Embed Size (px)
Citation preview

ARTHRITIS & RHEUMATISMVol. 46, No. 10, October 2002, pp 2695–2707DOI 10.1002/art.10552© 2002, American College of Rheumatology
Association of Autoantibodies to Nuclear Lamin B1 WithThromboprotection in Systemic Lupus Erythematosus
Lack of Evidence for a Direct Role of Lamin B1 in Apoptotic Blebs
Melanie Dieude,1 Jean-Luc Senecal,1 Joyce Rauch,2 John G. Hanly,3 Paul Fortin,4
Nathalie Brassard,1 and Yves Raymond1
Objective. To demonstrate the association be-tween autoantibodies to nuclear lamin B1 (aLB1) andprotection against thrombosis (“thromboprotection”) inpatients with systemic lupus erythematosus (SLE), andto elucidate the mechanism by which aLB1 cause throm-boprotection in vivo. Since a number of autoantigens inSLE have been localized specifically to the externalsurface of apoptotic blebs, it was hypothesized thatcirculating aLB1 may block the procoagulant effect ofapoptotic blebs by binding to LB1 displayed at theexternal bleb surface.
Methods. A cross-sectional study was performedusing serum samples obtained at first evaluation of 259English Canadian and French Canadian patients fromSLE registries at 3 hospitals. A case–control study wasperformed to analyze the relationship between aLB1and lupus anticoagulant (LAC) status and thromboticmanifestations between onset of disease and last fol-
lowup. Reactivity of aLB1 with Jurkat or endothelialcells, which had been induced to undergo apoptosis, wasdetermined by indirect immunofluorescence. Localiza-tion of LB1 in apoptotic cells and blebs was analyzed byconfocal microscopy and surface labeling of cell mem-brane proteins.
Results. High-titer aLB1 was restricted to a sub-set of SLE patients (46 patients), with an overallfrequency of 17.8% (range 11.6–24.3% in the 3 centers).LB1 antibodies were significantly associated with LACbut not with antibodies to cardiolipin (aCL) or �2-glycoprotein I (anti-�2GPI). The frequency of thrombo-sis differed markedly depending on aLB1 and LACstatus, as follows: presence of LAC and absence of aLB150%, presence of both LAC and aLB1 22.7%, absence ofboth LAC and aLB1 25.5%, absence of LAC and pres-ence of aLB1, 20.8%. Further subclassification of pa-tients based on aCL and anti-�2GPI status revealedthat, in the presence of LAC but in the absence of aCL,anti-�2GPI, and aLB1, the frequency of thrombosis was40%, whereas in the presence of aLB1, it decreasedstrikingly, to 9.1%. LB1 was found to be translocatedinto surface membrane blebs during apoptosis and to beentirely enclosed within the apoptotic bleb plasma mem-brane of Jurkat and endothelial cells.
Conclusion. The presence of aLB1 in SLE pa-tients with LAC essentially nullifies the strong pro-thrombotic risk associated with LAC. Hence, aLB1 isassociated with thromboprotection. Reactivity of aLB1with apoptotic blebs does not seem to play a direct rolein mediating this protection, since LB1 is buried withinapoptotic blebs and inaccessible to circulating aLB1.The mechanism by which aLB1 confers thromboprotec-tion in SLE remains to be elucidated.
Supported by grant MOP-36436 (to Drs. Senecal and Ray-mond) and grant MT-42391 (to Dr. Rauch) from the CanadianInstitutes of Health Research.
1Melanie Dieude, MSc, Jean-Luc Senecal, MD, FRCPC,FACP, Nathalie Brassard, BSc, Yves Raymond, PhD: Notre-DameHospital, Centre Hospitalier de l’Universite de Montreal, Montreal,Quebec, Canada; 2Joyce Rauch, PhD: Research Institute of the McGillUniversity Health Centre, Montreal, Quebec, Canada; 3John G.Hanly, MD, MRCPI, FRCPC: Queen Elizabeth II Health SciencesCentre and Dalhousie University, Halifax, Nova Scotia, Canada; 4PaulFortin, MD, MPH, FRCPC: University of Toronto, Toronto, Ontario,Canada.
Melanie Dieude and Dr. Senecal contributed equally to thiswork.
Address correspondence and reprint requests to Yves Ray-mond, PhD, Autoimmunity Research Laboratory, Notre-Dame Hos-pital, CHUM 1560 Sherbrooke Street East, Montreal, Quebec H2L4M1, Canada. E-mail: [email protected].
Submitted for publication February 22, 2002; accepted inrevised form June 18, 2002.
2695

The antiphospholipid syndrome is defined byrecurrent thrombotic events and pregnancy morbidity inassociation with the presence of antiphospholipid anti-bodies (aPL), such as lupus anticoagulant (LAC) (1).Systemic lupus erythematosus (SLE) is the main auto-immune disease associated with the antiphospholipidsyndrome. Of the various aPL observed in SLE, thepresence of LAC may be the strongest risk factor forboth arterial and venous thrombosis (2). LAC is presentin approximately one-third of SLE patients, and approx-imately half of these LAC-positive patients will developarterial or venous thrombosis during long-term followup(3,4). Thus, paradoxically, although LAC is a strongpredictor for thrombosis, half of the patients expressingLAC may not develop thrombotic episodes. In clinicalpractice, this causes a management dilemma. Acceptedpractice is to treat patients with LAC only when a firstserious thrombotic episode occurs, thus exposing un-treated patients to the risks of major end organ damagesuch as stroke. The question remains as to whetherlong-term prophylactic treatment such as oral anticoag-ulation, with associated side effects and costs, should beinstituted as soon as the presence of LAC is docu-mented, in the absence of a first thrombotic event. Thepressing need to resolve this issue has been emphasizedrecently (5). At present, there is no established methodto differentiate SLE patients with LAC who will developthrombotic episodes from those who will not.
The nuclear autoantigen lamin B1 (LB1) is apolypeptide forming part of the polymeric lamina mesh-work located between chromatin and the inner nuclearmembrane (for review, see ref. 6). Our earlier studies ina cohort of French Canadians with SLE showed thathigh titers of autoantibodies to LB1 (aLB1) were re-stricted to a subset of patients (7). Intriguingly, despite astrong association of aLB1 with LAC, SLE patients withLAC and aLB1 had a lower risk of thrombotic manifes-tations than those with LAC but without aLB1 (7).However, that study was restricted to a single center, andthe number of patients with aLB1 was small. Therefore,we conducted the present study to determine whetherthe correlation between the presence of aLB1 and anapparent reduced risk of thrombosis could be demon-strated in a large number of SLE patients from multiplecenters. As shown herein, we found that aLB1 wasindeed associated with a striking degree of protectionagainst thrombosis (“thromboprotection”) in SLE pa-tients with LAC.
In addition, we aimed to elucidate the mecha-nism by which aLB1 may cause thromboprotection invivo. Our hypothesis was that circulating aLB1 may
block the procoagulant effect of apoptotic blebs bybinding to LB1 displayed at the external bleb surface.The rationale for this hypothesis stemmed from 4 obser-vations. First, surface blebs of apoptotic cells are sites ofenhanced procoagulant activity, and this may be funda-mental to the pathogenesis of thrombotic events inpatients with aPL (8–10). Second, apoptotic cells andtheir resulting blebs are known sources of antigens thatare targeted by autoantibodies (9–13). Third, majorautoantigens of nuclear origin (e.g., La), of cytoplasmicorigin (e.g., Ro), and of granular origin (e.g., myeloper-oxidase) have been localized specifically to the externalsurface of apoptotic blebs, where their accessibility tocirculating autoantibodies may form the molecular basisof their pathogenic effects (14,15). Finally, LB1 is awell-characterized and early substrate of caspase 6 dur-ing apoptosis (16,17), suggesting that it might be presentin apoptotic blebs. Thus, we investigated whether LB1was present in apoptotic blebs. We found that it wasindeed present in apoptotic blebs but, in striking con-trast to other SLE autoantigens, it was entirely con-cealed within the apoptotic bleb plasma membrane andinaccessible to aLB1, thereby excluding the notion thatLB1 has a direct role in mediating thromboprotection.
PATIENTS AND METHODS
Patients and controls. A cross-sectional study of IgGaLB1 was performed with coded serum samples that wereconsecutively and unselectively obtained at the time of firstpatient evaluation by the authors (J-LS, JGH, PF) at theirrespective lupus clinics (Notre-Dame Hospital of the CentreHospitalier de l’Universite de Montreal [NDH], MontrealGeneral Hospital of McGill University Health Center [MGH],and Queen Elizabeth II Health Sciences Centre [QEIIHSC]).Serum samples were frozen at �70°C or �80°C. The studypopulation consisted of 259 adult SLE patients and 45 healthycontrols. The NDH cohort consisted of the original 94 patientsin the study that formed the basis for initiating the presentmulticenter study (7), plus 18 additional patients. All SLEpatients fulfilled the American College of Rheumatologycriteria for the disease (18). Criteria for inclusion in the controlgroup were as described previously (7,19). The proportion ofwomen in the control group was higher than in the generalpopulation because of the female predominance in SLE.
