Embed Size (px)
Citation preview

Marine NaturstoffeDOI: 10.1002/ange.200703172
Biologie und Chemie der Zoanthamin-Alkaloide**Douglas C. Behenna, Jennifer L. Stockdill und Brian M. Stoltz*
AngewandteChemie
Stichw�rter:Alkaloide · Biologische Eigenschaften ·Naturstoffe · Polycyclen ·Totalsynthesen
B. M. Stoltz et al.Aufs�tze
2400 www.angewandte.de � 2008 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2008, 120, 2400 – 2421

1. Einf�hrung
Marine Lebensformen sind eine wahre Fundgrube f�r dieIsolierung von Naturstoffen. Im Folgenden werden wir darausdie aus den marinen Zoanthiden isolierten Zoanthamin-Alkaloide vorstellen und deren biologische und chemischeEigenschaften besprechen. Die Ordnung der Zoanthariaumfasst eine komplexe Gruppe mariner Polypen, die anhandihrer Morphologie in mehr als zehn Gattungen zu unterteilenist. Spezies dieser Ordnung (Abbildung 1) finden sich in dengem-ßigten und tropischen K�stenzonen des IndischenOzeans, des Pazifiks und des Atlantiks. Diese pulsierendenWeichkorallen gelten gew1hnlich als aggressive Riffbesiedler,die sich sowohl geschlechtlich fortpflanzen als auch unge-schlechtlich vermehren k1nnen.[1] Durch eine Analyse dermitochondrialen DNA wurden vor wenigen Jahren die Ver-wandtschaftsbeziehungen zwischen diesen Lebensformenaufgekl-rt.[2]
Die Polypen haben einen symme-trischen r1hrenf1rmigen Rumpf, andessen Ende Tentakel angekn�pft
sind, die Nahrung zur zentralen Mund1ffnung f�hren. BeimVerdacht einer Bedrohung ziehen die Polypen ihre Tentakelein, und einige Spezies schießen einen Wasserstrom mitstarken Giften heraus, um Fressfeinde abzuwehren. Bei-spielsweise k1nnen die Zoanthidea, aus denen die Zoantha-mine erhalten wurden, eine stark augenreizende Substanzabsondern.[3] Viele Zoanthidea enthalten Mikroalgen alsSymbionten, deren Photosynthese ihnen zus-tzliche Energieliefert. Diesen Dinoflagellaten wird eine entscheidende Rollebei der Biosynthese von Sekund-rmetaboliten zugeschrieben,die aus Zoanthidea isoliert wurden.[4]
Verschiedenartige Naturstoffe wurden aus Spezies derOrdnung Zoantharia isoliert (Schema 1). Zoanthamin (1)z-hlt zu den Zoanthus-Alkaloiden und steht im Mittelpunktdieses Aufsatzes. Auch das Ecdysteroid Zoanthusteron (2)wurde aus einer Zoanthus-Spezies isoliert.[5] Prostaglandinewie PGA2 (3) aus Palythoa kochii stabilisieren die Mikrotu-buli in einer -hnlichen Weise wie Paclitaxel.[6] >ber einDutzend Naturstoffe mit demGer�st von Zoanthoxanthin (4)wurde aus Parazoanthus axinellae isoliert,[7] und das struktu-rell verwandte Parazoanthoxanthin A hemmt die Cholines-terase.[8] Die bekannteste Verbindung aus diesen marinenOrganismen ist aber Palytoxin (5), das aus einer vor Hawaiiheimischen Palythoa-Spezies stammt und zu den st-rkstenGiften �berhaupt z-hlt (LD50 = 15 mgkg�1 f�r M-use).[9] DieStruktur von Palytoxin wurde von Kishi, Uemura und Hirataet al. bestimmt, und Kishi und Suh gelang sp-ter auch dieSynthese dieser Verbindung.[10]
[*] Dr. D. C. Behenna, J. L. Stockdill, Prof. Dr. B. M. StoltzThe Arnold and Mabel Beckman Laboratories of Chemical SynthesisDivision of Chemistry and Chemical EngineeringCalifornia Institute of Technology1200 E. California Boulevard, MC 164-30, Pasadena, CA 91125(USA)Fax: (+1)626-564-9297E-Mail : [email protected]: http://www.stoltz.caltech.edu
[**] Eine Zusammenstellung wichtiger Abk@rzungen findet sich imAnhang.
Marine Naturstoffe spielen schon lange eine wichtige Rolle in derNaturstoffchemie und bei der Wirkstoffentwicklung. Die Artenvielfaltund das facettenreiche Wechselspiel im Lebensraum Meer spiegelnsich in den Eigenschaften der Substanzen wider, die aus marinen Le-bensformen isoliert werden: Sie zeigen unwahrscheinlich vielseitigeMolek,lstrukturen und biologische Aktivit-ten. Die Naturstoffe ausden Polypen der marinen Zoanthidea bilden darin keine Ausnahme.Die Zoanthamin-Alkaloide, deren erste Vertreter vor ,ber zwanzigJahren isoliert wurden, erregten besonderes Interesse als Syntheseziele,weil sie sich durch eine große Bandbreite an biologischen Aktivit-tenauszeichnen. Hier fassen wir die wichtigsten Beitr-ge zur Isolierungund strukturellen Charakterisierung der nat,rlichen Zoanthaminezusammen und f,hren Studien zu ihrer biologischen Aktivit-t undTotalsynthese an.
Aus dem Inhalt
1. Einf�hrung 2401
2. Die Naturstoffklasse derZoanthamine 2402
3. Biologische Aktivit�t derZoanthamin-Alkaloide 2406
4. Die Synthese vonZoanthaminen 2408
5. Zusammenfassung und Ausblick 2419
Abbildung 1. AusgewAhlte Zoanthidea.
NaturstoffsyntheseAngewandte
Chemie
2401Angew. Chem. 2008, 120, 2400 – 2421 � 2008 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim

2. Die Naturstoffklasse der Zoanthamine
2.1. Isolierung und Strukturbestimmung
Im Jahr 1984 vermeldeten Rao, Faulkner und Mitarbeiterdie Isolierung des Naturstoffs Zoanthamin (1) aus einerSpezies der Gattung Zoanthus, die an der K�ste des indischenVisakhapatnam zu finden ist.[3] Das Verkn�pfungsmuster und
die relative Konfiguration des zuvor unbekannten Alkaloid-ger�sts wurden durch R1ntgenbeugung am Einkristall er-mittelt.[3] Im Zuge dieser ersten Isolierung wurden �berdiesdie verwandten Naturstoffe Zoanthenamin (6) und Zoan-thenamid (7) erhalten, die 1985 beschrieben wurden.[11] 1989isolierten Rao undMitarbeiter dann 28-Desoxyzoanthenamin(8) und 22-epi-28-Desoxyzoanthenamin (9) aus einer Zoan-thus-Spezies aus dem Golf von Bengalen (Schema 2).[12] Die
Douglas C. Behenna wurde 1978 in Norris-town, Pennsylvania, geboren. Er erhieltseinen BA in Chemie an der University ofPennsylvania (2000), wo er dem Arbeitskreisvon Professor Dewey G. McCafferty angeh-r-te. Nach seiner Promotion unter Anleitungvon Professor Brian M. Stoltz am Caltech(2006, Hertz-Stipendium) forscht er zurzeitals Postdoktorand in der Gruppe von Profes-sor E. J. Corey an der Harvard University.Seine Interessen umfassen die Synthesekomplexer Naturstoffe und die Entwicklungneuer Syntheseverfahren.
Jennifer L. Stockdill wurde 1981 in Manka-to, Minnesota, geboren. 2003 erhielt sie denBSc in Chemie (Virginia Polytechnic Institu-te and State University). Nachdem sie dortunter Professor Felicia A. Etzkorn und Pro-fessor Alan R. Esker gearbeitet hatte, fertigtsie zurzeit als Novartis Women and Minori-ties Research Fellow in der Gruppe von Pro-fessor Brian M. Stoltz am Caltech ihre Dok-torarbeit an. Zu ihren Forschungsinteressenz<hlen die Totalsynthese von Naturstoffenmit wichtigen biologischen Eigenschaftensowie die Entwicklung neuer Reaktionen undmechanistische Untersuchungen.
Schema 1. Einige Naturstoffe, die aus Zoanthidea isoliert wurden.
B. M. Stoltz et al.Aufs�tze
2402 www.angewandte.de � 2008 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2008, 120, 2400 – 2421

Strukturen 6–9 wurden durch einen Vergleich der spektro-skopischen Daten mit denjenigen von Zoanthamin (1) abge-leitet. Bei diesen Forschungen stand zwar stets die Suche nachdemAugenreizstoff derZoanthus-Spezies imMittelpunkt, f�ralle f�nf isolierten Alkaloide wurde abweichend davon abernachgewiesen, dass sie die Phorbolmyristatacetat(PMA)-be-dingte Ohrentz�ndung bei M-usen hemmen.[3,11]
Im Jahr 1995 identifizierten Uemura und Mitarbeiter f�nfweitere Zoanthamine aus einer Zoanthus-Spezies, die vor derAyamaru-K�ste der Amami-Inseln s�dlich von Japan ge-sammelt worden war.[13] Die Strukturen dieser Isolate wei-chen deutlich voneinander ab (Schema 3): Manche Verbin-
dungen tragen an C19 keinen Substituenten, etwa Norzoan-thamin (10) und Norzoanthaminon (11), das �berdies an C11oxidiert ist. F�r Oxyzoanthamin (12) ist die Oxidation an C26charakteristisch. Die relative Konfiguration von Norzoan-thamin wurde durch R1ntgenbeugung best-tigt,[13] und die inSchema 3 gezeigte absolute Konfiguration wurde sp-teranhand einer NMR-spektroskopischen Analyse von MTPA-Derivaten ermittelt.[14] Zoanthaminon (13) enth-lt 30 Koh-lenstoffatome und ist an C11 oxidiert; eine R1ntgenstruk-turanalyse wurde von Clardy und Mitarbeitern beschrie-ben.[15] Cyclozoanthamin (14) und Epinorzoanthamin (15)zeichnen sich dadurch aus, dass die Enonfunktion des A-Rings modifiziert ist. Beide Strukturen wurden auf derGrundlage von NOE-Experimenten zugewiesen.[13]
Im Jahr 1996 isolierten Norte und Mitarbeiter eine Reihevon Zoanthamin-Alkaloiden mit interessanten Oxidations-mustern aus Zoanthidea der Kanarischen Inseln (Schema 4).Epioxyzoanthamin (16) ist einzigartig durch die Konfigura-tion an C19, die durch einen Vergleich mit den NMR-spek-troskopischen Daten von Oxyzoanthamin (12) zugeordnet
Brian M. Stoltz wurde 1970 in Philadelphia,Pennsylvania, geboren. W<hrend seines Stu-diums verbrachte er ein Jahr an der Ludwig-Maximilians-Universit<t in M>nchen. Er er-hielt den BSc in Chemie und den BA inDeutsch von der Indiana University of Penn-sylvania (1993) und promovierte 1997 unterder Anleitung von Professor John L. Wood ander Yale University. Im Anschluss an einPostdoktorat bei Professor E. J. Corey an derHarvard University (1998—2000, NIH-Sti-pendium) wechselte er ans Caltech, an demer nun eine Professur f>r Chemie innehat.
Seine Forschungsvorhaben drehen sich um das Design und die Anwendungneuer Synthesestrategien f>r Verbindungen mit komplexen Molek>lstruktu-ren und wichtigen biologischen Eigenschaften. Ein weiteres Gebiet ist derEinsatz von asymmetrischer Katalyse und Reaktionskaskaden in Synthesen.
Schema 2. Von den Gruppen von Rao und Faulkner isolierte Zoantha-mine. Im gesamten Aufsatz werden wir die Kohlenstoffatome undRinge des Zoanthaminger@sts so bezeichnen wie f@r Zoanthamin (1)gezeigt.
Schema 3. Von den Gruppen von Uemura und Clardy isolierte Zoan-thamine.
NaturstoffsyntheseAngewandte
Chemie
2403Angew. Chem. 2008, 120, 2400 – 2421 � 2008 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.de

