Embed Size (px)
Citation preview

R
Btp
Pa
b
a
ARRAA
KLBPMB
C
t1
1h
Journal of Molecular Catalysis A: Chemical 388–389 (2014) 2–34
Contents lists available at ScienceDirect
Journal of Molecular Catalysis A: Chemical
jou rna l h om epa ge: www.elsev ier .com/ locate /molcata
eview
iomimetic metalloporphines and metalloporphyrins as potentialools for delignification: Molecular mechanisms and applicationerspectives
aolo Zuccaa,b, Antonio Rescignob, Andrea C. Rinaldib, Enrico Sanjustb,∗
Consorzio UNO, Consortium University of Oristano, Oristano, ItalyDipartimento di Scienze Biomediche, Università di Cagliari, Monserrato, Italy
r t i c l e i n f o
rticle history:eceived 11 June 2013eceived in revised form 8 September 2013ccepted 11 September 2013vailable online 20 September 2013
eywords:
a b s t r a c t
Lignin is a recalcitrant polymer arising from addition polymerization of phenylpropanoid units via anoxidative, enzyme-catalyzed radical mechanism. Lignin removal is a serious technological challenge inwood-related industries such as pulping for paper production. In this review, some outstanding aspectsin lignin biosynthesis and structure are depicted; also the commonly used industrial protocols for pulpdelignification are described, with special emphasis on their molecular aspects. A discussion is presentedconcerning the known chemical mechanisms of enzyme-catalyzed delignification by white-rot fungi.
igniniodegradationeroxidase/peroxygenase emulationetalloporphyrins
iomimetic
Biomimetic and bioinspired synthetic metalloporphines show monooxygenase/peroxygenase-like cat-alytic activity, being quite more versatile catalysts than ligninolytic enzymes (being capable only ofone-electron oxidations). The advantages of this behavior are encompassed with an in-depth discussionabout the molecular aspects of their action mechanisms, the possible oxygen donors, and the knownoxidizable substrates. Limitations and perspectives about their practical use at an industrial scale indelignification processes are discussed.
© 2013 Elsevier B.V. All rights reserved.
ontents
1. Introduction: lignin removal poses serious technological and environmental issues . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32. Lignin: occurrence, structure, and properties . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3
2.1. Lignin structure is very heterogeneous . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32.2. Oxidative polymerization of monolignols . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
3. Chemical methods for lignin degradation/solubilization . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 43.1. The Kraft lignin . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 43.2. Sulfite pulping . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 73.3. Chlorine- and ozone-based treatments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 73.4. Peroxide-based treatments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7
4. Oxidative biodegradation of lignin . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 94.1. Laccase . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 94.2. Ligninolytic peroxidases . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9
4.2.1. Lignin peroxidase . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10
4.2.2. Manganese peroxidase . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 124.2.3. Versatile peroxidase. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 124.2.4. Peroxygenase . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 124.3. Other enzymatic activities are required for in vivo heme-peroxidase-based ligninolysis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
Abbreviations: LiP, lignin peroxidase, E.C. 1.11.1.14; MnP, manganese peroxidase, E.C. 1.11.1.13; TDCP, meso-tetrakis-(2,6-dichloro-phenyl)porphine; TFPP, meso-etrakis(pentafluorophenyl)porphine; TSPP, meso-tetrakis(4-sulfonatophenyl)porphine; TPP, meso-tetraphenylporphine; VA, veratryl alcohol; VP, versatile peroxidase, E.C..11.1.16.∗ Corresponding author at: Dipartimento di Scienze Biomediche, Complesso Universitario, I-09042 Monserrato, CA, Italy. Tel.: +39 0706754518; fax: +39 0706754527.
E-mail addresses: [email protected], [email protected] (E. Sanjust).
381-1169/$ – see front matter © 2013 Elsevier B.V. All rights reserved.ttp://dx.doi.org/10.1016/j.molcata.2013.09.010

P. Zucca et al. / Journal of Molecular Catalysis A: Chemical 388–389 (2014) 2–34 3
5. Learning from nature: from natural metalloporphyrins to synthetic metalloporphines. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 135.1. Natural metalloporphyrins are too unstable for in vitro applications . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 135.2. The development of more stable and active synthetic metalloporphines . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
6. Molecular aspects of oxidation/oxygenation reactions catalyzed by metalloporphines . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 156.1. Fe-porphines . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 156.2. Mn-porphines . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 186.3. Ru-porphines . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 206.4. The importance of the axial coordination . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
7. The reducing substrates . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 228. Product and/or catalyst recovery: heterogenized metalloporphines . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 239. Metalloporphines and lignin . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2410. Conclusion and perspectives . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30
. . . . . .
1a
bboasawbto–tcpr
narbsb
asqal
2
2
ivh(fl
ldhtpw
abundant in tracheae, whereas S concentration is higher in fibers[13].
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. Introduction: lignin removal poses serious technologicalnd environmental issues
After cellulose, lignin is the most abundant biopolymer in theiosphere [1] and is therefore of outstanding importance in theiogeochemical carbon cycle. Lignin itself, however, poses seri-us challenges to lignocellulosics exploitation [2] such as in pulpnd paper technology, as it is an insoluble, chemically resistantubstance hindering the complete utilization of cellulose and inddition producing huge amounts of environmentally-impactingastes. In fact, although lignocellulosics are natural products, the
iodegradation of their lignin component is very slow. Moreover,he close chemical and physical association between lignin and thether lignocellulosic components makes selective lignin removal
usually needed in order to exploit cellulose fibers – a very hardask, which is usually tackled through high-impacting technologi-al solutions. Thus, removed lignin is eventually found into highlyolluting wastes requiring further expensive treatments for theiremediation.
Despite many focused efforts by the scientific commu-ity, the economical feasibility of alternative, less expensivend more eco-friendly processes (mainly oxidative) for ligninemoval/degradation – is far to be achieved. For instance,ioethanol production from lignocellulosic wastes is not yet a largecale process, still requiring further significant enhancements toecome economically sustainable [3].
Besides the treatment of lignocellulosics, oxidative methods canlso find application in the removal of many other pollutant wastes,uch as olive mill wastewater [4,5] and textile dyes [6–8]. Conse-uently, the optimization of oxidative lignin removal processes hasn inherent importance that goes well beyond the specific field ofignocellulosics treatment.
. Lignin: occurrence, structure, and properties
.1. Lignin structure is very heterogeneous
Lignin is typical for vascular plants, and particularly abundantn wood stricto sensu as well as in other wood-like tissues. It exertsarious physiological key functions [9] such as rendering tissuesard and firm [10], conferring them high resistance to compressionthe high weight of the tree itself), and strictly regulating the waterux through the wood structure [11].
The highest lignin concentrations are found in the middleamella [12,13], followed by the secondary cell walls; however,ue to the much larger volume of the secondary cell wall, it
as the highest lignin content. Lignin composition and struc-ure vary to a certain extent with the plant species, with thearticular tissue considered within the same individual, andith the age of that tissue. Therefore, ‘lignins’ rather than. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31
‘lignin’ should be preferred when a general view on the topic isassumed.
Lignins are complex heteropolymers [14,15] mainly based onthree phenylpropanoid (C6C3) monomers (monolignols, Fig. 1),derived from plant cinnamate path: 4-hydroxycinnamyl alco-hol (p-coumaryl alcohol), 4-hydroxy-3-methoxycinnamyl alcohol(coniferyl alcohol), and 4-hydroxy-3,5-dimethoxycinnamyl alco-hol (sinapyl alcohol). These undergo one-electron oxidation [16]by H2O2-dependent peroxidases [17] or – it is debatable – byO2-dependent laccases [18], and the arising phenoxyl radicalstrigger monolignol polymerization by a polyaddition mechanism.The three monolignols give rise to three phenylpropanoid unitsp-hydroxyphenyl (coumaryl) H, guaiacyl G, and syringyl S, respec-tively, when incorporated in the lignin polymer [19,20]. Besides,the presence of hydroxycinnamate analogs has been also described,being particularly involved in covalent bonding with hemicellu-loses [21].
Generally speaking [22], angiosperm (hardwood) lignins(approximately accounting for 18 to 25% w/w of dry wood) con-tain mainly G and S units (G/S/H = 50/50/traces, with the extremecase of Eucalyptus globulus lignin where S units account for the82–86% [23]). G units largely prevail in gymnosperm (softwood)lignins (25–35% lignin content), where also low levels of H unitsare present (G/S/H = 96/traces/4 approximately). Although gym-nosperm lignin is chemically simpler, it is more abundant in wood[24]. Lignins from monocotyledon grasses (13–26% lignin con-tent) contain prevailing amounts of G, and more H units thandicotyledons (G/S/H = 70/25/5) [25,26]. There is clear evidence thatin angiosperms H incorporation in the growing lignin polymertakes place preferentially during the first steps of biosynthesis, fol-lowed by G units and finally by S units [12]. Also, G units are more
Fig. 1. The general structure and numbering of the three monolignols involved inlignin composition. R1 = R2 = H: p-coumaryl alcohol; R1 = H, R2 = OCH3: coniferylalcohol; R1 = R2 = OCH3: sinapyl alcohol.

4 atalys
2
ptwtbmvs[rcnvt[
httofaiafipeb
etiqaAccma49al2ba
sficwcttssppaimrs
P. Zucca et al. / Journal of Molecular C
.2. Oxidative polymerization of monolignols
The most accepted polymerization mechanism relies on the cou-ling of monolignols to the growing lignin polymer [11]; radicalransfer plays most probably a key role in the whole synthesis,here the same monolignols as well as p-hydroxycinnamates and
he Mn(II)/Mn(III) redox couple are deeply involved [16,27]. Ligniniosynthesis is a relatively slow process relying on a continuousonomer supply; monolignols concentrations remain constantly
ery low so that gradual growth of the tridimensional lignintructure sharply prevails over monolignol coupling/dimerization28–30]. Accordingly, great care should be exerted when infer-ing structural information from dehydrogenative polymerizatesoming from in vitro oxidation/dehydrogenation of monolig-ol mixtures [31], unless the reactant concentrations are keptery low for a long time, and the polymerization processakes place in the presence of lignin, acting as a template28].
A wide variety of different linkages among the monolignol unitsave been found in lignins [19,32,33], taking into the due accounthat position 3 in coniferyl alcohol is unavailable for coupling reac-ions and both 3 and 5 positions are also unavailable in the casef sinapyl alcohol. Fig. 2 shows the main substructures, commonlyound in natural lignins. Undoubtedly, �-O-4 linkages (�-aryl ether)re the commonest, followed by a number of different linkages,ncorporated in different substructures. �-Hydroxyls only seldomre involved in coupling reactions, therefore largely surviving in thenal polymeric structures, where they contribute to the slightlyolar character of lignin [34]. On the contrary, �-hydroxyls notngaged in inter-subunit linkages can take part in ether and esteronds to hemicelluloses [35,36].
However, the brown color of lignins is not justified by the pres-nce of the substructures cited above, so one could well hypothesizehat not all the reactive, transient quinone methides [30] involvedn the biosynthesis actually lose their quinonoid character: someuinonoid substructures survive in lignin (or can be well gener-ted by further oxidation) and these explain its coloration [37].lso some �-carbonyl groups or cinnamaldehyde-like end groupsould be usually detected in lignins [38]. On the whole, ligninsould be regarded as mainly non-phenolic polymers, being theajority of the 4-OH groups engaged in the �-O-4 inter-unit link-
ges. These �-O-4 linkages account in softwood lignin for almost5–50% of total inter-unit linkages, followed by 18–25% for 5–5,–12% for �-5, 7–10% for �-1, 6–8% for �-O-4, 4–8% for 4-O-5,nd only 3% for �–�. However, the occurrence of simple �-O-4inkages in native lignin has been recently questioned [39]. Also,0–30 phenolic hydroxyls per 100 phenylpropanoid units haveeen estimated [40], that explains the solubility of lignins in stronglkali.
More recently, an alternative hypothesis on lignin biosynthe-is has been developed and corroborated by some experimentalndings [41–44], based on the assumption that monolignol (radi-al) binding dirigent proteins should direct the biosynthesis, whichould therefore be not random, but rather template-based. The
ontroversy is quite fascinating [45] although not so relevant to theopic discussed here; in fact, the presence of well defined struc-ural domains in lignins such as the hexamers and pentamers ofinapyl alcohol found in E. globulus lignin [46] does not modify thetatistical occurrence of the linkages to be (bio)cleaved to accom-lish lignin degradation and solubilization. On the other hand, therecise identification of the genes, codifying for dirigent proteinslong lignin biosynthesis, could have an outstanding importance
n tuning that biosynthesis in selected crops toward modified,ore soluble and/or more degradable lignins [47,48], so that ligninemoval in pulp and paper industrial processes could be made sub-tantially easier.
is A: Chemical 388–389 (2014) 2–34
3. Chemical methods for lignin degradation/solubilization
The main aim of the pulp and paper industry is to obtain cellu-lose as pure as possible, i.e. ideally free from both hemicellulosesand lignins. Hemicellulose removal is a minor problem as thesepolysaccharides are relatively soluble in aqueous extractants. Thecase of lignins is quite different, as these are relatively hydrophobic,water-insoluble polymers, whose solubilization requires concen-trate reagents under quite harsh operative conditions. Aqueousacids are unable to dissolve lignins while hydrolyzing hemicellu-loses and, to a minor extent, cellulose. Concentrate, strong mineralacids rather tend to promote further condensation reactions amongthe C6C3 units. However, aqueous hydrochloric acid dissolvedin dioxane is effective in lignin solubilization and removal [49].Though useful at the laboratory scale, this is obviously void ofany practical significance at the industrial scale. Other lignin sol-vents, based on organic compounds such as acids and alcohols,and containing different amounts of water, mineral acids or bases(‘organosolv’ agents), have been tested about their efficiency inlignin solubilization and removal [50], even if they all share thedeep drawback of the huge amounts of organic solvents requiredfor the scaling up to industrial applications [51,52]. Even when rel-atively environmentally, low-impacting solvents are used such asethanol, the main disadvantage of the process is the need for solventrecovery [39]. The obtained preparations deeply differ from thenative lignin, since solubilization conditions promote substantialalteration through various chemical modifications [53].
On the whole, lignin solubilization along chemical pulping[54–56] is based on the use of various nucleophiles promoting elim-ination/addition reactions – involving very reactive intermediates– that have the final effect of breaking some inter-unit linkagesdown, in particular �-O-4 and especially �-O-4 ether bonds.
Nowadays, chemical lignin removal in the pulp and paper indus-tries is mainly achieved by the Kraft process [40] described below,although sulfite-based plants are still operating as they ensuresome features of the resulting cellulose fibers particularly welcomefor certain applications [57]. Other promising treatments are usedonly for a minor fraction of lignocellulosics to be pulped, but someshow the potential for a substantial expansion in the near future,such as that based on the combined action of hydrogen peroxideand sodium hypochlorite, leading to the very active singlet oxygen[58].
3.1. The Kraft lignin
The Kraft process is based on a strongly alkaline attack ofthe woody material, with a caustic liquor, containing concentratesodium hydroxide and sodium sulfide (the ‘white liquor’) underharsh temperature and pressure conditions [39]; hemicellulosesare rapidly dissolved and removed, whereas lignin degradation isnot complete and leads to partial fragmentation and solubilization[59]. Even harsher conditions could remove almost all lignin, but asignificant cellulose alteration with worsening of its technical prop-erties could be observed. Lignin fragmentation mainly proceedsthrough the cleavage of ether linkages; the obtained fragmentsbecome gradually more soluble as the generated free hydroxylgroups render them more hydrophilic [40]. The carbon–carbonlinkages tend to remain almost untouched by the treatment.
The targets of the alkaline attack are the �-aryl ether and �-aryl ether linkages (mainly �-O-4 and �-O-4), i.e. the linkagesjoining the C6C3 units together. Cleavage of an �-O-4 linkage isfavored when the corresponding phenolic hydroxyl is free; in that
case a quinomethide is formed concomitant with the expulsion ofthe �-OR− anion as the leaving group. This reaction is responsi-ble for lignin fragmentation but also to a certain extent for thebreakage of the main lignin–hemicellulose linkages. In any case,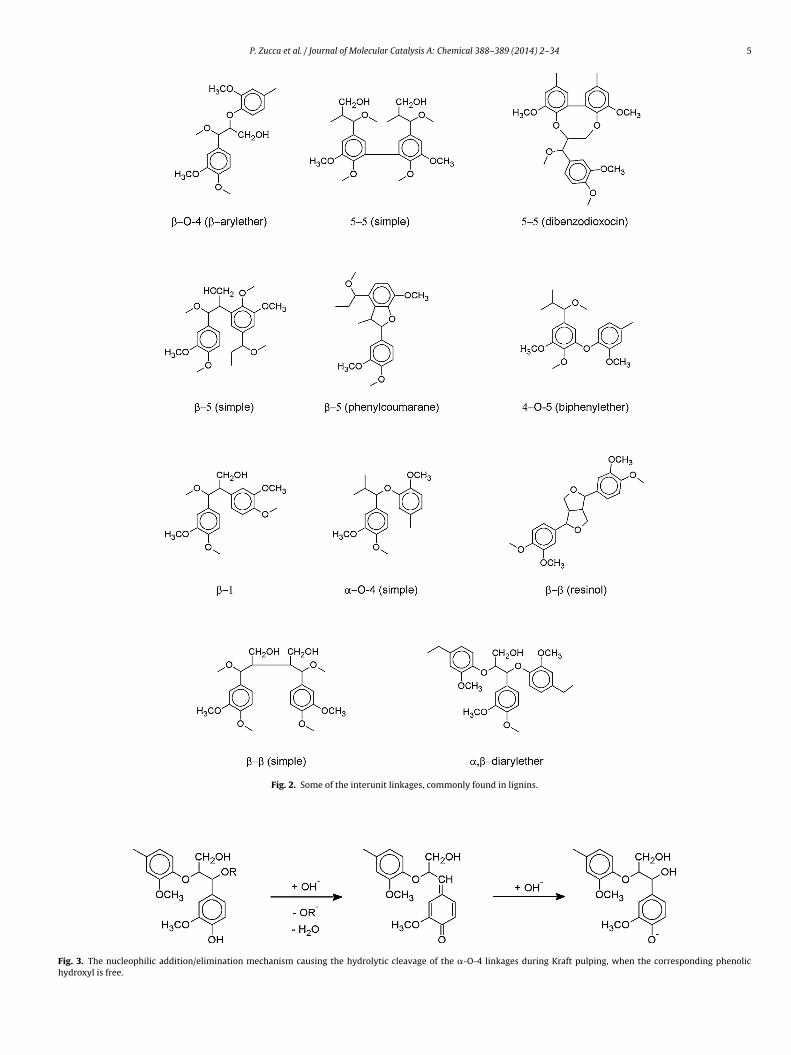
P. Zucca et al. / Journal of Molecular Catalysis A: Chemical 388–389 (2014) 2–34 5
Fig. 2. Some of the interunit linkages, commonly found in lignins.
Fig. 3. The nucleophilic addition/elimination mechanism causing the hydrolytic cleavage of the �-O-4 linkages during Kraft pulping, when the corresponding phenolichydroxyl is free.
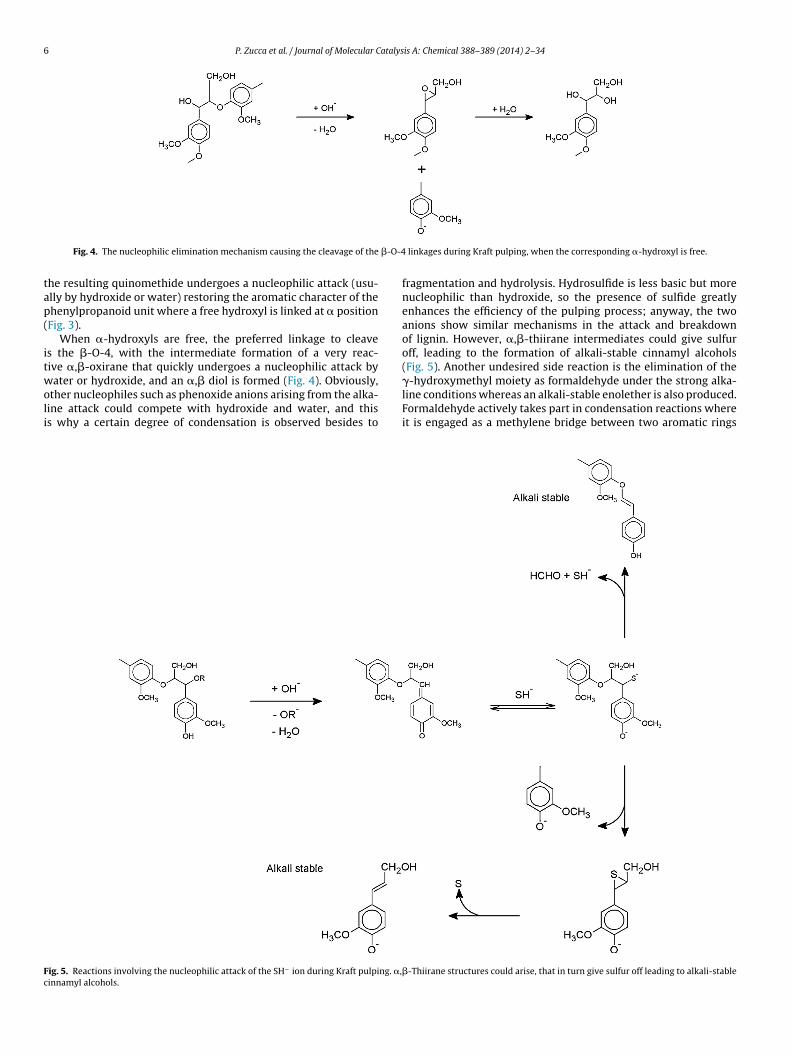
6 P. Zucca et al. / Journal of Molecular Catalysis A: Chemical 388–389 (2014) 2–34
�-O-4
tap(
itwoli
Fc
Fig. 4. The nucleophilic elimination mechanism causing the cleavage of the
he resulting quinomethide undergoes a nucleophilic attack (usu-lly by hydroxide or water) restoring the aromatic character of thehenylpropanoid unit where a free hydroxyl is linked at � positionFig. 3).
When �-hydroxyls are free, the preferred linkage to cleaves the �-O-4, with the intermediate formation of a very reac-ive �,�-oxirane that quickly undergoes a nucleophilic attack byater or hydroxide, and an �,� diol is formed (Fig. 4). Obviously,
ther nucleophiles such as phenoxide anions arising from the alka-ine attack could compete with hydroxide and water, and thiss why a certain degree of condensation is observed besides to
ig. 5. Reactions involving the nucleophilic attack of the SH− ion during Kraft pulping. �,innamyl alcohols.
linkages during Kraft pulping, when the corresponding �-hydroxyl is free.
fragmentation and hydrolysis. Hydrosulfide is less basic but morenucleophilic than hydroxide, so the presence of sulfide greatlyenhances the efficiency of the pulping process; anyway, the twoanions show similar mechanisms in the attack and breakdownof lignin. However, �,�-thiirane intermediates could give sulfuroff, leading to the formation of alkali-stable cinnamyl alcohols(Fig. 5). Another undesired side reaction is the elimination of the�-hydroxymethyl moiety as formaldehyde under the strong alka-
line conditions whereas an alkali-stable enolether is also produced.Formaldehyde actively takes part in condensation reactions whereit is engaged as a methylene bridge between two aromatic rings�-Thiirane structures could arise, that in turn give sulfur off leading to alkali-stable

atalys
[e
Kbado
fptkto
3
hsairclhgour
GpohasTtni
paMsral
firooiLr
3
baac
metal complexes [86].Sun et al. [83] have found a significant increase in carboxylic
P. Zucca et al. / Journal of Molecular C
40]. The occurrence of such condensation reactions has been how-ver recently questioned [39].