Thrombosis. A case–control design was used to com-pare LAC status and status of aLB1 and other antibodies inpatients with and those without thrombosis. Arterial andvenous thrombotic events were identified as previously de-scribed (7). Briefly, the charts of the 259 SLE patients werereviewed retrospectively, using a standardized protocol forthrombotic and embolic manifestations at any time betweendisease onset and last followup. Patients were categorized ashaving a history of thrombosis (cases) if they had experiencedat least 1 definite thrombotic event; other SLE patients were
2696 DIEUDE ET AL

categorized as thrombosis-free (controls). Among patientswith thrombosis, 33 had deep vein thrombosis, 29 had cerebralinfarction, 19 had myocardial infarction, 23 had other throm-botic events, and 13 had embolic events. One or more of theseevents had occurred in 81 patients. Patients with LAC butwithout a thrombotic event were not treated prophylacticallywith aspirin or anticoagulants.
Antiphospholipid antibodies. Plasma samples from thepatients (obtained between 1979 and 1998) were considered tohave LAC activity if the activated partial thromboplastin timewas prolonged by at least 8 seconds (compared with that innormal control plasma) and the prolongation was not cor-rected by a 1:1 dilution with normal plasma. LAC was consid-ered positive if present on at least 2 occasions at least 3 monthsapart. Antibodies to cardiolipin (aCL) and to �2-glycoproteinI (anti-�2GPI) were determined by enzyme-linked immunosor-bent assay (ELISA), using IgG- and IgM-specific conjugates aspreviously described (7).
Anti-LB1 ELISA. Immulon-2 high-binding ELISA plates(Dynatech, Alexandria, VA) were coated for 16 hours at 4°C with4 �g/ml of purified LB1 (7) in phosphate buffered saline (PBS)(pH 7.4, unless stated otherwise) or with PBS containing 0.2%normal goat serum (Gibco BRL, Grand Island, NY) and 0.04%casein to determine the nonspecific binding (no antigen) of eachserum. All other incubations were done at 25°C. After 4 washeswith PBS containing 0.5% Tween 20 (wash buffer), the plateswere blocked for 2 hours with PBS containing 2% casein and 10%normal goat serum (blocking buffer). After 4 washes, normal sera(for cutoff value calculation) and SLE sera diluted 1:500 inblocking buffer were added to duplicate wells and incubated for 1hour. The plates were then washed 4 times, and peroxidase-conjugated goat anti-human IgG (� chain specific; Jackson Im-munoResearch, Baltimore, MD) diluted 1:5,000 in blockingbuffer was added and incubated for 1 hour. Finally, after 4washes, 0.4 mg/ml of o-phenylenediamine (Sigma, St. Louis, MO)in 0.1M citrate buffer (pH 6.0) containing 0.003% hydrogenperoxide was added and the plates incubated for 10 minutes. Thereaction was stopped by addition of 2M sulfuric acid, and theplates were read at 490 nm on an MRX ELISA reader (Dynex,Chantilly, VA).
Each plate included duplicate wells with serum fromthe same negative control (healthy subject) and the samepositive control (SLE patient); the coefficient of variation was17.1% over 37 plates. For each serum, the optical density (OD)values of control wells (no antigen) were subtracted from theOD values obtained with the antigen-coated wells. The ODvalues of the control wells were very low in all cases. Resultsshown are the means of duplicate values for each serum. Thecutoff value for aLB1 IgG antibody positivity, based on themean � 4 SD of values in the 45 healthy subjects, was OD0.370.
Preparation and purification of LB1. In order toincrease the yield of LB1, the full-length coding DNA forhuman LB1 was synthesized and optimized for Escherichia colicodon preferences by Operon Technologies (Alameda, CA),and inserted into the T7 polymerase–based pET19b expressionvector (Novagen; Madison, WI). Transformation into E coliBL21 (DE3) bacteria (Stratagene, La Jolla, CA) was done forprotein expression. Production of LB1 was induced by additionof IPTG (Sigma). Inclusion bodies were extracted with B-PERreagent (Pierce, Rockford, IL). LB1 was purified by electro-
elution after sodium dodecyl sulfate (SDS)–polyacrylamide gelelectrophoresis. The overall yield of purified LB1 was �5 mgper liter of original bacterial culture, which is 5–10 times theyield obtained with the wild-type LB1 DNA sequence (7).
Cell culture, apoptosis induction, and cell fraction-ation. Jurkat cells (E6-1; American Type Culture Collection,Manassas, VA) were grown in RPMI 1640 medium (Wisent,St-Bruno, Quebec, Canada) supplemented with 20% fetalbovine serum (Wisent) and 100 �g/ml gentamycin (Wisent) in5% CO2 at 37°C. Human umbilical vein endothelial cells(HUVECs; Clonetics, San Diego, CA) were grown in EGM2MV medium (Clonetics) and were used at confluence and notbeyond the fourth passage. Cells were washed and resus-pended in RPMI 1640 without serum. Apoptosis was inducedwith either anti-Fas antibody (CH-11) (50 �g/ml; UpstateBiotechnology, Lake Placid, NY) or staurosporine (25 �g/ml;Sigma). After 16 hours of anti-Fas treatment, cells werepelleted at 1,500g and the supernatant was centrifuged at16,000g to pellet the blebs.
DNA fragmentation assay. DNA fragmentation wasanalyzed with the Cellular DNA Fragmentation ELISA(Roche; Laval, Quebec, Canada). Jurkat cells were incubatedwith the thymidine analog 5�-bromo-2�-deoxy-uridine (BrdU),which was incorporated into genomic DNA. This labelingallows for the identification of BrdU-labeled DNA fragmentsthat are released into the cytoplasm during apoptosis.
Gel electrophoresis and immunoblotting. Jurkat cellsand blebs were washed once in PBS, resuspended in SDSsample buffer (250 mM Tris buffer [pH 6.8], containing 8%SDS, 40% glycerol, 20% �-mercaptoethanol, and 0.005%bromphenol blue), and separated on 10% polyacrylamide gels.Samples were transferred onto nitrocellulose membranes (Bio-Rad, Hercules, CA) using semidry transfer or stained withCoomassie blue (20). Immunoblots were blocked overnightwith TBS-T (50 mM Tris, 150 mM NaCl, buffer [pH 7.4], with0.5% Tween 20) containing 5% nonfat dry milk and exposed toa guinea pig antiserum (GP5) raised against recombinanthuman LB1 or a human SLE serum that was monospecific forLa, followed by peroxidase-conjugated anti–guinea pig IgG(Zymed, Oxnard, CA) or peroxidase-conjugated anti-humanIgG (Jackson ImmunoResearch), respectively. Between incu-bations, the blots were washed 3 times with TBS-T. Antibodybinding was revealed using a chemiluminescence kit (Super-Signal West Pico; Pierce).
Immunofluorescence and confocal microscopy. Jurkatcells or HUVECs were washed twice in PBS and cytospun ontoglass slides at 1,500g for 2 minutes. Blebs were washed once,resuspended in PBS, and dried onto glass slides. Samples werefixed for 5 minutes in 1% paraformaldehyde at 4°C andpermeabilized for 30 seconds in ice-cold acetone. Sampleswere rehydrated with ice-cold PBS for 10 minutes prior toincubation at room temperature with either GP5, a humanaLB1-positive SLE serum, affinity-purified human aLB1 anti-bodies (see below), or fluorescein isothiocyanate (FITC)–conjugated annexin V (BD PharMingen, San Diego, CA).Detection of antibodies was performed by incubation withFITC- or tetramethylrhodamine isothiocyanate (TRITC)–conjugated anti–guinea pig IgG (Fc specific; Jackson Immu-noResearch) or FITC- or TRITC-conjugated anti-human IgG(Fc specific; Jackson ImmunoResearch). Slides were mountedwith Vectashield (Vector, Burlingame, CA) and examined with
AUTOANTIBODIES TO LAMIN B1 AND THROMBOPROTECTION IN SLE 2697
a Zeiss (Thorwood, NY) 510 confocal microscope. For eachsample, a minimum of 200–250 blebs were examined, and blebsrepresentative of the entire population were photographed.Due to the high magnification required for viewing andphotographing apoptotic blebs, only 1 or a few blebs can beshown in each photograph.
Biotinylation of cell surface protein and pure LB1.Jurkat cells and blebs were washed 3 times and resuspended inPBS adjusted to pH 8 in the presence of a biotinylating reagent(Sulfo-NHS-LC-biotin) (0.5 mg/ml; Pierce) for 30 minutes atroom temperature. Samples were washed 3 times with PBS(pH 8), resuspended in lysis buffer (PBS [pH 8] containing 1%Triton X-100, 1 mM phenylmethylsulfonyl fluoride, 1 �g/mlpepstatin, 2 �g/ml leupeptin, and 2 �g/ml aprotinin) and incu-bated on ice for 30 minutes. Lysates were centrifuged at 20,000g,and the supernatants were incubated with streptavidin-coatedbeads (ImmunoPure Immobilized streptavidin; Pierce) overnightat 4°C. Beads were washed 3 times with lysis buffer and resus-pended in SDS sample buffer. The beads were then boiled for 5minutes, pelleted (600g, 5 minutes), and the supernatant contain-ing the biotinylated proteins was stored at �20°C.