wurde.[16] 3-Hydroxyzoanthamin (17) und 30-Hydroxyzoan-thamin (18) sind an charakteristischen Stellen oxidiert, w-h-rend 11-Hydroxyzoanthamin (19) und 11-Hydroxynorzoan-thamin (20) vermutlich mit Zoanthaminon bzw. Norzoan-thaminon in Verbindung stehen. Zoanthenol (21) schließlichenth-lt einen außergew1hnlichen oxidierten, aromatischenA-Ring, sodass an C13 und C18 nicht l-nger Stereozentrenvorliegen. Wegen dieses Unterschieds waren ausf�hrlicheHMBC- und ROESY-Korrelationsexperimente erforderlich,um die Struktur und die relative Konfiguration abzusi-chern.[17]
2.2. Biosynthese der Zoanthamine
Obwohl die Zoanthamine schon vor �ber zwanzig Jahrenentdeckt wurden, ist vergleichsweise wenig �ber ihre Bio-synthese bekannt. Rao undMitarbeiter stellten 1984 fest, dassTeilstrukturen des 30-atomigen Kohlenstoffger�sts vonZoanthamin auf eine Triterpen-Zwischenstufe hindeuten, siekonnten darin aber keine der �blichen Kopf-Schwanz-Ver-kn�pfungen identifizieren.[3] Sp-ter postulierten Uemuraet al. , dass die Zoanthamine aus der Polyketid-Vorstufe 22entstehen k1nnten (Schema 5),[14,18] wobei die Polyketid-Biosynthese am C24 des Carboxylats beginnen sollte,[4] eineexakte Beschreibung des Biosynthesepfads blieben sie aberschuldig. Sicher ist jedoch, dass sich anhand des vorgeschla-genen Intermediats 22 die Oxygenierungsmuster der meistenZoanthamine erkl-ren lassen und dass es durch organischeStandardreaktionen mit etablierten Mechanismen in dieZoanthamin-Struktur �berf�hrt werden kann (Schema 6).
Die Umwandlung von 22 in 1 beginnt mit einer Tauto-merisierung, einer Elektrocyclisierung und einer Diels-Alder-
Reaktion zur Bildung der Zwischenstufe 23. Eine weitereTautomerisierung und eine Protonierung liefern dann 24, indem eine Carbonylgruppe aktiviert ist, sodass sie von derAlkoholfunktion unter 6-exo-Ringschluss und Bildung von 25angegriffen werden kann. Die Abspaltung von Wasser ergibtdaraufhin das Oxocarbeniumion 26, das durch einen Angriffder Aminfunktion und Protonentransfer in die Zwischenstufe27 �bergeht. In dem daraus entstehenden Iminiumion 28 wirdschließlich die Iminiumgruppe durch die Carbons-uregruppeangegriffen, und die abschließende Deprotonierung ergibtZoanthamin (1). Da alle Schritte beim Aufbau des DEFG-Ringsystems reversibel verlaufen, resultiert das thermody-namisch g�nstigste Produkt. Dies wird sich im Hinblick aufSyntheseans-tze noch als wichtig erweisen (siehe Ab-schnitt 4.2).
Uemura und Mitarbeiter schlugen nicht nur diesen Poly-ketidweg zu Zoanthaminen vor, sondern stellten dar�berhinaus auch Vermutungen zur Herkunft derjenigen Nor-zoanthamin-Alkaloide an, die keine Methylgruppe an C19tragen. Nach der Isolierung von Oxyzoanthamin (12) postu-lierten sie einen Mechanismus f�r die oxidative Demethylie-rung von Zoanthamin (Schema 7). Eine direkte Oxidationvon Zoanthamin an C26 w�rde demnach zu Oxyzoanthaminf�hren, das formal durch eine Retroaldolreaktion in Form-aldehyd und Norzoanthamin (10) zerfallen k1nnte.[13] R-t-selhaft bleibt, warum Uemura et al. nicht vom Einbau einerAcetateinheit anstelle der entsprechenden Propionateinheitausgingen. Eine solche Modifizierung der Struktur 22(Schema 5) w�rde einen direkten Zugang zu Zoanthamin-Derivaten ohne C26, etwa zu Norzoanthamin (10), er1ffnen,w-hrend die Oxidation an C26 weiterhin herangezogenwerden k1nnte, um die Bildung von Oxyzoanthamin (12) zuerkl-ren.
Bei Betrachtungen zur Biosynthese der Zoanthamin-Alkaloide muss außerdem die Rolle der symbiontischen Dino-flagellaten ber�cksichtigt werden, die h-ufig in Zoanthidealeben. Algen der Gattung Symbiodinium wurden aus Zo-anthidea der Gattung Zoanthus isoliert.[19] Solche symbionti-schen St-mme lassen sich nur schwer axenisch kultivieren,dennoch gelang es Nakamura und Mitarbeitern, nennens-werte Mengen zoanthidfreier Symbiodinium-Spezies zu er-halten.[4] (Von einer axenischen Kultur spricht man, wenndiese vollst-ndig frei von jedem anderen lebenden Organis-mus, einschließlich Kontaminationen oder, wie in diesem Fall,dem Zoanthid-Wirt, pr-pariert wird.) Diese Algen produ-
Schema 4. Von Norte et al. isolierte Zoanthamine.
Schema 5. Die hypothetische Polyketidvorstufe 22 f@r Zoanthamin.
B. M. Stoltz et al.Aufs�tze
2404 www.angewandte.de � 2008 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2008, 120, 2400 – 2421

zierten je nach Medium Metaboliten inunterschiedlichen Verteilungen, doch mitgeeigneten Kulturbedingungen konnteNakamuras Gruppe schließlich das neueC30-Alkaloid Zooxanthellamin (29) isolie-ren. Zooxanthellamin liegt im dynami-schen Gleichgewicht als Halbaminallactonund Iminiumcarboxylat vor (Schema 8).Die Struktur und die relative Konfigurati-on wurden durch NMR-spektroskopischeStudien abgesichert. Vergleiche der NMR-Daten f�r die MTPA-Ester zeigten, dassZooxanthellamin die gleiche absoluteKonfiguration hat wie die Zoanthamin-Alkaloide.[4]
Die bemerkenswerte Khnlichkeit vonZooxanthellamin und Zoanthamin ließZweifel daran aufkommen,[20] dass dieZoanthamine wirklich von Zoanthideaproduziert werden – ihnen k1nnte stattdessen nur eine untergeordnete Rolle inder Biosynthese zukommen, etwa dieEinstellung des Oxygenierungsgrads desfertigen Zoanthaminger�sts. Feine Ab-weichungen in der Alkaloidstruktur k1nn-ten durch Einfl�sse aus der Umgebungoder der Zoanthidea-Wirtspezies bedingtwerden. Alternativ ist es m1glich, dassunterschiedliche Algen an der Produktionverschiedener Zoanthamine beteiligt sind.
Bislang wurde nur ein Versuch be-schrieben, die Biogenese der Zoanthamineaufzukl-ren. Norte und Mitarbeiter ver-f�tterten isotopenmarkiertes Natriumace-tat, Glycin und Glucose an kleine Zoan-thus-Kolonien;[7] die markierten Atomewurden jeweils zu �ber 10% aufgenom-men, der Einbau schien aber zuf-llig.
Die vorgestellten Vorschl-ge zur Bio-genese dieser Verbindungen m�ssen letzt-lich anhand weiterer Studien �berpr�ftwerden. Ohne aussagekr-ftige experimen-telle Daten k1nnen sie weder als nachge-wiesen gelten noch verworfen werden.
2.3. Reaktivit�t von Norzoanthamin
Nach der Isolierung wurde Norzoan-thamin unter zahlreichen Reaktionsbedin-gungen umgesetzt. Das Ziel hierbei war es,Hypothesen f�r biologische Wirkmecha-nismen aufzustellen.
Ein dynamisches Gleichgewicht vonLacton- und Iminiumform – wie f�rZooxanthellamin (Schema 8) – wurde auchf�r einige weitere Zoanthamine beobach-tet (Schema 9). Norzoanthamin bildetunter sauren Bedingungen das Iminiumion
Schema 6. Mechanismus der Cyclisierung der hypothetischen Polyketidvorstufe 22 zu Zoan-thamin.
Schema 7. Vorschlag zur Biosynthese von Norzoanthamin.
NaturstoffsyntheseAngewandte
Chemie
2405Angew. Chem. 2008, 120, 2400 – 2421 � 2008 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.de

30 und kehrt beim Neutralisieren in den urspr�nglichen Zu-stand zur�ck.[18] In neutralem bis basischem Medium entstehtdurch eine Eliminierung das Enamin 31. Dass Norzoantha-min mit dem Enamin 31 im Gleichgewicht vorliegt, wurdedurch die Umwandlung in den Methylester 32 belegt, der beider Umsetzung mit Diazomethan binnenMinuten entsteht.[18]
NMR-Spektren von Norzoanthamin in D2O zeigen eineebenso schnelle, spezifische und vollst-ndige Deuterierung in
11b-Position zu 33.[16,17] Eine vergleichbare Deuterierungwurde f�r Zoanthenol, 3-Hydroxynorzoanthamin und 30-Hydroxynorzoanthamin nachgewiesen, dagegen bauen 11b-Hydroxyzoanthamine keine signifikanten Deuteriummengenein, wohl deshalb, weil die Eliminierung zum Enamin indiesem Fall nicht m1glich ist.[17] Dieses dynamische Verhaltenim w-ssrigenMedium bei physiologischen pH-Werten k1nnteden biologischen Aktivit-ten der Verbindungen zugrundeliegen.
Auch die Reduktion der Halbaminalfunktion der Zoan-thamin-Alkaloide nimmt einen bemerkenswerten Verlauf.Die Umsetzung von Norzoanthamin mit Natriumborhydriderzeugt zwei ungew1hnliche Produkte: das Enon 34 und denAllylalkohol 35 (Schema 10).[14,18] Die Bildung dieser Pro-dukte l-sst sich mit der Mffnung des Halbaminals zum Imi-niumion 36 erkl-ren. Eine Deprotonierung f�hrt zumEnamin, das den Lactonring in 37 intramolekular unter Bil-dung der Keto-Iminium-Zwischenstufe 38 angreifen sollte;Dehydratisierung liefert dann das Iminiumion 39, das zumEnon 34 und weiter zum Allylalkohol 35 reduziert wird.
3. Biologische Aktivit�t der Zoanthamin-Alkaloide
3.1.Wirkung gegen Osteoporose
Die wohl bekannteste biologische Aktivit-t der Zoan-thamin-Alkaloide ist ihre Wirkung gegen Osteoporose, dieerstmals von Uemura et al. im Jahr 1996 beschriebenwurde.[21] Als Osteoporose bezeichnet man eine Deminerali-sierung der Knochen, die oft darauf zur�ckzuf�hren ist, dassOsteoklasten das Knochengewebe schneller reabsorbieren,als es regeneriert wird.[22] In Versuchen an M-usen, denen dieEierst1cke entfernt worden waren, – einem pharmazeuti-schen Modellsystem f�r die Osteoporose nach der Meno-pause, weil solcherart pr-parierte M-use unter Mstrogen-mangel leiden und ihre Knochen dadurch schnell abgebautwerden – verhinderten Norzoanthamin und sein Hydrochlo-rid das Auftreten von Osteoporosesymptomen.[21] BeiM-usen, die Norzoanthamin-Hydrochlorid in Dosierungenvon 0.08–2.0 mgkg�1d�1 erhielten (p.o., vier Wochen langjeweils an f�nf Tagen), nahm das Gewicht des Oberschen-kelknochens statistisch weniger ab als bei entsprechendenM-usen ohne Medikation.[18,21] Bei einer Dosierung0.4 mgkg�1d�1 blieb die St-rke der Oberschenkelknochenvon M-usen ohne Eierst1cke in Bruchlasttests vergleichbarhoch wie bei einer Kontrollpopulation aus nichtamputiertenM-usen.[18,21] Mit Norzoanthamin-Hydrochlorid behandelteM-use hatten �berdies eine deutlich dickere kompakteKnochensubstanz als die Vergleichstiere.[23]
Analog zur Mstrogenersatztherapie f�r Frauen jenseitsder Menopause k1nnen M-use ohne Eierst1cke durch Ver-abreichung von 17b-Mstradiol vor Osteoporose bewahrtwerden. Die Behandlung mit 17b-Mstradiol verursacht inM-usen mit einer dosisabh-ngigen Zunahme des Uterusge-wichts aber eine Nebenwirkung, die bei der Behandlung mitNorzoanthamin-Hydrochlorid nicht auftritt.[23]
Die Wirkung von Norzoanthamin gegen Osteoporosek1nnte – wie bei Mstrogen[24] – auf einer Hemmung der In-
Schema 8. Die Struktur von Zooxanthellamin.
Schema 9. Lacton-Enamin-Gleichgewichte f@r Norzoanthamin.
B. M. Stoltz et al.Aufs�tze
2406 www.angewandte.de � 2008 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2008, 120, 2400 – 2421

