Embed Size (px)
Citation preview

ÇUKUROVA ÜNİVERSİTESİ FEN BİLİMLERİ ENSTİTÜSÜ
DOKTORA TEZİ
Şevket ŞİMŞEK
ABO3 (A=Ag, Na; B=Nb, Ta) TİPİ KRİSTALLERİN DİNAMİK ÖZELLİKLERİNİN Ab İNİTİO YÖNTEMİYLE İNCELENMESİ
FİZİK ANABİLİM DALI
ADANA, 2011

ÇUKUROVA ÜNİVERSİTESİ FEN BİLİMLERİ ENSTİTÜSÜ
ABO3 (A=Ag, Na ; B=Na, Ta) TİPİ KRİSTALLERİN DİNAMİK
ÖZELLİKLERİNİN Ab İNİTİO YÖNTEMİYLE İNCELENMESİ
Şevket ŞİMŞEK
DOKTORA TEZİ
FİZİK ANABİLİM DALI Bu Tez 16/12/2011 Tarihinde Aşağıdaki Jüri Üyeleri Tarafından Oybirliği/Oyçokluğu ile Kabul Edilmiştir. ……………….................... ………………………….. ……................................ Doç.Dr. Süleyman ÇABUK Prof.Dr.Amirrullah MAMEDOV Doç.Dr. Kasım KURT DANIŞMAN ÜYE ÜYE ...……………….................. ...……………………….. Doç. Dr. Ramazan BİLGİN Doç. Dr. Faruk KARADAĞ ÜYE ÜYE Bu Tez Enstitümüz Fizik Anabilim Dalında hazırlanmıştır. Kod No:
Prof. Dr. İlhami YEĞİNGİL Enstitü Müdürü
Bu Çalışma Ç. Ü. Araştırma Projeleri Birimi Tarafından Desteklenmiştir. Proje No: FEF2010D5 Not: Bu tezde kullanılan özgün ve başka kaynaktan yapılan bildirişlerin, çizelge ve fotoğrafların
kaynak gösterilmeden kullanımı, 5846 sayılı Fikir ve Sanat Eserleri Kanunundaki hükümlere tabidir.

I
ÖZ
DOKTORA TEZİ
ABO3 (A=Ag, Na ; B=Nb, Ta) TİPİ KRİSTALLERİN DİNAMİK ÖZELLİKLERİNİN Ab İNİTİO YÖNTEMİYLE İNCELENMESİ
Şevket ŞİMŞEK
ÇUKUROVA ÜNİVERSİTESİ FEN BİLİMLERİ ENSTİTÜSÜ
FİZİK ANABİLİM DALI
Danışman: Doç.Dr. Süleyman ÇABUK Yıl: 2011, Sayfa: 119
Jüri : Doç. Dr. Süleyman ÇABUK : Prof. Dr. Emirullah MEHMETOV : Doç.Dr. Kasım KURT : Doç.Dr. Ramazan BİLGİN : Doç.Dr. Faruk KARADAĞ
Bu tezde, ortorombik yapıdaki AgNbO3, NaTaO3, NaNbO3 ve rombohedral
yapıdaki AgTaO3 'ın yapısal, elektronik ve dinamik özellikleri ilk defa düzlem dalga pseudo potansiyel metodu, yoğunluk fonksiyonel teorisinin yerel yoğunluk yaklaşımıyla incelendi. Hesaplanan örgü parametreleri ve atomik pozisyonlar mevcut deneysel ve teorik sonuçlarla uyumludur. Brillouin bölgesinde AgNbO3 ve AgTaO3 her ikisi de dolaylı bir bant aralığına sahiptir, buna karşın, NaTaO3 ve NaNbO3 ise doğrudan bir bant aralığı vardır. Born etkin yükleri, dielektrik geçirgenlik tensörü ve elastik özellikler yoğunluk fonksiyonel perturbasyon teorisi çerçevesinde hesaplandı. Her bir iyonun Born etkin yük tensörleri, q→0 limitinde dinamik matrise analitik olmayan katkıyla belirlenir. Fonon dispersiyon eğrisi ve fonon durum yoğunluğu, direk metot kullanılarak elde edildi. LO / TO yarılması, süper hücre içinde bulunan fonon frekansları ortaya çıkardı. Gamma noktasındaki fonon hesaplamalarının negatif frekans vermesi AgNbO3, NaTaO3, NaNbO3 ve AgTaO3 yapılarının yarı kararlı olduğunu işaret etmektedir. Aynı zamanda literatürde mevcut olan deneysel ve teorik verilerle karşılaştırma yapıldı.
Anahtar Kelimeler: ABO3, Elektronik Özellikler, Elastik Sabitler, Born Efektif
Yükler, Fonon Dispersiyon Eğrisi

II
ABSTRACT
PhD THESIS INVESTIGATIN WITH AB INITIO METHOD OF DYNAMIC PROPERTIES
OF ABO3 (A=Ag, Na ; B=Nb, Ta) TYPE CRYSTALS
Şevket ŞİMŞEK
ÇUKUROVA UNIVERSITY INSTITUTE OF NATURAL AND APPLIED SCIENCES
DEPARMANT OF PHYSICS
Supervısor: Assoc. Prof. Süleyman ÇABUK Year: 2011, Pages: 119 Jury : Assoc. Prof. Süleyman ÇABUK
: Prof.Dr. Emirullah MEHMETOV : Assoc. Prof. Kasım KURT : Assoc. Prof. Ramazan BİLGİN : Assoc. Prof. Faruk KARADAĞ
In this dissertation, we have investigated the structural, electronic and dynamical properties of the AgNbO3, NaTaO3, NaNbO3 in the orthorhombic structures and AgTaO3 in the rhombohedral structure, by employing the plane wave pseudopotential method, local density approximation of the density functional theory for the first time. The calculated equilibrium lattice parameters and atomic position are in the agreement with the available experimental and theoretical results. AgNbO3 and AgTaO3 both have an indirect band gap, whereas, NaTaO3 and NaNbO3 both have a direct band gap in the Brillouin zone. Born effective charges, dielectric permittivity tensors and elastic properties have been calculated within the framework of density functional perturbation theory. Born effective charges tensors for each ion, which determine the non-analytic contribution to the dynamical matrix in the limit NaTaO3 and NaNbO3 q→0. The phonon dispersion curves and the phonon density of states are derived using the direct method. The LO / TO splitting was extracted from phonon frequencies found within the supercell. Gamma point phonon calculations give negative frequencies which indicates the AgNbO3, NaTaO3, NaNbO3 and AgTaO3 structures are metastable. We have also made comparisons with related experimental and theoretical data which is available. Key Words: ABO3, Electronic Properties, Elastic Constants, Born Effective
Charges, Phonon Dispersion Curve.

III
TEŞEKKÜR
Öncelikle, bu tezin yönetiminde ve oluşumunda aynı zamanda çalışmalarım
sırasında karşılaştığım sorunların çözümünde her türlü desteğini esirgemeyen,
değerli hocam Doç Dr. Süleyman ÇABUK’ a sonsuz saygı ve teşekkürlerimi
sunarım.
Tez çalışmam sırasında her türlü bilgi ve desteğini esirgemeyen sayın Prof.
Dr. Emirullah Mehmetov’a teşekkür ederim.
Çalışmalarım sırasında yardımlarını esirgemeyen ve daima bizlere destek
olan sayın Güzide ÜNLÜ’ye ve manevi desteklerinden dolayı Fizik Bölümü'ndeki
tüm öğretim üyelerine saygı ve teşekkürlerimi sunarım.
Tez çalışmam sırasında yardımlarını esirgemeyen değerli arkadaşlarım Ali
ÇETİNKAYA, Ayşe BOZDUMAN AKYOL, Kamuran KARA ve Erkan TETİK’e
teşekkürlerimi sunarım.
Böyle yoğun bir çalışma sürecinde beni sonuna kadar destekleyen ve hayatım
boyunca benim için her türlü fedakârlıkları gösteren değerli annem, babam,
kardeşlerime sonsuz saygı ve teşekkürlerimi sunarım.

IV
İÇİNDEKİLER SAYFA
ÖZ ........................................................................................................................ I
ABSTRACT ........................................................................................................ II
TEŞEKKÜR ...................................................................................................... III
İÇİNDEKİLER ............................................................................................ …..IV
ÇİZELGELER DİZİNİ ...................................................................................... VI
ŞEKİLLER DİZİNİ .......................................................................................... VII
SİMGELER VE KISALTMALAR .................................................................... IX
1. GİRİŞ .............................................................................................................. 1
2. ÖNCEKİ ÇALIŞMALAR ................................................................................ 7
3. MATERYAL VE METOD ............................................................................ 17
3.1. Tek Atomlu Örgü Titreşimleri ................................................................ 17
3.2. İki Atomlu Örgü Titreşimleri ................................................................. 21
3.3. Örgü Titreşimlerinin Kuantumlanması.................................................... 25
3.4. Dinamik Matris ...................................................................................... 29
3.5. Elastik Sabitler ....................................................................................... 33
3.6. Bulk Modülü .......................................................................................... 39
3.7.Born Effektif Yükler ............................................................................... 40
3.8. Yoğunluk Fonksiyonel Teorisi................................................................ 41
3.8.1. Çok Cisim Problemi ........................................................................ 42
3.8.2. Born-Oppenheimer Yaklaşımı ......................................................... 43
3.8.3. Hartree Yaklaşımı ........................................................................... 44
3.8.4. Hartree-Fock Yaklaşımı .................................................................. 45
3.8.5. Hohenberg-Kohn Teoremi .............................................................. 47
3.8.6. Çok Parçacık Sistemi: Kohn-Sham Denklemi ................................. 52
3.8.7. Değişim ve Korelasyon ................................................................... 54
3.8.8.Yerel Yoğunluk Yaklaşımı ............................................................... 55
3.8.9. Genelleştirilmiş Gradyent Yaklaşımı ............................................... 57
3.9. Düzlem Dalga Yaklaşımı ..................................................................... 59
3.10. Pseudo Potansiyel Yaklaşımı............................................................... 60

V
3.10.1. Norm-Koruyucu Pseudo Potansiyeller ........................................... 61
3.11. Toplam Enerji Hesabı .......................................................................... 67
3.12. ABINIT Programı ................................................................................ 67
3.13. SIESTA Programı ................................................................................ 68
3.14. PHONON Programı ............................................................................. 68
4. BULGULAR VE TARTIŞMA ...................................................................... 71
4.1. Hesaplama Metodu ................................................................................. 71
4.2. Örgü Parametreleri ................................................................................. 72
4.3. Elektronik Band Yapısı ve Durum Yoğunluğu ........................................ 74
4.3.1. AgNbO3 Kristali ............................................................................. 75
4.3.2. NaNbO3 Kristali.............................................................................. 77
4.3.3. AgTaO3 Kristali .............................................................................. 79
4.3.4. NaTaO3 Kristali .............................................................................. 81
4.4. Dinamik Özellikler ................................................................................. 83
4.4.1. Elastik Sabitler ve Bulk Modülü...................................................... 83
4.4.2. Born Efektif Yükler ve Optik Dielektrik Sabiti................................ 85
4.4.3. Fonon Dispersiyon Eğrileri ve Durum Yoğunluğu .......................... 90
4.4.3.1. AgTaO3 Kristali .................................................................. 93
4.4.3.2. NaTaO3 Kristali .................................................................. 96
4.4.3.3. AgNbO3 Kristali ................................................................. 99
4.4.3.4. NaNbO3 Kristali .................................................................103
5. SONUÇLAR VE ÖNERİLER .....................................................................107
KAYNAKLAR .................................................................................................111
ÖZGEÇMİŞ .....................................................................................................119

VI
ÇİZELGELER DİZİNİ SAYFA
Çizelge 4.1. AgNbO3, AgTaO3, NaNbO3 ve NaTaO3 kristallerinin örgü
parametreleri ........................................................................................ 72
Çizelge 4.2. Ortorombik yapıda AgNbO3 kristalinin atomik konumları ..................... 73
Çizelge 4.3. Ortorombik yapıda NaNbO3 kristalinin atomik konumları .................... 73
Çizelge 4.4. Ortorombik yapıda NaTaO3 kristalinin atomik konumları ..................... 73
Çizelge 4.5. Rombohedral yapıda AgTaO3 kristalinin atomik konumları .................. 73
Çizelge 4.6. AgNbO3 kristallinin yüksek simetri noktalarındaki doğrudan ve
dolaylı band aralıkları ........................................................................... 76
Çizelge 4.7. NaNbO3 kristallinin yüksek simetri noktalarındaki doğrudan ve
dolaylı band aralıkları ........................................................................... 77
Çizelge 4.8. AgTaO3 kristallinin yüksek simetri noktalarındaki doğrudan ve
dolaylı band aralıkları ........................................................................... 79
Çizelge 4.9. NaTaO3 kristallinin yüksek simetri noktalarındaki doğrudan ve
dolaylı band aralıkları ........................................................................... 81
Çizelge 4.10. Ortorombik yapıda hesaplanmış NaTaO3’ün elastik sabitleri ................ 84
Çizelge 4.11. Rombohedral yapıda hesaplanmış AgTaO3’ün elastik sabitleri ............. 85
Çizelge 4.12. Rombohedral yapıda hesaplanmış AgTaO3’ün born efektif yükleri ...... 86
Çizelge 4.13. Ortorombik yapıda hesaplanmış NaTaO3’ün born efektif yükleri ......... 86
Çizelge 4.14. Ortorombik yapıda hesaplanmış AgNbO3’ün born efektif yükleri ........ 87
Çizelge 4.15. Ortorombik yapıda hesaplanmış NaNbO3’ün born efektif yükleri......... 88
Çizelge 4.16. Rombohedral yapıda hesaplanmış AgTaO3’ün optik ve statik
dielektrik sabiti ..................................................................................... 89
Çizelge 4.17. Ortorombik yapıda hesaplanmış NaTaO3’ün optik ve statik
dielektrik sabiti ..................................................................................... 89
Çizelge 4.18. İlkel hücresinde S tane atom bulunan kristalin kip tipi ve sayısı ........... 92
Çizelge 4.19. AgTaO3 kristalinin k=0 da gamma noktasında hesaplanan titreşim
kiplerinin frekansı (THz) ...................................................................... 94
Çizelge 4.20. NaTaO3 kristalinin k=0 da gamma noktasında hesaplanan titreşim
kiplerinin frekansı (THz) ...................................................................... 97

VII
Çizelge 4.21. AgNbO3 kristalinin k=0 da gamma noktasında hesaplanan titreşim
kiplerinin frekansı (THz) ...................................................................... 100
Çizelge 4.22. NaNbO3 kristalinin k=0 da gamma noktasında hesaplanan titreşim
kiplerinin frekansı (THz) ...................................................................... 103

VIII
ŞEKİLLER DİZİNİ SAYFA
Şekil 1.1. ABO3 tipi perovskit oksitlerin kristal yapısı ............................................ 1
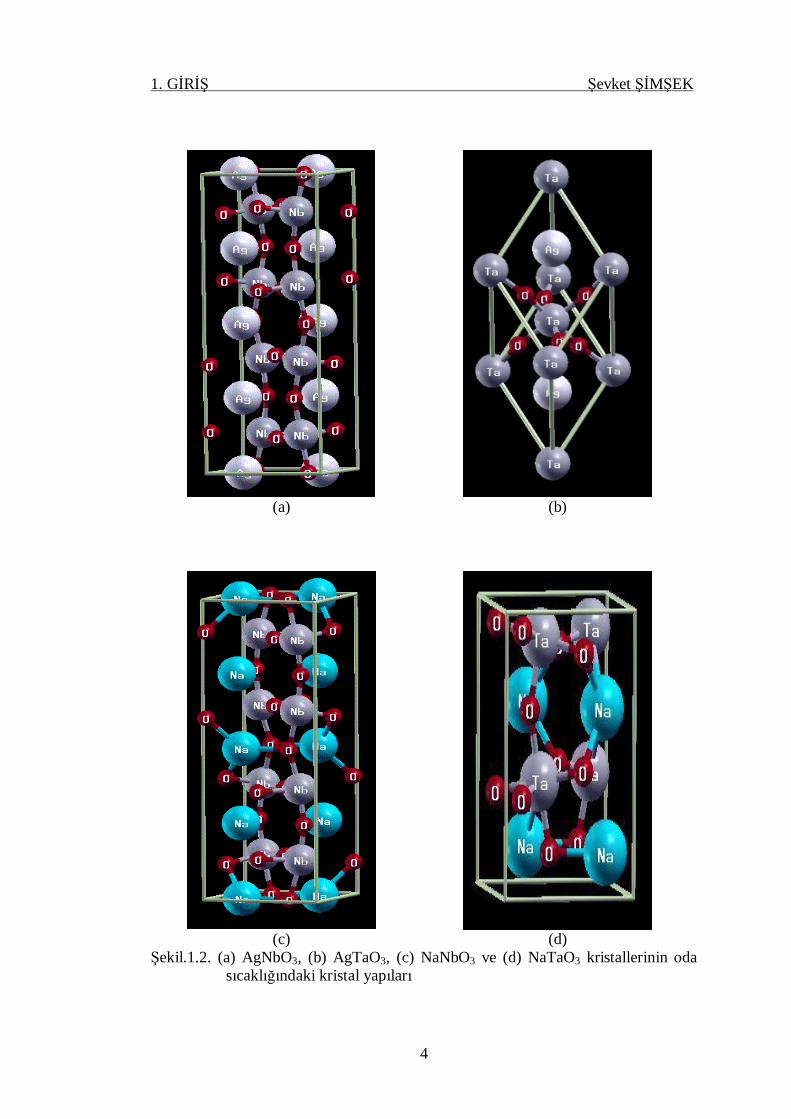
Şekil 1.2. (a) AgNbO3, (b) AgTaO3, (c) NaNbO3 ve (d) NaTaO3 kristallerinin
oda sıcaklığındaki kristal yapıları ........................................................... 4
Şekil 3.1. Boyuna bir dalga sırasında atom düzlemlerinin yer değiştirmesi ........... 17
Şekil 3.2. Enine bir dalga sırasında atom düzlemlerinin yer değiştirmesi .............. 18
Şekil 3.3. Atomların titreşim frekanslarının ( dalga vektörüne ( göre
değişim grafiği ..................................................................................... 20
Şekil 3.4. İki atomlu doğrusal örgünün dispersiyon bağıntısında optik ve
akustik dalgalar .................................................................................... 21
Şekil 3.5. Kütleleri M1 ve M2 olan ve düzlemler arası kuvvet sabiti C olan iki
atomlu bir kristal yapısı ........................................................................ 22
Şekil 3.6. İki atomlu doğrusal örgüde enine optik ve akustik dalgalar ................... 24
Şekil 3.7. Pseudo potansiyel, pseudo ve gerçek dalga fonksiyonları ..................... 61
Şekil 4.1. (a) Ortorombik yapıda, (b) Rombohedral yapıda Brilouin bölgesinin
yüksek simetri noktaları........................................................................ 74
Şekil 4.2. AgNbO3 kristalinin elektronik band yapısı............................................ 75
Şekil 4.3. AgNaO3 kristalinin parçalı (PDOS) ve toplam durum yoğunluğu
(DOS) .................................................................................................. 76
Şekil 4.4. NaNbO3 kristalinin elektronik band yapısı ............................................ 78
Şekil 4.5. NaNbO3 kristalinin parçalı (PDOS) ve toplam durum yoğunluğu
(DOS) .................................................................................................. 78
Şekil 4.6. AgTaO3 kristalinin elektronik band yapısı ............................................ 80
Şekil 4.7. AgTaO3 kristalinin parçalı (PDOS) ve toplam durum yoğunluğu
(DOS) .................................................................................................. 80
Şekil 4.8. NaTaO3 kristalinin elektronik band yapısı ............................................ 82
Şekil 4.9. AgTaO3 kristalinin parçalı (PDOS) ve toplam durum yoğunluğu
(DOS) .................................................................................................. 82
Şekil 4.10. AgTaO3 kristalinin fonon dispersiyon eğrisi ........................................ 95
Şekil 4.11. AgTaO3 kristalinin ve atomlarının fonon durum yoğunluğu ................. 95

IX
Şekil 4.12. NaTaO3 kristalinin fonon dispersiyon eğrisi ......................................... 98
Şekil 4.13. NaTaO3 kristalinin ve atomlarının fonon durum yoğunluğu .................. 98
Şekil 4.14. AgNbO3 kristalinin fonon dispersiyon eğrisi ...................................... 102
Şekil 4.15. AgNbO3 kristalinin ve atomlarının fonon durum yoğunluğu .............. 102
Şekil 4.16. NaNbO3 kristalinin fonon dispersiyon eğrisi ...................................... 105
Şekil 4.17. NaNbO3 kristalinin ve atomlarının fonon durum yoğunluğu .............. 105

X
SİMGELER VE KISALTMALAR
Ab-initio : Temel ilkelere dayanan
ABINIT : Yoğunluk fonksiyonel teorisine dayalı olarak pseudo potansiyel
yöntem kullanan ab-initio yazılımı
AKN : (Ag,K)Nb
ATN : Ag(Ta,Nb)
C : Kübik faz
DFT : Yoğunluk fonksiyoneli teorisi
DFPT : Yoğunluk fonksiyoneli pertürbasyon teorisi
DOS : Durum yoğunluğu
EFISPS : Elektrik alan-uyarılmış yüzey foto voltaj spektroskopisi
εxc : Değişim-korelasyon enerjisi
FE : Ferroelektrik faz
FHI98PP : Yoğunluk fonksiyoneli teorisine dayalı olarak pseudo potansiyel
üreten yazılım
GGA : Genelleştirilmiş gradyent yaklaşımı
ICP : Endüktif kuplajlı plazma
IR : Infared
KB : Kleinman-Bylander formülü
LA : Boyuna akustik
LCOA : Sıkı-bağlanma yöntemi
LDA : Yerel yoğunluk yaklaşımı
LO : Boyuna optik
M1, M2, M3: Monoklinik M1, M2, M3 fazları
O1, O2 : Ortorombik O1, O2 fazları
PDOS : Parçalı durum yoğunluğu
PE : Paraelektrik
PHONON : Yoğunluk fonksiyonel teorisine dayalı olarak pseudo potansiyel
yöntem kullanan ab-initio yazılımı
RM : Raman

XI
SIESTA : Yoğunluk fonksiyonel teorisine dayalı olarak pseudo potansiyel
yöntem kullanan ab-initio yazılımı
SPS : Yüzey foto voltaj spektroskopisi
T : Tetragonal faz
TA : Enine akustik
TO : Enine optik
UV : Ultra viola
VASP : Yoğunluk fonksiyonel teorisine dayalı olarak pseudo potansiyel
yöntem kullanan ab-initio yazılımı
WIEN97 : Yoğunluk fonksiyonel teorisine dayalı olarak pseudo potansiyel
yöntem kullanan ab-initio yazılımı

1. GİRİŞ Şevket ŞİMŞEK
1
1. GİRİŞ
CaTiO3 minerali 1839 yılında Ural dağlarında Rus yer bilimci Gustav Rose
tarafından keşfedildi ve saygın Rus maden bilimci Count Lev Alexevich von
Perovski onuruna perovskit ismi verildi. Bundan sonra perovskit ismi ABO3 formuna
sahip olan bileşiklerin geniş bir ailesini adlandırmak için kullanılmaktadır.
Perovskitler yer kabuğunda en çok bulunan bileşiklerden biridir.
ABO3 formundaki bileşikler perovskit ailesine aittir ve geçiş metal
oksitlerinin bir alt sınıfını oluştururlar. Burada A ve B, metal katyonlarını O ise
oksijen anyonunu ifade ediyor. Şekil 1.1' de gösterilen yapı beş atomlu birim hücresi
ile basit kübik bir yapıya sahiptir. Perovskit oksitlerin birçoğunun örgü sabiti (2a
uzaklığı), 4 Å civarındadır.
Şekil 1.1. ABO3 tipi perovskit oksitlerin kristal yapısı

1. GİRİŞ Şevket ŞİMŞEK
2
B katyonu Nb, Ta, Ni, Ti, Fe, Co, yada Mn gibi geçiş metal iyonlarıdan
oluşuyor ve sekiz yüzlü (octahedron) oksijen anyonlarının merkezine yerleşmiştir. B
konumu tam kübik nokta grubu (Oh ) simetrisine sahiptir. A katyonu tek değerlikli
(Ag+, Na+, K+, Li+), iki değerlikli (Sr+2, Ba+2, Ca+2) ya da üç değerlikli (La+3, Nd+3,
Pr+3) olabilir. Perovskit yapıdaki bu malzemeler A+2B+4 O-23, A+1 B+5 O-2
3, A+3 B+3
O-23 formunda bulunurlar. Eğer A iyonun yükü qA ise B iyonunun yükü qB=6-qA
şeklinde olacaktır. Buradaki 6 faktörü üç oksijen iyonuna olan katkıyı belirtiyor. A
iyonu eşit uzaklıktaki 12 oksijen iyonu ile sarmalanmıştır. A konumu da aynı
zamanda Oh nokta grubuna sahiptir.
Oksijen iyonları kübik nokta grubu simetrisinin konumlarında değildirler.
Şekil 1.1 de X ile belirtilmiş olan oksijen iyonun konumunun simetrisinin D4h olduğu
görülebilir.
Kübik perovskitlerin iyi bilinen örneklerinden bazıları BaTiO3, SrTiO3,
AgNbO3, AgTaO3, NaNbO3, NaTaO3, KNbO3, KTaO3.
Perovskit bileşiklerinin diğer bir sınıfı, BO3 birim hücre formuna sahip olan
pseudo perovskitlerdir. Perovskit yapıya sahip olan böyle bileşiklerin A
konumlarının boş olduğu kabul edilir. ReO3 ve WO3 pseudo perovskitlere örnek
olarak gösterilebilir. Bu bileşiklerin boş olan A konumlarına alkali iyonlarının
eklenmesi ile WO3’den perovskitlerin bir ara sınıfını oluşturmak mümkündür.
Tungsten bronzs olarak bilinen bu bileşikler AxWO3 birim hücre formuna sahiptir.
Burada x, 0' dan 1' e kadar değişir ve A konumundaki iyon H, Li, Na, K, Rb ya da Cs
olabilir. Bu sistemlerin kristal yapıları da genelde x' in değerine bağlı olarak değişir.
Perovskit oksitler çok geniş bir katı hal fenomeni sergiledikleri için oldukça
ilginç malzemelerdir. Bu materyaller yalıtkan, yarı iletken, metal ve süper iletkenlik
gibi ilginç fiziksel özellikler sergilerler. Bazıları delokalize olmuş enerji bandı
durumlarına, bazıları lokalize olmuş elektronlara sahiptirler. Bazıları da bu iki tip
davranış arasında geçişler sergilerler. Perovskitlerin çoğu manyetik olarak düzenlidir
ve manyetik yapının birçok çeşidini yapılarında bulundurabilirler.
Perovskitlerin elektronik özellikleri, A ve B konumlarındaki iyonların yerine
farklı tür iyonların yerleştirilmesiyle kontrol edilir bir biçimde değiştirilebilir.

1. GİRİŞ Şevket ŞİMŞEK
3
Perovskitler durum yoğunluğu, fermi yüzeyi, dielektrik fonksiyonu, fonon
spektrumları ve foto emisyon spektrumları gibi özelliklerde kendine özgü bir yapı
ortaya koyması ve iki boyutlu davranış sergilemesi bakımından elektronik enerji
bandları çok sıra dışıdır.
Perovskitler aynı zamanda birçok teknolojik alanda da önemlidir.
Fotokromatik, elektrokromatik ve görüntü depolama araçlarında kullanılmaktadır.
Perovskit malzemelerin ferroelektrik ve piezoelektrik özellikleri anahtarlama,
filtreleme ve yüzey akustik dalga sinyal işleme içeren diğer araç uygulamalarında
kullanılmaktadır.
Perovskitlerin birçoğu katalitik olarak aktiftir. Karbon monoksit ve
hidrokarbonların oksidasyonu ve nitrojen oksitlerinin azaltılması için perovskit
katalist sistemlerin geliştirilmesi öneriliyor. Perovskitler aynı zamanda hidrojen
üretmek için suyun foto elektrolizini içeren elektrokimyasal uygulamalarda da
kullanılmaktadır (Wolfram ve ark, 2006).
AgNbO3, AgTaO3, NaNbO3 ve NaTaO3 kristallerinin yapıları da ABO3 tipi
perovskit yapısındadır. AgNbO3 kristali yüksek sıcaklıkta (T > 852 K) kübik
prototype simetriye sahiptir.
AgNbO3 kristali yüksek bir sıcaklıktan soğutulursa yapıdaki faz değişimi,
CTOOMMMO KKKKKK →← →← →← →← →← →← 852660634626540340 2132)1(
şeklindedir. Burada M1, M2, M3: Monoklinik, O1, O2: Ortorombik,T: Tetragonal ve
C: Kübik yapıyı göstermektedir (Sciau ve ark, 2004). Monoklinik yapıda Ag katyonu
NbO3 oktehadralin eğilmesine bağlı olarak faklı monoklinik fazda farklı uzay
grubunda olabilir. Son yapılan çalışmlarda AgNbO3 kristalinin oda sıcaklığında
ortorombik yapıda ve uzay grubunun Pbcm (No:57) olduğu saptanmıştır (Levin ve
ark, 2009). AgNbO3 kristalinin oda sıcaklığındaki yapısı şekil.1.2 (a)’da
verilmektedir.
1. GİRİŞ Şevket ŞİMŞEK
4
(a) (b)
(c) (d)
Şekil.1.2. (a) AgNbO3, (b) AgTaO3, (c) NaNbO3 ve (d) NaTaO3 kristallerinin oda sıcaklığındaki kristal yapıları

1. GİRİŞ Şevket ŞİMŞEK
5
AgTaO3 kristali (T > 777 K) kübik yapıdadır. AgTaO3 kristali sıcaklığın
düşürülmesiyle,
CTMR KKK →← →← →← 777692663
faz geçişleri gösterir. Burada R rombohedral yapıyı göstermektedir. Oda
sıcaklığında AgTaO3 kristali rombohedral yapıdadır ve R3c (No:161) uzay grubuna
aittir (Francombe ve ark, 1958). AgTaO3’ün oda sıcaklığındaki yapısı
Şekil.1.2.(b)’de gösterildiği gibidir.
NaNbO3, (T > 914 K) ideal perovskit yapıya sahiptir. NaNbO3 kristali
soğutulduğunda,
CTOOOOR KKKKKK →← →← →← →← →← →← 914848793753646163 4321
faz değişimlerini gösterir. NaNbO3 rombohedral fazda ferroelektriktir ve diğer
fazlarda antiferroelektriktir. NaNbO3 oda sıcaklığında ortorombik yapıdadır ve uzay
grubu Pbcm (No:57) dir ( Mishra ve ark, 2007). NaNbO3 kristalinin oda
sıcaklığındaki ortorombik yapısı şekil.1.2.(c)’ de veriliyor.
NaTaO3 kristali yüksek sıcaklıklarda paralektrik fazda bulunur. Oda
sıcaklığında ise ortorombik fazda olup uzay grubu Pbnm (No:62) dir. Sıcaklığın
düşürülmesiyle, yapıdaki simetri bozularak sırasıyla,
CTOO KKK →← →← →← 893835720 21
faz geçişlerine uğrar. NaTaO3 kristalinin oda sıcaklığındaki ortorombik yapısı
şekil.1.2.(d)’ de verilmektedir.
Faz geçişi sonucu kristallerin elektronik yapısı, optik ve dinamik özellikleri
değişir. Katıların elastik sabitleri, kristalin mekaniksel ve dinamiksel davranışları
arasında bağlantı kurar. Ayrıca katıdaki doğal kuvvetler ve malzemenin sertliği ve
kararlılığı hakkında önemli bilgiler verir. Elastik sabitlerinin teorik ve deneysel

1. GİRİŞ Şevket ŞİMŞEK
6
değerlerinin karşılaştırılması, kullanılan potansiyelin güvenirliğinin testi için
önemlidir. Bu yüzden hesaplanan elastik sabitlerinin doğruluğu mevcut metodun
doğruluğu için de bir ölçüttür. AgNbO3, AgTaO3, NaNbO3 ve NaTaO3 materyali
üzerine yapılan elektronik ve dinamik çalışmalar yetersiz olup bu özelliklerinin
belirlenmesi hem temel fizik açısından hem de bu malzemelerin teknolojik
uygulamaları açısından oldukça önemlidir.
Bu tez çalışmasındaki amacımız, oda sıcaklığındaki AgNbO3, AgTaO3,
NaNbO3 ve NaTaO3 kristallerin yapısal, elektronik band yapısı, toplam durum
yoğunlukları (DOS), parçalı durum yoğunluğu (PDOS) ve dinamik özellikleri
(elastik sabitler, Born efektif yükler, fonon dispersiyon eğrileri, fonon durum
yoğunluğu ve optik dielektrik sabiti) DFT kullanılarak LDA altında
pseudopotansiyel yöntemiyle hesaplamaktır.

2. ÖNCEKİ ÇALIŞMALAR Şevket ŞİMŞEK
7
2. ÖNCEKİ ÇALIŞMALAR
AgTaO3’ ün faz geçişleri serisi Kugel ve arkadaşları (1987) tarafından, 10
K’den 770 K’e kadar geniş bir sıcaklık aralığında incelenmişdir. Kugel ve
arkadaşlarına (1987) göre, yüksek sıcaklıkta Rombohedral-Monoklinik ve
Monoklinik-Tetragonal fazları bir arada bulunmaktadır. 667 K’ de Rombohedral
yapıdan monoklinik yapıya, 694 K’ de monoklinik yapıdan tetragonal yapıya ve 770
K’ de Tetragonal yapıdan Kübik yapıya geçişin olduğu gözlenmektedir. Dielektrik
ölçümler, gümüş elektrotları kullanılarak yaklaşık 2×2×1 mm3 boyutunda tek
kristaller üzerinde gerçekleştirilmişdir. Kendiliğinden polarizasyonun değerini 80 K
için yaklaşık 0,02 µM/cm2 olarak bulmuşlardır.
Eyi ve ark (2010), değiş-tokuş korelasyon etkileri için yerel yoğunluk
yaklaşımlı (LDA) yoğunluk fonksiyoneli teorisi (DFT) kullanarak NaTaO3
kristalinin kübik (Pm3m), tetragonal (P4/mbm) ve ortorombik (Pcmn) fazda yapısal
(örgü parametreleri ve atomik pozisyonlar), elektronik band yapısı, durum yoğunluğu
(DOS) ve optik özelliklerini incelemişlerdir. NaTaO3’ün optik ve tetragonal fazdaki
elektronik özellikleri teorik olarak ilk defa Eyi ve ark (2010), tarafından
araştırılmıştır. Eyi ve ark (2010), göre band yapısı, Brillouin bölgesindeki (R-Γ)
noktasında 2,183 eV (kübik fazda), (Z- Γ ) noktasında 2,230 eV (tetragonal fazda)
dolaylı ve (Γ - Γ) noktasında 2,231 eV (ortorombik fazda) direkt bir band aralığı
göstermektedir. NaTaO3 kristalleri için foton-enerjisine bağlı olarak dielektrik
fonksiyonları ve soğurma katsayısı, enerji kayıp fonksiyonu ve yansıtıcılık gibi optik
özellikleri scissor yaklaşımı altında hesaplanmışlardır. Valans elektronlarının etkin
sayısı her üç faz için hesaplanmışlardır.
AgNbO3’ deki yapısal faz geçişleri, nötron kırınımı ve X-ışınımı kullanarak
araştırılmışdır. 295 K’ de M1 (monoklinik) fazı, 423 K’ de M2 (monoklinik) fazı, 773
K’ de M3 (monoklinik) fazı, 645 K2’ de O2 (ortorombik) fazı, 733 K’ de T
(tetragonal) fazı ve 903 K’ de C (kübik) fazın olduğu gözlenmişdir. Bu çalışmada O1
fazı, bu fazların olduğu bölgedeki sıcaklığa yakın olduğu için gözlenmemişdir
(Sciau, Kania, Dhkil, and Ratuszna, 2004).

