Embed Size (px)
Citation preview

doi:10.1006/mcne.2001.0970, available online at http://www.idealibrary.com on
Molecular and Cellular Neuroscience 17, 696–705 (2001)MCN
Apoptosis and ROS Detoxification EnzymesCorrelate with Cytochrome c Oxidase Deficiencyin Mitochondrial Encephalomyopathies1Simone Di Giovanni, Massimiliano Mirabella, Manuela Papacci,Francesca Odoardi, Gabriella Silvestri, and Serenella ServideiInstitute of Neurology, Catholic University, Largo A. Gemelli 8, 00168 Rome, Italy
sihdMrwd
fa
advm
The aim of this work was to investigate in muscle the roleof apoptosis and of oxidative stress in mitochondrial dis-orders with dysfunction of respiratory chain. In patientswith cytochrome c oxidase deficiency (COX) we found avariable number of myofibers with apoptotic nuclei thatmatched with the level of enzymatic reduction androughly correlated with muscle weakness. In parallel, apositive immunostaining for apoptosis-related proteinsand Mn and Cu/Zn superoxide dismutase (SOD) weremostly localized in COX-negative fibers. Moreover, gluta-thione peroxidase activity was increased in muscles withhigh number of SOD-positive myofibers and prominentapoptotic features. No signs of apoptosis were observedin patients with deficiencies of complexes I and II andwithout muscle weakness. These data suggest that apo-ptosis along with increased ROS production, revealed byanti-oxidant enzymes overexpression, may play an impor-tant role in the pathophysiology of mitochondrial diseasesassociated with COX deficiency.
INTRODUCTION
In physiologic conditions mitochondrial oxidativephosphorylation consumes about 90% of the inhaledoxygen and is an important source of reactive oxygenspecies (ROS); about 1–2% of oxygen reduced in mito-chondria is in fact converted to the toxic anion super-oxide (Richter, 1992). Both mitochondrial/peroxisomalMn superoxide dismutase (SOD) and cytoplasmicCu/Zn SOD convert anion superoxide to hydrogenperoxide, which itself is quite inert, but can be reducedto the highly toxic hydroxyl radical by the Fenton reac-tion. Before conversion to hydroxyl radical, hydrogen
1 Supported by Telethon (Grant 1121) and M.U.R.S.T. ex 40%.
696
peroxide is partially metabolized and eliminated byglutathione peroxidase (GSHPx).
In pathologic and experimental conditions with im-pairment of oxidative phosphorylation (OXPHOS) mi-tochondria increase the production of free radicals(Richter et al., 1995). Moreover, the inhibition of ubiqui-nol-cytochrome c reductase by antimycin or of cyto-chrome c oxidase by azide or hypoxia increases gener-ation of anion superoxide in cardiomyocytes(Wolvetang et al., 1994; Duranteau et al., 1998). Simi-larly, OXPHOS deficiency induced in mice lackingthe heart/muscle adenine nucleotide translocator(Ant1tm2Mgr) results in increased hydrogen peroxide as-sociated with overexpression of the ROS detoxificationenzymes SOD and GSHPx in both whole skeletal mus-cle homogenate and isolated muscle mitochondria (Es-posito et al., 1999). Furthermore, oxidative damage tokeletal muscle DNA and increased SOD immunostain-ng in cytochrome c oxidase (COX)-deficient myofibersave been detected in two patients with a mitochon-rial encephalomyopathy (Mitsui et al., 1996). Intensen and Cu/Zn SOD immunostaining has also been
eported in COX negative myofibers in various patientsith different mtDNA mutations and respiratory chainefects (Ohkoshi et al., 1995; Carrier et al., 2000).These evidences suggest a possible pathogenic role of
ree radicals and their antioxidant enzymes in patientsffected by mitochondrial encephalomyopathies.The capacity of oxidative stress to induce or amplify
poptotic cell death has been intensively studied andocumented, mainly under experimental conditions inarious cellular systems including rat skeletal muscleyoblasts (Slater et al., 1995; Stangel et al., 1996; Tan et
al., 1998; Anderson et al., 1999). Experimentally oxida-
tive stressed cardiomyocytes also undergo apoptosis1044-7431/01 $35.00Copyright © 2001 by Academic Press
All rights of reproduction in any form reserved.