terleukin-6(IL-6)-Produktion beruhen. IL-6 ist an der Sti-mulierung der Bildung von Osteoklasten beteiligt, die Kno-chengewebe reabsorbieren. Norzoanthamin und sein Hydro-chlorid 30 unterdr�cken in vitro die IL-6-Aussch�ttung ausPr-osteoblasten in Konzentrationen von 13 bzw.4.6 mgmL�1.[25] In-vitro-Studien zeigten f�r Norzoanthamin-Hydrochlorid allerdings keinen Einfluss auf die Osteoklas-tenbildung, und die Wirkung auf die IL-6-Aussch�ttungwurde noch nicht in vivo nachgewiesen.[23] Diese beidenPunkte und das Ausbleiben einer Gewichtszunahme desUterus in M-usen ohne Eierst1cke legen nahe, dass dieZoanthamin-Alkaloide nach einem eigenen Mechanismuswirken[26] und daher eine besser vertr-gliche Alternative zurMstrogenersatztherapie der Osteoporose jenseits der Meno-pause bieten k1nnten.
Die Suche nach Alternativen zur Mstrogenersatztherapieforcierte die Erforschung von Struktur-Wirkungs-Beziehun-gen (SAR) f�r Norzoanthamin. Durch Semisynthesen er-hielten Uemura undMitarbeiter eine Reihe von Zoanthamin-
Derivaten, die auf ihre Inhibitoraktivit-tgegen IL6 getestet wurden(Schema 11).[18,25] Bemerkenswerter-weise schr-nkten alle untersuchtenVerbindungen die IL-6-Produktiondeutlich weniger stark ein als Norzoan-thamin (h1here IC50-Werte).[27] Wenndie olefinische Doppelbindung fehlte(wie im Keton 41 oder im Diol 42),wurde eine Aktivit-tsabnahme beob-achtet, und auch ein Abbau der Lacton-Halbaminal-Funktion – in der Carbon-s-ure 43 und dem Ester 32 – senkte dieAktivit-t deutlich.[18]
Eine aktuellere SAR-Studie vonHirama und Mitarbeitern sollte zeigen,welche Strukturmerkmale der Zoan-thamine erforderlich sind, um dasWachstum IL-6-abh-ngiger MH-60-Zellen zu hemmen (Schema 12).[28] Indiesen Tests erzielten die Hydrochloridevon Norzoanthamin und Zoanthamin,30 bzw. 44, die gr1ßte Wirkung (IC50: 13bzw. 26 mm). Die Modellverbindungen45, f�r die „n1rdliche“, carbocyclischeTeilstruktur von Zoanthenol, und 46, f�rdie „s�dliche“, heterocyclische Region,waren kaum wirksam, die Aktivit-t desIminiumsalzes 47 hingegen kam derje-nigen der Zoanthamin-Hydrochloriden-her. Dieses Ergebnis best-tigt zweiBeobachtungen: 1) Die Hydrochloridevon Zoanthaminen hemmen die IL-6-Produktion gew1hnlich st-rker als dieentsprechenden Naturstoffe, und 2) dieheterocyclische Molek�lregion ist ver-mutlich ein wichtiger Bestandteil desPharmakophors f�r die IL-6-Hemmung.
3.2.Weitere biologische Aktivit�ten
F�r Zoanthamine wurden zahlreiche weitere biologischeAktivit-ten beschrieben. Wie in Abschnitt 2.1 erw-hnt,hemmen Zoanthamin (1), Zoanthenamin (6) und Zoanthen-amid (7) die PMA-induzierte Ohrentz�ndung im Maus-test.[3,11] Uemura und Mitarbeitern zufolge wirken Norzoan-thamin (10), Norzoanthaminon (11), Oxyzoanthamin (12),Cyclozoanthamin (14) und Epinorzoanthamin (15) gegenmurine P388-Leuk-miezellen (Tabelle 1);[13] die st-rkste Zy-totoxizit-t zeigten dabei Norzoanthaminon und Oxyzoan-thamin.
Auch die antibakterielle Wirkung von Zoanthamin undeinigen reduzierten Derivaten wurde untersucht.[29] In Sus-zeptibilit-tstests waren die Zoanthamin-Alkaloide gegenGram-negative und Gram-positive Bakterien aktiv (Tabel-le 2).
Zoanthamin-Alkaloide beeinflussen auch die Thrombo-zytenaggregation beim Menschen:[30] 11b-Hydroxyzoantha-
Schema 10. Reduktion von Norzoanthamin.
NaturstoffsyntheseAngewandte
Chemie
2407Angew. Chem. 2008, 120, 2400 – 2421 � 2008 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.de

min (19) und der Methylester 32 hemmten in Kon-zentrationen von 0.5 mm die durch Collagen, Arachi-dons-ure oder Thrombin induzierte Aggregation.Dagegen verhinderten die Inhibitoren Oxyzoantha-min (12) und Zoanthenol gezielt die Aggregation inGegenwart von Collagen bei Konzentrationen von0.5 mm ; im Fall von Arachidons-ure oder Thrombinzeigten sie fast keine Wirkung. Eine derart selektiveAktivit-t ist im Hinblick auf potenzielle Therapien f�rHerzkranzgef-ßerkrankungen interessant. Als Folgeeiner (anomalen) Thrombozytenaggregation kanneine Vene oder Arterie verstopft werden, sodass es zueinem Infarkt oder Schlaganfall kommt.[31] Die �bli-chen Antithrombotika f�r die Behandlung von Herz-kranzgef-ßerkrankungen sind nicht sehr effizient, daihre Wirksamkeit gering ist und/oder ernste Neben-wirkungen wie Blutungen infolge einer beeintr-ch-tigten H-mostase auftreten.[32] Experimentelle undklinische Resultate deuten an, dass ein selektiverCollagenrezeptor-Antagonist die H-mostase nur ge-ringf�gig beeinflussen w�rde und daher bei gleicherAktivit-t ein sicherer Wirkstoff sein k1nnte.[32]
4. Die Synthese von Zoanthaminen
4.1. Allgemeines
Die beeindruckende F�lle an biologischen Akti-vit-ten und die Funktionalisierungsdichte der Zoan-thamin-Strukturen haben viele Forschungsgruppen
Schema 11. IC50-Werte f@r die Hemmung der IL-6-Produktion aus der SAR-Studievon Uemura et al.
Schema 12. IC50-Werte f@r die Hemmung des Wachstums IL-6-abhAngi-ger Zellen aus der SAR-Studie von Hirama et al.
Tabelle 1: IC50-Werte f@r die Hemmung von murinen P388-LeukAmiezel-len.
Verbindung IC50
[mgmL�1]Verbindung IC50
[mgmL�1]
Norzoanthamin (10) 24.0 Cyclozoanthamin (14) 24.0Oxyzoanthamin (12) 1.0 Epinorzoanthamin (15) 2.6Norzoanthaminon (11) 7.0
Tabelle 2: Antibakterielle AktivitAten einiger Zoanthamin-Alkaloide an-gegeben als Durchmesser des Inhibitionsumkreises in mm.
Verbindung Gram-negativ Gram-positivS. typhimurium E. coli B. sphaericus S. aureus
1 6 6 8 748 12 6 8 1049 7 6 8 1050 6 7 7 9
B. M. Stoltz et al.Aufs�tze
2408 www.angewandte.de � 2008 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2008, 120, 2400 – 2421

angespornt, Strategien f�r m1gliche Totalsynthesen dieserAlkaloide zu entwerfen. Oft stand der Aufbau des tricycli-schen ABC-Ringsystems im Mittelpunkt, dessen große Zahlan Stereozentren hohe Anforderungen an die Synthesepla-nung stellt. Allein der C-Ring enth-lt drei quart-re Stereo-zentren – deren Aufbau bekanntlich immer eine Herausfor-derung ist – in vicinaler und nichtvicinaler Stellung zueinan-der. Erschwerend kommt der Raumbedarf ihrer Substituen-ten hinzu, der Probleme selbst bei Routineumwandlungen anbenachbarten Funktionalit-ten mit sich bringt. AndereGruppen konzentrierten sich auf die Synthese des heterocy-clischen DEFG-Ringsystems. Auf dem Weg zu dieser Teil-struktur m�ssen die Heterocyclen mit der richtigen Halb-aminal-Verkn�pfung und Konfiguration aufgebaut werden.
4.2.Miyashitas Norzoanthamin-Synthese
Zwanzig Jahre nach der Isolierung der ersten Zoantha-min-Alkaloide beschrieben Miyashita und Mitarbeiter dieerste und bislang einzige abgeschlossene Totalsynthese einerdieser Verbindungen.[33] Ihre Vorgehensweise bei der Syn-these von Norzoanthamin ist in Schema 13 veranschaulicht;sie umfasst kreative L1sungen zu einigen Problemen, die imZuge der 41-stufigen Synthese auftraten. Ihr Verfahren zumAufbau des ABC-Ringsystems von Norzoanthamin durcheine Diels-Alder-Reaktion wurde 2002 vorgestellt(Schema 14).[34]
Die Synthese begann mit der Addition des Cuprats 51 andas enantiomerenreine Enon 52 und der anschließenden Al-dolreaktion mit dem Aldehyd 53 zum Keton 54. Diese Re-aktionsfolge legte die absolute Konfiguration von C13 fest,auf der die �brigen Stereozentren aufbauten. Nach einigenvorbereitenden Schritten wurde das Furan 55 nach der Me-thode von Katsumura photochemisch oxidiert. Durch dieBildung eines Silylenolethers entstand das Diels-Alder-Sub-strat 56, das beim Erhitzen auf 240 8C haupts-chlich �ber denexo->bergangszustand 57 in ein Isomerengemisch der Sil-ylenolether 58 �berging. Nach Abspaltung der Silylgruppewurde das diastereomerenreine Keton 59 in 51% Ausbeute�ber die beiden Schritte isoliert. Diese Diels-Alder-Reaktionerzeugte die quart-ren Zentren an C12 und C22 von Nor-zoanthamin mit der erforderlichen absoluten Konfiguration.
Nun folgten einige Umwandlungen, um eine Homologi-sierung an C23 zu erm1glichen und das fehlende quart-reZentrum an C9 einzuf�hren (Schema 15). Die diastereose-lektive Reduktion beider Ketogruppen von 59 gelang durchK-Selectrid-Addition von der konvexen Molek�lseite aus,und die neue Hydroxyfunktion an C10 bildete anschließendauch wie gew�nscht das Lacton. Silylierung mit TBSOTf undanschließende Umwandlung des phenolischen Acetats in eineTES-Gruppe f�hrten zum gesch�tzten Lacton 60. Nach derReduktion zum Lactol ergab eine Wittig-Reaktion das di-deuterierte Olefin 61. Hydroborierung und Oxidation f�hrtenweiter zumKetoalkohol 62, in dem nun das quart-re Zentruman C9 eingerichtet werden konnte. Zu diesem Zweck wurde62 mit Dimethylcarbonat und Lithium-tert-butoxid acyliert.Dieser Schritt verlief vermutlich deswegen regioselektiv, weilzuerst die Hydroxygruppe acyliert wird und die anschlie-ßende C-Acylierung des Enolats dann den Lactonringschließt. Im Anschluss an diese Abfolge konnte durch Ab-fangen mit Methyliodid das Lacton 63 erhalten werden. Li-thium-tert-butoxid und Methyliodid in DMPU �berf�hrtendieses Lacton 63 durch C-Alkylierung in das quaternisierte d-Lacton 64. Diese schwierige Transformation lieferte in einereindrucksvollen Ausbeute von 83% ein einziges Diastereo-mer als Produkt.
Zur Synthese des „s�dlichen“ Molek�lteils von Nor-zoanthamin wurde zun-chst das Lacton-Kohlenstoffatom C8in eine Ethinylgruppe umgewandelt, an die anschließend die
Schema 13. Miyashitas Retrosynthese von Norzoanthamin.
Schema 14. Aufbau der ABC-Einheit durch eine Diels-Alder-Reaktionnach Miyashita et al.
NaturstoffsyntheseAngewandte
Chemie
2409Angew. Chem. 2008, 120, 2400 – 2421 � 2008 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.de