2. ÖNCEKİ ÇALIŞMALAR Şevket ŞİMŞEK
8
DFT kullanarak, piezoelektrik özellik gösteren perovskitlerde, A-konumunda
gümüş kullanımının fizibilitesini incelemek için AgNbO3 perovskitinin yerel yapısı
ve deformasyon örnekleri incelenmişdir. Gümüş atomlarının kısa kovalent Ag-O bağ
yaptığı ve bu yüzden oktahedral rotasyonların büyük B-O6 yokluğunda
ferroelektriksel olarak aktif oldukları bulunmuşdur. Yapılan hesaplamalar, 40 atomlu
AgNbO3 birim hücresinin düşük sıcaklıkta, saf gümüş niyobat’daki ferroelektrikliğin
varlığını kanıtlamıştır (Grinberg ve Rappe, 2003).
Ag(Ta, Nb)O3 tabanlı seramiklerde dielektrik geçirgenliğin sıcaklığa
bağımlılığı Valant ve ark (2001), tarafından araştırılmışdır. ATN tabanlı bileşimleri
katı hal reaksiyon metodu kullanarak sentezlemişlerdir. -20 ºC den +80 ºC sıcaklık
aralığında Ag(Nb0.5Ta0.5)O3, yaklaşık 1300 ppm/K olduğu aralıkta herhangi bir
noktada dielektrik geçirgenliğin sıcaklığa bağımlı maksimumu ile, yaklaşık 60 ºC’ de
geniş bir dielektrik geçirgenlik sergiler. Bu da ticari amaçlı uygulamalar için
yeterlidir (Valant ve ark, 2001).
AgTaO3-AgNbO3 karma seramiklerin yüksek frekans dielektrik spektraları
radyo dalga, mikro dalga, milimetre altı ve kızıl ötesi bölgelerde incelenmiştir.
Volkovt ve arkadaşlarına göre, AgTaO3-AgNbO3 seramiklerinin dielektrik
araştırmaları, 1 MHz den 3×1013 Hz frekans aralığında milimetre altı dalga
bölgesinde güçlü bir gevşeme modunun varlığını göstermektedir. Bu modun
sıcaklığa bağımlılığı, faz geçişi çevresindeki monoklinik M2 fazından M3 fazına
statik dielektrik geçirgenliğin geniş bir anormalliğini sergiliyor. Gevşeme frekansının
davranışı ve sıcaklığa karşı gücü düzenli-düzensiz yada yer değiştirmeli
ferroelektriklerde görülenden farklıdır. Bu sistemdeki dielektrik sabitine ana katkısını
teşkil eden böyle yüksek frekanslı bir dielektrik dağılımın oluşumu, Nb iyonlarının
varlığına bağlanıyor (Volkovt ve ark, 1995).
Kato ve ark (2002), AgTaO3 ve AgNbO3’ün band yapısı ve fotokatalistik
özelliklerinde Ag+ iyonlarının rollerini araştırmışlardır. Hesaplamaları CASTEP
programı içerisinde düzlem dalgaları baz alan yoğunluk fonksiyonu metodu ile
yapmışladır. Ag+ iyonlarının AgTaO3 ve AgNbO3’ de band aralıklarının azalmasına
neden olduğunu fakat uyarılmış enerjilerin yerel olmayan etkilerinin etkilemediğini
gözlemişlerdir. Kato ve arkadaşlarına (2002) göre, Ag 4d orbitalleri O 2p

2. ÖNCEKİ ÇALIŞMALAR Şevket ŞİMŞEK
9
orbitallerinden daha negatif seviyelerde valans bandları yapıyorlar. AgTaO3’ün band
aralığını 3.4 eV ve AgNbO3’ün band aralığını 2.8 eV olarak bulmuşlardır ( Kato,
Kobayashi ve Kudo, 2002).
AgTaO3 ve AgNbO3 (ATN) tozları, karma oksitli metot ile hazırlanarak
mikrodalga dielektrik özellikleri incelenmiştir. Bu çalışmanın amacı Ag(Ta,Nb)O3
kristallerini ayarlanabilir yüksek frekans araçlarda kullanmaktır. ATN hacimsel
seramiklerin ve ekrana basılmış kalın filmlerin geçirgenlik, kayıplar ve tunabilty’ si -
150 ºC den +120 ºC bir sıcaklık aralığında 1 MHz’ den küçük bir frekansta
karakterize edilmişdir (Zimmermann ve ark, 2004).
Ag1-xAxNbO3 ve AgNb1-xTaxO3 katı çözeltilerinin dielektrik özelliklerinin
sıcaklığa bağımlılığı 120 K’den 700 K’e kadar geniş bir sıcaklık aralığında
incelenmişdir. Katı çözelti seramiklerinin hepsi katıhal reaksiyon metodu ile elde
edilmişdir. Silver niobat ve katı çözeltilerinin dielektrik çalışmaları, dielektrik
dağılımın ferroelektrik M1 fazında meydana geldiğini göstermişdir. Kania’a (2000)
göre, dielektrik geçirgenliğin sıcaklığa bağımlılığı iki alt sistemin bir arada
bulunduğunu gösteriyor. İlk düzenli alt sistem yüksek sıcaklıklarda klasik
paraelektrik davranışlar ve keskin M3-O1 faz geçişlerinin oluşumundan sorumludur.
İkinci düzensiz alt sistem daha düşük sıcaklıklarda yüksek ve alçak frekans gevşeme
katkısının ortaya çıkmasından sorumludur (Kania, 2000).
VASP programı kullanarak AgNbO3 ve NaNbO3 deki fonon modlarının
karşılaştırmalı analizleri yapılmışdır. Bu kristallerin örgü dinamikleri ilk ilke
yöntemi baz alınarak hesaplanmışdır. Sodyum ve gümüş niyobat daki kararlı
olmayan modlarda oksijen oktahedra çarpıklığı ve ferroelektrik atomik yer
değiştirmelerin analizleri yapılmıştır. Kristal örgüdeki sodyum ve gümüşün atomik
konumlarının kararsızlıkları ile ilişkilendirilen ferroelektrik modların varlığı
belirlenmişdir. İlk prensip hesaplamalarına göre, (T=0) temel durumda (rombohedral
ferroelektrik fazda) hem gümüş hem de sodyum niyobat, ferroelektrik yer
değiştirmeler ve oksijen oktahedra donma eğilimi (frozen tilting) ile karakterize
edilmiştir. Bu ikisi arasındaki fark oksijen oktahedra eğilim modlarının, gümüş
niyobatın Brillouin bölgesinde ki M noktasına karşılık gelirken sodyum niyobatta
Birillouin bölgesinde ki R noktasına karşılık geliyor. Bu da gümüş atomlarıyla

2. ÖNCEKİ ÇALIŞMALAR Şevket ŞİMŞEK
10
kıyaslandığında sodyum atomlarının biraz daha küçük olan boyutu ile ilişkilendirilir
(Prosandeev, 2005).
AgNbO3’ün kristal yapısının sıcaklıkla değişimi oda sıcaklığndan 850 K’e
kadar geniş bir sıcaklık aralığında Ratuszna ve arkadaşları (2002) tarafından
incelenmiştir. Deformasyon tiplerini ve Pseudo-kübik perovskit hücrenin çeşitliliğini
ana kırınım çizgilerinin yarılmasından ve süper örğü (superlattice) piklerinden
belirlemişlerdir. Ratuszna ve arkadaşlarına (2002) göre, 873 K üzerinde AgNbO3
kristali kübik yapıya sahipitir. 873 K’de tetragonal ve 661 K altında ortorombik (O1)
yapıdadır. 629 K’de monoklinik (M3) fazı ve yaklaşık 300K’de de monoklinik (M1)
fazı meydana gelmektededir. Fakat bu çalışmada literatürde rapor edilen monoklinik
(M2) ve ortorombik (O2) fazlarını gözleyememişlerdir ( Ratuszna ve ark, 2002).
Desheng Fu ve ark(2009), (Ag1-xKx)NbO3 (AKN) katı çözeltilerindeki faz
değişimini x-ışını kırınımı, dielektrik ve ferroelektrik ölçümlerle araştırmışlarıdır.
Desheng Fu ve arkadaşlarına göre oada sıcaklığında ≈ 0,07, ≈ 0,20 ve ≈ 0,8 olmak üzere üç tip faz sınırı vardır. < olduğunda AKN, zayıf bir
ferroelektriklikle AgNbO3 –tipi ortorombik fazdan güçlü bir ferroelektrikle yeni bir
ortorombik faza dönüşüyor. Bu ferroelektrik faz < < için kararlıdır. Bu
ferroelektrik faz aynı zamanda 525 K bileşik-bağımsız Cruie noktası ve çok büyük
bir polarizasyon ( = 0,1 seramik numunesi için = 20,5 ⁄ ) gösteriyor. < olduğunda, AKN, KNbO3-tipi ortorombik yapı gösteriyor ve saf KNbO3’a
benzer faz geçişlerine uğramaktadır. Curie noktası = 0,80 için 633 K’den =1,00 için 696 K’e ile lineer olarak artıyor.
Desheng Fu ve ark (2011), (1 − ) − katı
çözeltilerindeki polarizasyon durumlarındaki değişimi ve elektromekaniksel
çiftlenimi dielektrik ve starin histeris eğrisi tekniği kullanarak gözlemlemişlerdir.
Polarizasyonun AgNbO3’daki ferrielektrik düzenden NaNbO3’daki ferroelektrik
düzene doğru değiştiğini gözlemlemişlerdir. Bu iki durum arasındaki geçit yeri ≈ 0,8 ile katı çözeltide meydana geliyor. Bu katı çözeltilerin seramiklerinde ~ % 20 bir strain seviyesi mevcuttur.
Desheng Fu ve ark(2007), AgNbO3’ün polorizasyon ve elektromekaniksel
tepkisi üzerine çalışmalar yapmışlardır. AgNbO3’ün poli kristallerinin (polycrystals)

2. ÖNCEKİ ÇALIŞMALAR Şevket ŞİMŞEK
11
büyük bir polarizasyona sahipi olduğunu ve polarizasyonun 52 ⁄ değerine
kadar çıkabildiğini rapor etmişlerdir. Deneyler aynı zamanda AgNbO3’daki büyük
içsel atom deformasyonlarının elektrik alanla çiftlendiğini göstermiştir ve AgNbO3’
de yüksek piezoelektrik performansın olduğu görülmüştür.
David Arney ve ark (2010), AgNbO3 parçacıklarının eriyik-tuz akı
sentezlerini 1-10 saat reaksiyon zamanı için 900 oC ye kadar ısıtıma ve tepkime
molar oranına 1:1, 2:1 ve 3:1 akı kullanarak Na2SO4’deki akıda gerçekleştirmişlerdir.
Dikdörtgensel biçimli parçacıklar 100 ’ den 5000 boyutunda ve toplam
yüzey alanı 0,16 den 0,65 aralığında homojen mikroyapılarla yüksek
saflıkta elde edilmiştir. David Arney ve arkadaşları tarafından en küçük parçacık
boyutu dağılımı ve en yüksek yüzey alanı, akının en büyük miktarı (3:1 oranı) ve en
kısa reaksiyon zamanı (1 saat) için elde edilmiştir.
M olarak adlandırılan AgNbO3’ün poli kristallerinin yapısal farklılıklar
kombine edilmiş yüksek çözünürlüklü X-ışını kırınımı, nötron toplam saçılması,
elektron kırınımı ve X-ışınımı soğurma ince-yapı ölçümleri kullanılarak Igor Levin
ve ark (2009) tarafından analiz edilmiştir. Bu poli kristallerin hepsi simetrisi
ile kristalleşmişlerdir. Igor Levin ve arkadaşlarının çalışmalarında, AgNbO3’deki Nb
katyonları, KNbO3’dekine benzer Nb-Nb-Nb zinciri boyunca ilişkili olan lokal
merkez dışı yer değiştirmeler göstermişdir. Yer değiştirmeler Nb katyonlarının
ortalama, ideal sabit kordinat konumlarında bağlı olduğu yüksek sıcaklık AgNbO3
poli kristalerinde bile olduğu görülüyor.
Suchanicz ve ark (2009), AgTaO3 tek kristallerinin geçirgenliklerinin
sıcaklığa bağımlılığını, 0-1000 bar’lık basınç aralığında çalışmışlardır. AgTaO3
dielektrik özelliklerinin uyğulanan basınca hassas olduğu bulunmuştur. Bu durum faz
geçişindeki değişikliği ve geçirgenlikteki (permittivity) artışı içeriyor. Basınç aynı
zamanda kristali daha etkin anormal geçirgen yapıyor. Bu etkiler, tek eksenli
(uniaxial) basınç etkisi altında, elektriksel iletkenlikdeki değişmeye, kusurların
yoğunluğundaki değişmeye, iç iyonik uzaklıkların değişmesine ve domein
yapısındaki değişmelere neden olabilmektedir.
Hwee Ping Soon ve ark (2010), AgTaO3 seramiklerinin hem dielektrik hemde
mikro-Raman ölçümlerini araştırmışlardır. 2,2 K ve 694 K arasında her hangi bir

2. ÖNCEKİ ÇALIŞMALAR Şevket ŞİMŞEK
12
dielektrik anormallik gözlenmemişdir. Daha önce önerilmiş olan 170 K’ de 3 uzay
grubunda 3 uzay grubuna bir faz geçişinin olduğu bu çalışmayı desteklemiyordu
fakat 3 simetrili kuantum paraelektrik temel durum ile uyum içindedir. Bununla
birlikte 667 K’ de rombohedral temel durumdan daha yüksek sıcaklıktaki monoklinik
duruma bir faz geçişinin olduğunu gözlemişleridir.
Matjaz Valant ve ark (2007), AgNbO3 ve AgTaO3’ün oluşmasını ve
ayrışmasını içeren ara işlemleri analiz etmek için termogravimetrik yöntem
(thermogravimetric method) kullanmışlardır. Oluşumun kinetiğini kontrol eden kritik
parametreler oksijen taşınmasıyla ilişkilidir. Nb2O5 kristal yapısı, başlangıç
karışımında var olan Ag2O’nın ayrışması boyunca değişiklik geçiren moleküler
oksijeni tutma kapasitesine sahiptir. Perovskit fazın oluşumu eş zamanlı olarak O2,
Ag ve Nb2O5/Ta2O5 gibi üç türün reaksiyonunu içermektedir.
Matthias tarafından ilk olarak NaTaO3 ün kristalin yaklaşık olarak 480 oC de
bir Curie noktasının altında ferroelektrik özelliğe sahip olduğu rapor edilmiştir (Jona
ve ark, 1962). Fakat daha sonra yapılan optik ölçümlerde oda sıcaklığındaki NaTaO3
kristalinin yapısı Pcmn (merkezi simetrik) uzay grubuna ait olduğu görülmüştür.
Uzay grubundan dolayı NaTaO3 oda sıcaklığında ferroelektrik değildir.
Mishra ve ark (2007), 12 K’ den 350 K sıcaklık aralığında NaNbO3 kristalinin
düşük sıcaklıklı faz geçişinin kompleks serisini anlamak için toz numuneleri
kullanarak nötron toz kırınmı çalışması yapmışladır. Kırınım verilerinin detaylı
analizleri, ferroelektriklikten antiferroelektrikliğe geçişin ısıtma uyğulandığında 245
K’ de oluşurken, tersine yaklaşık 73 K soğutulduğunda anti ferroelektriklikten
ferroelektrikliğe faz geçişinin olduğunu göstermiştir.
Cross (1956) tarafından yapılan ilk optik çalışmada, sıcaklığın artmasıyla
NaTaO3 kristali, ortorombik yapıdan başka bir ortorombik yapıya, sonra tetragonal
ve daha sonra da kübik yapıya yapısal faz geçişi yaptığını ileri sürmüştür. Smolenski
tarafından NaTaO3 kristali üzerinde yapılan çalışmalarda -180 0C ile 510 0C arasında
ısıl artışın dielektrik sabitinde bir anormallik olmadığı gözlemlenmiştir. Oda
sıcaklığında faz simetrisinin ortorombik olduğu rapor edilmiştir. Kay ve Miles
ortorombik fazda c örgü sabitinin kübik hücrenin örgü sabitinin iki katı olduğunu
bulmuştur. Ortorombik fazda örgü parametreleri a = 5,513 Å , b = 5,494 Å, c = 2 ×

2. ÖNCEKİ ÇALIŞMALAR Şevket ŞİMŞEK
13
3,875 Å değerlerine sahiptir. Aynı zamanda oda sıcaklığındaki c örgü sabiti sanki
kübik yapıdaki örgü parametresinin 4 katıymış gibi gözlendi.
Jonstone ve arkadaşları (2010), NaNbO3’ün polar fazını sol-gel tekniği
kullanarak sentezlemişlerdir. Bu fazın detaylı bir şekilde karakterizasyonu, ikinci
harmonik oluşumu ölçümleri ve yoğunluk fonksiyonel teorisi hesaplamaları ile
desteklenen 23Na çeşitli kuantum sihirli açılı savurma (magic-angle spinning, MAN),
nükleer manyetik rezonons (NMR) ve yüksek çözünürlüklü toz kırınımı (X-ışınımı
ve nötron) kullanılarak yapılmıştır. Aynı zamanda NaNbO3 numuneleri geleneksel
katı hal yöntemi kullanılarak sentezlenmiş ve NaNbO3’ün iki farklı poli kristallerinin
bir karışımını içermesi için rutin olarak gözlemlenmiştir. Karen ve arkadaşlarının
çalışmaları, NaNbO3’ün poli kristallerinin kristalografik olarak iki farklı Na konumu
içerdiğini göstermiştir.
Ahtee ve ark (1980), nötron toz kırınım yöntemiyle NaTaO3 yapısını
belirlemişlerdir. Oda sıcaklığında NaTaO3 ortorombik (Pcmn, a = 5,4842 Å, b =
7,7952Å, c = 5,5213 Å) yapıda olduğu belirlenmiştir. Sıcaklığın artışıyla 803 K’de
kübik-ortorombik (Bmmb, a = 7,8453 Å, b = 7,8541 Å, c = 7,8633 Å) yapıda, daha
sonra 893 K ’de tetragonal (P4/mbm, a = b = 5,5552 Å, c = 3,9338 Å) yapıya
geçtiğini gözlemlemişlerdir.
Hwee Ping Soon ve ark (2009), kuantum paraelektrik fazdaki AgTaO3
içerisine +Li iyonları yerleştirerek ferroelektriklik uyarılmıştır. Li katkılı AgTaO3’ ün
geçiş sıcaklığı lityum iyomlarının artışı ile artmakdadır. Çözünürlük sınırına yakın
olan AgTaO3’ün içerisine % 12 mol +Li iyonlarının yerleştrilmesiyle ve 258 ’
de ferroelektriklik harekete geçirilmiştir.
Kennedy ve ark (1999), nötron toz kırınımı yöntemini kullanarak oda
sıcaklığından 933 K kadar NaTaO3 kristalinde yapısal faz geçişini incelemişlerdir.
Bu sıcaklık aralığında üç faz geçişinin oluşumunu farklı ısıl analiz, toz kırınım
tekniği ve optik mikroskop kullanarak belirlemişlerdir. Yaptıkları bu çalışmada,
NaTaO3 oda sıcaklığında ortorombik yapıdan (Pcmn, a = 5,4768 Å, b = 5,5212 Å, c
= 7,7890 Å) 700 K civarında ortorombik (Cmcm, a = 7,83372 Å, b = ,8485 Å, c =
7,8552 Å ) yapıya, sonra 835 K’de tetragonal (P4/mbm, a = b = 5,503 Å, c = 3,9335
Å ), daha sonra 890 K üzerindeki sıcaklıkta kübik (Pm 3m, a = b = c = 3,9313 Å)

2. ÖNCEKİ ÇALIŞMALAR Şevket ŞİMŞEK
14
yapıya dönüştüğünü gözlemlediler. Bu çalışmadaki faz geçişleri daha sonra Ahtee ve
Darlington tarafından NaTaO3 üzerine yaptıkları çalışmalarla doğrulandılar.
Minseok Choi ve ark (2008), NaTaO3 deki örgü boşlukları, antisit (antisite)
kusurlar ve lantan atomunun katkılarının elektronik band yapısını ve oluşum
enerjilerini yoğunluk fonksiyonel teoriyi baz alan temel-ilke yöntemi kullanarak
araştırmışlardır.
Reznitchenko ve ark (2001), polarize olmuş NaNbO3 seramiklerinin farklı
bölümleri üzerinde piezoelektrik ve dielektrik ölçümler gerçekleştirmişlerdir.
Polarizasyonun bir sonucu olarak uzun zamandır var olan, yarı kararlı ferroelektrik
fazın ortaya çıktığı belirlendi. Bu fazın varlığı, dielektrik geçirgenliğin piezo
rezonans dağılımı ile beraber bir piezoelektrik etki olarak kendini gösteriyor.
NaTaO3 seramiklerinin d33 piezoelektrik katsayılarının büyüklükleri ve sıcaklığa
bağımlılıkları ilk kez ölçülmüştür.
Kübik NaTaO3’ün elektron yapısı Wang ve ark (2001) tarafından, lineer
genişletilmiş düzlem dalga metodu (Linearized Augmented Plane Wave Method)
kullanılarak araştırılmışdır. Değiş-tokuş etkisi için genelleştirilmiş gradyent
yaklaşımı (Generalized Gradient Approximation, GGA) kullanılmıştır.
Hesaplamalarını bu metotları içeren WİEN97 simulasyon programı kullanarak
yapmışlardır. Atomlar arasındaki etkileşimi anlayabilmek için durum yoğunluğu,
bant yapısı ve yük yoğunluğunun dağılımını hesaplamışlardır. Durum yoğunluğunun
analizinden Ta d durumu ile O p durumu arasında önemli bir hibritleşme olduğunu
gördüler. Bu durum, yük yoğunluk dağılımı ve bant yapısı analizlerinin tutarlı
olduğunu göstermiştir. NaTaO3 kristalde iki tür elektronik etkileşim vardır: Ta ile O
arasındaki etkileşim kovalent ve Na ile TaO3 arasındaki etkileşim iyoniktir.
Samuel ve ark (2005), NaTaO3 ve NaNbO3’ ün tek fazlarını saf ve çok küçük
hazırlamak için basit bir ortak çökeltme yöntemi kullamışlardır. Amonyum karbonat
ve amonyum hidroksidin alkolik çözeltisini ayrı ayrı karbonat ve hidroksit gibi temel
şartlar altında Na+, Nb+5 yada Ta+5 katyonlarını hazırlamak için kullanmışlarıdır. 700 oC’ ye ısıtıldığında bu uyğulamalar ayrı ayrı ürünler oluşturmaktadır. Aynı zamanda
kıyaslama amacıyla, NaTaO3 ve NaNbO3 tozları geleneksel katı hal metodu ile de
hazırlanmışdır. Fazların saflığı ve örğü parametreleri X-ışını kırınımı tozu ile

2. ÖNCEKİ ÇALIŞMALAR Şevket ŞİMŞEK
15
araştırılmışdır. Parçacık boyutları ve biçimleri taramalı elektron mikroskop (SEM)
ile çalışılmıştır.
Kato ve ark (2002), ortorombik (Pcmn) yapısındaki NaTaO3 üzerinde yaptığı
optik soğurma ölçümleri sonucunda band aralığını 4,0 eV olarak bulmuşlardır. Aynı
çalışmada DFT hesaplama metodunu kullanan CASTEP programını kullanarak
NaTaO3 ‘ün elektronik band yapısını ve durum yoğunluğunu hesaplamışlardır.
He ve ark (2004), de düşük sıcaklıkta nanoboyutta toz NaTaO3 kübik
morfolojide 120 0C de 12 saatte hidrotermal yöntemiyle sentezlediler. Nanoboyuttaki
NaTaO3’ün band aralığı 3,96 eV olarak buldular.
AgNbO3’ deki Raman çizgilerini yok olması ya da görünmesinin yapısal
bağlantılı oldukları gözlenmiş ve özellikle alçak sıcaklıkta X-ışını ölçümleri ile
belirlenemeyen ilave bir yapısal faz geçişin olduğu belirlenmişdir ( Kugel ve ark,
1987).
Xu ve ark (2005) de 600 0C sıcaklıkta toz NaTaO3’ü kimyasal yöntemle
sentezlediler. Toz NaTaO3’ün özellikleri XRD, SEM ve ultra viola spektrumununda
karakterize edildi. Kübik toz NaTaO3’ün band aralığını ~4,0 eV olarak buldular.
Lui ve ark (2007), perovskite yapıdaki toz alkali tantan ATaO3 ve alkali
niyobyum ANbO3 (A=Na ve K) hidrotermal metoduyla sentezlediler. Niyobyumlu
bileşiklerin tahmin edilen band aralığını, tantallı bileşiklerden küçük olduğunu
buldular. Kübik yapıdaki NaTaO3’ün band aralığını 3,96 eV olarak buldular.
Guoqiang Li ve ark (2009), AgNbO3’ün fotokataliz parçacıklarının yüzey
fotoelektrik özelliklerini, yüzey foto voltaj spektroskopi (SPS) ve elektrik alan-
uyarılmış yüzey foto voltaj spektroskopi (EFISPS) ile araştırmışlarıdr. SPS’ de ayrı
ayrı iki tepki piki görünmüş ve UV ışık bölgelerinde ortaya çıkmıştır. EFISPS
göstermiştir ki, görünür ışık bölgesindeki pik, ayrı ayrı pozitif ve negatif dış bias
kullanılarak artırılır ve azaltılabilir
Kübik (Pm3m) ve ortorombik (Pbnm ve Pcmn) NaTaO3’ün elektronik
yapısını ve optik soğurma özellikleri Li ve ark (2007) tarafından, DFT kullanılarak
incelendi. Elektronik band yapısı hesaplamalarında tam-potansiyel lineerleştirilmiş
muffin tin orbital metodu (full-potential linear-muffin-tin-orbital, FP-LMTO)
kullanarak değiş-tokuş etkisi için GGA ‘yı hesaba katmışlardır. Yaptıkları çalışmada

2. ÖNCEKİ ÇALIŞMALAR Şevket ŞİMŞEK
16
6x6x6 Monkhorst-Pack örgü ağı kullanmışlardır. NaTaO3 kübik yapısında (R-Γ)
noktasında dolaylı bant aralıklı (band aralığı 1,7 eV) yarıiletken olmasına rağmen
ortorombik yapıda (X) noktasında (band aralığı 2,1 eV) doğrudan bant aralıklı
yarıiletkendir. Doğrudan bant aralık özelliğinin ortorombik NaTaO3 için uzay
grubundan bağımsız olduğunu ileri sürdüler. Teorik olarak hesapladıkları soğurma
spektrumunun deneysel sonuçlarla uyumlu olduğunu söylemektedirler. Band
aralığının azalışını ve soğurma kenarının kaymasını ortorombik yapıdan kübik
yapıya geçmesinden kaynaklandığını buldular. NaTaO3 üzerinde yapılan çalışmaların
büyük bir kısmı kristal yapı ve kristal sentezleme üzerinde yoğunlaşmıştır.
Masatomo Yashima ve ark (2010) tarafından, ferroelektrik AgNbO3
kristalinin yapısı araştırılmışdır. AgNbO3 poli kristalleri O2 atmosferinde 6 saat için
1050 oC’ de katı hal reaksiyon metodu ile hazırlanmıştır. Kimyasal bileşim endüktif
kuplajlı plazma (ICP) analizleri ile doğrulanmıştır. AgNbO3 kristalinin nötron toz
kırınımı dataları 1,82646 Å nötron dalga boyunda 150 dedektör sistem HERMES
kullanarak 23 oC de ölçülmüşdür. Siklotron toz kırınımı ölçümleri 25,1 oC’ de
yapılmıştır. Daha önce Pbcm olarak önerilen AgNbO3 kristalin simetri grubu, bu
çalışmada Pmc21 olarak bulunmuştur. Birim hücre parametreleri a=15,647733 Å,
b=5,551991 Å, c=5,609081 Å ve açıları = = = 90 olarak bulumuştur.
Wang ve ark (2001), ilk prensip yöntemiyle kübik NaTaO3 kristalinin
elektronik, yük yoğunluğu özelliklerini, Li ve ark.-2007 ise kübik ve ortorombik
yapıdaki NaTaO3 ‘ün elektronik band yapısı ve optik soğurma özelliklerini
araştırmışlardır. Bu çalışmaların dışında elektronik band yapısı ve soğurma
özellikleri üzerinde başka teorik çalışma yoktur. Bu açıdan bakıldığında NaTaO3’ ün
kübik, tetragonal ve ortorombik yapının teorik olarak örgü parametresi ve atomik
pozisyonunun optimizasyonu, elektronik band yapısı, yük yoğunluğu ve optik
özelliklerinin çalışılması kristal içindeki atomların etkileşim doğasının anlaşılması
için çok önemlidir.

3. MATERYAL VE METOD Şevket ŞİMŞEK
17
3. MATERYAL VE METOD
3.1. Tek Atomlu Örgü Titreşimleri
İlkel hücresinde bir atom bulunan bir kristalin elastik titreşimlerini göz önüne
alalım. Amacımız elastik dalgaların frekansını, dalgayı belirleyen dalga vektörü ve
ortamın elastik sabitleri cinsinden bulmaktır.
Matematik çözümün en kolay olduğu durum kübik kristallerde [100], [110]
ve [111] ilerleme yönleridir. Bu yönler sırasıyla küp kenarı, yüzey köşegeni ve cisim
köşegeni yönüdür. Bir dalga, bu yönlerden birinde ilerlediğinde bir düzlemdeki
atomlar hep birlikte ve aynı fazda, ya dalga yönünde ya da dalga yönüne dik olarak
yer değiştirirler. s ile tanımlanan bir düzlemdeki tüm atomların denge konumlarından
yer değiştirmelerini tek bir us koordinatıyla gösterebiliriz. Bu durumda problem tek
boyutlu olur. Her dalga vektörü için biri boyuna titreşim ve diğer ikisi enine titreşim
olmak üzere üç ayrı titreşim kipi vardır. Şekil 3.1 ve şekil 3.2’de enine ve boyuna bir
dalga sırasında atom düzlemlerinin yer değiştirmesi gösterilmektedir. Şekil 3.1 ve
şekil 3.2’deki kesikli çizğiler atomların denğe konumlarını, tam çizgiler ise enine ve
boyuna bir dalga sırasında yer değiştiren atom düzlemlerini göstermektedir. u
koordinatı düzlemlerin yer değiştirme miktarını belirtmektedir.
Şekil 3.1. Boyuna bir dalga sırasında atom düzlemlerinin yer değiştirmesi (KITTEL)

3. MATERYAL VE METOD Şevket ŞİMŞEK
18
Şekil 3.2. Enine bir dalga sırasında atom düzlemlerinin yer değiştirmesi (KITTEL)
Kristalin elastik davranışının uygulanan kuvvetin lineer bir fonksiyonu
olduğunu varsayalım. Buna göre elastik enerji, kristaldeki iki noktanın bağıl yer
değiştirmesinin karesiyle orantılıdır. Enerjideki lineer terimler sıfır olur. Küçük
elastik deformasyonlar için kübik ya da daha yukarı dereceden terimler ihmal
edilebilirler, ancak yüksek sıcaklıklarda bu terimler önemli bir rol oynarlar.
Bu varsayımlara göre, s+p düzleminin yer değiştirmesi nedeniyle s düzlemine
etkiyen kuvvet us+p-us yer değiştirme farkına eşit olacaktır. Bu ilk yaklaşımda sadece
en yakın komşuları göz önüne alacak olursak p=±1 olur ve s düzlemi üzerindeki
toplam kuvvet s±1 düzlemlerinden kaynaklanır.
Fs=C us+1-us +C us-1-us (3.1)
Yer değiştirmelere lineer bağlı olan bu ifade Hook yasasıdır.
C sabiti en yakın komşu düzlemler arasındaki kuvvet sabiti olup enine ve
boyuna dalgalar için farklıdır. Buradan itibaren C katsayısını düzlemin bir atomu için
tanımlanmış olduğunu kabul edersek Fs kuvveti s düzlemindeki iki atoma etkiyen
kuvvet olur. s düzleminin hareket denklemini yazarsak,

3. MATERYAL VE METOD Şevket ŞİMŞEK
19
)2uuC(udt
udM s1s1s2s
2
−+= −+ (3.2)
Burada M bir atomun kütlesidir. Bu denklemin zamana ɯʂ ɺ (− ɳ ɾ) şeklinde bağımlı
çözümlerini arayalım. Buna göre ɮᴀɿ ɀ ɮɾ ᴀ = ᴀح ɿ ɀ olur ve denklem (3.2)
-Mω2us=C(us+1+us-1-2us) (3.3)
olup u yer değiştirmeleri için bir farklar denklemi yapısındadır. Bu denklemin
çözümleri ilerleyen dalga karakterinde olur.
us±1=u exp isKa exp(±iKa) (3.4)
Burada a düzlemler arası uzaklık ve K dalga vektörüdür. a için alınacak değerler K’
nın yönüne bağlı olacaktır. Bu ifade (3.3) denkleminde kullanılırsa
-ω2Muexp isKa = Cu{exp i s+ 1 Ka + exp i s-1 Ka − 2 exp(ɳɽ ɛ ɫ )} (3.5)
yazılıp iki taraf ɿ ɯʂ ɺ (ɳɽ ɛ ɫ ) ile sadeleştirilirse
ᴀɝ = − ɒ [ɯʂ ɺ ɳɛ ɫ + ɯʂ ɺ − ɳɛ ɫ − 2] (3.6)
olur.
2cosKa= exp iKa +exp(-iKa) (3.7)
özdeşliğini kullanarak ω ile K arasındaki dispersiyon bağıntısı aşağıdaki gibi
bulunur.
ω2= 2C Mح (1-cosKa) (3.8)
3. MATERYAL VE METOD Şevket ŞİMŞEK
20
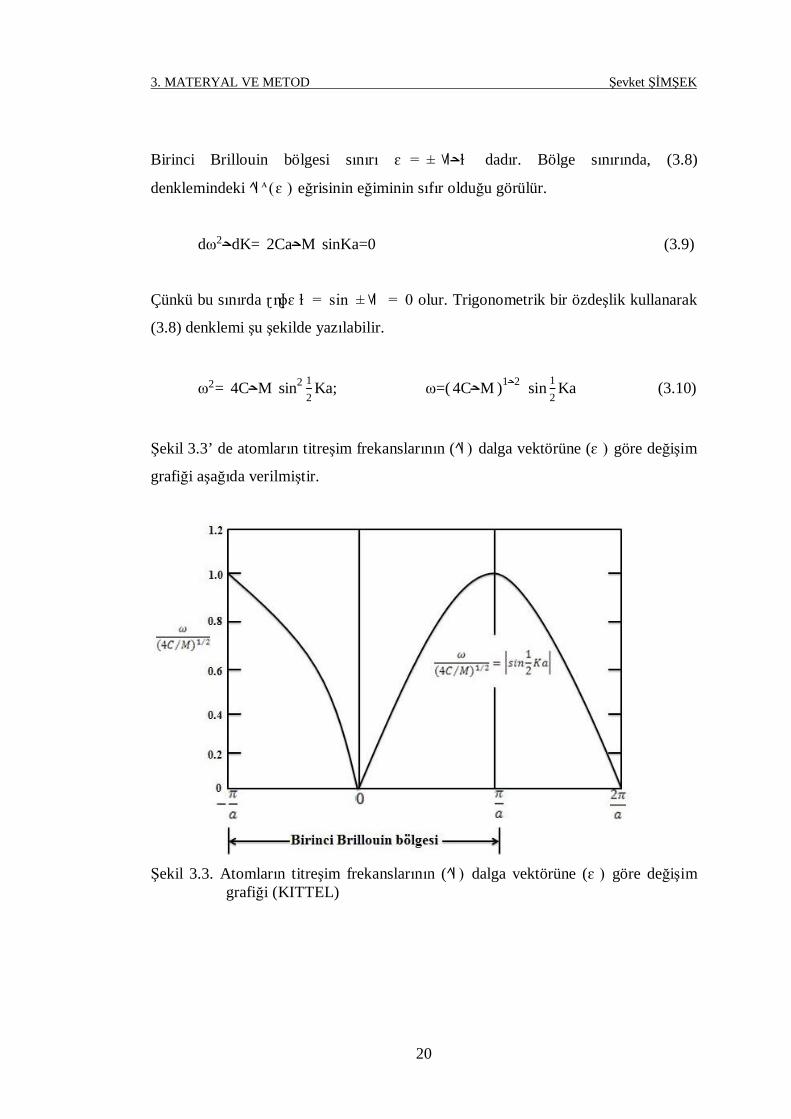
Birinci Brillouin bölgesi sınırı ɛ = ± ɫح dadır. Bölge sınırında, (3.8)
denklemindeki ᴀ (ɛ ) eğrisinin eğiminin sıfır olduğu görülür.
dω2 dK= 2Ca Mح sinKa=0ح (3.9)
Çünkü bu sınırda ɽ ɳɸɛ ɫ = sin ± = 0 olur. Trigonometrik bir özdeşlik kullanarak
(3.8) denklemi şu şekilde yazılabilir.
ω2= 4C Mح sin2 12
Ka; ω=( 4C Mح )1 ح2 sin 12
Ka (3.10)
Şekil 3.3’ de atomların titreşim frekanslarının ( ) dalga vektörüne (ɛ ) göre değişim
grafiği aşağıda verilmiştir.
Şekil 3.3. Atomların titreşim frekanslarının ( ) dalga vektörüne (ɛ ) göre değişim
grafiği (KITTEL)

3. MATERYAL VE METOD Şevket ŞİMŞEK
21
3.2. İki Atomlu Örgü Titreşimleri
Hücre bazında iki atom bulunan kristallerde fonon dispersiyon bağıntısı yeni
özellikler gösterir. Verilen bir yöndeki her titreşim kipinde ω(K) bağıntısı, biri
akustik diğeri optik denilen, iki dala ayrılır. Boyuna akustik (LA) veya enine akustik
(TA), boyuna optik (LO) veya enine optik (TO) denilen şekil 3.4’deki gibi titreşim
kipleri oluşur. Şekil 3.4’de optik ve akustik fonon dalları için, frekansın K=0 ve
K=Kmax= ∕a daki limit değerleri gösterilmiştir.
Şekil 3.4. İki atomlu doğrusal örgünün dispersiyon bağıntısında optik ve akustik
dallar (KITTEL)
Kübik bir kristalde şekil 3.5’deki gibi M1 kütleli atomların bir düzlemde, M2
kütleli atomların komşu düzlemde yer aldığını düşünelim. Kütlelerin farklı olması
önemli değildir; hücre bazında iki atom, eş değer örgü noktalarında iseler, ya kuvvet
sabiti yada kütlelerinin farklı olması gerekir. Ele alınan bu örgü düzlemlerine dik
olan yönde örgünün tekrarlandığı uzaklığa a diyelim. Sadece tek tip atomların yer
aldığı bir simetri düzlemi doğrultusunda ilerleyen bir dalga göz önüne alalım.

3. MATERYAL VE METOD Şevket ŞİMŞEK
22
Şekil 3.5. Kütleleri M1 ve M2 olan ve düzlemler arası kuvvet sabiti C olan iki atomlu kristal yapısı (KITTEL)
Hareket denklemlerini yazarken her düzlemin sadece en yakın komşu
düzlemle etkileştiğini ve bu düzlem çiftleri arasında kuvvet sabitlerinin aynı
olduğunu varsayarız.
)2uvC(vdt
udM s1ss2s
2
1 −+= −
)2vuC(udt
vdM ss1s2s
2
2 −+= + (3.11)
İlerleyen dalga yapısında bir çözüm için, ardışık düzlemlerde farklı u,v genlikleri
olur.
us=u exp isKa exp -iωt , vs=v exp isKa exp -iωt (3.12)
(3.12) denklemi (3.11) denkleminde kullanılırsa
-ω2M1u=Cv 1+ exp -iKa -2Cu
-ω2M2v=Cu 1+ exp iKa -2Cv (3.13)

3. MATERYAL VE METOD Şevket ŞİMŞEK
23
olur. Bu homojen denklem sisteminin çözümünün olması için, u,v bilinmeyenlerinin
katsayılar determinantı sıfır olmalıdır.
2C-M1ω2 -C[1+exp(-iKa)]-C[1+exp(iKa)] 2C-M2ω2
=0 (3.14)
veya
M1M2ω4-2C M1+M2 ω2+2C2 1-cosKa =0 (3.15)
Bu denklemin ᴀ için tam çözümü vardır; ancak, Ka≪1 limit durumu ve Ka=±a
bölge sınırı durumlarını incelemek daha yararlı olacaktır. Küçük Ka değerleri için
ɭ ɹ ɽ ɛ ɫ ≈ 1 − ϸᴀɛ ᴀ ɫ ᴀ yaklaşık ifadesi alınırsa iki kök bulunur.
ω2≈2C( 1M1
+ 1M2
) (optik dal) (3.16)
ω2≈12C
M1+M2K2a2 (akustik dal) (3.17)
Birinci Brillouin bölgesi - π aح ≤K≤ π aح aralığında olup a örgünün kendisini
tekrarladığı uzaklıktır. ɛ Ⱥ ͛ = ± ɫح olduğunda kökler
ω2= 2C M1; ω2= 2C M2ح ح (3.18)
olur. M1>M2 için ’ nın K’ ya bağımlılığı şekil 3.4’ de gösterilmiştir.
Enine akustik (TA) ve enine optik (TO) dallarda parçacıkların yer
değiştirmeleri şekil 3.6’ da gösteriliyor. Optik dallarda ɛ = 0 değeri için denklem
(3.16) denklem (3.13)’de kullanılırsa,

3. MATERYAL VE METOD Şevket ŞİMŞEK
24
1
2
MM
vu
−= (3.19)
olur. Farklı türdeki atomlar zıt fazda titreşirler, ancak kütle merkezi hareketsizdir.
Parçacıklar zıt elektrik yüklü ise, şekil 3.6’da olduğu gibi, bu tür hareketi bir ışık
dalgasının elektrik alanı ile uyarabiliriz; buna optik dal denmesinin nedeni budur.
Genel bir K değerinde ɿ ʀح oranı, denklem (3.13)’den, dolayı kompleks olacaktır.
Küçük K değerlerinde, denklem (3.17)’nın ɛ = 0 limiti olarak, genlikler için ɿ = ʀ
çözümü elde edilir. Atomlar (ve kütle merkezi), akustik titreşimlerde olduğu gibi,
birlikte hareket ederler ve akustik dal ismi buradan gelir. Şekil 3.6. İki atomlu
doğrusal örgüde enine optik ve akustik dalgalar gösterilmektedir.
Şekil 3.6. İki atomlu doğrusal örgüde enine optik ve akustik dalgalar (KITTEL)
Bazı frekanslarda, örneğin ( 2C M1ح )1 ح2 ile ( 2C M2ح )1 ح2 arasında dalga
çözümleri oluşmaz. Bu durum, çok atomlu örgülerde elastik dalgaların karakteristik
bir özelliğidir. Birinci Brillouin bölgesinin Kmax=± π aح sınırında bir frekans aralığı
oluşur. Bu aralıkta reel bir için çözümler arandığında, K dalga vektörü kompleks
olur ve sönümlü bir dalga çözümü çıkar.