The importance of molecular oxygen intervention along theraft pulping has been studied in detail [60]. The occurrence ofoth radical and ionic mechanisms has been underlined, as wells the arising of quinoid structures (the main responsible for theark color of both the black liquor mentioned just below and thebtained Kraft pulp).
In conclusion, the degraded and solubilized lignin is richer inree phenolic hydroxyls (responsible for its solubility in alkali) andoorer in �-O-4 and �-O-4 linkages; it could be recovered fromhe spent liquor (‘black liquor’) upon precipitation by acids and isnown as Kraft lignin. Conversely, remaining, undissolved lignin inhe pulp is poorer in phenolic hydroxyls and should be bleached tobtain white paper.
.2. Sulfite pulping
Sulfite pulping is based on the nucleophilic properties ofydrogenosulfite ion HSO3
− which is the prevailing ionic form ofulfite in a wide range of pH conditions, from moderately acidic tobout neutral [57]. Hydrogenosulfite is well known as a sulfonat-ng agent for certain organic electrophilic compounds (owing to aelatively high electron density on sulfur atom) and this featurean be well exploited to achieve lignin fragmentation and solubi-ization. Apart from the so-called labile sulfonate groups (readilyydrolysable by alkalis) arising from addition reactions to carbonylroups, the striking feature of the sulfite pulping is the introductionf C-linked sulfonate groups into the lignin backbone. In partic-lar, both �-O-4 and �-O-4 arylether linkages are broken withelease of phenolic hydroxyl groups whereas a sulfonate groupSO3
− is attached to the � or respectively � carbon atom [61].enerally speaking, sulfitolysis takes place at acidic pHs on non-henolic substructures, and at neutral or even slightly alkaline pHsn the phenolic ones [62]. Moreover, some further sulfonic acidsave been characterized in soluble lignosulfonates [63]. The over-ll result of hydrogenosulfite attack is lignin fragmentation andolubilization of the arising fragments, bearing sulfonate moieties.he obtained lignosulfonates have a number of technical applica-ions [64], such as plasticizers and dispersing agents. It is worthoting that a minor fraction of (poorly) sulfonated lignin is almost
nsoluble and remains in the obtained chemical pulp.Likewise Kraft process, sulfitolysis leads to a usually incom-
lete delignification, and for certain uses the remaining lignin is problem since it adversely affects some properties of the paper.oreover, remaining lignin tends to brown, becoming progres-
ively more colored with time due to oxidation reactions that alsoelease acids. These, when not neutralized by calcium carbonatedded to the paper paste, tend to promote cellulose hydrolysis thusowering the paper strength.
Therefore, a pulp-bleaching step usually follows the deligni-cation, to obtain white pulps and to stop the alteration of theemaining lignin. As opposed to pulping, bleaching is mainly basedn electrophilic reactions (sometimes followed by nucleophilicthers), although this classification becomes debatable when pulp-ng and bleaching are carried out at the same time [54,55].ignin-retaining bleaching is almost exclusively due to nucleophiliceactions.
.3. Chlorine- and ozone-based treatments
The use of elemental chlorine has been for a long time the
leaching treatment of choice for pulps, since the cheapnessnd effectiveness of its bleaching action. Unfortunately chlorinects through both oxidation and chlorination, and the arisinghlorolignins are highly recalcitrant to (bio)degradation and quiteis A: Chemical 388–389 (2014) 2–34 7
toxic [65]. Sodium chlorite and chlorine dioxide, although muchmore costly than chlorine, act mainly via oxidative reactions. How-ever, a noticeable degree of chlorination is unavoidable, as is acertain degree of cellulose damage [66–70].
Ozone as a bleaching agent gives only oxygen off, but must beproduced at the bleaching plant, is expensive and not very effective,so its use is limited [71,72].
3.4. Peroxide-based treatments
Completely different is the case of hydrogen peroxide. This com-pound is available in large amounts at a reasonable price, can beeasily stored and delivered, is very reactive as an oxidizing and/orbleaching agent, releasing only water and molecular oxygen asreaction/decomposition products. This is an extremely crucial issuefrom an environmental perspective. Therefore its use is increas-ingly growing in pulp and paper industries, and many molecularmechanisms of its complex action on lignin have been studiedand elucidated. Although the redox potential for H2O2 reductionis higher at acidic pHs (where it acts as a weak electrophile towardelectron-rich substrates), it is well known that hydrogen peroxide isgenerally much more effective as an oxidant for organic compoundsin alkaline environment. This has been confirmed also for delig-nification and bleaching of pulps [73,74], where acidic or neutralH2O2 solutions are substantially ineffective. An apparent, notice-able exception is the case of peroxyformic acid [73,75] generatedin situ by HCOOH and H2O2, which is a source of the virtual hydrox-ylium (OH+) ion, an extremely reactive electrophile.
The effectiveness of alkaline delignification or bleaching is dueto the formation of important HO2
− concentrations above pH 11,where this nucleophile can attack electrophilic positions in the oxi-dizable molecules. Even more important, H2O2 becomes unstablewithin the pH range 10–12, where H2O2 and HO2
–concentrationsare comparable, allowing reaction (1) to proceed. Overall, thedecomposition reactions are thus the following [76]:
H2O2 + HO2− → OH• + O2
•− + H2O (1)
OH• + O2•− → O2 + OH− (2)
2O2•− + H2O → 1O2 + HO2
− + OH– (3)
1O2 + O2•− → O2 + O2
•− (4)
The occurrence of reaction (3) explains why at least a fractionof formed molecular oxygen is in its singlet (1�g), very reactivestate. Undoubtedly, the transient superoxide and hydroxyl radi-cals arising from reaction (1) play a key role in oxidation reactionsby alkaline hydrogen peroxide. According to this hypothesis, themaximum effect of hydrogen peroxide toward lignin reaches itsmaximum just at pH ≈ 11.5 (pKa for H2O2 is 11.6). Gellerstedtand colleagues have worked thoroughly [76–78] on the topic oflignin degradation by alkaline hydrogen peroxide, and have elu-cidated some mechanisms by which selected lignin substructuresare attacked and cleaved. More recent work [79] has substantiallyconfirmed these early findings, and the understanding of thosemechanisms has later led to a growing interest not only towardlignin-retaining bleaching [80,81], but also toward optimizationof delignifying/bleaching treatments, alternative to conventionalKraft pulping [82–85], possibly under catalysis by redox-active
functions as the result of alkaline oxidation by hydrogen perox-ide. These acidic functions are responsible for the increased ligninsolubility in water. Omori and Dence [87,88], in a study series some-what paralleling that of Gellersted and colleagues, confirmed the

8 P. Zucca et al. / Journal of Molecular Catalysis A: Chemical 388–389 (2014) 2–34
F eroxidp
fiio�Tqmaadomw�iua
tmeeacip
htr
M
M
M
M
O
a
(
reaction (Fig. 6). Also in the case of cinnamaldehyde-type endgroups in lignins, the nucleophilic attack by HO2
− takes placeat the � carbon; in this case the intermediate �-hydroperoxide
ig. 6. The C�-phenyl cleavage of �-carbonyl motifs of lignin by alkaline hydrogen products.
ndings of the latter authors: hydrogen peroxide at pH = 10.5 andn the presence of diethylenetriaminepentaacetic acid as the per-xide stabilizing agent was substantially ineffective against the-O-4 lignin model compound guaiacylglycerol-�-guaiacylether.he slight consumption of the substrate was found to be the conse-uence of a slow Dakin-like reaction yielding the easily oxidizableethoxyquinol and the very reactive and non-isolable intermedi-
te �-O-(2-methoxyphenyl)glyceraldehyde. Guaiacol and glycoliccid were the only products isolated in reasonable yields after theescribed treatment. By contrast, when the stabilizing agent wasmitted from the alkaline peroxide reagent, the yields of the aboveentioned degradation products substantially arose, togetherith a noticeable increase in the extent of guaiacylglycerol--guaiacylether consumption. These observations suggest an
mportant contribution of hydrogen peroxide degradation prod-cts alongside the oxidative destruction of the model compound,lso involving radical species.
In fact, even if hydrogen peroxide is decidedly more expensivehan the white liquor of Kraft pulping, the reaction conditions are
uch milder and therefore less energy-consuming. Moreover theffluents can be disposed of or managed more easily. The knowl-dge of the possible action mechanisms by which H2O2 oxidizesnd solubilizes lignin is extremely important to compare reactiononditions and obtained products to those observed when work-ng with both biological and biomimetic/bioinspired H2O2-basedrocesses (vide infra).
The reactivity and mechanistic changes observed in the systemydrogen peroxide/lignin in the presence of redox-active transi-ion metal ions must be emphasized: in that case, the followingeactions take place [76]:
en+ + H2O2 → Me(n+1)+OH + OH• (5)
en+ + OH• → Me(n+1)+OH (6)
e(n+1)+OH + H2O2→+ + HO2• + H2O (7)
e(n+1)OH + HO2• → Men+ + O2 + H2O (8)
2•– + H2O → 1O2 + HO2
• + OH− (9)
These are the Fenton reactions, showing hydroxyl generationnd scavenging by a metal ion (usually Fe, Cu, or to a certain extent
e (Dakin reaction) gives both p-quinol and glyceric acid derivatives as the identified
Mn) engaged in a one-electron redox cycle. Regeneration of Men+ byexcess H2O2 leads to O2 evolution (in other words, H2O2 wasting)through transient superoxide.
In the presence of phenoxide ions (this discussion is referred tosharply alkaline reaction conditions), additional reactions can takeplace [76]:
PhO− + Me(n+1)+ → PhO• + Men+ (10)
PhO− + OH• → PhO• + OH− (11)
PhO• + O2•– → oxidativedegradation (12)
PhO− + 1O2 → oxidativedegradation (13)
Some important conclusions have been therefore drawn by thestudies on alkaline hydrogen peroxide/lignin interaction that areencompassed just below:
a) In the presence of H2O2-stabilizing agents such as sodiummetasilicate and heavy metal chelators, an efficient lignin-retaining bleaching of pulps could be achieved in alkalinesolution, together with a limited H2O2 consumption. Phenolicfunctions of lignin remain substantially unaffected under theseconditions. Bleaching is therefore due to degradation of car-bonyl chromophores, mainly �-carbonyl structures, quinonoidand cinnamaldehyde-like structures [89]. In the case of �-carbonyl structures the ketogroup is the target of a nucleophilicattack of the same hydroperoxide anion (Dakin-like reaction),leading to the cleavage of the phenyl-C� bond; a quinol deriva-tive and a glyceric acid derivative are the products of the
evolves with fission of the � � bond, and further action ofHO2
− leads to a benzaldehyde derivative and formic acid. Nosignificant release of lignin degradation products is observeddue to the low occurrence of the cited structures. Concomi-tant nucleophilic attack of the hydroperoxide anion HO2
− tothe methoxy-bearing carbon(s) of quinones derived from G and

atalysis A: Chemical 388–389 (2014) 2–34 9
(
(
clcascota–T
4
tesiraieItplor
4
1o(Poser
Fig. 7. The structure of the protoporphyrin-IX ferric complex (hemin, ferrriheme).This is the prosthetic group of peroxidases, peroxygenases, cytochromes P-450. Its
P. Zucca et al. / Journal of Molecular C
S units leads to methanol elimination and ring cleavage, withthe formation of colorless muconic acid derivatives.
b) In the absence of H2O2 stabilizers, and even more when cat-alytic amounts of certain heavy metal ions such as CuII, FeII/III, orMnII are added (efficiently triggering Fenton-like reactions, andmoreover promoting phenoxyl radicals formation), also phe-nolic substructures become targets for bleaching; in particularsuperoxide could attack phenoxyl radicals leading to aromaticring oxidative fission (reaction (9)). Contrarily to superoxide,singlet oxygen can easily attack and destroy also phenoxideanions (reaction (10)). In particular, MnIII species arising fromFenton-like reactions are per se strong oxidizing agents andcould in turn promote further oxidative degradation of lignin.
c) Redox-active metal ions under lignin-retaining bleaching couldlead to undesired hydrogen peroxide consumption. These ionscatalyze the above-mentioned Fenton-like H2O2 decomposi-tion and subsequent reactions wasting hydrogen peroxide asmolecular oxygen. So careful removal of redox active metal ionsprior to bleaching is advisable [90]. Otherwise, delignificationcould overcome bleaching, but the resulting, partially deligni-fied pulp would be still colored, and would therefore require anadditional (and expensive) bleaching step.
In conclusion, both nucleophilic and electrophilic treatmentsould achieve pulp delignification. In particular, the strongly alka-ine conditions of the widespread Kraft process lead to hydrolyticleavage of �-O-4 and �-O-4 interunit linkages. The same link-ges are broken under comparatively milder conditions duringulfitolysis. Chlorine, chlorite, chlorine dioxide, ozone are on theontrary very strong electrophiles, giving rise to rather non-specificxidative degradation. Hydrogen peroxide under alkaline condi-ions occupies a special place as it is able to act as a nucleophilend/or as a radical generator, leading – under suitable conditions
to extensive lignin fragmentation through Dakin-like reactions.hese facts are summarized in Table 1.
. Oxidative biodegradation of lignin
Lignin is among the most durable natural polymers; never-heless, in the biosphere, some classes of ligninolytic fungi canfficiently manage this potential energy source. In particular, theo-called white-rot fungi are the only class able to cause the typ-cal decay of wood, that becomes whitish due to the selectiveemoval of the lignin, usually not affecting cellulose (unlike soft-rotnd brown-rot fungi) [91,92]. White-rot fungi (mainly holobasid-omycetes) can be sub-classified on the ground of the differentnzymes they excrete to promote oxidative degradation of lignin.n reality, oxidative degradation of lignins by these fungi takes placehrough the cooperation of a number of enzymic and non-enzymicrocesses [93]. The use of fungal ligninolytic enzymes to exploit
ignin biotransformation to valuable products [94], as well the statef the art in biopulping by means of white-rot fungi [95], have beenecently discussed.
.1. Laccase
Laccase (p-diphenol:oxygen oxidoreductase, E.C. number.10.3.2) is a copper oxidase [96] catalyzing one-electron oxidationf phenolics, that are converted to the corresponding semiquinonephenoxyl) radicals while molecular oxygen is reduced to water.henoxyl radicals in turn can spontaneously evolve to dimers,
ligomers, polymers, quinones, depending on their particulartructure, on the concentration of molecular oxygen, on the pres-nce of compounds capable of participating into cross-couplingeactions and/or electron transfer reactions. The last cited featureferrous counterpart ferroheme (not shown) is the prosthetic group of myoglobinsand hemoglobins.
opens a very promising way for the use of laccase catalysis tooxidize nonphenolics by the means of the intermediacy of partic-ular substrates that are converted to their radical counterparts,capable in turn of oxidizing molecules that are not primary sub-strates. A huge number of studies exist about the redox mediatorsfor laccase that substantially widen the substrate specificity ofthe enzyme [97–99]. However, although laccase is essential forligninolysis by white-rot fungi [100], laccase alone is not capableof oxidatively depolymerizing/solubilizing lignin [101,102]. Someprocesses have been proposed [103] for both delignification andbleaching of pulps using laccase in the presence of suitable redoxmediators. However, experiments with soluble lignosulfonateshave confirmed that a substantial polymerization takes place, asshown by a noticeable increase of molecular weight. The analysisof the reaction products also showed that no significant action onthe lignin backbone takes place, being the laccase action confinedto peripheral functional groups [104]. Moreover, unexpected and– in this context – unwanted reactions could be observed whenlaccase acts on lignocellulosic components in the presence of redoxmediators, such as galactomannan cross-linking in the presence ofTEMPO [105].
4.2. Ligninolytic peroxidases
Many white-rot fungi excrete other ligninolytic enzymes,completely different from laccase, and belonging to the heme-peroxidases. These enzymes all contain as their prosthetic group,the ferriprotoporphyrin-IX or ferriheme (Fig. 7; the same prostheticgroup, but in the ferrous state of ferroprotoporphyrin IX or ferro-heme, is found in hemoglobins and myoglobins). All these enzymesshare the same catalytic cycle [106–111]. Briefly, the iron(III) of theferriheme interacts with hydrogen peroxide, to form a very labileiron(III)-hydroperoxide complex porphFe(III) O OH, known as theCompound zero (Cpd0) (Fig. 8). This rapidly evolves to a Compound I(CpdI), upon heterolytic scission of the O O bond that releases OH−
and formation of a ‘ferryl’ entity within a radical cation protopor-phyrin IX Porph•+ Fe(IV) O.
The indicated structure for CpdI is a somewhat formal one; theFe(V)-containing structure is of little importance but is sometimesused to underline the two oxidation equivalents present in com-parison with the resting state. On the contrary, the importance ofthe alternative structure PorphFe(IV) O•· explains the attitude of
CpdI of extracting an hydrogen atom from a C H bond, which is thefirst step in hydroxylations (vide infra, Section 4.2.4). CpdI is a rela-tively strong oxidizing species, but is relatively unstable and proneto oxidative self-destruction. It easily undergoes an one-electron
10 P. Zucca et al. / Journal of Molecular Catalysis A: Chemical 388–389 (2014) 2–34
Table 1Different approaches of delignification affect different lignin bonds, through various molecular mechanisms.
Delignificationmethod/catalyst
Lignin bonds broken Main molecular mechanisms
Kraft pulping �-O-4, �-O-4 • Nucleophilic attack by OH− and SH−
Sulfite pulping �-O-4, �-O-4 (with incorporation of sulfonate SO3−
group)• Nucleophilic attack by HSO3
−
Chlorine Non-specific oxidations and aromatic chlorinations • Electrophilic attackChlorine dioxide Non-specific oxidations • Electrophilic attackOzone Carbon–carbon double bonds • Electrophilic attackH2O2 Phenyl-C� (Dakin-like reaction, in the presence of
peroxide-stabilizers); phenolic ring oxidative fission(Fenton-like reaction, in the presence of transition metals)
• Nucleophilic attack by HO2−
• HO· production through Fenton-like reactions
Lignin peroxidase Mainly C�-C� cleavage; �-oxidation, C� hydroxylation • One-electron oxidationManganeseperoxidase
C�-C� cleavage • One-electron oxidation
Versatile peroxidase C�-C� cleavage • One-electron oxidationFe-porphines C�-C� cleavage (with subsequent quinonization); C1-C�
cleavage; phenolic ring oxidative fission; �-oxidation;condensation
• One-electron oxidation• Oxygenation through oxygen rebound from CpdII
�-oxid
ri(Car(act
rMPpTfitdan
4
itmt
Mn-porphines C�-C� cleavage (with aldehyde production);
(no quinonization or condensation)Ru-porphines No data available
eduction by the substrate, leading to another ferryl species, lack-ng the radical cation character of CpdI, and known as Compound IICpdII), PorphFe(IV) O. CpdII exhibits milder oxidizing power thanpdI, and is therefore considerably less reactive; however, it canccept one electron by the reducing substrate, thus reverting to theesting enzyme and closing the catalytic cycle (Fig. 8). Compound IIICpdIII) is formed (starting from CpdII) only in the presence of rel-tively high concentrations of peroxide; it is actually a superoxideomplex of the ferriheme, has no catalytic activity and is very proneo an irreversible bleaching upon reaction with peroxide [112].
Three main ligninolytic peroxidases have been found in white-ot fungi: (i) lignin peroxidase LiP, E.C. number 1.11.1.14; (ii)anganese Peroxidase MnP, E.C. number 1.11.1.13; (iii) Versatile
eroxidase VP, E.C. number 1.11.1.16. These all belong to the class IIeroxidases [113], and share a relatively high redox potential [114].heir occurrence varies among white-rot fungi that can be classi-ed just on the basis of their peroxidase pattern [93,115]. Moreover,heir excretion strongly depends on the particular cultural con-itions adopted, so laboratory results should be interpreted with
great care when inferring conclusions about ligninolysis underatural conditions.
.2.1. Lignin peroxidaseThe first discovered ligninolytic peroxidase, LiP [116,117], was
nitially described as an H2O2-requiring oxygenase because someransient radical species arising from its action could react with
olecular oxygen, giving rise to peroxy radicals R O O•. These inurn undergo fragmentation to the final products [118,119]. It has
Fig. 8. The peroxidase catalytic cycle. Legend: Cpd0: Compound zero
ation • One-electron oxidation• Direct oxygenation from CpdI• –
been studied mainly in the fungus Phanerochaete chrysosporium,and has been – and still is – the object of much work to under-stand its action mechanism(s) [120–124]. The enzyme follows ausual catalytic cycle [109] strictly resembling that of horseradishperoxidase, but with a particularly labile CpdI [125] and a very easyformation of CpdIII [126] in the presence of an even slight excessof hydrogen peroxide. This is quite prone to heme bleaching andsubsequent irreversible enzyme inactivation, unless a ‘protective’molecule such as veratryl alcohol (3,4-dimethoxybenzyl alcohol) ispresent [112,117]. The general sensitivity of heme-peroxidases tosuicide inactivation by their substrate hydrogen peroxide has beenreviewed [127], being a serious challenge in many industrial appli-cations of these enzymes. A continuous supply of veratryl alcohol(which is mainly oxidized to veratraldehyde) is required to achievereasonable oxidation rates.
The enzyme preferentially acts on nonphenolic units oflignin, by abstracting one electron from the aromatic ring of aphenylpropanoid unit. The resulting cationic radical undergoes ahomolytic cleavage of the C� C� linkage, leading to a neutral rad-ical at the � carbon atom whereas the positive charge remains onthe phenyl-C� fragment [128–130]. This cation stabilizes throughproton expulsion leading directly to the oxidation of the ben-zylic (�) carbon to the corresponding carbonyl function (Fig. 9).Therefore, substituted benzaldehydes arise, that are the obvious
precursors of the benzoic acids, found during lignin decay by white-rot fungi [131]. The radical fragment could non-enzymically reactwith molecular oxygen, so a number of oxidized products couldbe detected in the reaction mixtures [128–130,132,133]. Under. CpdI: Compound I. CpdII: Compound II. CpdIII: Compound III.

P. Zucca et al. / Journal of Molecular Catalysis A: Chemical 388–389 (2014) 2–34 11
Fig. 9. The most widely accepted homolytic cleavage mechanism of the C�–C� interunit linkage by LiP. This is only one among the possible different fragmentation pathwaysd on traa ce at tC
asns
agcespe
yowr
af[apfsb
epending on which ring is primarily oxidized. Moreover, an intramolecular electrction [134]. The homolytic cleavage explains why no 18O incorporation takes pla�–C� cleavage has been questioned [135].
naerobic conditions, the C�-centered radicals could dimerize, aseen in the case of properly designed models [128]. Such a mecha-ism would act also in the cleavage of ‘minor’ lignin substructuresuch as those involving �-1 linkages.