Seitenkette angesetzt wurde (Schema 16).Die Addition von Methyllithium und dieEinf�hrung einer Silylschutzgruppe �ber-f�hrten das Lacton 64 in das methylsub-stituierte Keton 65, das mit Trifluorme-thansulfons-ureanhydrid und DBU in dasAlkin 66 umgewandelt wurde. Bei dieserTransformation bildeten sich geringeMengen des Nebenprodukts 67 a. Wurdehingegen das nichtdeuterierte Substrateingesetzt, so entstand das entsprechendeNebenprodukt 67 b in 30% Ausbeute, unddas gew�nschte Alkin wurde nur noch in66% Ausbeute erhalten. Durch die Ad-dition des lithiierten Alkins 66 an denAldehyd 68 mit nachfolgender Oxidationzum Inon 69 wurde eine Zwischenstufeerreicht, die bereits alle Kohlenstoffatomedes Norzoanthamin-Ger�sts enthielt.
Um zur Zielverbindung zu gelangen,mussten zw1lf weitere Entsch�tzungs-,Redox- und Dehydratisierungsschritte
durchlaufen werden (Schema 17). Von 69 aus f�hrten dieReduktion der C-C-Dreifachbindung, Enolether- und Ace-talspaltung unter sauren Bedingungen, vollst-ndige Desily-lierung und eine Oxidation der sekund-ren Alkoholeinheitenzu 70. Die schrittweise Oxidation der prim-ren Alkohol-funktion ergab die Carbons-ure, die dann mit Trimethyl-silyldiazomethan verestert wurde. Eine Saegusa-Ito-Oxidati-on baute die Enonfunktion im A-Ring auf (!71). DieseVerbindung reagierte in heißer w-ssriger Essigs-ure durchCarbamatspaltung zum Iminiumion. In w-ssriger TFA bei110 8C addierte die Methylesterfunktion an die Iminium-gruppe, wobei das Trifluoracetat von Norzoanthamin ent-stand, das durch Einwirkung von basischem Aluminiumoxidin Methanol in den Naturstoff �berf�hrt wurde. Diese ein-drucksvolle Synthese best-tigte die absolute Konfigurationvon Norzoanthamin, die zuvor aus NMR-spektroskopischenExperimenten abgeleitet worden war.
4.3. Aufbau des ABC-Ringsystems von Zoanthamin mit der Diels-Alder-Strategie nach Tanner
Tanner und Mitarbeiter w-hlten zum Aufbau des ABC-Ringsystems ebenfalls einen Diels-Alder-Ansatz(Schema 18), wobei die Cyclisierungsvorstufe durch eineStille-Kupplung erzeugt werden sollte. Die Synthese ging vonPerillaalkohol aus, der in Form beider Enantiomere erh-ltlichist. Die Konfiguration aller �brigen Stereozentren sollte sich
Schema 15. Funktionalisierung des ABC-Frag-ments.
Schema 16. Ankn@pfung der Seitenkette f@r den „s@dlichen“ Strukturteil.
B. M. Stoltz et al.Aufs�tze
2410 www.angewandte.de � 2008 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2008, 120, 2400 – 2421

dann durch diastereoselektive Reaktionen in vorhersehbarerWeise festlegen lassen.
Tanners Synthese begann mit einer Sharpless-Epoxidie-rung von Perillaalkohol (72) mit nachfolgender Silylierungzum Epoxid 73 (Schema 19).[35] Die Methylgruppe an C15wurde durch eine diastereoselektive Addition von Gilman-Reagens an das Epoxid eingef�hrt. Nach der Desilylierungf�hrte die Umsetzung mit Bleitetraacetat zum gew�nschtenmethylsubstituierten Keton 74. Das kinetisch bevorzugteEnolat dieser Zwischenstufe wurde mit PhSeBr abgefangenund mit Wasserstoffperoxid zum Enon oxidiert. Bei der Re-duktion mit Lithiumaluminiumhydrid fiel der Allylalkohol 75als einziges Diastereomer an. Eine Alkylierung durch Clai-sen-Umlagerung wurde durch Triethylorthoacetat unterZusatz katalytischer Mengen an 2,4-Dinitrophenol eingelei-tet; darauf folgte die Verseifung mit LiOH zu 76. Die Iod-lactonisierung ergab eine Mischung aus f�nf- und sechs-gliedrigen Iodlactonen, die beim Erhitzen -quilibrierte. Beider Zugabe von DBU zur Reaktionsmischung wurde dasthermodynamisch bevorzugte Produkt durch HI-Eliminie-rung irreversibel abgefangen. Die diastereoselektive Alky-lierung dieses Enollactons mit MeI f�hrte zu 77, das mit Li-
thiobutadien zum Halbacetal 78 reagierte.Durch die Oxidation mit Mangandioxidentstand die Diels-Alder-Vorstufe 79, diebeim Erhitzen in [D8]Toluol quantitativden Tricyclus 80 ergab.
Diese erfolgreiche Diels-Alder-Cyclo-addition ermutigte die Arbeitsgruppe vonTanner, ausgehend von (�)-Carvon funk-tionalisierte Modellsubstrate herzustellen,um die Leistungsf-higkeit der Reaktion zupr�fen. Doch schon die Umuntersuchungdes erstenModellsubstrats (81, Schema 20)f�hrte zu einer ern�chternden Entde-ckung: Auch bei mehrt-gigem Erhitzen inToluol war keinerlei Umsetzung zu beob-achten. Als Grund wurde gefolgert, dassdie zus-tzliche Elektronendichte am Diender gew�nschten Diels-Alder-Reaktionmit inversem Elektronenbedarf entgegen-stand. Daher wurde das Diels-Alder-Sub-strat 83 (R=TBS) synthetisiert, das beimErhitzen zwei Produkte lieferte: das ge-w�nschte Diels-Alder-Addukt 84 und dasunerwartete Produkt 85. Wurde dieSchutzgruppe R entsprechend gew-hlt, so
entstand 85 gar als einziges Produkt. Dagegen gen�gte bereitsdie Verl-ngerung der Seitenkette an C22 um eine Methy-
Schema 17. Abschließende Stufen der Norzoanthamin-Synthese.
Schema 18. Tanners Retrosynthese von Zoanthamin.
Schema 19. Tanners Ansatz zur Synthese eines ABC-Modellring-systems. SAE=Sharpless-Epoxidierung.
NaturstoffsyntheseAngewandte
Chemie
2411Angew. Chem. 2008, 120, 2400 – 2421 � 2008 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.de

lengruppe (86), um die Bildung des entsprechenden Neben-produkts g-nzlich zu unterbinden und mit 66% Ausbeute zu87 zu gelangen, das die richtige Konfiguration f�r die Syn-these von Zoanthamin aufwies.
Diese abschließende Modifizierung best-tigte den inSchema 21 skizzierten Mechanismus. Durch eine [1,5]-sig-matrope Umlagerung des Diels-Alder-Substrats 83 enstehtdas erweiterte Enol 88, das durch Abspaltung von ROH (R=
TBS, TBDPS, Bn) in das terminale Olefin 89 �bergeht. Eine6p-Elektrocyclisierung ergibt dann das Pyran 90, dessen in-tramolekulare Diels-Alder-Reaktion zum Nebenprodukt 85f�hrt.
Mit diesem Wissen ausgestattet wurde Perillaalkohol indas Vinyliodid 91 �berf�hrt (Schema 22).[36] Die Stille-Kupplung von 91 mit dem Stannan 92 zum Diels-Alder-Sub-strat 93 gelang erst unter den Corey-Bedingungen mit be-friedigender Ausbeute.[37–39] Die Diels-Alder-Cyclisierungzum b,g-unges-ttigten Ester 94 verlief dann zwar hoch dia-
Schema 20. Cyclisierungen von Modellverbindungen, die ausgehendvon (�)-Carvon erhalten worden waren.
Schema 21. Mechanistischer Vorschlag zur Bildung des unerw@nschtenProdukts 85.
Schema 22. Tanners Ansatz f@r die Synthese des funktionalisiertenABC-Ringsystems.
B. M. Stoltz et al.Aufs�tze
2412 www.angewandte.de � 2008 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2008, 120, 2400 – 2421