tpttta
temcant
potv(e
baMat
p
ml
697Apoptosis and Oxidative Stress in Mitochondrial Diseases
following cytochrome c release and caspases activation(Turner et al., 1998; Von Harsdorf et al., 1999). The roleof mitochondria is emphasized by the evidence thatmitochondrial respiratory chain inhibitors may inducecell death along with ROS production. Mitochondriaalso have a part in apoptosis independently of oxidativestress. In fact, experimental studies in vitro have shownhat disruption of the mitochondrial transmembraneotential and the release of some mitochondrial pro-
eins (cytochrome c and apoptosis-inducing factor) intohe cytoplasm can trigger the apoptotic pathwayhrough caspase 3 activation (Crompton, 1999; Kroemernd Reed, 2000).We have recently reported the occurrence of apopto-
ic phenomena in muscle of patients with mitochondrialncephalomyopathies with specific mitochondrial DNAutations. The degree of the apoptotic changes in mus-
le correlated with the percentage of mutant genomesnd with the severity of the myopathy. Moreover, COX-egative myofibers harbored more apoptotic nuclei
han COX-positive myofibers (Mirabella et al., 2000).The aim of the present study was to correlate the
resence of apoptotic features with the expression ofxidative stress defensive mechanisms in muscle of pa-ients with mitochondrial encephalomyopathies and se-ere respiratory chain dysfunction, comparing COXcomplex IV) deficiency with other respiratory chainnzymes defects.
We investigated the presence of DNA fragmentationy TUNEL and electron microscopy, the expression ofpoptosis related proteins (Fas, caspase 1, caspase 3), ofn SOD and Cu/Zn SOD by immunocytochemistry
nd the biochemical activity of GSHPx by spectropho-ometry.
RESULTS AND DISCUSSION
Results
TUNEL-positive myonuclei were present in a vari-able number of myofibers in all patients with COXdeficiency, isolated or combined with impairment ofother enzymatic complex (Table 1). The degree of bio-chemical and histochemical COX reduction roughlycorrelated with the number of TUNEL-positive myofi-bers (Table 1). Patients with deficiencies of complexes Iand II had only rare myofibers with TUNEL-positivenuclei (Table 2). Rare or no apoptotic nuclei were de-tected in patients proven to be free from muscle disease.Immunohistochemistry for mitochondrial Mn SOD and
cytoplasmic Cu/Zn SOD showed a strong immunore- zactivity in all patients with COX deficiency in a variableamount of fibers not confined to ragged-red-fibers(RRF). Muscle biopsies from patients with otherOXPHOS defects and from nonmitochondrial controlswere negative for SOD.
In patients with COX deficiency histochemistry forCOX and immunohistochemistry for Mn SOD andCu/Zn SOD performed in serial muscle sections re-vealed a clear correlation between absence of COX re-action and positive immunosignal for Mn SOD andCu/Zn SOD (Fig. 1, Table 1). By contrast, only 2–3% ofnormal COX-positive fibers were weakly immunola-beled for SOD. By performing TUNEL after COX his-tochemistry we found that about 60% of the total num-ber of COX-negative fibers harbored single or multipleTUNEL-positive nuclei, while only 5% of COX-positivefibers had apoptotic nuclei (Fig. 1).
Immunostaining for apoptosis-related proteinscaspase 1, caspase 3, and Fas correlated with theamount of myofibers positive at TUNEL in almost allpatients. Caspase 1 and caspase 3 had an intense cyto-plasmic immunolabeling, while Fas staining wasmainly detected on the membrane (Fig. 2).
Electron microscopy confirmed the presence of apo-ptotic features showing multiple nuclei with highlycondensed chromatin in hypotrophic fibers often asso-ciated with abnormalities of number and structure ofmitochondria. There were no signs of necrosis in themuscle fibers with abnormal nuclei (Fig. 3).