This homolytic mechanism has been however questioned [135]nd an alternative heterolytic cleavage mechanism has been sug-ested, leading to the localization of the positive charge on the C�arbon, whereas the lone electron should be on the phenyl-C� moi-ty. In this view, the lack of oxygen incorporation in the arisingubstituted benzaldehydes is explained by a reaction where theutative peroxyl radical intermediate on the C� would supposedlyxpel HO2
•.As a point of fact, lignin oxidation by H2O2 under LiP catal-
sis follows therefore a pathway completely different from thatbserved in the case of H2O2 oxidation under alkaline conditions,hich mainly involves the breakage of the phenyl-C� (Dakin-like
eaction, see Section 3.4).Most of the information, available on the structural features
nd the modes of breakage of the lignin substructures comesrom synthetic model compounds, either phenolic or nonphenolic15,39,119,128,130,136] such as guaiacylglycerol-�-aryl ethersnd analogs (Fig. 10). An exhaustive presentation of model com-
ounds, used to study lignin oxidative (bio)degradation, can beound in the review of Zakzeski et al. [39], which also discusses thetructural features the model compounds should have to optimallyehave as useful tools to understand lignin (bio)degradationnsfer could take place, explaining the different products arising from the enzymehe �-carbonyl of the arising benzaldehyde. However, the homolytic nature of the
mechanisms. By using such models, a number of additionalLiP-catalyzed oxidative reactions have been ascertained, such as�-oxidation (to a ketone function) without C� C� cleavage, as theresult of heterolytic C� H scission of the radical cation, and subse-quent one electron oxidation (by LiP) of the resulting radical [134].C� hydroxylation (in �-methylene models) has also been observed[137]. Under aerobic conditions, ring cleavage could finally occur[15,132].
The compulsory role of the redox couple veratryl alco-hol/veratryl alcohol radical cation as an one-electron shuttlebetween the enzyme and lignin [138] had been most probablyoverestimated; in fact, the enzyme is capable of directly interac-ting with lignin as substrate [139], although in general, the higherthe molecular weight of the substrate, the lower the enzyme effi-ciency [134]. No general agreement exists about the specific aminoacid residues involved in the enzyme–substrate interaction and inthe long-range electron transfer from lignin and oxidized heme.However, an invariant tryptophan residue, which is irreversibly �-hydroxylated under turnover conditions [140] appears to be the‘bridge’ between the enzyme active site and bulky hydrophobicsubstrates such as lignin, whereas more polar, small substrates arepresumably oxidized at the heme exposed edge [125].
As it regards phenolic substrates such as ferulic acid, LiPs arevery active but very rapidly deactivated by the arising phenoxylradicals [141]. This finding could help explaining the alleged, gen-eral, apparent lack of activity of LiPs toward phenolics.

12 P. Zucca et al. / Journal of Molecular Catalysis A: Chemical 388–389 (2014) 2–34
F of LiP
aa
4
[itatttpapom(atMproco
ttucriwTt
or no activity is shown toward chloride. Their action mechanismcombines the peroxide shunt of P-450 enzymes with a ‘classical’
ig. 10. Some �-O-4 (a–d) and �-1 (e) models used to study the action mechanism
On the whole, despite of the relatively wide diffusion of LiPmong white-rot fungi, many of these simply lack the enzyme,lthough being very efficient lignin degraders [93].
.2.2. Manganese peroxidaseMnP is much more widespread than LiP among white-rot fungi
142] and has Mn2+ as its preferred oxidizable substrate. Accord-ngly, it lacks the invariant tryptophan residue typical for LiPs, andherefore it cannot directly interact with lignin. Instead, it has a neg-tively charged cluster, formed by some aminoacidic residues alonghe polypeptide chain, and also by a carboxylate anion belongingo one of the two carboxyethyl heme sidechains [129], devotedo transient Mn chelation during the catalytic cycle. That strictlyarallels those of other peroxidases, with the noticeable feature of
comparatively low oxidizing power of CpdII [143], which com-ulsorily requires Mn2+ for resting state regeneration [144]. Thene-electron oxidation of Mn2+ by H2O2 in the presence of MnP for-ally produces Mn3+. In fact, Mn(III) does not exist as the aqua ion
contrarily to Mn2+) but must be stabilized by chelation (by oxalicnd malonic acids, for example) to prevent quick disproportiona-ion to Mn2+ and Mn(IV) in the form of insoluble MnO2. Chelates of
n(III) are one-electron oxidizers, potentially capable of attackinghenols, aromatic amines, and also a number of aromatic, electron-ich nonphenolic substrates [145]. In the case of lignin models,xidation of the benzylic (�) carbon leads to the formation of aarbonyl function. The obtained ketone could be eventually furtherxidized with C� C� bond cleavage [146].
The intrinsic oxidizing power of Mn(III) is lowered upon chela-ion by dicarboxylic acids such as oxalic and malonic acids, andherefore only the relatively rare phenolic units in lignin couldndergo direct attack by Mn(III) chelates. However, the very oxalateould be oxidized by Mn(III) leading to CO2 and the formate anionadical. This latter in turn reacts with molecular oxygen converting
t to hydroperoxyl radical HO2• at the relatively low pH of decayedood, thus triggering an autocatalytic oxidative mechanism [147].
races of polyunsaturated fatty acids are also present in wood, andhese are oxidized to the corresponding free radicals by chelated
. There are both examples of phenolic (d) and nonphenolic (a, b, c, e) compounds.
Mn(III). The obtained radicals react with molecular oxygen, alsotriggering the oxidative degradation of lignin, regardless to thephenolic or nonphenolic nature of the involved units [133,148].Unfortunately, in vitro experiments have shown that the overallefficiency is low, provided that lignin was oxidized rather thancleaved/solubilized [129]. The enzyme seems to be more promisingas a tool for biodegradation and/or bioremediation of recalcitrant,low MW organic pollutants [149].
4.2.3. Versatile peroxidaseVersatile peroxidases (VPs) are a kind of ‘hybrid’ peroxidases
showing both the tryptophan residue required for the direct inter-action with large lignin fragments, and the aminoacidic residuesdevoted to manganese recognition [150,151]. VPs would seemtherefore the ‘ideal’ peroxidases for an efficient lignin biodegra-dation; however, it is worth noting that a typical VP producer,Pleurotus eryngii, degrades lignin very slowly.
4.2.4. PeroxygenaseAlthough only seldom emphasized, the role of another oxidative,
heme-containing enzyme produced by fungal organisms shouldbe taken into the due consideration: peroxygenase (E.C. 1.11.2.1).Fungal peroxygenases have been thoroughly studied by Hofrichterand colleagues and are extracellular hemoenzymes resemblingcytochromes P-450 as their ferriheme prosthetic group has acysteine thiolate anion as the proximal ligand [152–157]. Peroxy-genases show wide substrate specificity, being able to use hydrogenperoxide to oxygenate (epoxidize and/or hydroxylate) aromaticrings, benzylic carbons, and even recalcitrant heterocycles such aspyridine. They can also act as bromide peroxidases whereas little
heme peroxidase catalytic cycle. However, their importance in gen-eral oxidative ligninolysis is debatable, as until now peroxygenaseshave been found only as extracellular enzymes of a few ligninolyticfungal species.

atalysis A: Chemical 388–389 (2014) 2–34 13
4h
iiibte
tMatiii
1qbrpfwObo
nemipidtcoPnea
5t
5a
i
TS
P. Zucca et al. / Journal of Molecular C
.3. Other enzymatic activities are required for in vivoeme-peroxidase-based ligninolysis
As a matter of fact, none of the known ligninolytic peroxidasess able to efficiently promote ligninolysis in vitro – and the sames true also for mixtures of ligninolytic enzymes. Accordingly, its possible to state that all these enzymes are important in ligniniodegradation by white-rot fungi, but the process strictly requireshe additional intervention of other catalytic systems, possibly bothnzymatic and non-enzymatic.
The widespread flavoenzyme cellobiose:quinone oxidoreduc-ase (E.C. 1.1.5.1) was shown [158] to be able to reduce to soluble
n(II) and Mn(III) species the insoluble MnO2 that unavoidablyrises from the action of MnP in vivo. The same enzyme reduceshe quinones, arising from laccase action, to polyphenols that aren turn re-oxidized by laccase, in an apparently futile cycle, thats however a source of Reactive Oxygen Species (ROS), deeplynvolved in lignin oxidative degradation [159].
A NAD(P)H-dependent enzymatic membrane system (E.C..6.5.2) was characterized in P. chrysosporium [160] reducinguinones arising from lignin biodegradation. A similar activity haseen also found in Pleurotus sajor-caju [161]. Quinone reductiveemoval could be the key mechanism by which white-rot fungirevent condensation and/or re-polymerization of the quinonoidragments arising from oxidative ligninolysis, thus forcing thehole process toward lignin fragmentation and solubilization.bviously, also in this case, reoxidation of the arising polyphenolsy laccase and/or other oxidizers triggers ROS production and ligninxidative degradation.
In conclusion, laccase excretion appears to be a necessary butot sufficient condition for effective in vivo ligninolysis; in vitroxperiments have led to lignin polymerization rather than depoly-erization, unless suitable redox mediators are present. However
ts application perspectives are nowadays still uncertain. LiP isotentially a strong oxidation catalyst for lignin, but its efficiency
n practical applications is unsatisfactory; MnP triggers the pro-uction of the diffusible strong oxidant Mn(III), but it appears onhe whole as a lignin oxidizer rather than a lignin solubilizer; VPsould be rather disappointing if one judges their usefulness basedn the very low ligninolytic efficiency of their fungal producers.eroxygenases could play a fundamental role in industrial biodelig-ification in the future, although they appear as relatively rarenzymes among ligninolytic fungi. Some features of these enzymesre encompassed in Table 2.
. Learning from nature: from natural metalloporphyrinso synthetic metalloporphines
.1. Natural metalloporphyrins are too unstable for in vitro
pplicationsThe well-known peroxidase-like activity of hemin (ferriheme,ron(III) complex of protoporphyrin IX, Fig. 7) as well as the
able 2ome striking features of ligninolytic enzymes. For a more complete comparison, cytochr
Laccase Lignin peroxidase Mangperox
Ligninolytic enzyme Yes Yes Yes
Hemoenzyme No (Cu2+) Yes Yes
Heme proximal ligand – His His
Physiological substrates Phenolics Non-phenolicaromatics (mainly)
Mn2+
Oxidant O2 H2O2 H2O2
One-electron oxidations Yes Yes Yes
Direct oxygenation from CdpI state – No No
Fig. 11. The structure of the porphine macrocycle, showing the beta- and the meso-positions.
catalase-like activity of hemoglobin have triggered a huge num-ber of studies about the possibility of exploiting these features toobtain suitable oxidation catalysts. This task has been made moreand more easy as new synthetic ways to unnatural porphyrins andporphines became available at laboratory scale. Nowadays also sev-eral porphines and metalloporphines are commercially available atreasonable prices.
It is worth noting that, following the IUPAC directions [162],porphyrins are porphine derivatives where organic side chainsare substituted for all the eight hydrogen atoms in the porphinepyrrole rings (the positions). As a matter of fact, almost allthe unnatural, heme-inspired catalysts described so far are meso-tetraarylsubstituted porphines (Fig. 11), lacking any organic chainat the � positions, but are nevertheless and misleadingly referredusually to as porphyrins.
Porphyrin and porphine chemistry is quite complex, and dif-ferent metal complexes have been classified on the basis of anexhaustive spectroscopic analysis [163]. The most studied com-plexes (Fe(III) and Mn(III)) fall into the d-type hyper class [164]. It isworth noting that many oxidation numbers, assigned to the centralmetal ions in metalloporphines, have a largely formal value [165],due to attitude of the porphine macrocycle to accept some elec-tronic density from the metal ion into its �* (antibonding) orbitals.
Even if many metalloporphyrins and metalloporphines showa wide variety of noticeable (photo)catalytic activities [166,167],much interest has been focused on their redox catalytic activitiesin the presence of O2, H2O2, ClO− and a number of other oxidizingagents acting as single oxygen atom donors [168–170].
Some outstanding differences between the catalytic activitiesof heme-containing peroxidases, peroxygenases, and monooxyge-nases on one hand, and free metalloporphines on the other one,are quite not surprising: besides the diversity in kinetic efficiency,
the heme group in heme enzymes is more or less buried within theprotein scaffold. This ensures effective protection of the prostheticgroup against its bleaching by the oxidizing agent(s). Moreover,the apoenzyme drives the catalytic activity to a peroxidase orome P450 has been included.
aneseidase
Versatile peroxidase Peroxygenase Cytochrome P450
Yes Debated NoYes Yes YesHis Cys CysMn2+/non-phenolicaromatics (mainly)
Aromatics andhydro carbons
Hydro carbons
H2O2 H2O2 O2
Yes Yes NoNo Yes Yes

14 P. Zucca et al. / Journal of Molecular Catalysis A: Chemical 388–389 (2014) 2–34
Fig. 12. The structures of some among the most studied metalloporphines, whose metal complexes could exert noticeable peroxidase-like and/or monooxygenase-likec lorophm orphin2
a[
dcbpasmbtpmbmacd
dboase
5m
stopPtbc
m
atalytic activity. Legend: (1) meso-tetraphenylporphine; (2) meso-tetrakis(2,6-dicheso-tetrakis(pentafluorophenyl)porphine; (5) meso-tetrakis(4-sulfonatophenyl)p
-pyridinio)porphine.
lternatively to a monooxygenase (also, to a peroxygenase) one171].
On the other hand, the nature and shape of the active siteeeply controls the substrate specificity of each heme enzyme by aomplex interplay among sterical effects, hydrophilic/hydrophobicalance, electrostatic interactions, and so on. Free metallopor-hines are not subjected to the restrictions dictated by anypoenzyme, so their reactivity mainly depends on their particulartructures, i.e. meso and � substituents, and the nature of the centraletal ion. Consequently, very wide substrate specificity could well
e expected, together with a more or less pronounced sensitivityo the bleaching effects of the used oxidants. Last but not least, theotential coexistence of different action mechanisms for the sameetalloporphine could be reasonably anticipated, and is confirmed
y a huge mass of experimental findings [172,173]. Fortunately,odern methods in organic synthesis afford a vast number of met-
lloporphines, whose catalytic efficiency and chemical robustnessan be optimally balanced, overcoming natural metalloporphyrinsrawbacks.
In fact, free ferriheme (hemin), which is the most obvious candi-ate as a peroxidase emulator, is quite unsatisfactory being rapidlyleached by even low H2O2 concentrations, through the formationf meso-oxo-derivatives that in turn undergo macrocycle fissionnd further degradation [174]. However, some reports account for aatisfactory peroxidase-like catalytic activity of hemin, when prop-rly protected against oxidative bleaching [175,176].
.2. The development of more stable and active syntheticetalloporphines
The presence of four aryl substituents at the meso positions washown to be the key factor to render the porphine ring more resis-ant against oxidation. Thus metal complexes (mainly of Fe and Mn)f meso-tetraphenylporphine TPP (Fig. 12, 1) have been studied asotential emulators of heme-containing mono-oxygenases (such as-450 [177]). Although more stable and more active than ferriheme,hey are on the whole rather poor catalysts. These complexes have
een called by Meunier [178] the first generation metalloporphineatalysts.A second generation [178] was obtained by modifying theeso-phenyls by introducing electron-withdrawing substituents
enyl)porphine; (3) �-octacholoro-meso-tetrakis(2,6-dichlorophenyl)porphine; (4)e; (6) meso-tetrakis(N-methyl-4-pyridinio)porphine, (7) meso-tetrakis(N-methyl-
(such as in the case of tetrakis(pentafluorophenyl)porphine, TFPP,Fig. 12, 4) [172,179] or also two bulky substituents at the twoortho positions of the four meso-phenyls. Such bulky substituents,preventing coplanarity among the porphine macrocycle and thefour phenyls, hinder the meso carbons, that become less proneto attack and subsequent macrocycle fission [172]. However,meso-tetramesitylporphine derivatives are quite inefficient as oxi-dation catalysts, being also readily bleached by oxidants, becauseof the electron-donating attitude of the mesityl substituents[180]. When such bulky substituents are chlorine atoms (suchas in the case of meso-tetrakis-(2,6-dichloro-phenyl)-porphine,TDCP, 2 in Fig. 12), a sharp electron-withdrawing effect becomesimportant, rendering even more stable and more active the cor-responding Fe and Mn complexes as oxidation catalysts [181].The importance of avoiding coplanarity of the porphine ringwith the meso substituents has been shown by Liu and Su [182]by comparing the high activity and stability of meso-tetrakis(N-methyl-2-pyridinio)porphine-Mn(III) (7 in Fig. 12) with the isomericmeso-tetrakis(N-methyl-4-pyridinio)porphine-Mn(III) (6 in Fig. 12)which is unstable below pH 9. Only the latter could extensivelydelocalize the positive net charges of the N-methylpyridinium sub-stituents along the whole porphine macrocycle so causing the facilemetal ion loss, whereas in the former only a strong electron with-drawing effect is operating, that does not affect the stability ofthe metal complex and raises the redox potential of the cata-lyst.
Very recently, a comparative study [183], regarding someisomeric meso-substituted Mn-porphines as catalysts in alkeneepoxidation by tetrabutylammonium periodate, has shownthat noticeable differences exist in both chemical stabilityagainst bleaching and catalytic performances of the examinedcatalysts. In particular, among the three possible tetra(meso-pyridyl)porphine-Mn isomers, the one bearing the 4-pyridylsubstituent is by far both the most stable and the most activecatalyst when compared with the 2- and 3-pyridyl isomers(differently from that described for the N-methylpyridiniumcounterparts, Section 6.2: in that case, the 2-isomer is more
stable and also more inert than the 4-isomer). As expected,tetraphenyl and tetrakis(4-methoxyphenyl) substituents lead toboth poorer stability and poorer activity. Moreover, tetrakis(4-sulfonatophenyl) substituent substantially improved both stability
P. Zucca et al. / Journal of Molecular Catalysis A: Chemical 388–389 (2014) 2–34 15
Table 3Resume of the features of all generation of metalloporphines, following Meunier classification [178].
Metalloporphine Feature Advantages Disadvantages
Hemin Naturalferri-protoporphyrin IX
• Widespread natural molecule• Low cost
• Very unstable
First generation (i.e. Fe- and Mn-TPP) meso-Phenyl-substituents
• Quite higher stability than hemin• Not very expensive synthesis
• Low activity
Second generation (i.e. Fe- and Mn-TDCP) Electron-withdrawingsubstituents (i.e.halogens) in themeso-phenyl rings
• High stability• High catalytic activity
• High cost
• Onlycompoactivit
ait
sofl
throtodoac�vecTt[
afibmsamtspFeg[
lps(sgofoti
Third generation Further insertion ofhalogens at �-positions
nd catalytic performances, whereas, somewhat surprisingly,t showed comparatively poorer stability and activity thanetrakis(meso-4-methoxy-3-sulphonatophenyl)porphine-Mn.
Avoiding coplanarity is also important to prevent undesired sub-tituent activation at the para position of the phenyl moieties, asbserved by Kadish and colleagues [184] in the case of TFPP. In fact,uorine atoms are not bulky enough to avoid coplanarity.
A third generation of metalloporphine catalysts stems fromhe idea that a complete substitution at the � positions withalogen atoms could still improve activity and stability of theesulting macrocycles. However it was early shown that �-ctachloro- and �-octabromo-metalloporphines were less stablehan their non-halogenated counterparts: an inactive, dechloro-xo-derivative at a � position was quickly formed and precipitatesuring catalysis in the presence of hydrogen peroxide [185]. More-ver, � chlorine and especially bromine substituents could cause
decrease in catalytic activity due to excessive crowding. Byontrast, the Fe(III) complex of meso-tetrakis(pentafluorophenyl)--octafluoroporphine was shown to be both very active andery stable [186]. Such perfluoroporphines are however not veryasy to be synthesized, which prevents extensive technologi-al applications of their metal complexes. Nitration of TDCP orFPP with fuming nitric acid afforded �-poly-nitro-derivativeshat are both very stable and very efficient epoxidation catalysts187].
Metalloporphines are per se rather hydrophobic and thereforelmost insoluble in water. However, this drawback, that would con-ne their use within non-aqueous systems, has been overcomey introducing highly polar and hydrophilic substituents at theeso-phenyls or also at the � positions. Electrically charged sub-
tituents have the advantage of preventing stacking/aggregationnd formation of dimers, where an oxo-bridge between the twoetal ions exists [188,189]. Moreover, these substituents make
he compounds suitable for ion exchange techniques. In particular,ulfonation of the meso-phenyls provides anionic, water-solubleorphines (meso-tetrakis(4-sulfonatophenyl)porphine, TSPP, 5 inig. 12) [190] that can be easily metallated. Sulfonation of thelectron-deficient TFPP at its � positions requires oleum andives rise to a robust, water-soluble, easily metallable porphine191].
In conclusion, in this chapter we analyze the structural featureseading from the iconic natural metalloporphine, ferriprotopor-hyrin IX (ferriheme) to the first generation (meso-tetraphenylubstituted metalloporphines), then to the second generationsterically hindered and/or electron-withdrawing aromatic mesoubstituents), and finally to the third generation (as the secondeneration, with halogen- or nitro-substituents at the �-positions)f bioinspired and often biomimetic metalloporphines. The dif-
erent behavior of these compounds has been discussed in termsf stability, reactivity, and catalytic efficiency in oxidative reac-ions. The striking features of the three generations are resumedn Table 3.perfluoro or polynitrounds show very highy and stability
• Typically, lower stability than 2ndgeneration• Higher costs than 2nd generation
6. Molecular aspects of oxidation/oxygenation reactionscatalyzed by metalloporphines
Many redox-active metalloporphines easily react with suitableoxygen donors (N-oxides, iodosyl compounds, hydroperoxides,peracids, and others) giving rise to oxo derivatives [177,181,192],more or less resembling the common key intermediate of per-oxidases, peroxygenases, and heme-containing monooxygenasesbelonging to the P-450 family: the CpdI. For the sake of clarity,a synoptic presentation of the catalytic pathways of the threeenzyme classes cytochromes P-450, peroxidases, and peroxyge-nases is shown in Fig. 13. It is worth noting that peroxidases areusually incapable of performing direct oxygen transfer reactions,for the redox reactions take place at the heme edge [171], thusrather away from the iron center. A different path occurs in thecase of haloperoxidases, peroxygenases, and cytochromes P-450,since the substrate could closely approach the high-valent oxoironintermediates. All these enzymes share the proximal coordinationof the iron by a thiolate anion instead of the imidazole ring presentin the peroxidases stricto sensu.
Emulation of P-450 enzymes has drawn much attention owingto the outstanding synthetic relevance of monooxygenation and inparticular epoxidation reactions. A detailed discussion about theelectronic structure of the high-valent oxoferryl derivatives of P-450 enzymes, i.e. CpdI and CpdII, is presented in a review [193].Although P-450 enzymes are not involved in biological ligninolysis,they play a central role in understanding the catalytic mechanismsunderlying the activities of heme-containing peroxidases, peroxy-genases, monooxygenases, due to their versatility [194].
In a quite parallel fashion, synthetic metalloporphines provedto be able to follow several catalytic oxidation paths [178]. Inparticular, two main oxidation mechanisms were described (mono-electronic oxidation, or alternatively oxygenation), depending onthe reaction media, the axial ligand, the oxidant nature and so on.Each of these paths allows to obtain different intermediates and tooxidize different substrates. Accordingly, a careful investigation onthese aspects is crucial, in order to lead to the proper (and best)way for the oxidation of such a recalcitrant compound as lignin.
6.1. Fe-porphines
The metal ion present in nature as the central metal in metal-loporphyrins is almost invariably iron: the huge number of studies[177,195–198] focused on iron-porphines is therefore not surpris-ing.
