, J.I. and Kaplan, S. (2002) The incidence of congenital heartdisease. J. Am. Coll. Cardiol., 39, 1890–1900.
2. Brun, J., Wright, M., Goodship, J. and Ling, L. (2002) The aetiology ofcongenital heart disease. In Anderson, R.H., Baker, E.J., Macartney, F.J.,Rigby, M.L., Shinebourne, E.A. and Tynan, M. (eds), PaedriaticCardiology. Churchill Livingstone, London, Vol. 1, pp. 141–165.
3. Ferencz, C. (1993) Epidemiology of Congenital Heart Disease: TheBaltimore–Washington Infant Study 1981–1989, Futura Publishings,Mount Kisco, NY.
4. Ferencz, C., Neill, C.A., Boughman, J.A., Rubin, J.D., Brenner, J.I. andPerry, L.W. (1989) Congenital cardiovascular malformations associatedwith chromosome abnormalities: an epidemiologic study. J. Pediatr., 114,79–86.
5. Nora, J.J. (1993) Causes of congenital heart diseases: old and new modes,mechanisms, and models. Am. Heart J., 125, 1409–1419.
6. Ching, Y.H., Ghosh, T.K., Cross, S.J., Packham, E.A., Honeyman, L.,Loughna, S., Robinson, T.E., Dearlove, A.M., Ribas, G., Bonser, A.J.et al. (2005) Mutation in myosin heavy chain 6 causes atrial septal defect.Nat. Genet., 37, 423–428.
7. Carniel, E., Taylor, M.R., Sinagra, G., Di Lenarda, A., Ku, L., Fain, P.R.,Boucek, M.M., Cavanaugh, J., Miocic, S., Slavov, D. et al. (2005)Alpha-myosin heavy chain: a sarcomeric gene associated with dilated andhypertrophic phenotypes of cardiomyopathy. Circulation, 112, 54–59.
8. Hershberger, R.E., Norton, N., Morales, A., Li, D., Siegfried, J.D. andGonzalez-Quintana, J. (2010) Coding sequence rare variants identified inMYBPC3, MYH6, TPM1, TNNC1 and TNNI3 from 312 patients with
familial or idiopathic dilated cardiomyopathy. Circ. Cardiovasc. Genet.,3, 155–161.
9. Niimura, H., Patton, K.K., McKenna, W.J., Soults, J., Maron, B.J.,Seidman, J.G. and Seidman, C.E. (2002) Sarcomere protein genemutations in hypertrophic cardiomyopathy of the elderly. Circulation,105, 446–451.
10. Abu-Daya, A., Sater, A.K., Wells, D.E., Mohun, T.J. and Zimmerman,L.B. (2009) Absence of heartbeat in the Xenopus tropicalis mutationmuzak is caused by a nonsense mutation in cardiac myosin myh6. Dev.Biol., 336, 20–29.
11. Berdougo, E., Coleman, H., Lee, D.H., Stainier, D.Y. and Yelon, D.(2003) Mutation of weak atrium/atrial myosin heavy chain disrupts atrialfunction and influences ventricular morphogenesis in zebrafish.Development, 130, 6121–6129.
12. Rutland, C., Warner, L., Thorpe, A., Alibhai, A., Robinson, T., Shaw, B.,Layfield, R., Brook, J.D. and Loughna, S. (2009) Knockdown of alphamyosin heavy chain disrupts the cytoskeleton and leads to multiple defectsduring chick cardiogenesis. J. Anat., 214, 905–915.
13. Kinose, F., Wang, S.X., Kidambi, U.S., Moncman, C.L. and Winkelmann,D.A. (1996) Glycine 699 is pivotal for the motor activity of skeletalmuscle myosin. J. Cell. Biol., 134, 895–909.
14. Resnicow, D.I., Deacon, J.C., Warrick, H.M., Spudich, J.A. andLeinwand, L.A. Functional diversity among a family of human skeletalmuscle myosin motors. Proc. Natl Acad. Sci. USA, 107, 1053–1058.
15. Wang, Q., Moncman, C.L. and Winkelmann, D.A. (2003) Mutations inthe motor domain modulate myosin activity and myofibril organization.J. Cell Sci., 116, 4227–4238.
16. Pierpont, M.E., Basson, C.T., Benson, D.W. Jr., Gelb, B.D., Giglia, T.M.,Goldmuntz, E., McGee, G., Sable, C.A., Srivastava, D. and Webb, C.L.(2007) Genetic basis for congenital heart defects: current knowledge: ascientific statement from the American Heart Association CongenitalCardiac Defects Committee, Council on Cardiovascular Disease in theYoung: endorsed by the American Academy of Pediatrics. Circulation,115, 3015–3038.