Glutathione peroxidase activity was significantly in-creased in the COX deficient muscles that showed nu-merous myofibers with SOD positive immunostainingand apoptotic features (Table 1) and did not strictlycorrelate with mitochondrial proliferation (evaluatedby the number of RRF and the activity of citrate syn-thase). The analysis of variance between COX activitiesand GSHPx activities values (Table 1) demonstrated ahigh statistical significance (P 5 2.3213). Complex I-and II-deficient muscles had GSHPx activities compa-rable to normal muscles controls (Table 2). Mean val-ues 6 SD of enzymatic activity (mmol/min/mg of non-collagen proteins) in COX-deficient muscles (a),complex I and II (b), and nonmitochondrial controls (c)were the following: 97.42 (6 46.4)(a); 47.45 (6 9.5)(b);and 55.29 (6 9.2) (c). Statistical significance was reachedbetween a and b (P 5 0.0072) and between a and c(P 5 0.011). GSHPx activity by “box and whiskers
lot” is represented in Fig. 4.In patients 6 and 13, who died from a fatal cardio-yopathy, the cardiac muscle specimens showed a
evel of apoptotic features and ROS detoxifying en-
ymes even more impressive than in the skeletal muscle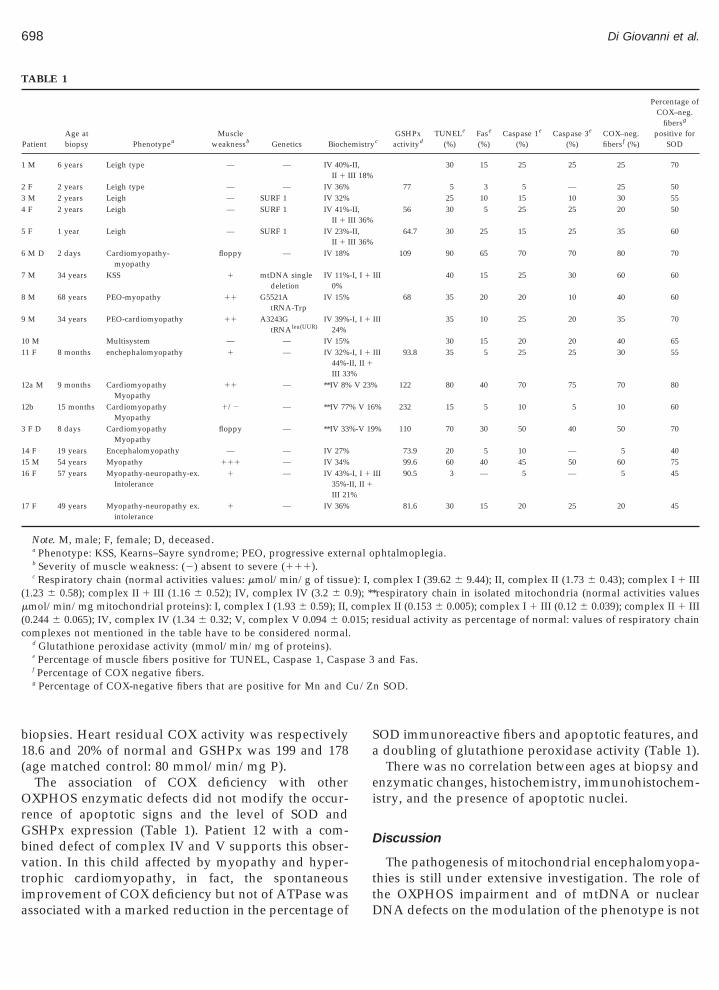
(
(c
u/Z
698 Di Giovanni et al.
biopsies. Heart residual COX activity was respectively18.6 and 20% of normal and GSHPx was 199 and 178(age matched control: 80 mmol/min/mg P).
