Embed Size (px)
Citation preview

MOLECULAR PHYSICS, 1997, VOL. 91, NO. 3, 537± 550
Calculation of the hyper® ne constants of phosphorus-containingradicals
By MINH THO NGUYEN1, STEVEN CREVE1, LEIF A. ERIKSSON2 andLUC G. VANQUICKENBORNE1
1 Department of Chemistry, University of Leuven, Celestijnenlaan 200F, B-3001Leuven, Belgium
2 Department of Physics, Stockholm University, Box 6730, 113 85 Stockholm,Sweden
(Received 18 October 1996, revised version accepted 12 December 1996)
The hyper® ne coupling constants of some small radicals containing phosphorus (PH, PH2,PF2, PCl2, PH+
3 , PH4, H3PF, HPF3) and for which experimental values are available areinvestigated using ab initio MO and DFT methods. Geometries were obtained at theUMP2, UQCISD and B3LYP levels, in combination with the 6-311G(d,p) basis set. Theisotropic hyper® ne coupling constants were calculated using the above methods with a varietyof basis sets, in particular for the B3LYP method. For the smallest radicals (PH, PH2, PH+
3and PH4), UQCISD(T) calculations also were performed using a ® nite ® eld method.Anistropic coupling constants were calculated using the PWP density functionals. The in¯ u-ence of geometry is discussed, pointing out that UMP2 yields geometries closer to theUQCISD than the B3LYP for this type of molecules. It is shown again that DFT, whenused in conjunction with purposely tailored basis sets, is a very economic alternative to highlycorrelated MO methods for computing the hyper® ne properties of phosphorus-containingradicals. Such a basis set, which is being introduced in this work, seems to provide a bettercancellation of errors and is small enough to be used in large molecular computations whilestill giving reliable hyper® ne coupling constants. The improved basis set is then applied to thestudy of three somewhat larger radicals, namely PF4, PCl4 and P2H
+6 .
1. Introduction
In recent years the theoretical computation of hyper-® ne coupling constants (HFCCs) has received muchattention [1, 2], with the aim of understanding theelectronic properties and interpreting electron spinresonance (ESR) spectra of open-shell species. The inter-action between magnetic nuclei and unpaired electronsis represented by the hyper® ne tensor, which can bedivided into an anisotropic and an isotropic part. Theanisotropic (dipolar) part can be derived from theclassical expression for interacting magnetic dipolesand depends mainly upon the global spin distribution.Owing to its global nature, calculated values for aniso-tropic constants are rather less method-dependent andoften in reasonable agreement with experiment. The iso-tropic part, on the other hand, depends on the fermicontact interaction with the nucleus under considera-tion, and therefore is determined by the spin density atthe nucleus position. Available results show that it ismuch more di� cult to compute isotropic HFCCs inquantitative agreement with experiment, as these arehighly sensitive to several factors including geometry,electron correlation and one-electron basis set. In fact,
massively correlated ab initio molecular orbital meth-ods, such as multireference con® guration interaction(MRCI), quadratic con® guration interaction (QCI)or coupled cluster (CC) techniques are imperativelyneeded to describe adequately the hyper® ne structure(HFS) of radicals. Moreover, extremely large basis setsneed to be used to achieve a good description of the spindensity at the nucleus. A new approach has been takenrecently by Rassolov and Chipman [3], who introducednew operators for electronic density calculations. Appli-cation of these new operators to hydrogen, ® rst-rowatoms and ® rst-row diatomic hydrides was found tohave superior performance [4] with respect to the usualdelta function formulation and the Hiller± Sucher±Feinberg operator [5]. Nevertheless, the calculatedresults remain crucially dependent on the wavefunctionquality.
It is evident that correlated ab initio approachesrapidly become intractable when dealing with largemolecules. Hence, not surprisingly, much e� ort hasbeen spent recently on using density functional theory(DFT) to calculate the HFCCs of radicals [1, 6, 7]. As inmany other areas, DFT has two appealing features: it
0026± 8976/97 $12.00 Ñ 1997 Taylor & Francis Ltd.

partly incorporates electron correlation through theexchange± correlation functional and it is applicablereadily to large systems, owing to its favourable scalingas compared with correlated MO methods. The suc-cess of non-local, gradient corrected density functionalshas been proved by several groups [7± 9], as well asthe importance of inclusion of some Hartree± Fockexchange [6]. In particular, the B3LYP functional hasbeen shown to perform well in predicting HFCCs [6, 7].The Perdew± Wang± Perdew combination of functionalsalso yields satisfactory results in this area [1].
In spite of the current interest in calculating HFCCsusing DFT, little has been known on phosphorus-con-taining radicals. Concerning MO methods, a systematicstudy on 25 phosphorus-containing radicals has beenperformed by Cramer et al. [10], suggesting that theUMP2/6-311G(d,p) method based on UHF/6-31G(d,p)geometries, rather than UMP2/6-31G(d,p) geometries,yields acceptable results. The fact that less accurate geo-metries provide better HFCCs arises presumably from alarger cancellation of errors. Nevertheless, the errorsresulting from this approach remain too large to besatisfactory. In the framework of a continuing theore-tical study of low-coordinated phosphorus compounds[11], we report in this work the performance of somerepresentative MO and DFT methods, namely UMP2,UQCISD and B3LYP, in predicting the HFCCs of asample of small phosphorus compounds (PH, PH2,PF2, PCl2, PH+
3 , PH4, H3PF and HPF3) for whichexperimental data are well established. On the smallestsystems (PH, PH2, PH+
3 and PH4), some UQCISD(T)calculations of the spin density will be performed aswell, using a ® nite ® eld method. Particular attention ispaid to the one-electron basis functions. Thus anattempt is made to improve a DFT basis set, which isthen applied to the PF4, PCl4 and P2H
+6 species in order
to test its reliability.
2. Computational details
Geometries of the species considered were optimizedusing the UMP2, UQCISD and B3LYP [12]methods inconjunction with the 6-311G(d,p) basis set. Throughoutthe UMP2 and UQCISD calculations, all core electronswere included in the correlation treatment. Subse-quently, single point calculations were performed atthe obtained geometries, using a variety of basis sets.The isotropic coupling for nucleus N is obtained fromthe following expression:
aiso(N) = (4p /3)ggN b b N k sz l - 1q (N), (1)
where g is the electronic g-factor, here set to the freeelectron value 2.0023, b is the Bohr magneton, and gN
and b N are the analogues for nucleus N. q (N) is the
Fermi contact integral corresponding to the spin densityat nucleus N, and is given by
q (N) = å¹ t
Pa - b¹ n k u ¹ (rkN)|d (rkN)|u t (rkN) l , (2)
where Pa - b¹ u is an element of the one-electron spin den-
sity matrix and {u } designates the atomic basis func-tions.
The anisotropic components of the hyper® ne tensorare given by
T ij(N) =12
ggN b b N k Sz l - 1 å¹ t
Pa - b¹ t
´ k u ¹ (rkN)|r- 5kN(r2
kN d ij - 3rkN,irkN,j)|u t (rkN) l (3)The split valence plus polarization 6-311G(d,p) basis setwas used for both geometry optimizations and HFCCcalculations. Note that the 6-311G notation speci® es the6-311G bases for ® rst-row atoms, but the McLean±Chandler bases for second-row atoms [13], namely a(621111/52111) basis. The 6-311G(d,p) set has beenextended by adding several extra polarization functionsand di� use functions (6-311 + G(2df,p)) in the HFCCcalculations. A further modi® cation of the 6-311 +G(2df,p) basis is provided by either fully decontractingthe s functions (referred to hereafter by the `us’ pre® x) orfully decontracting both s and p functions (`usp’ pre® x).The double zeta (DZ), triple zeta (TZ) and their polar-ized counterpart (DZP and TZP) were considered inDFT calculations to emphasize the e� ects of polariza-tion functions and the description of the core electrons.The TZ basis set consists of a (12s9p)/[7s5p]contraction.The loosely contracted IGLO-III basis set [14]was usedalso in view of its previous success in the calculation ofmagnetic properties [1, 9, 14]. A further decontractedform of the IGLO-III basis set is denoted as IGLO-IIIus, in which the core s orbitals on phosphorus havebeen decontracted in order to yield a more ¯ exibledescription of the core region. A valence triple zetaplus polarization basis, TZVP [15], optimized in theframework of a local spin density approximation(LSDA) was employed also. Both the IGLO-III andTZVP bases were modi® ed further by adding a tight sfunction in the innermost s orbital. They are referred toas IGLO-III + 1s and TZVP+ 1s, respectively. In com-bination with the B3LYP method, ® nally, we employedthe fairly large cc-pVDZ, cc-pVTZ and aug-cc-pVTZbasis sets. Similarly, the s functions of cc-pVTZ havebeen fully decontracted, yielding a basis set denoted asuscc-pVTZ. The concept of adding tight s functionsor decontracting basis sets has been applied in recentstudies [6, 16].