17. Oldfors, A. (2007) Hereditary myosin myopathies. Neuromuscul. Disord.,17, 355–367.
18. Nagy, E. and Maquat, L.E. (1998) A rule for termination-codon positionwithin intron-containing genes: when nonsense affects RNA abundance.Trends Biochem. Sci., 23, 198–199.
19. Holbrook, J.A., Neu-Yilik, G., Hentze, M.W. and Kulozik, A.E. (2004)Nonsense-mediated decay approaches the clinic. Nat. Genet., 36, 801–808.
20. Jensen, L.R., Amende, M., Gurok, U., Moser, B., Gimmel, V., Tzschach,A., Janecke, A.R., Tariverdian, G., Chelly, J., Fryns, J.P. et al. (2005)Mutations in the JARID1C gene, which is involved in transcriptionalregulation and chromatin remodeling, cause X-linked mental retardation.Am. J. Hum. Genet., 76, 227–236.
21. Kerr, T.P., Sewry, C.A., Robb, S.A. and Roberts, R.G. (2001) Longmutant dystrophins and variable phenotypes: evasion ofnonsense-mediated decay? Hum. Genet., 109, 402–407.
22. Bateman, J.F., Freddi, S., Nattrass, G. and Savarirayan, R. (2003)Tissue-specific RNA surveillance? Nonsense-mediated mRNA decaycauses collagen X haploinsufficiency in Schmid metaphysealchondrodysplasia cartilage. Hum. Mol. Genet., 12, 217–225.
23. Resta, N., Susca, F.C., Di Giacomo, M.C., Stella, A., Bukvic, N., Bagnulo,R., Simone, C. and Guanti, G. (2006) A homozygous frameshift mutationin the ESCO2 gene: evidence of intertissue and interindividual variation inNmd efficiency. J. Cell. Physiol., 209, 67–73.
24. Sohn, R.L., Vikstrom, K.L., Strauss, M., Cohen, C., Szent-Gyorgyi, A.G.and Leinwand, L.A. (1997) A 29 residue region of the sarcomeric myosinrod is necessary for filament formation. J. Mol. Biol., 266, 317–330.
25. Kintses, B., Gyimesi, M., Pearson, D.S., Geeves, M.A., Zeng, W.,Bagshaw, C.R. and Malnasi-Csizmadia, A. (2007) Reversible movementof switch 1 loop of myosin determines actin interaction. EMBO J., 26,265–274.
26. Holmes, K.C., Angert, I., Kull, F.J., Jahn, W. and Schroder, R.R. (2003)Electron cryo-microscopy shows how strong binding of myosin to actinreleases nucleotide. Nature, 425, 423–427.
27. Bejsovec, A. and Anderson, P. (1990) Functions of the myosin ATP andactin binding sites are required for C. elegans thick filament assembly.Cell, 60, 133–140.
28. Yang, Y., Gourinath, S., Kovacs, M., Nyitray, L., Reutzel, R., Himmel,D.M., O’Neall-Hennessey, E., Reshetnikova, L., Szent-Gyorgyi, A.G.,
Human Molecular Genetics, 2010, Vol. 19, No. 20 4015
at Periodicals Departm
ent , Hallw
ard Library, U
niversity of Nottingham
on Novem
ber 29, 2011http://hm
g.oxfordjournals.org/D
ownloaded from

Brown, J.H. et al. (2007) Rigor-like structures from muscle myosinsreveal key mechanical elements in the transduction pathways of thisallosteric motor. Structure, 15, 553–564.
29. Coureux, P.D., Wells, A.L., Menetrey, J., Yengo, C.M., Morris, C.A.,Sweeney, H.L. and Houdusse, A. (2003) A structural state of the myosinV motor without bound nucleotide. Nature, 425, 419–423.
30. Cammarato, A., Dambacher, C.M., Knowles, A.F., Kronert, W.A.,Bodmer, R., Ocorr, K. and Bernstein, S.I. (2008) Myosin transducermutations differentially affect motor function, myofibril structure, and theperformance of skeletal and cardiac muscles. Mol. Biol. Cell., 19, 553–562.
31. Himmel, D.M., Gourinath, S., Reshetnikova, L., Shen, Y., Szent-Gyorgyi,A.G. and Cohen, C. (2002) Crystallographic findings on the internallyuncoupled and near-rigor states of myosin: further insights into themechanics of the motor. Proc. Natl Acad. Sci. USA, 99, 12645–12650.
32. Houdusse, A., Kalabokis, V.N., Himmel, D., Szent-Gyorgyi, A.G. andCohen, C. (1999) Atomic structure of scallop myosin subfragment S1complexed with MgADP: a novel conformation of the myosin head. Cell,97, 459–470.