The association of COX deficiency with otherOXPHOS enzymatic defects did not modify the occur-rence of apoptotic signs and the level of SOD andGSHPx expression (Table 1). Patient 12 with a com-bined defect of complex IV and V supports this obser-vation. In this child affected by myopathy and hyper-trophic cardiomyopathy, in fact, the spontaneousimprovement of COX deficiency but not of ATPase was
TABLE 1
PatientAge atbiopsy Phenotypea
Muscleweaknessb Genetics Bioche
1 M 6 years Leigh type — — IV 40%II 1 I
2 F 2 years Leigh type — — IV 36%3 M 2 years Leigh — SURF 1 IV 32%4 F 2 years Leigh — SURF 1 IV 41%
II 1 I5 F 1 year Leigh — SURF 1 IV 23%
II 1 I6 M D 2 days Cardiomyopathy-
myopathyfloppy — IV 18%
7 M 34 years KSS 1 mtDNA singledeletion
IV 11%0%
8 M 68 years PEO-myopathy 11 G5521AtRNA-Trp
IV 15%
9 M 34 years PEO-cardiomyopathy 11 A3243GtRNAleu(UUR)
IV 39%24%
10 M Multisystem — — IV 15%11 F 8 months enchephalomyopathy 1 — IV 32%
44%-III 33
12a M 9 months CardiomyopathyMyopathy
11 — **IV 8%
12b 15 months CardiomyopathyMyopathy
1/2 — **IV 77
3 F D 8 days CardiomyopathyMyopathy
floppy — **IV 33
14 F 19 years Encephalomyopathy — — IV 27%15 M 54 years Myopathy 111 — IV 34%16 F 57 years Myopathy-neuropathy-ex.
Intolerance1 — IV 43%
35%-III 21
17 F 49 years Myopathy-neuropathy ex.intolerance
1 — IV 36%
Note. M, male; F, female; D, deceased.a Phenotype: KSS, Kearns–Sayre syndrome; PEO, progressive exterb Severity of muscle weakness: (2) absent to severe (111).c Respiratory chain (normal activities values: mmol/min/g of tissu
1.23 6 0.58); complex II 1 III (1.16 6 0.52); IV, complex IV (3.2 6 0mmol/min/mg mitochondrial proteins): I, complex I (1.93 6 0.59); II,0.244 6 0.065); IV, complex IV (1.34 6 0.32; V, complex V 0.094 6 0.omplexes not mentioned in the table have to be considered normal.
d Glutathione peroxidase activity (mmol/min/mg of proteins).e Percentage of muscle fibers positive for TUNEL, Caspase 1, Caspf Percentage of COX negative fibers.g Percentage of COX-negative fibers that are positive for Mn and C
associated with a marked reduction in the percentage of
SOD immunoreactive fibers and apoptotic features, anda doubling of glutathione peroxidase activity (Table 1).
There was no correlation between ages at biopsy andenzymatic changes, histochemistry, immunohistochem-istry, and the presence of apoptotic nuclei.
Discussion
The pathogenesis of mitochondrial encephalomyopa-thies is still under extensive investigation. The role ofthe OXPHOS impairment and of mtDNA or nuclear
cGSHPx
activitydTUNELe
(%)Fase
(%)Caspase 1e
(%)Caspase 3e
(%)COX–neg.fibersf (%)
Percentage ofCOX–neg.
fibersg
positive forSOD
30 15 25 25 25 70
77 5 3 5 — 25 5025 10 15 10 30 55
56 30 5 25 25 20 50
64.7 30 25 15 25 35 60
109 90 65 70 70 80 70
III 40 15 25 30 60 60
68 35 20 20 10 40 60
III 35 10 25 20 35 70
30 15 20 20 40 65III 93.8 35 5 25 25 30 55
122 80 40 70 75 70 80
% 232 15 5 10 5 10 60
% 110 70 30 50 40 50 70
73.9 20 5 10 — 5 4099.6 60 40 45 50 60 75
III 90.5 3 — 5 — 5 45
81.6 30 15 20 25 20 45
phtalmoplegia.
complex I (39.62 6 9.44); II, complex II (1.73 6 0.43); complex I 1 IIIrespiratory chain in isolated mitochondria (normal activities valueslex II (0.153 6 0.005); complex I 1 III (0.12 6 0.039); complex II 1 IIIesidual activity as percentage of normal: values of respiratory chain
and Fas.
n SOD.
mistry
-II,II 18%
-II,II 36%-II,II 36%
-I, I 1
-I, I 1
-I, I 1
II, II 1
%V 23%
% V 16
%-V 19
-I, I 1
II, II 1
%
nal o
e): I,.9); **comp015; r
ase 3
DNA defects on the modulation of the phenotype is not

cCiega
D.