In principle, a ferriheme analog could behave as a peroxidase-like entity, or as a mono-oxygenase-like one, since the sameprosthetic group is present in ligninolytic peroxidases, peroxyge-
nases, and cytochrome P-450 family [177]. In any case, an analog ofthe CpdI (arising from the resting state of the catalyst upon reactionwith a suitable oxygen donor) must be the key intermediate in thecatalytic cycle. This could revert to the iron(III)-based resting state
16 P. Zucca et al. / Journal of Molecular Catalysis A: Chemical 388–389 (2014) 2–34
les of
tita
tatoC([eC
Fig. 13. A synopsis of the (simplified) catalytic cyc
hrough electron abstraction from the reducing substrate (perox-dase character), or alternatively by donating one oxygen atom tohe substrate (monooxygenase character, typical for peroxygenasesnd cytochrome P-450) [193,199].
It is worthy of note that a formally identical CpdI arises duringhe catalytic cycle of cytochrome P-450: after substrate bindingnd subsequent, specific, NAD(P)H-dependent enzymatic reduc-ion, the arising ferrous form of the enzyme reacts with molecularxygen leading – after a further one-electron reduction – to apdI, via a Cpd0 intermediate. This is alternatively formed upon
slow) reaction of the P-450 resting state with hydroperoxides177]. In the case of peroxygenases, the ferric, resting state of thenzyme efficiently reacts with hydroperoxides, again leading to apdI (peroxidase-like behavior). This latter then monooxygenatescytochrome P-450, peroxygenase, and peroxidase.
the substrate (P-450-like behavior) [200]. In other words, peroxy-genases could be regarded as a kind of ‘hybrid’ enzymes betweenperoxidases and monooxygenases. Admittedly, ferriporphines tendto behave usually rather as peroxygenase than P-450 emulators(vide infra).
In fact, the practical application of the interaction betweenmolecular oxygen and Fe(II)-porphines is usually prevented bysome hindrances: (i) stoichiometric amounts of a sacrificialreductant should be used to afford the ferrous form of the met-alloporphine [178]; (ii) unreactive and – as a rule – useless
�-oxodimers of ferriporphine [201] are formed almost instan-taneously upon reaction between ferroporphines and molecularoxygen. So, oxygen donors such as hydroperoxides are requiredto afford a Cpd0 analog eventually evolving to CpdI (vide infra).
P. Zucca et al. / Journal of Molecular Catalysis A: Chemical 388–389 (2014) 2–34 17
F roxylao ing s
Aytcocthvorrwact
smfaw
raosotrmga
Gdb
hih
ig. 14. Oxygen rebound versus direct transfer mechanisms: (a) the proposed hydxygen transfer in the case of the conversion of an organic sulfide to the correspond
lternatively, oxygen donors such iodosyl derivatives can directlyield CpdI analogs by direct oxygen transfer. However, under rela-ively harsh experimental conditions such as high temperaturesombined with high substrate concentrations, reasonable yieldsf epoxides, alcohols and/or ketones starting from certain hydro-arbon substrates (see for example [202]) could be obtained. Inhese cases, ferriporphine reduction to its ferrous state by theydrocarbon substrate occurs, followed by molecular oxygen inter-ention by a radical mechanism. Such reaction conditions arebviously unsuitable in the field of delignification. However, recenteports describe the use of molecular oxygen under quite mildereaction conditions [203]. As expected, porphines bearing electron-ithdrawing substituents are most suitable for the use of dioxygen
s the oxidizing agent, owing to the increased attitude of theorresponding ferriporphines to be reduced to their ferrous coun-erparts.
When mild and/or biomimetic reaction conditions are applied,uch as for example, room temperature, aqueous reaction environ-ent, and nonvolatile substrates, proper oxygen donors, different
rom molecular oxygen, are required, directly leading to the CpdInalog (eventually through the intermediacy of a Cpd0 analog,hen a hydroperoxide is used).
This key intermediate could proceed to the oxygenationeactions of substrates, by following in principle two distinct mech-nisms. In the true direct oxygen atom transfer mechanism, thexygen atom moves ‘directly’ from the CpdI analog to the acceptingubstrate (path b in Fig. 14) [204,205]. Alternatively, the transfer ofne electron takes place from the substrate to the CpdI analog. Then,he formed CpdII analog donates its oxygen atom to the substrateadical to afford the final product (the so-called oxygen reboundechanism [206,207], path a in Fig. 14). This latter mechanism has
ained credit in time, owing to a vast number of both theoreticalnd practical studies, and is nowadays widely accepted [177].
The formation of a CpdI analog was demonstrated early byroves et al. [168] by using iodosylbenzene as a single oxygen atomonor. A stronger, but costlier oxygen donor is pentafluoroiodosyl-enzene [208].
As the physiological oxidant for heme peroxidases is usuallyydrogen peroxide, much work has been carried out to test its abil-
ty to act as a single oxygen donor, accordingly to the following
eterolytic mechanism [209]:PorphFe(III) OH + H2O2 → PorphFe(III) OOH →Porph+•Fe(IV) O + OH−
tion mechanism for a generic hydrocarbon substrate. (b) The hypothesized directulfoxide.
Here, the other axial ligand of the metal (usually H2O or OH−,when water is not excluded from the reaction mixture) is omittedfor the reasons of the clarity. Low pH values favor this heterolyticmechanism, as water is much better as leaving group than hydrox-ide.
Contrarily to what is observed in hemoenzymes, the Cpd0 ana-log [210–212] shows a noticeable attitude to follow an alternative,homolytic pathway [213], i.e.:
PorphFe(III) OOH → PorphFe(III) O• + OH•
In fact, PorphFe(III) O• is the high-spin form of the more usualPorphFe(IV) O [214], the relatively less reactive CpdII analog [215].This mode of decomposition of Cpd0 somewhat resembles the firststep of a Fenton reaction, and this is why it is sometimes called aFenton-like decomposition. Although not biomimetic, this way toOH• production would have been quite useful for lignin (and forother organic pollutants) oxidative degradation. Sadly, the sameOH• is able to rapidly destroy the very metalloporphine [174]. In aquite similar way, organic hydroperoxides R OOH give rise to R O•
radicals, as shown by Almarsson and Bruice [189] who found mainlyacetone and methanol as the degradation products arising from t-butyloxy radical. R O• radicals are somewhat less reactive thanOH• are, and are on the whole much less destructive toward themetalloporphine. On the other hand, they can abstract a hydrogenatom from the hydrocarbon (hc) substrate:
R O• + hc H → R OH + hc•
hc• could in turn possibly rearrange, prior to reaction with molecu-lar oxygen when in the presence of air, that explains the formationof a number of different reaction products. Many oxidations withalkyl hydroperoxides, catalyzed by ferriporphines or more gener-ally by metalloporphines, mainly depend on the formation of R O•
radicals rather than on high-valent oxoferryl intermediates [216].As expected, acyl hydroperoxides show a definite tendency
toward heterolytic scission, as carboxylate anions are much betteras leaving groups than alkoxide anions are. Even more inter-esting, the monopersulfate anion SO5
2− shows a sharp attitudeto give rise to the CpdI analog by heterolytic cleavage of thetransient Cpd0 analog (see Section 7). Evidence has been pre-
sented suggesting that electron-withdrawing substituents on theporphine ring enhance the oxidizing power of the correspond-ing Porph+•Fe(IV) O intermediate, whereas its oxidizing poweris weakened by electron-donating substituents [179,217]. CpdI
1 atalys
at
P
c
ss
t[
P
gcp
ghmccgrps
P
P
P
trifah2rttk
ithgHvotdAfweactwth
8 P. Zucca et al. / Journal of Molecular C
nalogs deriving from electron-rich ferriporphines show a sharpendency to speed up reaction with excess hydroperoxide:
orph+•Fe(IV) O + ROOH → PorphFe(IV) O + ROO• + H+
By this way, the reactive CpdI analog is replaced by its CpdIIounterpart.
Heterolytic cleavage of the Cpd0 analog is favored by proticolvents [179,218], whereas aprotic media promote homolytic scis-ion of the peroxide O O linkage [189].
The CpdII analog could also arise, without OH• formation, inhe case of aggregating ironporphines, from a comproportionation219] such as:
orph+•Fe(IV) O + PorphFe(III) OH → 2PorphFe(IV) O + H+
This unwanted reaction occurs especially when the oxy-en donor is a peracid (acyl-hydroperoxide) such as m-hloroperbenzoic acid, whereas it cannot take place in the case ofolyionic metalloporphines owing to electrostatic repulsion [201].
Detailed studies [220,221] have been conducted on oxy-enation reactions of cyclohexane and cyclohexene by t-butyl-ydroperoxide in a CH3CN/H2O medium, under catalysis by someeso-tetrakis-(polyfluorophenyl)porphine-Fe(III) complexes. The
onclusion was reached that under the adopted experimentalonditions, no epoxidation of cyclohexene was seen, whereasood yields of 2-cyclohexen-1-ol were obtained. The absoluteequirement of PorphFe(IV) O+• for the hydroxylation reaction wasostulated, while PorphFe(IV) O is not reactive enough to performuch a reaction (Eqs. (14)–(16)).
orphFe(III) OH + t-BuOOH → PorphFe(III) OO-t-Bu + H2O (14)
orphFe(III) OO-t-Bu → PorphFe(IV) = O + t-Bu O•(slow) (15)
orphFe(IV) O → + t-Bu O• → Porph+•Fe(IV) O + t-Bu O− (16)
Reaction (16) producing the reactive CpdI analog is favored inhe indicated solvent and no significant concentrations of the lesseactive PorphFe(IV) O (the CpdII analog) could be formed. Accord-ngly, in other solvent systems such as CH2Cl2/CH3OH that do notavor reaction (16), only marginal hydroxylation of cyclohexanend cyclohexene was found, whereas significant amounts of cyclo-exene were converted to the corresponding epoxide. Moreover,-cyclohexen-1-one arises among the products of cyclohexeneeaction, as significant concentrations of t-Bu O• come from reac-ion (15) in the presence of the above mentioned solvent mixtures,hat promote oxidation of 2-cyclohexen-1-ol to the correspondingetone. These findings are encompassed in Fig. 15.
At any rate, only the CpdI analog is capable of perform-ng hydroxylation, by following a pathway quite similar tohat proposed (oxygen rebound mechanism) for P-450-catalyzedydroxylations [206]. The topic of iron porphine-catalyzed oxy-enation of hydrocarbons by different peroxides in the presence of2
18O has been studied by Lee and Nam [222]. They found that atery low temperatures the intermediate Cpd0 is capable of directlyxygenating the hydrocarbon substrates, when an acyl peroxidehat stabilized the Cpd0 was used as the oxidant. Under these con-itions, no 18O was found within the cyclooctene epoxide produced.t room temperature, the oxygenating agent is the CpdI arising
rom Cpd0 by expulsion of a RO− anion. In this case, some 18Oas incorporated into the product, in agreement with a slow redox
xchange between labeled water and oxygen of the CpdI at thexial positions of the metalloporphine [223]. Addition of a strongomplexing agent to the reaction mixture abolished 18O incorpora-
ion, quite paralleling that found in P-450-promoted oxygenationshere a thiolate group from a cysteine residue firmly complexeshe iron center of the enzyme. However, Nam and colleagues [224]ave later found that under similar conditions as above also the
is A: Chemical 388–389 (2014) 2–34
CpdII analog arising from a strongly electron-deficient ferripor-phine is capable of epoxidizing alkenes. Indeed, plain epoxidationof an alkene by a CpdII analog should produce a ferrous por-phine complex. Such Fe(II)-containing metalloporphines have beentrapped under proper experimental conditions as the correspond-ing CO complexes by Cui and colleagues [225]. Fe(II)-porphines canarise also by reaction of ferriporphine with superoxide, formedby substrate radicals reacting with molecular oxygen. Somewhatsurprisingly, the obvious formation of a transient Fe(II)-containingspecies when the CpdII analog undergo a two-electron reductionreaction has received no much attention so far.
It is worth noting that the role of a Cpd0 analog obtainedupon H2O2 treatment on ferriporphines has been recently re-evaluated and emphasized [214], by following the studies ofValentine and colleagues [226]. These researchers have under-lined the nucleophilic character of the Cpd0 analog in its anionicform PorphFe(III) OO−, as shown by the quick epoxidation reactionon menadione (2-methyl-1,4-naphthoquinone), whereas a com-paratively electron-rich alkene such as cyclohexene is unreactive[227,228]. However, the role of Cpd0 as the active oxidizing agentin ferriporphine chemistry has been recently questioned [229] onthe basis of both thermodynamic considerations and experimentaldata.
6.2. Mn-porphines
Generally speaking, simple Mn(III) species are rather unstable,with a very high tendency to dismutation in Mn(II) and Mn(IV) (usu-ally producing the insoluble and poorly reactive MnO2). By contrast,Mn(III)-porphines are quite stable and so they have been and stillare the subject of numerous studies [230–233].
These complexes can be compared to their Fe(III)-containingcounterparts in general chemical behavior [213,234]. Some aspectsabout the catalytic cycle need however to be pointed out to under-stand differences in reactivity, catalytic specificity and efficiency.In fact, the CpdI analog arising from PorphMn(III) derivatives – upontreatment with hydroperoxides or other oxygen donors such asiodosylbenzene and so on – is not a Porph•+Mn(IV) O but the ratherelusive PorphMn(V)+ O [230,235,236]. This latter has been unam-biguously detected and even isolated later for some Mn-porphinesunder suitable experimental conditions, being able to transferdirectly its oxygen atom to alkenes to afford the correspondingepoxides, without the intermediacy of the well-known CpdII ana-log, PorphMn(IV) O. This difference in the electronic structure ofthe CpdI analog is most probably responsible for the higher stabil-ity of Mn-porphines along their catalytic cycle as compared withtheir Fe-containing counterparts [237], when the reaction mix-tures contain a relatively high concentration of the oxygen donorand a relatively low concentration of the oxidizable/oxygenablesubstrate. Deep studies on the properties and reactivity of Mn-porphines have shown that the Mn-containing Cpd0 analogs showa comparatively low tendency to undergo homolytic cleavage.This could contribute to the relatively high resistance of Mn-porphines against bleaching by hydrogen peroxide [233,234,238].Moreover, such a poor attitude to undergo homolytic cleavage –generating the highly destructive hydroxyl radical – could con-tribute to explain why no low pH values are needed for theoptimal catalytic activity in the case of Mn-porphines (contrarilyto that generally observed in the case of Fe-porphines, see Section6.1).
Surprisingly, the meso-tetrakis-2-N-methylpyridinioporphine-
Mn(V)+ O is unusually stable and poorly reactive. This effect, notobserved for the meso-tetrakis-4-N-methylpyridinio isomer, hasbeen ascribed to the low-spin, d2 electronic configuration of theoxoMn(V) derivative of the 2-isomer [239]. It is worth comparing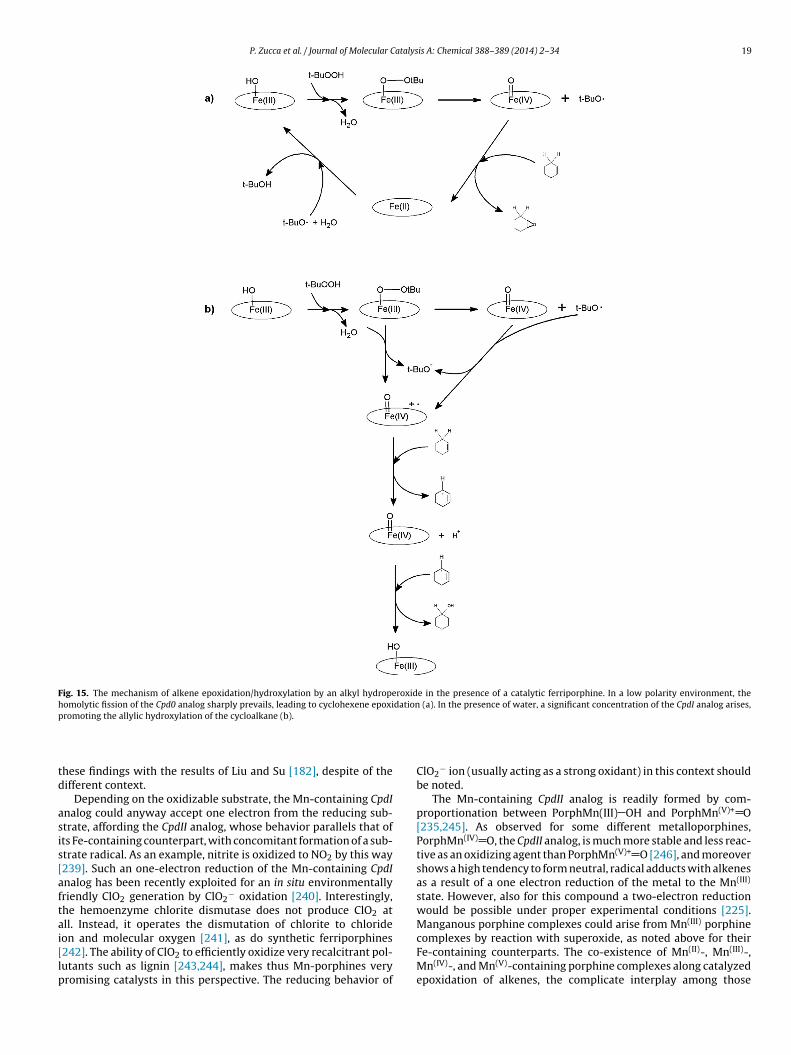
P. Zucca et al. / Journal of Molecular Catalysis A: Chemical 388–389 (2014) 2–34 19
Fig. 15. The mechanism of alkene epoxidation/hydroxylation by an alkyl hydroperoxide in the presence of a catalytic ferriporphine. In a low polarity environment, thehomolytic fission of the Cpd0 analog sharply prevails, leading to cyclohexene epoxidation (a). In the presence of water, a significant concentration of the CpdI analog arises,p
td
asis[aftai[lp
romoting the allylic hydroxylation of the cycloalkane (b).
hese findings with the results of Liu and Su [182], despite of theifferent context.
Depending on the oxidizable substrate, the Mn-containing CpdInalog could anyway accept one electron from the reducing sub-trate, affording the CpdII analog, whose behavior parallels that ofts Fe-containing counterpart, with concomitant formation of a sub-trate radical. As an example, nitrite is oxidized to NO2 by this way239]. Such an one-electron reduction of the Mn-containing CpdInalog has been recently exploited for an in situ environmentallyriendly ClO2 generation by ClO2
− oxidation [240]. Interestingly,he hemoenzyme chlorite dismutase does not produce ClO2 atll. Instead, it operates the dismutation of chlorite to chloride
on and molecular oxygen [241], as do synthetic ferriporphines242]. The ability of ClO2 to efficiently oxidize very recalcitrant pol-utants such as lignin [243,244], makes thus Mn-porphines veryromising catalysts in this perspective. The reducing behavior ofClO2− ion (usually acting as a strong oxidant) in this context should
be noted.The Mn-containing CpdII analog is readily formed by com-
proportionation between PorphMn(III) OH and PorphMn(V)+ O[235,245]. As observed for some different metalloporphines,PorphMn(IV) O, the CpdII analog, is much more stable and less reac-tive as an oxidizing agent than PorphMn(V)+ O [246], and moreovershows a high tendency to form neutral, radical adducts with alkenesas a result of a one electron reduction of the metal to the Mn(III)
state. However, also for this compound a two-electron reductionwould be possible under proper experimental conditions [225].Manganous porphine complexes could arise from Mn(III) porphine
complexes by reaction with superoxide, as noted above for theirFe-containing counterparts. The co-existence of Mn(II)-, Mn(III)-,Mn(IV)-, and Mn(V)-containing porphine complexes along catalyzedepoxidation of alkenes, the complicate interplay among those
2 atalys
dewn
cmPeaitafwPa
eewftsarsfiobnetsaiowvP
tfdaPpPcao
deNtssbhdcpe
R
0 P. Zucca et al. / Journal of Molecular C
ifferent species based on comproportionation/disproportionationquilibria, and moreover the reaction of Mn(II)-porphine complexith molecular oxygen under aerobic conditions, had been alreadyoted [247].
In a fashion quite paralleling that of the PorphMn(V)+ Oomplexes, PorphCr(V)+ O is a relatively efficient hydroxylating/ono-oxygenating agent for alkanes and alkenes. However,
orphCr(IV) O is only able to transfer its oxygen atom to triph-nylphosphine and is quite unreactive as regards alkane andlkene hydroxylation/mono-oxygenation. Very little or no activ-ty is shown also by PorphTi(IV) O, PorphV(IV) O, PorphNb(IV) O,rans-PorphOs(VI)(=O)2 and cis-PorphMo(VI)(=O)2 [192]. A compar-tive study, examining the differences in electronic configurationsor a series of PorphM(IV) O complexes had already explainedhy PorphV(IV) O and PorphCr(IV) O are stable entities whereas
orphMn(IV) O and especially PorphFe(IV) O are rather unstablend highly reactive species [248].
At any rate, the CpdII analog of Mn-porphines is reactivenough to efficiently attack variously substituted alkenes [249]. Asxpected, electron-donating substituents made the alkene some-hat more reactive, in agreement with a mechanism involving the
ormation of a PorphMn(III) OCC• radical intermediate adduct, inurn arising from a charge-transfer complex with a little chargeeparation. The postulated radical intermediate explains why trans-lkenes are slightly more reactive than the cis- ones; such aadical is long-lived enough to undergo isomerization to the moretable trans form, which explains the large loss of stereospeci-city along the reaction. Moreover, in the presence of molecularxygen, a peroxy radical is formed that in turn evolves by C Cond fission to yield aldehydes or ketones. The hypothesis of aeutral radical following a charge transfer complex is also strength-ned by the observation that similar products are observed whenreating alkenes with simple neutral radicals such as OH•, whatuggests a similar reaction pathway, again preferring trans-alkeness the substrates. The PorphMn(III) OCC• radical intermediate eas-ly undergoes a further reaction leading for example to a mixturef cis- and trans-stilbene epoxide when starting from cis-stilbene,ith concomitant production of PorphMn(II). This latter low-
alent manganese porphine cannot regenerate the CpdII analogorphMn(IV) O upon reaction with molecular oxygen [249].
By contrast, PorphMn(V)+ O acts on alkenes with preference forhe cis-isomers, that is explained by sterical reasons and by a dif-erent mechanism involving a ‘direct’ oxygen transfer [173]. Such airect transfer is also in agreement with the higher epoxide yieldsnd noticeable stereospecificity. The reaction pathways by whichorphMn(V)+ O and PorphMn(IV) O react with alkenes are encom-assed in Fig. 16. The stereospecificity of alkene epoxidation by theorphMn(V)+ O complex is lower than that observed with its Fe-ontaining counterpart. This has been interpreted as the proof of
higher contribution of a radical-based mechanism in the case ofxomanganyl complexes [250,251].