3. MATERYAL VE METOD Şevket ŞİMŞEK
25
3.3. Örgü Titreşimlerinin Kuantumlanması
Kristal örgüdeki atomların titreşim hareketleri, kuantize olmuş
elektromanyetik dalgalardaki fotonlara benzetilerek, fonon olarak isimlendirilmiştir.
Kristallerdeki elastik dalgalar fononlardan oluşmuştur. Yine kristallerdeki ısısal
titreşimler, ısısal olarak uyarılmış fononlardır. açısal frekanslı elastik bir kipin
enerjisi;
ω)21(nE h+= (3.20)
şeklinde olup, burada n, kipte yer alan fononların sayısı ve ℏ Planck sabitidir.
1 ح 2 terimi sıfır enerji düzeyine karşılık gelir. u kristalde bir hacim elemanının
yer değiştirmesi olmak üzere,
)cos()cos(0 tKxuu ω= (3.21)
şeklinde bir kip düşünelim. Bilindiği gibi, bir titreşicinin toplam enerjisinin zamana
göre ortalaması alındığında yarısı kinetik enerji diğer yarısı da potansiyel enerjidir.
kütle yoğunluğu olmak üzere, kinetik enerji yoğunluğu,
2)tuρ(
21
∂∂ (3.22)
olur. Kristalin hacmi V ise, bütün hacim boyunca kinetik enerji, yukarıdaki terimin
integrali alınarak,
20
2uρVω41 (3.23)

3. MATERYAL VE METOD Şevket ŞİMŞEK
26
bulunur. Zamana göre kinetik enerjinin ortalaması denklem (3.20)’den elde edilecek
olan ortalama enerji değerine eşitlenirse,
18ρVω2u0
2= 12
(n+ 12
) ω (3.24)
ve genliğin karesi,
u02= 4(n+ 1 ح2 ) (ρVω)ح (3.25)
olacaktır. Son denklem incelediğimiz kip için, yer değiştirmenin kipteki n fonon
sayısına bağımlılığını verir. Burada , (-) veya (+) olabilir, ancak fonon enerjisi
daima (+) olduğundan ’ yı (+) olarak almak daha uygun olur.
K, fononun dalga vektörü olmak üzere fononun momentumunu, de Broglie
bağıntısını kullanarak ortaya koyabiliriz;
Kλhp h== (3.26)
Bu momentum değeri ile fonon, aynı elektronlar, nötronlar veya fotonlar gibi diğer
parçacıklarla etkileşmeye girebilir. Ancak fononlar, (3.26) denkleminde verilen
momentum büyüklüğü ile etkileşime girebilir diye düşünmemize karşın, fiziksel
anlamda momentum taşımazlar. Bunun açıklaması fononun koordinatları ile ilgilidir.
Fonon koordinatları atomun relatif koordinatlarına bağlıdır. Mesala, bir H2
molekülünde, çekirdeğin titreşim koordinatı r1-r2 relatif bir koordinattır ve çizgisel
momentum taşımazlar. Buna karşın kütle merkezi koordinatı 1 2 (ɼϸ + ɼᴀ)ح düzgün
dağılımlı bir kipe karşılık gelir ve çizgisel momentumu vardır. Bir kristalin fiziksel
momentumu,
∑= su)dtdM(p (3.27)

3. MATERYAL VE METOD Şevket ŞİMŞEK
27
şeklindedir. Bilindiği gibi, ʂ ɀ şeklinde bir terimin 0 ile N-1 aralığındaki toplamı,
( ) ( )x1/x1x1N
0s
Ns −−=∑−
=
(3.28)
şeklinde yazabiliriz. Kristalde bir K fononu varsa, N atom için toplam momentum,
( ) ( )iKaiNKa
s
isKa e1/e1dtduMe
dtduMp −−
=
= ∑ (3.29)
olacaktır. Burada K sınır şartları ile belirlenir ve
K = ± 2πp Naح (3.30)
değerlerini alır. Bu yüzden,
eiNKa=e±2πp=1 (3.31)
ve denklem (3.29)’daki momentum eşitliğinden,
0edtduMp
s
isKa =
= ∑ (3.32)
şeklinde kristal momentumu sıfır bulunur. Bu durumun gözlenmediği tek örnek
düzgün dağılımlı K=0 için olandır. Burada, bütün us değerleri u’ya eşit olur ve
)dtduNM(p = (3.33)
bulunur.

3. MATERYAL VE METOD Şevket ŞİMŞEK
28
Fononun taşıdığını düşündüğümüz ɛ Ө momentumu çoğu zaman kristal
momentumu olarak adlandırılır.
Kristalde dalga vektörü seçim kuralları vardır ve bu seçim kuralı kuantum
durumları arasındaki izinli geçişlerle ilişkilidir. Bir kristal tarafından elastik olarak
saçılan bir X-ışınları fotonu için dalga vektörü seçim kuralı,
k' Ө=k Ө+G Ө (3.34)
şeklinde verilebilir. Burada, ɖӨ ters örgü vektörü, ɵ Ө gelen fotonun dalga vektörü, ɵ Ө
de saçılan fotonun dalga vektörüdür. Yansıma sırasında kristal bütünü ile bir − ɖ
momentumuna sahip olacak şekilde geri tepilir. (3.34) denklemi, periyodik bir
örgüde, etkileşen dalgaların toplam dalga vektörünün bir ters örgü vektörü ile
korunduğunun gösterir. Sistemin gerçek momentumu kesinlikle korunmaktadır.
Fotonun saçılımı, bir fonon ortaya çıkaracak şekilde elastik değilse, fononun
dalga vektörü ɛ Ө olmak üzere, dalga vektörü seçim kuralı,
k' Ө+K Ө=k Ө+G Ө (3.35)
olur ve bu olay sırasında K fononu emilmiş ise, denklem
k' Ө=k Ө+K Ө+G Ө (3.36)
şeklini alır.
Kristallerdeki ısı iletimi, elektron saçılımı ve diğer birçok fiziksel olaylar
sırasında fononlar ortay çıkabilir veya var olan fononlar yok olabilir. Bütün bu
olaylarda örgü titreşimlerinin parçacık karakteri, dalga karakteri kadar önemlidir.

3. MATERYAL VE METOD Şevket ŞİMŞEK
29
3.4. Dinamik Matris
Tüm atom-atom etkileşimleri üzerinden bir toplam alınarak örgü enerjisi
aşağıdaki denklemle tanımlanabilir.
= ∑ '
'
ll,jj lljj
21W
''
ϕ (3.37)
Burada j, l. birim hücredeki bir atomu belirtiyor. φ, (ɴɶ) ve (ɴ ɶ ) atom çiftleri için
etkileşim enerjisidir. Harmonik yer değiştirme enerjisi matris formunda
( ) ( ) ( ) ( )''βαβ
ll',jj' αβα
''
ll',jj'
Tharm ljuΦjlu21'lj.ΦΦ.jlu
21E ∑∑∑ == (3.38)
şeklindedir. Burada 3×1’lik, ɿ (ɴɶ) yer değiştirme matrisi
u jl = ux(jl) uy(jl) uz(jl)
(3.39)
olarak tanımlanıyor ve ɿ ɿ ,لایر ’ nun transpozudur. Kuvvet sabiti matrisi Φ,
)l'(j'u(jl)uW
ll'jj'
Φβα
2
αβ ∂∂∂
=
(3.40)
elemanları ile 3×3’lük bir matrisidir. ve β alt indisleri x, y ve z kartezyen vektörü
bileşenlerini belirtiyor. Kristal enerjisinin Taylor açılımındaki matris formu
( ) ( )[ ] ( ) ( )[ ]'''
'
ll',jj'
T''harm ljujlulljj
Φljujlu41E −
−= ∑ (3.41)

3. MATERYAL VE METOD Şevket ŞİMŞEK
30
şeklindedir. Burada Φ matrisi,
)l'(j'u(jl)ull'jj'
Φ
ll'jj'
Φβα
2
αβ ∂∂
∂
=
(3.42)
elemanlarına sahiptir. (3.41) denklemi ileri ve geri çift saymadan dolayı fazladan bir ϸᴀ faktörü içeriyor. Tüm terimlerin açılmasıyla (3.41) denkleminin (3.38) denklemini
ile uyumlu olduğu görülebilir.
+
−=
∑ ''
''
'l''j'αβll'jj''
'
αβ
'
αβ lljj
φδδlljj
φll'jj
Φ (3.43)
(3.43) denklemindeki ikinci terim kristalin geri kalan kısmı ile her hangi bir ɴɶ
atomunun etkileşiminden meydana geliyor ve öz (self) terim olarak biliniyor.
Bununla birlikle, genel durumda l. birim hücredeki j. atom için hareket denklemi
matris formunda
mju jl, t =-u(j'l',t) (3.44)
(3.44) denklemi ile veriliyor. Burada ɷᵫ, j. atomun kütlesidir. Şimdi yer
değişim vektörü ɿ (ɴɶ, ɾ)’ ye zaman da dahil edilmiş oldu. ɿ (ɴɶ, ɾ) için çözüm, v ile
belirtilmiş modun ve farklı k dalga vektörünün hareketli harmonik dalgalarının bir
lineer süper pozisyonu olacaktır.
( ) ( ) ( ) ( )[ ]( )tυk,ωjlk.riexpυk,j,Utjl,uυk,
−= ∑ (3.45)
Burada ɼ ɴɶ iki nicelikten herhangi biri olarak alınabilir. ɼ ɴɶ , ɴɶ
atomunun denge konumu olarak alınabilir ya da ɶ birim hücresinin orijini olarak

3. MATERYAL VE METOD Şevket ŞİMŞEK
31
alınabilir. Fark, ɥ (ɴ,ɵ ,ʀ)’nın fazına transfer edildiği için seçim yapılması önemli
değildir ve hesaplanmış mod frekansları etkilenmemiştir. Yer değiştirme vektörü
olarak da bilinen ɥ (ɴ,ɵ ,ʀ) genlik vektörü, komşu birim hücreler arasındaki,
hareketlerdeki farklar, tamamen exponansiyel faz faktörü ile belirlendiği için ɶ’ den
bağımsızdır. Dalga denklemini (3.45)’ i, (3.44)’deki hareket denkleminde yerine
koyduğumuzda, denklem (3.46)’ daki standart hareket denklemini elde ederiz.
mjω2 k,v U j,k,v =
Φ∑ '0
'
'' ljj
ljU j',k,v exp(ik[r j'l' -r(j0)]) (3.46)
Burada birim hücredeki referans atom ɶ= 0’ dır. ʀ , ile belirtilen tek bir çözüm için
hareket denklemleri vektör formunda ifade edilebilir ve sonuç olarak,
ω2 k,v e k,v =D k e(k,v) (3.47)
şeklinde yazılabilir. Sütun vektörü ɯ(ɵ ,ʀ) , atomik kütlenin karekökü ile ağırlıklı yer
değişim vektöründen oluşuyor, bu yüzden 3ɸ tane elamana sahiptir. Burada ɸ, birim
hücredeki atomların sayısıdır.
e k,v =
Dž
DŽDŽDŽǃm1Ux(1,k,v)ضm1Uy(1,k,v)ضm1Uz(1,k,v)ضm2Ux(2,k,v)ض
mnUz(n,k,v)Ljض
LJLJLJdž
(3.48)
ɓ ɵ , burara 3ɸ × 3ɸ’lik dinamiksel bir matristir. Dinamiksel matrisi ɓ ɵ , 3 × 3’
lük blok matrisine göre yazalım. Her blok ɴ ve ɴ′ ile indekslenmiş atom çiftlerine
karşılık geliyor ve her bloğun elemanları ayrı ayrı olarak ʂ , ʃ , ʄ karetzyen
koordinatları ifade eden , = 1,2, 3 indislerine sahiptir. Tam dinamiksel matris
ɓ ɵ , daha küçük 3 × 3’lük matrislerin ɸ × ɸ’lik dizilimlerinden oluşuyor.
Dinamiksel matrisin küçük 3 × 3 bloklarının elemanları

3. MATERYAL VE METOD Şevket ŞİMŞEK
32
r(j0)]))l''exp(ik[r(j0l'jj'
Φ)m(m
1k),(jj'Dl'
αβ21j'j
αβ −
= ∑ (3.49)
olarak veriliyor. Burada ɶ= 0, referans birim hücreyi belirtiyor. Tam dinamiksel
matris deki bu elemanın konumları açıkça,
3 j-1 +α; 3 j'-1 +β (3.50)
şeklindedir.
(3.47) denklemi 3ɸ tane bileşene sahip olduğu için, dispersiyon grafiğinde 3ɸ
tane moda karşılık gelen 3ɸ tane çözüm olacaktır. 3ɸ × 3ɸ ɯ(ɵ) matrisi oluşturmak
için, açısal frekansların karelerinin diyagonal matrisi olarak Ω(ɵ) frekans matrisini
tanımlayalım ve ɯ(ɵ ,ʀ) sütun vektörlerini dahil ederek denklemlerimizi
düzenleyebiliriz.
Ω k =
Dž
DŽDŽDŽǃ
ω2(k,1)ω2(k,2)
ω2(k,3).
.ω2(k,3n)Lj
LJLJLJdž
(3.51)
e k Ω k =D k e(k) (3.52)
Dinamiksel matris hermityendir. Yani,
D(k)=(D*(k))T (3.53)
Dinamiksel matrisin öz değerleri, farklı titreşim modlarının açısal frekanslarının
kareleridir ve öz vektörler, her titreşim modu ile ilişkili olan atomların rölatif yer
değiştirmelerini verir. Hareket denklemleri genlik hakkında bilgi içermez. Bu

3. MATERYAL VE METOD Şevket ŞİMŞEK
33
prosedürle hesaplanmış olan öz vektörler (3.54) denklemindeki gibi normalize
olurlar.
e k T. e k
*= e k
T.e -k =1 (3.54)
ɯ(ɵ ,ʀ) vektörüne polarizasyon vektörü denir.
Dinamiksel matris, hem her hangi bir atom-atom etkileşimi için kuvvet
sabitlerini hem de her hangi bir dalganın atomik hareketi için faz faktörünü içeriyor.
Bu yüzden (3.47) denklemi hareket denklemini ifade ediyor. Kütle ağırlıklı
değişkenlerin kullanımı bize, ɷ ᴀ ’den ziyade ᴀ için çözümler bulma olanağı
sağlıyor. Ayrıca, her dalga ile ilişkili olan atomik hareketlerin tam bir setini
oluşturuyor. Bir dalga ile ilişkili olan hareketlerdeki her hangi biri için hareketleri
oluşturmadığından dolayı, bu hareketler lineer olarak bağımsızdır. Bu
e k,v T.e -k,v' =δvv' (3.55)
olarak ifade edilebilir. Bunlara normal modlar denir ve temel titreşim harekeleridir.
Frekanslar reel olmasına rağmen, hareketler genelde komplekstir ve her atomun
hareketinin reel ve sanal kısımlarının rölatif fazları yoluyla basitçe ifade edilebilir.
3.5. Elastik Sabitler
Elastik sabitler katıların birçok özelliklerinin anlaşılması için gerekli bilgilere
sahip olan, katıların önemli karakteristikleridir. Elastik sabitler özellikle kristallerin
esnekliklerini ve mekaniksel kararlılığını belirleyen parametrelerdir. Ayrıca, elastik
sabitler dışarıdan uygulanan bir kuvvete karşı kristalin sertliğini belirler ve kristalin
mekaniksel ve dinamiksel davranışları arasında bir bağlantı kurar.
Bir katının esneklik özellikleri zor (stres) ve zorlanma (starin) nicelikleriyle
ifade edilebilirler. Zor, bozulmayı meydana getiren kuvvetle orantılı bir niceliktir.
Zorlanma ise, bozulma derecesinin bir ölçüsüdür. Zor, zorlanmayla doğru orantılıdır

3. MATERYAL VE METOD Şevket ŞİMŞEK
34
ve bu orantı katsayısına elastik katsayılar veya sertlik katsayıları denir. Zor ve
zorlanma tensörü arasındaki ilişki
σαβ= ط Cαβ,γδεγδγδ σαβ= γδγδ
γδαβ ε∑ ,C (3.56)
şeklinde veriliyor. Elastik enerji o zaman,
Uharm= 12 γδγδαβ
αβγδαβ εε ,C∑ (3.57)
olarak ifade edilir. Elastik özellikler dört indeksli bir tensör ile karakterize edilirler.
Ancak, hem zor hem de zorlanma tensörleri simetrik oldukları için
Cv ᵜ ,ᵝ ᵞ = Cᴥ ᵛ ,ᵝ ᵞ = Cv ᵜ ,ᵞ ᵝ = Cβ ᵞ ,ᵛ ᵜ (3.58)
şeklinde ifade edilebilir. Kristalin simetrisine bağlı olarak tensör elemanları arasında
başka ilişkilerde olabilir. Dört indeksli elastik sabitlerin yerine, literatürde genelde
Voigt elastik sabitleri kullanılıyor. Bu ifadeler simetrik zorlanma tensörü elemanları
için
εxx=ε1 , εyy=ε2 , εzz=ε3,
2εyz=ε4 , 2εzx=ε5 , 2εxy=ε6, (3.59)
notasyonu kullanılarak elde edilirler. Bu nicelikler kullanılarak, elastik enerji
yoğunlu için homojen kuadratik ifade,
Uharm= 12 ji
jiijC εε∑
=
6
1,
, (3.60)

3. MATERYAL VE METOD Şevket ŞİMŞEK
35
olur. Burada ɒϻᵫ katsayıları Voigt elastik sabitleridir. Zor tensörü elemanları içinde
benzer bir notasyon kullanarak,
σxx=σ1 , σyy=σ2 , σzz=σ3,
2σyz=σ4 , 2σzx=σ5 , 2σxy=σ6, (3.61)
şeklinde ifade edilirler. Zor ve zorlanma tensörü arasında bağlantı kuran,
genelleştirilmiş Hook yasası
∑=
=∂
∂=
6
1jjij
i
harmi CU
εε
σ (3.62)
şeklini alır. Elastik katsayıları matrisi simetrik bir matristir ve en genel durumda
triklinik kristallerin elastik özellikleri 21 bağımsız elastik sabit ile karakterize
edilirler.
Daha yüksek simetrili kristallerde bağımsız elastik sabitlerin sayısı daha
azdır. Örneğin, monoklinik kristal sisteminde z-ekseni iki katlı dönme ekseni olarak
seçildiğinde, x→-x, y→-y, z→z dönüşümü altında elastik enerji değişmez
kalmalıdır. Böyle bir dönmede ᴂ ve ᴃ işaret değiştirir fakat zorlanma tensörünün
diğer dört elamanı işaret değiştirmez.
C12=C21, C13=C31, C15=C51, C23=C32, C25=C52, C35=C53, C46=C64 (3.63)
olur. Bundan dolayı monoklinik kristallerde 13 tane bağımsız elastik sabit vardır.
Monoklinik kristal sistemi için elastik sabitleri matrisi denklem (3.64)’de

3. MATERYAL VE METOD Şevket ŞİMŞEK
36
Cij =
Dž
DŽDŽǃ
C11 C12 C13 0 C15 0C12 C22 C23 0 C25 0C13 C23 C33 0 C35 00 0 0 C44 0 C46
C15 C25 C35 0 C55 00 0 0 C46 0 C66Lj
LJLJdž
(3.64)
şeklinde veriliyor.
Ortorombik kristal sisteminde üç eksenin her biri etrafında, 180° dönme
altında elastik enerji değişmez kalır.
C12=C21, C13=C31, C23=C32 (3.65)
Ortorombik kristal sisteminin elastik davranışı 9 tane elastik sabit ile karakterize
edilir. Ortorombik kristal sisteminde bütün kristal sınıfları için elastik katsayıları
matrisi denklem (3.66)’de veriliyor.
Cij =
Dž
DŽDŽǃ
C11 C12 C13 0 0 0C12 C22 C23 0 0 0C13 C23 C33 0 0 00 0 0 C44 0 00 0 0 0 C55 00 0 0 0 0 C66Lj
LJLJdž
(3.66)
Tetragonal kristallerde, z-ekseni etrafında 90° dönülmesinden dolayı yeni bir
simetri ortaya çıkar. x→y, y→-x, z→z dönüşümü altında elastik enerji
değişmezliği,
C11=C22, C12=C21, C13=C31=C23=C32, C44=C55 (3.67)
olmasını gerektiriyor. O zaman elastik enerji için ifade,
Uharm= 12
C11 εxx2 +εyy
2 + 12
C33εzz2 +C12εxxεyy

3. MATERYAL VE METOD Şevket ŞİMŞEK
37
+C13 εxxεyy+εyyεzz +2C44 εyz2 +εzx
2 +2C66εxy2 (3.68)
olur. Tetragonal kristal sisteminde 4ɷɷ , 4ⱨ2ɷ , 422 ve 4 ɷɷɷح simetrisine sahip
kristal sınıfları için elastik katsayıları matrisi,
Cij =
Dž
DŽDŽǃ
C11 C12 C13 0 0 0C12 C11 C13 0 0 0C13 C13 C33 0 0 00 0 0 C44 0 00 0 0 0 C44 00 0 0 0 0 C66Lj
LJLJdž
(3.69)
olur. Bu durum için bağımsız elastik katsayılarının sayısı 6 dır. Tetragonal kristal
sisteminde 4, 4 Ⱪ ve 4 ɷح simetrilerine sahip kristal sınıfları için elastik katsayıları
matrisi,
C11=C22, C12=C21, C44=C55, C13=C31=C23=C32, C13=C31=C23=C32 (3.70)
Cij =
Dž
DŽDŽDŽǃ
C11 C12 C13 0 0 C16
C12 C11 C13 0 0 -C16C13 C13 C33 0 0 00 0 0 C44 0 00 0 0 0 C44 0
C16 -C16 0 C46 0 C66 Lj
LJLJLJdž
(3.71)
ve bağımsız elastik katsayılarının sayısı 7 dir.
Kübik kristallerde x ve y-eksenleri etrafında 90° dönülmesi
(x→x, y→z, z→-y ve y→y, x→z, z→-x) simetriktir. Bundan dolayı,
C33=C11, C13=C12, C66=C44 (3.71)
olmalıdır. Geriye kalan 3 elastik sabite göre elastik enerji,

3. MATERYAL VE METOD Şevket ŞİMŞEK
38
Uharm= 12
C11 εxx2 +εyy
2 +εzz2 +C12 εxxεyy+εxxεzz+εyyεzz
+2C44 εxy2 +εyz
2 +εzx2 (3.72)
olarak ifade edilir. Kübik kristal sistemi için elastik katsayıları matrisi,
C11=C22=C33, C44=C55=C66,
C12=C21=C13, C31=C23=C32, (3.73)
Cij =
Dž
DŽDŽǃ
C11 C12 C12 0 0 0C12 C11 C12 0 0 0C12 C12 C11 0 0 00 0 0 C44 0 00 0 0 0 C44 00 0 0 0 0 C44Lj
LJLJdž
(3.74)
Trigonal kristal sisteminde 3 ve 3ⱨ simetrilerine sahip kristal sınıfları için,
C11=C22, C44=C55, C13=C31= C23=C32,
C14= −C24=C56=C41= −C42,
−C15=−C51=C25=C52=C46=C64 (3.75)
Cij =
Dž
DŽDŽDŽDŽǃ
C11 C12 C13 C14 -C25 0C12 C11 C13 -C14 C25 0C13 C13 C33 0 0 0C14 -C14 0 C44 0 C25
-C25 C25 0 0 C44 C14
0 0 0 C25 C1412
(C11-C12)Lj
LJLJLJLJdž
(3.76)

3. MATERYAL VE METOD Şevket ŞİMŞEK
39
ve bağımsız elastik katsayılarının sayısı 7 dir. Trigonal kristal sisteminde 32, 3ⱨɷ ve
3ɷ simetrilerine sahip kristal sınıfları için ,
C11=C22, C44=C55, C12=C21,
C13=C31=C23=C32, C14=C41=-C24=-C42=C56=C65, (3.76)
Cij =
Dž
DŽDŽDŽDŽǃ
C11 C12 C13 C14 0 0C12 C11 C13 -C14 0 0C13 C13 C33 0 0 0C14 -C14 0 C44 0 00 0 0 0 C44 C14
0 0 0 0 C1412
(C11-C12)Lj
LJLJLJLJdž
(3.77)
ve elastik katsayılarının sayısı 6 dır.
Hegzagonal kristal sistemindeki bütün kristal sınıfları için,
C11=C22, C12=C21, C44=C55, C13=C31=C23=C32, (3.78)
Cij =
Dž
DŽDŽDŽǃ
C11 C12 C13 0 0 0C12 C11 C13 0 0 0C13 C13 C33 0 0 00 0 0 C44 0 00 0 0 0 C44 00 0 0 0 0 1
2(C11-C12)Lj
LJLJLJdž
(3.79)
ve bağımsız elastik sabitlerinin sayısı 5 dir.
3.6. Bulk Modülü
Bulk modülü, bir malzemeye bir dış basınç uygulandığında malzemenin ne
kadar sıkıştırılabileceğini belirten bir niceliktir. Yani bir deformasyon oluşturmak
için verilmesi gereken enerjinin büyüklüğünü gösterir. Bulk modülü,

3. MATERYAL VE METOD Şevket ŞİMŞEK
40
VΔVΔPB −= (3.80)
olarak tanımlanıyor. Bulk modülünün tersine malzemenin sıkışabilme özelliği denir
ve,
B1χ = (3.81)
şeklinde tanımlanır.
3.7. Born Efektif Yükler
Born efektif yükler, yalıtkan kristallerin örgü dinamiğinde önemli bir rol
oynamaktadır. Born efektif yükü Z*, iyonik yer değiştirmelerden dolayı elektronik
polarizasyondaki değişimin bir ölçüsüdür. Aynı zamanda born efektif yükü, çekirdek
ve boyuna (LO) ve enine (TO) optik fonon modları arasındaki uzun erimli Coulomb
etkileşiminin genliğini etkilediği için yalıtkan kristallerin örgü dinamiği
çalışmalarında önemli bir nicelik olarak dikkate alınıyor. Periyodik sistemler için,
genel olarak alt örgüsüne ait olan çekirdeğin Born efektif yük tensörü ɪ ݒ ,ݔ∗ , sıfır
makroskobik elektrik alan şartı altında birim hücre hacmi ΩϷ kere, yönü boyunca
atomlarının toplu nükleer yer değiştirmeleri ve yönü boyunca makroskobik
polarizasyondaki Ѿݒ değişimiyle ilişkili orantı katsayı olarak tanımlanır.
Zκ,αβ* =Ω0
∂Pβ
∂τκα ε=0
(3.82)
Bununla birlikte termodinamiksel eşitlik, makroskobik polarizasyonu elektrik
entalpisinin ɔ türevine ilişkilendirir ve diğer bir ilişki çekirdeğinin üzerindeki ɕݔ
kuvveti ile elektrik entalpisinin türevi arasında bir bağlantı kurulabilir. Bundan
dolayı ɪ ݒ ,ݔ∗ , alternatif olarak aşağıdaki gibi tanımlanabilir.

3. MATERYAL VE METOD Şevket ŞİMŞEK
41
Zκ,αβ* =- ∂2E
∂εβ∂τκα= ∂Fκ,α
∂εβ τκα=0
(3.83)
Bu ilişkilerden born efektif yükü ɪ ݒ ,ݔ∗ ,
1. Sıfır elektrik alan şartı altında, atomlarının toplu yer değiştirmesiyle
uyarılmış polarizasyondaki değişim olarak düşünülebilir.
2. Elektrik entalpisinin karma ikinci türevi olarak düşünülebilir.
3. Sıfır atomik yer değiştirmede, homojen efektif elektrik alan ݒ tarafından
çekirdeği üzerinde uyarılmış olan kuvvetin türevi olarak düşünülebilir.
Bu üç tanım birbirine eşdeğerdir, ancak ilk prensip hesaplamalarında ɪ ݒ ,ݔ∗ ’nin
hesaplanması farklı algoritmalara neden olmaktadır. En geniş bir şekilde kullanılan
yaklaşımlar arasında, Sternheimer denklemini temel alan lineer tepki
hesaplamalarından ɪ ݒ ,ݔ∗ ‘nin belirlenmesini öneren ilk güçlü ve sistematik prosedür
Baroni, Giannozzi ve Tesla tarafından ortaya atılmıştır. Daha sonra varyasyonel
prensibi baz alan farklı bir algoritma Gonze, Allan ve Teter tarafından rapor
edilmiştir. ɪ ݒ ,ݔ∗ , aynı zamanda polarizasyondaki sonlu bir farktan da doğrudan
hesaplanabilir.
3.8. Yoğunluk Fonksiyonel Teorisi
1920’lerde Thomas ve Fermi’nin çalışmalarını baz alan Hohenberg-Kohn
(1964) teoremleri ve onun devamı olan ve Kohn-Sham (1965) teoremleri DFT’nin
temellerini oluşturmuştur. Yoğunluk fonksiyonel teorisinin ana fikri, etkileşen bir
elektronlar sistemini çok-cisim dalga fonksiyonları yoluyla değil elektron yoğunluğu
olarak tanımlamaktır. Yani DFT, çok elektronlu sistemlerin taban durum özelliklerini
belirlemek için elektron yük yoğunluğu )(rrρ ’yi temel değişken olarak kabul
etmektedir. DFT, metaller, yarıiletkenler ve yalıtkanların temel durum özelliklerini
belirlemek için oldukça başarılı bir yaklaşımdır. DFT’ nin başarısı sadece bulk
hacimli malzemelerle sınırlı olmasından değil aynı zamanda protein ve karbon nano
tüpler gibi kompleks materyallere de uygulanabilir olmasından kaynaklanmaktadır.

3. MATERYAL VE METOD Şevket ŞİMŞEK
42
3.8.1. Çok Cisim Problemi
Bir katıdaki elektronların ve iyonların davranışları göz önünde bulundurarak
Ne tane elektron ve Ni tane çekirdekten oluşan bir sistemin Hamiltoniyeni
∑∑
∑∑∑∑∑∑
=
== ===
−+
−+
−−∇−∇−=
e i
e ie ie
N
1i
N
ij JI
JI
N
1i
N
ij ji
N
1i
N
1i Ii
iNi
1i
2I
I
N
1i
2i
RRZZ
rr1
RrZ
2M1
21H
f
f
rr
rrrr
(3.84)
şeklinde verilir. Burada MI kütle, ZI çekirdeklerin atom numarası irr
ve IRr
ise elektron
ve çekirdeğin koordinatlarıdır. Birinci ve ikinci terimler; sırasıyla elektron ve
çekirdeğin kinetik enerjileri, üçüncü terim; çekirdek ve elektronlar arasındaki
Coulomb çekim alanıdır. Dördüncü terim elektronlar arasındaki ve beşinci terim ise
çekirdekler arasındaki Coulomb itme etkileşimidir. Yukarıda tanımlanan sistemin
taban durumu özellikleri zamandan bağımsız Schrödinger denkleminin çözümüyle
belirlenir:
{ } { }( ) { } { }( )iiii R,rEΨR,rHΨrrrr
= (3.85)
Burada { } { }( )ii R,rΨrr , çok cisimli sistemin dalga fonksiyonu ve E sistemin enerjisidir.
Eş. 3.84’ün karmaşıklığından dolayı çözümü kolaylaştırmak için bazı yaklaşımlar
yapmak gerekir. Bu yaklaşımlardan bir tanesi katıhal fiziği ve atom ve molekül
fiziğinde çok kullanılan Born-Oppenheimer yaklaşımıdır. Bu yaklaşımla elektron ve
çekirdeklerin hareketleri ayrı ayrı incelenir.

3. MATERYAL VE METOD Şevket ŞİMŞEK
43
3.8.2. Born-Oppenheimer Yaklaşımı
Bir sistem içerisindeki elektronların oluşturduğu çok parçacık sisteminin
Schrödinger denklemini çözmek için çeşitli yaklaşımlar yapmak gerekir. Born-
Oppenheimer yaklaşımı, bir veya iki elektrondan daha fazlasına sahip olan
sistemlerin Schrödinger denklemini çözmeye çalışan yaklaşımlardan biridir. Bu
yaklaşımdaki temel düşünce, çekirdeğin kütlesinin elektron kütlesinden daha fazla
olması nedeniyle çekirdekleri sabit ve hareketsiz olarak kabul ediyor. Bu nedenle
çekirdeğin sabit olduğu bir alanda elektronları hareket halinde düşünebiliriz. Bu
yaklaşımı dikkate aldığımızda (3.84) denklemindeki ikinci terim yani çekirdeğin
kinetik enerjisi ihmal edilebilir. Ayrıca çekirdekler arasındaki Coulomb itme
etkileşmesi sabit düşünülerek son terimde ihmal edilebilir. Bu durumda
∑∑∑ ∑∑== = = −
+−
−∇−=e ie e i N
1i
N
ij Ii
N
1i
N
1i
N
1I Ii
I2ie rr
1Rr
Z21H
frrrr (3.86)
denklemi elde edilir. Bu denklem eN tane elektronun, iN tane çekirdeğin alanında
hareketini tanımlayan elektronik Hamiltonyen ifadesine dönüşür.
( )Rr,ΨΨ ee = (3.87)
( )Rεε ee = (3.88)
(3.87) denklemi elektronların hareketini, (3.88) denklemi ise elektronların enerjisini
gösterir. Nükleer itme ile beraber toplam enerji
( ) ∑∑= −
+=N N
RRZZ
Rε(R)εi i
1α αβ βα
βαetot
f
(3.89)

3. MATERYAL VE METOD Şevket ŞİMŞEK
44
şeklinde ifade edilir. Bu ifade potansiyel enerji yüzeyini oluşturur. Born-
Oppenheimer yaklaşımında, elektronik problemin çözülmesiyle çekirdek bir
potansiyel enerji yüzeyinde hareket eder. Bu yaklaşım da elektron ve çekirdeğin
hareketi birbirinden ayrılmadığında geçersizdir.
3.8.3. Hartree Yaklaşımı
Bir katı içindeki elektronların kuantum mekaniksel davranışlarını açıklamak
için, sistemin çok elektronlu dalga fonksiyonunu hesaplamak gerekir. Prensip olarak
bu, zamandan bağımsız Schrödinger denkleminden elde edilebilir. Fakat pratikte
potansiyel, katı içerisindeki diğer elektronların davranışlarıyla belirlenir. Gerçekte
birbirlerine yakın elektronlar, uzak olan elektronlardan daha güçlü etkileşmeler
içindedir. Tüm elektronların Schrödinger denklemini çözebilmek için aynı anda 1023
civarında diferansiyel denklemi çözmek gerekir.
Problemi çözmek için ilk adım Hartree tarafından atılmıştır. Hartree
yaklaşımında çok elektronlu sistemin dalga fonksiyonu, tek elektron dalga
fonksiyonlarının çarpımı olarak yazılır. Bu durumda dalga fonksiyonu,
∏=)r,...,r,rΨ( N21rrr
(3.90)
)(rΨ)r,...,r,rΨ( i
N
1iiN21 ∏
=
=rrr (3.91)
şeklinde ifade edilir. Burada i. elektrona etkiyen potansiyel
)r(V)r(V(r)V Hiyonirr
+= (3.92)
denklemi ile verilir. Bu potansiyel, iyon ve Hartree potansiyelinin toplamı
şeklindedir. (3.90) denklemi yardımıyla iyonV ve HartreeV potansiyelleri,

3. MATERYAL VE METOD Şevket ŞİMŞEK
45
∑ −−=
α α
αiyon rr
Z)r(V rrr
, ∫ −−=
'rr)rr(rd)r(VHartree rr
rrr (3.93)
şeklinde elde edilir. i. elektrona etkiyen Hartree potansiyelindeki yoğunluk terimi
2
jij )'r(Ψ)'rρ( ∑
≠
=rr
(3.94)
şeklinde verilir.
)r(V21H i
N
1i
2i
r+∇−= ∑
=
∧
(3.95)
şeklinde ifade edilen Hamiltonyenin (3.91) denklemi ile alınan beklenen değerini
(toplam enerjiyi) en küçük yapan tek elektron dalga fonksiyonları Hartree denklemi
ile verilir. Bu denklem
)r(Ψε)r(Ψ'rr)'r(Ψ
rd)r(Ψ)r(V21
iiiij
2
jiiyon
2 rrrr
rrrr
=−
+
+∇− ∑∫
≠
(3.96)
şeklinde ifade edilir. (3.96) denklemi, orbitaller için öz uyumlu çözüldüğünde (3.91)
ile sistemin dalga fonksiyonu elde edilmiş olacaktır. Hartree yaklaşımında değiş-
tokuş ve korelasyon etkileri hesaba katılmadığı için günümüzde çok az
kullanılmaktadır.
3.8.4. Hartree-Fock Yaklaşımı
Hartre-Fock yaklaşımı, etkileşmeyen elektron orbitallerine karşı gelen dalga
fonksiyonlarını temsil etmek için kullanılan bir yaklaşımdır. Sistemin dalga
fonksiyonu, asimetri özelliğini sağlayacak şekilde seçilir. Elektronlardan oluşan

3. MATERYAL VE METOD Şevket ŞİMŞEK
46
sistemin dalga fonksiyonu, Pauili dışarlama ilkesi gereği, sistemdeki iki elektronun
yer değiştirmesi altında asimetrik olmalıdır.
,...)r,...,rΨ(...,,...)r,...,rΨ(..., ijjirrrr
−= (3.97)
(3.197) denklemini sağlayan en basit dalga fonksiyonu Slater determinantı ile verilir
ve
D(r⃗1,r⃗2,…r⃗N)= Ψ1(r⃗1) Ψ1(r⃗2)Ψ2(r⃗1) Ψ2(r⃗2) ⋯ Ψ1(r⃗N)⋯ Ψ2(r⃗N)⋮ ⋮ΨN(r⃗1) ΨN(r⃗2) ⋮ ⋮⋯ ΨN(r⃗N) (3.98)
şeklinde ifade edilir. (3.96) denklemine benzer olan Hartree-Fock denklemi de enerji
beklenen değerini minimum yapan (3.98) denklemindeki tek elektron dalga
fonksiyonlarını verir ve
( ) ( ) ( ) )r(ΨεrΨrr
rΨrΨrdδ
)r(Ψ'rr)'r(Ψ
rd)r(Ψ)r(V21
iijj
'
'i
'j'
σi.σj
ij
2
jiiyon
2
rrrr
rrr
rrr
rrrr
=−
−−
+
+∇−
∑ ∫
∑∫∗
(3.99)
şeklinde ifade edilir. Buradan son terim değiş tokuş terimidir ve
ile spinleri aynı olduğunda sıfırdan farklıdır.
Bu yaklaşımın avantajı tek elektron dalga fonksiyonunu içeren bir Slater
determinantı kullanması, varyasyonel olması ve toplam enerjiyi minimize eden bir
deneme dalga fonksiyonu kullanmasıdır. Fakat Hartree-Fock metodu elektronlar
arasındaki korelasyonu (ilişkiyi) göz önüne almaz. Bunun yanında değiş tokuş terimi
yerel olmadığından Hartree-Fock denkleminin çözümü oldukça zordur ve
hesaplanması da yoğunluk fonksiyonel teorisine göre oldukça zordur.