Pure LB1 (55 �g) was incubated with caspase 6 (25 �g;BD PharMingen) in 20 mM PIPES, 1 mM EDTA, 0.1% CHAPS,10% sucrose, 10 mM NaCl (pH 7.2) for 3 hours at 37°C.Biotinylating reagent (2 �g) was added (molar ratio of biotiny-lating reagent to LB1 44:1) for 30 minutes at room temperature,followed by incubation with streptavidin-coated beads overnightat 4°C. The beads were processed as described above.
Affinity purification of human aLB1. Pure LB1 (75 �g)was dried onto a 1-cm2 nitrocellulose membrane, and themembrane was washed with water and blocked overnight withTBS-T. The blocked membrane was incubated with aLB1-positive SLE patient serum (1:100) for 2 hours and washed 3times with TBS-T. Anti-LB1 was eluted by overnight incuba-tion of the membrane in 6M GuHCl in 0.5 mM Tris HCl (pH7.5) (21). The eluate was renatured by dialysis against 0.5 mM
Tris HCl (pH 7.5) with 5 changes over a 48-hour period. Thesample was concentrated in PBS with an Ultrafree centrifugalfilter device (Biomax-30K; Millipore, Bedford, MA) andstored at 4°C. The monospecificity of each sample for LB1 wasassayed by immunoblotting on Jurkat cell lysates.
Statistical analysis. Chi-square analysis was performedfor frequency comparison among groups (using Fisher’s2-tailed exact test for 2 � 2 comparisons and the chi-squaretest for trend for larger contingency tables) (InStat3; Graph-Pad Software, San Diego, CA). The t-test was used forcomparison of group means. P values less than or equal to 0.05were considered significant.
RESULTS
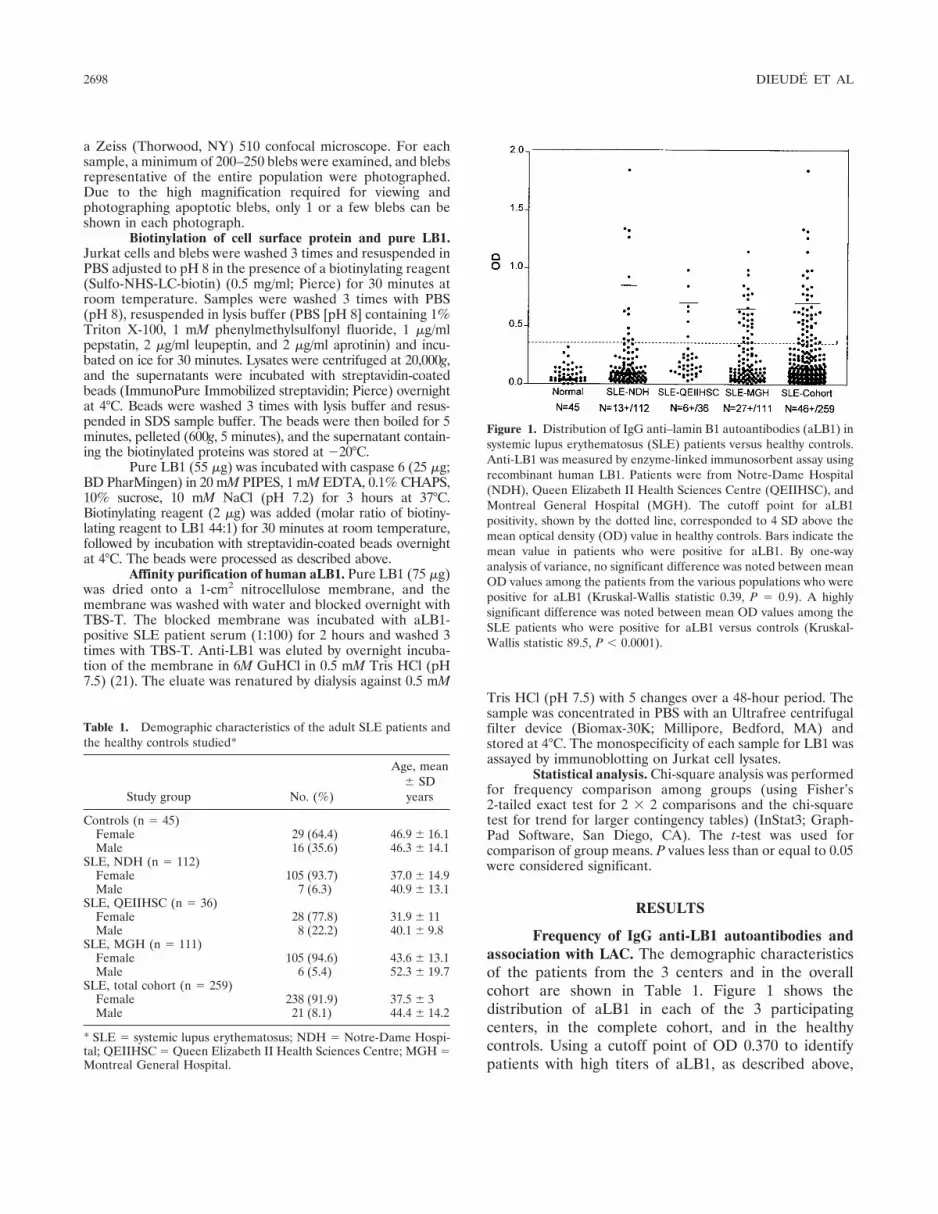
Frequency of IgG anti-LB1 autoantibodies andassociation with LAC. The demographic characteristicsof the patients from the 3 centers and in the overallcohort are shown in Table 1. Figure 1 shows thedistribution of aLB1 in each of the 3 participatingcenters, in the complete cohort, and in the healthycontrols. Using a cutoff point of OD 0.370 to identifypatients with high titers of aLB1, as described above,
Figure 1. Distribution of IgG anti–lamin B1 autoantibodies (aLB1) insystemic lupus erythematosus (SLE) patients versus healthy controls.Anti-LB1 was measured by enzyme-linked immunosorbent assay usingrecombinant human LB1. Patients were from Notre-Dame Hospital(NDH), Queen Elizabeth II Health Sciences Centre (QEIIHSC), andMontreal General Hospital (MGH). The cutoff point for aLB1positivity, shown by the dotted line, corresponded to 4 SD above themean optical density (OD) value in healthy controls. Bars indicate themean value in patients who were positive for aLB1. By one-wayanalysis of variance, no significant difference was noted between meanOD values among the patients from the various populations who werepositive for aLB1 (Kruskal-Wallis statistic 0.39, P � 0.9). A highlysignificant difference was noted between mean OD values among theSLE patients who were positive for aLB1 versus controls (Kruskal-Wallis statistic 89.5, P � 0.0001).
Table 1. Demographic characteristics of the adult SLE patients andthe healthy controls studied*
Study group No. (%)
Age, mean� SDyears
Controls (n � 45)Female 29 (64.4) 46.9 � 16.1Male 16 (35.6) 46.3 � 14.1
SLE, NDH (n � 112)Female 105 (93.7) 37.0 � 14.9Male 7 (6.3) 40.9 � 13.1
SLE, QEIIHSC (n � 36)Female 28 (77.8) 31.9 � 11Male 8 (22.2) 40.1 � 9.8
SLE, MGH (n � 111)Female 105 (94.6) 43.6 � 13.1Male 6 (5.4) 52.3 � 19.7
SLE, total cohort (n � 259)Female 238 (91.9) 37.5 � 3Male 21 (8.1) 44.4 � 14.2
* SLE � systemic lupus erythematosus; NDH � Notre-Dame Hospi-tal; QEIIHSC � Queen Elizabeth II Health Sciences Centre; MGH �Montreal General Hospital.
2698 DIEUDE ET AL
high titers of aLB1 were observed in SLE patients butnot in healthy or disease controls (ref. 7 and data notshown). The frequency of high-titer aLB1 in the overallSLE patient group was 17.8% (NDH 11.6%, QEIIHSC16.7%, MGH 24.3%), and the mean aLB1 levels (ODvalues) for the positive patients from each center weresimilar (Kruskal-Wallis statistic 0.39, P � 0.9). Anti-LB1was found to be associated with LAC. Twenty-two (47.8%)of the 46 patients with aLB1 were positive for LAC,whereas 68 (31.9%) of the 213 patients without aLB1 wereLAC positive (OR 1.95, 95% confidence interval [95% CI]1.024–3.73, P � 0.04 by chi-square analysis).
However, there was no association between aLB1and aCL or anti-�2GPI. Sixteen (34.8%) of the 46patients with aLB1 were positive for any aCL (i.e., ofIgG and/or IgM isotype), whereas 87 (41.8%) of 208patients without aLB1 were aCL positive (OR 0.7, 95%CI 0.4–1.4, P � 0.3). Similarly, 6 (13%) of 46 patientswith aLB1 were positive for any anti-�2GPI, whereas 38(18%) of 211 patients without aLB1 were anti-�2GPIpositive (OR 0.7, 95% CI 0.3–1.7, P � 0.4).