Apart from hydroperoxides, another important single oxygenonor, very useful when working with Mn-porphines, is undoubt-dly hypochlorite. The commonest and cheapest hypochlorite isaClO, industrially obtained as a strongly alkaline solution, con-
aining large NaOH excess, by controlled chlorination of aqueousodium hydroxide. Hypochlorite ion ClO− is a relatively strong base,o noticeable amounts of hypochlorous acid HClO are detectableelow pH 9, and at pH 5 almost all hypochlorite is present asypochlorous acid. In relatively high concentrations, HClO reactsirectly and sometimes vigorously with oxidizable substrates;hloro-organics are often the final products of its action. For exam-
le, alkenes give the corresponding chlorohydrins through anlectrophilic addition:CH CH R′ + HClO → R CHCl CHOH R′
is A: Chemical 388–389 (2014) 2–34
Sadly, many metalloporphines are quickly and irreversiblybleached [252] by sodium hypochlorite under various experimen-tal conditions. However, the action mechanism operating in alkeneoxidation by sodium hypochlorite in the presence of some metal-loporphines, has been deeply investigated [253,254]. In particular,TPP-Mn(III) acetate was found as by far the most active catalystwhen compared with its Cr(III), Fe(III), and Co(III) analogs. Sharplybasic conditions are required to see the catalytic activity, whereasbelow pH 8 the bleaching of the catalyst was found concomi-tant with a partial alkene conversion (styrene was used along thatstudy). A mechanism involving the transient formation of a metal-loporphine/hypochlorite complex has been suggested:
TPPMn(III) OH + ClO− → TPPMn(III) OCl + OH−
TPPMn(III) OCl → Cl− + TPPMn(V)+ O
Another study [255] has shown that under certain conditions theoxidative O-dealkylation of complex alkenes such as isosafrole maycompete with epoxidation by hypochlorite under Mn-porphinecatalysis.
6.3. Ru-porphines
Quite interesting, and also promising for technological appli-cations, is the family of ruthenoporphines and their high-valentoxo-derivatives that are under study since many years, in compari-son with other metalloporphine catalysts [169,256–260]. Since thecatalytic activity of meso-(tetrakismesityl)porphine-Ru(VI)-trans-( O)2 in oxygenation of alkenes by molecular oxygen – withoutthe intervention of any co-reductant – was discovered [261,262],much interest has arisen around this topic. In fact, catalytic aer-obic oxidations/oxygenations show the substantial advantage ofusing the inexpensive molecular oxygen (in most cases, simplyair) instead of other comparatively costly oxygen donors. Inter-estingly, tuning the reactivity of Ru-porphines by the peripheralsubstituents on the porphine ring is much more important in com-parison with Fe- and Mn-porphines. Moderately electron-deficientsystems presumably use a reactive Porph-Ru(VI)( O)2 intermediateas a monooxygenating species; the arising intermediate Porph-Ru(IV) O regenerates by fast disproportionation the high-valentintermediate Porph-Ru(VI)( O)2 and a Porph-Ru(II) species whichin turn easily reacts with O2 and closes the catalytic cycle. How-ever, in strongly electron-deficient systems such as perhalogenatedruthenoporphines, the reactivity of the Porph-Ru(II) species withdioxygen is substantially lowered, while that of Porph-Ru(VI)( O)2with reducing substrates is only slightly increased [263]. So, analternative catalytic cycle, involving a redox couple based on Ru(III)
and Ru(V) species, has been suggested [264]: by this view, therather inert carbonyl complex of a porph-Ru(II) species under-goes a relatively slow one-electron oxidation (by Fe(ClO4)3) to thecorresponding radical cation. This slow ‘activation’ of the rutheno-porphine explains a sort of lag time observed in such oxygenationreactions. The Ru(III)-containing carbonyl complex loses CO andreacts with the oxygen donor, evolving through an internal elec-tron transfer to the reactive porph-Ru(III)/oxygen donor complex,which is the parent compound of the final oxidizing/oxygenatingspecies, porph-Ru(V)+ O. In fact, this Ru(V)-containing complexcould be regarded as the ruthenium-based CpdI analog, rather thanthe Ru(VI)-trans-dioxo complex. The various suggested pathwaysfor ruthenoporphine catalysis are depicted in Fig. 17.
It is worth noting that, apart from aerobic oxida-tions/oxygenations using molecular oxygen, N-oxides of
nitrogen-containing heteroaromatics, such as 2,6-dichloropyridineN-oxide or certain pyrazine N-oxides, are the oxygen donors com-monly used along the majority of the published experimentalwork. This is obviously a serious drawback when envisaging a
P. Zucca et al. / Journal of Molecular Catalysis A: Chemical 388–389 (2014) 2–34 21
Fig. 16. (a) Alkene epoxidation by an oxomanganese(V)porphine; (b) alkene epoxidation/fragmentation by an oxomanganese(IV)porphine. When an oxomanganese(IV)porphinep e com
mabp
htlcdcstir
erforms alkene oxidations, it is reduced to the corresponding manganous porphin
assive use of such catalysts for lignocellulose delignificationt plant scale, as the N-oxides of nitrogenous heteroaromaticases are rather costly reagents. Interestingly, case studies about aeroxidase-like behavior of ruthenoporphines are lacking.
On the whole, significant differences among metalloporphinesave been shown, indicating that Fe-porphines show a noticeableendency to homolytic cleavage of the corresponding Cpd0. Thiseads to a radical mechanism often triggering the formation ofomplex product mixtures and sometimes causing catalyst auto-estruction. Mn-porphines on the contrary prefer a heterolyticleavage of the corresponding Cpd0 due to a comparatively higher
tability of the corresponding CpdI, often leading to direct oxygenransfer. Ru-porphines show as their outstanding feature the abil-ty of using molecular oxygen to promote catalytic oxygenationeactions. These facts are encompassed in Tables 4 and 5.plex, which cannot be regenerated directly by the oxygen donor.
6.4. The importance of the axial coordination
It is well known that the properties of hemoproteins largelydepend on their ‘fifth’ axial ligand anchoring the heme moiety toits protein counterpart. This ligand is the imidazole nitrogen atomof a specific histidine residue in myoglobins, hemoglobins, andthe majority of peroxidases, whereas a tyrosine residue – actingthrough its phenolic hydroxyl – is present in catalases. A sulfur atomfrom a cysteine residue is typical for P-450 cytochromes, perox-ygenases and haloperoxidases [155,265,266]. Oxygen-containingproximal ligands lead to reduced oxygenase activity while enhanc-
ing dismutase-like activity such as in catalase [267].Quite paralleling this physiological interaction, early studieshave given prominence to the deep effect observed on bothreactivity and action mechanism(s) of metalloporphine-promoted

22 P. Zucca et al. / Journal of Molecular Catalysis A: Chemical 388–389 (2014) 2–34
F thenoc x of th( theno
oclhaoah([aotChattr
TSO
ig. 17. (a) The catalytic cycle of aerobic alkene epoxidation under catalysis by a ruatalysis by a ruthenoporphine. The preliminary activation of the carbonyl compleO.D.) under catalysis by a rutheno(III)porphine. The preliminary activation of the ru
xidations/oxygenation in the presence of suitable ligands of theentral metal ions in the reaction mixtures. Too strong ligandsead to axial bis-ligated metalloporphines, whose attitude to formigh-valent oxo species is substantially lowered if not totallybolished. On the other hand, too feeble ligation implies the usef large ligand excess [181]. Imidazole as the added axial ligand isn obvious biomimetic choice, capable of substantially improvingydroxylation/mono-oxygenation yields when hydroperoxidesalkyl-, acyl-hydroperoxides and H2O2) are the oxygen donors230,268–270]. The beneficial effect of imidazole – an electron-richromatic heterocycle – depends on its attitude to shift the cleavagef the O O bond in the Cpd0 analog from a homolytic mechanismo a heterolytic one, thus favoring the formation of the highly activepdI analog. In the case of Mn-porphines, almost no reaction withydroperoxide is observed under many reaction conditions unless
suitable nitrogen base such as imidazole is present [271]. Unfor-
unately imidazole is susceptible to destructive oxidation alonghe course of the catalysis, so a continuous supply is necessary thatenders practical applications unfeasible [272].able 4ummary of reaction factors able to promote heterolytic and hemolytic scission of-O bond in Cpd0.
Factors promoting theheterolytic scission ofCpd0
Factors promoting thehomolytic scission of Cpd0
• Polar solvent• Protic solvent• Low pH• Electron-withdrawing
substituents• Acyl hydroperoxide• SO5
2−
• Non-polar solvent• Aprotic solvent• High pH• Electron-donating
substituents• Hydrogen peroxide• Alkyl hydroperoxides
(II)porphine; (b) alkene epoxidation by meta-chloroperbenzoic acid (MCPBA) undere rutheno(II)porphine is shown; (c) alkene epoxidation by a generic oxygen donor(II)porphine carbonyl complex by a ferric salt is shown.
Pyridine is not biomimetic as a ligand for metalloporphinesbut is bioinspired, as it can axially bind the metal ion with thesame fashion as imidazole, with its lone electron pair on thenitrogen atom. Contrarily to imidazole, pyridine is a typicalelectron-deficient heteroaromatic ring, very resistant againstoxidative destruction, so it has been proposed as a chemicallyrobust and efficient axial ligand for metalloporphine-catalyzed oxi-dations/monooxygenations [253,255]. In fact, imidazole, pyridineor thiolate ion provide the � electron density to the antibondingO O orbital through the metal center, therefore weakening theO O bond. Protonation of the distal oxygen in this Cpd0 analogfavors the heterolytic scission leading to the required CpdI analog.Although imidazole is a stronger electron-donating agent [273],pyridine has a higher �-backbonding attitude, therefore stabilizingthe lower oxidation states of the central metal ion; this rendersmore reactive the high-valent metal-oxo species by increasingtheir redox potential. So, on the whole it displays a better catalyticefficiency when compared with imidazole [274]. Unfortunately,large-scale application of pyridine for example in industrial plantsis prevented by its volatile, malodorous and highly toxic nature.
Various attempts have been made to directly fix the requiredaxial ligand such as imidazolyl [270], pyridyl [275] or thiolate [276]moieties to the porphine ring. As already pointed out by Rocha-Gonsalves and Pereira [181] no substantial advantages arise fromthe use of these all-in-one constructs: their syntheses are oftenlaborious, whereas only a little increase in activity in compari-son with ‘simple’ metalloporphines in the presence of ‘free’ axialligands is observed.
7. The reducing substrates
A huge variety of oxidizable/oxygenable substrates have beenstudied as targets of the oxygen-containing high-valent states

P. Zucca et al. / Journal of Molecular Catalysis A: Chemical 388–389 (2014) 2–34 23
Table 5Striking features of catalytic pathways for Fe-, Mn- and Ru-porphines.
Fe-porphines Mn-porphines Ru-porphines
• CpdI is Porph+•Fe(IV) O • CpdI is PorphMn(V)+ O • Oxygenating species can be both Porph-Ru(VI)( O)2
and Porph-Ru(V)+ O• Hydroperoxides are the preferred
oxidants• Hydroperoxides and hypochlorite are the preferredoxidants
• Various generic oxygen donor (including N-oxides,hydroperoxides and O2) are able to performoxygenations
• Oxygenation through reboundmechanism (CpdII intermediate)
• Direct oxygenation from CpdI • Three peculiar mechanisms for oxygenation
• Low pH required • Active at neutral/basic pH• Tendency to both homolytic and
heterolytic cleavage of Cpd0• Low tendency to homolytic cleavage of Cpd0
• Higher stereospecificity in alkene • Lower stereospecificity in alkene epoxidations
osaitrfid
cesbmo
tawmtiit
ppao
io[lottc
opacaasaguw
epoxidations• More stable than Fe-counterparts
f metalloporphines, and several studies have focused on newynthetic procedures, in particular in the fields of alkane andrene hydroxylations, and alkene epoxidations [277–279]. In manynstances, stereospecific reactions have given substantial contribu-ion to understand the reaction mechanism(s) (generally speaking,adical- versus non-radical pathways, homolytic versus heterolyticssion of the O O bonds of hydroperoxides acting as oxygenonors, and so on [280–283]).
However, quite paralleling the metabolic versatility ofytochrome P-450 family and of fungal peroxygenases, a huge vari-ty of substrates, different from simple hydrocarbons, have beenhown to be oxidized by biomimetic/bioinspired metalloporphine-ased catalytic systems [284]. Hydroxylation, epoxidation,ono-oxygenation and/or deeper demolition reactions have been
bserved.In a comparative study [285] some well-known drugs, belonging
o different chemical families (such as phencyclidine, diclofenac,minopyrine, tiagabine, nicotine, and others, Fig. 18), were treatedith suitable oxidants such as NaClO in the presence of differentetalloporphines. Isolated degradation products are almost iden-
ical in structure and relative yields to those metabolites observedn vivo in humans and experimental animals. Interestingly, signif-cant differences were observed in some instances depending onhe central metal ion (Fe as opposed to Mn).
A catalytic system formed by TPPMnOAc and pyridine efficientlyromotes the oxidation of a large variety of differently substitutedrimary and secondary alcohols to the corresponding aldehydesnd ketones, using tetrabutylammonium monopersulfate as thexidant [286].
The oxidation of organic sulfides to the corresponding sulfox-des or eventually to their sulfone counterparts in the presencef Fe and Mn porphines has been recently studied in some detail265,287–289], showing the influence of the nitrogenous axialigand on the efficiency of the catalysis. Also the positive influencef the electron-withdrawing substituents at the meso positions onhe metalloporphine ring has been assessed [274] in the case ofhe oxidation of aliphatic, alicyclic and benzylic alcohols to theorresponding carbonyl compounds.
Different azo dyes have been tested as substrates for catalyticxidative degradation in the presence of suitable metallopor-hines [290,291]. In particular, Fe and Mn porphines have shown
promising activity in azo dye cleavage, although a compli-ate mixture of products usually arises [291]. No N-oxidation tozoxy compounds was observed in the presence of the best cat-lyst tested, FeTDCP. Instead, as a general rule, the oxidationtarts with a one-electron abstraction from the more activated
romatic ring (better, from an amino or hydroxy substituent,iving rise to a delocalized radical cation, which in turn canndergo some different reaction pathways (Fig. 19), as observedith certain peroxidases [292]. In particular, 4-phenylazoaniline[291] gave p-benzoquinone, ammonia, benzene and molecularnitrogen. Besides, azobenzene, bis(p-phenylazo)benzene, pheny-lazonitrobenzene, 1,4′-bis(phenylazo)azobenzene were found assecondary products, thus showing that condensation side reac-tions involving transient nitroso-compounds and the unchangedsubstrate, take also place. These latter compounds are not fur-ther degraded and represent a serious drawback when planningthe use of metalloporphine catalysts in remediation of amino-azo dyes. On the other hand, azo dyes bearing phenolic hydroxysubstituent are more easily degraded under these biomimetic con-ditions [291]: obviously, no more condensed azocompounds canarise from their oxidative degradation. In this perspective, theiruse could be encouraged, as they should be more environment-friendly than their amino counterparts are. More recently, alsomore complex azodyes have been tested as substrates for oxidativedegradation in the presence of soluble Mn-containing porphinesand hydrogen peroxide in aqueous solution [290].
Both halogenations [293] and de-halogenations [294,295] ofvarious substrates under metalloporphine catalysis have beenreported. In particular, a radical mechanism together with a cyclingof the manganese porphine between the Mn(V) and Mn(IV) oxi-dation numbers has been proposed, where the hypochlorite ionis both the oxidizing agent for the manganese porphine and thesource of chlorine atom found in the halogenated product. Bythis way, a number of useful alkyl chlorides have been preparedwith moderate to good yields. With concern to dehalogenations,these are of potential interest toward biomimetic remediation oftoxic and/or recalcitrant chloroaromatics. Even co-polymerizationof polyalogenated phenols has been recently described [296].
Fe- and Mn-porphines also catalyze peroxynitrite decompo-sition yielding nitrite and molecular oxygen; however, in thepresence of phenols, catalytic nitration occurs at the o- and p-positions of the phenol ring [297].
Not surprisingly, very little interest arose about ‘classical’ per-oxidase substrates such as phenols and aromatic amines, mainlybecause of the large availability of inexpensive enzymes suchas horseradish peroxidase. However some studies exist, helpingunderstanding both kinetics and mechanism(s) of such peroxidase-like activity of certain metalloporphines [209,298,299].
8. Product and/or catalyst recovery: heterogenizedmetalloporphines
In the field of metalloporphine-based catalysis, the usefulnessof catalyst heterogenization is well established [300]. The synthe-sis of metalloporphines is a quite laborious and challenging task in
several instances, and the obtained products are therefore expen-sive, especially when precious metals such as ruthenium give thecentral ion of the complex catalyst. The toxicological propertiesof many metalloporphines are not yet known, so contamination
24 P. Zucca et al. / Journal of Molecular Catalysis A: Chemical 388–389 (2014) 2–34
e Fe-
oresetcAcotao
ciVpp
emcpiuoic
itapocpl[os
ac[
Fig. 18. Some drugs, not chemically related to lignin that are oxidized by som
f industrial products or effluents by these catalysts should beeduced to a minimum. Last but not least, the recycle of the het-rogeneous catalysts along potential industrial applications couldubstantially contribute to significantly lower the costs. Heterog-nization is in principle advisable when the metalloporphine andhe reaction product(s) are both soluble in the same solvent(s), aircumstance that makes their separation quite tedious and costly.lso, a relative stabilization of the immobilized catalyst by physi-al hindrance against oxidative bleaching could be forecast. On thether hand, heterogenized metalloporphines will be subjected tohe mass transfer limitations affecting all heterogenized catalysts,nd therefore an increase in substrate constant Kd and a decreasef turnover number should be expected.
Heterogenization of porphine or metalloporphine catalystsould be achieved by entrapment and or encapsulation within 3Dnert and insoluble networks or within hollow spheres or fibers.ery recently [301–304], preparations of such catalysts have beenroposed for the photoremediation of polluted wastewaters or ashotodynamic therapeutic agents.
Adsorption – sometimes not so easy to distinguish fromncapsulation [305–312] – is a very profitable technique foretalloporphine heterogenization as it does not usually require
omplicate functionalization of the supports and/or tedious cou-ling procedures. Nanoparticles obtained from suitable organic or
norganic materials could be effective for therapeutic purposespon intravenous infusion, and in general heterogeneous, robustxidation/oxygenation catalysts could be obtained. Obviously,n-depth toxicological studies are required prior to practical appli-ation of such preparations in medicine.
Covalent immobilization [313] of metalloporphines is usuallyrreversible and therefore warrants that no unwanted release ofhe catalyst could take place during use, washing, and regener-tion. Covalent immobilization could be achieved with variousrocedures, requiring a suitably reactive moiety at the peripheryf the porphine macrocycle to form a stable linkage with a properounterpart onto the insoluble support [314,315]. Exploiting the-carboxyphenyl meso substituents allows the preparation of cova-ently bound metalloporphines on amino-functionalized supports316–319]. Also the reactive fluorine atoms at the para positionsf the pentafluorophenyl rings of metallated TFPP could form aecondary amine linkage with aminated supports [320].
A substantial step toward biomimesis is achieved when a suit-ble axial ligand is properly bound to the support, and is thereforeapable of forming a coordinative bond with the metalloporphine321–324]. This immobilization technique also helps protecting the
and Mn-containing porphines in the presence of different oxidizing agents.
covalently bound axial ligand against oxidative degradation, as thevery metalloporphine shields the axial ligand and prevents to acertain extent the approach of the oxygen donor to the surfaceof the support. However, this technique is not always applicable,and different interactions could prevent immobilization. This wasobserved early [323] in the case of the Mn(III) derivative of thecationic meso-tetra(N-methyl-4-pyridinio)-porphine, that did notreact with an imidazole-functionalized silica gel whereas it wasadsorbed by plain silica gel owing to the acidic character of thesilanol groups, leading to an ionic interaction.
Zucca and colleagues have studied the biomimetic or bioin-spired immobilization of TSPPMn(III) and TFPPFe(III) on imidazole-[325], pyridine- [326], and thiol-functionalized [327] hydrophilicsupports, mimicking in the former case the active site of LiP andin the latter the active site of cytochrome P-450 or, better, thatof peroxygenase. The versatile-peroxidase-like activity of suchadducts toward different substrates such as phenolic and non-phenolic aromatics, eventually related to lignin, was assessed.These catalysts showed both LiP-like and peroxygenase-like activ-ity. Also some industrially relevant organic dyes were bleachedusing aqueous hydrogen peroxide as the oxidant. In some cases,when the preparations acted on electron-deficient cationic dyessuch as phenosafranine, the ‘direct’ transfer of an oxygen atomfrom the Cpd I analog to the dye was suggested. This approachled to an efficient and stable loading onto the support, compa-rable with covalent linking. Moreover the favorable effect of theaxial ligand was also exploited, preventing the need of continuoussupply of free ligands [325–327]. However, an excessive crowd-ing of ligand moieties on the surface of the support should beavoided, in order to prevent the formation of inactive bis-ligatedmetalloporphines.
In conclusion, different immobilization approaches for met-alloporphines have been reviewed, showing advantages anddrawbacks, typical for each immobilization method. These areencompassed in Table 6.
9. Metalloporphines and lignin
Being assessed the ability of synthetic metalloporphines tobehave as P-450/peroxygenase mimics, the possibility of usingthese compounds also as ligninolytic peroxidase emulators was
explored. The finding that ferriheme in ligninolytic peroxidasesis per se capable of performing oxidations of high-potentiallignin substructures [328] has triggered further studies in thefield [225,328–331]. Water-soluble or hydrophilized [327,332]
P. Zucca et al. / Journal of Molecular Catalysis A: Chemical 388–389 (2014) 2–34 25
F d by Fb pount
rirmptscorccrvb
ig. 19. (a) The catalytic oxidation mechanism of hydroxy-azocompounds mediatey FeTDCPP; (c) Main products identified after catalytic oxidation of amino-azocomhe hydroxylamino-azobenzene is only hypothesized but not assessed.
edox-active synthetic metalloporphines have great potentialnterest in delignification of lignocellulosics in aqueous envi-onment, in the presence of hydrogen peroxide, potassiumonopersulfate or other water-soluble oxygen donors such as
eracetic acid or t-butyl hydroperoxide. They could overcomehe problems arising from the high costs and the low operationaltability of the ligninolytic peroxidases. Moreover, their dualatalytic nature as both peroxidase and peroxygenase mimicspens the way to manage structural motifs and molecules that areesistant against the commonest ligninolytic enzymes such as lac-ases, LiP, and MnP. In fact, many redox-active metalloporphines
ould oxidatively attack chemical functionalities and linkages thatemain untouched by the ligninolytic enzymes (vide infra). Theersatility of redox-active metalloporphines is further widenedy the fact that they are not subjected to the sterical constraintseTDCPP; (b) the catalytic oxidation mechanism of amino-azocompounds mediatedds mediated by FeTDCPP. The direct condensation reaction between benzene and
at the active sites that limit the applicability of the ligninolyticperoxidases, as already noted in Section 4.2.
Since lignin is insoluble in water and most organic solvents, animpressive number of model compounds soluble in water and/ororganic solvents have been developed, bearing some of the typi-cal linkages and/or structural motifs present in true lignins [39].Among these, a wide variety of lignin model compounds showingdifferent chemical complexity have been proposed as substratesto test the ligninolytic ability of redox-active metalloporphyrins. Itis worth noting that among such model compounds many are thesame used to test LiP, MnP, and VP activities, owing to the poorly
defined chemical structures of lignins. Soluble lignosulfonates(Section 3.2), although attractive for their high solubility in water,low cost and wide availability, draw little interest as lignin mod-els because the sulfonate groups significantly affect the reactivity
26 P. Zucca et al. / Journal of Molecular Catalysis A: Chemical 388–389 (2014) 2–34
Table 6Advantages and disadvantages of the various immobilization approaches proposed for metalloporphines heterogenization.