In order to calculate isotropic hyper® ne coupling con-stants using the UQCISD(T) method, the Fermi contact
538 Minh Tho Nguyen et al.

term is treated as a perturbation, associated with ® nite® eld ¹ :
H1N = ¹ q (N). (4)
Hence, the derivative of the energy with respect to the® eld yields the spin density on nucleus N under consid-eration. In practice this derivative is calculated by acentral ® nite di� erence approximation,
¶ E¶ ¹
=(E¹ - E- ¹ )
2¹. (5)
In order to minimize the in¯ uence of the neglect ofhigher order terms, small values of ¹ need to bechosen; in this work a value of 10- 4 was used. Theconvergence criterion to the energy was set to 10- 8.
All geometry optimizations and calculations of iso-tropic HFCCs were performed by the Gaussian 94 [17]suite of electronic structure programs. To calculate ani-sotropic HFCCs using DFT methods, the deMon pro-gram [18] was employed in conjunction with a recentlyreported ESR subroutine [8]. It should be noted that thee� ects of vibrational averaging on the calculatedHFCCs are ignored in this study.
3. Results and discussion
3.1. GeometriesFigure 1 shows the optimized geometrical parameters
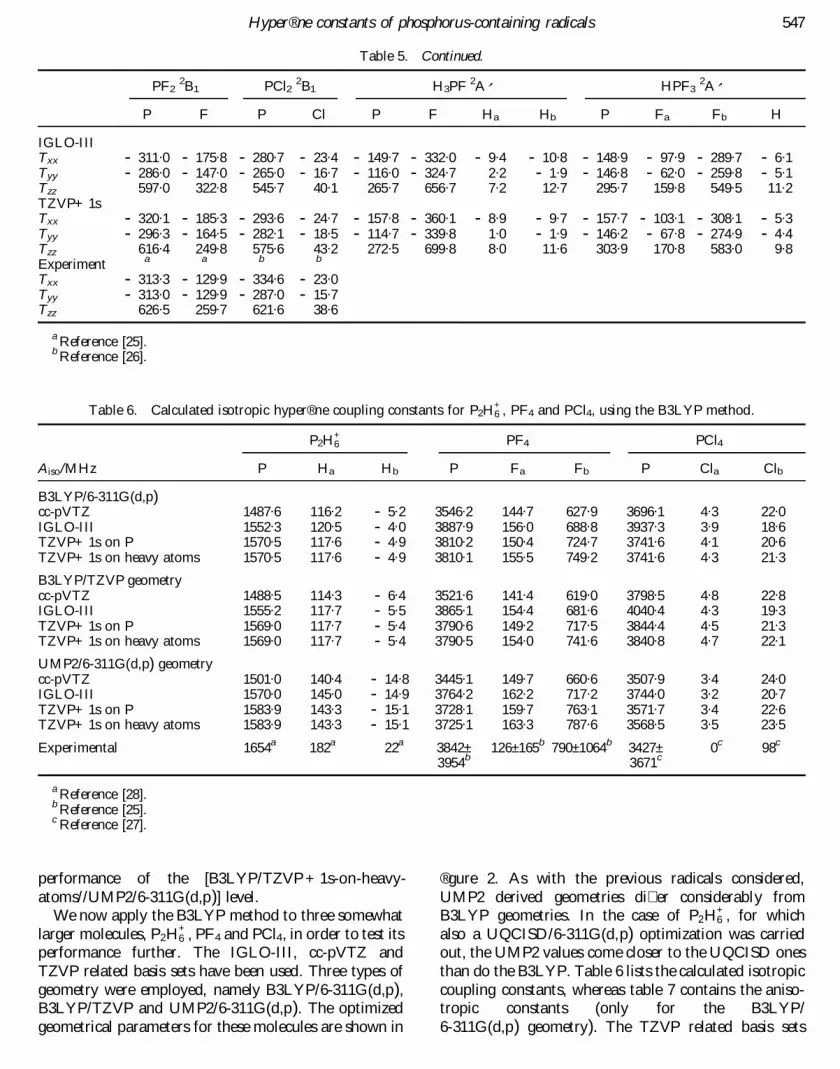
obtained at the di� erent levels of theory, for the test setof six small phosphorus-containing radicals. Due to thelack of experimental geometries, the UQCISD/6-311G(d,p) parameters represent the best availabledata. While UMP2 generally underestimates the bonddistances relative to the UQCISD values, B3LYP over-estimates them. For PCl2, this overestimation evenamounts up to 0.03 AÊ . It is noted that in most casesthe UMP2 values agree considerably better withUQCISD than do the B3LYP. This ® nding is in linewith a recent study of Schaefer et al. [19] pointing outthat for molecules containing second-row elementsB3LYP yields bond lengths signi® cantly too long ascompared with experiment, while UMP2 results com-pare more favourably with experiment. It was shownalso, however, that this problem can be partly curedby using very large basis sets [6-31 + G(3df,2p)] in com-bination with B3LYP [19]. For the bond angles nogeneral trends can be detected, except for the fact thatagain UMP2 agrees better with UQCISD than doesB3LYP. One should bear this deviation in mind, sincethe geometries employed are of considerable importancewhen calculating the hyper® ne structure of molecules.The in¯ uence of geometries on calculated HFCCs willbe discussed in a later section.
3.2. Isotropic hyper® ne coupling constantsTables 1 and 2 contain the isotropic hyper® ne coup-
ling constants computed at the various levels of theory,and the available experimental data. Also listed is theground electronic state of the radicals considered. Foreach method, calculations were carried out using thecorresponding 6-311G(d,p) geometry. Some calculationson molecules containing F or Cl could not be carried outdue to their large computational cost. Table 3 shows themean absolute deviations of the computed values withrespect to the experimental ones, which is a measure ofthe accuracy of the method considered. In what follows,we will discuss the performance of each method, ® rstguided by the type of basis sets used, then followed bythe in¯ uence of the geometry in calculating HFCCs.Hereafter we will use also the concepts of direct andindirect contributions to the spin density. The spin den-sity at a given centre can be split into two contributions.The ® rst part is proportional to the spin density of thesingly occupied molecular orbital (SOMO) at the centreunder consideration, and it will be called the `directcontribution’ . The di� erence between the direct contri-bution and the total spin density at the given centre isthen summarized as the indirect contribution’ . Hence,indirect contributions originate from spin polarizationof the other electrons, caused by the unpaired electron.
3.2.1. Split valence basis setsConcerning the UMP2 computed HFCCs, it is clear
that in general they do not compare well with experi-mental values. It has been shown, however, that some-what better results could be obtained when using UHFgeometries instead of UMP2 geometries [10], presum-ably due to a cancellation of opposite e� ects. As seenin table 1, di� use functions induce only small changes,while additional d-type polarization functions exert amuch larger e� ect. The e� ect of enlarging the polariza-tion space, however, is not consistent: in going from(d,p) to (2d,p) it is noted that for the PH radical theadditional d function enlarges the HFCC drastically,while for PF it induces the opposite behaviour. Evenaddition of a third d function modi® es the P HFCCsconsiderably. An extra f function lowers the phosphorusHFCCs in most of the cases. It is remarkable that theUMP2 method, combined with this type of basis set,underestimates the P HFCCs of those radicals whereonly indirect contributions (i.e., pure spin polarizatione� ects) are responsible for the spin density at the nucleus(PH, PH2, PF2 and PCl2). For the radicals where directcontributions also are important (PH+
3 , PH4, H3PF andHPF3), a certain overestimation of the P HFCCs is seen.Note that the calculated hydrogen HFCCs agree reason-ably well with experiment, irrespective of the bases.
Hyper® ne constants of phosphorus-containing radicals 539
While the split valence basis sets perform poorly inconjunction with the UMP2 method, they yield a moreconsistent set of results when combined with the moreadvanced UQCISD treatment of electron correlation.Table 3 shows that a sequential addition of d polariza-tion functions improves the HFCCs in a more consistentway. Addition of a di� use function lowers the PHFCCs, whereas in¯ uence of an f function dependsrather on the radicals: where only indirect contributionsare active, it increases the P HFCCs, whereas in othercases (direct contributions as well) it lowers them. TheUQCISD hydrogen HFCCs also are improved as com-pared with the UMP2 values. It turns out that both us6-311 + G(2df,p) and usp6-311 + G(2df,p) bases performvery poorly with UQCISD, except for PH4, by mark-edly overestimating the P HFCCs. Because the uncon-
tracted basis tends to approach the Hartree± Fock limit,such a poor performance is somewhat intriguing,pointing towards either an inherent insu� ciency of theUQCISD treatment or an imbalance in the decontractedbasis. In this context, better performance using con-tracted bases can be seen as resulting from another can-cellation of errors. One of the errors involved herepresumably is due to the fact that, in split valencebases, the core electrons are described by only one con-tracted orbital, while valence electrons are described bymore than one orbital. A certain imbalance in the core±valence contribution could occur and induce an errorwhich, in the UQCISD case, turns out to be a bene® cialcompensation.