Influence of IgG anti-LB1 and LAC on the fre-quency of thrombotic manifestations. To determinewhether higher levels of aLB1 were more strongly
associated with protection against thrombosis, we com-pared the mean OD370 values for aLB1 in patients withthrombosis (n � 81) versus those without thrombosis(n � 178). No significant difference was found (mean �SD OD370 0.193 � 0.259 in patients with thrombosisversus 0.228 � 0.271 in those without thrombosis; P �0.3 by 2-tailed t-test). We then compared the meanOD370 values among patients with high titers of aLB1and thrombosis (n � 10) versus those with aLB1 withoutthrombosis (n � 36). Again, no significant differencewas found (0.786 � 0.316 in patients with thrombosisversus 0.675 � 0.305 in those without thrombosis; P �0.3 by 2-tailed t-test).
Because LAC is known to be associated with thethrombotic manifestations of the antiphospholipid syn-drome in SLE (2), we first determined whether thisassociation also occurred in our study cohort. Thepresence of LAC at first evaluation was indeed associ-ated with thrombosis during the disease course: LACwas found in 39 (48.1%) of 81 patients with thrombosisand in 51 (28.7%) of 178 patients without thrombosis(OR 2.24, 95% CI 1.3–3.85, P � 0.004).
Since LAC were also associated with aLB1, asdiscussed above, we then determined whether aLB1were themselves associated with thrombosis. In contrastto the findings with LAC, this was not the case. Throm-bosis occurred in 10 (21.7%) of 46 aLB1-positive pa-tients versus 71 (33.3%) of 213 aLB1-negative patients(OR 0.555, 95% CI 0.26–1.18, P � 0.16). Finally, weexamined the relationship between the presence ofaLB1 and the frequency of thrombosis in patients withLAC. As shown in Table 2, the frequency of thrombosiswas not significantly different in patients with both LACand aLB1 in comparison with all other SLE patients inthe cohort (OR 0.62, 95% CI 0.22–1.75, P � 0.47). Instriking contrast, the frequency of thrombosis in patientswho were LAC positive but aLB1 negative was signifi-cantly greater than in other SLE patients (OR 3.06, 95%
Table 2. Frequency of thrombosis in SLE patients with LAC: effect of the presence of aLB1*
Thrombosis No thrombosisOdds ratio(95% CI) P†
Patients with both LAC 5 17and aLB1 0.62 (0.22–1.75) 0.47
All other patients 76 161
Patients with LAC and 34 34absence of aLB1 3.06 (1.71–5.46) 0.0002
All other patients 47 144
* SLE � systemic lupus erythematosus; LAC � lupus anticoagulant; aLB1 � anti–lamin B1; 95% CI �95% confidence interval.† By Fisher’s 2-tailed exact test.
Table 3. Frequency of thrombosis according to LAC and aLB1status in 259 patients with SLE*
Thrombosis,no.†
No thrombosis,no.†
Frequency ofthrombosis, %
Presence of LAC andabsence of aLB1
34 34 50
Presence of bothLAC and aLB1
5 17 22.7
Absence of bothLAC and aLB1
37 108 25.5
Absence of LAC andpresence of aLB1
5 19 20.8
* See Table 2 for definitions.† P � 0.0004 by chi-square test for trend (thrombosis group versus nothrombosis group).
AUTOANTIBODIES TO LAMIN B1 AND THROMBOPROTECTION IN SLE 2699

CI 1.71–5.46, P � 0.0002) (Table 2). Similar results wereobtained when each of the 3 participating centers wasanalyzed separately (data not shown). In patients with
aLB1 and thrombosis, thrombotic events occurred bothin arterial and/or in venous beds.
The relationship between thrombosis, LAC, and
Figure 2. Close synchrony of the occurrence of lamin B1 (LB1) cleavage and DNA fragmentation during apoptosis in Jurkat cells. a, Detection ofLB1 fragments in immunoblots of lysates of Jurkat cells treated for the indicated periods of time with anti-Fas antibody (50 �g/ml) (left panel) orstaurosporine (25 �g/ml) (right panel) by an antiserum directed against LB1 (GP5). Intact LB1 migrated as a 67-kd band, while apoptotic N- andC-terminal fragments migrated as 25- and 47-kd bands, respectively. b, Quantification of DNA fragmentation by an enzyme-linked immunosorbentassay detecting 5�-bromo-2�-deoxy-uridine (BrdU)–labeled DNA fragments in Jurkat cells treated for the indicated periods of time with anti-Fasantibody (50 �g/ml) (left panel) or staurosporine (25 �g/ml) (right panel). OD � optical density.
Table 4. Frequency of thrombosis according to LAC, aCL, anti-�2GPI, and aLB1 status in 253 patientswith SLE*
LAC and other antibody statusThrombosis,
no.†No thrombosis,
no.†Frequency of
thrombosis, %
Presence of LACPresence of aCL/anti-�2GPI and absence
of aLB115 9 62.5
Presence of both aCL/anti-�2GPI andaLB1
4 7 36.4
Absence of both aCL/anti-�2GPI and aLB1 16 24 40.0Absence of aCL/anti-�2GPI and presence
of aLB11 10 9.1
Absence of LACPresence of aCL/anti-�2GPI and absence
of aLB116 33 32.7
Presence of both aCL/anti-�2GPI andaLB1
2 6 25.0
Absence of both aCL/anti-�2GPI and aLB1 21 73 22.3Absence of aCL/anti-�2GPI and presence
of aLB13 13 18.8
* aCL/anti-�2GPI � anticardiolipin and/or anti–�2-glycoprotein I of the IgG and/or IgM isotype (seeTable 2 for other definitions).† P � 0.0002 for all 8 subsets; P � 0.007 for LAC-positive subsets; P 0.1 for LAC-negative subsets, bychi-square test for trend (thrombosis group versus no thrombosis group).
2700 DIEUDE ET AL

aLB1 was further explored, as shown in Table 3. In thepresence of LAC and absence of aLB1 at first evalua-tion, the frequency of thrombosis during the diseasecourse was 50%. However, when LAC and aLB1 werepresent simultaneously, the frequency of thrombosisdecreased to 22.7%. The latter frequency was similar tothat in patients expressing neither autoantibody (25.5%)or aLB1 only (20.8%). The frequency of thrombosis wassignificantly different (P � 0.0004 by chi-square test fortrend) among these 4 groups of patients (Table 3). Todetermine if the lower frequency of thrombosis in pa-tients with aLB1 could be related to a shorter durationof followup in these patients, we compared the durationof followup in patients with and those without aLB1.The mean followup period was not significantly differentin aLB1-positive versus aLB1-negative patients for anyof the centers (NDH 8.46 years versus 8.01 years [P � 0.7by t-test]; QEIIHSC 6.62 years versus 5.99 years [P � 0.7];MGH 16.92 years versus 15.79 years [P � 0.3]). In thecohort as a whole, the duration of followup was longer foraLB1-positive patients, but the difference was not statisti-cally significant (13.19 versus 10.79 years [P � 0.065]).
Interrelationships between IgG anti-LB1, LAC,aCL, anti-�2GPI, and the frequency of thrombotic man-ifestations. Because aCL and anti-�2GPI may occur withLAC and are associated with the thrombotic manifesta-tions of the antiphospholipid syndrome in SLE (3), wewished to determine whether the frequency of thrombo-sis was influenced by a relationship between theseantibodies and aLB1 status. We found that the presenceof any aCL (i.e., of IgG and/or IgM isotype) or anyanti-�2GPI was indeed strongly associated with throm-bosis in our cohort. Positivity for aCL was noted in 42(53.8%) of 78 patients with thrombosis and 61 (34.7%)of 176 patients without thrombosis (OR 2.2, 95% CI1.28–3.8, P � 0.004). Similarly, anti-�2GPI was positivein 24 (30%) of 80 patients with thrombosis versus 20(11.3%) of 177 patients without thrombosis (OR 3.3,95% CI 1.7–6.5, P � 0.0002). Second, we determined ifaLB1 was associated with aCL or anti-�2GPI. In contrastto LAC, no such association was found: aCL was presentin 16 (34.8%) of 46 patients with aLB1 versus 87 (41.8%)of 208 patients without aLB1 (OR 0.7, 95% CI 0.4–1.4,P � 0.3). Similarly, anti-�2GPI was present in 6 (13%) of46 patients with aLB1 versus 38 (18%) of 211 patientswithout aLB1 (OR 0.7, 95% CI 0.3–1.7, P � 0.4).