33. Dominguez, R., Freyzon, Y., Trybus, K.M. and Cohen, C. (1998) Crystalstructure of a vertebrate smooth muscle myosin motor domain and itscomplex with the essential light chain: visualization of the pre-powerstroke state. Cell, 94, 559–571.
34. Patterson, B., Ruppel, K.M., Wu, Y. and Spudich, J.A. (1997)Cold-sensitive mutants G680V and G691C of Dictyostelium myosin IIconfer dramatically different biochemical defects. J. Biol. Chem., 272,27612–27617.
35. Koppole, S., Smith, J.C. and Fischer, S. (2007) The structural couplingbetween ATPase activation and recovery stroke in the myosin II motor.Structure, 15, 825–837.
36. Mesentean, S., Koppole, S., Smith, J.C. and Fischer, S. (2007) Theprincipal motions involved in the coupling mechanism of the recoverystroke of the myosin motor. J. Mol. Biol., 367, 591–602.
37. Iwai, S. and Chaen, S. (2007) Mutation in the SH1 helix reduces theactivation energy of the ATP-induced conformational transition ofmyosin. Biochem. Biophys. Res. Commun., 357, 325–329.
38. Sasaki, N., Ohkura, R. and Sutoh, K. (2003) Dictyostelium myosin IImutations that uncouple the converter swing and ATP hydrolysis cycle.Biochemistry, 42, 90–95.
39. Bandman, E., Arrizubieta, M.J., Wick, M., Hattori, A., Tablin, F., Zhang,S. and Zhang, Q. (1997) Functional analysis of the chicken sarcomeric
myosin rod: regulation of dimerization, solubility, and fibrillogenesis. Cell
Struct. Funct., 22, 131–137.
40. Tripet, B., Wagschal, K., Lavigne, P., Mant, C.T. and Hodges, R.S. (2000)Effects of side-chain characteristics on stability and oligomerization stateof a de novo-designed model coiled-coil: 20 amino acid substitutions inposition ‘d’. J. Mol. Biol., 300, 377–402.
41. Matsson, H., Eason, J., Bookwalter, C.S., Klar, J., Gustavsson, P.,Sunnegardh, J., Enell, H., Jonzon, A., Vikkula, M., Gutierrez, I. et al.
(2008) Alpha-cardiac actin mutations produce atrial septal defects. Hum.
Mol. Genet., 17, 256–265.
42. Budde, B.S., Binner, P., Waldmuller, S., Hohne, W., Blankenfeldt, W.,Hassfeld, S., Bromsen, J., Dermintzoglou, A., Wieczorek, M., May, E.et al. (2007) Noncompaction of the ventricular myocardium is associatedwith a de novo mutation in the beta-myosin heavy chain gene. PLoS ONE,2, e1362.
43. Monserrat, L., Hermida-Prieto, M., Fernandez, X., Rodriguez, I., Dumont,C., Cazon, L., Cuesta, M.G., Gonzalez-Juanatey, C., Peteiro, J., Alvarez,N. et al. (2007) Mutation in the alpha-cardiac actin gene associated withapical hypertrophic cardiomyopathy, left ventricular non-compaction, and
septal defects. Eur. Heart J., 28, 1953–1961.
44. Hove, J.R., Koster, R.W., Forouhar, A.S., Acevedo-Bolton, G., Fraser,S.E. and Gharib, M. (2003) Intracardiac fluid forces are anessential epigenetic factor for embryonic cardiogenesis. Nature, 421,
172–177.
45. Bartman, T., Walsh, E.C., Wen, K.K., McKane, M., Ren, J., Alexander, J.,Rubenstein, P.A. and Stainier, D.Y. (2004) Early myocardial function
affects endocardial cushion development in zebrafish. PLoS Biol., 2,E129.
46. Liu, W., Smith, D.I., Rechtzigel, K.J., Thibodeau, S.N. and James, C.D.(1998) Denaturing high performance liquid chromatography (DHPLC)
used in the detection of germline and somatic mutations. Nucleic. Acids
Res., 26, 1396–1400.
47. Armour, J.A., Sismani, C., Patsalis, P.C. and Cross, G. (2000)Measurement of locus copy number by hybridisation with amplifiable
probes. Nucleic. Acids Res., 28, 605–609.
48. Rayment, I., Holden, H.M., Sellers, J.R., Fananapazir, L. and Epstein,N.D. (1995) Structural interpretation of the mutations in the beta-cardiac
myosin that have been implicated in familial hypertrophiccardiomyopathy. Proc. Natl Acad. Sci. USA, 92, 3864–3868.
4016 Human Molecular Genetics, 2010, Vol. 19, No. 20
at Periodicals Departm
ent , Hallw
ard Library, U
niversity of Nottingham
on Novem
ber 29, 2011http://hm
g.oxfordjournals.org/D
ownloaded from





