stereoselektiv, aber nur mit 14% Ausbeute. AlsHauptprodukt wurde das Tetrahydrofuran 95 in31% Ausbeute isoliert.
Beim Vergleich dieses von Perillaalkoholabgeleiteten Substrats mit einem entsprechendenSubstrat, das aus Carvon erhalten worden war,stellten Tanner und Mitarbeiter fest, dass dieMOM-gesch�tzte Allylalkohol-Einheit in derDiels-Alder-Vorstufe 93 pseudo-axial orientiertist, im Modellsubstrat 96 hingegen eine pseudo--quatoriale Stellung einnimmt (Schema 23).Dieser Unterschied sollte bei 93 einen dissozia-tiven SN1-Mechanismus und eine Cyclisierungzum Tetrahydrofuran 95 erm1glichen.
Daher wurde das Substrat 97 entworfen,[40]
dessen Diels-Alder-Reaktion in Toluol bei 205 8C�ber einen exo->bergangszustand glatt und in85% Ausbeute zum Tricyclus 98 verlief(Schema 24). Auf die Einf�hrung einer MOM-Ether-Schutzgruppe an C20 (98!99) folgte eineOxidation unter Bildung des Enons 100. An-schließend wurde durch Spaltung des PMB-Ethers und Oxidation der Aldehyd 101 erhalten,der dann zu der mit tert-Butyllithium vorbehan-delten Seitenkettenvorstufe 102 gegeben wurde.Der resultierende Alkohol wurde schließlich zu103 oxidiert.
Mit dieser Diels-Alder-Strategie gelingt es,das quart-re Zentrum an C12 einzuf�hren, sieverschiebt aber den schwierigen Aufbau der vi-cinalen quart-ren Stereozentren C9 und C22 aufein sp-teres Synthesestadium. Die Arbeitsgrup-pe um Tanner vertraut zu diesem Zweck auf eineMichael-Addition und eine Alkylierung. Sollte esgelingen, diese quart-ren Zentren einzuf�hren,verblieben nur noch die Oxidation an C24 und
die Cyclisierung der Seitenkette zum DEFG-Ringsystem, umdie Totalsynthese von Norzoanthamin abzuschließen.
4.4. Aufbau des ABC-Ringsystems von Norzoanthamin nachUemura
Uemura und Mitarbeiter haben k�rzlich eine Synthese-strategie vorgestellt, die auf ihrem Vorschlag zur Biosyntheseberuht (siehe Abschnitt 2.2). Dabei sollen die Zoanthamin-Alkaloide aus einem linearen Polyketidger�st durch eineReihe pericyclischer Reaktionen entstehen.[41] Um dieseHypothese zu st�tzen, versuchten sie, das Polyen 104 als einem1gliche Zwischenstufe auf dem Weg zum Naturstoff zusynthetisieren und zu cyclisieren (Schema 25).
Schema 23. Mechanismus, der die Bildung des Neben-produkts 95 erklArt.
Schema 24. Diels-Alder-Cyclisierung und Modifizierung des Cycloaddukts nach Tanner et al.
NaturstoffsyntheseAngewandte
Chemie
2413Angew. Chem. 2008, 120, 2400 – 2421 � 2008 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.de
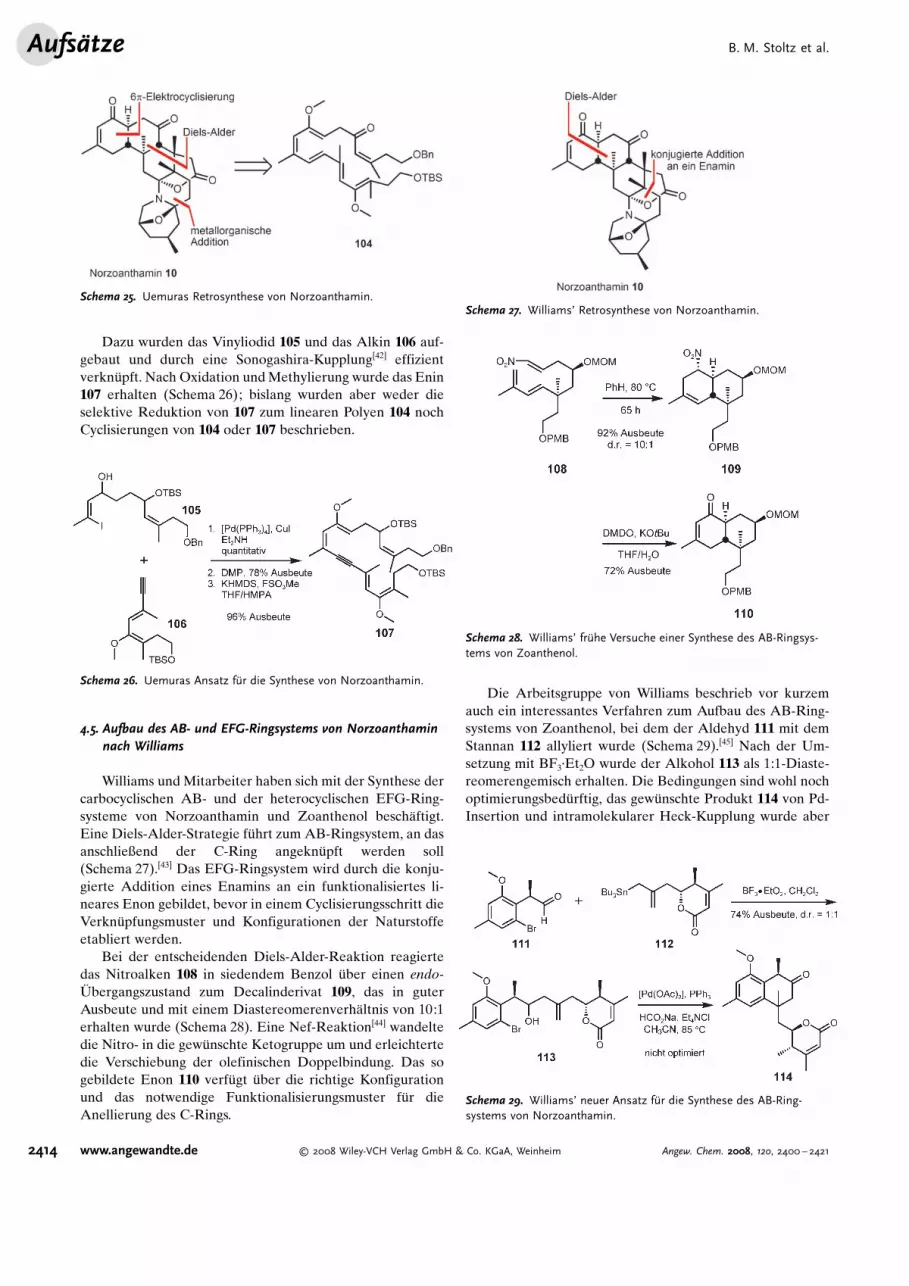
Dazu wurden das Vinyliodid 105 und das Alkin 106 auf-gebaut und durch eine Sonogashira-Kupplung[42] effizientverkn�pft. Nach Oxidation undMethylierung wurde das Enin107 erhalten (Schema 26); bislang wurden aber weder dieselektive Reduktion von 107 zum linearen Polyen 104 nochCyclisierungen von 104 oder 107 beschrieben.
4.5. Aufbau des AB- und EFG-Ringsystems von Norzoanthaminnach Williams
Williams und Mitarbeiter haben sich mit der Synthese dercarbocyclischen AB- und der heterocyclischen EFG-Ring-systeme von Norzoanthamin und Zoanthenol besch-ftigt.Eine Diels-Alder-Strategie f�hrt zum AB-Ringsystem, an dasanschließend der C-Ring angekn�pft werden soll(Schema 27).[43] Das EFG-Ringsystem wird durch die konju-gierte Addition eines Enamins an ein funktionalisiertes li-neares Enon gebildet, bevor in einem Cyclisierungsschritt dieVerkn�pfungsmuster und Konfigurationen der Naturstoffeetabliert werden.
Bei der entscheidenden Diels-Alder-Reaktion reagiertedas Nitroalken 108 in siedendem Benzol �ber einen endo->bergangszustand zum Decalinderivat 109, das in guterAusbeute und mit einem Diastereomerenverh-ltnis von 10:1erhalten wurde (Schema 28). Eine Nef-Reaktion[44] wandeltedie Nitro- in die gew�nschte Ketogruppe um und erleichtertedie Verschiebung der olefinischen Doppelbindung. Das sogebildete Enon 110 verf�gt �ber die richtige Konfigurationund das notwendige Funktionalisierungsmuster f�r dieAnellierung des C-Rings.
Die Arbeitsgruppe von Williams beschrieb vor kurzemauch ein interessantes Verfahren zum Aufbau des AB-Ring-systems von Zoanthenol, bei dem der Aldehyd 111 mit demStannan 112 allyliert wurde (Schema 29).[45] Nach der Um-setzung mit BF3·Et2O wurde der Alkohol 113 als 1:1-Diaste-reomerengemisch erhalten. Die Bedingungen sind wohl nochoptimierungsbed�rftig, das gew�nschte Produkt 114 von Pd-Insertion und intramolekularer Heck-Kupplung wurde aber
Schema 25. Uemuras Retrosynthese von Norzoanthamin.
Schema 26. Uemuras Ansatz f@r die Synthese von Norzoanthamin.
Schema 27. Williams’ Retrosynthese von Norzoanthamin.
Schema 28. Williams’ fr@he Versuche einer Synthese des AB-Ringsys-tems von Zoanthenol.
Schema 29. Williams’ neuer Ansatz f@r die Synthese des AB-Ring-systems von Norzoanthamin.
B. M. Stoltz et al.Aufs�tze
2414 www.angewandte.de � 2008 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2008, 120, 2400 – 2421