Method of immobilization Advantages Disadvantages
Adsorption • No chemical/sterical modification of the catalyst• Easy to be performed
• Low strength of the binding• Catalyst leakage• No real emulation of peroxidases active site
Encapsulation/entrapment • No chemical/sterical modification of the catalyst • Low strength of the binding• Catalyst leakage• No real emulation of peroxidases active site
Covalent binding • Strength of the binding• Minimization of catalyst leakage
• Possibility of chemical/sterical modification of thecatalyst• No real emulation of peroxidases active site
Ion exchange • No chemical/sterical modification of the catalyst • Low strength of the binding• Catalyst leakage• No real emulation of peroxidases active site
Axial coordination • Real emulation of peroxidases active site• Increased stability
ng tow
• Possible of axial bis-ligation of the catalysts
oywdaacpiatvclp5atoao
LotMcmTgapp
Fp
• Increased activity• If the ligand is imidazole, shifticleavage
f their organic backbone. The cumbersome assay based on eth-lene generation from 2-keto-4-methylthiobutyric acid [116,118]as soon abandoned in favor of the rapid and easy photometricetermination at 310 nm of veratraldehyde arising from veratryllcohol [333] (although ferriporphines tend to promote veratryllcohol oxidation yielding other products, vide infra). This commer-ially available, inexpensive reagent has the advantage on otherotential substrates of being insensitive to both ‘classical’ perox-
dases and laccases, as it emulates to a certain extent the benzyllcohol (�-hydroxy) motif and the nonphenolic �-O-4 substruc-ures of lignin [334]. Some studies have been focused on the fate oferatryl alcohol upon oxidative treatments under metalloporphineatalysis [335–337] and have shown that these reactions paral-el those found by using LiP or also Ce(IV) salts [338]: two mainroducts were identified, mainly veratraldehyde and 2-methoxy--hydroxymethyl-1,4-benzoquinone [326]. Fe-porphines are mostctive at low pH values while Mn-porphines preferred almost neu-ral environment. The similarity between the product patternsbtained with selected metalloporphines, LiP, and ceric salts, isnother proof in favor of a one-electron abstraction mechanismperating in these reactions.
In particular, interesting findings have been presented byabat and Meunier [336] who studied the effect of severalxidants. Potassium hydrogen monopersulfate KHSO5 provedo be much more efficient than H2O2 when various Fe- and
n-porphines catalyzed the oxidation of the lignin modelompounds veratryl alcohol and 1-(3,4-dimethoxyphenyl)-2-(2-ethoxyphenoxy)-propane-1,3-diol (Fig. 20) in aqueous media.
his is most probably due to the sharp attitude of SO42− as leaving
–
roup when compared to OH , that renders the heterolytic cleav-ge of the O O bond of the Cpd0 analog easier in the case of theersulfate anion, as already noted in Section 6.1. On the whole, Fe-orphines are more active at acidic pH values (protonation of Cpd0ig. 20. The cleavage reactions of a lignin model by aqueous monopersulfate in theresence of the tetrasulfonic derivative of FeTDCPPS.
ard heterolytic
gives rise to H2O elimination while lowering its tendency towardhomolytic cleavage of the O O bond) whereas Mn-porphines prefernearly neutral conditions, under the described experimental con-ditions. Clearly, the putative Cpd0 analog stemming from Mn por-phines has a very high tendency to evolve to the Mn(V)-containingCpdI analog. The same authors also found that veratraldehyde wasthe preferred product of veratryl alcohol oxidation in the presenceof Mn-porphines, whereas the 2-methoxy-5-hydroxymethyl-1,4-benzoquinone was the major product under Fe-porphine catalysis.
In general, MnTDCP is more stable against oxidative bleachingthan FeTDCP; among different oxidants [336], H2O2 is the mostdestructive, most probably for the high tendency of its Cpd0 ana-log to undergo homolytic cleavage with concomitant OH• release.The group of Cui and Dolphin [335,339,340] has worked for someyears about Fe- and Mn-porphines as emulators of LiP. They haveshown that, contrarily to that observed with LiP, the sulfonic acidderivative of FeTDCP is capable of degrading phenylcoumaran sub-structures: sidechain cleavage and oxidation, phenyl ring cleavage,and also oxidative destruction of the very coumaran nucleus (whichremains untouched upon LiP treatment) were observed (Fig. 21). Onthe whole, the Authors observed a noticeable resemblance betweenLiP-catalyzed and iron-porphine-catalyzed oxidative degradationof some simple model compounds, such as veratryl alcohol, ver-atryl acetate, �-O-4, �-1, and phenylcoumaran dimers. Furtherstudies have been focused on 4-ethoxy-3-methoxyphenyl-propaneand -propene models, since these units have been identified ear-lier as common chain termini of the lignin polymer [341]. On thewhole, �-phenylpropane and �-phenylpropene units represent avery small percentage of the C6C3 units, but they play an importantrole due to their position along the polymer. While 4-ethoxy-3-methoxypropane is oxidized by the tetrasulfonated Fe-TDCPP to adead-end product, a C1 carbinol, its alkene counterpart is deeplycleaved as the double bond was converted to a glycol functionalityupon biomimetic oxidation under the same experimental condi-tions. Such a glycol functionality is well prone to further catalyticoxidation and C C bond cleavage (Fig. 22).
Moreover, the Authors have confirmed that �-perchloro-metalloporphines are more reactive as oxidation catalysts but arealso rapidly bleached by the oxidant, so they are on the whole lessefficient in comparison with their non-�-halogenated counter-parts [172]. Generally speaking, also in this context Mn-porphinesare more stable than their Fe-containing counterparts; among thevarious oxidants studied, H2O2 was sadly the most destructive
toward the catalysts [225], for the reasons stated above. Finally,the same Authors have tested some halogenated Fe- and Mn-porphines dissolved in dimethylformamide as catalysts for Kraftsoftwood lignin oxidative degradation by peracetic acid. Sound
P. Zucca et al. / Journal of Molecular Catalysis A: Chemical 388–389 (2014) 2–34 27
uOOH
sd
twp[
tfbftetcF
Fig. 21. The oxidative cleavage of phenylcoumaran substructures by t-B
pectroscopic evidence was found in favor of a deep oxidativeegradation of the substrate.
The fate of some lignin-related o-dimethoxyarenes and in par-icular of 1-(3′,4′-dimethoxyphenyl)-2-phenyl-1-propanol (Fig. 23)hen oxidized under catalysis by some electron-deficient Fe-orphines has been studied in detail by Artaud and colleagues180].
They have found that such Fe-porphines catalyzed the forma-ion of four product classes: (i) substituted benzaldehydes arisingrom benzylic carbon oxidation and �,� fission of the C� C�ond (when present); (ii) substituted o-benzoquinones, arisingrom the C1 C� cleavage; (iii) p-benzoquinones, still bearinghe propanoid sidechain; (iv) substituted muconic dimethyl
sters. Both quinones and muconic esters derived from the oxida-ive attack on the o-dimethoxyphenyl moieties of the modelompounds. The same authors also compared LiP and somee-porphines as catalytic agents under similar experimentalFig. 22. The oxidative transformation of phenylpropane and phenylp
in the presence of FeTDCPPS catalyst, a reaction not observed with LiP.
conditions on 1-(3′,4′-dimethoxyphenyl)-2-phenyl-1-propanol,and found that similar product patterns arose. However, only underaerobic conditions could LiP produce quinones and/or muconicdiesters, whereas in the absence of oxygen veratraldehyde was byfar the main reaction product. In fact, the substrate radical formedby LiP is generated far from the ferryl moiety of the enzyme, so it canreact with molecular oxygen, producing new peroxy radicals that inturn give rise to the final products. In the case of the Fe-porphines,oxygenation of the first substrate-derived radical is accomplishedby the CpdII analog (an ‘oxygen rebound’ mechanism), and there-fore the presence of molecular oxygen had not significant effect onthe product pattern. On the whole, 1-(3′,4′-dimethoxyphenyl)-2-phenyl-1-propanol underwent both C� C� cleavage and oxidation
of the dimethoxyphenyl moiety, with proportions depending onthe chosen solvent (protic versus aprotic environment), on theparticular Fe-porphine, on the particular oxidant (hydrogenperoxide or magnesium monoperoxyphthalate). In more detail,ropene units by t-BuOOH in the presence of FeTDCPPS catalyst.
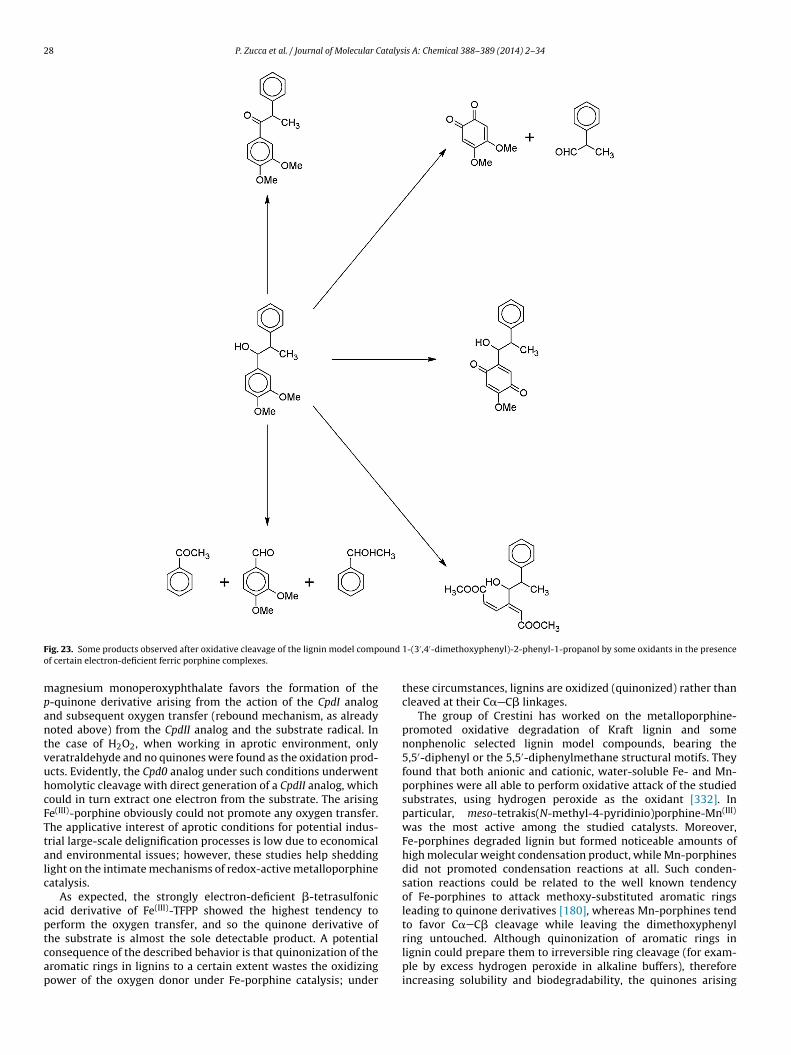
28 P. Zucca et al. / Journal of Molecular Catalysis A: Chemical 388–389 (2014) 2–34
F ound
o
mpantvuhcFTtalc
aptcap
ig. 23. Some products observed after oxidative cleavage of the lignin model compf certain electron-deficient ferric porphine complexes.
agnesium monoperoxyphthalate favors the formation of the-quinone derivative arising from the action of the CpdI analognd subsequent oxygen transfer (rebound mechanism, as alreadyoted above) from the CpdII analog and the substrate radical. Inhe case of H2O2, when working in aprotic environment, onlyeratraldehyde and no quinones were found as the oxidation prod-cts. Evidently, the Cpd0 analog under such conditions underwentomolytic cleavage with direct generation of a CpdII analog, whichould in turn extract one electron from the substrate. The arisinge(III)-porphine obviously could not promote any oxygen transfer.he applicative interest of aprotic conditions for potential indus-rial large-scale delignification processes is low due to economicalnd environmental issues; however, these studies help sheddingight on the intimate mechanisms of redox-active metalloporphineatalysis.
As expected, the strongly electron-deficient �-tetrasulfoniccid derivative of Fe(III)-TFPP showed the highest tendency toerform the oxygen transfer, and so the quinone derivative of
he substrate is almost the sole detectable product. A potentialonsequence of the described behavior is that quinonization of theromatic rings in lignins to a certain extent wastes the oxidizingower of the oxygen donor under Fe-porphine catalysis; under1-(3′ ,4′-dimethoxyphenyl)-2-phenyl-1-propanol by some oxidants in the presence
these circumstances, lignins are oxidized (quinonized) rather thancleaved at their C� C� linkages.
The group of Crestini has worked on the metalloporphine-promoted oxidative degradation of Kraft lignin and somenonphenolic selected lignin model compounds, bearing the5,5′-diphenyl or the 5,5′-diphenylmethane structural motifs. Theyfound that both anionic and cationic, water-soluble Fe- and Mn-porphines were all able to perform oxidative attack of the studiedsubstrates, using hydrogen peroxide as the oxidant [332]. Inparticular, meso-tetrakis(N-methyl-4-pyridinio)porphine-Mn(III)
was the most active among the studied catalysts. Moreover,Fe-porphines degraded lignin but formed noticeable amounts ofhigh molecular weight condensation product, while Mn-porphinesdid not promoted condensation reactions at all. Such conden-sation reactions could be related to the well known tendencyof Fe-porphines to attack methoxy-substituted aromatic ringsleading to quinone derivatives [180], whereas Mn-porphines tendto favor C� C� cleavage while leaving the dimethoxyphenyl
ring untouched. Although quinonization of aromatic rings inlignin could prepare them to irreversible ring cleavage (for exam-ple by excess hydrogen peroxide in alkaline buffers), thereforeincreasing solubility and biodegradability, the quinones arising
P. Zucca et al. / Journal of Molecular Catalysis A: Chemical 388–389 (2014) 2–34 29
Fig. 24. The oxidative cleavage of apocynol and of its nonphenolic counterpart in the presence of meso-tetrakis(N-methyl-4-pyridinio)porphine-Mn(III) , immobilized withinmontmorillonite.
Fig. 25. The complicated oxidation/cleavage pattern of two model dilignols, differing for their phenolic or respectively nonphenolic character.

3 atalys
fctirnc
mm(adsttto
tsouspCiihl
lwatTemmMparMrmofirtc5pctiw
bpt[
sma
0 P. Zucca et al. / Journal of Molecular C
rom the action of Fe-porphines in turn would be responsible forondensation and/or addition reactions. This difference in oxida-ive behavior between Fe- and Mn-porphines is of the highestmportance when envisaging an industrial application of theseedox-active catalysts. Generally speaking, the more stable ando-quinone-producing Mn-porphines would be the candidates ofhoice to perform industrial delignification.
The same authors also tested the ability of meso-tetrakis(N-ethyl-4-pyridinio)porphine-Mn(III), immobilized within mont-orillonite, to promote oxidative degradation of apocynol
�-(4-hydroxy-3-methoxyphenyl)ethanol) and of its methylethert the phenolic hydroxyl [342]. The tested substrate was oxi-ized by hydrogen peroxide in the presence of the catalyst:idechain oxidation and/or cleavage were observed in the case ofhe phenolic substrate, whereas only oxidation of the � hydroxylo the carbonyl function took place with its nonphenolic coun-erpart (Fig. 24). Somewhat surprisingly, no quinonization wasbserved.
Two dilignols, differing only for the presence or absence ofhe free phenolic hydroxyl, were also tested as substrates for theame catalytic system. Interestingly, a sharply different pattern ofxidation products arose. In particular, only the phenolic modelnderwent quinonization following demethylation; moreover, nopecies arising from �,� cleavage were found. In contrast, its non-henolic counterpart gave no quinonization products, whereas� C� cleavage took place. Also, a ring hydroxylation product was
dentified (Fig. 25). A similar approach was shown to be effective,n the presence of suitable redox mediators such as veratryl alco-ol or 1-hydroxy-benzotriazole, also for oxidative degradation of
ignin [343].Zucca and colleagues [326] have shown that properly immobi-
ized FeTFPP has a pH optimum around 3 like a LiP-like system,hereas at pH 7 it behaved as a MnP-like system, capable of using
Mn(II)-malonate complex as a redox mediator for the oxida-ive degradation of various phenolic and nonphenolic aromatics.his pH optimum shift of Fe-porphines when they act as MnPmulators is unprecedented, and one could speculate about theolecular reasons of this behavior. In principle, two alternativeechanisms could be envisaged: (a) direct electron transfer fromn(II) to CpdI analog, leading to Mn(III) and CpdII analog, as it hap-
ens in MnP; (b) homolytic cleavage of Cpd0 analog that is favoredt neutral pH values, with direct formation of CpdII analog. Theeleased OH• could well undergo the one electron transfer fromn(II), thus releasing Mn(III) and OH–. Preliminary studies of the
eaction in the presence of hydroxyl radical scavengers suggest aechanism such as (a), excluding the formation and participation
f OH• [326]. This could be of practical interest in industrial deligni-cation, for the destructive action of hydroxyl radicals against theedox metalloporphines is well known. It is also worth noting thathe immobilized FeTFPP described by Zucca and colleagues [326]atalyzed the oxidation of veratryl alcohol to 2-hydroxymethyl--methoxy-1,4-benzoquinone as the main oxidation product: theresence of an axial ligand such as a pyridyl moiety does not appre-iably influence the well known tendency of Fe-porphines to attackhe dimethoxyphenyl ring leading to demethylation and quinon-zation. Veratraldehyde was on the other hand the main product
hen working with immobilized MnTSPP [325].Very recently, an oxidative delignification protocol of sawdust
y hydrogen peroxide in the presence of water-soluble Fe- and Mn-orphines has been proposed, although no quantitative data abouthe efficiency of the delignification process have been presented344].
Overall, unfortunately until now no published study identifieduch an efficiently oxidation/solubilization/depolymerization/ineralization process of lignin allowing its real practical
pplication.
is A: Chemical 388–389 (2014) 2–34
However, on the whole metalloporphines (if compared withother delignification methods, see Table 1) are able to cleave dif-ferent patterns of lignin bonds. In fact, their double nature of bothperoxidase and peroxygenase mimics enables them to promotedegradation reaction that are on the whole more profound thanthat catalyzed by ligninolytic peroxidases, and for example some-times aromatic hydroxylations have been observed, contyributingto increase both water solubility and reactivity of the substrates.In Table 1 a complete summary is reported of the outstandingcatalytic properties of the commonest metalloporphine classesin comparison with the others approaches toward delignification(both chemical and enzymatic).
10. Conclusion and perspectives
Many redox-active metalloporphines have been thoroughlystudied with particular attention to their reaction mechanisms,their substrate specificity, their resistance against oxidative bleach-ing. For a number of sound reasons, such as environmental andsafety issues as well of economical concerns, the massive useof organic solvents in practical industrial applications should beavoided in favor of water or suitable aqueous buffers. Indeed, whenworking in aqueous environment, these intriguing compoundsshow a dual nature of both peroxidases and peroxygenases, andtherefore could carry out both one-electron oxidations and directoxygen transfer reactions. As emphasized in Section 9, this versatilebehavior leads to a widened oxidative degradation potential withrespect to ‘natural’ systems, as certain structures, well representedin lignins, are out of the possible action of ligninolytic peroxidases,whereas the same are efficiently degraded by some redox-activemetalloporphines.
In spite of such favorable premises, no current industriallyrelevant delignification process is based on the use of any metallo-porphine as an oxidative degradation catalyst. This is due mainlyto the insoluble nature of lignin, which requires the use of a solu-ble (ideally, water soluble) catalyst to minimize the mass transferissues preventing any efficient chemical action on this insoluble andrelatively inert substrate. However, as noted above, soluble (ide-ally, again, water soluble) metalloporphines have to be efficientlyseparated from the lignin degradation products, and recoveredto be reused. Possible strong adsorption of the catalysts onto thelignocellulose particles should also be taken into the due consid-eration. This circumstance could lead to both heavy losses of the(costly) catalyst and undesired contamination of the material to bedelignified. In fact, only properly immobilized metalloporphinesin the presence of suitable redox mediators will be in perspectivethe catalysts of choice for eco-friendly delignification processes,provided that a chemically resistant, nontoxic, and inexpensivemolecule is found. Veratryl alcohol is reasonably inexpensive, butsadly it is rapidly converted into veratraldehyde and into a num-ber of quinonoid degradation products, so it should be continuallysupplied, rendering unfeasible the process. N-hydroxy compoundssuch as hydroxybenzotriazole, hydroxysuccinimide, or hydroxyph-thalimide are toxic, and also are unstable enough during their redoxcycle that their use is anyway indefensible. Rather, Mn(II) simplesalts such as sulfate, acetate or up to a certain extent chloride (chlo-rination side reactions possible?) in the presence of dicarboxylicacids or hydroxyacids could represent a very promising redoxmediator system, joining together a relatively harmless nature, avery low price, and high efficiency at mild pH values. The unavoid-ably arising insoluble MnO2 could be easily recycled to soluble
Mn(II) salts upon treatment with excess H2O2 under mild acidicconditions. Also, exploiting density differences or, better, usingmagnetic supports could achieve the separation between the woodpulp and the immobilized catalyst. Alternatively, sharply anionic
atalys
opotmpftbtilu
ppps
R
P. Zucca et al. / Journal of Molecular C
r cationic metalloporphines could be used profitably in solution,rovided that they could easily recovered and recycled by meansf ionic exchange techniques. Since many products, arising fromhe oxidative degradation of lignins are carboxylic acids, cationic
etalloporphines should be reasonably the catalyst of choice forractical applications, as they could be very selectively recoveredrom the solutions by means of cation-exchange procedures. Onhe other hand, one should consider that lignocellulosics tend toecome more and more acidic (anionic) as their oxidation proceeds;his could render the catalyst recovery a very hard task due to ionicnteractions. However, the question about the need of proper axialigands, required to stabilize and activate the catalysts, remainsnanswered.
In conclusion, still much work is needed to build up industrialrocesses based on oxidative delignification under metallopor-hine catalysis. Nevertheless, the enormous interest in suchrocesses will certainly drive many efforts to this desired goal untiluccessful achievements.
eferences
[1] A.M. Boudet, J. Grima-Pettenati, Mol. Breed. 2 (1996) 25.[2] A.T.W.M. Hendriks, G. Zeeman, Bioresource Technol. 100 (2009) 10.[3] Y. Sun, J. Cheng, Bioresource Technol. 83 (2002) 1.[4] L. Ayed, N. Assas, S. Sayadi, M. Hamdi, Lett. Appl. Microbiol. 40 (2005) 7.[5] M. Hamdi, Bioprocess Eng. 8 (1993) 208.[6] P. Zucca, C. Vinci, F. Sollai, A. Rescigno, E. Sanjust, J. Mol. Catal. A: Chem. 288
(2008) 97.[7] P. Zucca, C. Vinci, A. Rescigno, E. Dumitriu, E. Sanjust, J. Mol. Catal. A: Chem.
321 (2010) 27.[8] P. Zucca, A. Rescigno, M. Pintus, A.C. Rinaldi, E. Sanjust, Chem. Central J. 6
(2012).[9] A.M. Anterola, N.G. Lewis, Phytochemistry 61 (2002) 221.