Due to limited computing resources, we have beenable to perform UQCISD(T) calculations only on the
540 Minh Tho Nguyen et al.
Table 1. Calculated isotropic hyper® ne couplings using UMP2 and UQCISD methods.
PH 3 S - PH22B1 PH+
32A1 PH4
2A1
Aiso /MHz P H P H P H P Ha Hb
UMP2a
6-311G(d,p) 46.8 - 55.6 160.1 - 57.0 1263.3 - 8.1 1430.0 - 25.5 500.26-311G(2d,p) 64.6 - 59.6 168.0 - 59.4 1265.7 - 6.5 1384.7 - 23.7 498.26-311+ G(2d,p) 61.4 - 59.8 161.2 - 59.6 1258.6 - 6.9 1388.0 - 23.8 497.06-311+ G(3d,p) 45.5 - 59.7 150.7 - 58.8 1248.3 - 5.6 1400.0 - 23.8 500.76-311+ G(2df,p) 65.4 - 57.9 161.7 - 56.3 1232.9 - 4.3 1357.2 - 21.9 497.9
UQCISDb
6-311G(d,p) 66.8 - 50.5 170.7 - 54.1 1245.8 - 3.8 1477.8 - 28.3 502.16-311G(2d,p) 120.2 - 50.3 206.6 - 53.2 1246.5 - 1.0 1432.0 - 25.5 498.46-311+ G(2d,p) 116.7 - 50.3 200.2 - 53.3 1239.7 - 1.3 1434.0 - 25.5 497.36-311+ G(3d,p) 109.4 - 49.4 198.0 - 52.1 1233.0 - 0.2 1446.0 - 25.2 499.96-311+ G(2df,p) 127.1 - 44.9 209.9 - 48.2 1216.7 0.9 1399.9 - 24.5 497.7us6-311+ G(2df,p) on P 188.8 - 45.2 267.9 - 48.4 1240.4 0.1 1355.3 - 24.5 496.3usp6-311+ G(2df,p) on P 194.5 - 45.2 273.6 - 48.4 1246.9 0.1 1356.9 - 24.4 496.6IGLO-III 134.5 - 48.4 222.9 - 50.7 1260.6 3.0 1541.3 - 27.7 506.6IGLO-IIIus on P 124.8 - 48.4 212.1 - 50.7 1230.0 3.0 1512.5 - 27.7 506.7IGLO-III+ 1s on P 137.8 - 48.4 228.2 - 50.7 1290.0 3.0 1577.2 - 27.7 506.6TZVP 166.1 - 49.1 235.6 - 52.9 1219.0 - 2.8 1296.6 - 30.0 530.6TZVP+ 1s on P 171.4 - 49.1 243.3 - 52.9 1259.5 - 2.8 1339.9 - 30.0 530.7
UQCISD(T)b
6-311+ G(2df,p) 131.8 205.9 1198.7 1394.7us6-311+ G(2df,p) on P 165.2 236.6 1203.7 1362.5usp6-311+ G(2df,p) on p 170.2 241.5 1209.5 1365.0IGLO-III 137.4 218.3 1242.5 1537.3IGLO-IIIus on P 127.6 207.5 1212.3 1508.6
k S2 l c 2.027 0.769 0.757 0.778
Experimental 125.4d - 48.9d 217e - 49e 1176 f 1459.2g - 16.9g 558.3g
a Using the UMP2/6-311G(d,p) geometry.b Using the UQCISD/6-311G(d,p) geometry.c Expectation values of the spin operator from UHF-references, using the 6-311+ G(2df,p) basis.d Reference [29].e Reference [23].f Reference [28].g Reference [24].

smallest radicals (PH, PH2, PH+3 and PH4). Using 6-
311 + G(2df,p) and its uncontracted counterparts, us6-311 + G(2df,p) and usp6-311 + G(2df,p), the computedUQCISD(T) values for 31P HFCCs follow the samegeneral trend as the corresponding UQCISD values(table 1). It should be noted, however, that inclusionof triple excitations reduces the computed HFCCsnoticeably in all cases. Although this results in betteragreement with experiment, the absolute error remainsappreciable, in particular for the PH4 radical,amounting to 100 MHz.
A di� erent situation emerges for B3LYP. Withoutpolarization functions (DZ and TZ), the behaviour ofcalculated values is irregular; in some cases, they areeven qualitatively incorrect (table 2). Incorporating ofpolarization functions in DZP and TZP improves theHFCCs somewhat. The results employing split-valencebasis sets are unacceptable. It is seen, however, that theobtained set of HFCCs shows less ¯ uctuations than theUMP2 values. Thus, for the property under considera-tion, DFT methods also are less basis-set dependentthan MO methods. Decontraction of the s part of 6-
311 + G(2df,p) results in a considerable enhancementof the B3LYP values. A further decontraction of the ppart has, in contrast, no in¯ uence. Similar to the UMP2method, there is a systematic underestimation of the PHFCCs in PH, PH2, PF2 and PCl2 which have onlyindirect contributions to the spin density at the Pnucleus (table 2).
3.2.2. IGL O basis setsOnly UQCISD, UQCISD(T) and B3LYP calculations
have been performed using the IGLO basis sets. Themain di� erence between split valence and IGLO-typebasis sets consists in the contraction scheme: theIGLO bases are loosely contracted, thus allowing formore ¯ exibility in the core and inner-valence regions.Three IGLO bases are used: the standard IGLO-IIIbasis which corresponds to a (3111/11) basis on H, a(5111111/211111/11) basis on ® rst-row atoms, and a(51111111/2111111/111) basis on the second-rowelements. The IGLO-IIIus corresponds to the standardbasis, but with the outermost s function of the coredecontracted on P. In the IGLO-III + 1s basis, 1s
Hyper® ne constants of phosphorus-containing radicals 541
Table 1. Continued.
PF22B1 PCl2 2B1 H3PF2A Â HPF3
2A ÂAiso/MHz P F P Cl P F Ha Hb P Fa Fb H
UMP2a
6-311G(d,p) 219.6 106.7 95.4 2.8 2079.4 865.9 309.6 - 41.9 2939.6 97.8 574.0 67.16-311G(2d,p) 153.7 81.8 83.5 1.4 2074.5 865.4 300.5 - 40.8 2982.0 96.5 568.9 74.86-311+ G(2d,p) 120.6 65.7 79.9 1.3 2060.0 866.0 301.9 - 40.9 2962.8 100.0 564.9 78.46-311+ G(3d,p) 96.0 63.7 56.7 1.2 2070.3 866.3 301.7 - 40.3 2980.3 101.4 566.4 81.46-311+ G(2df,p) 115.7 56.4 69.6 1.2 2065.5 855.7 303.7 - 39.8 2936.2 97.3 560.9 77.9
UQCISDb
6-311G(d,p) 281.1 115.7 165.7 3.0 2098.5 897.9 322.4 - 42.9 2928.9 88.2 603.9 62.66-311G(2d,p) 260.1 104.4 209.4 2.1 2091.5 895.6 314.7 - 40.5 2968.7 88.0 600.0 71.36-311+ G(2d,p) 232.3 91.5 201.7 1.9 2075.0 900.6 316.3 - 40.66-311+ G(3d,p) 224.5 93.4 2086.0 899.2 315.0 - 39.86-311+ G(2df,p) 250.1 91.8 2043.4 894.0 317.7 - 40.0us6-311+ G(2df,p) on Pusp6-311+ G(2df,p) on PIGLO-III 261.4 97.0 2143.4 942.3 324.1 - 39.5IGLO-IIIus on P 249.6 97.0 2099.3 942.3 324.2 - 39.5IGLO-III+ 1s on P 267.6 97.0 2192.2 942.3 341.1 - 39.5TZVP 341.5 127.8 1955.2 1033.4 341.8 - 44.5TZVP+ 1s on P 352.8 127.8 2020.4 1033.4 341.8 - 44.5
k S2 l c 0.782 0.775d 0.765 0.757
Experimental 237.7e 91.3e 191.4 f - 1.12 f 2021.4g 973.0g 364.6g - 35.3g 2888.8g 98g 635.6g 107.9g
a Using the UMP2/6-311G(d,p) geometry.b Using the UQCISD/6-311G(d,p) geometry.c Expectation values of the spin operator from UHF-references, using the 6-311+ G(2df,p) basis.d Using 6-311+ G(2d,p).e Reference [25].f Reference [26].g Reference [30].