3. MATERYAL VE METOD Şevket ŞİMŞEK
47
3.8.5. Hohenberg-Kohn Teoremi
Hohenberg ve Kohn (1964’de), bir ( )rv r dış potansiyeli içinde etkileşen bir
elektron gazının temel durumunun çalışıldığı bir makaleyi Physical Review’ de
yayınladılar. Temel değişken olarak dikkate alınan elektron yoğunluğunun, yük
yoğunluğunun bir fonksiyonu olarak ifade edilebileceğini kanıtladılar. Bu toplam
enerji fonksiyonunun minimum değeri sistemin temel durum enerjisine karşılık
gelmektedir. Ayrıca bu minimum değere neden olan yük yoğunluğu, tam tek
parçacık temel durum yoğunluğudur.
Herhangi bir kuantum mekaniksel problemin çözümü ψ dalga fonksiyonunun
belirlenmesine bağlıdır. Dalga fonksiyonu, sistem hakkında bilinebilen tüm bilgilere
geçiş sağladığı için merkezi bir niceliktir. Bir katı gibi geniş bir sistem için, dalga
fonksiyonunu belirlemenin bazı problemleri vardır. Dalga fonksiyonu çok karmaşık
bir niceliktir: Deneysel olarak ölçülemez ve N elektronun her biri için bir spin
değişkeni ve üç uzaysal değişken olmak üzere N4 değişkene bağlıdır. Katıhal
sistemlerin çoğu, çok sayıda elektron ve iyonlar içerdiği için herhangi bir dalga
fonksiyonuna dayalı davranışı inanılmaz derecede hesaplama gücü gerektirmektedir.
Bu durum çözümü zorlaştırmakla kalmıyor aynı zamanda sistemi tanımlamayı da
karmaşık hale getirmektedir. Diğer taraftan ( )rrρ elektron yoğunluğunun değişken
bir fonksiyon olduğu temel durum enerjisi için tam formal bir prensip geliştirmek
mümkündür. Bu yoğunluk fonksiyonu ( )rrρ , sadece üç uzaysal koordinata bağlıdır
ve dolayısıyla üç boyutlu ( D3 ) reel uzayındaki amaç, Schrödinger denklemini
çözümüne ulaşmaktır.
DFT’ nin altında yatan düşünce, temel değişken olarak ( )rrρ yoğunluğunun
seçmektir. Herhangi bir atomik veya moleküler sistemin Hamiltonyen operatörü,
elektronların sayısı )(N , çekirdek yükleri )( kZ ve uzaydaki çekirdeğin konumu
)( kR ile tanımlanıyor. M çekirdek ve N elektrondan oluşan bir sistem için, atomik
birimler ( )1=== eme h cinsinden temel Hamiltonyen,

3. MATERYAL VE METOD Şevket ŞİMŞEK
48
çekel HHH += (3.100)
olarak yazılır. Burada
∑∑∑∑∑== ==
+−∇−=N
1i
N
ij ij
N
1i
M
1k ik
kN
1i
2iel r
1rZ
21H (3.101)
∑∑∑==
+∇−=M
1k
M
kl kl
lk2k
M
1k kçek R
ZZM1
21H (3.102)
dır. Eğer, elektron ve çekirdeğin kütleleri arasındaki farklar bir avantaj olarak alınırsa
Hamiltonyen basitleştirilebilir. Bütün çekirdeklerin en hafif bile, yani proton bir
elektrondan 1800 kez daha ağırdır. Böylece çekirdek, elektronlarla kıyaslandığında
daha yavaş hareket eder. Bu nedenle elektronlar sabit çekirdek alanında hareket
ediyor olarak düşünülebilir. Bu Born-Oppenheimer yaklaşımı olarak bilinir. Eğer
çekirdek uzayda hareketsiz ise kinetik enerjileri sıfırdır ve çekirdek-çekirdek
itmelerinden dolayı potansiyel enerjileri sadece bir sabittir. Sonuç olarak (3.100)
denklemindeki Hamiltonyen, elH elektronik Hamiltonyene indirgenir. Böylece
sistemin çözümü sadece elektronik dalga fonksiyonudur. Bu durum öz değer
problemine dönüşür.
elelelel ψEψH = (3.103)
Bu yaklaşımdan sonra sistemin toplam enerjisi
çekeltot EEE += (3.104)
ile verilir. Burada çekE , (3.102) denklemindeki ikinci terimdir ve bir sabit olarak
görülür.

3. MATERYAL VE METOD Şevket ŞİMŞEK
49
Sonuç olarak herhangi bir atomik ya da moleküller sistem için toplam
Hamiltonyen, çekirdek yükleri )( kZ , çekirdek konumları )( kR ve elektronların
sayısı )(N ile tanımlanabilir. Burada basit olması için sadece dejenere olmayan
temel durumdaki durumlarla ilgileniyoruz. Diğer taraftan Hamiltonyen, elektron
yoğunluğuna göre ifade edilebilir. Hamiltonyen ile ilişkili olan üç önemli özellik
vardır. Bunlar;
1) Elektron yoğunluğu ( )rrρ , sistemdeki parçacıkların toplam sayısına normalize
edilir.
( ) Nrρrd =∫rr
(3.105)
2) Doğal olarak ( )rrρ , iyon merkezlerinde )( kRr
maksimuma sahiptir.
3) ( )rrρ , çekirdek koordinatlarında )( kRr
, nükleer yük )( kZ hakkında bilgi içerir.
( ) ( )0ρ2Zrρr k
0rk
kk
−=∂∂
=r
rr (3.106)
Burada krr
, k indeksi ile belirtilmiş iyon korlarından olan radyal uzaklıktır ve ( )krr
ρ
aynı iyon çevresindeki yük yoğunluğunun küresel ortalamasıdır.
Bu nedenle özel bir Hamiltonyen sistemi için ( )rrρ tek değişken olarak
seçilebilir ve tüm moleküller özelliklerin tanımlanabilmesi için ( )rrρ yeterlidir.
Elektron yoğunluğu ( )rrρ , tüm sistemin fiziksel özelliklerini ortaya çıkarmak için
kullanılabilir. Temel durumda ( )rrρ ;
( ) ( ) N21
2
N21 x...dxddsx,...,x,xΨNrρ rrrrr∫= (3.107)
= ( ) ( )ΨrψrψΨ el*el
rr

3. MATERYAL VE METOD Şevket ŞİMŞEK
50
olarak tanımlanır.
İlk Hohenberg-Kohn teoremi sistemin tüm özelliklerini ve Hamiltonyen
operatörünü belirleyen elektron yoğunluğunu sağlamaktadır. Hohenberg ve Kohn
(1964’de) makalelerinde çok basit bir ispat verdiler. Bir dış ( )rv r potansiyeli etkisi
altındaki bir elektron gazını dikkate aldılar. Hamiltonyeni aşağıdaki gibi verdiler,
UVTH ++= (3.108)
Burada
( ) ( ) rdrψrψ21T * rrrrr
∇∇= ∫ (3.109)
( ) ( ) ( ) rdrψrψruV * rrrr∫= (3.110)
( ) ( ) ( ) ( )rψ'rψ'rψrψ'rr'rdrd
21U * rrrr
rrrr
∫ −= (3.111)
(3.107) denkleminde tanımlandığı gibi ( )rrρ , ( )rv r’ nin bir fonksiyonu olduğu
görülmektedir. Sonra aynı yük yoğunluğu ( )rrρ ’ ye neden olan ( )rv r ve ( )'rv r
iki dış
potansiyel dikkate aldılar. Bu düşünce, (3.107) denkleminde verilen dalga
fonksiyonundan nasıl bir yoğunluk formülü inşa edilmesi gerektiğini göstermektedir.
Ψ temel durumundaki daha önceki sonuçlar ve 'Ψ neden olan daha sonraki
durumlar dejenere olmayan elektron sistemi ile ilişkilidir. Gerçek Hohenberg-Kohn
ispatının dejenere olmayan temel durumlarına getirilen kısıtlama daha sonra
kaldırılacaktır. Potansiyeller en az bir sabitle birbirinden farklı olmasaydı, farklı
Schrödinger denklemlerini sağlamadıkları için 'Ψ , Ψ ‘e eşit olamazdı. ( )rv r dış
potansiyeline karşılık gelen temel durum enerjisi

3. MATERYAL VE METOD Şevket ŞİMŞEK
51
ΨH'ΨΨ'H'Ψ'E' <=
= ΨVV'HΨ −+ (3.112)
olarak yazılabilir. Burada doldurulmuş nicelikler 'Ψ ile karakterize edilen sisteme
aittir ve doldurulmamış niceliklerde Ψ aittir. Hamiltonyen sadece dış potansiyeller
içinde farklı olduğu için,
( ) ( )[ ] ( ) rdrρrvrv'EE'rrrr
∫ −+= (3.113)
doldurulan ve doldurulmayan niceliklerde değişme aşağıdaki aynı işlemle bulunur.
( ) ( )[ ] ( ) rdrρrv'rvE'Errrr
∫ −+=
(3.114)
(3.113) ve (3.114) denklemlerinin toplamı bir tutarsızlığa neden oluyor.
E'EE'E +<+ (3.115)
Bu yüzden aynı temel durum elektron yoğunluğunu sağlayan iki farklı potansiyel
olmayabilir. Ayrıca ( )rv r, bir sabit içinde ( )rrρ ’nin bir fonksiyonudur.
İkinci teorem, değişken teorem (variational teorem) olarak adlandırılıyor.
Değişken teorem, tüm parçacık temel durum enerjisi ( )rrρ ’nin bir fonksiyonu olduğu
gerçeğinden ortaya çıkıyor ve değişken teorem, kinetik enerji ve elektron-elektron
etkileşim enerjisidir. Bu nedenle temel durum enerjisi )(E , ( )rrρ temel yük
yoğunluğu terimine bağlı olarak iki kısma ayrılabilir.
[ ] ( ) ( ) [ ]ρFrdrρrvρE += ∫rrr
(3.116)
Burada,

3. MATERYAL VE METOD Şevket ŞİMŞEK
52
[ ] ΨUTΨρF +≡
(3.117)
Bu ifadede [ ]ρF Hohenberg-Kohn fonksiyonu olarak adlandırılır ve elektronların
sayısı )(N , nükleer koordinatlar )( kRr
ve çekirdek yükü )( kZ ’den bağımsızdır. Yani
[ ]ρF , herhangi bir dış potansiyel ve herhangi bir parçacıkların sayısı için geçerli
olan genel bir fonksiyondur.
Giriş yoğunluğu sadece doğru temel durum yoğunluğu ise ve en düşük
enerjiyi belirtiyorsa, Hohenberg-Kohn fonksiyonu [ ]ρF , sistemin temel durum
enerjisini ortaya çıkarır. Doğru temel durum enerjisini bulmak için değişken prensip
kullanılabilir. Bu nedenle (3.116) eşitliğindeki ifade bir değişken problem olarak ele
alınabilir. Değişken metodun uygulanabilir olmasında bazı kısıtlamalar vardır. İlk
olarak bu metot sistemin en düşük enerji durumunu ifade ettiği için temel duruma
sınırlandırılıyor. İkincisi, deneme yoğunluğu ρ pozitif olmalı ve denklem (3.105) da
verildiği gibi parçacıkların sayısı N ’ yi integre etmelidir.
Herhangi bir deneme yoğunluğu ρ~ , kendi Hamiltonyeni H~ ve kendi dalga
fonksiyonu Ψ~ tanımlıyor. Şimdi bu dalga fonksiyonu gerçek dış potansiyel ( )rv r
’den türetilen Hamiltonyen için deneme dalga fonksiyonu olarak alınabilir (Mete,
2003).
3.8.6. Çok Parçacık Sistemi: Kohn-Sham Denklemi
Kohn ve Sham, çok parçacık problemini, öz-uyumlu tek-elektron
denklemlerinin eşdeğer bir seti ile değiştirilebileceğini gösterdiler. Kohn ve Sham
(1965’de) Hamiltonyen denklemini çok basit bir formda yer alan değişken
yaklaşımdan türetilebileceğini kanıtladılar. Kohn-Sham olarak bilinen denklem,
elektron yoğunluğunun bir fonksiyonu olarak ifade edilen elektronlar tarafından
oluşturulan potansiyel dışında, zamandan bağımsız Schrödinger denklemine benzer
bir formdadır. Elektron –iyon etkileşiminden gelen katkıya ek olarak, elektron-
elektron etkileşim potansiyeli, kolaylık olması için Hartree potansiyeli ve değişim-

3. MATERYAL VE METOD Şevket ŞİMŞEK
53
korelasyon potansiyeline ayrılır. Gerçek hayat da bu teoremin uygulanması ciddi
hesaplama gücü gerektirmektedir.
Kohn ve Sham, daha iyi bir kinetik enerji elde etmek için, problemin içerisine
yoğunluk yerine etkileşmeyen orbitalleri yeniden dahil ettiler. Tam olarak etkileşen
sistemin temel durum yoğunluğuna tamamen eşit olan temel durum yoğunluğu için
etkileşmeyen bir referans sistemi kullanarak, her hangi bir N -temsil edilebilir
yoğunluğun orbitallere ayrışabileceğini göstermeyi başardılar. Bunlara Kohn-Sham
orbitalleri denir ve bu orbitaller kullanılarak kinetik enerji operatörünün beklenen
değeri, etkileşimsiz kinetik enerjidir. Yük yoğunluğu için bir baz olarak ortonormal
Kohn-Sham orbitallerinin bir setinin kinetik enerji terimi,
( ) ( ) 2
ii rψ2rρ ∑=
rr (3.118)
şeklindedir. Burada ( )rir
ψ , Kohn-Sham orbitalleridir. Bu orbitaller sadece yük
yoğunluğu açılımı için bir araç olarak kullanılıyor ve tek-parçacık durumları olarak
yorumlanamaz. Kohn-Sham orbitalleri ile ifade edilen kinetik enerji T ;
( )[ ] ( ) ( ) rdrψrψrρT i2
i
*i
rrrr∇−= ∑∫ (3.119)
olarak yazılabilir.
Çift olarak işgal edilmiş iψ elektronik durumları bir seti için Kohn-Sham
toplam enerji fonksiyonu,
{ }[ ] ( ) ( ) ( ) ( )
( )[ ] { }( )kçekXC
ii
2*ii
RErρE
'rdrd'rr'rρrρ
21rdrρrvrdψψψE
rr
rrrrrrrrrr
++
−++∇−= ∫∫∑∫
(3.120)

3. MATERYAL VE METOD Şevket ŞİMŞEK
54
olarak yazılabilir. Burada çekE , kRr
konumlarındaki çekirdekler arasında Coulomb
etkileşim enerjisi, ( )rv r toplam elektron-iyon etkileşim potansiyeli ve ( )[ ]rEXC
rρ
değiş tokuş-korelasyon enerji fonksiyonudur.
Kohn-Sham enerji fonksiyonunun minimum değeri sistemin temel durum
enerjisine eşittir. Bundan başka bir fiziksel anlamı yoktur. Bu nedenle Kohn-Sham
enerji fonksiyonunu minimize ettiği için ( )rir
ψ orbitalleri mutlaka belirtilmelidir.
Bunlar Kohn-Sham denklemlerine öz-uyumu çözümler olarak elde edilebilir.
( ) ( ) ( ) ( ) ( )rψεrψrVrVrv21
iiiXCH2 rrrrr
=
+++∇− (3.121)
Burada HV Hartree potansiyeli olup
( ) rd'rr
rρVHr
rrr
∫ −= (3.122)
olarak tanımlanır (Mete, 2003).
3.8.7. Değişim ve Korelasyon
Coulomb etkileşiminden dolayı, elektronların hareketleri birbirleriyle ilişkili
olduğu için fiziksel olarak birbirinden kaçmaya çalışırlar. Bu durum, çok-cisim dalga
fonksiyonlarının, etkileşmeyen bir durumdaki tek-elektron orbitallerinin basit bir
kombinasyonu olarak yazılmasını engeller.
Elektronlar fermiyonlar oldukları için, çok-parçacık sistemi aynı zamanda
herhangi iki elektronun değişimine uygun asimetrik bir dalga fonksiyonuna sahip
olmalıdır. Bunun anlamı aynı spinli elektronlar Pauli dışarlama ilkesinden dolayı
zorunlu olarak birbirlerinden kaçınırlar.

3. MATERYAL VE METOD Şevket ŞİMŞEK
55
Bu iki etkiyle birlikte Coulomb etkileşimi yoluyla yoğunluğun basit bir
etkileşiminden kolayca çıkarmayı beklediğimiz enerjiyle kıyaslanan sistemin enerjisi
daha düşüktür (Hartree yaklaşımı).
(3.111) denklemindeki elektron-elektron etkileşim terimi iki kısma ayrılır.
Hartree potansiyeli ve değişim-korelasyon potansiyeli;
[ ] ( ) ( ) [ ]ρE'rdrd'rr'rρrρ
21ρE XCee +
−= ∫
rrrrrr
(3.123)
Burada ilk terim Hartree enerjisidir. İkinci terim ise değişim korelasyon enerjisidir.
Malesef, çok –cisim dalga fonksiyonu çok karmaşık olduğu için bu sonuçları
tayin etmek çok zordur. Bununla birlikte katı ve moleküllerin nicel analizine izin
veren değişik yöntemler vardır. DFT, Hohenberg-Kohn tarafından doğrulanan bir
işlem ile yoğunluk üzerinde değişim ve korelasyonun toplam etkileri ile ilgilenir
(Mete, 2003).
3.8.8. Yerel Yoğunluk Yaklaşımı
Yerel yoğunluk yaklaşımı (LDA), bir molekül veya katıdaki her bir noktanın
belirli bir elektron yoğunluğuna sahip olduğu kabul edilir ve her noktadaki
elektronun, çevresindeki aynı yoğunluklu diğer elektronlarla aynı çok cisim
etkileşmesine maruz kaldığı kabul edilir. O zaman tüm moleküllerin veya bir katını
toplam değiş tokuş korelasyon enerjisi, bütün hacim elemanları üzerinden alınacak
katkıların integrali olarak verilir. LDA’da değiş-tokuş korelasyon enerjisi,
E [n] = ∫ d rn(r) e n(r) (3.124)
şeklinde verilir. Buradaki ( ) terimi, uzaysal olarak düzgün bir yoğunluğuna
sahip olan elektron gazındaki parçacık başına düşen değiş-tokuş korelasyon
enerjisidir. Bu enerji Monte-Carlo hesaplamalarından elde edilebilir.

3. MATERYAL VE METOD Şevket ŞİMŞEK
56
Bir hacim elemanından gelen katkı, yerel elektron yoğunluğuna bağlı olarak farklı
olabilir. (3.124) denkleminde yer alan ’yi hesaplamak için literatürde farklı
yöntemler mevcuttur. Bunlar arasında en çok kullanılan yaklaşım Ceperley-Alder
(1980) yaklaşımıdır. iki kısma ayrıldığında
EXC=EX+EC (3.125)
şeklinde yazılabilir. Buradaki ve Hartree biriminde
sXE
τ4582,0
= (3.126)
EC= -0,0480+0.03111lnτs, τs≥1, -0.0116τs+0.0020τslnτs, τs<1,
(3.127)
şeklindedir. Eşitliklerde yer alan yoğunlukla ilişkili olup, bu ilişki =
şeklindedir. Değiş tokuş-korelasyon potansiyeli ise,
s
XCsXCXC dτ
dE3τEV −= (3.128)
şeklinde ifade edilir.
LDA yaklaşımı genel olarak, bağ enerji değerleri ve bulk modülü deneysel
değerlerden büyük; örgü sabiti değerleri ise deneysel değerlerden küçük sonuçlar
vermektedir. Ayrıca LDA yüzey ve ara yüzey hesaplamalarında ve dinamik
hesaplamalar için fonon dispersiyon bağıntısı hesaplamalarında iyi sonuçlar
vermektedir. Bunun yanında dielektrik sabitleri ve buna bağlı büyüklüklerin
hesaplamalarında, ayrıca zayıf bağlarda ve Hidrojen bağlarında çok iyi sonuçlar
vermemektedir. Ayrıca, saf metaller sabit elektron yoğunluğuna sahip oldukları için
kesin ve doğru sonuçlar verdiği halde, değişen elektron yoğunluğuna sahip
sistemlerde daha az doğru sonuçlar vermektedir.

3. MATERYAL VE METOD Şevket ŞİMŞEK
57
3.8.9. Genelleştirilmiş Gradyent Yaklaşımı
Bazı malzemelerde yoğunluk gradyenti büyük değerlere sahip olabilir.
Yoğunluğun uzaysal değişimini hesaba katan yaklaşımlara Genelleştrilmiş Gradyent
yaklaşımı (GGA) denir.
GGA, her türlü sistemlerde LDA’dan daha iyi sonuçlar üretmez fakat pek çok
sistem için bağ uzunlukları ve toplam enerjiyi daha tahmin ettiği gösterilmiştir.
Genelleştirilmiş Gradyent Yaklaşımı sayesinde, daha da geliştirilmiş
fonksiyonellerin araştırılmasında önemli ilerlemeler kaydedilmiştir. GGA’ da spin
polarizesiz sistemler için değiş-tokuş korelasyon enerjisi:
E [n] = ∫ d rf n(r),∇n(r) (3.129)
şeklinde verilir. LDA’ da inputu tek olmasına rağmen, GGA’ da fonksiyonu
tek değildir ve pek çok farklı formlar önerilmiştir.
Son yıllarda GGA doğrultusundaki gelişmeler devam etmektedir ve hatta yeni
bir sınıf olan “meta-GGA” fonksiyonelleri önerilmiştir. meta-GGA fonksiyonelleri,
yoğunluk ve birinci mertebeden gradyente bağlılığın yanında,
∑ ∇=OCC
i
2i (r)2
1τ(r) ϕ (3.130)
şeklinde ifade edilen Kohn-Sham orbitallerinin kinetik enerji yoğunluğuna da
bağlıdır. Bu durumda meta-GGA fonksiyoneli,
EXCMGGA[n]=∫ d3rg n(r),τ(r) (3.131)
şeklinde olmaktadır. Yeni bir değişkenin tanımlanması ile fonksiyonel formda
kazanılan ilave esneklik, yaklaşıma daha fazla özelliğin dahil edilmesine olanak
verir. Bu yolla, diğer sonuçları etkilemeden bazı fiziksel özellikler için GGA’nın
doğruluğunu artırmak mümkün olabilmiştir.

3. MATERYAL VE METOD Şevket ŞİMŞEK
58
LDA ve GGA açıkca yoğunluğun fonksiyoneli olmasına karşın, meta-
GGA’lar açıkça Kohn-Sham orbitallerine de bağlıdır. Bu hali ile hala DFT bölgesi
içinde bulunduğumuzu belirtmek önemlidir; çünkü Kohn-Sham denkleminde
orbitaller Kohn-Sham potansiyelinin fonksiyonelidir ve Hohenberg-Kohn teoremi
nedeni ile de ayrıca yoğunluğun fonksiyonelleridir.
Bu tür orbital fonksiyonelleri veya gerçek yoğunluk fonksiyonelleri aktif ve
yoğun araştırma alanları olmuştur. Belkide en iyi bilinen orbital fonksiyoneli
aşağıdaki “tam değiş-tokuş enerji” fonksiyonelidir.
∑∫ ∫ ∑ ′−
′′−=
σ
OCC
ij
iσ*
iσ*
iσ33Exxx rr
(r))r((r)xrdrd21[n]E ϕϕϕ (3.132)
Değiş-tokuş fonksiyoneli kullanılıp korelasyon ihmal edilirse, oluşacak
toplam enerji fonksiyoneli tam olarak Hartree-Fock fonksiyoneli olacaktır. Fakat tam
değiş-tokuş, Hartree-Fock orbitallerinden ziyade Khon-Sham denklemi ile
hesaplanır. Ayrıca Hartree-Fock orbitalleri, bir sınırlama olmaksızın (ortonormalite
hariç) minimizasyondan elde edilir. Bu orbitaleler bizi, yerel olmayan Hartree-Fock
potansiyeline götürür. Diğer taraftan tam değiş-tokuş orbitalleri, ek sınırlamalara tabi
tutarak aynı fonksiyoneli minimize ederek elde edilir ve bu durumda tek-parçacık
orbitalleri yerel potansiyelden gelir. Bu nedenle, toplam Hartree-Fock enerjisi daima
tam değiş-tokuş toplam enerjisinden daha azdır fakat bu enerji farkı çok azdır. Ancak
orbitalleri ve orbital öz değerleri gibi tek parçacık özellikleri her iki yaklaşımda da
oldukça farklıdır. Örneğin, Khon-Sham tam enerji spektrumları, deneysel sonuçlara
Hartree-Fock spektrumlarından daha yakındır. Bu da tam değiş-tokuş DFT’nin
korelasyonu dahil etmek açısından, Hartree-Fock spektrumundan daha iyi bir çıkış
noktası olduğu anlamındadır.
Katılarda ve moleküllerde GGA hesaplarında bağ uzunlukları ve örgü
sabitleri deneysel sonuçlardan büyük, bulk modülü ise küçük çıkmaktadır. GGA’nın
en iyi başarısı demirin manyetik momenti üzerinde olmuştur. GGA, bcc demirin
taban durumunu ferromanyetik olarak önerdiği halde, LDA taban-durumu fcc ve
manyetik olmadığını önermiştir. Ayrıca GGA yaklaşımı özellikle hidrojen bağlarının

3. MATERYAL VE METOD Şevket ŞİMŞEK
59
tanımlanmasında önemli sonuçlar vermiştir. Yüzey enerji hesaplamalarında ise
LDA’dan daha düşük sonuçlar vermektedir. Bunun nedeni ise muhtemelen GGA’nın
zayıf bağlar öngörmesinden kaynaklamaktadır. Ayrıca kohesif enerji
hesaplamalarındaki doğruluk GGA’da LDA’dan biraz daha iyidir.
LDA ve GGA basit metallerin bant yapılarını deneysel sonuçlara çok yakın
bulmasına rağmen, yarı iletkenlerde hem LDA hem de GGA uygulandığın da bant
aralığı deneysel değerlerden daha düşük hesaplamaktadır. Bunun sebebinin DFT’nin
kendisinde olduğu iddia edilmesine rağmen DFT’nin tam çözümünün yapılıp
yapılamayacağı da açık bir sorudur. Eğer tam çözüm yapılabilirse daha doğru
sonuçlar verebilecektir. Bant aralığı hesaplamalarında, tam değişim ve zamana bağlı
DFT (ZB-DFT) ve Green Fonksiyonu (GW) metotları daha iyi sonuçlar vermektedir.
Bu metotlar uygulama aşamasında pahalı bilgisayar sistemleri gerektirmektedir. Bu
eksiklik, meta-GGA veya hibrid fonksiyoneller gibi “tam değiş-tokuş fonksiyoneli”
kullanılarak giderilebilir (Deliğöz, 2007).
3.9. Düzlem Dalga Yaklaşımı
Periyodik bir süper hücrenin göz önüne alınmasıyla, sadece elektronların
sonlu bir sayısı ile ilgilenilebilinir ve sonlu bir baz seti kullanarak Kohn-Sham
denklemeleri çözülebilir.
En yaygın olarak kullanılan baz seti düzlem dalga bazlarıdır. Bloch teoremine göre,
elektronik dalga fonksiyonları düzlem dalgaların toplamı olarak yazılabilir,
∑ ++=G
G)ri(knnk GekC(r)ψ (3.133)
Burada ters örgü vektörü, Brillioun bölgesine aittir ve bantlar için tam sayı
indeksidir. Prensipte düzlem dalga baz setleri sonsuzdur, fakat pratikte toplam,
kesilim enerjisi ( )’den daha düşük enerjilere sahip olan düzlem dalgaların sonlu
bir seti ile yer değiştiriyor yani,

3. MATERYAL VE METOD Şevket ŞİMŞEK
60
12
|k+G|2≤Ecut (3.134)
şeklinde ifade ediliyor. Düzlem dalga baz setinin kesilimi, fazladan hesaplamaya
dayalı hatalar ortaya çıkarıyor. Hataların diğer bir kaynağı Brillioun bölgesi
örneklemesinden (sampling) geliyor. Brillioun bölgesi örneklemesi, uzayının bir
bölgesini ifade eden noktalarının sonlu bir sayısı üzerinden bir toplam ile Brillioun
bölgesi üzerinden integrasyonun yer değiştirdiği bir yaklaşımdır. Ancak bu
yaklaşımdan kaynaklanan hatalar, ince bir -nokta ağı( finer k-point mesh)
kullanarak ve kesilim enerjisi ( ) artırılarak azaltılabilir. Bununla birlikte kor
bölgesindeki sıkı bağlı kor durumlarını ve valans elektronlarını tanımlamak için, hızlı
değişen osilasyonları ile elektronik dalga fonksiyonlarını açmak düzlem dalga baz
fonksiyonlarının çok büyük bir sayısını gerektirecektir. Pratikte bu sorun donmuş kor
(frozen-core) yaklaşımı ile yada daha belirgin bir biçimde pseudo potansiyel
yaklaşımı ile çözülüyor. Ayrıca tüm elektron hesaplamalarında kullanılabilen
LABW, LMTO gibi diğer baz setleri de vardır.
3.10. Pseudo Potansiyel Yaklaşımı
Pseudo potansiyel, verilen bir yarıçap, kor yarıçapı )( cr olarak alınarak
gerçek potansiyel gibi inşa edilir. Benzer şekilde, her bir pseudo dalga fonksiyonu
şekil 3.7’de gösterildiği gibi bu uzaklığın ötesinde karşılık gelen dalga fonksiyonuna
uymalıdır. Ayrıca kor bölgesi dışında elde edilen yük yoğunlukları gerçek yük
yoğunluklarına özdeş olmalıdır. Böylece kor bölgesi üzerinde gerçek ve pseudo
dalga fonksiyonlarının genliklerinin karesinin integrali özdeş olmalıdır. Bu şart
norm-koruma olarak biliniyor. Bu tip yerel ve yerel olmayan pseudo potansiyellerin
geçirebilme (transferability) olarak işaret edilen bir özellik olan bir çeşit atomik
ortamlardaki iyon korlarından dolayı saçılmayı tarif edebilir olduğu bilinmektedir.
Pseudo potansiyeller, ab-initio prosedürü kullanılarak inşa edilir. Gerçek
dalga fonksiyonları, tüm-elektron DFT yaklaşımı kullanılarak yalıtkan bir atom için
hesaplanır. Valans dalga fonksiyonları, norm-koruma kısıtlamasına uyarken

3. MATERYAL VE METOD Şevket ŞİMŞEK
61
titreşimleri kaldırmak için kor bölgesinde değiştirilir. Daha sonra Schrödinger
denklemi, pseudo dalga fonksiyonlarını yeniden üretilecek olan Pseudo potansiyelleri
bulmak için tersine çevrilir. Bu prosedür, geniş çapta değişken sistemler arasında
transfer edilebilen pseudo potansiyelleri üretir. Bu, belirli atomik çevreleri
belirlemede kullanılan yarı-ampirik potansiyeli karşılar ve farklı ortamlara kolayca
transfer edilemez (Mete, 2003).
Şekil 3.7. Pseudo potansiyel, Pseudo ve gerçek dalga fonksiyonları
3.10.1. Norm-Koruyucu Pseudo Potansiyeller
‘’FHI98PP’’ paket yazılımı, ABINIT için norm-koruyucu pseudo
potansiyelleri türetmek için kullanılıyor. Bu yazılım aynı zamanda türetilen pseudo
potansiyellerin test edilmesini ve değerlendirmesini kolaylaştırmaktadır(Fuchs ve
Scheffler, 1999).
Kompleks çok atomlu sistemlerin ab-initio elektronik yapı hesaplamalarını
doğru ve verimli olmasını sağlayan norm-koruyucu pseudo potansiyeller DFT içinde
uygulanır ve türetilir. Bu potansiyeller aşağıdaki özelliklere sahiptir.
1) Fiziksel ve kimyasal özelliklerin çoğu valans elektronlarından ortaya çıktığı için,
kor atomik durumlar donmuş (frozen) olarak dikkate alınır. Buna ‘’frozen core
yaklaşımı’’ denir.

3. MATERYAL VE METOD Şevket ŞİMŞEK
62
2) Pseudo potansiyel çekirdek etrafında kor bölgeleri içinde çok daha düz iken kor
yarıçapı dışında gerçek tüm elektron potansiyeline eşit olmalıdır. Başka bir deyişle,
pseudo potansiyel kor bölgesi dışında gerçek valans dalga fonksiyonlarına eşdeğer
olan düz pseudo dalga fonksiyonları üzerinde hareket ediyor, fakat radyal
düğümlerden kaçınmakla gerçek valans ve kor orbitallerinin ortogonalliğini kırıyor.
Bu yüzden pseudo potansiyel, pseudo dalga fonksiyonlarının hesaplama yükünü
kolaylaştıran düzlem dalgalar gibi tam ortonormal setler olarak açılmasına izin
veriyor. Bu, özellikle karmaşık sistemler için Poission ve Schrödinger
denklemlerinin nümerik çözümlerinde önemlidir.
3) Bu pseudo potansiyeller kor bölgesi dışında tüm-elektron tanımlayıcıları gibi aynı
davranan norm-koruyucu kısıtlamalar ile sınırlandırılıyor. Norm-koruma, uygun bir
potansiyel tasarımıyla doğru ve güvenilir hesaplamalar yapar.
Bu özelliklerle birlikte, potansiyelin niteliğini belirleyen bir potansiyel inşa
edildiğinde yapısında karşılaşılan iki önemli kriter vardır.
1) Pseudo potansiyelinin geçirebilirliği (transferability) yerine getirilmelidir. Bu
farklı atomik, moleküller ve katı hal ortamlarda valans elektronlarını doğru bir
şekilde tanımlamak için potansiyelin gücüdür. Öz uyumlu toplam enerji
hesaplamalarında valans durumları, kimyasal bağların oluştuğu yerlerde özellikle kor
bölgesi dışında uygun elektrostatik ve değişim-korelasyon potansiyelleri sağlayan
uygun bir şekilde normalize edilmiş elektron dağılımlarına yol açar ve uygun
enerjilere sahiptirler.
2) Pseudo potansiyelin verimi dikkate alınmalıdır. Bu mümkün olduğu kadar soft
potansiyel üretmek içindir. ‘’soft potansiyel’’ terimi elektron yoğunlukları ve dalga
fonksiyonlarını açmak için birkaç baz fonksiyonu gerektirdiği için kullanılıyor. Bu
da hesaplama yükünü azaltmaya yardımcı oluyor.
Aslında, norm-koruyucu pseudo potansiyeller yerel ve yerel olmayan kısımlara sahip
olmaları için inşa ediliyorlar.
( ) ( ) ( ) ( ) ( ) ( ) ( )'rlm2PSlr
l
0l
l
lm
*lmloc
PS ΩYr
'rrδrΔVΩY'rrδrV'r,rVmax
rr
rrrrrrrr −+−= ∑ ∑
= −= (3.135)

3. MATERYAL VE METOD Şevket ŞİMŞEK
63
Burada ( )rVlocr
yerel terimdir ve yarı yerel bileşenler ( ) ( ) ( )rVrVrΔV locPSl
PSl
rrr−= kor
bölgesine sınırlandırılıyor ve sonunda maxl ötesinde yok oluyorlar. cr kor yarı çapı
dışında büyük radyal uzaklıklar için uzun erimli yerel terim etkindir ve genel
potansiyeli iyonik potansiyele indirger.
Pseudo potansiyel inşa etmek için ilk adım tek atom Hamiltonyenine karşılık
gelen çözüm ile temel durumda seçilen bir değişim-korelasyon şekillenimi ile DFT
kullanarak tüm-elektron potansiyelini hesaplamaktır. Sonra perdelenmiş (veya ara)
pseudo potansiyel hem Humann (1989) hem de Troullier-Martiens ‘in (1991)
aşağıdaki tarifi ile oluşturulur. Bu perdelenmiş pseudo potansiyeller valans durumları
üzerinde hareket ederler,
( ) ( ) ( )rlmPSl
PSl
PSlm ΩYr,εu
r1rψ =
r (3.136)
Burada radyal kısım rölâtivistlik olmayan Schrödinger denklemini sağlıyor.
( ) ( ) ( ) 0r,εuεrV2r
1lldrd
21 PS
lPSl
PSl
SCRPS,l22
2
=
−+
++−
r (3.137)
(3.137) denkleminin tersine çevrilmesi, karşılık gelen dalga fonksiyonları olarak
perdelenmiş pseudo potansiyelleri verir.
( ) ( )( ) ( )ru
drd
r2u1
2r1llεrV PS
l2
2
PSl
2PSl
SCRPS,l
rr+
+−=
(3.138)
Hem Humann hem de Troullier-Martiens kurulum şeması aşağıdaki
kısıtlamarı içeriyor:
1) Pseudo ve gerçek öz değerler denk olmalıdır. nlPSl εε ≡

3. MATERYAL VE METOD Şevket ŞİMŞEK
64
2) Geçirebilirliği (transferability) göz önünde bulundurmak için, pseudo ve tüm-
elektron radyal dalga fonksiyonlarının logaritmik türevleri kor kesilme yarıçapı clr ,
ötesinde karşılaşırlar,
( ) ( )r,εlnudrdr,εlnu
drd
nlnlPSl
PSl → c
lrr > için (3.139)
3) Genlikler , clr ötesinde eşit ve normalizedirler.
( ) ( )r,εur,εu nlnlPSl
PSl → c
lrr > için (3.140)
4) Norm-koruma kısıtlaması empoze edilmelidir,
( ) ( ) drεudrr,εu2r'
0rnl,nl
2r'
0
PSl
PSl ∫∫ = c
lrr >' için (3.141)
5) Radyal pseudo dalga fonksiyonlarında radyal düğümler yoktur. Bu yüzden orjinde
düzenli davranan sürekli potansiyeli sağlamak için iki kez differansiyellenebilir
olmalıdır.
( ) 1lPSl0r
rrulim +
→∝
r (3.142)
Bu kısıtlamalara ek olarak Troullier-Martiens şekillenimi ekstra kısıtlamalar içeriyor.
1) Pseudo potansiyelin eğriliği orjinde kayboluyor.
( ) 0rVdrd
0r
SCRPS,l2
2
==
r (3.143)
2) Pseudo ve tüm-elektron dalga fonksiyonlarının ilk dört türevleri clr de uyuşmalıdır.