[10] L. Jones, A.R. Ennos, S.R. Turner, Plant J. 26 (2001) 205.[11] K.V. Sarkanen, C.H. Ludwig, Lignins: Occurence, Formation, Structure, and
Reactions, Wiley Interscience, New York, 1971.[12] L.A. Donaldson, Phytochemistry 57 (2001) 859.[13] S. Saka, D.A.I. Goring, in: T. Higuchi (Ed.), Biosynthesis and Biodegradation of
Wood Components, Academic Press, Orlando, 1985, p. 51.[14] V.L. Chiang, Environ. Chem. Lett. 4 (2006) 143.[15] T. Umezawa, M. Shimada, T. Higuchi, K. Kusai, FEBS Lett. 205 (1986) 287.[16] H. Onnerud, L. Zhang, G. Gellerstedt, G. Henriksson, Plant Cell 14 (2002) 1953.[17] M. Elfstrand, F. Sitbon, C. Lapierre, A. Bottin, S. Von Arnold, Planta 214 (2002)
708.[18] P. Ranocha, M. Chabannes, S. Chamayou, S. Danoun, A. Jauneau, A.M. Boudet,
D. Goffner, Plant Physiol. 129 (2002) 145.[19] W. Boerjan, J. Ralph, M. Baucher, Annu. Rev. Plant Biol. 54 (2003) 519.[20] K. Fukushima, J. Plant Res. 114 (2001) 499.[21] J. Ralph, Phytochem. Rev. 9 (2010) 65.[22] H. Wei, Q. Xu, L.E. Taylor Ii, J.O. Baker, M.P. Tucker, S.Y. Ding, Curr. Opin.
Biotechnol. 20 (2009) 330.[23] D.V. Evtuguin, C.P. Neto, A.M.S. Silva, P.M. Domingues, F.M.L. Amado, D.
Robert, O. Faix, J. Agric. Food Chem. 49 (2001) 4252.[24] B.J. Zobel, J.P. van Buijtenen, Wood Variation. Its Causes and Control, Springer-
Verlag, Heidelberg, 1989.[25] K. Bauer, D. Garbe, Common Fragrance and Flavor Materials, VCH, Weinheim,
1985.[26] D.W.S. Wong, Appl. Biochem. Biotechnol. 157 (2009) 174.[27] R. Hatfield, J. Ralph, J.H. Grabber, Planta 228 (2008) 919.[28] E. Adler, Wood Sci. Technol. 11 (1977) 169.[29] K. Syrjanen, G. Brunow, J. Chem. Soc. Perkin Trans. 1 (2000) 183.[30] J.H. Grabber, Crop Sci. 45 (2005) 820.[31] Y. Matsushita, T. Sekiguchi, R. Ichino, K. Fukushima, J. Wood Sci. 55 (2009)
344.[32] L. Zhang, G. Gellerstedt, Chem. Commun. (2001) 2744.[33] J. Ralph, K. Lundquist, G. Brunow, F. Lu, H. Kim, P.F. Schatz, J.M. Marita, R.D.
Hatfield, S.A. Ralph, J.H. Christensen, W. Boerjan, Phytochem. Rev. 3 (2004)29.
[34] A. Holmgren, M. Norgren, L. Zhang, G. Henriksson, Phytochemistry 70 (2009)147.
[35] R. Sun, J.M. Lawther, W.B. Banks, Ind. Crops Prod. 6 (1997) 1.[36] H. Trajano, N. Engle, M. Foston, A. Ragauskas, T. Tschaplinski, C. Wyman,
Biotechnol. Biofuels 6 (2013) 110.
[37] L. Hemra, K. Lundquist, Acta Chem. Scand. 27 (1973) 365.[38] M. Kihara, M. Takayama, H. Wariishi, H. Tanaka, Spectrochim. Acta – Part A:Mol. Biomol. Spectrosc. 58 (2002) 2213.[39] J. Zakzeski, P.C.A. Bruijnincx, A.L. Jongerius, B.M. Weckhuysen, Chem. Rev. 110
(2010) 3552.[40] F.S. Chakar, A.J. Ragauskas, Ind. Crops Prod. 20 (2004) 131.
is A: Chemical 388–389 (2014) 2–34 31
[41] V. Burlat, M. Kwon, L.B. Davin, N.G. Lewis, Phytochemistry 57 (2001) 883.[42] L.B. Davin, N.G. Lewis, Curr. Opin. Biotechnol. 16 (2005) 398.[43] Y.-R. Chen, S. Sarkanen, Phytochemistry 71 (2010) 453.[44] N. Lourith, T. Katayama, T. Suzuki, J. Wood Sci. 51 (2005) 370.[45] R. Hatfield, W. Vermerris, Plant Physiol. 126 (2001) 1351.[46] D.V. Evtuguin, F.M.L. Amado, Macromol. Biosci. 3 (2003) 339.[47] R. Vanholme, K. Morreel, J. Ralph, W. Boerjan, Curr. Opin. Plant Biol. 11 (2008)
278.[48] N.D. Bonawitz, C. Chapple, Curr. Opin. Biotechnol. 24 (2013) 336.[49] B. Monties, A.W. Willis, T.K. Scott, Methods in Enzymology, Academic Press,
1988, pp. 31.[50] R. El Hage, N. Brosse, L. Chrusciel, C. Sanchez, P. Sannigrahi, A. Ragauskas,
Polym. Degradation Stab. 94 (2009) 1632.[51] K.V. Sarkanen, in: K.V. Sarkanen, D.A. Tillman (Eds.), Progress in Biomass
Conversion, Academic Press, New York, 1980, p. 127.[52] A.L. Macfarlane, R. Prestidge, M.M. Farid, J.J.J. Chen, Chem. Eng. J. 148 (2009)
15.[53] T. Yokoyama, Y. Matsumoto, Holzforschung 62 (2008) 164.[54] J. Gierer, Wood Sci. Technol. 19 (1985) 289.[55] J. Gierer, Wood Sci. Technol. 20 (1986) 1.[56] G.J. Kubes, B.I. Fleming, J.M. MacLeod, H.I. Bolker, Wood Sci. Technol. 14 (1980)
207.[57] J.R.G. Bryce, in: J.P. Casey (Ed.), Pulp and Paper: Chemistry and Chemical
Technology, John Wiley & Sons, New York, 1980, p. 291.[58] Y.J. Lee, C.H. Chung, D.F. Day, Bioresource Technol. 100 (2009) 935.[59] G. Gellerstedt, A. Majtnerova, L. Zhang, C. R. Biol. 327 (2004) 817.[60] K. Kratzl, P. Claus, W. Lonsky, J.S. Gratzl, Wood Sci. Technol. 8 (1974) 35.[61] A.P. Marques, D.V. Evtuguin, S. Magina, F.M.L. Amado, A. Prates, J. Wood Chem.
Technol. 29 (2009) 337.[62] G. Gellersted, J. Gierer, Acta Chem. Scand. 24 (1970) 1645.[63] G. Gellersted, J. Gierer, Acta Chem. Scand. 31B (1977) 729.[64] S.E. Lebo, J.D. Gargulak, T.J. McNally, Kirk-Othmer Encyclopedia Chem. Tech-
nol. (2001) 1.[65] P. Axegard, L. Renberg, Chemosphere 19 (1989) 661.[66] C.A. Hubbell, A.J. Ragauskas, Bioresource Technol. 101 (2010) 7410.[67] R. Kumar, G. Mago, V. Balan, C.E. Wyman, Bioresource Technol. 100 (2009)
3948.[68] J.J. Kolar, B.O. Lindgren, B. Pettersson, Wood Sci. Technol. 17 (1983) 117.[69] T. Lehtimaa, V. Tarvo, S. Kuitunen, A.S. Jääskeläinen, T. Vuorinen, J. Wood
Chem. Technol. 30 (2010) 19.[70] T. Lehtimaa, V. Tarvo, S. Kuitunen, A.S. Jääskeläinen, T. Vuorinen, J. Wood
Chem. Technol. 30 (2010) 1.[71] J. Quesada, F. Teffo-Bertaud, J.P. Croué, M. Rubio, Holzforschung 56 (2002)
32.[72] T. Kreetachat, M. Damrongsri, V. Punsuwon, P. Vaithanomsat, C. Chiemchaisri,
C. Chomsurin, J. Hazard. Mater. 142 (2007) 250.[73] R. Sun, J. Tomkinson, W. Zhu, S.Q. Wang, J. Agric. Food Chem. 48 (2000)
1253.[74] R.C. Sun, J.M. Fang, J. Tomkinson, Ind. Crops Prod. 12 (2000) 71.[75] D. Haverty, K. Dussan, A.V. Piterina, J.J. Leahy, M.H.B. Hayes, Bioresource Tech-
nol. 109 (2012) 173.[76] R. Agnemo, G. Gellersted, Acta Chem. Scand. 33B (1979) 337.[77] R. Agnemo, G. Gellersted, E.-V. Lindfors, Acta Chem. Scand. 33B (1979) 154.[78] G. Gellersted, H.-L. Hardell, E.-V. Lindfors, Acta Chem. Scand. 34B (1980)
669.[79] G. Gellersted, I. Petterson, J. Wood Chem. Technol. 2 (1982) 231.[80] F. Lopez, M.J. Diaz, M.E. Eugenio, J. Ariza, A. Rodriguez, L. Jimenez, Bioresource
Technol. 87 (2003) 255.[81] C.S.R. Freire, A.J.D. Silvestre, C. Pascoal Neto, D.V. Evtuguin, Bioresource Tech-
nol. 97 (2006) 420.[82] N. Curreli, M. Agelli, B. Pisu, A. Rescigno, E. Sanjust, A. Rinaldi, Process Biochem.
37 (2002) 937.[83] R.C. Sun, X.F. Sun, P. Fowler, J. Tomkinson, Eur. Polym. J. 38 (2002) 1399.[84] X.F. Sun, F. Xu, H. Zhao, R.C. Sun, P. Fowler, M.S. Baird, Bioresource Technol.
96 (2005) 1342.[85] Y. Yamashita, M. Shono, C. Sasaki, Y. Nakamura, Carbohydr. Polym. 79 (2010)
914.[86] Z. Li, C. Chen, E. Hegg, D. Hodge, Biotechnol. Biofuels 6 (2013) 119.[87] S. Omori, C.W. Dence, Wood Sci. Technol. 15 (1981) 113.[88] S. Omori, C.W. Dence, Wood Sci. Technol. 15 (1981) 67.[89] G. Gellersted, R. Agnemo, Acta Chem. Scand. 34B (1980) 275.[90] D.G. Mancosky, W. Ban, L.A. Lucia, Ind. Eng. Chem. Res. 44 (2005) 1652.[91] K.-E.L. Eriksson, R.A. Blanchette, P. Ander, Microbial and Enzymatic Degrada-
tion of Wood and Wood Components, Springer-Verlag, Berlin, 1990.[92] A. Leonowicz, A. Matuszewska, J. Luterek, D. Ziegenhagen, M. Wojtas-
Wasilewska, N.S. Cho, M. Hofrichter, J. Rogalski, Fungal Genet. Biol. 27 (1999)175.
[93] A. Hatakka, FEMS Microbiol. Rev. 13 (1994) 125.[94] G. Sena-Martins, E. Almeida-Vara, J.C. Duarte, Ind. Crops Prod. 27 (2008) 189.[95] P. Singh, O. Sulaiman, R. Hashim, P. Rupani, L.C. Peng, Rev. Environ. Sci.
Biotechnol. 9 (2010) 141.
[96] S. Riva, Trends Biotechnol. 24 (2006) 219.[97] E.D. Babot, A. Rico, J. Rencoret, L. Kalum, H. Lund, J. Romero, J.C. del, R. Ão, T.Ã.Martinez, A. Gutierrez, Bioresource Technol. 102 (2011) 6717.[98] S. Camarero, D. Ibarra, A.T. Martinez, J. Romero, A. Gutierrez, J.C. del Rio,
Enzyme Microb. Technol. 40 (2007) 1264.
3 atalys
2 P. Zucca et al. / Journal of Molecular C[99] P. Zucca, A. Rescigno, A. Olianas, S. Maccioni, F. Sollai, E. Sanjust, J. Mol. Catal.B: Enzym. 68 (2011) 216.
[100] C. Eggert, U. Temp, K.E.L. Eriksson, FEBS Lett. 407 (1997) 89.[101] P. Bajpai, Biotechnol. Prog. 15 (1999) 147.[102] A.I. Canas, S. Camarero, Biotechnol. Adv. 28 (2010) 694.[103] D. Singh Arora, R. Kumar Sharma, Appl. Biochem. Biotechnol. 160 (2010) 1760.[104] E. Nugroho Prasetyo, T. Kudanga, L. Ostergaard, J. Rencoret, A. Gutierrez, J.C.
del Rio, J. Ignacio Santos, L. Nieto, J. Jimenez-Barbero, A.T. Martinez, J. Li,G. Gellerstedt, S. Lepifre, C. Silva, S.Y. Kim, A. Cavaco-Paulo, B. SeljebakkenKlausen, B.F. Lutnaes, G.S. Nyanhongo, G.M. Guebitz, Bioresource Technol.101 (2010) 5054.
[105] M. Lavazza, C. Formantici, V. Langella, D. Monti, U. Pfeiffer, Y.M. Galante, J.Biotechnol. 156 (2011) 108.
[106] D.L. Harris, Curr. Opin. Chem. Biol. 5 (2001) 724.[107] J. Everse, Free Radic. Biol. Med. 24 (1998) 1338.[108] S. Nagano, M. Tanaka, Y. Watanabe, I. Morishima, Biochem. Biophys. Res.
Commun. 207 (1995) 417.[109] A.T. Smith, N.C. Veitch, Curr. Opin. Chem. Biol. 2 (1998) 269.[110] R. Silaghi-Dumitrescu, J. Biol. Inorg. Chem. 9 (2004) 471.[111] M.T. Green, J. Am. Chem. Soc. 128 (2006) 1902.[112] H. Wariishi, M.H. Gold, J. Biol. Chem. 265 (1990) 2070.[113] D. Koua, L. Cerutti, L. Falquet, C.J.A. Sigrist, G. Theiler, N. Hulo, C. Dunand,
Nucleic Acids Res. 37 (2009) D261.[114] L. Banci, I. Bertini, P. Turano, M. Tien, T.K. Kirk, Proc. Natl. Acad. Sci. U.S.A. 88
(1991) 6956.[115] C. Srinivasan, T.M. D’Souza, K. Boominathan, C.A. Reddy, Appl. Environ. Micro-
biol. 61 (1995) 4274.[116] J.K. Glenn, M.A. Morgan, M.B. Mayfield, M. Kuwahara, M.H. Gold, Biochem.
Biophys. Res. Commun. 114 (1983) 1077.[117] M. Tien, T. Kent Kirk, Science 221 (1983) 661.[118] M. Kuwahara, J.K. Glenn, M.A. Morgan, M.H. Gold, FEBS Lett. 169 (1984)
247.[119] P.J. Kersten, M. Tien, B. Kalyanaraman, K. Kirk, J. Biol. Chem. 260 (1985) 2609.[120] M. Saritha, A. Arora, L. Nain, Bioresource Technol. 104 (2012) 459.[121] K. Pakshirajan, S. Jaiswal, R.K. Das, J. Sci. Ind. Res. 70 (2011) 987.[122] S.Y. Mirzaakhmedov, Z.F. Ziyavitdinov, Z.R. Akhmedova, A.B. Saliev, D.T.
Ruzmetova, S.T. Azizov, D. Fessas, S. Iametti, Chem. Nat. Compd. 43 (2007)682.
[123] F. Vahabzadeh, A. Mogharei, M. Mehranian, Iran. J. Chem. Chem. Eng. 21 (2002)126.
[124] J. Fu, X. Li, W. Gao, H. Wang, A. Cavaco-Paulo, C. Silva, Biocatal. Biotransform.30 (2012) 141.
[125] W.A. Doyle, W. Blodig, N.C. Veitch, K. Piontek, A.T. Smith, Biochemistry 37(1998) 15097.
[126] M. Mylrajan, K. Valli, H. Wariishi, M.H. Gold, T.M. Loehr, Biochemistry 29(1990) 9617.
[127] B. Valderrama, M. Ayala, R. Vazquez-Duhalt, Chem. Biol. 9 (2002) 555.[128] K.E. Hammel, B. Kalyanaraman, T.K. Kirk, Proc. Natl. Acad. Sci. U.S.A. 83 (1986)
3708.[129] K.E. Hammel, D. Cullen, Curr. Opin. Plant Biol. 11 (2008) 349.[130] K.E. Hammel, K.A. Jensen Jr., M.D. Mozuch, L.L. Landucci, M. Tien, E.A. Pease,
J. Biol. Chem. 268 (1993) 12274.[131] M.G.S. Chua, C.L. Chen, T.K. Kirk, Holzforschung 36 (1982) 165.[132] K. Miki, V. Renganathan, M.B. Mayfield, M.H. Gold, FEBS Lett. 210 (1987) 199.[133] K.E. Hammel, FEBS Lett. 354 (1994) 297.[134] E. Baciocchi, C. Fabbri, O. Lanzalunga, J. Org. Chem. 68 (2003) 9061.[135] R. ten Have, P.J.M. Teunissen, Chem. Rev. 101 (2001) 3397.[136] L. Banci, S. Ciofi-Baffoni, M. Tien, Biochemistry 38 (1999) 3205.[137] M. Tien, K. Kirk, Proc. Natl. Acad. Sci. U.S.A. 81 (1984) 2280.[138] P.J. Harvey, H.E. Schoemaker, J.M. Palmer, FEBS Lett. 195 (1986) 242.[139] T. Johjima, N. Itoh, M. Kabuto, F. Tokimura, T. Nakagawa, H. Wariishi, H.
Tanaka, Proc. Natl. Acad. Sci. U.S.A. 96 (1999) 1989.[140] K. Piontek, A.T. Smith, W. Blodig, Biochem. Soc. Trans. 29 (2001) 111.[141] G. Ward, Y. Hadar, C.G. Dosoretz, Enzyme Microb. Technol. 29 (2001) 34.[142] M. Hofrichter, Enzyme Microb. Technol. 30 (2002) 454.[143] K. Kishi, H. Wariishi, L. Marquez, H.B. Dunford, M.H. Gold, Biochemistry 33
(1994) 8694.[144] H. Wariishi, L. Akileswaran, M.H. Gold, Biochemistry 27 (1988) 5365.[145] A. Hatakka, in: M. Hofrichter, A. Steinbuchel (Eds.), Biopolymers. Vol. 1-Lignin,
Humic Substances and Coal, Wiley-VCH, Weinheim, Germany, 2001, p. 129.[146] E. Srebotnik, K. Messner, R. Foisner, Appl. Environ. Microbiol. 54 (1988) 2608.[147] J.L. Popp, B. Kalyanaraman, T. Kent Kirk, Biochemistry 29 (1990) 10475.[148] M. Enoki, T. Watanabe, S. Nakagame, K. Koller, K. Messner, Y. Honda, M.
Kuwahara, FEMS Microbiol. Lett. 180 (1999) 205.[149] W. Fritsche, K. Scheibner, A. Herre, M. Hofrichter, in: J.C. Spain, J.B. Hughes,
H.J. Knackmuss (Eds.), Nitroaromatic Compounds and Explosives, Lewis Pub-lishers, Boca Raton, FL, 2000, p. 213.
[150] S. Camarero, S. Sarkar, F.J. Ruiz-Dueñas, M.J. MartÃ-nez, A.T. MartÃ-nez, J.Biol. Chem. 274 (1999) 10324.
[151] M. Perez-Boada, F.J. Ruiz-Duenas, R. Pogni, R. Basosi, T. Choinowski, M.J. Mar-tinez, K. Piontek, A.T. Martinez, J. Mol. Biol. 354 (2005) 385.
[152] H.A. Dau, R. Ullrich, D. Benndorf, A. Svatos, A. Muck, M. Hofrichter, Appl.Environ. Microbiol. 73 (2007) 5477.
[153] M. Kinne, C. Zeisig, R. Ullrich, G. Kayser, K.E. Hammel, M. Hofrichter, Biochem.Biophys. Res. Commun. 397 (2010) 18.
[154] R. Ullrich, C. Dolge, M. Kluge, M. Hofrichter, FEBS Lett. 582 (2008) 4100.
is A: Chemical 388–389 (2014) 2–34
[155] R. Ullrich, J. Nüske, K. Scheibner, J. Spantzel, M. Hofrichter, Appl. Environ.Microbiol. 70 (2004) 4575.
[156] M. Hofrichter, R. Ullrich, M.J. Pecyna, C. Liers, T. Lundell, Appl. Microbiol.Biotechnol. 87 (2010) 871.
[157] A. Yarman, G. Gröbe, B. Neumann, M. Kinne, N. Gajovic-Eichelmann, U. Wol-lenberger, M. Hofrichter, R. Ullrich, K. Scheibner, F.W. Scheller, Anal. Bioanal.Chem. 402 (2012) 405.
[158] B.P. Roy, M.G. Paice, F.S. Archibald, S.K. Misra, L.E. Misiak, J. Biol. Chem. 269(1994) 19745.
[159] V. Gomez-Toribio, A.B. Garcia-Martìn, M.J. Martìnez, Ã.T. Martìnez, F. Guillén,Appl. Environ. Microbiol 75 (2009) 3944.
[160] J.D. Stahl, S.J. Rasmussen, S.D. Aust, Arch. Biochem. Biophys. 322 (1995) 221.[161] N. Curreli, A. Rescigno, A. Rinaldi, B. Pisu, F. Sollai, E. Sanjust, Mycol. Res. 108
(2004) 913.[162] The Merck Index, 12th edition, Merck and Co. Inc., Whitehouse Station, NJ,
1996.[163] M. Gouterman, The Porphyrins, Academic Press, New York, 1978, pp. 1.[164] K.S. Suslick, R.A. Watson, New J. Chem. 16 (1992) 633.[165] R. Weiss, V. Bulach, A. Gold, J. Terner, A.X. Trautwein, J. Biol. Inorg. Chem. 6
(2001) 831.[166] Y.S. Kim, R. Song, D.H. Kim, M.J. Jun, Y.S. Sohn, Bioorg. Med. Chem. 11 (2003)
1753.[167] W. Zhou, B. Hu, Z. Liu, Appl. Catal. A: Gen. 358 (2009) 136.[168] J.T. Groves, T.E. Nemo, R.S. Myers, J. Am. Chem. Soc. 101 (1979) 1032.[169] J.T. Groves, J. Porphyrins Phthalocyanines 4 (2000) 350.[170] C.M. Dicken, T.C. Woon, T.C. Bruice, J. Am. Chem. Soc. 108 (1986) 1636.[171] P.R.O. De Montellano, Acc. Chem. Res. 20 (1987) 289.[172] D. Dolphin, T.G. Traylor, L.Y. Xie, Acc. Chem. Res. 30 (1997) 251.[173] D. Ostovic, T.C. Bruice, Acc. Chem. Res. 25 (1992) 314.[174] C.K. Chang, M.S. Kuo, J. Am. Chem. Soc. 101 (1979) 3412.[175] G. Díaz-Díaz, M.C. Blanco-López, M.J. Lobo-Castanón, A.J. Miranda-Ordieres,
P. Tunón-Blanco, J. Mol. Catal. A: Chem. 353–354 (2012) 117.[176] G. Díaz-Díaz, M. Celis-García, M.C. Blanco-López, M.J. Lobo-Castanón, A.J.
Miranda-Ordieres, P. Tunón-Blanco, Appl. Catal. B: Environ. 96 (2010) 51.[177] D. Mansuy, C. R. Chim. 10 (2007) 392.[178] B. Meunier, Chem. Rev. 92 (1992) 1411.[179] Y.M. Goh, W. Nam, Inorg. Chem. 38 (1999) 914.[180] I. Artaud, K. Ben-Aziza, D. Mansuy, J. Org. Chem. 58 (1993) 3373.[181] A.M.A. Rocha-Gonsalves, M.M. Pereira, J. Mol. Catal. A: Chem. 113 (1996) 209.[182] M.H. Liu, Y.O. Su, J. Chem. Soc. Chem. Commun. (1994) 971.[183] S. Zakavi, A.G. Mojarrad, S. Rayati, J. Mol. Catal. A: Chem. 363–364 (2012)
153.[184] K.M. Kadish, C. Araullo-McAdams, B.C. Han, M.M. Franzen, J. Am. Chem. Soc.