stands for an additional tight core s function on P,having the exponent a = 699 400, in order to provide abetter description of the 1s cusp. The latter exponent hasbeen optimized with respect to the electronic energy.
As seen from table 3, in general the IGLO basis setsdo not behave similarly to the split valence basis sets,when utilized in combination with UQCISD. However,decontraction of the core s orbital on P yields a signi® -cant improvement. Addition of a tight s function in theP core orbital enlarges in all cases the correspondingHFCC, yielding a mean absolute deviation of 36 MHz(table 3), due to a serious overestimation of the PHFCCs in PH+
3 and PH4. The UQCISD approachhence seems to bene® t more from using a ¯ exible basisset (IGLO-IIIus) than from ameliorating the descriptionof the core orbitals.
Concerning the IGLO-III and IGLO-IIIus bases, thetrend in UQCISD(T) results again is very similar to thatof their UQCISD counterparts, but the former aresmaller in absolute values. With the B3LYP method,IGLO basis sets perform considerably better than the
split valence bases. Decontracting the 1s shell results ina loss of spin density at the P nucleus, and a seriousunderestimation of the P HFCCs. Adding a tight s func-tion into the core has the same e� ect as with theUQCISD method, i.e., it enlarges the electron densityat the nucleus and hence increases the P HFCCs. Itshould be noted that the standard IGLO-III basis per-forms similarly to the uncontracted split valence basesus6-311 + G(2df,p) and usp6-311 + G(2df,p). The equalperformance of the IGLO-III and uncontracted splitvalence bases in computing HFCCs was noted also inan earlier work on radicals consisting of ® rst-row ele-ments only [16]. On the other hand, its deviation fromexperiment is much smaller when using the UQCISDmethod instead of the B3LYP functional.
3.2.3. cc-pV XZ basis setsCorrelation consistent basis sets were employed only
in B3LYP calculations. While a valence double-zetaquality (cc-pVDZ) yields no improvement over theother basis sets considered so far, a valence triple-zeta
542 Minh Tho Nguyen et al.
Table 2. Calculated isotropic hyper® ne couplings using B3LYP.
PH 3S - PH22B1 PH+
32A1 PH4
2A1
Aiso/MNz P H P H P H P Ha Hb
B3LYPa
6-311G(d,p) 31.4 - 30.9 115.7 - 36.8 1082.4 14.0 1513.2 - 18.5 533.26-311G(2d,p) 40.1 - 30.5 127.8 - 36.5 1092.4 15.1 1492.3 - 18.9 526.86-311+ G(2d,p) 40.7 - 30.1 126.5 - 36.1 1088.3 15.4 1498.8 - 19.2 524.06-311+ G(3d,p) 45.3 - 29.8 134.6 - 35.6 1088.4 15.8 1497.3 - 19.7 525.16-311+ G(2df,p) 40.7 - 29.6 128.6 - 34.8 1084.2 15.9 1493.1 - 19.5 523.4us6-311+ G(2df,p) on P 77.8 - 29.1 163.7 - 34.1 1095.0 16.6 1460.7 - 19.1 524.3usap6-311+ G(2df,p) on P 78.9 - 29.0 164.9 - 34.1 1096.3 16.8 1460.4 - 18.8 524.8DZb - 19.7 - 44.8 40.3 - 50.6 1075.9 - 3.7 1532.0 - 11.8 588.6DZ(d,p)c 2.8 - 35.0 89.5 - 41.2 1037.3 12.4 1423.4 - 19.4 544.2TZ 13.8 - 39.8 81.2 - 45.3 1132.8 0.7 1658.3 - 12.0 559.6TZ(d,p)c 49.4 - 31.2 136.8 - 37.5 1101.0 13.3 1535.1 - 19.6 532.2IGLO-III 72.4 - 29.4 160.9 - 35.0 1126.7 18.3 1571.4 - 21.7 542.4IGLO-IIIus on P 58.2 - 29.5 145.6 - 35.2 1089.4 18.6 1545.2 - 22.0 541.7IGLO-III+ 1s on P 76.7 - 29.1 166.5 - 34.9 1152.6 17.9 1603.7 - 21.3 544.0cc-pVDZ 155.9 - 33.1 215.2 - 38.9 1094.9 10.7 1370.8 - 16.6 484.9cc-pVTZ 135.3 - 28.8 215.6 - 33.8 1103.7 16.2 1421.6 - 19.8 530.6ayg-cc-pVTZ 130.8 - 29.1 210.6 - 33.9 1086.8 15.0 1402.2 - 18.1 528.8uscc-pVTZ on P 50.8 - 28.8 143.6 - 33.9 1080.7 16.2 1499.1 - 19.2 531.6TZVP 152.2 - 29.4 214.9 - 35.3 1136.1 15.6 1400.4 - 20.0 571.8TZVP+ 1s on P 157.1 - 29.4 221.9 - 35.3 1173.2 15.5 1440.4 - 19.7 572.4TZVP+ 1s on heavy atoms 157.1 - 29.4 221.9 - 35.3 1173.2 15.5 1440.4 - 19.7 572.4
Experimental 125.4d - 48.9d 217e - 49e 1176 f 1459.2g - 16.9g 558.3g
a Using the B3LYP/6-311G(d,p) geometry.b Dunning’s D95 basis set.c Polarization functions from 6-311G(d,p).d Reference [29].e Reference [23].f Reference [28].
basis (cc-pVTZ) leads to HFCCs in better agreementwith experiment. Including di� use functions (aug-cc-pVTZ) results in all cases in a slight decrease in the PHFCCs as compared with cc-pVTZ. The uncontracteduscc-pVTZ basis underestimates the P HFCCs drastic-ally for those radicals where only indirect contributionsdetermine the spin density at the P nucleus. For theother radicals, which exhibit both direct and indirectcontributions, a reverse trend happens (except forPH+
3 ), i.e., the PHFCCs are considerably overestimated.Thus it is seen that this basis set is insu� ciently balancedfor use in conjunction with B3LYP HFCC calculations:while B3LYP underestimates valence polarization forsecond-row elements, uscc-pVTZ allows for too muchcore polarization (which has the opposite sign) henceresulting in a serious underestimation of the HFCCsfor those radicals where only indirect contributions tothe spin density at the nucleus under consideration arepresent. Overall, the contracted triple-zeta correlationconsistent basis sets perform slightly better for thepresent systems than the IGLO basis sets, probablydue to their better description of the core orbitals.
3.2.4. TZV P basis setsThe TZVP basis set [15], optimized within the frame-
work of the local spin density approach (LSDA), con-sists of a (311/1) basis for H, a (7111/411/1) basis for® rst row elements, and a (73111/6111/1) basis forsecond-row elements. It turns out that the B3LYP/TZVP method consistently yields results closer toexperiment than any of the previous bases. We havefound that further improvements can be achievedwhen adding a tight s function to the core orbital. Inthe basis set denoted as TZVP+ 1s, an s function withan exponent a = 435 485 has been optimized for P. Themean absolute deviation from experiment now amountsto 26 MHz. Note that the TZVP+ 1s basis does notshow any further improvement on other atoms, as com-pared with TZVP. With an extra tight s function on allheavy atoms (denoted TZVP+ 1s-on-heavy-atoms) afurther improvement to only 24 MHz as MAD occurs.Thus, it can be seen that purposely tailoring basis setscan improve B3LYP calculations of hyper® ne structuresconsiderably. However, one must be aware of the factthat this approach inherently includes some cancellation
Hyper® ne constants of phosphorus-containing radicals 543
Table 2. Continued.