Lastly, we determined whether aCL and anti-�2GPI status modified the impact of aLB1 and LAC onthrombosis. As is clear from the data in Table 4, theimpact of aLB1 on thrombosis was specifically linked tothe presence of LAC. In the absence of LAC, the fre-
quency of thrombosis was similar in all aCL/anti-�2GPIsubsets, regardless of aLB1 status, whereas in the presenceof LAC, the frequency between subsets differed signifi-cantly. Specifically, in the presence of both LAC and aCLand/or anti-�2GPI but in the absence of aLB1, the fre-quency of thrombosis was 62.5%. This frequency decreasedto 36.4% in the presence of aLB1 (Table 4). In thepresence of LAC but in the absence of aCL and/oranti-�2GPI and of aLB1, the frequency of thrombosis was40%. Again, in the presence of aLB1, the frequency ofthrombosis was strikingly decreased, to 9.1%. In contrast,in the absence of LAC but in the presence of aCL and/oranti-�2GPI, the frequency of thrombosis was similar inpatients without aLB1 (32.6%) and in patients with aLB1(25%). Finally, in the absence of all 4 antibodies, thefrequency of thrombosis was 22.3%, similar to the 18.8%observed in patients with aLB1 only. Thus, the frequenciesof thrombotic episodes were significantly different acrossLAC-positive subsets (P � 0.007) but they were notsignificantly different in LAC-negative subsets (P 0.1,chi-square test for trend) (Table 4).
Cleavage of LB1 and DNA fragmentation occurin close synchrony during apoptosis in Jurkat cells.During apoptosis, a series of nuclear modificationsoccurs, including DNA fragmentation and cleavage ofspecific proteins by proteases of the caspase family.Cleavage of LB1 by caspase 6 generates 2 fragments (47kd and 25 kd; for review, see ref. 16), and this event iscommonly used as a marker for apoptosis. Apoptosis wasinduced in Jurkat cells, the prototypical cell line used forapoptosis studies, with anti-Fas antibody, via the deathreceptor pathway, or with staurosporine, via the mitochon-drial pathway, for various periods of time. Cell lysates wereanalyzed by immunoblotting with an antiserum directedagainst LB1 (GP5). LB1 apoptotic fragments were firstdetected after 3 hours of treatment with anti-Fas and afteronly 1 hour of treatment with staurosporine (Figure 2a),thus confirming the occurrence of LB1 cleavage under the2 principal pathways of apoptosis.
DNA fragmentation in Jurkat cells treated withanti-Fas or staurosporine for different periods of timewas then studied. Using an ELISA to quantitate BrdU-labeled DNA fragments, DNA fragmentation in Jurkatcells was first detected after 4 hours of treatment withanti-Fas antibody and after 2 hours of treatment withstaurosporine (Figure 2b). Hence, LB1 cleavage andDNA fragmentation occur in close synchrony in Jurkatcells during apoptosis, with LB1 cleavage preceding thatof DNA, as expected (22).
AUTOANTIBODIES TO LAMIN B1 AND THROMBOPROTECTION IN SLE 2701
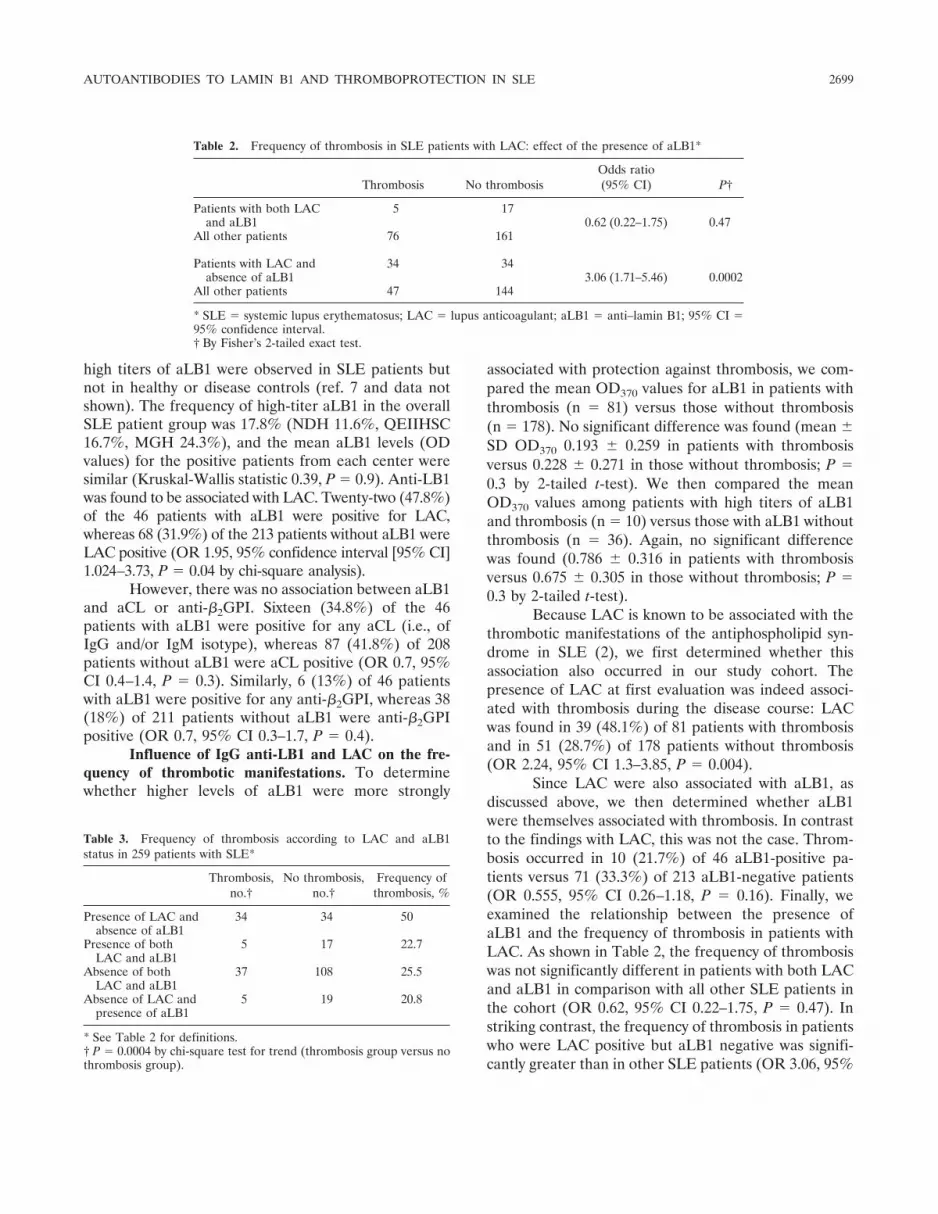
Presence of LB1 in blebs of apoptotic Jurkatcells. In order to localize LB1 during apoptosis, Jurkatcells were treated with staurosporine for various periodsof time, stained with GP5 or aLB1-positive SLE serum(serum 9176), and examined by confocal microscopy. Fig-ure 3 shows the results of a time course study duringstaurosporine treatment. The fluorescence pattern ob-served with GP5 in untreated Jurkat cells (0 hours) was analmost continuous rim at the nuclear periphery, represent-ing the nuclear lamina that is exclusively located at theinner face of the nuclear envelope (6). After 4 hours oftreatment, there was no evident change in the fluorescencepattern observed. However, after 8 hours of treatment,significant changes were detected: gaps appeared in thefluorescence pattern, presumably due to localized disorga-nization of the lamina meshwork. After 12 hours, someLB1 fluorescence was located at the periphery of theoriginal nucleus, while a circular fluorescence patternappeared in a vesicle budding from the main body of thecell. Thus, at least part of LB1 appeared to be redistributedfrom the lamina network to vesicles representing apoptoticblebs. After 16 hours, the original lamina became unde-
Figure 3. Relocalization of LB1 into blebs during late apoptosis in Jurkat cells. Time course analysis of Jurkat cells treated with staurosporine (25�g/ml) for the indicated periods of time. Cells were double-stained with an antiserum directed against LB1 (GP5) (green) and an aLB1-positivehuman SLE serum 9176 (red) and examined by confocal microscopy. Images of fluorescent signals (green or red) or phase-contrast images are shownindividually or after all 3 digitalized images were overlaid by computer processing (Merge). Bars � 5 �m. See Figure 1 for definitions.