112 (1990) 8364.[185] A.M. Rocha Gonsalves D’A, R.A.W. Johnstone, M.M. Pereira, J. Shaw, A.J.F.N.
Sobral do, Tetrahedron Lett. 32 (1991) 1355.[186] S. Tsuchiya, M. Seno, Chem. Lett. 18 (1989) 263.[187] J.F. Bartoli, P. Battioni, W.R. De Foor, D. Mansuy, J. Chem. Soc. Chem. Commun.
(1994) 23.[188] M.F. Zipplies, W.A. Lee, T.C. Bruice, J. Am. Chem. Soc. 108 (1986) 4433.[189] Ö. Almarsson, T.C. Bruice, J. Am. Chem. Soc. 117 (1995) 4533.[190] S.M.S. Chauhan, B.B. Sahoo, Bioorg. Med. Chem. 7 (1999) 2629.[191] I. Artaud, K.B. Aziza, C. Chopard, D. Mansuy, J. Chem. Soc. Chem. Commun.
(1991) 31.[192] J.L. McLain, J. Lee, J.T. Groves, in: B. Meunier (Ed.), Biomimentic Oxidations
Catalyzed by Transition Metal Complexes, Imperial College Press, London,2000, p. 91.
[193] B. Meunier, S.P. de Visser, S. Shaik, Chem. Rev. 104 (2004) 3947.[194] E.G. Hrycay, S.M. Bandiera, Arch. Biochem. Biophys. 522 (2012) 71.[195] C. Fabbri, C. Aurisicchio, O. Lanzalunga, Central Eur. J. Chem. 6 (2008) 145.[196] D. Kumar, B. Karamzadeh, G.N. Sastry, S.P. De Visser, J. Am. Chem. Soc. 132
(2010) 7656.[197] B. Fontaine, A. Nuzzo, R. Spaccini, A. Piccolo, J. Geochem. Exploration 129
(2012).[198] F. Sannino, R. Spaccini, D. Savy, A. Piccolo, J. Hazard. Mater. 261C (2013) 55.[199] A.W. Munro, H.M. Girvan, K.J. McLean, Nat. Prod. Rep. 24 (2007) 585.[200] X. Wang, S. Peter, M. Kinne, M. Hofrichter, J.T. Groves, J. Am. Chem. Soc. 134
(2012) 12897.[201] Ö. Almarsson, H. Adalsteinsson, T.C. Bruice, J. Am. Chem. Soc. 117 (1995) 4524.[202] Y. Kokubo, X.W. Wu, Y. Oshima, S. Koda, J. Supercrit. Fluids 30 (2004) 225.[203] A. Aggarwal, S. Singh, J. Samson, C.M. Drain, Macromol. Rapid Commun. 33
(2012) 1220.[204] Y. Goto, T. Matsui, S.I. Ozaki, Y. Watanabe, S. Fukuzumi, J. Am. Chem. Soc. 121
(1999) 9497.[205] F. Van Rantwijk, R.A. Sheldon, Curr. Opin. Biotechnol. 11 (2000) 554.[206] J.T. Groves, Y.-Z. Han, in: P.R.O. De Montellano (Ed.), Cytochrome P450: Struc-
ture, Mechanism, and Biochemistry, Plenum Press, New York, 1995.[207] K. Auclair, Z. Hu, D.M. Little, P.R. Ortiz de Montellano, J.T. Groves, J. Am. Chem.
Soc. 124 (2002) 6020.[208] P.S. Traylor, D. Dolphin, T.G. Traylor, J. Chem. Soc. Chem. Commun. (1984) 279.[209] A. Brausam, S. Eigler, N. Jux, R. Van Eldik, Inorg. Chem. 48 (2009) 7667.
[210] H.K. Back, H.E. Van Wart, Biochemistry 28 (1989) 5714.[211] K. Kamaraj, D. Bandyopadhyay, J. Am. Chem. Soc. 119 (1997) 8099.[212] K. Kühnel, E. Derat, J. Terner, S. Shaik, I. Schlichting, Proc. Natl. Acad. Sci. U.S.A.104 (2007) 99.[213] T.C. Bruice, Acc. Chem. Res. 24 (1991) 243.
atalys
[
[[[[[
[
[
[[[
[
[
[[[[
[
[[[[[[
[[[[
[[[[
[
[[
[[[[
[
[[
[[
[[
[[[
[
[
[[[[
[[[[
[
P. Zucca et al. / Journal of Molecular C
214] S.L.H. Rebelo, M.M. Pereira, M.M.Q. SimoÌfes, M.G.P.M.S. Neves, J.A.S. Cav-aleiro, J. Catal. 234 (2005) 76.
215] S.P. De Visser, J. Am. Chem. Soc. 132 (2010) 1087.216] G.X. He, T.C. Bruice, J. Am. Chem. Soc. 113 (1991) 2747.217] S.J. Yang, W. Nam, Inorg. Chem. 37 (1998) 606.218] T.G. Traylor, C. Kim, W.P. Fann, C.L. Perrin, Tetrahedron 54 (1998) 7977.219] P.N. Balasubramanian, J.R. Lindsay Smith, M.J. Davies, T.W. Kaaret, T.C. Bruice,
J. Am. Chem. Soc. 111 (1989) 1477.220] A. Agarwala, D. Bandyopadhyay, Chem. Commun. (Cambridge) (2006)
4823.221] A. Singh, A. Agarwala, K. Kamaraj, D. Bandyopadhyay, Inorg. Chim. Acta 372
(2011) 295.222] K.A. Lee, W. Nam, J. Am. Chem. Soc. 119 (1997) 1916.223] J. Bernadou, B. Meunier, Chem. Commun. (1998) 2167.224] W. Nam, S.E. Park, I.K. Lim, M.H. Lim, J. Hong, J. Kim, J. Am. Chem. Soc. 125
(2003) 14674.225] F. Cui, D. Dolphin, T. Wijesekera, R. Farrell, P. Skerer, in: T.K. Kirk, H.M.
Chang (Eds.), Applications of Biotechnology of Pulp and Paper Manufacture,Butterworth-Heinmann, Boston, 1990, p. 491.
226] M.F. Sisemore, M. Selke, J.N. Burstyn, J.S. Valentine, Inorg. Chem. 36 (1997)979.
227] D.L. Wertz, J.S. Valentine, Struct. Bonding 97 (2000) 37.228] S.P. De Visser, J.S. Valentine, W. Nam, Angew. Chem. Int. Ed. 49 (2010) 2099.229] C. Fertinger, A. Franke, R.V. Eldik, J. Biol. Inorg. Chem. 17 (2012) 27.230] P. Battioni, J.P. Renaud, J.F. Bartoli, M. Reina-Artiles, M. Fort, D. Mansuy, J. Am.
Chem. Soc. 110 (1988) 8462.231] J.F. Hull, D. Balcells, E.L.O. Sauer, C. Raynaud, G.W. Brudvig, R.H. Crabtree, O.
Eisenstein, J. Am. Chem. Soc. 132 (2010) 7605.232] Z. Solati, M. Hashemi, L. Ebrahimi, Catal. Lett. 141 (2011) 163.233] N. Jin, D.E. Lahaye, J.T. Groves, Inorg. Chem. 49 (2010) 11516.234] J.T. Groves, Y. Watanabe, Inorg. Chem. 25 (1986) 4808.235] J.T. Groves, J. Lee, S.S. Marla, J. Am. Chem. Soc. 119 (1997) 6269.236] A. Ghosh, P.R. Taylor, Curr. Opin. Chem. Biol. 7 (2003) 113.237] I.D. Cunningham, T.N. Danks, J.N. Hay, I. Hamerton, S. Gunathilagan, C. Janczak,
J. Mol. Catal. A: Chem. 185 (2002) 25.238] N. Jin, M. Ibrahim, T.G. Spiro, J.T. Groves, J. Am. Chem. Soc. 129 (2007) 12416.239] N. Jin, J.T. Groves, J. Am. Chem. Soc. 121 (1999) 2923.240] T.P. Umile, D. Wang, J.T. Groves, Inorg. Chem. 50 (2011) 10353.241] A.Q. Lee, B.R. Streit, M.J. Zdilla, M.M. Abu-Omar, J.L. DuBois, Proc. Natl. Acad.
Sci. U.S.A. 105 (2008) 15654.242] M.J. Zdilla, A.Q. Lee, M.M. Abu-Omar, Inorg. Chem. 48 (2009) 2260.243] Y. Ni, G.J. Kubes, A.R.P. van Heiningen, Wood Sci. Technol. 29 (1995) 87.244] Q. Wang, K. Chen, J. Li, J. Xu, S. Liu, Bioresources 6 (2011) 1868.245] R. De Paula, M.M.Q. Simões, M.G.P.M.S. Neves, J.A.S. Cavaleiro, J. Mol. Catal. A:
Chem. 345 (2011) 1.246] R. Latifi, L. Tahsini, B. Karamzadeh, N. Safari, W. Nam, S.P. De Visser, Arch.
Biochem. Biophys. 507 (2011) 4.247] J.T. Groves, M.K. Stern, J. Am. Chem. Soc. 110 (1988) 8628.248] R.S. Czernuszewicz, Y. Oliver Su, K.A. Macor, D. Kirn, J.T. Groves, T.G. Spiro, J.
Chem. Soc. 110 (1988) 4158.249] R.D. Arasasingham, G.X. He, T.C. Bruice, J. Am. Chem. Soc. 115 (1993) 7985.250] J.T. Groves, W.J. Kruper, R.C. Haushalter, J. Am. Chem. Soc. 102 (1980) 6375.251] C.L. Hill, B.C. Schardt, J. Am. Chem. Soc. 102 (1980) 6374.252] A.M.D.A. Rocha Gonsalves, M.M. Pereira, A.C. Serra, R.A.W. Johnstone, M.L.P.G.
Nunes, J. Chem. Soc. Perkin Trans. 1 (1994) 2053.253] B. Meunier, E. Guilmet, M.E.D. Carvalho, R. Poilblanc, J. Am. Chem. Soc. 106
(1984) 6668.254] V. Maraval, J.E. Ancel, B. Meunier, J. Catal. 206 (2002) 349.255] L.B. Chiavetto, G. Guglielmetti, C. Querci, M. Ricci, Tetrahedron Lett. 37 (1996)
1091.256] H. Ohtake, T. Higuchi, M. Hirobe, Heterocycles 40 (1995) 867.257] R. Babakhania, F. Bahadoran, N. Safari, J. Porphyrins Phthalocyanines 11
(2007) 95.258] C.M. Che, J.S. Huang, Chem. Commun. (2009) 3996.259] S.N. Dhuri, S.S. Mi, Y.M. Lee, H. Hirao, Y. Wang, W. Nam, S. Shaik, Angew. Chem.
Int. Ed. 47 (2008) 3356.260] P. Fackler, S.M. Huber, T. Bach, J. Am. Chem. Soc. 134 (2012) 12869.261] J.T. Groves, R. Quinn, J. Am. Chem. Soc. 107 (1985) 5790.262] C. Abebrese, Y. Huang, A. Pan, Z. Yuan, R. Zhang, J. Inorg. Biochem. 105 (2011)
1555.263] E.R. Birnbaum, J.A. Labinger, J.E. Bercaw, H.B. Gray, Inorg. Chim. Acta 270
(1998) 433.264] J.T. Groves, M. Bonchio, T. Carofiglio, K. Shalyaev, J. Am. Chem. Soc. 118 (1996)
8961.265] D. Kumar, G.N. Sastry, S.P. De Visser, Chem. Eur. J. 17 (2011) 6196.266] T.C. Bruice, Ann. N.Y. Acad. Sci. 471 (1986) 83.267] A. Robert, B. Loock, M. Momenteau, B. Meunier, Inorg. Chem. 30 (1991) 706.268] J.P. Renaud, P. Battioni, J.F. Bartoli, D. Mansuy, J. Chem. Soc. Chem. Commun.
(1985) 888.269] A. Takahashi, T. Kurahashi, H. Fujii, Inorg. Chem. 48 (2009) 2614.270] Y. Miyazaki, A. Satake, Y. Kobuke, J. Mol. Catal. A: Chem. 283 (2008) 129.
271] C.L. Hill, J.A. Smegal, T.J. Henly, J. Org. Chem. 48 (1983) 3277.272] A.M.D.A. Rocha Gonsalves, R.A.W. Johnstone, M.M. Pereira, J. Shaw, J. Chem.Soc. Perkin Trans. 1 (1991) 645.273] C.D. Sohl, J. Lee, S.S. Alguindigue, M.A. Khan, G.B. Richter-Addo, J. Inorg.
Biochem. 98 (2004) 1238.
is A: Chemical 388–389 (2014) 2–34 33
[274] A. Rezaeifard, M. Jafarpour, H. Raissi, E. Ghiamati, A. Tootoonchi, Polyhedron30 (2011) 592.
[275] S. Banfi, F. Montanari, G. Pozzi, S. Quici, Tetrahedron 50 (1994) 9025.[276] Y. Cai, Y. Liu, Y. Lu, G. Gao, M. He, Catal. Lett. 124 (2008) 334.[277] D. Kumar, G.N. Sastry, S.P. De Visser, J. Phys. Chem. B 116 (2012) 718.[278] C.M. Che, V.K. Lo, C.Y. Zhou, J.S. Huang, Chem. Soc. Rev. 40 (2011) 1950.[279] A. Rezaeifard, M. Jafarpour, H. Kavousi, M. Alipour, H. Stoeckli-Evans, Polyhe-
dron 30 (2011) 2303.[280] E. Baciocchi, T. Boschi, L. Cassioli, C. Galli, L. Jaquinod, A. Lapi, R. Paolesse, K.M.
Smith, P. Tagliatesta, Eur. J. Org. Chem. (1999) 3281.[281] M. Tavarès, R. Ramasseul, J.C. Marchon, B. Bachet, C. Brassy, J.P. Mornon, J.
Chem. Soc. Perkin Trans. 2 (1992) 1321.[282] J.C. Marchon, R. Ramasseul, J. Chem. Soc. Chem. Commun. (1988) 298.[283] G. Simonneaux, P.L. Maux, Y. Ferrand, J. Rault-Berthelot, Coord. Chem. Rev.
250 (2006) 2212.[284] M.M.Q. Simoes, C.M.B. Neves, S.M.G. Pires, M. Graca, M.S. Neves, J.A.S. Cav-
aleiro, Pure Appl. Chem. 85 (2013) 1671.[285] G.T. Balogh, G.M. Keseru, Arkivoc 2004 (2004) 124.[286] A. Rezaeifard, M. Jafarpour, G.K. Moghaddam, F. Amini, Bioorg. Med. Chem.
15 (2007) 3097.[287] E. Baciocchi, M.F. Gerini, O. Lanzalunga, A. Lapi, M.G. Lo Piparo, Org. Biomol.
Chem. 1 (2003) 422.[288] E. Baciocchi, M.F. Gerini, A. Lapi, J. Org. Chem. 69 (2004) 3586.[289] A. Ghaemi, S. Rayati, S. Zakavi, N. Safari, Appl. Catal. A: Gen. 353 (2009) 154.[290] T.K. Saha, H. Frauendorf, M. John, S. Dechert, F. Meyer, ChemCatChem 5 (2013)
796.[291] F.L. Emmert Iii, J. Thomas, B. Hon, A.J. Gengenbach, Inorg. Chim. Acta 361
(2008) 2243.[292] M. Chivukula, J.T. Spadaro, V. Renganathan, Biochemistry 34 (1995) 7765.[293] W. Liu, J.T. Groves, J. Am. Chem. Soc. 132 (2010) 12847.[294] G. Labat, J.L. Seris, B. Meunier, Angew. Chem. Int. Ed. Engl. 29 (1990) 1471.[295] S. Hasan, Appl. Biochem. Biotechnol. – Part A: Enzyme Eng. Biotechnol. 63–65
(1997) 845.[296] B. Fontaine, A. Piccolo, Environ. Sci. Pollut. Res. 19 (2012) 1485.[297] G.G.A. Balavoine, Y.V. Geletii, D. Bejan, Nitric Oxide – Biol. Chem. 1 (1997) 507.[298] R. Song, A. Robert, J. Bernadou, B. Meunier, Inorg. Chim. Acta 272 (1998) 228.[299] J.R.L. Smith, P.N. Balasubramanian, T.C. Bruice, J. Am. Chem. Soc. 110 (1988)
7411.[300] T. Tatsumi, M. Nakamura, H.o. Tominaga, Catal. Today 6 (1989) 163.[301] M. Trytek, M. Majdan, A. Lipke, J. Fiedurek, J. Catal. 286 (2012) 193.[302] R. Rychtarikova, S. Sabata, J. Hetflejs, G. Kuncova, J. Sol-Gel Sci. Technol. 61
(2012) 119.[303] M. Silva, M.E. Azenha, M.M. Pereira, H.D. Burrows, M. Sarakha, C. Forano, M.F.
Ribeiro, A. Fernandes, Appl. Catal. B: Environ. 100 (2010) 1.[304] M.A. García-Sánchez, V. De La Luz, M.I. Coahuila-Hernández, F. Rojas-
González, S.R. Tello-Solís, A. Campero, J. Photochem. Photobiol. A: Chem. 223(2011) 172.
[305] G. Huang, S.Y. Liu, A.P. Wang, Y.A. Guo, H. Zhou, Catal. Commun. 8 (2007)1183.
[306] M. Kanehisa, S. Asayama, H. Kawakami, Desalination Water Treat. 17 (2010)31.
[307] Z. Tong, T. Shichi, K. Takagi, Mater. Lett. 57 (2003) 2258.[308] P. Battioni, J.P. Lallier, L. Barloy, D. Mansuy, J. Chem. Soc. Chem. Commun.
(1989) 1149.[309] F. Farzaneh, M. Poorkhosravani, M. Ghandi, J. Mol. Catal. A: Chem. 308 (2009)
108.[310] H. Sharghi, M.H. Beyzavi, M.M. Doroodmand, Eur. J. Org. Chem. (2008) 4126.[311] S. Nakagaki, A.R. Ramos, F.L. Benedito, P.G. Peralta-Zamore, A.J.G. Zarbin, J.
Mol. Catal. A: Chem. 185 (2002) 203.[312] K. Nazari, S. Shokrollahzadeh, A. Mahmoudi, F. Mesbahi, N.S. Matin, A.A.
Moosavi-Movahedi, J. Mol. Catal. A: Chem. 239 (2005) 1.[313] A. Molinari, A. Maldotti, A. Bratovcic, G. Magnacca, Catal. Today 161 (2011)
64.[314] S.L.H. Rebelo, A.R. Gonc alves, M.M. Pereira, M.M.Q. Simoes, M.G.P.M.S. Neves,
J.A.S. Cavaleiro, J. Mol. Catal. A: Chem. 256 (2006) 321.[315] J. Nakazawa, B.J. Smith, T.D.P. Stack, J. Am. Chem. Soc. 134 (2012) 2750.[316] M. Fukushima, S. Shigematsu, S. Nagao, Chemosphere 78 (2010) 1155.[317] M. Ghiaci, F. Molaie, M.E. Sedaghat, N. Dorokstar, Catal. Commun. 11 (2010)
694.[318] B. Gao, R. Wang, Y. Zhang, J. Appl. Polym. Sci. 112 (2009) 2764.[319] X. Guo, D.H. Shen, Y.Y. Li, M. Tian, Q. Liu, C.C. Guo, Z.G. Liu, J. Mol. Catal. A:
Chem. 351 (2011) 174.[320] A.L. Faria, T.O.C. Mac Leod, V.P. Barros, M.D. Assis, J. Brazil. Chem. Soc. 20
(2009) 895.[321] O. Leal, D.L. Anderson, R.G. Bowman, F. Basolo, R.L. Burwell Jr., J. Am. Chem.
Soc. 97 (1975) 5125.[322] P.R. Cooke, J.R. Lindsay Smith, Tetrahedron Lett. 33 (1992) 2737.[323] P.R. Cooke, J.R.L. Smith, J. Chem. Soc. Perkin Trans. 1 (1994) 1913.[324] A. Nuzzo, A. Piccolo, J. Mol. Catal. A: Chem. 371 (2013) 8.[325] P. Zucca, G. Mocci, A. Rescigno, E. Sanjust, J. Mol. Catal. A: Chem. 278 (2007)
220.
[326] P. Zucca, F. Sollai, A. Garau, A. Rescigno, E. Sanjust, J. Mol. Catal. A: Chem. 306(2009) 89.[327] P. Zucca, A. Rescigno, E. Sanjust, Chin. J. Catal. 32 (2011) 1663.[328] P.J. Kersten, B. Kalyanaraman, K.E. Hammel, B. Reinhammars, T.K. Kirk,
Biochem. J. 268 (1990) 475.

3 atalys
4 P. Zucca et al. / Journal of Molecular C[329] M. Shimada, T. Habe, T. Higuchi, T. Okamoto, B. Panijpan, Holzforschung 41(1987) 277.
[330] M. Shimada, T. Habe, T. Umezawa, T. Higuchi, T. Okamoto, Biochem. Biophys.Res. Commun. 122 (1984) 1247.
[331] N. Rajapakse, B.R. James, D. Dolphin, in: G. Centi, F. Trifiro (Eds.), New Devel-opments in Selective Oxidation, Elsevier, Amsterdam, 1990.
[332] C. Crestini, R. Saladino, P. Tagliatesta, T. Boschi, Bioorg. Med. Chem. 7 (1999)
1897.[333] M. Tien, T.K. Kirk, C. Bull, J.A. Fee, J. Biol. Chem. 261 (1986) 1687.[334] C. Crestini, P. Tagliatesta, in: R. Guilard, K.M. Kadish, K.M. Smith (Eds.), Hand-
book of Porphyrins and Phtalocyanines II, Elsevier, Amsterdam, 2003.[335] F. Cui, D. Dolphin, Can. J. Biochem. 70 (1992) 2314.
is A: Chemical 388–389 (2014) 2–34
[336] G. Labat, B. Meunier, J. Org. Chem. 54 (1989) 5008.[337] A. Kumar, N. Jain, S.M.S. Chauhan, Synlett (2007) 411.[338] S.D. Haemmerli, H.E. Schoemaker, H.W.H. Schmidt, M.S.A. Leisola, FEBS Lett.
220 (1987) 149.[339] F. Cui, D. Dolphin, Bioorg. Med. Chem. 2 (1994) 735.[340] F. Cui, T. Wijesekera, D. Dolphin, R. Farrell, P. Skerer, J. Biotechnol. 30 (1993)
15.
[341] A. Sakakibara, in: T. Higuchi, H.M. Chang, T.K. Kirk (Eds.), Recent Advances inLignin Biodegradation Research, Uni Publishers, Tokio, 1983, p. 12.[342] C. Crestini, A. Pastorini, P. Tagliatesta, J. Mol. Catal. A: Chem. 208 (2004) 195.[343] C. Crestini, A. Pastorini, P. Tagliatesta, Eur. J. Inorg. Chem. 22 (2004) 4477.[344] A. Barbat, V. Gloaguen, V. Sol, P. Krausz, Bioresource Technol. 101 (2010) 6538.





