PF22B1 PCl2 2B1 H3PF 2A Â HPF3
2A ÂAiso/MHz P F P Cl P F Ha Hb P Fa Fb H
B3LYPa
6-311G(d,p) 171.0 85.1 73.8 1.9 2151.9 761.8 338.1 - 31.1 2895.8 74.4 471.2 91.56-311G(2d,p) 150.1 80.0 97.9 1.4 2140.1 777.4 332.0 - 30.8 2935.1 75.9 484.9 93.46-311+ G(2d,p) 136/8 72.5 94.4 1.3 2134.5 791.9 335.4 - 30.9 2932.7 82.7 494.0 97.76-311+ G(3d,p) 145.5 73.2 97.6 1.4 2130.5 789.5 334.8 - 30.9 2937.9 83.9 495.4 99.46-311+ G(2df,p) 144.1 70.6 101.0 1.4 2129.0 797.3 335.0 - 31.3 2923.7 81.7 501.0 97.2ys6-311+ G(2df,p) on P 175.3 70.9 134.5 1.5 2089.6 794.6 335.4 - 30.5 2867.2 81.4 500.1 96.4usp6-311+ G(2df,p) on P 176.5 70.8 136.1 1.5 2091.3 794.9 335.3 - 31.4 2870.4 81.3 500.3 96.2DZb 122.3 133.1 6.2 2.6 2182.0 820.2 393.4 - 40.8 2853.3 79.2 472.5 64.6DZ(d,p)c 130.4 89.7 58.5 1.7 2077.9 819.0 346.6 - 32.6 2772.7 85.7 493.3 132.6TZ 146.3 126.4 38.5 3.3 2269.2 915.4 375.2 - 37.4 2939.0 84.6 516.5 64.3TZZ(d,p)c 172.8 99.6 98.5 2.3 2176.0 884.6 345.7 - 32.2 2936.6 87.5 559.6 95.1IGLO-III 178.0 83.4 134.8 1.9 2191.9 855.9 349.2 - 31.0 2964.6 87.3 536.3 102.3IGLO-IIIus on P 162.0 83.2 119.2 1.9 2144.8 850.5 349.2 - 31.0 2904.4 89.8 539.5 102.7IGLO-III+ 1s on P 178.9 83.1 136.6 2.0 2243.0 850.5 349.2 - 31.0 3036.4 90.0 539.7 102.7cc-pVDZ 264.2 129.3 202.5 3.3 1946.9 817.7 313.9 - 28.7 2585.5 90.2 547.8 86.3cc-pVTZ 244.9 60.4 198.9 3.2 2020.3 767.2 344.8 - 32.3 2737.8 74.7 485.3 91.0ayg-cc-pVTZ 231.1 48.5 193.1 3.2 1996.1 754.8 341.7 - 31.5 2733.1 74.3 467.9 93.1uscc-pVTZ on P 163.3 60.7 114.8 3.2 2119.3 765.7 344.3 - 32.1 2899.7 74.6 484.7 91.6TZVP 272.9 100.7 168.3 3.0 2095.6 907.3 367.7 - 34.0 2844.8 89.4 573.0 93.8TZVP+ 1s on P 281.9 100.7 173.8 3.0 2161.4 907.4 368.8 - 34.2 2937.8 89.5 573.5 92.9TZVP+ 1s on heavy atoms 281.9 103.7 173.8 3.1 2162.8 940.4 369.0 - 34.2 2932.1 92.5 591.8 92.5
Experimental 237.7d 91.3d 191.4e - 1.12e 2021.4 f 973.0 f 364.6f - 35.3 f 2888.8 f 98 f 635.6 f 107.9 f
a Using the B3LYP/6-311G(d,p) geometry.b Dunning’s D95 basis set.c Polarization functions from 6-311G(d,p).d Reference [25].e Reference [26].f Reference [30].

of errors, for it is known that B3LYP underestimates thevalence polarization of second-row elements.
In the case of UQCISD, however, this basis set per-forms quite poorly. UQCISD/TZVP+ 1s generallyovershoots the P HFCCs (except for PH4). This erro-neous behaviour indicates that the TZVP basis setindeed allows for too little core polarization, whichwould lower the P HFCCs. It should be stressed againthat the TZVP basis was not developed for ab initio MOcalculations, and thus the contraction scheme might notbe appropriate at all.
As a ® nal remark on basis set performance, we men-tion the recent work of Cohen and Chong [20], in whichvarious functionals and basis sets were tested. Theseauthors have studied eleven radicals containing only® rst-row elements. Using the B3LYP/TZVP model incombination with UMP2/D2 derived geometries(where D2 consists of the following contractionscheme: (52111111/311111/111) on C± F and (511111/111) on H), they obtained a mean absolute deviationfrom experimental values of 22.6 MHz. The lattervalue is to be compared with our MAD of 27 MHz forB3LYP/TZVP//UMP2/6-311G(d,p).
3.3. In¯ uence of geometry on calculated isotropichyper® ne coupling constants
It is known that the isotropic hyper® ne coupling con-stant is extremely sensitive to a number of factors, notonly the wavefunction quality but also geometry. Thelatter will be discussed in this section. Considerable dif-ferences usually exist between geometrical parametersobtained by the various levels of theory (® gure 1); there-fore it is sensible to investigate the in¯ uence of geometryon the HFCCs. B3LYP calculations using the IGLO-IIIand di� erent TZVP bases have been carried out, basedon three di� erent geometries, namely B3LYP/6-311G(d,p), UMP2/6-311G(d,p) and UQCISD/6-311G(d,p). Tables 4 and 3 show the calculated isotropicHFCCs and the mean absolute deviations (MAD) fromexperiment for these methods, respectively. The B3LYP/6-311G(d,p) derived geometries are not su� cient to pro-vide accurate HFCCs. It turns out that [B3LYP/TZVP+ 1s - on - heavy - atoms//UMP2/6 - 311G(d,p)]method yields the most accurate results, having a MADof only 16 MHz. The systematic improvement of theresults on going from TZVP to TZPV+ 1s andTZVP+ 1s on heavy atoms is obvious from table 3. It
544 Minh Tho Nguyen et al.
Table 3. Mean absolute deviations from experimental values (MAD) for calculated isotropic hyper® ne coupling constants.
Method/basis setMADMHz
Method/basis setMADMHz
UMP2/ . . . //UMP2/6-311G(d,p) B3LYP/ . . . //B3LYP/6-3111G(d,p)6-311G(d,p) 42 6-311G(d,p) 596-311G(2d,p) 49 6-311G(2d,p) 576-311+ G(2d,p) 50 6-311+ G(2d,p) 576-311+ G(3d,p) 54 6-311+ G(3d,p) 556-311+ G(2df,p) 49 6-311+ G(2df,p) 55
us6-311+ G(2df,p) on P 43UQCISD/ . . . //UQCISD/6-311G(d,p) usp6-311+ G(2df,p) on P 436-311G(d,p) 35 DZ 746-311G(2d,p) 30 DZ(d,p) 626-311+ G(2d,p) 24 TZ 676-311+ G(3d,p) 27 TZ(d,p) 486-311+ G(2df,p) 24 IGLO-III 48us6-311+ G(2df,p) on P 44 IGLO-IIIus on P 46usp6-311+ G(2df,p) on P 46 IGLO-III+ 1s on P 53IGLO-III 34 cc-pVDZ 54IGLO-IIIus on P 25 cc-pVTZ 40IGLO-III+ 1s on P 43 aug-cc-pVTZ 45TZVP 43 uscc-pVTZ on P 53TZVP+ 1s on P 40 TZVP 26
TZVP+ 1s on P 26TZVP+ 1s on heavy atoms 24
B3LYP/ . . . //UMP2/6-311G(d,p) B3YP/ . . . //UQCISD/6-311G(d,p)IGLO-III 34 IGLO-III 36TZVP 27 TZVP 28TZVP+ 1s on P 19 TZVP+ 1s on P 21TZVP+ 1s on heavy atoms 16 TZVP+ 1s on heavy atoms 18
is not yet clear why using a higher level of theory for thegeometry [UQCISD/6-311G(d,p)] results in slightlyworse values for the HFCCs (which has also beennoted in an earlier study [16]). This could be dueeither to another cancellation of errors, or to the factthat higher levels do not necessarily provide better geo-metries! However, the di� erences between the resultsbased on UMP2 and UQCISD geometries are relativelysmall, and hence might disappear if one would considera larger set of molecules.
3.4. Anisotropic hyper® ne coupling constantsWe turn now to the anisotropic constants whose
results are presented in table 5, along with availableexperimental data. For this property, we have employedonly the deMon program and a functional availablethere which has been shown to yield the most accurateHFCCs, namely the PWP86 functional [21, 22]. Threedi� erent basis sets have been chosen, including IGLO-III, TZVP and TZVP+ 1s. The results of the two latterbasis sets are nearly identical and therefore only theTZVP+ 1s results are shown in table 5. Calculationswere carried out using the B3LYP/6-3111G(d,p) opti-mized geometries. The agreement with experiment isreasonable overall, and there are no unacceptable di� er-ences between the various basis sets mentioned above.This is in agreement with the notion that anisotropichyper® ne couplings are relatively less sensitive to one-electron basis set changes. As noted earlier [1, 8, 9], theyshould not be sensitive to the functional form either. As
Hyper® ne constants of phosphorus-containing radicals 545
Figure 1. Optimized geometrical parameters of the eightradicals considered at various levels of theory. Bondlengths are given in angstroÈ ms and bond angles in degrees.
Table 4. B3LYP calculated isotropic hyper® ne coupling constants, using various basis sets and geometries.