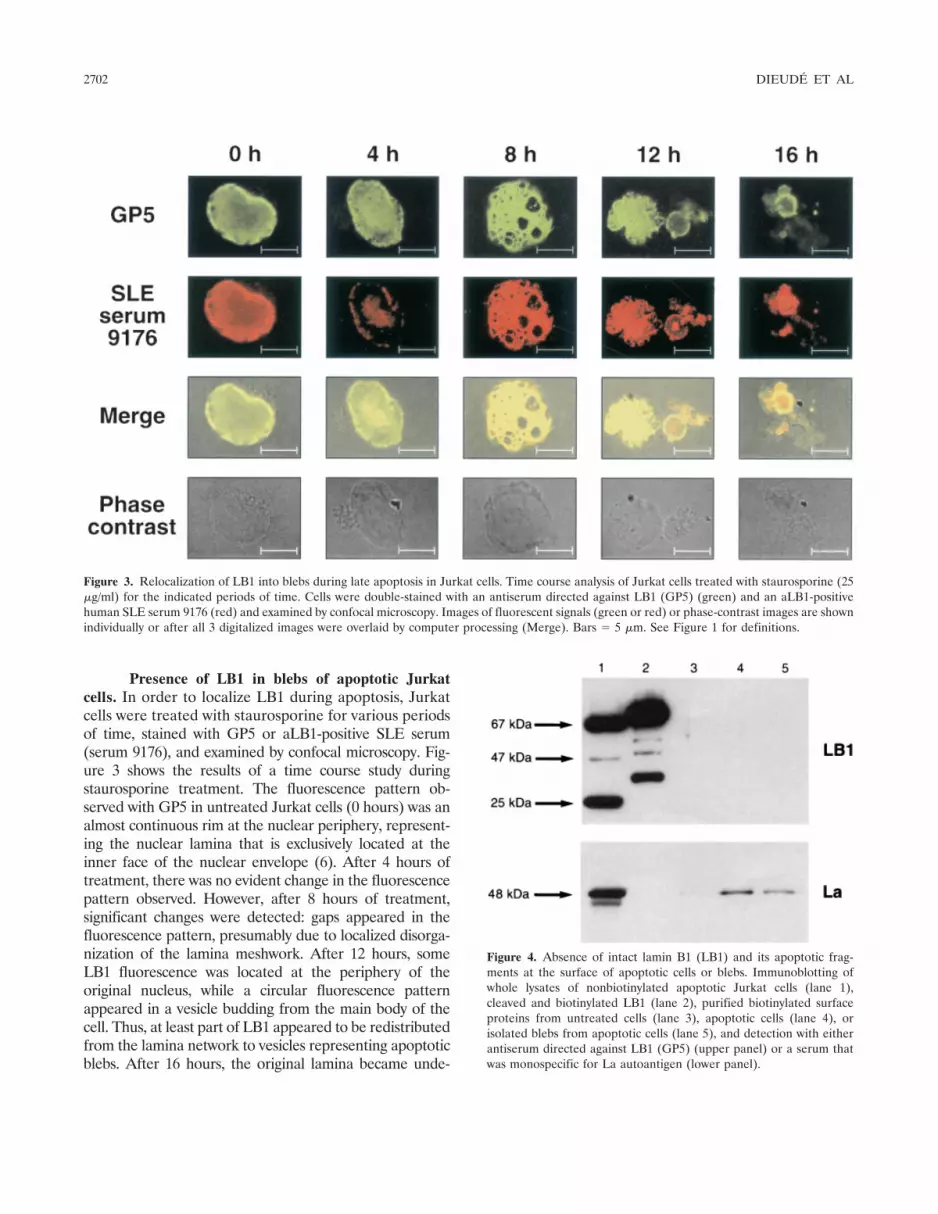
Figure 4. Absence of intact lamin B1 (LB1) and its apoptotic frag-ments at the surface of apoptotic cells or blebs. Immunoblotting ofwhole lysates of nonbiotinylated apoptotic Jurkat cells (lane 1),cleaved and biotinylated LB1 (lane 2), purified biotinylated surfaceproteins from untreated cells (lane 3), apoptotic cells (lane 4), orisolated blebs from apoptotic cells (lane 5), and detection with eitherantiserum directed against LB1 (GP5) (upper panel) or a serum thatwas monospecific for La autoantigen (lower panel).
2702 DIEUDE ET AL

tectable and LB1 was completely redistributed in blebswith a circular pattern.
Serum 9176 from an aLB1-positive SLE patientand GP5 showed complete colocalization at all timepoints, with the exception that, at 12 hours and 16 hours,a circular area of fluorescence at the center of the blebswas detected only with the SLE serum (Figure 3).Similar patterns of LB1 redistribution in late apoptoticblebs were observed in Jurkat cells treated with anti-Fasantibody (results not shown).
Sequestration of LB1 inside apoptotic blebs. Todetermine the accessibility of LB1 at the surface ofapoptotic Jurkat cells or blebs, surface proteins of
untreated cells and of apoptotic cells and blebs werebiotinylated and purified. As a positive control for GP5reactivity, purified LB1 was digested with caspase 6,biotinylated, and analyzed in parallel with the purifiedbiotinylated surface proteins. Unlabeled and biotinyl-ated LB1 and its apoptotic fragments were detected withGP5, but neither LB1 nor its fragments were detected inthe purified surface proteins of untreated cells, apopto-tic cells, or isolated blebs (Figure 4). As a positivecontrol for the efficiency of the surface biotinylationtechnique, a similar immunoblot of Jurkat cell surfaceproteins was probed with an SLE patient serum that wasmonospecific for the La autoantigen, which has previ-
Figure 5. Lamin B1 (LB1) sequestration in Jurkat apoptotic blebs. a, Staining of a fixed Jurkat bleb with antiserum directed against LB1 (GP5)(green), examined by confocal microscopy. b, Horizontal optical sections of a fixed Jurkat bleb double-stained with GP5 (red) and annexin V (green),examined by confocal microscopy. Bars � 2.5 �m. See Figure 3 for explanation of “merge.”
AUTOANTIBODIES TO LAMIN B1 AND THROMBOPROTECTION IN SLE 2703

ously been shown to be accessible at apoptotic cell andbleb surfaces (14). As expected, La was detected in thebiotinylated surface proteins of apoptotic cells and blebs,but not at the surface of intact untreated cells (Figure 4).LB1 was detected in the unlabeled whole cell lysate ofapoptotic Jurkat cells. The difference in mobility be-tween unlabeled and biotinylated LB1 fragments is dueto the added biotin moieties. Taken together, theseresults show that, unlike other autoantigens such as La,LB1 was not present at the surface of apoptotic cells orblebs.
To demonstrate these results at the microscopiclevel, isolated and fixed Jurkat blebs were examined byconfocal microscopy. Representative examples areshown in Figure 5a. Fluorescence due to GP5 detectionof LB1 was completely enclosed within the bleb plasmamembrane, as evidenced in the merged fluorescent andphase-contrast images. This finding was confirmed byexamination of horizontal optical sections of a typicalbleb, double-stained for LB1 and anionic phospholipidsby GP5 and annexin V, respectively (Figure 5b). GP5showed a circular pattern that was smaller in the firstand last sections and larger in the middle sections,indicating that LB1 is associated with a continuousperipherally stained structure within the blebs, as clearlyevidenced in the merged fluorescent and phase-contrastimages. Annexin V, in these blebs that had been perme-abilized to allow for GP5 penetration, stained not onlythe external bleb surface, but also the inner face of theplasma membrane. Nevertheless, GP5 staining wasclearly enclosed within the bulk of annexin V staining,with only minimal overlap at the interface of the 2, dueto their proximity and to the resolution of these opticalmicroscopy images. The results shown in Figure 5 arerepresentative of 8 independent experiments performedusing either staurosporine or anti-Fas antibody as induc-ers of apoptosis. Blebs containing LB1 were �2 �m indiameter, while smaller blebs of �1.5 �m were stainedby annexin V, but not by GP5.