3. MATERYAL VE METOD Şevket ŞİMŞEK
65
Troullier-Martiens şekillenimi geçiş metallerinin 3d, 4d, 5d valans
elektronları ve birinci sıra elementlerinin 2p valans durumları için ‘’softer’’ pseudo
potansiyeller türetir. Bu oksijen ve geçiş metalleri için tercih edilebilir
olmasındandır.
Pseudo potansiyeli oluşturmadaki bir sonraki adım perdelemedir. Bu adım da
çok atomlu sistemlerde elektron-iyon etkileşimini ifade eden son iyonik potansiyeli
verir. Bu valans elektronlarından dolayı elektrostatik ve değişim-korelasyon
bileşenlerinin kaldırılmasıyla tamamlanır.
( ) ( ) XCHSCRPS,l
PSl VVrVrV −−=
rr (3.144)
Kleinman-Bylander (1982), pseudo potansiyelin yerel olmadığı (3.135)
denklemindeki gibi açısal kısımlara sınırlandırılmazsa, aynı zamanda radyal
potansiyel bir projeksiyon operatörü ile yer değiştirirse elektronik yapı
hesaplamalarındaki hesaplama gücünde önemli ölçüde bir azalmanın
sağlanabileceğini gösterdiler.
( ) KBlm
KBlm
KBl
PSl XXErΔV ⇒
r (3.145)
Yarı yerel kısmın hesaplama etkinliğini artırmak için, tamamen yerel
olmayan KB formülü olarak yeniden yazılabilir.
( ) 'rXEXrr'rVr'rVr KBlm
KBl
l
0l
l
lm
KBlmloc
PSmax rrrrr ∑ ∑= −=
+=
(3.146)
Burada
( ) ( )( )
( )rlm2
1PSlll
PSl
PSllPS
lm ΩYuΔVΔVur
rurΔVr1Xr
rrr= (3.147)

3. MATERYAL VE METOD Şevket ŞİMŞEK
66
ve KBlE ;
PSl
PSl
PSl
PSl
PSl
PSl
PSlKB
luΔVu
uΔVΔVuE = (3.148)
ile verilen KB enerjisidir.
Tamamen ayrılabilir yerel olmayan KB formülü yarı yerel pseudo
potansiyellerin bellek maliyetini önemli ölçüde azaltır. Bu potansiyeller yerel
olmayan projektörlere bağımlı açısal momentum ve yerel bir potansiyelden inşa
ediliyor. Fourier uzayında, projektör, ( )',GGV değişimiyle ( ) ( )'. GWGW olarak ifade
edilebilir. Bu pseudo potansiyel, bellek boyutunu 2N den N indiriyor.
Yerel olmayan projektörlerin dahil edilmesi ile, KB formülünün valans
durumlarının enerjileri yakınında yada altında ortaya çıkan fiziksel olmayan tayf
durumlarına neden olmadığından emin olmak gerekir. Bu tayf durumları pseudo
potansiyelin geçirebilirliğini yok ediyor. Bu kusur Wronskian teoremini hesaba
katmayan KB Hamiltonyeninden dolayıdır. Wronskian teoremi enerji olarak l açısal
momentum kuantum sayısıyla düzenlenen atomik öz fonksiyonları içerir. Verilen bir
l enerjilerinin değerleri düğümlerin sayısı ile artar. Bu teorem KB şekillenimi için
geçerli olmadığından, tayf durumları sıfır düğüm durumu altında bile görülebilir
(Gonze, Käckell ve Scheffler, 1990).
l açısal momentum kanalı için tayf durumlarının elimine edilmesi pseudo
potansiyelin geçirebilirliğini ortadan kaldırılmaksızın biraz değişiklikle
başarılabilinir. Bu değişiklikler, hem yerel potansiyel olarak farklı bir yarı yerel
potansiyel bileşenini kullanmak için olabilir hem de açısal momentum kanalına
karşılık gelen clr kor kesme yarıçapını ayarlamak için olabilir. Bu kesme yarıçapı
artışı ile geçirebilme kayıp maliyetinde softer potansiyeller elde edilebilir. Bundan
dolayı her hangi bir tayf durumunun meydana gelmesinden kaçınılırken optimize
edilmelidir (Mete, 2003).

3. MATERYAL VE METOD Şevket ŞİMŞEK
67
3.11. Toplam Enerji Hesabı
Bir katının elektronik ve yapısal özelliklerini çalışmak sistemin toplam
enerjisini değerlendirilmesini gerektirir. Fiziksel özelliklerin çoğu toplam enerjiye ya
da bu enerjiler arasındaki farklara bağlı olduğu için, her ab initio hesabı için hayati
öneme sahiptir. Örnek olarak, bir kristalin örgü parametresi toplam enerjiyi minimize
eden parametredir. Örgü sabitini tahmin etmek için toplam enerji hesaplamalarının
bir serisine ihtiyaç vardır. Enerjiye karşı örgü sabiti eğrisi bu hesaplamalardan
çıkarılır. Teorik örgü parametresi, enerjinin minimum değerine karşılık geldiği
noktadaki değeridir.
Katının toplam Hamiltonyeni, çekirdek ve elektronların koordinatlarına bağlı
olarak değiştiğinden, elektronik ve nükleer koordinatlar yoluyla minimize edilir.
3.12. ABINIT Programı
ABINIT yazılım projesi 1997 yılında başladı. İlk hedef, materyal özelliklerinin
ab inito yöntemi kullanarak hesaplayan bilgisayar programının oluşturmak ve bu
programı serbest yazılım lisansı altında dağıtımını sağlamaktır. Aralık 2000’de
ABINIT’in ilk halka açık kullanılabilir versiyonu resmen yayımlandı. Bundan sonra,
ABINIT yazılımının geliştirici ve kullanıcı sayısı hızla arttı. ABINIT programı
malzemelerin özellikleriyle ilgili geniş bir hesaplama olanağı sunmaktadır. ABINIT,
metal, yalıtkan ve yarıiletken malzemelerin örgü parametresi, atomların konumları,
elastik özellikler, fonon, dielektrik ve piezoelektrik özellikler, lineer ve lineer olmayan
optik özellikler, manyetik özellikler, termodinamik özellikler (entropi, serbest enerji,
özısı), elektronik özellikler (metal/yalıtkan karakterizasyonu), vb. özelliklerini hesaplar.
ABINIT’in ana programı Yoğunluk Fonksiyoneli Teorisine dayanmaktadır.
Titreşimler, dielektrik ve piezoelektrik özellikler gibi tepki fonksiyonlarını hesaplamak
için DFT’nin gelişmiş versiyonu olan Yoğunluk Fonksiyoneli Pertürbasyon Teorisi
(DFPT) kullanır. Kristalin örgü dinamikleri ve bağ özellikleri üzerine DFPT’den elde
edilen sonuçlar araştırma makalesinde (Baroni ve ark, 2001), görülebilir. ABINIT, değiş-
tokuş etkisini, enerji fonksiyonun hesaplamalarında değişik yaklaşıklıkları kullanarak
hesaplayabilir. Bu yaklaşıklıklar LDA, GGA ve bunların farklı çeşitleri olabilir. Bu

3. MATERYAL VE METOD Şevket ŞİMŞEK
68
yaklaşıklıklar ile bağ uzunlukları ve açıları daha az hata ile tahmin edilebilir. ABINIT,
periyodik sınır şartları altında bir kutudaki sistemin periyodik gösterimi ile elektronik
dalga fonksiyonlarının bir düzlem dalga genişletilmiş baz seti alınarak oluşturulmuştur.
Bu gösterim özellikle kristal çalışmaları için uygundur: Kutu ilkel birim hücre olarak
alınır. Eğer ilkel olmayan hücre ( ya da süper-hücre) alınırsa öteleme simetrisini
azaltarak program sistemin çalışmasına izin verir.
ABINIT periyodik tablodaki elementler için norm-korunumlu
pseudopotansiyellerin geniş bir kütüphanesine sahiptir. Pseudopotansiyeller, ABINIT
paket programı içerisinde ve www.abinit.org web sayfasında mevcuttur. Ayrıca farklı
değiş-tokuş fonksiyonu, relativistik ya da relativistik olmayan durumlar içinde
pseudopotansiyel oluşturmaya imkân veren birçok yazılım programları vardır.
Oluşturulan bir pseudopotansiyel test edildikten sonra kullanılmalıdır (Eyi, 2009).
3.13. SIESTA Programı
SIESTA (Spanish Initiative for Electronic Simulations with Thousands of
Atoms) (Artacho ve ark, 2009), katı ve moleküllerin elektronik yapı hesaplamalarını
ve moleküler dinamik simülasyonlarını gerçekleştirmek için, hem bir metot hem de
bir bilgisayar uygulamasıdır. SIESTA, van der Waals etkileşimlerini içeren yerel
olmayan fonksiyonların yanında, yerel yoğunluk (LDA-LSD) ve genelleştirilmiş
gradyent (GGA) yaklaşımları içinde standart Kohn-Sham öz uyumlu yoğunluk
fonksiyonel metodunu kullanır. SIESTA programıyla, toplam ve parçalı enerjiler,
atomik kuvvetler, stress tensörü, elektrik dipol momenti, elektron yoğunluğu, atomik,
orbital ve bağ popülâsyonu gibi özellikler hesaplanabilir. Ayrıca geometri
optimizasyonu, band yapı, dielektrik polarizasyonu, titreşim (fonon) özellikleri gibi
birçok özellik de SIESTA programıyla hesaplanabilmektedir.
3.14. PHONON Programı
PHONON (Parlinski, 2011), fonon dispersiyon eğrilerini ve kristallerin fonon
yoğunluk spektrumlarını, ya kuvvet sabitleri yolu ile ya da ab initio programları ile

3. MATERYAL VE METOD Şevket ŞİMŞEK
69
hesaplanan kuvvetleri kullanarak hesaplayan bir yazılımdır. Yazılım, 230 adet
kristalografik uzay grubundan birini kullanarak kristal yapıyı kurar, komşuluk
listesini ve kuvvet sabitlerinin matrisini bulur ve dinamik matrisi kurar. Fonon
frekanslarını ve fonon simetrilerini (Γ-noktasında) ve fonon durumlar yoğunluğunu
üretir. Bu tür hesaplamalar, ya her hangi sayıda koordinasyon kabukları içinde ya da
periyodik sınır şartları uygulanmış herhangi bir paralelkenar şekilli süper hücre
içindeki komşuluk listesi için yapılabilir. Bir veya birçok atom denge konumlarından
ayrıldıklarında ortaya çıkan kuvvet sabitleri bir ab initio programı ile oluşturulan
Hellmann-Feynman kuvvetlerinden hesaplanabilir.
PHONON, bazı fonon frekanslarını, değişik şekle sahip süper hücrelerden,
“linear response” hesaplamalarından veya deneyden elde edilen değerlere fit ederek
kuvvet sabitlerini iyileştirebilir. Polar (kutuplu) kristaller için Born efektif yükler ve
optik dielektrik sabiti bilinirse, dış makroskopik alanın sebep olduğu optik mod
yarılmasını da hesaplayabilir. Program, serbestlik dereceleri azaltılmış ve/veya rigid
atomlardan oluşan modeller için de fononları hesaplayabilir. PHONON, süper hücre
yaklaşımı ve bir ab initio programı kullanarak, bulk kristallerin, yüzeylerin, ince
filmlerin, boşluklara sahip kristallerin, ara yer safsızlıklarının fononlarını
hesaplayabilir. Aynı zamanda, “harmonik yaklaşım”ı kullanarak termodinamik
fonksiyonu, iç enerjiyi, entropiyi ve ısı kapasitesini de hesaplar. Hatta yer
değiştirmelerin karelerinin ortalamasını temsil eden Debye-Waller çarpanlarını,
koherent nötron saçılmasının yapı faktörünü vs. de hesaplayabilir (Deligöz, 2007 ).

3. MATERYAL VE METOD Şevket ŞİMŞEK
70

4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
71
4. BULGULAR VE TARTIŞMA
4.1. Hesaplama Metodu
Bu tez çalışmasında yoğunluk fonksiyoneli yöntemleri kullanılarak AgNbO3,
AgTaO3, NaNbO3 ve NaTaO3 kristallerinin oda sıcaklığında elektronik band yapısı,
parçalı (PDOS) ve toplam durum yoğunlukları (DOS) ve dinamik özellikleri (elastik
sabitlerini, Born efektif yükünü, optik dielektrik sabiti ve fonon dispersiyon eğrileri)
yerel yoğunluk yaklaşımı (LDA) altında pseudo potansiyel yöntemle hesaplandı.
Hesaplamalarda değiş-tokuş ve korelasyon etkileri, LDA altında valans
elektronları, çekirdek ve kor elektronları arasındaki etkileşimi Troullier-Martiens
(1991) formatında üretilen Fritz-Haber-Institute (FHI) fonksiyonelleri kullanılarak
hesaba katıldı. Elektronik dalga fonksiyonları için düzlem dalga baz setleri
kullanıldı. Düzlem dalga baz setleri için kinetik enerji kesme değeri AgNbO3 için 70
Hartree, AgTaO3 için 42 Hartree, NaNbO3 için 68 Hartree ve NaTaO3 içinde 40
Hartree olarak alındı. Hesaplamalarda Ag atomu için 110 54 sd , Nb atomu için 14 54 sd , Na atomu için 13s , Ta atomu için 32 56 ds ve O atomu için de 42 22 ps
elektronları gerçek valans elektronları olarak alındı. Kohn-Sham denklemlerinin
çözümleri, ‘’conjugate gradient minimization method’’ kullanılarak ABINIT
yazılımı ile yerine getirildi. Brillouin bölgesinde özel k noktalarının üretimi için
Monkhorst-Pack (1976), yöntemi ile ABO3 kristalleri için 8x8x8 Monkhorst-Pack
örgü ağı kullanıldı.
ABO3 tipi kristalinin örgü dinamiği çalışmalarında lineer tepki yaklaşımları
kullanıldı. Örgü dinamiği ve elastik özellikler, yoğunluk fonksiyoneli pertürbasyon
teorisi (DFPT) çatısı altında hesaplandı. Kristallerin örgü dinamiğinin
karakterizasyonu yapılarak her bir iyonun Born efektif yük tensörü, optik dielektrik
sabiti, elastik sabitler ve fonon dispersiyon eğrisi belirlendi.
ABINIT yazılım programı yardımıyla sözü edilen kristallerin örgü sabiti,
atomik pozisyonları, elektronik band yapısı, DOS, PDOS, Born efektif yükleri, optik
dielektrik sabiti ve elastik sabitleri hesaplandı.
4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
72
Fonon disperisyon eğrileri ve fonon durum yoğunluğunu hesaplamak için
PHONON programı kullanıldı. ABO3 kristallerinin fonon dispersiyon eğrilerini ve
fonon durum yoğunluğunu hesaplamak için Hellman-Feyman kuvvet sabitleri
SIESTA programıyla hesaplandı. Hesaplama ile ilgili ayrıntılar kesim 4.4.3 de
verilmiştir. PHONON bu kuvvet sabitlerini kullanarak dispersiyon eğrilerini ve
durum yoğunluklarını hesapladı.
4.2. Örgü Parametreleri
Hesaplamalarda ilk adım olarak kristalin toplam enerjisinin kristalin
hacmine oranı minimize edilerek, kristalin örgü parametresi hesaplandı. Hesaplanan
bu değerler Çizelge 4.1 de gösterilmiştir. Bulunan sonuçlar deneysel ve mevcut
teorik örgü sabitleri ile kıyaslandı. Çizelge 4.1 den de görüldüğü gibi bizim
kullandığımız yöntemlerle hesapladığımız örgü parametrelerinin, deneysel sonuçlarla
uyum içinde olduğu görülmektedir. Aynı zamanda ABO3 kristallerinin atomik
pozisyonlarını optimize ettik. ABO3 kristallerinin optimize edilmiş konumları
sırasıyla çizelge 4.2, çizelge 4.3, çizelge 4.4 ve çizelge 4.5 de verilmiştir. Tüm
hesaplamalarda optimize edilmiş örgü parametrelerini ve atomik pozisyonları
kullandık.
Çizelge 4.1. ABO3 kristallerinin örgü parametreleri Bu çalışmada (Å) Deneysel değer (Å) Yüzde hata(%) a b c a b c a b c
AgNbO3 5,4999 5,5554 15,5041 5,5516a 5,6080a 15,6503a 0,94a 0,94a 0,94a 5,5468b 5,6038b 15,6421b 0,85b 0,87b 0,89b
AgTaO3 5,5751 5,5751 5,5751 5,5756c 5,5756c 5,5756c 0,02c 0,02c 0,02c - - - - - -
NaNbO3 5,4374 5,5002 15,2190 5,5010d 5,5645d 15,3970d 1,16d 1,16d 1,16d
- - - - - -
NaTaO3 5,4974 7,7036 5,3885 5,4842e 7,7952e 5,5213e 0,24e 1,18e 2,46f
5,4768f 7,7890f 5,5212f 0,37f 1,11f 2,46f
aLevin-2009, bSciau-2004, cFrancombe-1958, dMishra-2007, eKennedy-1999 , fSiozaki-2002
4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
73
Çizelge 4.2. Ortorombik yapıda AgNbO3 kristalinin atomik konumları Atom Tipi Wyckoff Atomik pozisyonlar
x y z Ag1 4c 0,772117 0,195975 0,750000 Ag2 4d 0,757945 0,250000 0,500000 Nb 8e 0,740034 0,700060 0,624645 O1 4d 0,685998 0,770718 0,750000 O2 4c 0,819279 0,750000 0,500000 O3 8e 0,442891 0,539703 0,607750 O4 8e 0,945994 0,435108 0,641172
Çizelge 4.3. Ortorombik yapıda NaNbO3 kristalinin atomik konumları
Atom Tipi Wyckoff Atomik pozisyonlar x y z
Na1 4c 0,257864 0,750000 0,000000 Na2 4d 0,258410 0,793783 0,250000 Nb 8e 0,244169 0,271159 0,125318 O1 4d 0,319874 0,250000 0,000000 O2 4c 0,177542 0,221109 0,250000 O3 8e 0,543380 0,039405 0,143587 O4 8e 0,951913 0,452279 0,107410
Çizelge 4.4. Ortorombik yapıda NaTaO3 kristalinin atomik konumları
Atom Tipi Wyckoff Atomik pozisyonlar x y z
Na 4c 0,036432 0,750000 0,492267 Ta 4a 0,000000 0,000000 0,000000 O1 4c 0,521203 0,250000 0,415092 O2 8d 0,293731 0,956044 0,204409
Çizelge 4.5. Rombohedral yapıda AgTaO3 kristalinin atomik konumları
Atom Tipi Wyckoff Atomik pozisyonlar x y z
Na 2a 0,249999 0,249999 0,249999 Ta 2a 0,000000 0,000000 0,000000 O 6b 0,820741 -0,320742 0,249999

4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
74
4.3. Elektronik Band Yapısı ve Durum Yoğunluğu
Brillouin bölgesinin yüksek simetri yönlerinde ortorombik yapıdaki
AgNbO3, NaNbO3, NaTaO3 kristallerinin ve rombohedral yapıdaki AgTaO3
kristalinin elektronik band yapıları, parçalı ve toplam durum yoğunlukları
hesaplandı. Fermi seviyesi sıfır enerji seviyesi olarak seçildi ve şekillerde sıralı
noktalar olarak gösterildi. Ortorombik yapıdaki AgNbO3, NaNbO3, NaTaO3
kristallerinin ters örgüdeki yüksek simetri noktaları Г (0, 0, 0); X (1/2, 0, 0); S (1/2,
1/2, 0); R (1/2, 1/2, 1/2); T (0, 1/2, 1/2); Y (0, 1/2, 0); Z (0, 0, 1/2); U (1/2, 0, 1/2 ) ve
rombohedral yapıdaki AgTaO3 kristalinin ters örgüdeki yüksek simetri noktaları Г
(0, 0, 0); X (-1/3, 1/6, 1/6); A (1/2, 0, 0); Z (1/2, 1/2, 1/2); D (1/2, 1/2, 0) şeklindedir.
Ortorombik ve rombohedral yapıdaki Brillouin bölgeleri Şekil 4.1 de verilmiştir.
(a) (b)
Şekil 4.1. (a) Ortorombik yapıda, (b) Rombohedral yapıda Brillouin bölgesinin yüksek simetri noktaları
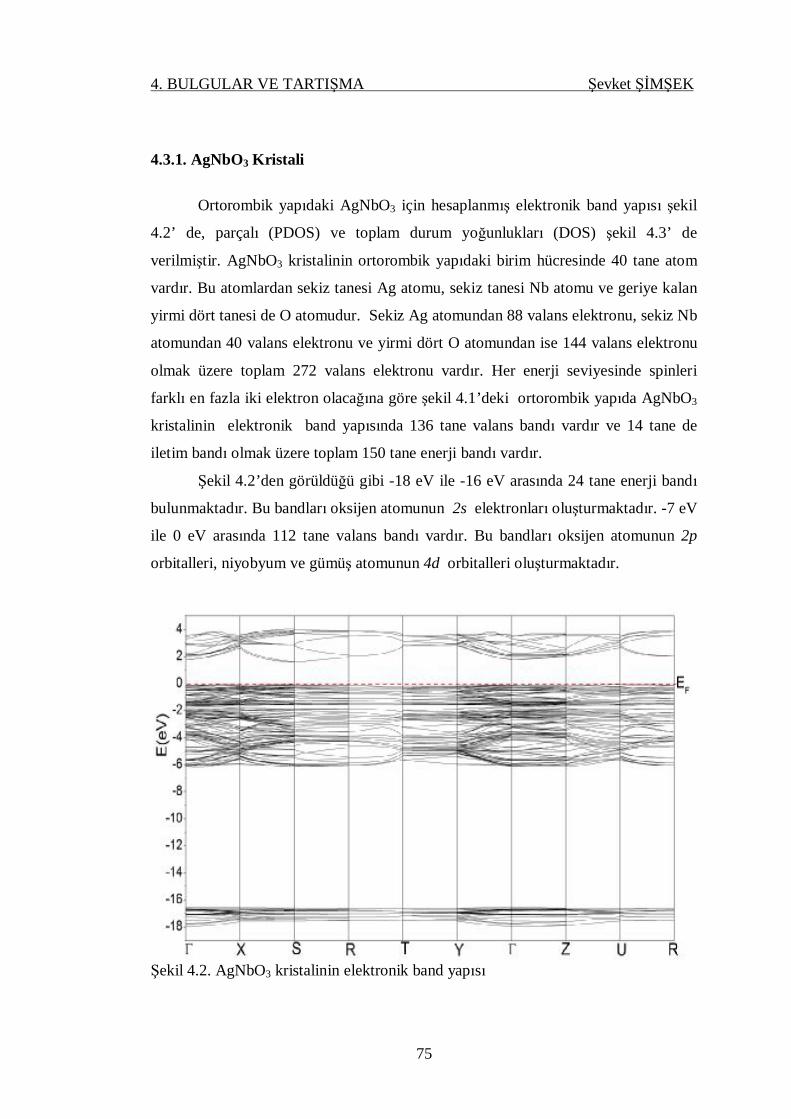
4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
75
4.3.1. AgNbO3 Kristali
Ortorombik yapıdaki AgNbO3 için hesaplanmış elektronik band yapısı şekil
4.2’ de, parçalı (PDOS) ve toplam durum yoğunlukları (DOS) şekil 4.3’ de
verilmiştir. AgNbO3 kristalinin ortorombik yapıdaki birim hücresinde 40 tane atom
vardır. Bu atomlardan sekiz tanesi Ag atomu, sekiz tanesi Nb atomu ve geriye kalan
yirmi dört tanesi de O atomudur. Sekiz Ag atomundan 88 valans elektronu, sekiz Nb
atomundan 40 valans elektronu ve yirmi dört O atomundan ise 144 valans elektronu
olmak üzere toplam 272 valans elektronu vardır. Her enerji seviyesinde spinleri
farklı en fazla iki elektron olacağına göre şekil 4.1’deki ortorombik yapıda AgNbO3
kristalinin elektronik band yapısında 136 tane valans bandı vardır ve 14 tane de
iletim bandı olmak üzere toplam 150 tane enerji bandı vardır.
Şekil 4.2’den görüldüğü gibi -18 eV ile -16 eV arasında 24 tane enerji bandı
bulunmaktadır. Bu bandları oksijen atomunun 2s elektronları oluşturmaktadır. -7 eV
ile 0 eV arasında 112 tane valans bandı vardır. Bu bandları oksijen atomunun 2p
orbitalleri, niyobyum ve gümüş atomunun 4d orbitalleri oluşturmaktadır.
Şekil 4.2. AgNbO3 kristalinin elektronik band yapısı
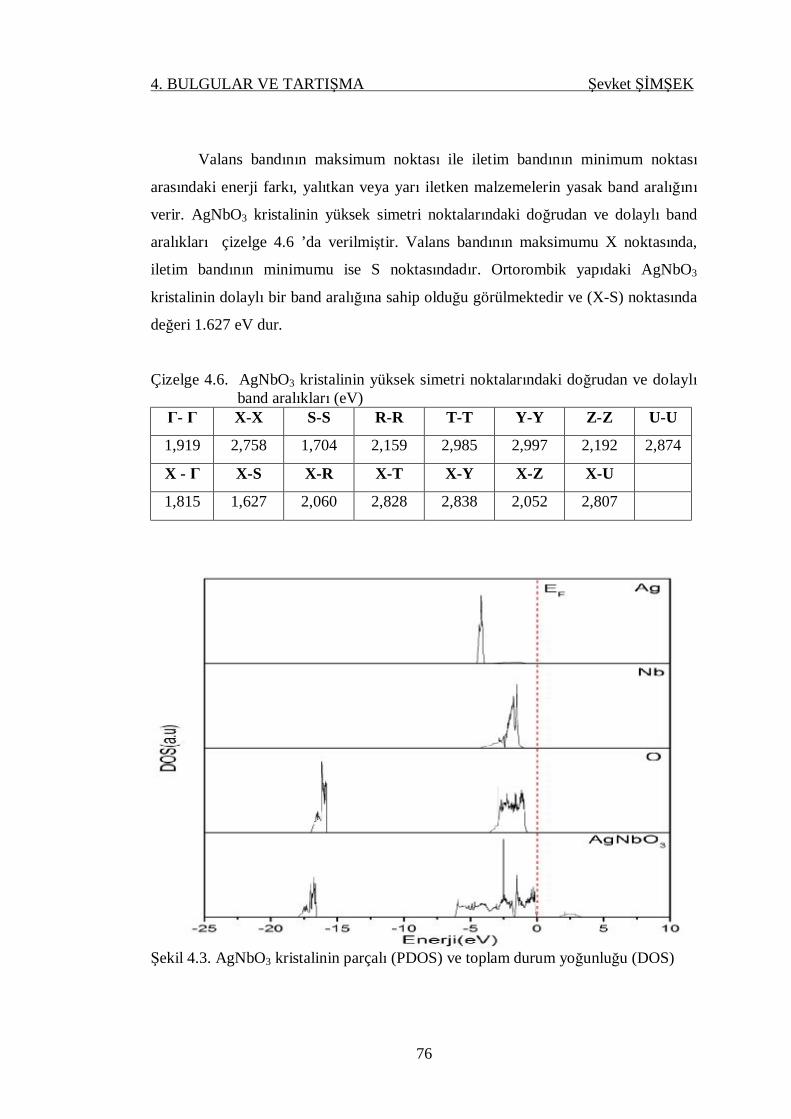
4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
76
Valans bandının maksimum noktası ile iletim bandının minimum noktası
arasındaki enerji farkı, yalıtkan veya yarı iletken malzemelerin yasak band aralığını
verir. AgNbO3 kristalinin yüksek simetri noktalarındaki doğrudan ve dolaylı band
aralıkları çizelge 4.6 ’da verilmiştir. Valans bandının maksimumu X noktasında,
iletim bandının minimumu ise S noktasındadır. Ortorombik yapıdaki AgNbO3
kristalinin dolaylı bir band aralığına sahip olduğu görülmektedir ve (X-S) noktasında
değeri 1.627 eV dur.
Çizelge 4.6. AgNbO3 kristalinin yüksek simetri noktalarındaki doğrudan ve dolaylı band aralıkları (eV)
Г- Г X-X S-S R-R T-T Y-Y Z-Z U-U
1,919 2,758 1,704 2,159 2,985 2,997 2,192 2,874
X - Г X-S X-R X-T X-Y X-Z X-U
1,815 1,627 2,060 2,828 2,838 2,052 2,807
Şekil 4.3. AgNbO3 kristalinin parçalı (PDOS) ve toplam durum yoğunluğu (DOS)

4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
77
4.3.2. NaNbO3 Kristali
NaNbO3 kristali için elektronik band yapısı, PDOS ve DOS sırasıyla şekil
4.4 ve Şekil 4.5’de verilmiştir. NaNbO3 kristalinin ortorombik yapıdaki birim
hücresinde 40 tane atom vardır. Bu atomlardan sekiz tanesi Na atomu, sekiz tanesi
Nb atomu ve geriye kalan yirmi dört tanesi de O atomudur. Şekil 4.4’deki NaNbO3
kristalinin elektronik band yapısında 110 tane band vardır. Bunlardan 96 tanesi
valans bandı ve 14 tanesi de iletim bandıdır. -19 eV ile -15 eV arasında 24 tane
enerji bandı bulunmaktadır. Bu bandları oksijen atomunun s2 elektronları
oluşturmaktadır. -7 eV ile 0 eV arasında 72 tane valans bandı vardır. Bu bandları
oksijen atomunun 2p, niyobyum atomun 4d ve 5s elektronları oluşturmaktadır.
Şekil 4.4’den görüldüğü gibi NaNbO3 kristalinin valans bandının maksimum
ve minimum noktaları Г-Г simetri noktasındadır. NaNbO3 kristalinin elektronik
band yapısı AgNbO3 kristalinin elektronik band yapısına benzemektedir fakat
ortorombik yapıdaki NaNbO3 kristali, ortorombik AgNbO3 kristalinin aksine
doğrudan band aralığına sahip bir kristal olduğu görülmektedir. Г- Г simetri
noktasındaki değeri 1.586 eV dur ve AgNbO3 kristalinden daha düşük bir yasak band
aralığına sahiptir. NaNbO3 kristalinin yüksek simetri noktalarındaki doğrudan ve
dolaylı band aralıkları çizelge 4.7’de verilmiştir.
Çizelge 4.7. NaNbO3 kristalinin yüksek simetri noktalarındaki doğrudan ve dolaylı band aralıkları (eV)
Г- Г X-X S-S R-R T T Y-Y Z-Z U-U
1,586 2,743 1,915 2,323 3,289 3,130 1,744 3,041
Г-X Г -S Г-R Г-T Г-Y Г-Z Г-U
2,670 1,843 2,228 2,976 2,853 1,734 2,908
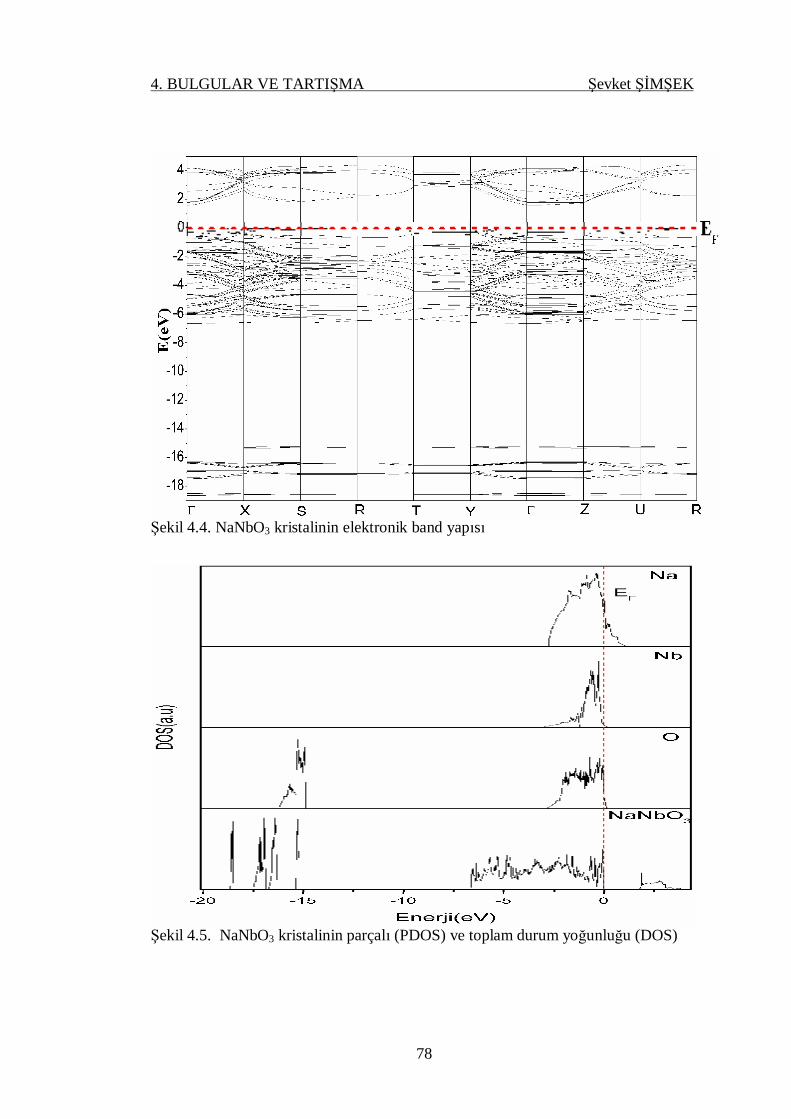
4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
78
Şekil 4.4. NaNbO3 kristalinin elektronik band yapısı
Şekil 4.5. NaNbO3 kristalinin parçalı (PDOS) ve toplam durum yoğunluğu (DOS)
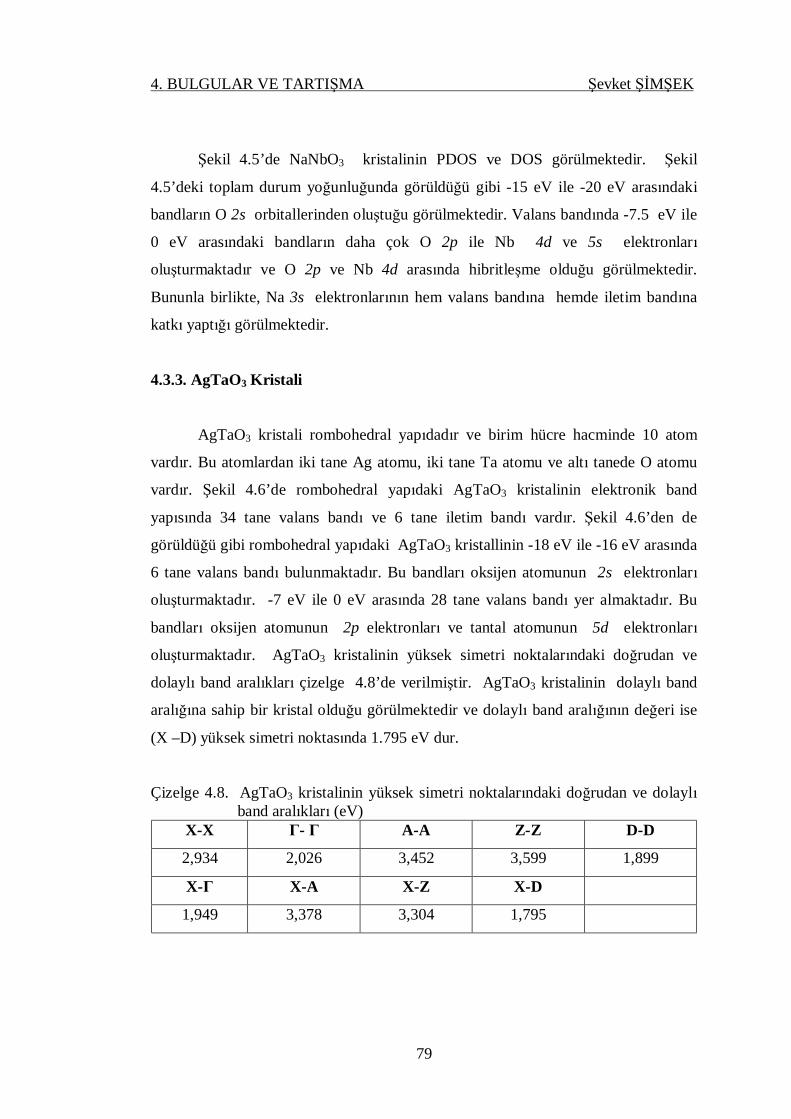
4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
79
Şekil 4.5’de NaNbO3 kristalinin PDOS ve DOS görülmektedir. Şekil
4.5’deki toplam durum yoğunluğunda görüldüğü gibi -15 eV ile -20 eV arasındaki
bandların O 2s orbitallerinden oluştuğu görülmektedir. Valans bandında -7.5 eV ile
0 eV arasındaki bandların daha çok O 2p ile Nb 4d ve 5s elektronları
oluşturmaktadır ve O 2p ve Nb 4d arasında hibritleşme olduğu görülmektedir.
Bununla birlikte, Na 3s elektronlarının hem valans bandına hemde iletim bandına
katkı yaptığı görülmektedir.
4.3.3. AgTaO3 Kristali
AgTaO3 kristali rombohedral yapıdadır ve birim hücre hacminde 10 atom
vardır. Bu atomlardan iki tane Ag atomu, iki tane Ta atomu ve altı tanede O atomu
vardır. Şekil 4.6’de rombohedral yapıdaki AgTaO3 kristalinin elektronik band
yapısında 34 tane valans bandı ve 6 tane iletim bandı vardır. Şekil 4.6’den de
görüldüğü gibi rombohedral yapıdaki AgTaO3 kristallinin -18 eV ile -16 eV arasında
6 tane valans bandı bulunmaktadır. Bu bandları oksijen atomunun 2s elektronları
oluşturmaktadır. -7 eV ile 0 eV arasında 28 tane valans bandı yer almaktadır. Bu
bandları oksijen atomunun 2p elektronları ve tantal atomunun 5d elektronları
oluşturmaktadır. AgTaO3 kristalinin yüksek simetri noktalarındaki doğrudan ve
dolaylı band aralıkları çizelge 4.8’de verilmiştir. AgTaO3 kristalinin dolaylı band
aralığına sahip bir kristal olduğu görülmektedir ve dolaylı band aralığının değeri ise
(X –D) yüksek simetri noktasında 1.795 eV dur.
Çizelge 4.8. AgTaO3 kristalinin yüksek simetri noktalarındaki doğrudan ve dolaylı band aralıkları (eV)
X-X Г- Г A-A Z-Z D-D
2,934 2,026 3,452 3,599 1,899
X-Г X-A X-Z X-D
1,949 3,378 3,304 1,795
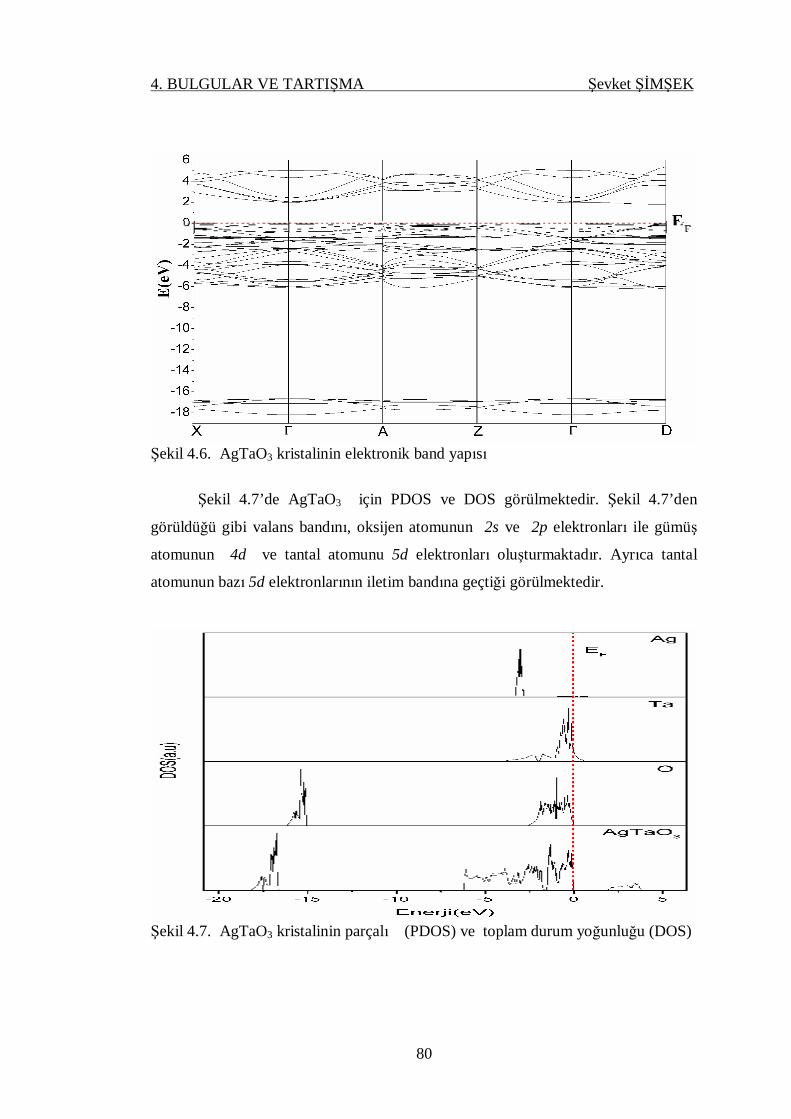
4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
80
Şekil 4.6. AgTaO3 kristalinin elektronik band yapısı
Şekil 4.7’de AgTaO3 için PDOS ve DOS görülmektedir. Şekil 4.7’den
görüldüğü gibi valans bandını, oksijen atomunun 2s ve 2p elektronları ile gümüş
atomunun 4d ve tantal atomunu 5d elektronları oluşturmaktadır. Ayrıca tantal
atomunun bazı 5d elektronlarının iletim bandına geçtiği görülmektedir.
Şekil 4.7. AgTaO3 kristalinin parçalı (PDOS) ve toplam durum yoğunluğu (DOS)
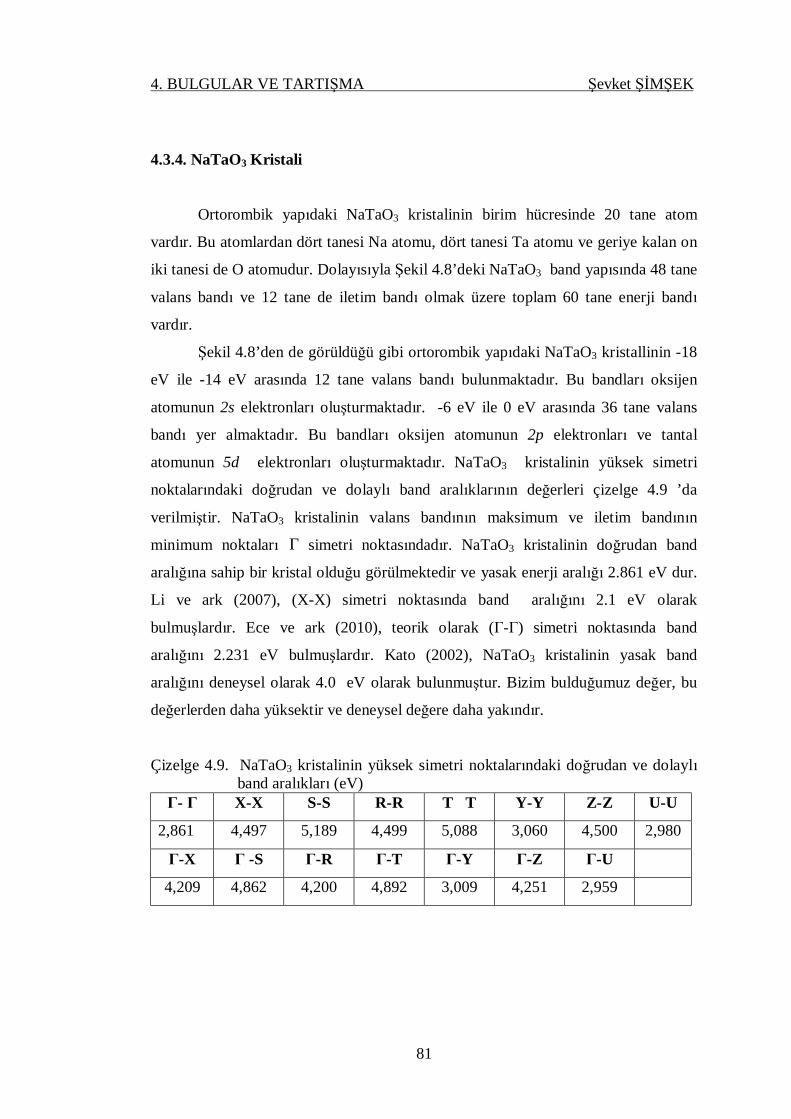
4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
81
4.3.4. NaTaO3 Kristali
Ortorombik yapıdaki NaTaO3 kristalinin birim hücresinde 20 tane atom
vardır. Bu atomlardan dört tanesi Na atomu, dört tanesi Ta atomu ve geriye kalan on
iki tanesi de O atomudur. Dolayısıyla Şekil 4.8’deki NaTaO3 band yapısında 48 tane
valans bandı ve 12 tane de iletim bandı olmak üzere toplam 60 tane enerji bandı
vardır.
Şekil 4.8’den de görüldüğü gibi ortorombik yapıdaki NaTaO3 kristallinin -18
eV ile -14 eV arasında 12 tane valans bandı bulunmaktadır. Bu bandları oksijen
atomunun 2s elektronları oluşturmaktadır. -6 eV ile 0 eV arasında 36 tane valans
bandı yer almaktadır. Bu bandları oksijen atomunun 2p elektronları ve tantal
atomunun 5d elektronları oluşturmaktadır. NaTaO3 kristalinin yüksek simetri
noktalarındaki doğrudan ve dolaylı band aralıklarının değerleri çizelge 4.9 ’da
verilmiştir. NaTaO3 kristalinin valans bandının maksimum ve iletim bandının
minimum noktaları Γ simetri noktasındadır. NaTaO3 kristalinin doğrudan band
aralığına sahip bir kristal olduğu görülmektedir ve yasak enerji aralığı 2.861 eV dur.
Li ve ark (2007), (X-X) simetri noktasında band aralığını 2.1 eV olarak
bulmuşlardır. Ece ve ark (2010), teorik olarak (Г-Г) simetri noktasında band
aralığını 2.231 eV bulmuşlardır. Kato (2002), NaTaO3 kristalinin yasak band
aralığını deneysel olarak 4.0 eV olarak bulunmuştur. Bizim bulduğumuz değer, bu
değerlerden daha yüksektir ve deneysel değere daha yakındır.
Çizelge 4.9. NaTaO3 kristalinin yüksek simetri noktalarındaki doğrudan ve dolaylı band aralıkları (eV)
Г- Г X-X S-S R-R T T Y-Y Z-Z U-U
2,861 4,497 5,189 4,499 5,088 3,060 4,500 2,980
Г-X Г -S Г-R Г-T Г-Y Г-Z Г-U
4,209 4,862 4,200 4,892 3,009 4,251 2,959
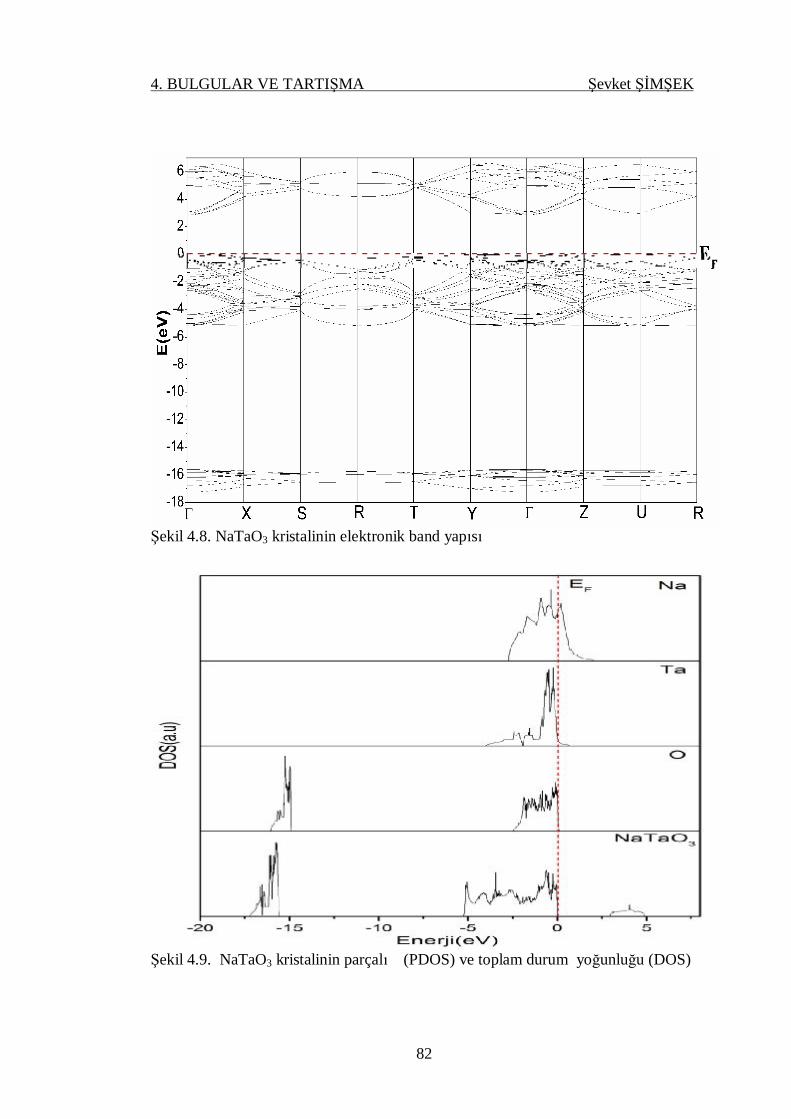
4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
82
Şekil 4.8. NaTaO3 kristalinin elektronik band yapısı
Şekil 4.9. NaTaO3 kristalinin parçalı (PDOS) ve toplam durum yoğunluğu (DOS)