bereits isoliert, und es gelang, das unverbrauchte Ausgangs-material zur�ckzugewinnen.
Weiterhin stellten Williams et al. eine effiziente Strategievor, um das Fragment C1–C8 (f�r die Ringe E, F und G) andas ABC-Ringsystem anzuh-ngen und das quart-re ZentrumC9 stereospezifisch aufzubauen.[46] Beim Erhitzen in Gegen-wart von Zink(II)-chlorid tautomerisiert das chirale Imin 115teilweise zum Enamin 116 (Schema 30). Das resultierende
Gleichgewicht wird durch die von der b-Seite erfolgendekonjugierte Addition des Enamins 116 an das Enon 117 nachrechts verschoben (wie in der energieminimierten Konfor-mation 116b gezeigt).[47] Das Enon 117 war in enantiome-renangereicherter Form mithilfe eines chiralen Evans-Oxazolidinons erh-ltlich.[48] Die Hydrolyse der Iminium-Zwischenstufe ergab das Diketon 118 mit hervorragenderDiastereoselektivit-t (22:1), und eine Staudinger-Reduktionder Azidgruppe f�hrte zum Imin 119. Die Spaltung des Silyl-ethers mit TBAF setzte dann die Alkoholfunktion frei, die dieImingruppe angriff. Das resultierende Amin kondensiertedaraufhin mit der Ketogruppe zum Modell-Enamin 120 f�rdas EFG-Ringsystem.
4.6. Aufbau des ABC-Ringsystems von Norzoanthamin durchAnellierung nach Theodorakis
Theodorakis und Mitarbeiter haben eine Strategie vor-geschlagen, bei der an den B-Ring nacheinander die Ringe Cund A angekn�pft werden.[49] Beide Schritte bedienen sichder Robinson-Anellierung (Schema 31).
Zu Beginn der Synthese wurde dasmeso-Diketon 121 mitdem Ketoester 122 in Gegenwart von Kaliumfluorid zu 123kondensiert (Schema 32).[50] Reduktion mit Natriumborhy-
Schema 30. Williams’ Synthese eines EFG-Modellringsystems.
Schema 31. Theodorakis’ Retrosynthese von Norzoanthamin.
Schema 32. Theodorakis’ Ansatz f@r die Synthese des ABC-Ring-systems.
NaturstoffsyntheseAngewandte
Chemie
2415Angew. Chem. 2008, 120, 2400 – 2421 � 2008 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.de
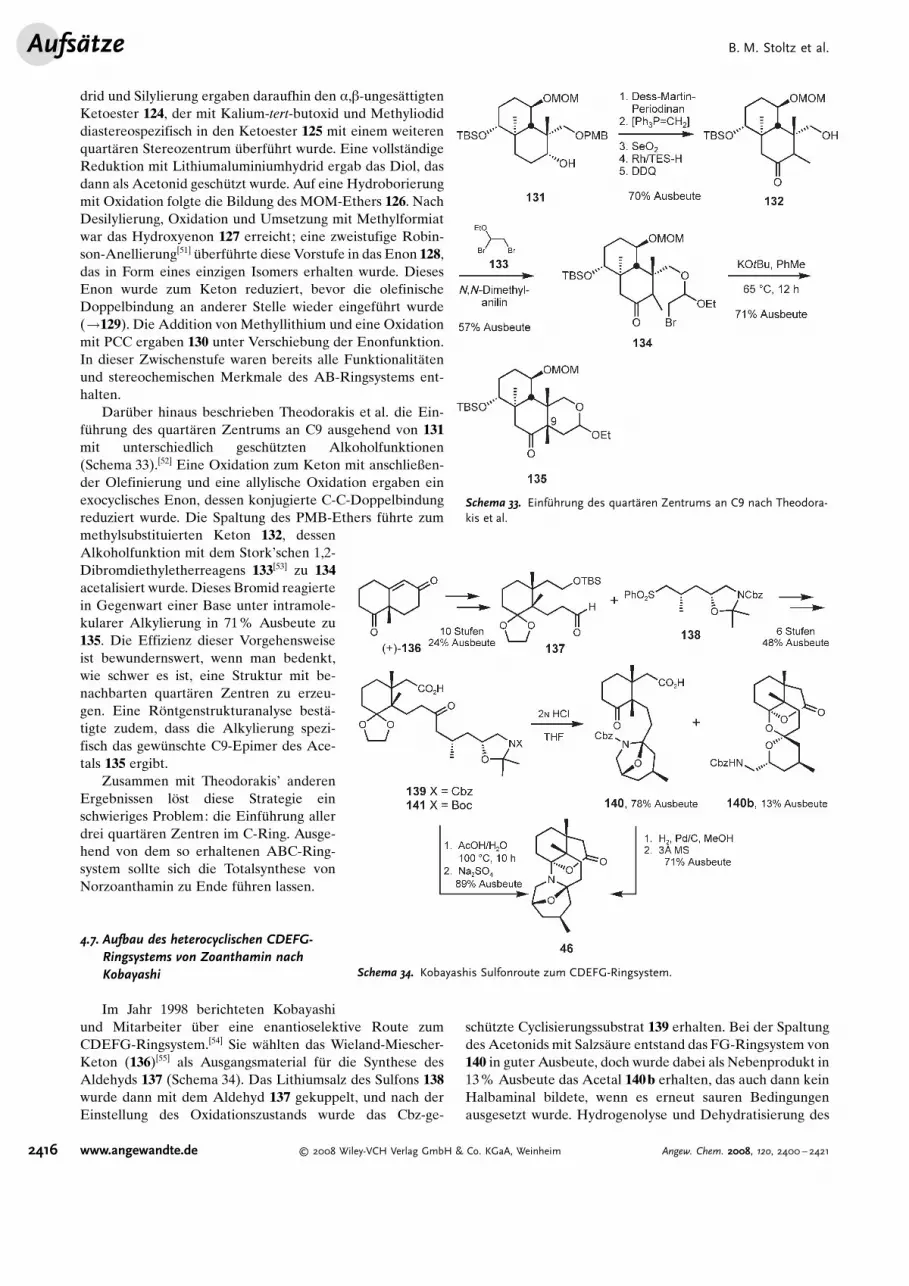
drid und Silylierung ergaben daraufhin den a,b-unges-ttigtenKetoester 124, der mit Kalium-tert-butoxid und Methyliodiddiastereospezifisch in den Ketoester 125 mit einem weiterenquart-ren Stereozentrum �berf�hrt wurde. Eine vollst-ndigeReduktion mit Lithiumaluminiumhydrid ergab das Diol, dasdann als Acetonid gesch�tzt wurde. Auf eine Hydroborierungmit Oxidation folgte die Bildung des MOM-Ethers 126. NachDesilylierung, Oxidation und Umsetzung mit Methylformiatwar das Hydroxyenon 127 erreicht; eine zweistufige Robin-son-Anellierung[51] �berf�hrte diese Vorstufe in das Enon 128,das in Form eines einzigen Isomers erhalten wurde. DiesesEnon wurde zum Keton reduziert, bevor die olefinischeDoppelbindung an anderer Stelle wieder eingef�hrt wurde(!129). Die Addition von Methyllithium und eine Oxidationmit PCC ergaben 130 unter Verschiebung der Enonfunktion.In dieser Zwischenstufe waren bereits alle Funktionalit-tenund stereochemischen Merkmale des AB-Ringsystems ent-halten.
Dar�ber hinaus beschrieben Theodorakis et al. die Ein-f�hrung des quart-ren Zentrums an C9 ausgehend von 131mit unterschiedlich gesch�tzten Alkoholfunktionen(Schema 33).[52] Eine Oxidation zum Keton mit anschließen-der Olefinierung und eine allylische Oxidation ergaben einexocyclisches Enon, dessen konjugierte C-C-Doppelbindungreduziert wurde. Die Spaltung des PMB-Ethers f�hrte zummethylsubstituierten Keton 132, dessenAlkoholfunktion mit dem StorkSschen 1,2-Dibromdiethyletherreagens 133[53] zu 134acetalisiert wurde. Dieses Bromid reagiertein Gegenwart einer Base unter intramole-kularer Alkylierung in 71% Ausbeute zu135. Die Effizienz dieser Vorgehensweiseist bewundernswert, wenn man bedenkt,wie schwer es ist, eine Struktur mit be-nachbarten quart-ren Zentren zu erzeu-gen. Eine R1ntgenstrukturanalyse best--tigte zudem, dass die Alkylierung spezi-fisch das gew�nschte C9-Epimer des Ace-tals 135 ergibt.
Zusammen mit TheodorakisS anderenErgebnissen l1st diese Strategie einschwieriges Problem: die Einf�hrung allerdrei quart-ren Zentren im C-Ring. Ausge-hend von dem so erhaltenen ABC-Ring-system sollte sich die Totalsynthese vonNorzoanthamin zu Ende f�hren lassen.
4.7. Aufbau des heterocyclischen CDEFG-Ringsystems von Zoanthamin nachKobayashi
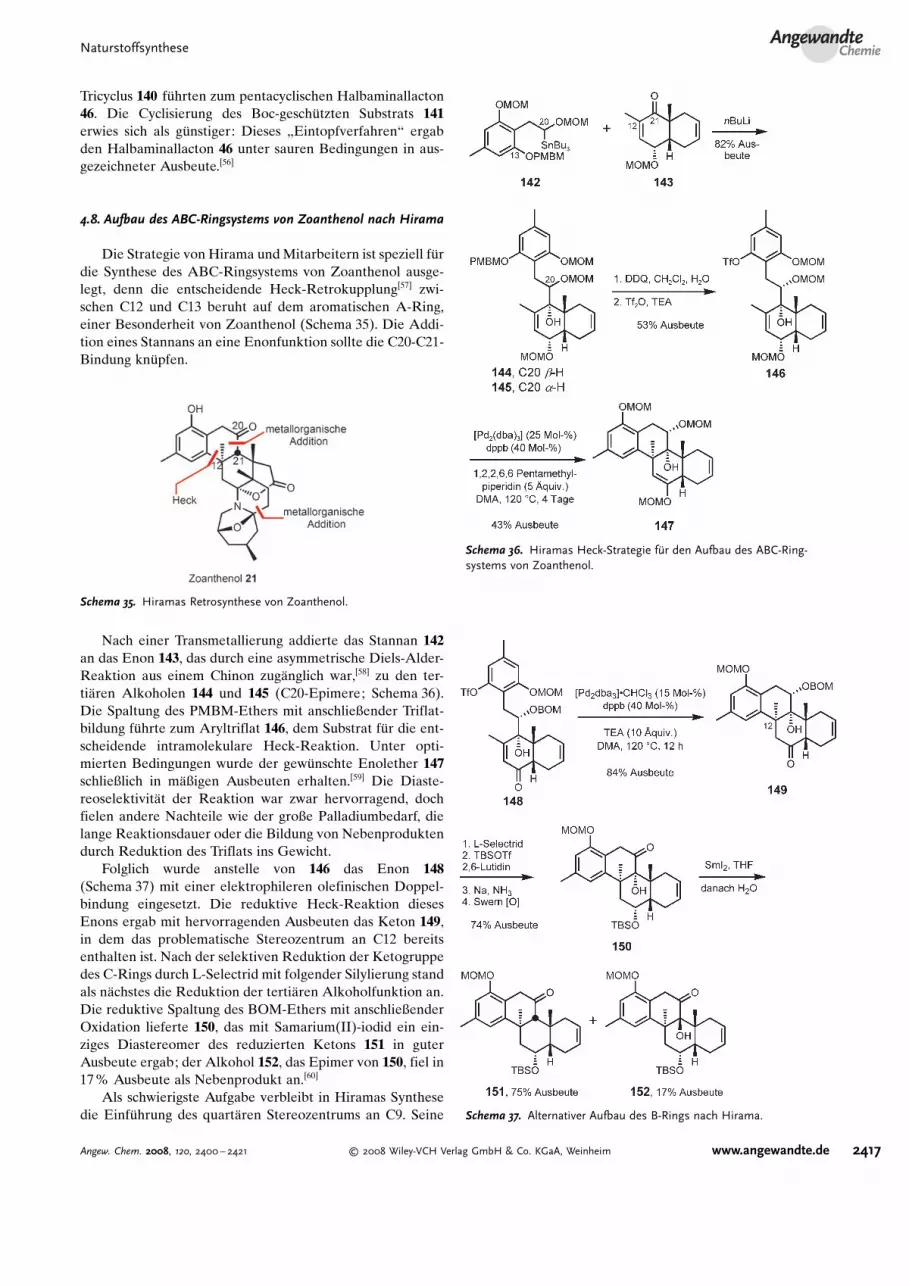
Im Jahr 1998 berichteten Kobayashiund Mitarbeiter �ber eine enantioselektive Route zumCDEFG-Ringsystem.[54] Sie w-hlten das Wieland-Miescher-Keton (136)[55] als Ausgangsmaterial f�r die Synthese desAldehyds 137 (Schema 34). Das Lithiumsalz des Sulfons 138wurde dann mit dem Aldehyd 137 gekuppelt, und nach derEinstellung des Oxidationszustands wurde das Cbz-ge-
sch�tzte Cyclisierungssubstrat 139 erhalten. Bei der Spaltungdes Acetonids mit Salzs-ure entstand das FG-Ringsystem von140 in guter Ausbeute, doch wurde dabei als Nebenprodukt in13% Ausbeute das Acetal 140 b erhalten, das auch dann keinHalbaminal bildete, wenn es erneut sauren Bedingungenausgesetzt wurde. Hydrogenolyse und Dehydratisierung des
Schema 33. Einf@hrung des quartAren Zentrums an C9 nach Theodora-kis et al.
Schema 34. Kobayashis Sulfonroute zum CDEFG-Ringsystem.
B. M. Stoltz et al.Aufs�tze
2416 www.angewandte.de � 2008 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2008, 120, 2400 – 2421
Tricyclus 140 f�hrten zum pentacyclischen Halbaminallacton46. Die Cyclisierung des Boc-gesch�tzten Substrats 141erwies sich als g�nstiger: Dieses „Eintopfverfahren“ ergabden Halbaminallacton 46 unter sauren Bedingungen in aus-gezeichneter Ausbeute.[56]
4.8. Aufbau des ABC-Ringsystems von Zoanthenol nach Hirama
Die Strategie von Hirama undMitarbeitern ist speziell f�rdie Synthese des ABC-Ringsystems von Zoanthenol ausge-legt, denn die entscheidende Heck-Retrokupplung[57] zwi-schen C12 und C13 beruht auf dem aromatischen A-Ring,einer Besonderheit von Zoanthenol (Schema 35). Die Addi-tion eines Stannans an eine Enonfunktion sollte die C20-C21-Bindung kn�pfen.
Nach einer Transmetallierung addierte das Stannan 142an das Enon 143, das durch eine asymmetrische Diels-Alder-Reaktion aus einem Chinon zug-nglich war,[58] zu den ter-ti-ren Alkoholen 144 und 145 (C20-Epimere; Schema 36).Die Spaltung des PMBM-Ethers mit anschließender Triflat-bildung f�hrte zum Aryltriflat 146, dem Substrat f�r die ent-scheidende intramolekulare Heck-Reaktion. Unter opti-mierten Bedingungen wurde der gew�nschte Enolether 147schließlich in m-ßigen Ausbeuten erhalten.[59] Die Diaste-reoselektivit-t der Reaktion war zwar hervorragend, dochfielen andere Nachteile wie der große Palladiumbedarf, dielange Reaktionsdauer oder die Bildung von Nebenproduktendurch Reduktion des Triflats ins Gewicht.
Folglich wurde anstelle von 146 das Enon 148(Schema 37) mit einer elektrophileren olefinischen Doppel-bindung eingesetzt. Die reduktive Heck-Reaktion diesesEnons ergab mit hervorragenden Ausbeuten das Keton 149,in dem das problematische Stereozentrum an C12 bereitsenthalten ist. Nach der selektiven Reduktion der Ketogruppedes C-Rings durch L-Selectrid mit folgender Silylierung standals n-chstes die Reduktion der terti-ren Alkoholfunktion an.Die reduktive Spaltung des BOM-Ethers mit anschließenderOxidation lieferte 150, das mit Samarium(II)-iodid ein ein-ziges Diastereomer des reduzierten Ketons 151 in guterAusbeute ergab; der Alkohol 152, das Epimer von 150, fiel in17% Ausbeute als Nebenprodukt an.[60]
Als schwierigste Aufgabe verbleibt in Hiramas Synthesedie Einf�hrung des quart-ren Stereozentrums an C9. Seine
Schema 35. Hiramas Retrosynthese von Zoanthenol.
Schema 36. Hiramas Heck-Strategie f@r den Aufbau des ABC-Ring-systems von Zoanthenol.
Schema 37. Alternativer Aufbau des B-Rings nach Hirama.
NaturstoffsyntheseAngewandte
Chemie
2417Angew. Chem. 2008, 120, 2400 – 2421 � 2008 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.de