PH PH2 PH+3 PH4
Aiso/MHz P H P H P H P Ha Hb
B3LYP/6-311G(d,p) geometryIGLO-III 72.4 - 29.4 160.9 - 35.0 1126.7 18.3 1571.4 - 21.7 542.4TZVP 152.2 - 29.4 214.9 - 35.3 1136.1 15.6 1400.4 - 20.0 571.8YZVP+ 1s on P 157.1 - 29.4 221.9 - 35.3 1173.2 15.5 1440.4 - 19.7 572.4572.4TZVP+ 1s on heavy atoms 157.1 - 29.4 221.9 - 35.3 1173.2 15.5 1440.4 - 19.7 572.4
UMP2/6-311G(d,p) geometryIGLO-III 76.0 - 28.7 164.3 - 34.4 1120.3 18.3 1515.1 - 19.5 519.5TZVP 153.1 - 28.9 216.5 - 34.8 1133.5 14.9 1329.6 - 16.6 550.2TZVP+ 1s on P 158.0 - 28.9 223.5 - 34.8 1171.3 14.9 1374.2 - 16.6 550.2TZVP+ 1s on heavy atoms 158.0 - 28.9 223.5 - 34.8 1171.3 14.9 1374.2 - 16.6 550.2
UQCISD/6-311G(d,p) geometryIGLO-III 72.3 - 29.3 158.0 - 34.9 1123.7 17.2 1526.4 - 20.0 526.6TZVP 152.7 - 29.1 215.9 - 35.0 1132.4 14.9 1348.6 - 17.5 555.6TZVP+ 1s on P 158.0 - 28.9 223.5 - 34.8 1171.3 14.9 1374.2 - 16.6 550.2550.2TZVP+ 1s on heavy atoms 158.0 - 28.9 223.5 - 34.8 1171.3 14.9 1374.2 - 16.6 550.2
Experimental 125.4a - 48.9a 217b - 49b 1176c 1459.2d - 16.9d 558.3d
a Reference [29]. b Reference [23]. c Reference [28]. d Reference [24].
a consequence, it will be rather di� cult to improve thecalculated values for this property.
3.5. Spin contaminationThe quality of an unrestricted wavefunction may be
measured by the expectation value of the S2 operator. Intable 1 we have included the values of k S2 l for the unre-stricted references calculated using 6-311 + G(2df,p) andTZVP+ 1s on P, respectively, in MO calculations. For
the set of molecules under consideration, the spin con-tamination of UHF-MO references is quite low. It istherefore plausible to assume that it induces only amarginal error on the HFCCs.
3.6. Calculations on P2H+6 ,PF4 and PCl4
In summary, the calculated results obtained for thetest set of eight molecules for which experimental dataare well established allow us to emphasize the best
546 Minh Tho Nguyen et al.
Table 4. Continued.
PF2 PCl2 H3PF HPF3
Aiso/MHz P F P Cl P F Ha Hb P Fa Fb H
B3LYP/6-311G(d,p) geometryIGLO-III 178.0 83.4 134.8 1.9 2191.9 855.9 349.2 - 31.0 2964.6 87.3 536.3 102.3TZVP 272.9 100.7 168.3 3.0 2095.6 907.3 367.7 - 34.0 2844.8 89.4 573.0 93.8TZVP+ 1s on P 281.9 100.7 173.8 3.0 2161.4 907.4 368.4 - 34.2 2937.8 89.5 573.5 92.9TZVP+ 1s on heavy atoms 281.9 103.7 173.8 3.1 2162.8 940.4 369.0 - 34.2 2932.1 92.5 591.8 92.5
UMP2/6-311G(d,p) geometryIGLO-III 175.5 84.1 141.5 2.0 2072.9 880.5 335.8 - 31.1 2896.8 97.3 564.6 94.4TZVP 272.7 101.7 176.1 3.1 1981.0 941.7 356.0 - 34.5 2782.8 96.5 600.4 87.4TZVP+ 1s on P 281.6 101.6 181.8 3.1 2048.1 942.1 356.2 - 35.0 2878.0 96.4 600.9 85.7TZVP+ 1s on heavy atoms 281.6 104.7 181.8 3.2 2047.3 973.9 356.0 - 34.5 2876.0 99.7 620.6 87.3
UQCISD/6-311G(d,p) geometryIGLO-III 177.0 84.4 139.3 2.0 2086.1 864.1 341.7 - 32.1 2884.0 94.2 558.7 96.3TZVP 271.7 101.4 173.2 3.0 1992.3 921.5 361.2 - 34.5 2770.4 93.4 594.2 89.3TZVP+ 1s on P 281.6 101.6 181.8 3.1 2059.8 921.9 361.3 - 35.0 2865.2 93.3 594.7 87.6TZVP+ 1s on heavy atoms 280.7 104.4 178.8 3.1 2059.0 953.0 361.2 - 34.5 2865.5 96.3 614.8 87.4
Experimental 237.7a 91.3a 191.4b - 1.12b 2021.4c 973.0c 364.6c - 35.3c 2888.8c 98c 635.6c 107.9c
a Reference [25].b Reference [26].c Reference [30].
Table 5. Anisotropic hyper® ne coupling constants (in MHz), calculated using the PWP86 functional and B3LYP/6-311G(d,p)geometries.
PH 3S g PH22B1 PH+
32A1 PH4
2A1
Aiso/MHz P H P H P H P Ha Hb
IGLO-IIITxx 146.2 - 7.7 - 279.0 - 18.5 - 294.5 - 16.1 - 104.5 - 7.6 - 3.2Tyy 146.6 - 7.7 - 298.1 2.6 - 294.5 - 0.5 - 40.0 - 2.8 1.2T zz - 292.8 15.3 577.1 15.9 589.1 16.6 144.5 10.3 2.0
TZVP+ 1sTxx 152.1 - 7.7 - 291.3 - 17.7 - 309.9 - 15.4 - 109.7 - 7.2 - 3.5Tyy 152.5 - 7.7 - 307.5 2.2 - 309.9 - 0.3 - 35.3 - 2.4 0.7T zz - 304.6 15.3 598.8 15.5 619.8 15.8 145.0 9.6 2.8Experiment a a b b c
Txx - 303 - 3 - 254Tyy - 318 - 3 - 254T zz - 304 10.2 621 5 504
a Reference [29].b Reference [23].c Reference [28].
performance of the [B3LYP/TZVP+ 1s-on-heavy-atoms//UMP2/6-311G(d,p)] level.
We now apply the B3LYP method to three somewhatlarger molecules, P2H
+6 , PF4 and PCl4, in order to test its
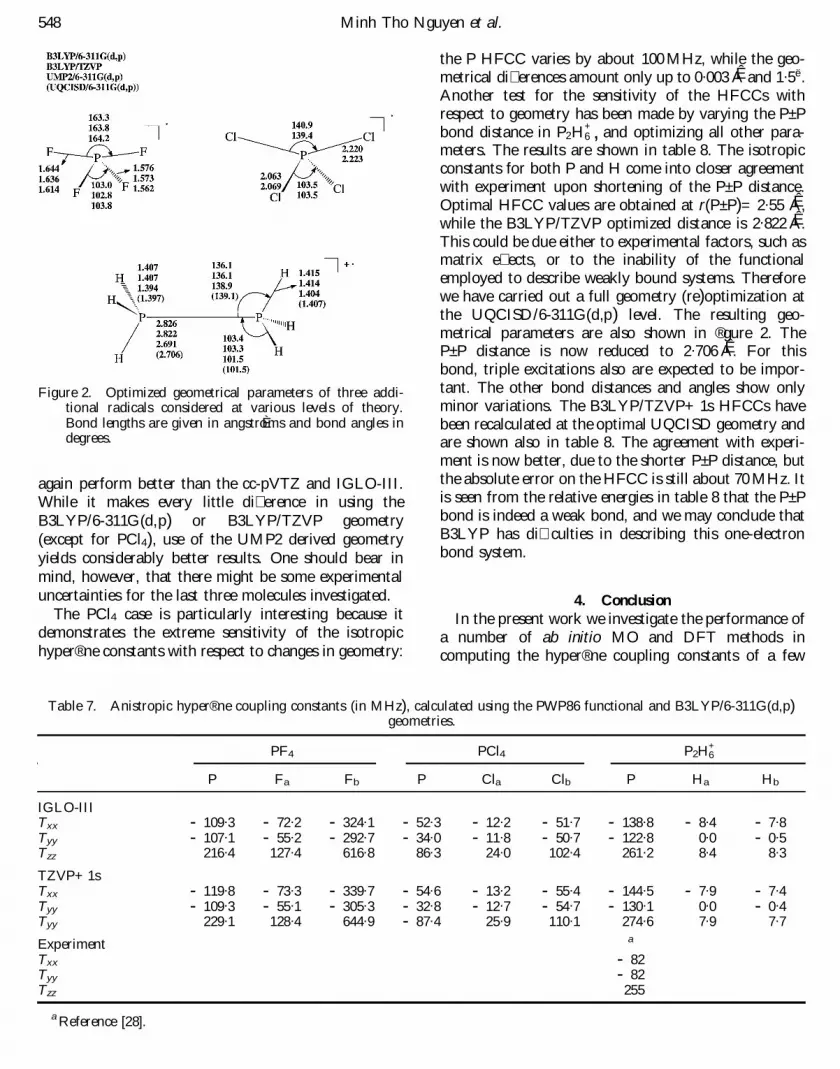
performance further. The IGLO-III, cc-pVTZ andTZVP related basis sets have been used. Three types ofgeometry were employed, namely B3LYP/6-311G(d,p),B3LYP/TZVP and UMP2/6-311G(d,p). The optimizedgeometrical parameters for these molecules are shown in
® gure 2. As with the previous radicals considered,UMP2 derived geometries di� er considerably fromB3LYP geometries. In the case of P2H
+6 , for which
also a UQCISD/6-311G(d,p) optimization was carriedout, the UMP2 values come closer to the UQCISD onesthan do the B3LYP. Table 6 lists the calculated isotropiccoupling constants, whereas table 7 contains the aniso-tropic constants (only for the B3LYP/6-311G(d,p) geometry). The TZVP related basis sets
Hyper® ne constants of phosphorus-containing radicals 547
Table 5. Continued.