4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
83
Şekil 4.9’ da DOS görüldüğü gibi valans bandının genişliği -5,786 – 0 eV
arasındadır. Valans bandını Ta 5d ile O 2p elektronları oluşturmaktadır. Ta nın
PDOS şeklinden bazı 5d elektronlarının valans bandına geçtiği görülmektedir.
Böylece valans bandında Ta 5d ile O 2p arasında hibritleşme olmaktadır. Na’nın
PDOS ‘una bakılırsa, -5,1 – 0 eV arasında küçük pikler vardır. Bu pikler bazı Na
3s elektronlarının TaO3 yapısına girdiğini göstermektedir ve Ta ile O arasındaki
bağlarda Na yer almaktadırlar.
Sonuç alarak AgNbO3, NaNbO3, AgTaO3 ve NaTaO3 kristallerinin elektronik
band yapıları ve durum yoğunluklarının birbirine benzediği görülmektedir. Fakat
AgTaO3 ve NaTaO3 kristallerinin aksine, AgNbO3 ve NaNbO3 kristallerinin
elektronik band yapısında daha fazla enerji bandı olduğu görülmektedir. Bunum
nedeni, bu kristallerin birim hacimde farklı sayıda atom içermesi ve birim hacimdeki
atomların hesaplamayı dahil edilen valans elektronlarının sayısının farklı olmasından
kaynaklanmaktadır. Diğer taraftan, AgNbO3 ve AgTaO3 kristallerinin aksine
NaNbO3 ve NaTaO3 kristallerinin doğrudan band aralığına sahip kristaller olduğu
görülmektedir. Ayrıca hesaplanan band aralıklarının deneysel değerlerden küçük
olduğu görülmektedir. Bunun nedeni DFT hesaplamalarında değiş-tokuş enerjisinin
süreksizliğinden kaynaklanmaktadır.
4.4. Dinamik Özellikler
4.4.1. Elastik Sabitler ve Bulk Modülü
Elastik sabitler dışarıdan uygulanan bir strain’e karşı kristalin sertliğini
belirler. Ayrıca katıların mekaniksel ve dinamiksel özellikleri hakkında ilginç bilgiler
verir ve kristallerin dinamiksel ve mekaniksel davranışları arasında bir bağlantı
kurar. İlk prensip hesaplamalarından materyallerin elastik katsayılarının
hesaplamanın birçok yolu vardır. Doğrudan ya da dolaylı metotlar kullanılarak
elastik sabitler hesaplanabilir. Doğrudan yaklaşımda stres (zor), her starin (zorlanma)
şartı altında optimize olmuş içsel koordinatlar ile strain’in bir fonksiyonu olarak
hesaplanır. Dolaylı yaklaşımda ise elastik sabitler, elektronik özellikle ilgili olarak

4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
84
sıkıştırılmış iyon kısmında ve alt örgülerin rölatif yer değiştirmesiyle ilgili olarak
içsel strain kısmında bir fark alarak ele alınır. Sonuç olarak her iki yöntemle de aynı
sonuçlar elde edilir.
Ortorombik yapıdaki NaTaO3 ve rombohedral yapıdaki AgTaO3 kristalleri
için ilk defa bu çalışmada elastik sabitleri ve bulk modülleri hesaplandı. NaTaO3 ve
AgTaO3 kristalleri için hesaplanan elastik sabitler ve bulk modülleri sırasıyla çizelge
4.10 ve çizelge 4.11’de verilmiştir.
Ortorombik kristal sisteminin elastik davranışı 9 tane elastik sabit ile
karakterize edilir. NaTaO3 kristali için bulk modülü, çizelge 4.10 ’daki elastik
sabitler kullanılarak denklem (4.1)’den hesaplandı (Wu ve ark, 2007).
= [ + + + 2( + + )] (4.1)
Çizelge 4.10. Ortorombik yapıda hesaplanmış NaTaO3’ün elastik sabitleri (GPa) B
268 310 243 85 135 88 77 95 30 136
Hesaplanan elastik sabitler ortorombik yapı için (C11+C22-2C12)>0, (C11+C33-2C13)>0, (C22+C33-2C23)>0, C11>0, C22>0, C33>0, C44>0, C55>0, C66>0 ve (C11+C22+C33+2C12+2C13+2C23)>0 şeklinde ifade edilen mekanik denge koşullarını (Wu ve ark, 2007) sağladığı görülmektedir. Ayrıca bulk modülü için ifade edilen ( + + ) < < ( + + ) mekanik denge
koşulunu sağlamaktadır.
Rombohedral yapıdaki AgTaO3 kristalinin bağımsız elastik katsayılarının
sayısı 7 dir. NaTaO3 kristali için bulk modülü, Çizelge 4.11 ’deki elastik sabitler
kullanılarak denklem 4.2’den hesaplandı (Wang ve ark, 2010).
= [2 + 2 + 4 + ] (4.2)
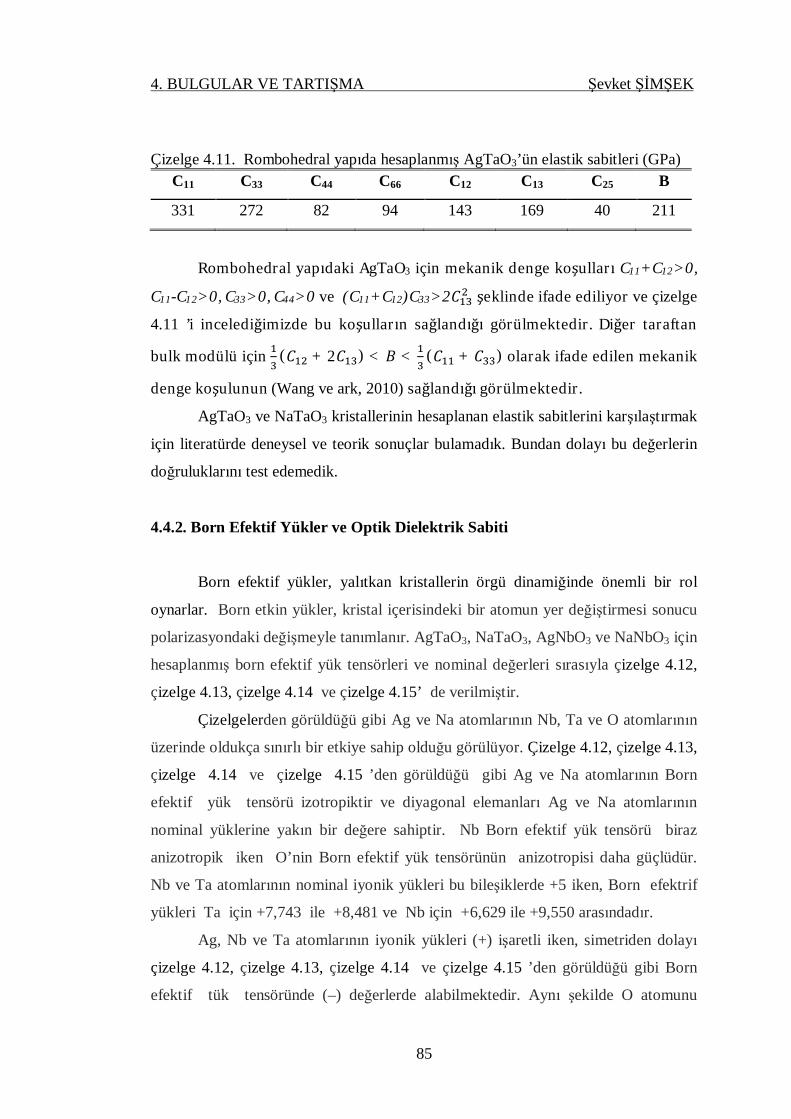
4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
85
Çizelge 4.11. Rombohedral yapıda hesaplanmış AgTaO3’ün elastik sabitleri (GPa) C11 C33 C44 C66 C12 C13 C25 B
331 272 82 94 143 169 40 211
Rombohedral yapıdaki AgTaO3 için mekanik denge koşulları C11+C12>0, C11-C12>0, C33>0, C44>0 ve (C11+C12)C33>2 şeklinde ifade ediliyor ve çizelge
4.11 ’i incelediğimizde bu koşulların sağlandığı görülmektedir. Diğer taraftan bulk modülü için ( + 2 ) < < ( + ) olarak ifade edilen mekanik denge koşulunun (Wang ve ark, 2010) sağlandığı görülmektedir. AgTaO3 ve NaTaO3 kristallerinin hesaplanan elastik sabitlerini karşılaştırmak
için literatürde deneysel ve teorik sonuçlar bulamadık. Bundan dolayı bu değerlerin
doğruluklarını test edemedik. 4.4.2. Born Efektif Yükler ve Optik Dielektrik Sabiti
Born efektif yükler, yalıtkan kristallerin örgü dinamiğinde önemli bir rol
oynarlar. Born etkin yükler, kristal içerisindeki bir atomun yer değiştirmesi sonucu
polarizasyondaki değişmeyle tanımlanır. AgTaO3, NaTaO3, AgNbO3 ve NaNbO3 için
hesaplanmış born efektif yük tensörleri ve nominal değerleri sırasıyla çizelge 4.12,
çizelge 4.13, çizelge 4.14 ve çizelge 4.15’ de verilmiştir.
Çizelgelerden görüldüğü gibi Ag ve Na atomlarının Nb, Ta ve O atomlarının
üzerinde oldukça sınırlı bir etkiye sahip olduğu görülüyor. Çizelge 4.12, çizelge 4.13,
çizelge 4.14 ve çizelge 4.15 ’den görüldüğü gibi Ag ve Na atomlarının Born
efektif yük tensörü izotropiktir ve diyagonal elemanları Ag ve Na atomlarının
nominal yüklerine yakın bir değere sahiptir. Nb Born efektif yük tensörü biraz
anizotropik iken O’nin Born efektif yük tensörünün anizotropisi daha güçlüdür.
Nb ve Ta atomlarının nominal iyonik yükleri bu bileşiklerde +5 iken, Born efektrif
yükleri Ta için +7,743 ile +8,481 ve Nb için +6,629 ile +9,550 arasındadır.
Ag, Nb ve Ta atomlarının iyonik yükleri (+) işaretli iken, simetriden dolayı
çizelge 4.12, çizelge 4.13, çizelge 4.14 ve çizelge 4.15 ’den görüldüğü gibi Born
efektif tük tensöründe (–) değerlerde alabilmektedir. Aynı şekilde O atomunu

4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
86
iyonik yükünün işareti (–) iken, Born efektif tük tensöründe (+) değerler
alabilmektedir.
Çizelge 4.12. Rombohedral yapıda hesaplanmış AgTaO3’ün born efektif yük tensörü Atom
Tipi Wyckoff Nominal Değerler Born Efektif Yük Tensörü
Ag 2a 1 1,417 0 00 1,417 00 0 1,328
Ta 2a 5 8,393 −0,726 00,726 8,393 00 0 8,481
O 6b -2 −4,543 0 2,0560 −1,996 01,955 0 −3,270
Çizelge 4.13. Ortorombik yapıda hesaplanmış NaTaO3’ün born efektif yük tensörü Atom
Tipi Wyckoff Nominal Değerler Born Efektif Yük Tensörü
Na 4c 1 1,036 0 −0,0290 1,049 0−0,063 0 1,021
Ta 4a 5 7,959 0,296 −0,888−0,011 7,850 1,2250,886 −1,128 7,743
O1 4c -2 −1,541 0 0,1410 −5,709 0−0,127 0 −1,619
O2 8d -2 −3,727 0,020 −2,0270,033 −1,595 −0,020−2,090 −0,017 −3,572
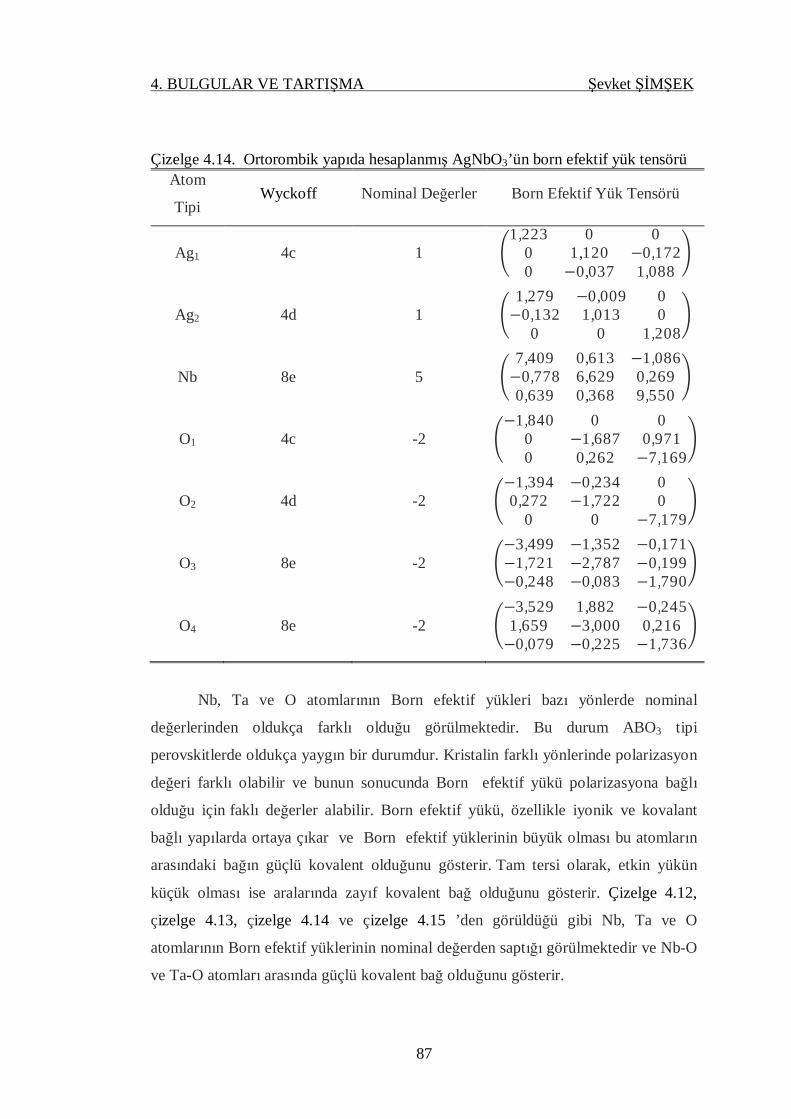
4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
87
Çizelge 4.14. Ortorombik yapıda hesaplanmış AgNbO3’ün born efektif yük tensörü Atom
Tipi Wyckoff Nominal Değerler Born Efektif Yük Tensörü
Ag1 4c 1 1,223 0 00 1,120 −0,1720 −0,037 1,088
Ag2 4d 1 1,279 −0,009 0−0,132 1,013 00 0 1,208
Nb 8e 5 7,409 0,613 −1,086−0,778 6,629 0,2690,639 0,368 9,550
O1 4c -2 −1,840 0 00 −1,687 0,9710 0,262 −7,169
O2 4d -2 −1,394 −0,234 00,272 −1,722 00 0 −7,179
O3 8e -2 −3,499 −1,352 −0,171−1,721 −2,787 −0,199−0,248 −0,083 −1,790
O4 8e -2 −3,529 1,882 −0,2451,659 −3,000 0,216−0,079 −0,225 −1,736
Nb, Ta ve O atomlarının Born efektif yükleri bazı yönlerde nominal
değerlerinden oldukça farklı olduğu görülmektedir. Bu durum ABO3 tipi
perovskitlerde oldukça yaygın bir durumdur. Kristalin farklı yönlerinde polarizasyon
değeri farklı olabilir ve bunun sonucunda Born efektif yükü polarizasyona bağlı
olduğu için faklı değerler alabilir. Born efektif yükü, özellikle iyonik ve kovalant
bağlı yapılarda ortaya çıkar ve Born efektif yüklerinin büyük olması bu atomların
arasındaki bağın güçlü kovalent olduğunu gösterir. Tam tersi olarak, etkin yükün
küçük olması ise aralarında zayıf kovalent bağ olduğunu gösterir. Çizelge 4.12,
çizelge 4.13, çizelge 4.14 ve çizelge 4.15 ’den görüldüğü gibi Nb, Ta ve O
atomlarının Born efektif yüklerinin nominal değerden saptığı görülmektedir ve Nb-O
ve Ta-O atomları arasında güçlü kovalent bağ olduğunu gösterir.
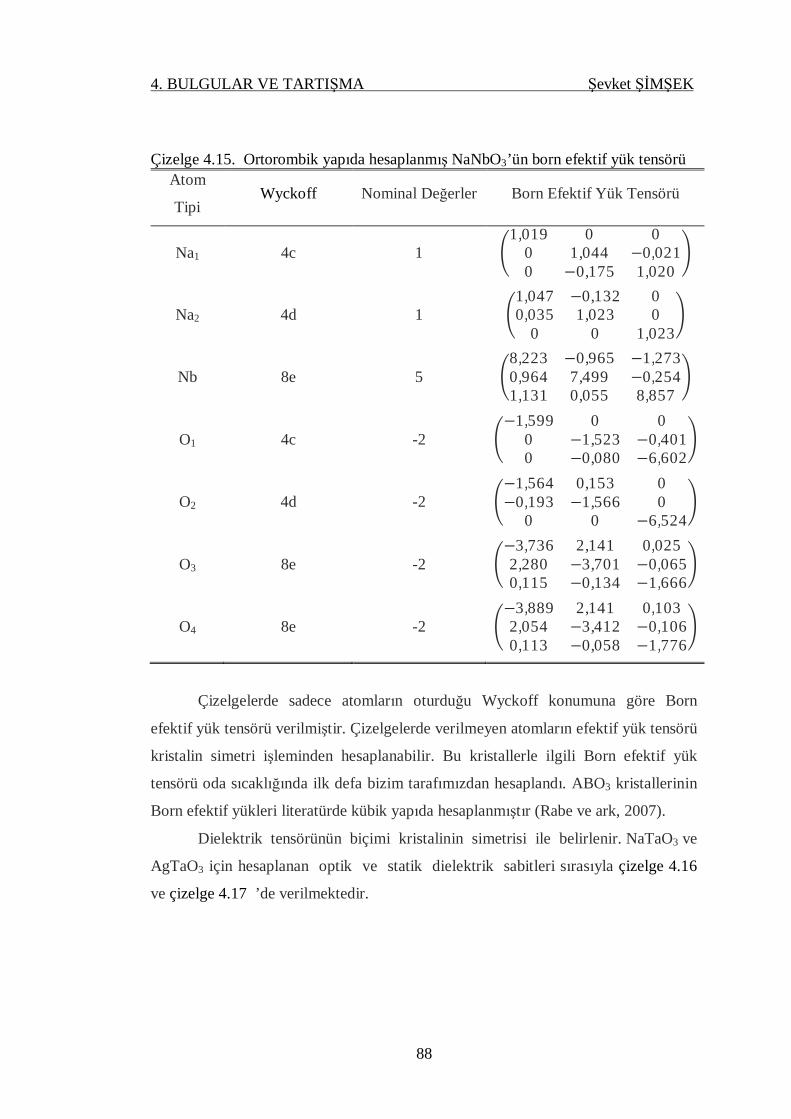
4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
88
Çizelge 4.15. Ortorombik yapıda hesaplanmış NaNbO3’ün born efektif yük tensörü Atom
Tipi Wyckoff Nominal Değerler Born Efektif Yük Tensörü
Na1 4c 1 1,019 0 00 1,044 −0,0210 −0,175 1,020
Na2 4d 1 1,047 −0,132 00,035 1,023 00 0 1,023
Nb 8e 5 8,223 −0,965 −1,2730,964 7,499 −0,2541,131 0,055 8,857
O1 4c -2 −1,599 0 00 −1,523 −0,4010 −0,080 −6,602
O2 4d -2 −1,564 0,153 0−0,193 −1,566 00 0 −6,524
O3 8e -2 −3,736 2,141 0,0252,280 −3,701 −0,0650,115 −0,134 −1,666
O4 8e -2 −3,889 2,141 0,1032,054 −3,412 −0,1060,113 −0,058 −1,776
Çizelgelerde sadece atomların oturduğu Wyckoff konumuna göre Born
efektif yük tensörü verilmiştir. Çizelgelerde verilmeyen atomların efektif yük tensörü
kristalin simetri işleminden hesaplanabilir. Bu kristallerle ilgili Born efektif yük
tensörü oda sıcaklığında ilk defa bizim tarafımızdan hesaplandı. ABO3 kristallerinin
Born efektif yükleri literatürde kübik yapıda hesaplanmıştır (Rabe ve ark, 2007).
Dielektrik tensörünün biçimi kristalinin simetrisi ile belirlenir. NaTaO3 ve
AgTaO3 için hesaplanan optik ve statik dielektrik sabitleri sırasıyla çizelge 4.16
ve çizelge 4.17 ’de verilmektedir.

4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
89
Çizelge 4.16. Rombohedral yapıda hesaplana AgTaO3’ün optik ve statik dielektrik sabiti
εxx εyy εzz
Optik Dielektrik Sabiti 6,735 6,735 6,578
Statik Dielektrik Sabiti 36,842 36,842 43,548
Çizelge 4.17. Ortorombik yapıda hesaplanan NaTaO3’ün optik ve statik dielektrik sabiti εxx εyy εzz
Optik Dielektrik Sabiti 5,033 4,970 4,905
Statik Dielektrik Sabiti 30,364 31,257 26,872
Ortorombik yapıda NaTaO3 ve rombohedral yapıda AgTaO3 kristallerinin
dielektrik tensörünün sadece x, y ve z yönlerinde bileşenleri vardır diğer yönlerdeki
bileşenleri sıfırdır. Çizelge 4.16 ve çizelge 4.17’den de görüldüğü gibi her iki
kristalin optik ve statik dielektrik sabitleri arasında oldukça büyük bir fark olduğu
görülmektedir. Çizelgelerden de görüldüğü gibi AgTaO3 ve NaTaO3 kristallerinin
optik dielektrik sabiti, statik dielektrik sabitten oldukça küçüktür. Bu durum, değişen
bir elektrik alanda, farklı polarizasyon mekanizmalarının ne kadar hızla etki ettiği
göz önüne alınarak açıklanabilir. Elektronik polarizasyon, elektronun hareketinden
kaynaklanmaktadır. Elektron, dış elektrik alandaki herhangi bir değişikliğe hızlıca
tepki gösterir ve bu mekanizmalar optik frekanslarda da devam etmektedir. Diğer
polarizasyon tipi olan iyonik polarizasyonda ise, iyonlar elektronlardan daha büyük
kütleye sahip olduklarından elektrik alanın hızlı değişimlerini takip edemezler.
Pratikte, polarizasyona dipolar ve iyonik katkı sırasıyla 1011 Hz ve 1013 Hz
üzerindeki frekanslarda hızla düşmektedir. Oysa elektronik polarizasyon 1016 Hz
kadar uzanabilir. Sonuç olarak, optik frekanslarda dielektrik sabite katkının yalnızca
elektronik polarizasyondan, düşük frekanslarda ise iyonik ve dipolar mekanizmaların
katkıda bulunduğunu söyleyebiliriz. AgTaO3 ve NaTaO3 kristallerinin optik
dielektrik sabiti ve statik dielektrik sabitlerini kıyaslayacağımız literatürde deneysel
ve teorik değerler bulunmamaktadır.

4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
90
AgNbO3 ve NaNbO3 kristallerinin oda sıcaklığındaki ortorombik yapısının
birim hücresinde 40 atom bulunmaktadır. Bu kristallerin optik ve dielektrik sabiti
ve elastik sabitlerini tüm uğraşlarımıza rağmen ABINIT programında
hesaplayamadık.
4.4.3. Fonon Dispersiyon Eğrileri ve Durum Yoğunluğu
Fonon dispersiyon bağıntıları yarı-klasik yaklaşım ve kuantum mekaniksel
yaklaşım olmak üzere iki değişik yolla hesaplanabilir (Hamann D ve ark, 1979).
Kuantum mekaniksel yaklaşımda doğrusal tepki (linear response), küçük yer
değiştirme (small displacement) ( Johnston ve ark, 1996) ve direk metot (frozen-
zone) (Schreiber ve ark, 1973) yaygın olarak kullanılmaktadır. Örgü dinamiğinin
temel problemi kristaldeki titreşiminin normal kiplerini bulmaktır. Başka bir deyişle
örgü dinamiği fonon enerjilerini veya frekanslarını (ω) dalga vektörlerinin (k) bir
fonksiyonu olarak hesaplar ( Parlinski ve ark, 1997). ω ve k arasındaki bağıntıya
dispersiyon (dağınım) bağıntısı denir. PHONON programı hesaplamalarda direk
metodu kullanır ve süper hücre yöntemini kullanarak hesaplama yapar. Program,
230 adet kristalografik uzay grubundan birini kullanarak kristal yapıyı oluşturur,
komşuluk listesini ve kuvvet sabitlerinin matrisini bulur ve dinamik matrisi oluşturur.
Komşu listesi bir parçacığın süper hücre denilen bir paralelkenar etrafındaki alanı
kapatarak seçilebilir.
AgTaO3, AgNbO3, NaTaO3 ve NaNbO3 kristallerinin fonon dispersiyon ve
durum yoğunlukları, yoğunluk fonksiyonel teorisindeki ab initio metodu ile
incelendi. Hesaplamalarda ab initio kod olarak PHONON yazılım programı
kullanıldı. İlk önce kristallerin ait olduğu uzay grupları, optimize edilmiş örgü
parametreleri ve atomik pozisyonları programda ilgi yerlerine kaydedildi. Sonra,
optimize edilmiş olan optik dielektrik sabiti (ε∞) ve Born efektif yükleri kristaller için
PHONON programına girildi. Hellman-Feynman kuvvetleri hesaplamak için
atomların 0.03 Å’luk yer değiştirmelere izin verildi. Ayrıca x, y ve z yönlerinde
pozitif ve negatif yer değiştirmeler dikkate alındı. Kristal yapılar için PHONON
programında l x m x n’ lik süper hücreler oluşturuldu. Burada l, m ve n birer

4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
91
tamsayıdır. Süper hücre oluştururken, hücrenin olabildiğince kübik şekilde olması
tercih edilir. Direkt metot da Hellmann–Feynman kuvvetleri, süper hücredeki
atomlar arasında oluşan süper hücrenin kuvvet sabitlerini hesaplamak amacı ile
kullanılır. Süper hücre kuvvet sabitleri, klasik kuvvet sabitlerinden çok farklıdır.
Çünkü bunlar süper hücrenin periyodik sınır şartlarını hesaba katar. Hellmann–
Feynman kuvvetleri periyodik sınır şartlarındaki süper hücreden hesaplanır.
Kristaller için Hellman-Feynman kuvvetleri hesaplamak için bir ab initio program
olan SIESTA yazılımı kullanıldı. Hellman-Feynman kuvvetleri hesaplamak için
LDA yaklaşımı alındı. Pseudopotansiyeller olarak Troullier-Martins’in norm
conserving pseudopotansiyeli kullanıldı. Atomların elektron dağılımı Ag: 5s1 4d10,
Na: 3s1, Ta: 6s25d3, Nb: 5s14d4 ve O: 2s2 2p4 seklinde alındı. Kor yarıçapları s, p
ve d orbitalleri için Ag: 2,52 au, Na: 2,83 au, Ta: 2,78 au (s), 2,51 au (d), Nb: 2,89
au (s), 2,55 au (d) ve O: 1,47 au alındı. Hesaplamalarda AgTaO3, AgNbO3, NaTaO3
ve NaNbO3 için mesh-cut off değeri sırasıyla 400 Ry, 270 Ry, 300 Ry ve 300
Ry yeterli görülmüştür. POA.EnergyShift değerleri AgTaO3 için 280 meV,
AgNbO3 ve NaTaO3 için 260 meV, NaNbO3 için ise 300 meV olarak alınmıştır.
Brillouin bölgesinde özel k noktalarının üretimi için Monkhorst-Pack yöntemi ile
kristal yapılar için 12 x 12 x 12 Monkhorst-Pack örgü ağı kullanılmıştır. SIESTA
bilgisayar programıyla hesaplanan Hellman-Feynman kuvvetleri PHONON
programına yerleştirilerek fonon dispersiyon ve durum yoğunlukları hesaplandı.
Metalik olmayan kristallerde, etkin iyonik yükler, kırmızı altı da etkin olan kipleri
boyuna optik (LO) ve enine optik (TO) bileşenlere ayırır. Brillouin bölgesinin
merkezinin k=0 daki dalga vektörü Γ noktasında LO ve TO bileşenlerine ayrılmadan
önce bütün indirgenemez gösterimler TO olarak bulunur (Parlinski, 2007). Süper
hücre kristalin simetrisini azaltmazsa, indirgenemez gösterim kristalin nokta
grubunun terimiyle verilir.
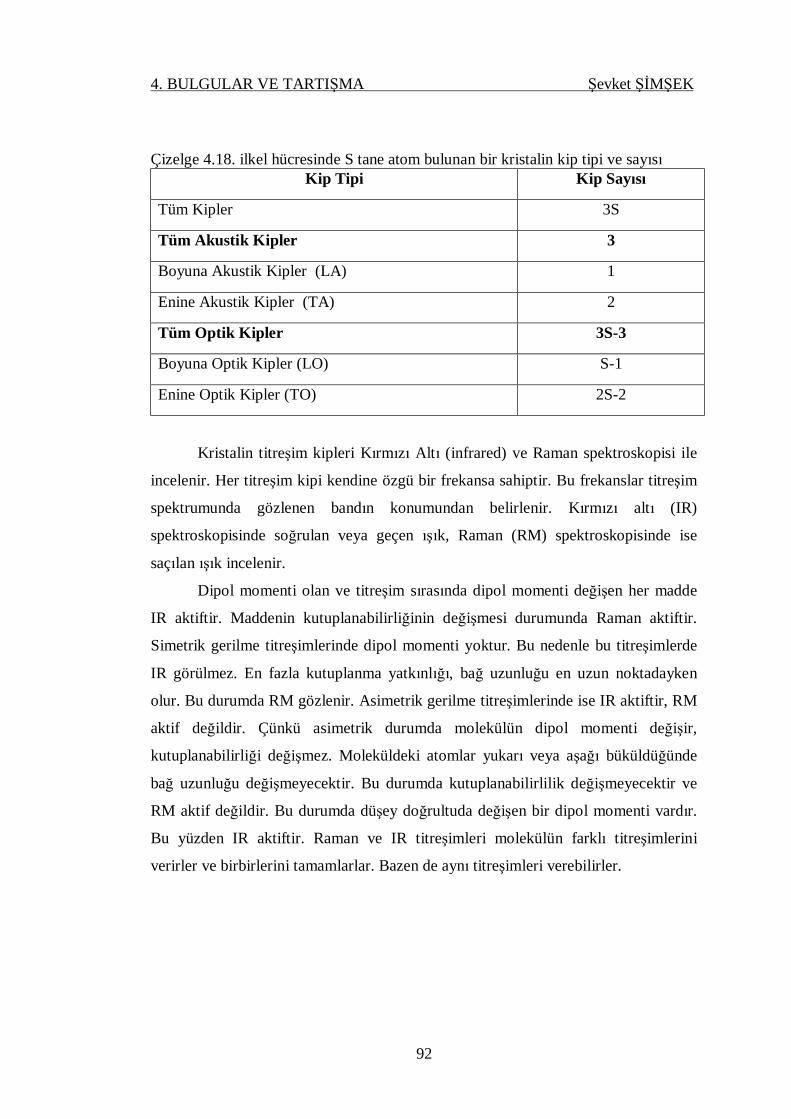
4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
92
Çizelge 4.18. ilkel hücresinde S tane atom bulunan bir kristalin kip tipi ve sayısı Kip Tipi Kip Sayısı
Tüm Kipler 3S
Tüm Akustik Kipler 3
Boyuna Akustik Kipler (LA) 1
Enine Akustik Kipler (TA) 2
Tüm Optik Kipler 3S-3
Boyuna Optik Kipler (LO) S-1
Enine Optik Kipler (TO) 2S-2
Kristalin titreşim kipleri Kırmızı Altı (infrared) ve Raman spektroskopisi ile
incelenir. Her titreşim kipi kendine özgü bir frekansa sahiptir. Bu frekanslar titreşim
spektrumunda gözlenen bandın konumundan belirlenir. Kırmızı altı (IR)
spektroskopisinde soğrulan veya geçen ışık, Raman (RM) spektroskopisinde ise
saçılan ışık incelenir.
Dipol momenti olan ve titreşim sırasında dipol momenti değişen her madde
IR aktiftir. Maddenin kutuplanabilirliğinin değişmesi durumunda Raman aktiftir.
Simetrik gerilme titreşimlerinde dipol momenti yoktur. Bu nedenle bu titreşimlerde
IR görülmez. En fazla kutuplanma yatkınlığı, bağ uzunluğu en uzun noktadayken
olur. Bu durumda RM gözlenir. Asimetrik gerilme titreşimlerinde ise IR aktiftir, RM
aktif değildir. Çünkü asimetrik durumda molekülün dipol momenti değişir,
kutuplanabilirliği değişmez. Moleküldeki atomlar yukarı veya aşağı büküldüğünde
bağ uzunluğu değişmeyecektir. Bu durumda kutuplanabilirlilik değişmeyecektir ve
RM aktif değildir. Bu durumda düşey doğrultuda değişen bir dipol momenti vardır.
Bu yüzden IR aktiftir. Raman ve IR titreşimleri molekülün farklı titreşimlerini
verirler ve birbirlerini tamamlarlar. Bazen de aynı titreşimleri verebilirler.