Arbeitsgruppe konnte aber schon eine hoch diastereoselek-tive Methylierung des Silylenolethers 153 als Modellreaktionf�r die Methylierung an C9 ausarbeiten (Schema 38).[61] EineSamarium(II)-iodid-vermittelte Cyclopropanierung und eineRing1ffnung unter sauren Bedingungen ergaben das me-thylsubstituierte Keton 155 und dessen C9-Epimer im Ver-h-ltnis 3:1 zugunsten des gew�nschten Produkts.
K�rzlich haben Hirama und Mitarbeiter eine alternativeRoute zum ABC-Ringsystem von Zoanthenol vorgestellt, beider die Bindungen des B-Rings in der umgekehrten Reihen-folge gebildet werden (Schema 39).[62] Eine Suzuki-Kupplung
verkn�pfte den A-Ring-Baustein 156 (Aryltriflat) und den C-Ring-Baustein 157 (Boran) �ber die C12-C13-Bindung zumBiaryl 158. Entfernen der BOM-Gruppe und Oxidation er-gaben das Chinon 159, das durch Erhitzen mit Butadien in 160�berf�hrt wurde. Im weiteren Verlauf k1nnte ausgehend vomDiels-Alder-Addukt 160 die fehlende Bindung des B-Rings,C20-C21, aufgebaut werden: entweder durch Addition einesOrganometallreagens (-hnlich wie in der Synthese der ter-ti-ren Alkohole 144/145 und 148) oder durch eine Pinakol-Kupplung nach Einf�hrung einer Aldehydfunktion an C20.
4.9. Aufbau des carbocyclischen Systems von Zoanthenol nachStoltz
Die Gruppe um Stoltz hat vor kurzem einen Zugang zumABC-Ringsystem von Zoanthenol beschrieben,[63] bei demdas quart-re Zentrum an der Verkn�pfungsstelle von B- undC-Ring anders als bei den �brigen Strategien durch eines-urevermittelte Friedel-Crafts-Alkylierung aufgebaut wird.Eine selektive Grignard-Addition soll den A- mit dem C-Ring verbinden, und der „s�dliche“Molek�lteil soll durch dieMichael-Addition an ein Enon angesetzt werden, das sichdurch sein Funktionalisierungsmuster f�r die thermodyna-misch kontrollierte Cyclisierung zum DEFG-Ringsystemeignet (Schema 40).
Eine enantiokonvergente Allylierung �berf�hrte denracemischen b-Ketoester 161 in das Keton (�)-162 mit einemquart-ren Zentrum in a-Stellung (Schema 41);[64] die Reak-
tion ergab hohe Ausbeuten und konnte bis in den 25-mmol-Maßstab �bertragen werden. Die olefinische Doppelbindungwurde anschließend oxidativ gespalten, und die resultierendeCarbons-ure wurde mit Boc2O in den tert-Butylester (+)-163umgewandelt. Deprotonieren mit LDA und Abfangen mitMethyliodid lieferten ein enantiomerenangereichertes Dia-stereomerengemisch des methylsubstituierten Ketons (+)-164. Das Keton wurde dann enolisiert und als Triflat 165abgefangen, dessen reduktive Carbonylierung unter opti-
Schema 38. Einf@hrung der Methylgruppe an C9 nach Hirama.
Schema 39. Hiramas modifizierte Strategie.
Schema 40. Stoltz’ Retrosynthese von Zoanthenol.
Schema 41. Enantioselektive Allylierung zum Aufbau des C-Ring-Bau-steins.
B. M. Stoltz et al.Aufs�tze
2418 www.angewandte.de � 2008 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2008, 120, 2400 – 2421

mierten Bedingungen das Enal 166 ergab (Schema 42). DieAddition des Grignard-Reagens 167 an 166 f�hrte in hoherAusbeute und mit ausgezeichneter Selektivit-t zum ge-w�nschten Diastereomer des Alkohols 168.
Durch die s-urevermittelte Cyclisierung dieser Zwi-schenstufe wollten Stoltz und Mitarbeiter den B-Ring vonZoanthenol aufbauen und das quart-re Stereozentrum anC12 einrichten. Der Allylalkohol 168 wurde dazu in reinerTrifluoressigs-ure auf 50 8C erhitzt und anschließend mitTBAF behandelt, um verbliebenen TBS-Ether zu spalten.Nach diesem Prozess konnte das gew�nschte Diastereomerdes Tricyclus 169 in 49% Ausbeute isoliert werden
(Schema 43). Diese Reaktion verl-uft erstaunlicherweise als6-endo-Cyclisierung nach einem SN’-Mechanismus. EineDoppelbindungsverschiebung zum endocyclischen C-Ring-Enon mit anschließender 6-exo-Cyclisierung und Dehydrati-sierung wurde nicht beobachtet.
Der Tricyclus 169 wurde verestert und nach Bildung desAryltriflats in Gegenwart von PdII zum Methylester 170 re-duziert. Beim Aufbau des Tricyclus 169 wurde C20 zwardesoxygeniert, an dieser Stelle konnte nun aber eine Keto-gruppe glatt wieder eingef�hrt werden: Verseifung des Me-thylesters, Ketalisierung und eine iodierende Lactonisierungf�hrten zu 171, und nach der Methanolyse des Lactonrings,Epoxidierung und einer 1,2-Hydridverschiebung wurde dasKeton 172 erhalten. Durch Spaltung des Ketals gelangte manzum Diketon 173, das r1ntgenkristallographisch analysiertwurde, um die stereochemische Zuordnung f�r die neu ge-bildeten quart-ren Zentren an C12 und C22 zu best-tigen.[63]
5. Zusammenfassung und Ausblick
Die Zoanthamin-Alkaloide sind Naturstoffe mit einzig-artigen Strukturen. Diese Sekund-rmetaboliten wurden zwaraus Weichkorallen der Ordnung Zoantharia isoliert, es istjedoch nicht auszuschließen, dass symbiontischen Algen einewichtige Rolle bei ihrer Biosynthese zukommt. Die Biosyn-these k1nnte nach einem Polyketidweg verlaufen, Detailshierzu sind aber ebensowenig bekannt wie der Verwen-dungszweck der komplexen Naturstoffe in den produzieren-den Organismen. Die Isolierung verschiedener Zoanthamin-Alkaloide aus Spezies des Indischen, Pazifischen und Atlan-tischen Ozeans legt allerdings nahe, dass sie eine wichtige
Funktion erf�llen.Einige Zoanthamine wirken gegen
Osteoporose, antibiotisch, entz�ndungs-hemmend oder zytotoxisch, sodass einInteresse an der Synthese dieser Verbin-dungen aufkam. Die Synthese der Zoan-thamin-Alkaloide fordert die verf�gbarenMethoden und f1rdert die Entwicklungneuer Reaktionen. Gew1hnlich vergehennach der Isolierung eines Naturstoffs mitinteressanter Struktur oder biologischerWirkung nur noch ein bis zwei Jahre biszur Totalsynthese, die Isolierung vonZoanthamin und Miyashitas Totalsynthe-se von Norzoanthamin im Jahr 2004 tren-nen hingegen zwanzig Jahre. Jede erfolg-reiche Synthese eines dieser Alkaloideerfordert ausgezeichnete Kenntnisse inCarbo- wie Heterocyclenchemie. DerAufbau des carbocyclischen ABC-Ring-systems wird durch die große Zahl anstereochemischen Merkmalen in diesemMolek�labschnitt – namentlich die dreiquart-ren Zentren des C-Rings – er-schwert. Um diese Aufgabe zu meistern,wurde eine Reihe kreativer Anellie-rungsstrategien entwickelt, bei denen
Schema 42. Verkn@pfung von A- und C-Ring durch eine diastereoselek-tive Grignard-Addition.
Schema 43. Stoltz’ Synthese der carbocyclischen Region von Zoanthenol.
NaturstoffsyntheseAngewandte
Chemie
2419Angew. Chem. 2008, 120, 2400 – 2421 � 2008 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.de