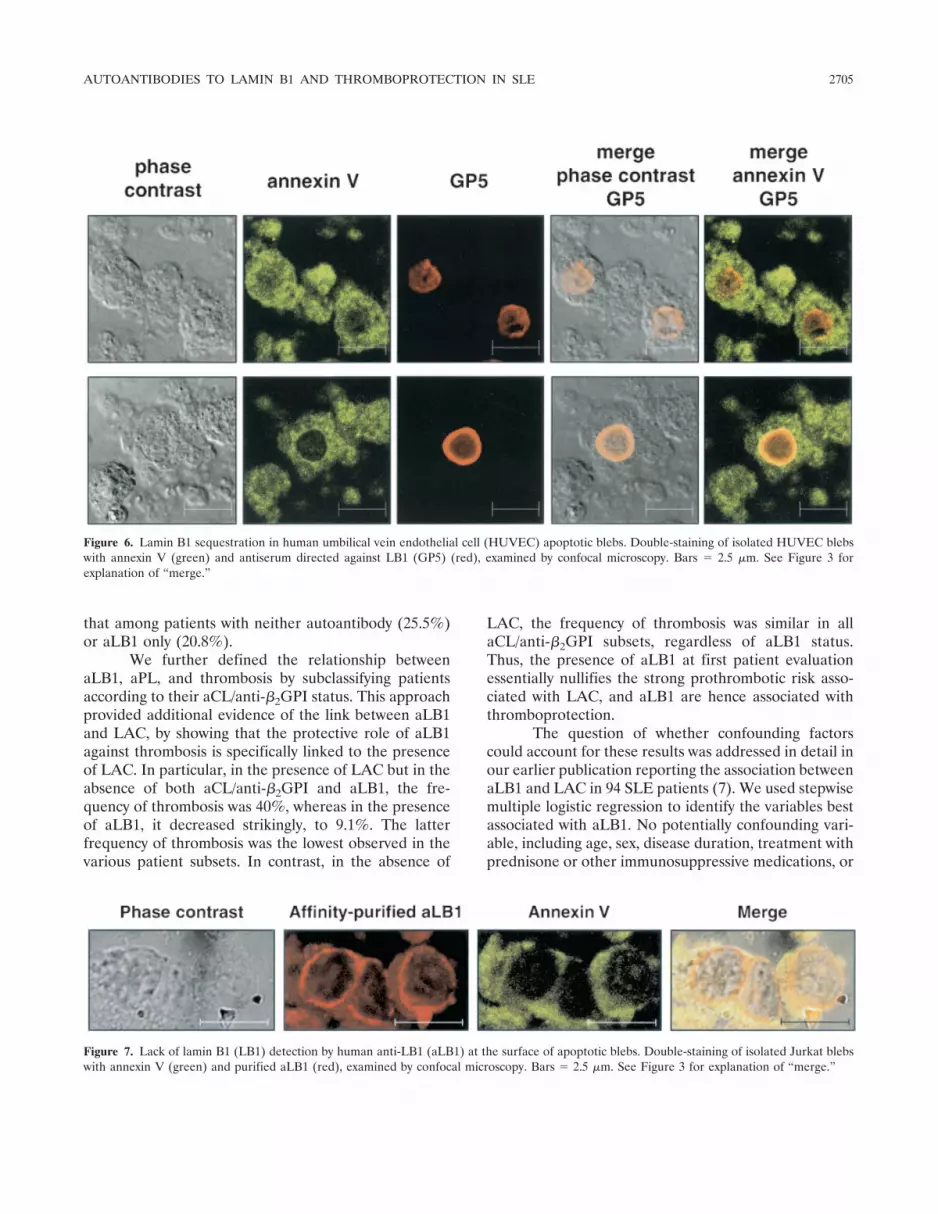
Since the most likely in vivo target for circulatingaLB1 would be endothelial cells, it was important todetermine whether LB1 was localized in HUVEC apo-ptotic blebs in a manner similar to that seen with Jurkatcell blebs. Hence, HUVECs were induced into apopto-sis, and blebs were collected and examined by phase-contrast microscopy. Representative examples areshown in Figure 6. Blebs from HUVECs were largerthan those from Jurkat cells, due to their larger cyto-plasmic compartment. Again however, it could clearly beseen that LB1 was restricted to a circular structurecompletely enclosed within the bleb plasma membrane,
as evidenced by the merged fluorescent and phase-contrast images, either in tangential sections or in mid-dle sections (Figure 6). Membrane staining by annexin Vin these permeabilized blebs was also shown to com-pletely enclose the LB1 pattern. These results confirmthat LB1 is enclosed within apoptotic blebs.
LB1 is not accessible to human aLB1 at thesurface of apoptotic blebs. Anti-LB1 immunoglobulinsfrom 4 high-titer SLE patient sera were affinity purified.Double-staining of isolated Jurkat blebs with annexin Vand purified aLB1 was performed, and the results ana-lyzed by confocal microscopy. Figure 7 shows represen-tative results obtained with these antibodies. Anti-LB1showed a circular pattern enclosed within the annexin Vmembrane staining profile, similar to the pattern ob-served with GP5 (Figure 5). Purified aLB1 did notrecognize any other target at the surface of blebs. Thus,LB1 is sequestered and is unlikely to be accessible tocirculating human aLB1 in apoptotic blebs. The purifiedaLB1 used in Figure 7 were isolated from SLE patientserum 9176. The apparent discrepancy observed be-tween the staining patterns of SLE serum 9176 and thatof GP5 at the 12-hour and 16-hour time points (Figure3) was not observed with aLB1 that was affinity purifiedfrom the same serum (Figure 7), suggesting that theadditional staining was due to antibodies directedagainst a target other than LB1.
DISCUSSION
We have demonstrated an association betweenthe presence of aLB1 in SLE patients and protectionagainst thrombosis (“thromboprotection”) in a large,multicenter study. The results of previous work from ourgroup supported such an association, but that cohort ofpatients was small and of limited ethnic diversity. Incontrast, the power of the present study was greatlyenhanced, since the total number of SLE patients stud-ied was increased from 94 to 259 and the number ofpatients expressing aLB1 from 8 to 46. Furthermore, thepresent multicenter study encompassed 3 lupus clinicpopulations including English Canadian and FrenchCanadian SLE patients from 3 different university hos-pitals in 2 geographic locations (Montreal [Quebec,Canada] and Halifax [Nova Scotia, Canada]). The ob-served frequency of thrombosis in the presence of LACwithout aLB1 was 50%, consistent with the findings ofother studies (for review, see refs. 3 and 4). However,when both LAC and aLB1 were present, there was amarked and highly significant reduction in the frequencyof thrombosis, to 22.7%. This frequency was similar to
2704 DIEUDE ET AL
that among patients with neither autoantibody (25.5%)or aLB1 only (20.8%).
We further defined the relationship betweenaLB1, aPL, and thrombosis by subclassifying patientsaccording to their aCL/anti-�2GPI status. This approachprovided additional evidence of the link between aLB1and LAC, by showing that the protective role of aLB1against thrombosis is specifically linked to the presenceof LAC. In particular, in the presence of LAC but in theabsence of both aCL/anti-�2GPI and aLB1, the fre-quency of thrombosis was 40%, whereas in the presenceof aLB1, it decreased strikingly, to 9.1%. The latterfrequency of thrombosis was the lowest observed in thevarious patient subsets. In contrast, in the absence of
LAC, the frequency of thrombosis was similar in allaCL/anti-�2GPI subsets, regardless of aLB1 status.Thus, the presence of aLB1 at first patient evaluationessentially nullifies the strong prothrombotic risk asso-ciated with LAC, and aLB1 are hence associated withthromboprotection.
The question of whether confounding factorscould account for these results was addressed in detail inour earlier publication reporting the association betweenaLB1 and LAC in 94 SLE patients (7). We used stepwisemultiple logistic regression to identify the variables bestassociated with aLB1. No potentially confounding vari-able, including age, sex, disease duration, treatment withprednisone or other immunosuppressive medications, or
Figure 6. Lamin B1 sequestration in human umbilical vein endothelial cell (HUVEC) apoptotic blebs. Double-staining of isolated HUVEC blebswith annexin V (green) and antiserum directed against LB1 (GP5) (red), examined by confocal microscopy. Bars � 2.5 �m. See Figure 3 forexplanation of “merge.”
Figure 7. Lack of lamin B1 (LB1) detection by human anti-LB1 (aLB1) at the surface of apoptotic blebs. Double-staining of isolated Jurkat blebswith annexin V (green) and purified aLB1 (red), examined by confocal microscopy. Bars � 2.5 �m. See Figure 3 for explanation of “merge.”
AUTOANTIBODIES TO LAMIN B1 AND THROMBOPROTECTION IN SLE 2705

prednisone dosage, emerged as being associated withaLB1 (7). Therefore it appears unlikely that confound-ing factors would explain the lower risk of thrombosis inpatients with aLB1 and LAC.
To our knowledge, there has been no otherexample of autoantibodies in systemic autoimmune dis-eases that are associated with a “thromboprotective”role, although other autoantibodies with protective ef-fects have been described (23). To date, aLB1 appearunique in this regard. These antibodies are also the firstidentified marker for a subset of LAC-positive SLEpatients with a better prognosis, and their presence mayexplain why some patients with LAC do not developthrombosis (3). Thus, this finding could have implica-tions with regard to treatment. It may be of value todetermine the aLB1 and aCL/anti-�2GPI status of newlyevaluated patients with SLE and LAC who have not hada first thrombotic episode (5). Given that the risk ofthrombosis in aLB1-positive, aCL/anti-�2GPI-negativepatients is low, prophylactic anticoagulation treatmentmay not be warranted in these patients. Conversely,patients who have LAC and aCL/anti-�2GPI in theabsence of aLB1 are at high risk for thrombosis, andprophylactic anticoagulation may be warranted. Thisapproach should be formally studied in a randomizedcontrolled trial.
We next investigated the mechanism by whichaLB1 may cause thromboprotection in vivo. Becausesurface blebs on apoptotic cells are procoagulant (8),which may be fundamental to the pathogenesis ofthrombotic events in patients with aPL (8–10), andbecause major SLE autoantigens are displayed at theexternal surface of these blebs, where their accessibilityto circulating autoantibodies may form the molecularbasis of their pathogenic effects (14,15), our mechanistichypothesis was that circulating aLB1 may block theprocoagulant effect of apoptotic blebs by binding toLB1 displayed at the external bleb surface. A furtherrationale for this hypothesis was that LB1 is an earlysubstrate of caspase 6 during apoptosis (16,17), suggest-ing that LB1 epitopes might be present in apoptoticblebs.