4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
93
4.4.3.1. AgTaO3 Kristali
AgTaO3 kristalinin birim hücresinde 10 atom bulunduğundan, PHONON
programında 2 x 2 x 2’lik süper hücre (80 atom) kuruldu. AgTaO3 kristalinin her bir
dalga vektörü için 30 adet fonon kipi bulunmaktadır. Bunlardan 1 tanesi LA, 2
tanesi TA, 9 tanesi LO ve 18 tanesi TO kipidir. Kristalin yüksek simetri noktaları
yönlerinde hesaplanan fonon dispersiyon eğrileri ve durum yoğunluğu eğrileri şekil
4.10 ve şekil 4.11 ‘de gösterilmektedir.
Gamma noktasındaki fononların titreşim kipleri R3c (#161) nokta grubunun
indirgenemez gösterimiyle sınıflandırılır. Grup teorisine göre bu nokta gruplarının
kiplerinin simetrisi (Kroumova ve ark, 2003),
Γ(R3c) = 5A1 (IR+RM) + 5A2 + 10E (IR+RM)
olarak verilmektedir. Gamma noktasında A1 ve E kipleri RM ve IR de aktiftir. A2 ise
IR ve RM de gözlenmez. Gamma noktasında hesaplanan fonon kiplerinin frekansları
ve simetrileri çizelge 4.19 da verilmiştir. Sanal frekans değerleri negatif olarak
görülmektedir. Yumuşak kip (soft mod), sanal fonon frekanslarını tanımlar.
Yumuşak kiplerin işaret ettiği yer değiştirmelerden dolayı kristal kararsızdır.
Ferroelektrik faz geçişlerini kararlı olmayan fonon kipleri yönetir. Tek kristal
AgTaO3 üzerine yapılan deneysel Raman çalışmalarında Kugel ve arkadaşları (
Kugel ve ark, 1987) oda sıcaklığında kip frekansları 65 cm-1 (1,95 THz), 100 cm-1
(3,0 THz), 200 cm-1 (6,0 THz), 420 cm-1 (12,6 THz) ve 590 cm-1 (17,7 THz)
gözlemişlerdir. Çizelge 4.19 daki hesapladığımız kip frekanslarıyla karşılaştırırsak
deneysel değerler E simetri kipinin 1,84, 2,554, 5,818, 12,563 ve 17,192 THz frekans
değerlerine karşılık gelmektedir. Hesapladığımız fonon kip frekanslarının deneysel
değerlerle oldukça uyumlu olduğu görülmektedir. Bu çalışmadan başka deneysel ve
teorik sonuçlar olmadığından dolayı detaylı karşılaştırma yapılamamıştır.
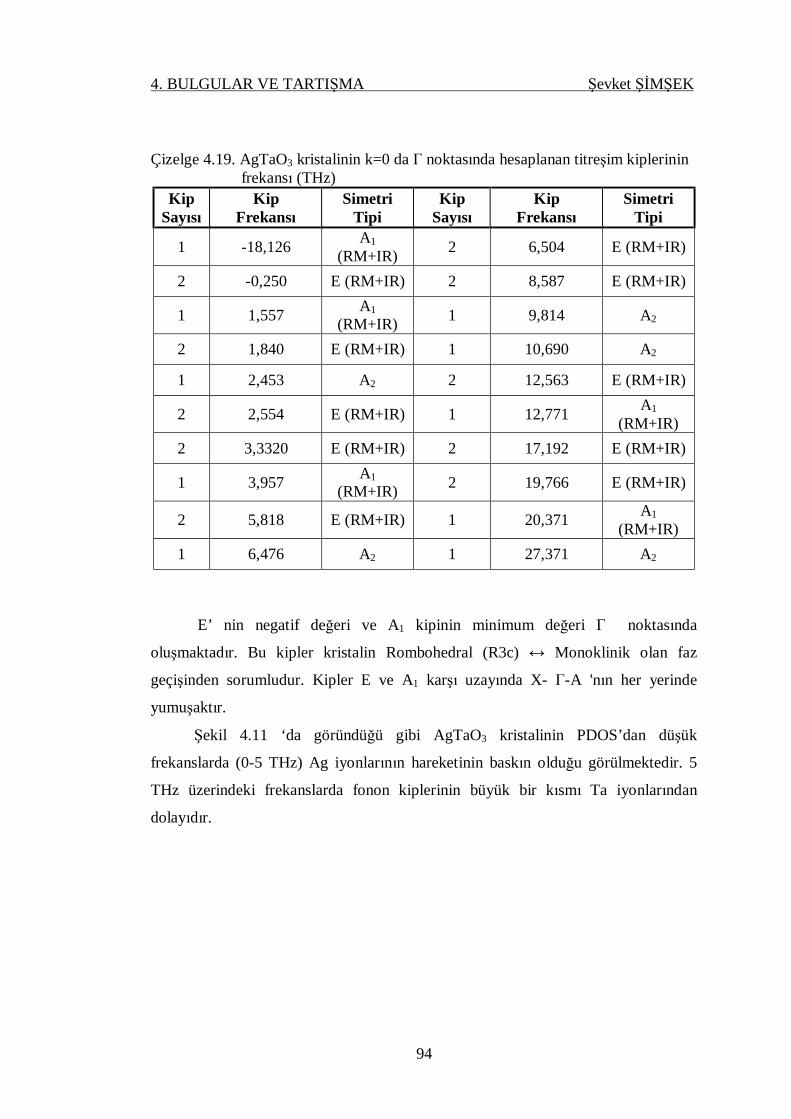
4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
94
Çizelge 4.19. AgTaO3 kristalinin k=0 da Γ noktasında hesaplanan titreşim kiplerinin frekansı (THz)
Kip Sayısı
Kip Frekansı
Simetri Tipi
Kip Sayısı
Kip Frekansı
Simetri Tipi
1 -18,126 A1 (RM+IR) 2 6,504 E (RM+IR)
2 -0,250 E (RM+IR) 2 8,587 E (RM+IR)
1 1,557 A1 (RM+IR) 1 9,814 A2
2 1,840 E (RM+IR) 1 10,690 A2
1 2,453 A2 2 12,563 E (RM+IR)
2 2,554 E (RM+IR) 1 12,771 A1 (RM+IR)
2 3,3320 E (RM+IR) 2 17,192 E (RM+IR)
1 3,957 A1 (RM+IR) 2 19,766 E (RM+IR)
2 5,818 E (RM+IR) 1 20,371 A1 (RM+IR)
1 6,476 A2 1 27,371 A2
E’ nin negatif değeri ve A1 kipinin minimum değeri Γ noktasında
oluşmaktadır. Bu kipler kristalin Rombohedral (R3c) ↔ Monoklinik olan faz
geçişinden sorumludur. Kipler E ve A1 karşı uzayında X- Γ-A 'nın her yerinde
yumuşaktır.
Şekil 4.11 ‘da göründüğü gibi AgTaO3 kristalinin PDOS’dan düşük
frekanslarda (0-5 THz) Ag iyonlarının hareketinin baskın olduğu görülmektedir. 5
THz üzerindeki frekanslarda fonon kiplerinin büyük bir kısmı Ta iyonlarından
dolayıdır.
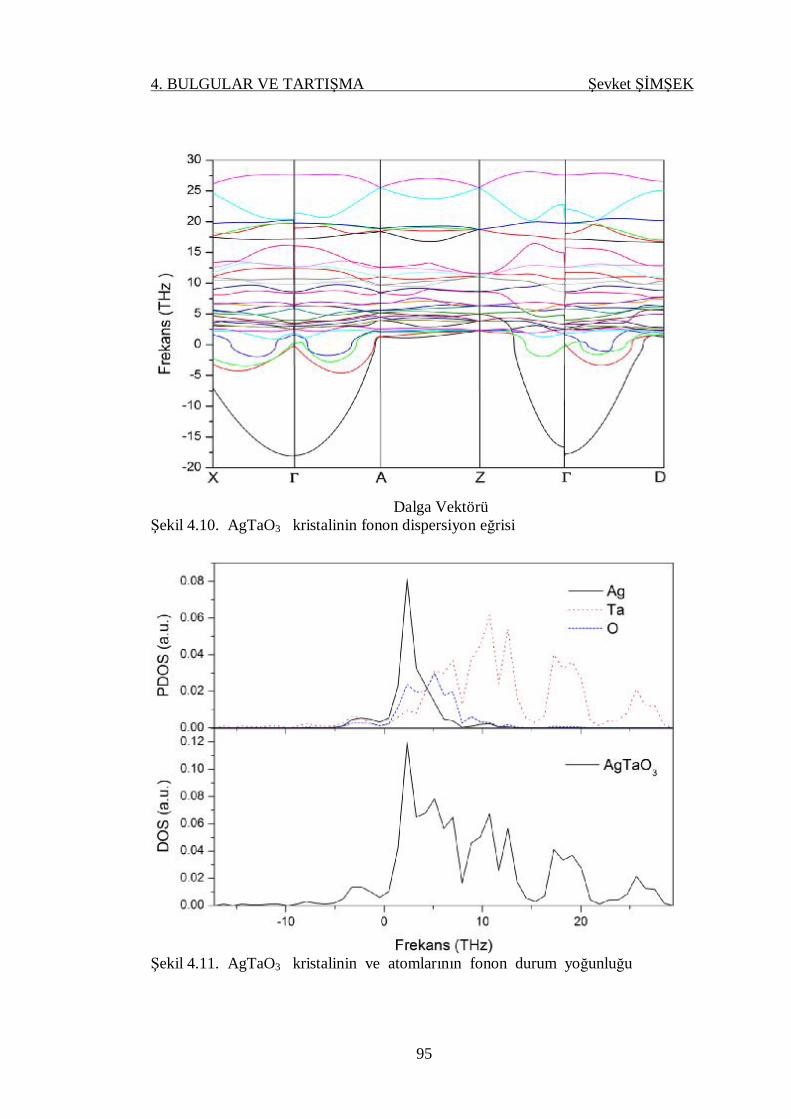
4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
95
Şekil 4.10. AgTaO3 kristalinin fonon dispersiyon eğrisi
Şekil 4.11. AgTaO3 kristalinin ve atomlarının fonon durum yoğunluğu
Dalga Vektörü

4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
96
4.4.3.2. NaTaO3 Kristali
NaTaO3 kristalinin birim hücresinde 20 atom bulunduğundan, her bir
dalga vektörü için 60 adet fonon kipi bulunmaktadır. Bunlardan 1 tanesi LA, 2
tanesi TA, 19 tanesi LO ve 38 tanesi TO kipidir. PHONON programında 3 x 2 x 3’
lik süper hücre (360 atom) kuruldu. Temel simetri yönlerinde hesaplanan fonon
dispersiyon eğrileri ve durum yoğunluğu eğrileri şekil 4.12 ve şekil 4.13’ de
gösterilmektedir.
Gamma noktasındaki fononların titreşim kipleri Pnma (#62) nokta grubunun
indirgenemez gösterimiyle sınıflandırılır. Gamma noktasında kiplerinin simetrisi
(Kroumova ve ark, 2003),
Γ(Pnma) = 7Ag (RM) + 8Au + 5B1g(RM) + 10 B1u (IR) + 7 B2g (RM)
+8 B2u (IR) + 5 B3g (RM) + 10 B3u (IR)
olarak verilmektedir. Gamma noktasında Ag, B1g, B2g ve B3g kipleri RM ve B1u,
B2u ve B3u IR de aktiftir. Au ise IR ve RM de gözlenmez. Gamma noktasında
hesaplanan fonon kiplerinin frekansları ve simetrileri çizelge 4.20’ de verilmiştir.
Ag, Au, B1u, B2u ve B3u negatif kip değeri Γ noktasında oluşmaktadır.
Bu kipler kristalin Ortorombik (Pnma) ↔ Ortorombik (Cmnm) olan faz geçişinden
sorumludur. Kipler Ag, Au, B1u, B2u ve B3u karşı uzayında X- Γ ve Y- Γ-Z’ nin
her yerinde yumuşaktır. Shanker ve ark (2009), NaTaO3 nano kristalinin üzerinde
100-1000 cm-1 arasında yaptıkları Raman spektroskopisi ölçümlerinden elde ettikleri
kip frekansları 100 cm-1 (3,0 THz), 250 cm-1 (7,50 THz), 440 cm-1 (13,20 THz), 500
cm-1 (15,0 THz), 600 cm-1 (18,0 THz), ve 810 cm-1 (24,30 THz) dir. Nano kristalin
fonon kipleri bizim değerlerle kısmen uyuşmaktadır. Şekil 4.13’ den göründüğü
gibi NaTaO3 kristalinin PDOS ’dan düşük frekanslarda (-5- 2 THz) O iyonlarının
hareketinin, (2-10 THz) Na ve (10-25) THz arasında Ta iyonun hareketinin baskın
olduğu görülmektedir.
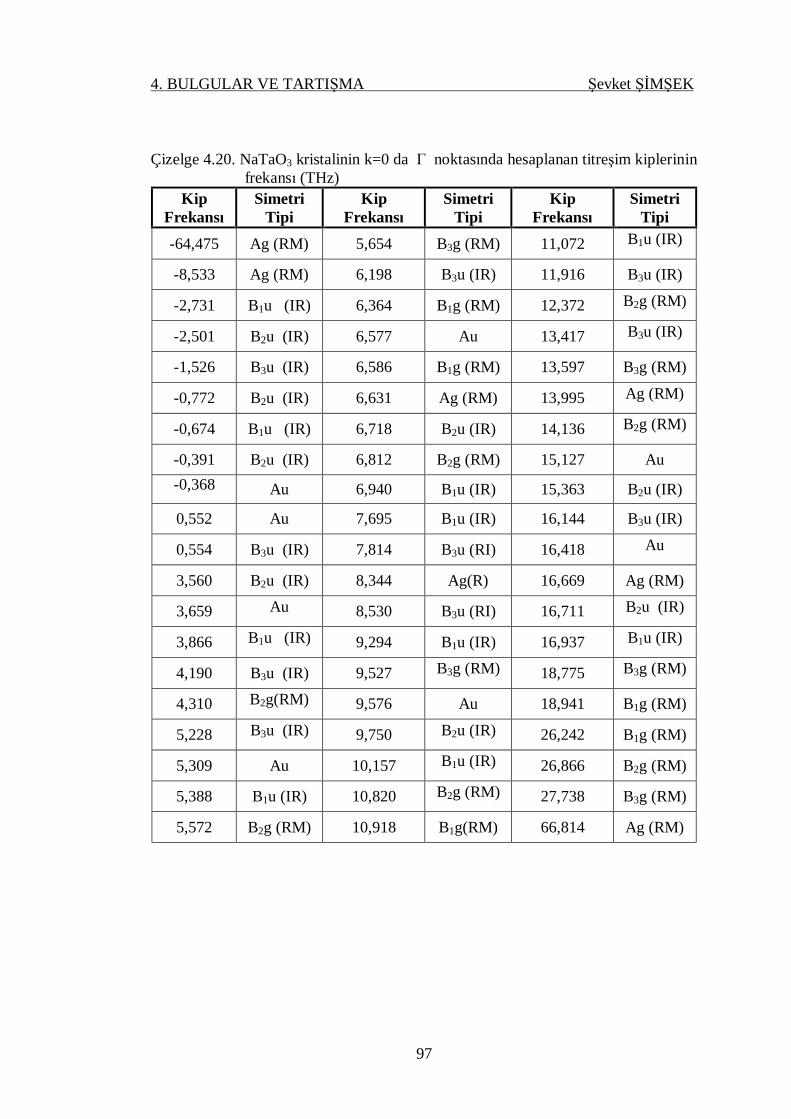
4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
97
Çizelge 4.20. NaTaO3 kristalinin k=0 da Γ noktasında hesaplanan titreşim kiplerinin frekansı (THz)
Kip Frekansı
Simetri Tipi
Kip Frekansı
Simetri Tipi
Kip Frekansı
Simetri Tipi
-64,475 Ag (RM) 5,654 B3g (RM) 11,072 B1u (IR)
-8,533 Ag (RM) 6,198 B3u (IR) 11,916 B3u (IR)
-2,731 B1u (IR) 6,364 B1g (RM) 12,372 B2g (RM)
-2,501 B2u (IR) 6,577 Au 13,417 B3u (IR)
-1,526 B3u (IR) 6,586 B1g (RM) 13,597 B3g (RM)
-0,772 B2u (IR) 6,631 Ag (RM) 13,995 Ag (RM)
-0,674 B1u (IR) 6,718 B2u (IR) 14,136 B2g (RM)
-0,391 B2u (IR) 6,812 B2g (RM) 15,127 Au -0,368 Au 6,940 B1u (IR) 15,363 B2u (IR)
0,552 Au 7,695 B1u (IR) 16,144 B3u (IR)
0,554 B3u (IR) 7,814 B3u (RI) 16,418 Au
3,560 B2u (IR) 8,344 Ag(R) 16,669 Ag (RM)
3,659 Au 8,530 B3u (RI) 16,711 B2u (IR)
3,866 B1u (IR) 9,294 B1u (IR) 16,937 B1u (IR)
4,190 B3u (IR) 9,527 B3g (RM) 18,775 B3g (RM)
4,310 B2g(RM) 9,576 Au 18,941 B1g (RM)
5,228 B3u (IR) 9,750 B2u (IR) 26,242 B1g (RM)
5,309 Au 10,157 B1u (IR) 26,866 B2g (RM)
5,388 B1u (IR) 10,820 B2g (RM) 27,738 B3g (RM)
5,572 B2g (RM) 10,918 B1g(RM) 66,814 Ag (RM)
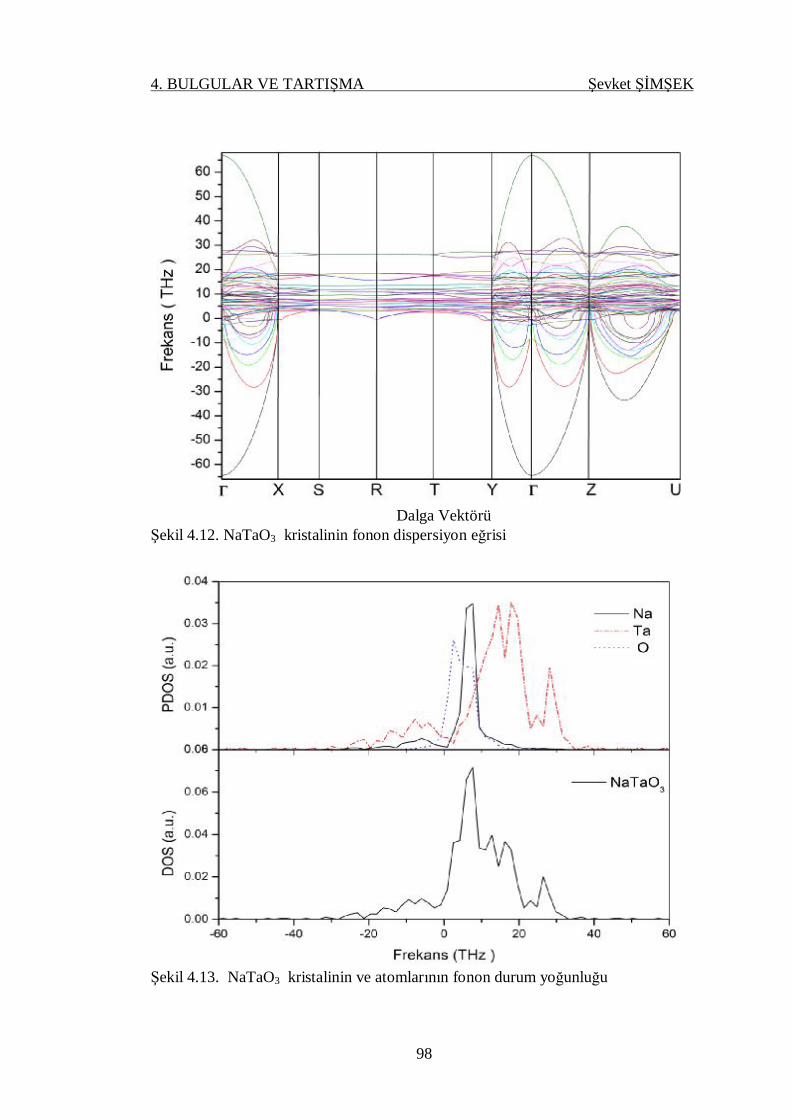
4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
98
Şekil 4.12. NaTaO3 kristalinin fonon dispersiyon eğrisi
Şekil 4.13. NaTaO3 kristalinin ve atomlarının fonon durum yoğunluğu
Dalga Vektörü

4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
99
4.4.3.3. AgNbO3 Kristali
AgNbO3 kristalinin birim hücresinde 40 atom bulunduğundan, PHONON
programında 2 x 2 x 1’lik süper hücre (160 atom) kuruldu. AgNbO3 kristalini her
bir dalga vektörü için 120 adet fonon kipi bulunmaktadır. Bunlardan 1 tanesi
LA, 2 tanesi TA, 39 tanesi LO ve 78 tanesi TO kipidir. Brilluoin bölgesindeki
yüksek simetri noktaları boyunca hesaplanan fonon dispersiyon eğrileri ve durum
yoğunluğu eğrileri şekil 4.14 ve şekil 4.15 ‘da gösterilmektedir.
Gamma noktasındaki fononların titreşim kipleri Pbcm (#57) nokta grubunun
indirgenemez gösterimiyle sınıflandırılır. Bu nokta grubunun kiplerinin simetrisi
(Kroumova ve ark, 2003),
Γ(Pbcm) = 15Ag (RM) + 13Au + 17 B1g (RM) + 15B1u (IR) + 15 B2g(RM)
+17 B2u (RM) + 13 B3g (RM) + 15B3u (IR)
olarak verilmektedir. Gamma noktasında Ag, B1g, B2g ve B3g kipleri RM ve
B1u, B2u ve B3u IR de aktiftir. Au ise IR ve RM de gözlenmez. Gamma
noktasında hesaplanan fonon kiplerinin frekansları ve simetrileri çizelge 4.21’ de
verilmiştir.
Fononların dispersiyon eğrisinden R - T yönünde 30 fonon kip görülmektedir.
Bunun nedeni bu bölgede dejenereliğin yoğun olmasındandır. X - S - R -T -Y
bölgelerinde yumuşak kipler de yoktur ve bu bölgelerde kristalin kararlı
olduğunu söyleyebiliriz. Γ - X [1 0 0], Y - Γ [ 0 1 0] ve Γ- Z [0 0 1]
doğrultularında ve Z - U yönlerinde yumuşak kipler görülmektedir. R -T bölgesinin
dışında Born efektif yükünün büyük olmasından dolayı dallar arasında yarılmalar
net olarak görülmektedir. İyonik kristallerin önemli bir özelliği, Brillouin
bölgesinin merkezinde LO / TO fonon kiplerinin büyük yarılmalar göstermesidir.
AgNbO3’ nin yüksek frekanslarda optik fonon kipleri arasında boşluklar oluştuğu
dikkat çekmektedir. Bu boşluğun nedeni anyon ve katyon arasındaki kütle farkının
etkin olmasındadır. AgNbO3 kristalinin PDOS 'dan görülebileceği gibi -5 - 0 THz
aralığında Ag ve Nb atomları oksijene göre daha baskındır. Bu durum kristal
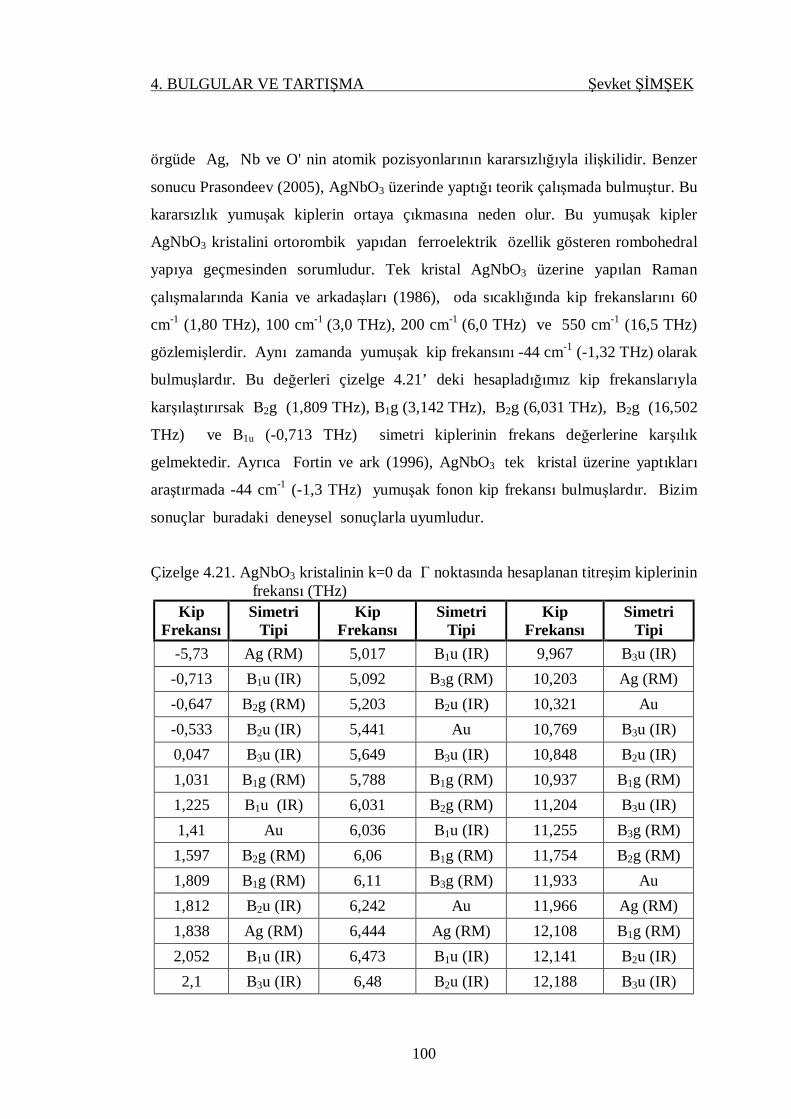
4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
100
örgüde Ag, Nb ve O' nin atomik pozisyonlarının kararsızlığıyla ilişkilidir. Benzer
sonucu Prasondeev (2005), AgNbO3 üzerinde yaptığı teorik çalışmada bulmuştur. Bu
kararsızlık yumuşak kiplerin ortaya çıkmasına neden olur. Bu yumuşak kipler
AgNbO3 kristalini ortorombik yapıdan ferroelektrik özellik gösteren rombohedral
yapıya geçmesinden sorumludur. Tek kristal AgNbO3 üzerine yapılan Raman
çalışmalarında Kania ve arkadaşları (1986), oda sıcaklığında kip frekanslarını 60
cm-1 (1,80 THz), 100 cm-1 (3,0 THz), 200 cm-1 (6,0 THz) ve 550 cm-1 (16,5 THz)
gözlemişlerdir. Aynı zamanda yumuşak kip frekansını -44 cm-1 (-1,32 THz) olarak
bulmuşlardır. Bu değerleri çizelge 4.21’ deki hesapladığımız kip frekanslarıyla
karşılaştırırsak B2g (1,809 THz), B1g (3,142 THz), B2g (6,031 THz), B2g (16,502
THz) ve B1u (-0,713 THz) simetri kiplerinin frekans değerlerine karşılık
gelmektedir. Ayrıca Fortin ve ark (1996), AgNbO3 tek kristal üzerine yaptıkları
araştırmada -44 cm-1 (-1,3 THz) yumuşak fonon kip frekansı bulmuşlardır. Bizim
sonuçlar buradaki deneysel sonuçlarla uyumludur.
Çizelge 4.21. AgNbO3 kristalinin k=0 da Γ noktasında hesaplanan titreşim kiplerinin frekansı (THz)
Kip Frekansı
Simetri Tipi
Kip Frekansı
Simetri Tipi
Kip Frekansı
Simetri Tipi
-5,73 Ag (RM) 5,017 B1u (IR) 9,967 B3u (IR) -0,713 B1u (IR) 5,092 B3g (RM) 10,203 Ag (RM) -0,647 B2g (RM) 5,203 B2u (IR) 10,321 Au -0,533 B2u (IR) 5,441 Au 10,769 B3u (IR) 0,047 B3u (IR) 5,649 B3u (IR) 10,848 B2u (IR) 1,031 B1g (RM) 5,788 B1g (RM) 10,937 B1g (RM) 1,225 B1u (IR) 6,031 B2g (RM) 11,204 B3u (IR) 1,41 Au 6,036 B1u (IR) 11,255 B3g (RM) 1,597 B2g (RM) 6,06 B1g (RM) 11,754 B2g (RM) 1,809 B1g (RM) 6,11 B3g (RM) 11,933 Au 1,812 B2u (IR) 6,242 Au 11,966 Ag (RM) 1,838 Ag (RM) 6,444 Ag (RM) 12,108 B1g (RM) 2,052 B1u (IR) 6,473 B1u (IR) 12,141 B2u (IR) 2,1 B3u (IR) 6,48 B2u (IR) 12,188 B3u (IR)
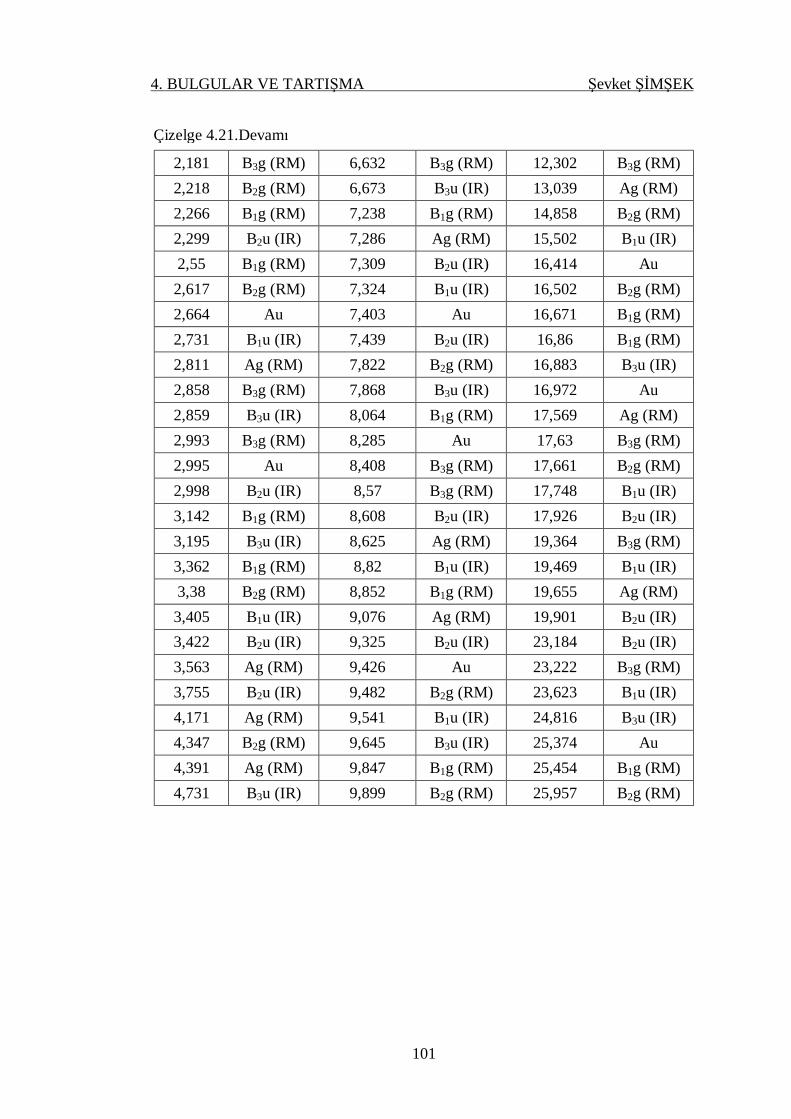
4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
101
2,181 B3g (RM) 6,632 B3g (RM) 12,302 B3g (RM) 2,218 B2g (RM) 6,673 B3u (IR) 13,039 Ag (RM) 2,266 B1g (RM) 7,238 B1g (RM) 14,858 B2g (RM) 2,299 B2u (IR) 7,286 Ag (RM) 15,502 B1u (IR) 2,55 B1g (RM) 7,309 B2u (IR) 16,414 Au 2,617 B2g (RM) 7,324 B1u (IR) 16,502 B2g (RM) 2,664 Au 7,403 Au 16,671 B1g (RM) 2,731 B1u (IR) 7,439 B2u (IR) 16,86 B1g (RM) 2,811 Ag (RM) 7,822 B2g (RM) 16,883 B3u (IR) 2,858 B3g (RM) 7,868 B3u (IR) 16,972 Au 2,859 B3u (IR) 8,064 B1g (RM) 17,569 Ag (RM) 2,993 B3g (RM) 8,285 Au 17,63 B3g (RM) 2,995 Au 8,408 B3g (RM) 17,661 B2g (RM) 2,998 B2u (IR) 8,57 B3g (RM) 17,748 B1u (IR) 3,142 B1g (RM) 8,608 B2u (IR) 17,926 B2u (IR) 3,195 B3u (IR) 8,625 Ag (RM) 19,364 B3g (RM) 3,362 B1g (RM) 8,82 B1u (IR) 19,469 B1u (IR) 3,38 B2g (RM) 8,852 B1g (RM) 19,655 Ag (RM) 3,405 B1u (IR) 9,076 Ag (RM) 19,901 B2u (IR) 3,422 B2u (IR) 9,325 B2u (IR) 23,184 B2u (IR) 3,563 Ag (RM) 9,426 Au 23,222 B3g (RM) 3,755 B2u (IR) 9,482 B2g (RM) 23,623 B1u (IR) 4,171 Ag (RM) 9,541 B1u (IR) 24,816 B3u (IR) 4,347 B2g (RM) 9,645 B3u (IR) 25,374 Au 4,391 Ag (RM) 9,847 B1g (RM) 25,454 B1g (RM) 4,731 B3u (IR) 9,899 B2g (RM) 25,957 B2g (RM)
Çizelge 4.21.Devamı
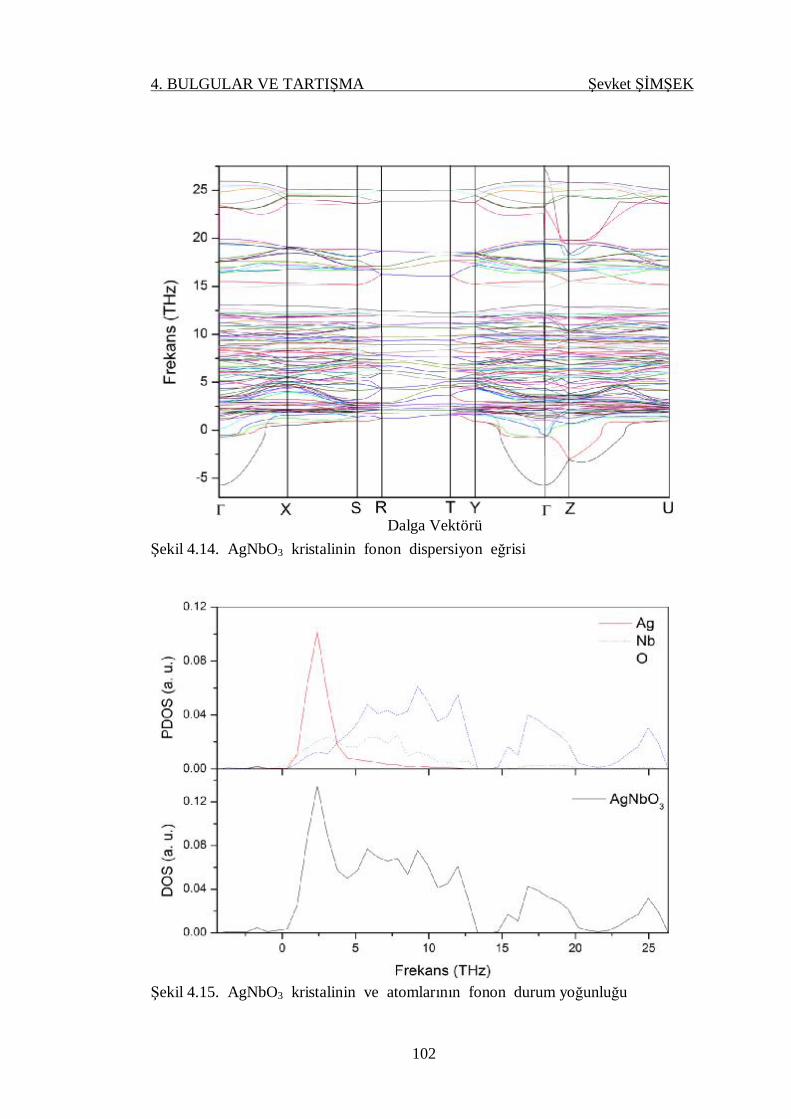
4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
102
Şekil 4.14. AgNbO3 kristalinin fonon dispersiyon eğrisi
Şekil 4.15. AgNbO3 kristalinin ve atomlarının fonon durum yoğunluğu
Dalga Vektörü
4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
103
4.4.3.4. NaNbO3 Kristali
NaNbO3 kristalinin birim hücresinde 40 atom bulunduğundan, PHONON
programında 2 x 2 x 1’ lik süper hücre (160 atom) kuruldu. NaNbO3 kristalinin her
bir dalga vektörü için 120 adet fonon kipi bulunmaktadır. Bunlardan 1 tanesi
LA, 2 tanesi TA, 39 tanesi LO ve 78 tanesi TO kipidir. Temel simetri yönlerinde
hesaplanan fonon dispersiyon eğrileri ve durum yoğunluğu eğrileri şekil 4.16 ve
şekil 4.17 ‘de gösterilmektedir. NaNbO3 kristali Pbcm (#57) nokta grubu
simetrisinde olduğundan gamma noktasındaki fononların titreşim kipleri AgNbO3
kristalinin indirgenemez gösterimiyle aynıdır. Gamma noktasında Ag, B1g, B2g ve
B3g kipleri RM ve B1u, B2u ve B3u IR de aktiftir. Au ise IR ve RM de
gözlenmez. Gamma noktasında hesaplanan fonon kiplerinin frekansları ve
simetrileri çizelge 4.22’ de verilmiştir.
Çizelge 4.22. NaNbO3 kristalinin k=0 da Γ noktasında hesaplanan titreşim kiplerinin
frekansı (THz) Kip
Frekansı Simetri Tip Kip Frekansı Simetri Tip Kip
Frekansı Simetri Tip
-72,180 Ag (RM) 5,669 B1u (IR) 11,644 B3u (IR)
-2,531 B1u (IR) 5,687 B2u (IR) 11,671 B1g (RM)
-1,011 B2u (IR) 5,690 Ag (RM) 11,720 Ag (RM)
-0,465 Ag (RM) 5,723 B3u (IR) 12,002 Ag (RM)
-0,225 B3u (IR) 5,898 B3g (RM) 12,114 B1g (RM)
0,025 B2g (RM) 5,954 B2g (RM) 12,269 B2u (IR)
0,081 B1u (IR) 6,112 B1g (RM) 12,605 B3u (IR)
1,142 B1g (RM) 6,207 B3u (IR) 12,774 B3g (RM)
1,832 B1u (IR) 6,220 Ag (RM) 12,815 Au
1,872 Au 6,359 B2u (IR) 12,832 B2g (RM)
2,024 B1g (RM) 6,470 B1u (IR) 12,998 B3u (IR)
2,285 B2g (RM) 6,660 Ag (RM) 13,051 B1g (RM)
2,388 B3g (RM) 6,703 B1g (RM) 13,117 Ag (RM)
2,672 B2u (IR) 7,014 B1g (RM) 13,251 B3g (RM)
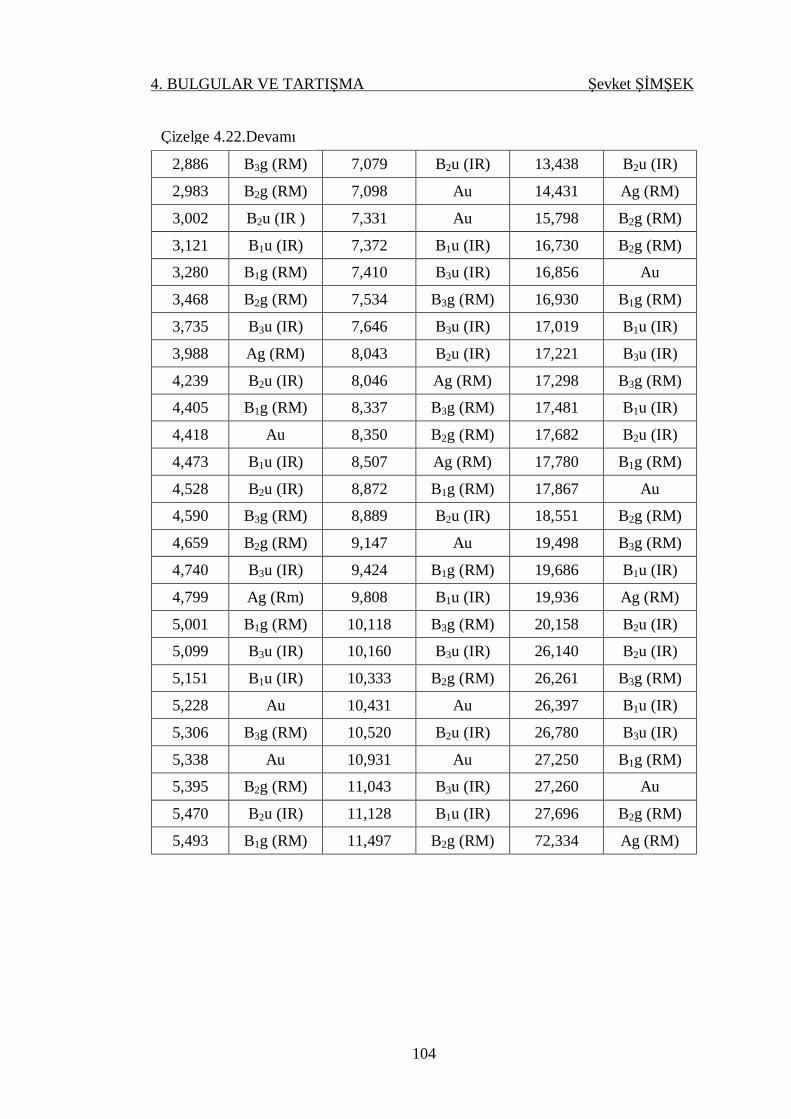
4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
104
2,886 B3g (RM) 7,079 B2u (IR) 13,438 B2u (IR)
2,983 B2g (RM) 7,098 Au 14,431 Ag (RM)
3,002 B2u (IR ) 7,331 Au 15,798 B2g (RM)
3,121 B1u (IR) 7,372 B1u (IR) 16,730 B2g (RM)
3,280 B1g (RM) 7,410 B3u (IR) 16,856 Au
3,468 B2g (RM) 7,534 B3g (RM) 16,930 B1g (RM)
3,735 B3u (IR) 7,646 B3u (IR) 17,019 B1u (IR)
3,988 Ag (RM) 8,043 B2u (IR) 17,221 B3u (IR)
4,239 B2u (IR) 8,046 Ag (RM) 17,298 B3g (RM)
4,405 B1g (RM) 8,337 B3g (RM) 17,481 B1u (IR)
4,418 Au 8,350 B2g (RM) 17,682 B2u (IR)
4,473 B1u (IR) 8,507 Ag (RM) 17,780 B1g (RM)
4,528 B2u (IR) 8,872 B1g (RM) 17,867 Au
4,590 B3g (RM) 8,889 B2u (IR) 18,551 B2g (RM)
4,659 B2g (RM) 9,147 Au 19,498 B3g (RM)
4,740 B3u (IR) 9,424 B1g (RM) 19,686 B1u (IR)
4,799 Ag (Rm) 9,808 B1u (IR) 19,936 Ag (RM)
5,001 B1g (RM) 10,118 B3g (RM) 20,158 B2u (IR)
5,099 B3u (IR) 10,160 B3u (IR) 26,140 B2u (IR)
5,151 B1u (IR) 10,333 B2g (RM) 26,261 B3g (RM)
5,228 Au 10,431 Au 26,397 B1u (IR)
5,306 B3g (RM) 10,520 B2u (IR) 26,780 B3u (IR)
5,338 Au 10,931 Au 27,250 B1g (RM)
5,395 B2g (RM) 11,043 B3u (IR) 27,260 Au
5,470 B2u (IR) 11,128 B1u (IR) 27,696 B2g (RM)
5,493 B1g (RM) 11,497 B2g (RM) 72,334 Ag (RM)
Çizelge 4.22.Devamı
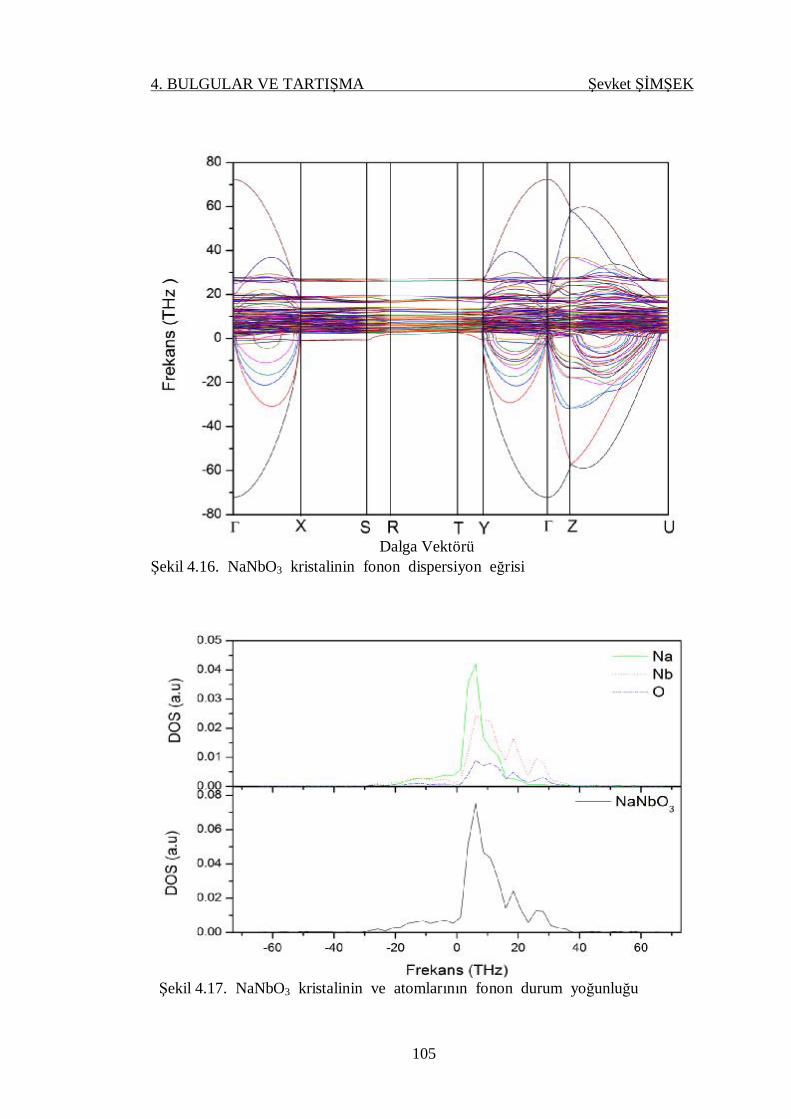
4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
105
Şekil 4.16. NaNbO3 kristalinin fonon dispersiyon eğrisi
Şekil 4.17. NaNbO3 kristalinin ve atomlarının fonon durum yoğunluğu
Dalga Vektörü