Diels-Alder-, Heck- und Friedel-Crafts-Reaktionen sowieRobinson-Anellierungen zum Einsatz kamen. Die hetero-cyclische DEFG-Region ist topographisch komplex und ent-h-lt einige empfindliche funktionelle Gruppen. Erste Syn-thesen dieses heterocyclischen Molek�lteils bewiesen, dassunterschiedliche Cyclisierungsstrategien gangbar sind.
Seit �ber zwanzig Jahren liefern die neuartigen biologi-schen Aktivit-ten und die Synthese der Zoanthamine nunStoff f�r wissenschaftliche Untersuchungen. Es sind nochviele Fragen offen, und so wird das Interesse an diesen Al-kaloiden wohl weiterhin zunehmen.
Abk#rzungen
Boc tert-ButoxycarbonylCbz Benzyloxycarbonyldba trans,trans-DibenzylidenacetonDBU 1,8-Diazabicyclo[5.4.0]undec-7-enDMA N,N-DimethylacetamidDMDO DimethyldioxiranDME 1,2-DimethoxyethanDMF N,N-DimethylformamidDMP Dess-Martin-Periodinand.r. Diastereomerenverh-ltnisHMBC (Heterokern)korrelation �ber mehrere
BindungenHMDS Hexamethyldisilazid oder Hexamethyldi-
silazanHMPA HexamethylphosphoramidIC50 inhibitorische Konzentration f�r 50% einer
VersuchspopulationIL-6 Interleukin 6LD50 letale Dosis f�r 50% einer Versuchspopu-
lationMH-60 murine MyelohybridomzellenMOM MethoxymethylMTPA a-Methoxy-a-(trifluormethyl)phenylessig-
s-ureMVK MethylvinylketonNOE Kern-Overhauser-EffektPCC PyridiniumchlorochromatPG ProstaglandinPMA PhorbolmyristatacetatPMB para-MethoxybenzylPMBM para-Methoxybenzyloxymethylp.o. per os (orale Anwendung)py PyridinROESY rotating-frame Overhauser effect spectros-
copySAR Struktur-Aktivit-ts-BeziehungTEA TriethylaminTBS tert-ButyldimethylsilylTf TrifluormethansulfonylTIPS TriisopropylsilylTs para-Toluolsulfonyl
Wir danken www.zoaid.com f,r die Bilder von Zoanthideaund Teresa Barth f,r Unterst,tzung bei der graphischen Ge-staltung des Beitrags und des Vortitelbildes.
Eingegangen am 16. Juli 2007Online ver1ffentlicht am 28. Februar 2008>bersetzt von Dr. Volker Jacob, Mannheim
[1] a) S. Ono, J. D. Reimer, J. Tsukahara, Zool. Sci. 2005, 22, 247 –255; b) J. S. Ryland, Invert. Rep. Develop. 1997, 31, 177 – 188.
[2] F. Sinniger, J. I. Montoya-Burgos, P. ChevaldonnT, J. Pawlowski,Mar. Biol. 2005, 147, 1121 – 1128.
[3] C. B. Rao, A. S. R. Anjaneyulu, N. S. Sarma, Y. Venkateswarlu,R. M. Rosser, D. J. Faulkner, M. H. M. Chen, J. Clardy, J. Am.Chem. Soc. 1984, 106, 7983 – 7984.
[4] H. Nakamura, Y. Kawase, K. Maruyama, A. Muria, Bull. Chem.Soc. Jpn. 1998, 71, 781 – 787.
[5] a) A. Suksamrarn, A. Jankam, B. Tarnchompoo, S. Putchakarn,J. Nat. Prod. 2002, 65, 1194 – 1197; b) H. Shigemori, Y. Sato, T.Kagata, J. Kobayashi, J. Nat. Prod. 1999, 62, 372 – 374.
[6] C. Han, J. Qi, X. Shi, Y. Sakagami, T. Shibata, K. Uchida, M.Ojika, Biosci. Biotechnol. Biochem. 2006, 70, 706 – 711.
[7] J. J. FernWndez, M. L. Souto, A. H. Daranas, M. Norte,Curr. Top.Phytochem. 2000, 4, 106 – 119.
[8] K. Sepcic, T. Turk, P. Macek, Toxicon 1998, 36, 937 – 940.[9] R. E. Moore, P. J. Scheuer, Science 1971, 172, 495 – 498.[10] Strukturaufkl-rung: J. K. Cha, W. J. Christ, J. M. Finan, H. Fu-
jioka, Y. Kishi, L. L. Klein, S. S. Ko, J. Leder, W. W. McWhorter,K.-P. Pfaff, M. Yonaga, D. Uemura, Y. Hirata, J. Am. Chem. Soc.1982, 104, 7369 – 7371; Totalsynthese: E. M. Suh, Y. Kishi, J. Am.Chem. Soc. 1994, 116, 11205 – 11206.
[11] C. B. Rao, A. S. R. Anjaneyula, N. S. Sarma, Y. Venkatateswarlu,R. M. Rosser, D. J. Faulkner, J. Org. Chem. 1985, 50, 3757 – 3760.
[12] C. B. Rao, D. V. Roa, V. S. N. Raju, Heterocycles 1989, 28, 103 –106.
[13] S. Fukuzawa, Y. Hayashi, D. Uemura, A. Nagatsu, K. Yamada,Y. Ijuin, Heterocycl. Commun. 1995, 1, 207 – 214.
[14] M. Kuramoto, K. Hayashi, Y. Fujitani, K. Yamaguchi, T. Tsuji,K. Yamada, Y. Ijuin, D. Uemura, Tetrahedron Lett. 1997, 38,5683 – 5686.
[15] Atta-ur-Rahman, K. A. Alvi, S. A. Abbas, M. I. Choudhary, J.Clardy, Tetrahedron Lett. 1989, 30, 6825 – 6828.
[16] A. H. Daranas, J. J. FernWndez, J. A. GavXn, M. Norte, Tetrahe-dron 1998, 54, 7891 – 7896.
[17] A. H. Daranas, J. J. FernWndez, J. A. GavXn, M. Norte, Tetrahe-dron 1999, 55, 5539 – 5546.
[18] a) M. Kuramoto, K. Hayashi, K. Yamaguchi, M. Yada, T. Tsuji,D. Uemura, Bull. Chem. Soc. Jpn. 1998, 71, 771 – 779; b) D.Uemura, Chem. Rec. 2006, 6, 235 – 248.
[19] R. K. Trench, Pure Appl. Chem. 1981, 53, 819 – 835.[20] In dieser Studie wurde mit Zooxanthellatoxin auch eine Ver-
bindung isoliert, die Palytoxin sehr -hnlich ist; siehe Lit. [4] f�rDetails.
[21] M. Kuramoto, H. Arimoto, K. Hayashi, I. Hayakawa, D.Uemura, T. Chou, K. Yamada, T. Tsuji, K. Yamaguchi, K.Yazawa, Symposium Papers, 38th Symposium on the Chemistryof Natural Products 1996, 79 – 84.
[22] T. J. Chambers, J. Cell Sci. 1982, 57, 247 – 260.[23] K. Yamaguchi, M. Yada, T. Tsuji, M. Kuramoto, D. Uemura,
Biol. Pharm. Bull. 1999, 22, 920 – 924.[24] P. V. N. Bodine, H. A. Harris, B. S. Komm, Endocrinology 1999,
140, 2439 – 2451.[25] K. Kuramoto, K. Yamaguchi, T. Tsuji, D. Uemura inDrugs from
the Sea (Hrsg.: N. Fusetani), Karger, Basel, 2000, S. 98 – 106.[26] Weitere Beschreibungen dieser Aktivit-t: a) M. Kuramoto, H.
Arimoto, D. Uemura, J. Synth. Org. Chem. Jpn. 2003, 61, 59 – 65;
B. M. Stoltz et al.Aufs�tze
2420 www.angewandte.de � 2008 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2008, 120, 2400 – 2421

b) M. Kuramoto, H. Arimoto, D. Uemura, Mar. Drugs 2004, 1,39 – 54; c) M. Kita, D. Uemura, Chem. Lett. 2005, 34, 454 – 459;d) K. Yamada, M. Kuramoto, D. Uemura, Recent Res. Dev. PureAppl. Chem. 1999, 3, 245 – 254.
[27] Die IC50-Werte beziehen sich auf die Inhibition der IL-6-Pro-duktion in pr-osteoblastischen MC3T3-E1-Zellen, die mit Pa-rathyroidhormon stimuliert wurden; siehe Lit. [25].
[28] G. Hirai, H. Oguri, M. Hayashi, K. Koyama, Y. Koizumi, S. M.Moharram, M. Hirama, Bioorg. Med. Chem. Lett. 2004, 14,2647 – 2651.
[29] Y. Venkateswarlu, N. S. Reddy, P. Ramesh, P. S. Reddy, K. Jamil,Heterocycl. Commun. 1998, 4, 575 – 580.
[30] R. M. Villar, J. Gil-Longo, A. H. Daranas, M. L. Souto, J. J.FernWndez, S. Peixinho, M. A. Barral, G. SantafT, J. RodrXguez,C. JimTnez, Bioorg. Med. Chem. 2003, 11, 2301 – 2306.
[31] N. L. Benowitz, Prog. Cardiovasc. Dis. 2003, 46, 91 – 111.[32] S. P. Jackson, S. M. Schoenwalder, Nat. Rev. 2003, 2, 1 – 15.[33] M. Miyashita, M. Sasaki, I. Hattori, M. Sakai, K. Tanino, Science
2004, 305, 495 – 499; M. Miyashita, Pure Appl. Chem. 2007, 79,651 – 665.
[34] M. Sakai, M. Sasaki, K. Tanino, M. Miyashita, Tetrahedron Lett.2002, 43, 1705 – 1708.
[35] D. Tanner, P. G. Anderson, L. Tedenborg, P. Somfai, Tetrahedron1994, 50, 9135 – 9144.
[36] a) D. Tanner, L. Tedenborg, P. Somfai, Acta Chem. Scand. 1997,51, 1217 – 1223; b) T. E. Nielsen, D. Tanner, J. Org. Chem. 2002,67, 6366 – 6371.
[37] T. E. Nielsen, S. Le Quement, M. Juhl, D. Tanner, Tetrahedron2005, 61, 8013 – 8024.
[38] X. Han, B. M. Stoltz, E. J. Corey, J. Am. Chem. Soc. 1999, 121,7600 – 7605.
[39] M. Juhl, T. E. Nielsen, S. Le Quement, D. Tanner, J. Org. Chem.2006, 71, 265 – 280.
[40] M. Juhl, R.Monrad, I. Søtofte, D. Tanner, J. Org. Chem. 2007, 72,4644 – 4654.
[41] T. Irifune, T. Ohashi, T. Ichino, E. Sakia, K. Suenaga, D. Uemura,Chem. Lett. 2005, 34, 1058 – 1059.
[42] K. Sonogashira, Y. Tohdo, N. Hagihara, Tetrahedron Lett. 1975,16, 4467 – 4470.
[43] D. R. Williams, T. A. Brugel, Org. Lett. 2000, 2, 1023 – 1026.[44] >bersicht zur Nef-Reaktion: H. W. Pinnick, Org. React. 1990,
38, 655 – 792.[45] D. R. Williams, D. C. Ihle, T. A. Brugel, S. Patanaik, Hetero-
cycles 2006, 70, 77 – 82.[46] D. R. Williams, G. A. Cortez, Tetrahedron Lett. 1998, 39, 2675 –
2678.
[47] a) J. dSAngelo, D. DesmaZle, F. Dumas, A. Guingant, Tetrahe-dron: Asymmetry 1992, 3, 459 – 505; b) P. W. Hickmott, Tetra-hedron 1984, 40, 2989 – 3051.
[48] >bersicht zur Evans-Aldolmethode: D. J. Ager, I. Prakash,D. R. Schaad, Aldrichimica Acta 1997, 30, 3 – 12.
[49] S. Ghosh, F. Rivas, D. Fischer, M. A. GonzWlez, E. A. Theodo-rakis, Org. Lett. 2004, 6, 941 – 944.
[50] a) T. Ling, C. Chowdhury, B. A. Kramer, B. G. Vong, M. A.Palladino, E. A. Theodorakis, J. Org. Chem. 2001, 66, 8843 –8853; b) T. Ling, B. A. Kramer, M. A. Palladino, E. A. Theo-dorakis, Org. Lett. 2000, 2, 2073 – 2076.
[51] >bersicht zu Robinson-Anellierungen: R. E. Gawley, Synthesis1976, 777 – 794.
[52] F. Rivas, S. Ghosh, E. A. Theodorakis, Tetrahedron Lett. 2005,46, 5281 – 5284.
[53] Stork et al. setzten das Dibromid 133 in radikalischen Cyclisie-rungen ein: a) G. Stork, R. Mook, S. A. Biller, S. D. Rychnovsky,J. Am. Chem. Soc. 1983, 105, 3741 – 3742; b) G. Stork, P. M. Sher,J. Am. Chem. Soc. 1983, 105, 6765 – 6766.
[54] a) N. Hikage, H. Furukawa, K. Takao, S. Kobayashi, TetrahedronLett. 1998, 39, 6237 – 6240; b) N. Hikage, H. Furukawa, K. Takao,S. Kobayashi, Tetrahedron Lett. 1998, 39, 6241 – 6244.
[55] a) P. Wieland, K. Miescher, Helv. Chim. Acta 1950, 33, 2215 –2228; b) enantioselektive Synthese: P. Buchschacher, A. F�rst, J.Gutzwiller, Org. Synth. 1985, 63, 37 – 43.
[56] N. Hikage, H. Furukawa, K. Takao, S. Kobayashi, Chem. Pharm.Bull. 2000, 48, 1370 – 1372.
[57] a) R. F. Heck, J. P. Nolley, J. Am. Chem. Soc. 1968, 90, 5518 –5526; b) R. F. Heck, Acc. Chem. Res. 1979, 12, 146 – 151; c) R. F.Heck, Org. React. 1982, 27, 345 – 390.
[58] S. M. Moharram, G. Hirai, K. Koyama, H. Oguri, M. Hirama,Tetrahedron Lett. 2000, 41, 6669 – 6673.
[59] G. Hirai, H. Oguri, S. M. Moharram, K. Koyama, M. Hirama,Tetrahedron Lett. 2001, 42, 5783 – 5787.
[60] G. Hirai, Y. Koizumi, S. M. Moharram, H. Oguri, M. Hirama,Org. Lett. 2002, 4, 1627 – 1630.
[61] G. Hirai, H. Oguri, M. Hirama, Chem. Lett. 1999, 141 – 142.[62] S. M. Moharram, H. Oguri, M. Hirama, Egypt. J. Pharm. Sci.
2003, 44, 177 – 193.[63] D. C. Behenna, J. L. Stockdill, B. M. Stoltz, Angew. Chem. 2007,
119, 4155 – 4158; Angew. Chem. Int. Ed. 2007, 46, 4077 – 4080.[64] a) D. C. Behenna, B. M. Stoltz, J. Am. Chem. Soc. 2004, 126,
15044 – 15045; b) J. T. Mohr, D. C. Behenna, A. M. Harned,B. M. Stoltz, Angew. Chem. 2005, 117, 7084 – 7087; Angew.Chem. Int. Ed. 2005, 44, 6924 – 6927; c) R. M. McFadden, B. M.Stoltz, J. Am. Chem. Soc. 2006, 128, 7738 – 7739.
NaturstoffsyntheseAngewandte
Chemie
2421Angew. Chem. 2008, 120, 2400 – 2421 � 2008 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.de





