PF22B1 PCl2 2B1 H3PF 2A Â HPF3
2A ÂP F P Cl P F Ha Hb P Fa Fb H
IGLO-IIITxx - 311.0 - 175.8 - 280.7 - 23.4 - 149.7 - 332.0 - 9.4 - 10.8 - 148.9 - 97.9 - 289.7 - 6.1Tyy - 286.0 - 147.0 - 265.0 - 16.7 - 116.0 - 324.7 2.2 - 1.9 - 146.8 - 62.0 - 259.8 - 5.1T zz 597.0 322.8 545.7 40.1 265.7 656.7 7.2 12.7 295.7 159.8 549.5 11.2TZVP+ 1sTxx - 320.1 - 185.3 - 293.6 - 24.7 - 157.8 - 360.1 - 8.9 - 9.7 - 157.7 - 103.1 - 308.1 - 5.3Tyy - 296.3 - 164.5 - 282.1 - 18.5 - 114.7 - 339.8 1.0 - 1.9 - 146.2 - 67.8 - 274.9 - 4.4T zz 616.4 249.8 575.6 43.2 272.5 699.8 8.0 11.6 303.9 170.8 583.0 9.8Experiment a a b b
Txx - 313.3 - 129.9 - 334.6 - 23.0Tyy - 313.0 - 129.9 - 287.0 - 15.7T zz 626.5 259.7 621.6 38.6
a Reference [25].b Reference [26].
Table 6. Calculated isotropic hyper® ne coupling constants for P2H+6 , PF4 and PCl4, using the B3LYP method.
P2H+6 PF4 PCl4
Aiso/MHz P Ha Hb P Fa Fb P Cla Clb
B3LYP/6-311G(d,p)cc-pVTZ 1487.6 116.2 - 5.2 3546.2 144.7 627.9 3696.1 4.3 22.0IGLO-III 1552.3 120.5 - 4.0 3887.9 156.0 688.8 3937.3 3.9 18.6TZVP+ 1s on P 1570.5 117.6 - 4.9 3810.2 150.4 724.7 3741.6 4.1 20.6TZVP+ 1s on heavy atoms 1570.5 117.6 - 4.9 3810.1 155.5 749.2 3741.6 4.3 21.3
B3LYP/TZVP geometrycc-pVTZ 1488.5 114.3 - 6.4 3521.6 141.4 619.0 3798.5 4.8 22.8IGLO-III 1555.2 117.7 - 5.5 3865.1 154.4 681.6 4040.4 4.3 19.3TZVP+ 1s on P 1569.0 117.7 - 5.4 3790.6 149.2 717.5 3844.4 4.5 21.3TZVP+ 1s on heavy atoms 1569.0 117.7 - 5.4 3790.5 154.0 741.6 3840.8 4.7 22.1
UMP2/6-311G(d,p) geometrycc-pVTZ 1501.0 140.4 - 14.8 3445.1 149.7 660.6 3507.9 3.4 24.0IGLO-III 1570.0 145.0 - 14.9 3764.2 162.2 717.2 3744.0 3.2 20.7TZVP+ 1s on P 1583.9 143.3 - 15.1 3728.1 159.7 763.1 3571.7 3.4 22.6TZVP+ 1s on heavy atoms 1583.9 143.3 - 15.1 3725.1 163.3 787.6 3568.5 3.5 23.5
Experimental 1654a 182a 22a 3842±3954b
126± 165b 790± 1064b 3427±3671c
0c 98c
a Reference [28].b Reference [25].c Reference [27].
again perform better than the cc-pVTZ and IGLO-III.While it makes every little di� erence in using theB3LYP/6-311G(d,p) or B3LYP/TZVP geometry(except for PCl4), use of the UMP2 derived geometryyields considerably better results. One should bear inmind, however, that there might be some experimentaluncertainties for the last three molecules investigated.
The PCl4 case is particularly interesting because itdemonstrates the extreme sensitivity of the isotropichyper® ne constants with respect to changes in geometry:
the P HFCC varies by about 100 MHz, while the geo-metrical di� erences amount only up to 0.003 AÊ and 1.5ë .Another test for the sensitivity of the HFCCs withrespect to geometry has been made by varying the P± Pbond distance in P2H
+6 , and optimizing all other para-
meters. The results are shown in table 8. The isotropicconstants for both P and H come into closer agreementwith experiment upon shortening of the P± P distance.Optimal HFCC values are obtained at r(P± P)= 2.55 AÊ ,while the B3LYP/TZVP optimized distance is 2.822 AÊ .This could be due either to experimental factors, such asmatrix e� ects, or to the inability of the functionalemployed to describe weakly bound systems. Thereforewe have carried out a full geometry (re)optimization atthe UQCISD/6-311G(d,p) level. The resulting geo-metrical parameters are also shown in ® gure 2. TheP± P distance is now reduced to 2.706 AÊ . For thisbond, triple excitations also are expected to be impor-tant. The other bond distances and angles show onlyminor variations. The B3LYP/TZVP+ 1s HFCCs havebeen recalculated at the optimal UQCISD geometry andare shown also in table 8. The agreement with experi-ment is now better, due to the shorter P± P distance, butthe absolute error on the HFCC is still about 70 MHz. Itis seen from the relative energies in table 8 that the P± Pbond is indeed a weak bond, and we may conclude thatB3LYP has di� culties in describing this one-electronbond system.
4. Conclusion
In the present work we investigate the performance ofa number of ab initio MO and DFT methods incomputing the hyper® ne coupling constants of a few
548 Minh Tho Nguyen et al.
Figure 2. Optimized geometrical parameters of three addi-tional radicals considered at various levels of theory.Bond lengths are given in angstroÈ ms and bond angles indegrees.
Table 7. Anistropic hyper® ne coupling constants (in MHz), calculated using the PWP86 functional and B3LYP/6-311G(d,p)geometries.
PF4 PCl4 P2H+6
P Fa Fb P Cla Clb P Ha Hb
IGLO-IIITxx - 109.3 - 72.2 - 324.1 - 52.3 - 12.2 - 51.7 - 138.8 - 8.4 - 7.8Tyy - 107.1 - 55.2 - 292.7 - 34.0 - 11.8 - 50.7 - 122.8 0.0 - 0.5T zz 216.4 127.4 616.8 86.3 24.0 102.4 261.2 8.4 8.3
TZVP+ 1sTxx - 119.8 - 73.3 - 339.7 - 54.6 - 13.2 - 55.4 - 144.5 - 7.9 - 7.4Tyy - 109.3 - 55.1 - 305.3 - 32.8 - 12.7 - 54.7 - 130.1 0.0 - 0.4Tyy 229.1 128.4 644.9 - 87.4 25.9 110.1 274.6 7.9 7.7
Experiment a
Txx - 82Tyy - 82T zz 255
a Reference [28].

phosphorus-containing radicals. The UMP2, UQCISD,UQCISD(T) and B3LYP methods have been consideredwith a variety of basis sets for calculating the isotropicHFCCs of PH, PH2, PF2, PCl2, PH+
3 , PH4, H3PF andHPF3. it is pointed out that split-valence basis sets, whileperforming rather well in combination with UQCISD,yield unacceptable results in conjunction with theB3LYP and UMP2 methods. The IGLO-III basis set,which has been developed especially for the calculationof magnetic properties, performs very similarly to theuncontracted split-valence basis sets with B3LYP, andis even more accurate when used in combination withUQCISD. Furthermore, correlation consistent cc-pVXZbasis sets have been tested with B3LYP and they seem toperform better than the previous bases, probably due totheir better description of the core region for phos-phorus. Decontraction of the cc-pVTZ basis yieldsworse results, indicating that the actual contractionscheme and balancing are important factors, especiallyin conjunction with B3LYP. Finally, a TZVP basis setwhich has been constructed for DFT calculations yieldsgood isotropic HFCCs with B3LYP, but does not per-form well in combination with UQCISD.