4. BULGULAR VE TARTIŞMA Şevket ŞİMŞEK
106
NaNbO3 kristalinin dispersiyon eğrisi AgNbO3 kristaline oldukça
benzemektedir. X - S - R - T - Y yönünde fonon kiplerini birbirinden ayırmak çok
güçtür. Bu bölgede efektif yüklerin çok küçük olmasından dolayı dispersiyon
eğrisinde LO / TO yarılması yok gibidir. Bu bölgede yumuşak kipler yoktur. Γ
-X [1 0 0], Y - Γ [ 0 1 0] ve Γ- Z [0 0 1] doğrultularında ve Z - U yönlerinde
yumuşak kipler görülmektedir. Z noktasında optik kiplerin dejenere olduğu
görülmektedir. Aynı zamanda bu bölgelerde Born efektif yükünün büyük olmasından
dolayı dallar arasında yarılmalar gözlenmektedir.
Mishra ve ark (2010), Raman spektroskopisi kullanarak NaNbO3 üzerinde
yaptıkları 30-950 cm-1 dalga sayısı aralığındaki deneysel ölçümlerde kip frekanslarını
60,1 cm-1 (1,803 THz), 72,1 cm-1 (2,163 THz), 92,2 cm-1 (2,766 THz), 123,6 cm-1
(3,708 THz), 142,4 cm-1 (4,272 THz), 153,7 cm-1 (4,611 THz), 174,8 cm-1 (5,244
THz), 182,8 cm-1 (5,484 THz), 201,5 cm-1 (6,045 THz), 220,2 cm-1 (6,606
THz), 253,8 cm-1 (7.614 THz), 274,5 cm-1 (8,265 THz), 293,4 cm-1 (8,802 THz),
378,6 cm-1 (11,358 THz), 433,7 cm-1 (13,011 THz), 555,9 cm-1 (16,677 THz),
601,7 cm-1 (18,051 THz), 674 cm-1 (20,22 THz), 871,3 cm-1 (26,139 THz) olarak
bulmuşlardır. Bazı Raman kipler bizim hesapladığımız kiplerle uyuşmaktadır.
Shanker ve ark (2009), NaNbO3 nano kristalleri üzerinde 100-1000 cm-1
arasında yaptıkları Raman spektroskopisi ölçümlerinden elde ettikleri kip frekansları
240 cm-1 (7,20 THz), 600 cm-1 (18,0 THz) ve 875 cm-1 (26,25 THz) dir. Nano
kristalin fonon kipleri bizim değerlerle kısmen uyuşmaktadır.
Machado ve ark (2011), WIEN2K bilgisayar programıyla kübik NaNbO3
kristallerinin fonon kiplerini hesaplamışlardır ve kübik yapıda yumuşak kiplerin
olduğunu göstermişlerdir. Bu da ortorombik yapıdaki yumuşak kiplerin
olabileceğinin açık kanıtıdır. NaNbO3 kristalinin PDOS 'dan görülebileceği gibi -
10 - 0 THz aralığında Na atomları baskındır. Bu durum kristal örgüde Na'nın atomik
pozisyonlarının kararsızlığıyla ilişkilidir. Benzer kararsızlık Nb ve O atomlarında da
vardır. Fakat oran olarak Na den küçüktürler. Benzer sonucu Prasondeev (2005),
NaNbO3 üzerinde yaptığı teorik çalışmada gözlemiştir. Bu kararsızlık yumuşak
kiplerin ortaya çıkmasına neden olur. Bu yumuşak kipler NaNbO3 kristalinin
ortorombik yapıdan rombohedral yapıya geçmesinden sorumludur.

5. SONUÇLAR Şevket ŞİMŞEK
107
5. SONUÇLAR VE ÖNERİLER
Bu tez çalışmasında yoğunluk fonksiyoneli yöntemleri kullanılarak ilk defa
oda sıcaklığında AgNbO3, AgTaO3, NaNbO3 ve NaTaO3 kristallerinin elektronik ve
dinamik özellikleri temel prensiplere dayanan ABINIT, SIESTA ve PHONON
yazılım programları kullanılarak aşağıdaki çalışmalar yapılmıştır:
1. AgNbO3, NaTaO3, NaNbO3 kristallerinin ortorombik yapıda ve AgTaO3
kristalininin rombohedral yapıda örgü parametreleri hesaplanmıştır. Bu dört
kristalinde örgü parametreleri deneysel sonuçlar ile uyum içinde olduğu görülmüştür.
2. AgNbO3, NaTaO3, NaNbO3 kristallerinin ortorombik yapıda ve AgTaO3
kristalininin rombohedral yapıda toplam ve parçalı durum yoğunlukları hesaplandı.
DOS ve PDOS’un enerjiyle değişimi incelendi.
3. AgNbO3, NaTaO3, NaNbO3 kristallerinin ortorombik yapıda ve AgTaO3
kristalininin rombohedral yapıda elektronik band yapısı hesaplanmıştır. AgNbO3
kristalinin valans bandını oksijen atomunun 2s ve 2p orbitalleri ve gümüş atomunun
4d orbitalleri tarafından oluşturulmuştur. İletim bandı ise Nb atomunun 4d ve gümüş
atomunun 5s orbitalleri tarafından oluşmaktadır. AgTaO3 için valans bandını O’nin
2s ve 2p ve Ag’nin 4d orbitalleri tarafından oluşturulurken iletim bandını Ta
atomunun 5d ve Ag atomunun 5s orbitalleri tarafından oluşturulduğu gözlenmiştir.
NaTaO3 kristalinin valans bandını bandını O’nin 2s ve 2p ve Ta atomunun 5d
elektronları oluşturmaktadır. Ta atomunun 5d elektronlarının bazılarının iletim
bandına geçtiği görülmektedir. NaNbO3 için valans bandını oksijen atomunun 2s ve
2p orbitalleri oluştururken iletim bandı ise Nb atomunun 4d ve Na atomunun 3s
orbitalleri tarafından oluşmaktadır.
4. AgNbO3 ve AgTaO3 kristallerinin dolaylı band aralığına sahip kristaller olduğu
görülmüştür. Ortorombik yapıdaki AgNbO3 ve rombohedral yapıda AgTaO3 için
dolaylı band aralığı değerleri sırasıyla 1,627 eV ve 1,795 eV dir. NaNbO3 ve NaTaO3
kristallerinin, AgNbO3 ve AgTaO3 kristallerinin aksine doğrudan band aralığına
sahip olduğu görülmüştür ve doğrudan band aralığı değerleri sırasıyla 1,586 eV ve
2,861 eV olarak bulundu.

5. SONUÇLAR Şevket ŞİMŞEK
108
5. AgNbO3, AgTaO3, NaNbO3 ve NaTaO3 kristallerinin Born efektif yük tensörleri
hesaplandı. Bu kristallerdeki atomların yerleştiği Wyckoff konumlarına göre Born
efektif yükleri nominal değerlerden tensörün göz önüne alınan elemanına bağlı
olarak büyük ve küçük değerler almaktadır. Bunun nedeni katyon ve anyonlar
arasındaki kovalent bağın büyüklüğü ile ilgilidir. Buna ek olarak, valans bandındaki
hibritleşmeden dolayı bandlar arasındaki yük alış verişleri Born efektif yükün
değerini değiştirmektedir.
6. Ortorombik yapıdaki NaTaO3 ve rombohedral yapıda AgTaO3 kristallerinin elastik
sabitleri ve bulk modülü hesaplandı. Elastik sabitlerin ve bulk modüllerin mekanik
denge koşullarını sağladığı görüldü.
7. AgTaO3 ve NaTaO3 kristallerinin statik ve optik dielektrik sabitleri hesaplandı.
Optik dielektrik sabitinin değeri, statik dielektrik sabitinin değerinden küçük olduğu
görülmüştür. Bunun sebebi optik bölgede polarizasyona sadece elektronik
polarizasyonun katkı vermesidir. Optik bölgede iyonik ve dipolar polarizasyon
kaybolmaktadır.
8. AgTaO3 kristali için 2x2x2x (80 atom), NaTaO3 için 2x3x2 (240 atom), AgNbO3
ve NaNbO3 için ise 2x2x1 (160 atom) süper hücreler oluşturuldu. Bu süper
hücrelerde kristallerin fonon dispersiyon eğrileri ve fonon durum yoğunlukları
hesaplandı. Teorik olarak bu çalışmalar ilk defa yapılmaktadır.
9. Gamma noktasında sözü edilen kristallerin kip sayısı, kip frekansı ve simetri
tipleri belirlendi. Gamma noktasındaki hangi simetri kiplerinin RM ve IR aktif olup
olmadıkları belirlendi.
10. Araştırdığımız bu kristallerin oda sıcaklığında yumuşak kiplere sahip olduğunu
gözlemledik. Yumuşak kiplerin işaret ettiği yer değiştirmelerden dolayı kristaller
karasızdır. Faz geçişlerinde bu yumuşak kipler sorumludur. Bir çok ABO3 tipi
perovskitlerde (SrTiO3, LiNbO3, BaTiO3, v.b) bu yumuşak kipler gözlenmektedir.
11. AgTaO3 kristalinin dispersiyon eğrisinden tüm simetri noktalarında LO/TO
ayrışması oluşmaktadır. AgNbO3, NaNbO3 ve NaTaO3 yapılarında ise Γ-X, Y-Γ-Z ve
Z-U simetri noktaları boyunca LO/TO ayrışması gözlendi. Fakat diğer simetri
noktalarında ise LO/TO ayrışması çok çok zayıftır. Bunun sebebi Born efektif

5. SONUÇLAR Şevket ŞİMŞEK
109
yüklerinin bu simetri noktalarında çok küçük olmasından kaynaklandığını
düşünmekteyiz.
12. AgTaO3’nın fonon PDOS eğrisinde düşük frekanslarda Ag atomlarının yüksek
frekanslarda Ta atomlarının durum yoğunluğunun baskın olduğu gözlendi. NaNbO3
ve NaTaO3 kristalleri ortorombik yapıda olduklarından PDOS’ları birbirine çok
benzemektedir. NaNbO3 0-10 THz arasında oksijen atomlarının durum yoğunlukları
baskın olmaktadır. NaTaO3 ise aynı aralıkta Na atomlarının durum yoğunluğu
baskındır. Oksijen atomları ise 10-20 THz de çok daha yoğun olarak
bulunmaktadırlar. AgNbO3 kristalinde ise -5 -0 THz aralığında Ag ve Nb atomları
oksijene göre daha baskındır.
Ortorombik yapıda AgNbO3 ve NaNbO3 kristallerinin birim hacimde 40 atom
bulunmasından dolayı elastik sabitleri ve bulk modülü incelenememiştir. Bu
özellikler yüksek RAM kapasitesi ve işlemcisi olan bilgisayarlarda hesaplanabilir.
Bunun yanında bu dört kristalin elektronik band yapısı ve dinamik özelliklerinin
basınçla değişimi incelenebilir. Ayrıca bu kristallerin oda sıcaklığında lineer
olmayan optik özellikleri de araştırılabilir
Yapılan bu tez çalışmasının sunulduğu toplantılar:
1. Ş. Şimşek, S. Çabuk, İlk ilke Yöntemiyle NaTaO3 Kristalinin Elektronik
ve Dinamik Özelliklerinin İncelenmesi: Yoğunluk fonksiyoneli teorisinin
uygulaması, 18. Yoğun Madde Fiziği Ankara Toplantısı, 25 Kasım 2011.
2. Ş. Şimşek, S. Çabuk, Oda sıcaklığında AgTaO3 kristalinin elektronik ve
dinamik özelliklerinin incelenmesi: Ab initio hesabı, 18. Yoğun Madde
Fiziği Ankara Toplantısı, 25 Kasım 2011.

5. SONUÇLAR Şevket ŞİMŞEK
110

111
KAYNAKLAR
AKKUŞ, H., 2007. SbSI Kristalinin Elektronik Ve Optik Özellikleri: Yoğunluk
Fonksiyonel Teorisinin Uyğulanması, ADANA, 113s.
ABDULLAH, N, A, 2004, Finite-temperature properties of complex perovskites
from first princibles calculations, University of Arkansas, 145s.
ARNEY, D., HARDY, C., GREVE, B., MAGGARD., P.A., 2010. Flux synthesis of
AgNbO3: Effect of particle surfaces and sizes on photocatalytic Activity.
Journal of Photochemistry and Photobiology A: Chemistry. 214:54-60
ARTACHO, E., GALE, J.D., GARCİA, A., JUNQUERA, J., ORDEJON, P.,
SANCHEZ-PORTAL., D., SOLER,J.M., 2009. S I E S T A trunk-301. http://www.uam.es/siesta
AULBUR,WG, JONHSON, L., AND WILKINS, JW., 2000. Qualisiparticle
Calculations İn Solids. Solid State Physics. 54: 1–231.
CEPERLEY, D. M, AND ALDER, B. J.,1980. Ground State of the Electron Gas by a
Stochastic Method, Phys. Rev. Lett. 45: 566 – 569.
COHEN,R,E., KRAKAUER, H., 1990. Lattice dynamics and origin of
ferroelectricity in BaTiO3: Linearized-augmented-plane-wave total-energy
calculations. Phys. Rev. B 42: 6416 – 6423.
CHOİ, M., OBA, F., TANAKA, I., 2008. First-principles study of native defects and
lanthanum impurities in NaTaO3. Phys. Rev. B 78: 014115
DURLU, T.N., 1996. Katıhal Fiziğine Giriş, ANKARA, 311s.
EYİ, E.E., CABUK, S., 2010. Ab initio study of the structural, electronic and optical
properties of NaTaO3. Philosophical Magazine. 90: 2965–2976
FRANCOMBE, M.H., AND LEWIS, G., 1958. Structural and Electrical Properties
Of Silver Niobate and Silver Tantalite. Acta Cryst, 11: 175–178.
FU, D., ENDO, M., TANİGUCHİ, H., TANİYAMA, T., ITOH, M., 2007. AgNbO3:
A lead-free material with large polarization and electromechanical response.
Applied Physics Letters 90: 252907

112
FU, D., ITOH, M., AND KOSHİHARA, S., 2009. Dielectric, ferroelectric, and
piezoelectric behaviors of AgNbO3–KNbO3 solid solution. Journal of
Applied Physics. 106: 104104
FU, D., ARİOKA, T., TANİGUCHİ, H., TANİYAMA, T., ITOH,M., 2011.
Ferroelectricity and electromechanical coupling in (1-x)AgNbO3-xNaNbO3
solid solutions. Applied Physics Letters. 99: 012904
FUCHS, M., SCHEFFLER, M., 1999. Ab initio pseudopotentials for electronic
structure calculations of poly-atomic systems using density-functional theory.
Comput. Phys. Commun., 119: 67-98.
GİANNOZZİ P., GİRONCOLİ S. D, PAVONE P. AND BARONİ S., 1991.Ab initio
calculation of phonon dispersions in semiconductors. Phys. Rev. B, 43:7231-
7242.
GONZE, X, KÄCKELL, P and SCHEFFLER, M., 1990. Ghost states for separable,
norm-conserving, Iab initioP pseudopotentials, Phys. Rev. B 41: 12264 –
12267.
GONZE, X., ALLAN, D. C. AND TETER, M. P., 1992. Dielectric tensor, effective
charges, and phonons in _-quartz by variational density-functional perturbation
theory, Phys. Rev. Lett., 68: 3603-3606.
GONZE, X., BEUKEN, M., CARACAS, R., DETRAUX, F., FUCHS, M.,
RIGNANESE, G. M., SINDIC, L., VERSTRATE, M., ZERAH, G., JOLLET,
F., TORRENT, M., ROY, A., MIKAMI, M., GHOSEZ, P., RATY, J, Y.,
ALLAN, D.C., 2002. First-princibles computation of material properties: the
ABINIT software Project. Computational Materials Science, 25: 478–492.
URL http://www.abinit.org
GHOSEZ, P AND XAVİER GONZE, X., 2000. Band-by –band decompositions of
the Born effective charges. J. Phys.:Condens. Matter 12: 9179-9188
GRINBERG, I and ROPPE, A.M., 2003. Ab initio study of silver niobate.
Fundemental Physics of Ferroelectrics. 667: 130–138.
HAMANN, D. R.,1989. Generalized Norm-Conserving Pseudopotentials, Phys. Rev.
B 40, 2980 – 2987.

113
HAMANN D. R., SCHLÜTER M. AND CHİANG C., 1979. Norm-conserving
Pseudopotentials. Phys. Rev. Lett., 43:1494-1428.
HARTREE D. R., 1928. The wave mechanics of an atom with a non-Coulomb
central field. Part I. Theory and methods. Proc. Cambridge Philos. Soc., 24,
89-110
HOHENBERG, P, AND KOHN, SHAM.1964. Inhomogeneous Electron Gas, Phys.
Rev. 136, B864 - B871.
İNTERNET., 2007. Dokuz Eylül Üniversitesi ”Yogunluk fonksiyoneli teorisi,
http://kisi.deu.edu.tr/umit.akinci/kmc/node1.html
JONA, F and SHIRANE, G., 1993. Ferroelectric Crystals. Dover Publications, Inc. 397s.
JOHNSTON, K.E., TANG, C.C., PARKER, J.E., KNİGHT, K.S., LİGHTFOOT, P.,
ASHBROOK, S.E., 2010. The Polar Phase of NaNbO3: A Combined Study
by Powder Diffraction, Solid-State NMR, and First-Principles Calculations. J.
Am. Chem. Soc. 132: 25
JOHNSTON, I., KEELER, G., ROLLİNS, R., SPİCKLEMİRE S., 1996. Solid State
Physics Simulations, The Consortium for Upper-Level Physics Software,
John Wiley, New York, 45-59.
KANIA, A., 2001. Dielectric properties of Ag1-xAxNbO3(A:K, Na and Li) and
AgNb1-xTaxO3 solid solutions in the vicinity of diffuse phase transtions.
J.Phys.D:Apply.Phys., 34: 1447-1455.
KANIA, A., ROLEDER, K and LUKASZEWSKİ, M., 1984. The Ferroelectric Phase
in AgNbO3. Ferroelectrics. 52: 265–269.
KANIA, A and ROLEDER, K., 1984. Ferroelectricity in AgNbl-xTaxO3 solid
solutions. Ferroelekctric lett. 2: 51–54.
KATO, H., KOBAYASKI, H., and KUDO, A., 2002. Role of the Ag+ in the Band
Structures and Perovskite Structure. J.Phys. Chem. B, 106: 12441–12447.
KENNEDY, B.J., PRODJOSANTOSO, A.K., and HOWARD, C. J, 1999. Powder
neurton diffraction study of the high temperature phase transitions in
NaTaO3. J.Phys: Condens. Matter., 11: 6319–6327.
KİTTEL, C., 1996. Katıhal Fiziğine Giriş

114
KLEINMAN, L, AND BYLANDER, D. M., 1988. Efficacious Form for Model
Pseudopotentials. Phys. Rev. Lett. 48: 1425 – 1428. KOHN, S, AND SHAM,L.J.,1965. Self-Consistent Equations Including Exchange
and Correlation Effects. Phys. Rev. 140: A1133 - A1138. KRAYZMAN, V., LEVİN, I., 2010. Reverse Monte Carlo refinements of local
displacive order in perovskites: AgNbO3 case study. J. Phys: Condens.
Matter. 22: 404201
KROUMOVA E., AROYO M. I, PEREZ MATO J. M., KİROV A., CAPİLLAS C.,
IVANTCHEV S. VE WONDRATSCHEK H., 2003. Bilbao Crystallographic
Server: useful databases and tools for phase transitions studies. Phase
Transitions, 76:155-170.
KUGEL, G. E., FANTANO, M. D., HAFID, M., ROLEDAR, K., KANIA, A. and
PAWELCZYK, M., 1987. A Raman Study Of Silver Tantalate (AgNbO3) and
its Structurel Phase Transtions Sequence. J.Phys. C: Solid state Phys, 20:
1217–1230.
LAASONEN K., CSAJKA F. AND PARRİNELLO M., 1992. Water dimer properties in
the gradient-corrected density functional theory, Chemical Physics Letters,
194:172-174.
LEVİN, I., KRAYZMAN, V., WOİCİK,J.C., KARAPETROVA, J., PROFFEN, T.,
TUCKER, M.G., REANEY, I.M., 2009. Structural changes underlying the
diffuse dielectric response in AgNbO3. Physical Review B 79: 104113
Lİ, G., BAİ, Y., LİU, X., ZHANG, W.F., 2009. Surface photoelectric properties of
AgNbO3 photocatalyst. J. Phys. D: Appl. Phys. 42: 235503
LINES, M. E., GLASS, A. M., 1977. Princibles and Applications of Ferroelctrics and
Related Materials. Clarendon Pres, Oxford, 680s.
METE, E, 2003, ELECTRONIC PROPERTIES OF TRANSTION METAL
OXIDES, ANKARA, 80s.
MİSHRA, S.K., CHOUDHURY, N., CHAPLOT, S.L., KRİSHNA, P.S.R.,
MİTTAL, R., 2007. Competing antiferroelectric and ferroelectric interactions
in NaNbO3: Neutron diffraction and theoretical studies. Phys. Rev. B, 76:
024110

115
MONKHORTST, H. J., AND PACK, J. D., 1976. Special Points For Brillouin-Zone
İntegrations. Phys. Rev. B, 13: 5188–5192.
NYE, J. F., 1957. Physical Properties of Crystals. Clarendon Pres, Oxford, 315s.
PAYNE, M. C., TETER, M. P., ALLAN, D. C., ARIAS, T. A., and
JOANNOPOULOS, J. D., 1992. Iterative Minimization Techniques for ab
initio Total-Enerji Calculations: Moleculer Dynamics and Conjugate
Gradients. Rev. Mod. Phys., 64:1045-1097.
PARLİNSKİ, K., 2007. Phonon software “Phonon Manual”
http.//wolf.ifj.edu.pl/phonon/
PARLİNSKİ K., Lİ Z. Q., KAWAZOE Y.,1997.First-Principles Determination of the
Soft Mode in Cubic ZrO2, Phys. Rev. Lett., 78: 4063-4066.
PAWELCZYK, M, 1987. Phase transitions in AgTaxNb1-xO3 solid solutions, phase
transtions, 8: 273 – 292.
PERDEW, J. P., WANG, Y., 1992. Accurate and simple analytic representation of
the electron-gas correlation enerji. Phys. Rev. B, 45: 13244–13249.
PINES, D., 1963. Elemantary Excitations in Solids, Benjamin Inc., NY-Amsterdam,
299s.
PROSANDEEV, S, A.,2005. Compartive Analysis Of The Phonon Modes İn
AgNbO3. Physics Of Solid State, 47: 2130–2134.
RABE, K.M., AHN, C.H., TRISCONE, J-M., 2007 Physics of Ferroelectrics: A
Modern Perspective. Springer Link.Com. Berlin, Heidelberg, New York. 385
s.
RATUSZNA, A., PAWLUK, J., AND KANIA, A., 2003. Temperature Evolution of
the Crystal Structure of AgNbO 3, Phase Transitions, 76: 611-620.
REZNİTCHENKO, L.A., TURİK, A.V., KUZNETSOVA, E.M., SAKHNENKO,
V.P., 2001. Piezoelectricity in NaNbO3 ceramics.J. Phys.: Condens. Matter,
13: 3875–3881
SAMUEL, V., GAIKWAD, A.B., RAVI, V., 2006, A coprecipitation technique to
prepare NaNbO3 and NaTaO3, Bull. Mater. Sci, 29: 123–125
SAI, N, 2002, First-Prencibles Modeling of Structural and Electronic Properties in
Ferroelectric Compounds, New Brunswick, New Jersey, 112s.

116
SCHREİBER, E., ANDERSON, O.L., SOGA, N., 1973. Elastic Constants and their
Measurements, McGraw-Hill, New York, 102-105
SCIAU, PH., KANIA, A., DKHIL, B., SUARD, E AND RATUSZNA, A., 2004.
Structurel İnvestigation Of AgNbO3 Phases Using X-Ray and Neutron
Diffraction. J.Phys: Condens Matter, 16: 2795–2810.
SOON, H.P., TANİGUCHİ, H., ITOH, M., 2009. Ferroelectricity triggered in the
quantum paraelectric AgTaO3 by Li-substitution. Applied Physics Letters.
95: 242904
SOON, H.P., TANİGUCHİ, H., ITOH, M., 2010. Dielectric and soft-mode behaviors
of AgTaO3. Phys. Rev. B, 81: 104105
SUCHANICZ, J., KANIA, A., STOPA, G., BUJAKIEWICZ-KORONSKA, R.,
2007. Influence Of Axial Pressure On Dielectric Properties Of AgNbO3
Singel Crystals and Ceramics. Phase Transtions., 80: 123-129.
SUCHANICZ1, J., KANIA, A., 2009. Uniaxial Pressure Effect on Dielectric
Properties of AgTaO3 Single Crystals. Ferroelectrics, 393: 21–26
TROULLIER, N., AND MARTIENS, J. L., 1991. Efficient Pseudo Potantials for
Plane-Wave Calculations. Phys. Rev B, 43: 1993–2006.
______, 1990. A straightforward method for generating soft transferable
pseudopotentials. Solid State Commun., 74: 613-616.
VALANT, M., SUVOROV, D.,HOFFMANN, C., SOMMARIVA, H., 2001.
Ag(Nb,Ta)O3-Based Ceramics with Suppressed Temperature Dependence Of
Permittivity. Jounal Of European Ceramic Society, 21: 2647–2651.
VALANT, M., AXELSSON, A.K., ZOU, B., ALFORD, N., 2007.Oxygen transport
during formation and decomposition of AgNbO3 and AgTaO3. J. Mater.
Res., 22: 6
VERWERFT, M., VAN DYCK, D., BRABERS, V. A. M., VAN LAYDUNT, J.,
AMELINCKX, S., 1989. Electron Microscopic Study of The Phase
Transformations in AgNbO3. Phys. Status Solidi (a), 112: 451–466.
VOLKOV, A, A., GORSHUNOV, B, P., KOMANDIN, G., FORTIN, W., KUGEL,
G, E., KANIA, A and Grigas, J., 1995. High-Frequency Dielectric Spectra Of
AgTaO3-AgNbO3 Mixed Ceramics. J. Phys: Condens. Matter, 7: 785–793.

117
WANG, C. S., and KLEIN, B. M., 1981. First-princibles electronic structure of Si,
Ge, GaP, GaAs, ZnS, and ZnSe. II. Optical Properties. Phys. Rev. B, 24:
3417–3429.
WANG, J.J., MENG, F.Y., MA, X.Q., XU, M.X., CHEN, L.Q., 2010. Lattice,
elastic, polarization, and electrostrictive properties of BaTiO3 from first-
principles. Journal of Applied Physics, 108:034107.
WANG Y.X., ZHONG W.L., WANG C.L., ZHANG P.L., 2001. First-principles
study of the electronic structure of NaTaO3. Solid State Communications
120:137-140
WOLFRAM, T., ELLİALTİOGLU, Ş., 2006. Elektronik and Optical Properties of
D-Band Perovskites. Cambridge University Press, New York, 315 s.
WU, Z., ZHAO, E., XİANG, H., HAO, X., LİU, X., MENG, J., 2007. Crystal
structures and elastic properties of superhard IrN2 and IrN3 from first
principles. Physical Review B 76:054115
XU, J., 2000, The Low Temperature Synthesıs, Characterızatıon And Propertıes Of
Ferroelectrıc, Unıted States, 238s.
XU J., XUE D., YAN C., 2005. Chemical synthesis of NaTaO3 powder at
lowtemperature. Materials Letters 59: 2920–2922.
YASHİMA, M., MATSUYAMA, S., SANO, R., ITOH, M., TSUDA, K., FU, D.,
2011. Structure of Ferroelectric Silver Niobate AgNbO3. Chem. Mater. 23:
1643–1645
ZIMMERMANN, F., MENESKLOU, W., IVERS-TIFFEE, E., 2004. İnvestigation
Of Ag(Ta,Nb)O3 as Tunable Microwave Dielektrik. Journal Of European
Ceramic Society, 24: 1811–1814.

118

119
ÖZGEÇMİŞ
1981 yılında Adana’nın Kozan ilçesinde doğdu. Kozan 60.Yıl İlkokulu’ nu
bitirdikten sonra Ortaokulu ve Liseyi Kozan 50.Yıl lisesinde 1998 yılında tamamladı.
2000 yılında Erciyes Üniversitesi Yozgat Fen-Edebiyat Fakültesi Fizik bölümünü
kazandı. 2004 yılında Erciyes Üniversitesi Yozgat Fen-Edebiyat Fakültesi Fizik
bölümünden mezun oldu. 2008 yılında Çukurova Üniversitesi Fen Bilimleri
Enstitüsü Fizik anabilim dalında yüksek lisans öğrenimini tamamladı. 2008 yılında
Çukurova Üniversitesi Fen Bilimleri Enstitüsü Fizik anabilim dalında doktora
öğrenimine başladı.















![9[1].Handover ta](https://img.dokumen.tips/doc/110x75/631bd2bd3e8acd9977059cca/91handover-ta.jpg)

![Synthesis and characterisation of the uranium pyrochlore betafite [(Ca,U)₂(Ti,Nb,Ta)₂O₇]](https://img.dokumen.tips/doc/110x75/636239939b985d7ef5024a36/synthesis-and-characterisation-of-the-uranium-pyrochlore-betafite-cautinbtao.jpg)