Therefore, we evaluated whether LB1 is ex-pressed in Jurkat and endothelial cells that have under-gone apoptosis. LB1 cleavage products were detected incells induced to undergo apoptosis by treatment witheither anti-Fas antibody or staurosporine. Cleavage ofLB1 preceded DNA fragmentation, a characteristic fea-ture of cells undergoing apoptosis. Confocal microscopy,using antilamin antibodies and SLE patient–derivedaLB1, demonstrated that LB1 redistributed from the
nuclear lamina to apoptotic blebs during the process ofapoptosis. Finally, we evaluated whether LB1 waspresent on the surface of apoptotic blebs, using biotiny-lation of surface proteins on apoptotic cells and blebs.We found that LB1 was not present on the surface of theblebs, although other autoantigens, such as La, werepresent. Confocal microscopy confirmed that LB1 wasburied within apoptotic blebs and inaccessible to circu-lating aLB1. These results disprove our hypothesis andsuggest that a direct role of LB1 in mediating thrombo-protection is unlikely. The mechanism by which aLB1confers thromboprotection in patients with SLE remainsto be elucidated. Studies are now in progress in ourlaboratory to determine whether aLB1 may exert itsthromboprotective effect by interacting with solublecoagulation factors or by modulating cells involved incoagulation.
The novel finding that LB1, either complete or ascaspase-cleaved fragments, was indeed present withinapoptotic blebs provides insights into the mechanisms bywhich a nuclear envelope protein, such as LB1, couldbecome immunogenic. As with several other autoanti-gens accumulated in apoptotic blebs, processing of LB1by antigen-presenting cells may give rise to aLB1 insusceptible hosts (i.e., patients with abnormalities in therecognition, uptake, processing, and presentation ofantigen, or with defects in the mechanism for apoptoticbleb clearance) (for review, see refs. 11, 24, and 25).Direct experimental evidence for the production ofautoantibodies following immunization with apoptoticcells has been obtained (26–28).
Our finding that, by both biochemical and mor-phologic criteria, LB1 was not detected at the surface ofapoptotic blebs is in striking contrast with the findingthat cytoplasmic autoantigens targeted by anti-Ro andantinuclear cytoplasmic antibodies (10,15), and the nu-clear autoantigen La (14), have been localized to theexternal surface of apoptotic blebs. In the latter cases,accessibility to circulating autoantibodies may explainthe pathogenic effects of these autoantibodies. In con-trast, our results suggest that, although apoptotic blebsmay serve as an important physiologic target for someautoantibodies (9–13), this cannot be generalized to allautoantigens. Even in the case of the nuclear autoanti-gen La, localization of this autoantigen within the cellchanges from nuclear to cytoplasmic as caspase cleavageproceeds during apoptosis (29). Hence, our results raisethe question as to whether any autoantigen of strictnuclear origin may be translocated to the externalsurface of apoptotic blebs.
2706 DIEUDE ET AL

ACKNOWLEDGMENTS
We thank Dr. Lisa R. Sammaritano for providingpatient sera and data in the preliminary phase of this study,and Pierrette Rego for typing the manuscript. We are gratefulto Mr. Fernand Locas and Mrs. Gisele Sarrazin-Locas for theirgenerous donations in support of our laboratory.
REFERENCES
1. Wilson WA, Gharavi AE, Koike T, Lockshin MD, Branch DW,Piette J-C, et al. International consensus statement on preliminaryclassification criteria for definite antiphospholipid syndrome: re-port of an international workshop. Arthritis Rheum 1999;42:1309–11.
2. Horbach DA, von Oort E, Donders RCJM, Derksan RHWM, deGroot PG. Lupus anticoagulant is the strongest risk factor for bothvenous and arterial thrombosis in patients with systemic lupuserythematosus. Thromb Haemost 1996;76:916–24.
3. Asherson RA, Cervera R, Lie JT. The antiphospholipid syn-dromes. In: Lahita RG, editor. systemic lupus erythematosus. NewYork: Academic Press; 1999. p. 829–70.
4. Petri M. Epidemiology of the antiphospholipid antibody syn-drome. J Autoimmun 2000;14:145–51.
5. Ruis-Irastorza G, Khamashta MA, Castellini G, Hughes GRV.Systemic lupus erythematosus. Lancet 2001;357:1027–32.
6. Hutchison CJ, Alvarez-Reyes M, Vaughan OA. Lamins in disease:why do ubiquitously expressed nuclear envelope proteins give riseto tissue-specific disease phenotypes? J Cell Sci 2001;114:9–19.
7. Senecal J-L, Rauch J, Grodzicky T, Raynauld J-P, Uthman I, NavaA, et al. Strong association of autoantibodies to human nuclearlamin B1 with lupus anticoagulant antibodies in systemic lupuserythematosus. Arthritis Rheum 1999;42:1347–53.
8. Casciola-Rosen L, Rosen A, Petri M, Schlissel M. Surface blebs onapoptotic cells are sites of enhanced procoagulant activity: impli-cations for coagulation events and antigenic spread in systemiclupus erythematosus. Proc Natl Acad Sci U S A 1996;93:1624–9.
9. Levine JS, Koh JS, Subang R, Rauch J. Apoptotic cells asimmunogen and antigen in the antiphospholipid syndrome. ExpMol Pathol 1999;66:82–98.
10. Rosen A, Casciola-Rosen L. Autoantigens as substrates for apo-ptotic proteases: implications for the pathogenesis of systemicautoimmune disease. Cell Death Differ 1999;6:6–12.
11. Grodzicky T, Elkon KB. Apoptosis in rheumatic diseases. Am JMed 2000;108:73–82.
12. Navratil JS, Ahearn JM. Apoptosis and autoimmunity: comple-ment deficiency and systemic lupus erythematosus revisited. CurrRheumatol Rep 2000;2:32–8.
13. Rovere P, Sabbadini MG, Fazzini F, Bondanza A, ZimmermannVS, Rugarli C, et al. Remnants of suicidal cells fostering systemicautoaggregation. Arthritis Rheum 2000;43:1663–72.
14. Miranda-Carus ME, Askanase AD, Clancy RM, Di Donato F,
Chou TM, Libera MR, et al. Anti-SSA/Ro and anti-SSB/Laautoantibodies bind the surface of apoptotic fetal cardiocytes andpromote secretion of TNF-alpha by macrophages. J Immunol2000;165:5345–51.
15. Gilligan HM, Bredy B, Brady HR, Hebert MJ, Slayter HS, Xu Y,et al. Antineutrophil cytoplasmic autoantibodies interact withprimary granule constituents on the surface of apoptotic neutro-phils in the absence of neutrophil priming. J Exp Med 1996;184:2231–41.
16. Earnshaw WC, Martins LM, Kaufmann SH. Mammalian caspases:structure, substrates, and functions during apoptosis. Annu RevBiochem 1999;68:383–424.
17. Cohen M, Lee KK, Wilson KL, Gruenbaum Y. Transcriptionalrepression, apoptosis, human disease and the functional evolutionof the nuclear lamina. Trends Biochem Sci 2001;26:41–7.
18. Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, RothfieldNF, et al. The 1982 revised criteria for the classification of systemiclupus erythematosus. Arthritis Rheum 1982;25:1271–7.
19. Senecal JL, Raymond Y. Autoantibodies to major and minorlamins are not restricted to autoimmune diseases. Clin ImmunolImmunopathol 1992;63:115–25.
20. Wong C, Sridhara S, Bardwell JC, Jakob U. Heating greatly speedsCoomassie blue staining and destaining. Biotechniques 2000;28:426–8.
21. Mayer JL, Beardsley DS. Varicella-associated thrombocytopenia:autoantibodies against platelet surface glycoprotein V. Pediatr Res1996;40:615–9.
22. Rao L, Perez D, White E. Lamin proteolysis facilitates nuclearevents during apoptosis. J Cell Biol 1996;135:1441–55.
23. Sharp GC, Irvin WS, May CM, Holman HR, McDuffie FC, HessEV, et al. Association of antibodies to ribonucleoprotein and Smantigens with mixed connective tissue disease, systemic lupuserythematosus and other rheumatic diseases. N Engl J Med1976;295:1149–54.
24. Lipsky PE. Systemic lupus erythematosus: an autoimmune diseaseof B cell hyperactivity. Nat Immunol 2001;2:764–6.
25. Marrack P, Kappler J, Kotzin BL. Autoimmune disease: why andwhere it occurs. Nat Med 2001;7:899–905.
26. Mevorach D, Zhou JL, Song X, Elkon KB. Systemic exposure toirradiated apoptotic cells induces autoantibody production. J ExpMed 1998;188:387–92.
27. Levine JS, Subang R, Koh JS, Rauch J. Induction of anti-phospholipid autoantibodies by �-2-glycoprotein I bound to apo-ptotic thymocytes. J Autoimmun 1998;11:413–24.
28. Gensler TJ, Hottelet M, Zhang C, Schlossman S, Anderson P, UtzPJ. Monoclonal antibodies from BALB/c mice immunized withapoptotic Jurkat cells recognize known autoantigens. J Autoim-mun 2001;16:59–69.
29. Ayukawa K, Taniguchi S, Masumoto J, Hashimoto S, SarvothamH, Hara A, et al. La antigen is cleaved in the COOH terminus andloses the nuclear localization signal during apoptosis. J Biol Chem2000;275:34465–70.
AUTOANTIBODIES TO LAMIN B1 AND THROMBOPROTECTION IN SLE 2707





