Various e� ects of adding and decontracting basisfunctions also are discussed. While adding d functionshas large but unpredictable e� ects on UMP2 calcula-tions, di� use and f functions show less in¯ uence. Inthe case of UQCISD, addition of d functions gives asystematic improvement of the HFCCs; the e� ect ofdi� use and f functions remains relatively small. Similarbehaviour is found also for B3LYP in combination withsplit-valence basis sets, but the overall agreement with
experiment is not satisfactory. Decontracted split-valence basis sets, on the other hand, while they enhancethe B3LYP values, are useless in combination withUQCISD. This indicates a certain imbalance to be pre-sent in the decontracted split valence basis set. Althoughpartly decontracted IGLO bases enhance the quality ofUQCISD results considerably, little e� ect is observedfor B3LYP calculations. Adding a tight s function toIGLO-III, however, worsens both B3LYP andUQCISD results. A tight s function added to TZVPon the other hand (TZVP+ 1s on heavy atoms), bringsthe B3LYP values in satisfactory agreement with experi-ment.
For the basis sets under consideration (6-311 +G(2df,p) and its us and usp counterparts, as well asIGLO-III and its uncontracted version), UQCISD(T)values follow the same trends as the correspondingUQCISD values. However, inclusion of triple excita-tions, UQCISD(T), generally lowers the computed 31Pisotropic HFCCs, often resulting in better agreementwith experimental data.
The in¯ uence of the geometry was investigated also,leading to the conclusion that B3LYP/6-311G(d,p)derived geometries are not su� ciently accurate for thecalculation of hyper® ne properties, at least for this typeof compound. The use of UMP2/6-311G(d,p) orUQCISD/6-311g(d,p) geometries instead enhances theaccuracy of B3LYP HFCC values considerably. It maybe concluded that the B3LYP functional, when used incombination with purposely tailored basis sets, mayyield good results, even with medium-sized sets such asthe TZVP+ 1s. However, it appears that none of thethree methods employed is intrinsically appropriate forcomputing hyper® ne parameters. The good performanceof the TZVP basis set with B3LYP is probably due to afortunate cancellation of errors originating from thetreatment of core and valence polarization. Given thefact that an appropriate method is not yet available, thisapproach can be used with certain con® dence. More-over, it can be extended easily to much larger molecules,owing to the favourable time scaling of DFT methods.Nevertheless, much caution should be taken when usingcurrently available DFT methods to treat non-classicalstructures such as one-electron radical cations.
The authors dedicate this paper to Professor JohnPople on his 70th birthday and honour his outstandingcontribution to modern quantum chemistry.
The authors are grateful to the Belgian National Fundfor Scienti® c Research (FWO), the K.U.-LeuvenResearch Council (GOA) and the Swedish NationalSciences Research Council (NFR) for continuingsupport.
Hyper® ne constants of phosphorus-containing radicals 549
Table 8. Variation of the isotropic constants of P2H+6 with
P± P distance.
Aiso/MHzb
P± P distancea P Ha Hb energy/kJ mol- 1
2.3 1566.0 282.0 - 26.4 31.72.4 1605.1 243.9 - 25.6 18.02.5 1619.4 208.9 - 23.0 9.12.55 1619.5 192.5 - 21.1 6.12.6 1615.7 177.0 - 18.9 3.82.7 1600.4 148.1 - 13.7 1.02.822 1572.1 116.3 - 6.1 0.02.9 1552.2 97.0 - 0.4 0.33.0 1530.0 69.7 8.9 1.52.706c 1583.7 142.5 - 14.1 1.6
Exp.d 1654 182 - 22
a At each distance, the other geometrical parameters areoptimized at the B3LYP/TZVP level.
b At the B3LYP/TZVP+ 1s level.c UQCISD/6-311G(d,p) optimized geometry.d Reference [28].
Relative

References[1] Malkin, V. G., Malkina , O. L., Eriksson, L. A., and
Salahub, D. R., 1995, Theoretical and ComputationalChemistry, Vol. 2, edited by J. M. Seminario and P.Politzer (Amsterdam: Elsevier).
[2] Lund, A., Lindgren, M., Lunell, S., and Maruani, J.,1989, Molecules in Physics, Chemistry and Biology, Vol. 3,edited by J. Maruani (Dordrecht: Kluwer).
[3] Rassolov, V. A., and Chipman, D. M., 1996, J. chem.Phys., 105, 1470.
[4] Rassolov, V. A., and Chipman, D. M., 1996, J. chem.Phys., 105, 1479.
[5] Hiller, J., Sucher, J., and Feinberg, G., 1978, Phys.Rev. A, 18, 2399.
[6] Barone, V., 1994, J. chem. Phys., 101, 6834.[7] Barone, V., 1996, Recent Advances in Density Functional
Methods, Vol. 1, edited by D. P. Chong (Singapore:World Scienti® c).
[8] Austen, M. A., Eriksson, L. A., and Boyd, R. J., 1994,J. Can. Chem., 72, 695.
[9] Eriksson, L. A., Wang, J., Boyd , R. J., and Lunell, S.,1994, J. chem. Phys., 98, 792.
[10] Cramer, C. J., and Lim, M. H., 1994, J. phys. Chem., 98,5024.
[11] Nguyen, M. T., Creve, S., and Vanquickenborne,L. G., 1996, J. chem. Phys. , 105, 1922.
[12] Becke, A. D., 1993, J. phys. Chem., 98, 5648.[13] McLean, A. D., and Chandler, G. S., 1980, J. chem.
Phys., 72, 5639.[14] Kutz elnigg, W., Fleischer, U., and Schindler , M.,
1990, NMR: Basic Principles and Progress, Vol. 23(Berlin: Springer-Verlag) p. 165.
[15] Godbout, N., Salahub, D. R., Andz elm, J., andWimmer, E., 1992, Can. J. Chem. , 70, 560.
[16] Gauld, J. W., Eriksson, L. A., and Radom, L., 1997,J. phys. Chem., 101, 1352.
[17] Frisch, M. J., Trucks, G. W., Schlegel, H. B., Gill,P. M. W., Johnson, B. G., Wong, M. W., Foresman,J. B., Robb, M. A., Head-Gordon, M., Replogle,E. S., Gomperts, R., Andres, J. L., Raghavachari,K., Binkley, J. S., Gonz alez , C., Martin, R. L., Fox,D. J., Defrees, D. J., Baker, J., Stewart, J. J. P., andPople, J. A., 1993, Gaussian 94, Revision C.3(Pittsburgh, PA: Gaussian, Inc.).
[18] St-Amant, A., and Salahub, D. R., 1990, Chem. Phys.L ett. , 169, 387.
[19] Ma , B., Lii, J.-H., Schaefer III, H. F., and Allinger,N. L., 1996, J. phys. Chem., 100, 8763.
[20] Cohen, M. J., and Chong, D. P., 1995, Chem. Phys.L ett. , 234, 405.
[21] Perdew , J. P., 1986, Phys. Rev. B, 33, 8822; 34, 7406.[22] Perdew , J. P., and Wang, Y., 1986, Phys. Rev. B, 33,
8800.[23] Davies, P. B., Russell, D. K., and Thrush, B. A., 1979,
Chem. Phys. L ett. , 44, 421.[24] Colussi, A. J., Morton, J. R., and Preston, K. F.,
1975, J. chem. Phys., 62, 2004.[25] Nelson, W., Jackel, G., and Gordy, W., 1970, J. chem.
Phys., 52, 4572.[26] Bonaz z ola , L., Michaut, J. P., and Roncin, J., 1981,
J. chem. Phys., 75, 4829.[27] Mishra, S. P., and Symons, C. R. S., 1976, J. Chem. Soc.
Dalton Trans, 139.[28] Knight Jr ., L. B., Tyler, D. J., Kudelko, P., Lyon,
J. B., and McKinley, A. J., 1993, J. chem. Phys., 99,
7384.[29] Davies, P. B., Russell, D. K., Smith, D. R., and
Trush, B. A., Can. J. Phys., 57, 522.[30] Coulossi, A. J., Morton, J. R., and Preston, K. F.,
1975, J. phys. Chem., 79, 1855.
550 Hyper® ne constants of phosphorus-containing radicals





