699Apoptosis and Oxidative Stress in Mitochondrial Diseases
fully elucidated. Heteroplasmy and threshold effect,site, percentage, inter and intratissue distribution ofspecific mtDNA mutations contribute to the phenotype,but do not explain the overall clinical heterogeneity thatis also present within the same genetic defect (Di Mauroand Bonilla, 1997). The final common step is a defect ofenergy production due to the respiratory chain impair-ment.
We have recently documented the presence of apo-ptotic features in muscle biopsies from patients withmitochondrial encephalomyopathies who carried mito-chondrial DNA defects. Apoptotic changes were notfound in all patients and depended upon the type of themutation (being present with tRNA points mutations orsingle deletions) and the percentage of the mutant ge-nomes above a given threshold. Moreover, COX-nega-tive myofibers appeared to have more apoptotic fea-tures than COX-positive fibers (Mirabella et al., 2000).
Muscle COX-negative fibers together with RRF areonsidered the hallmarks of mitochondrial diseases.OX deficiency is the most common enzymatic defect
n mitochondrial patients independently of the pres-nce or absence of a known mitochondrial or nuclearene mutation. In the present work we demonstrated
TABLE 2
PatientAge atbiopsy Phenotype
Muscleweaknessa Genetics Biochemistr
1 M 60 years Myalgia-ex. intoleranceparaparesis
1/2 — I, I 1 III 8%
2 F 5 years Encephalomyopathy 1/2 — I, I 1 III 31%3 F 55 years Ex. intolerance-
cataracta-ageing— — I, I 1 III 26%
4 M 67 years Ex. intolerance — — I, I 1 III 38%5 M 18 years Encephalopathy — — I, I 1 III 43%-
II 1 III 20%6 F 18 years Encephalomyopathy 1/2 — I, I 1 III 38%-
II 1 III 31%7 M 85 years Myopathy 1 — I, I 1 III 30%-
II 1 III 43%8 F 4 months Encephalopathy — — II, II 1 III 28%9 M 3 years Encephalomyopathy 1/2 — II, II 1 III 35%10 F 81 years Neuromyopathy 1 — II, II 1 III 26%11 M 8 months Hypotonia-
hyperlattacidemia1/2 — II, II 1 III 32%
Note. M, male; F, female.a Severity of muscle weakness: (—) absent to severe (111);b Respiratory chain (normal activities values: mmol/min/mg of prot
(1.23 6 0.58); complex II 1 III (1.16 6 0.52); IV, complex IV (3.2 6respiratory chain complexes not mentioned in the table have to be co
c Glutathione peroxidase activity (mmol/min/mg of proteins).d Percentage of muscle fibers positive for TUNEL, Caspase 1, Caspe Percentage of COX-negative muscle fibers.f Percentage of COX-negative fibers positive for Mn and Cu/Zn SO
poptotic features and overexpression of antioxidant
enzymes SOD and GSHPx in muscle of patients withmitochondrial disorders and severe COX deficiency butnot in patients with other respiratory chain defects.
The contemporary presence in numerous COX-nega-tive fibers of multiple apoptotic nuclei and positiveimmunostaining for Mn and Cu/Zn SOD (Fig. 1) indi-cates a linkage between COX defect, apoptosis, andoverexpression of antioxidant mechanisms.
The strong immunoreactivity for these enzymes indeficient fibers may imply an attempt to compensatefree radical overproduction. This hypothesis is vali-dated by the observation that GSHPx activity is ele-vated in muscles with numerous SOD-positive myofi-bers. The correlation between TUNEL-positive nucleiand apoptosis-related proteins Fas, caspase 1, andcaspase 3 further supports the specificity of the findingsobtained by TUNEL.
The significance of apoptosis in muscle pathology isstill on debate. In mitochondrial myopathies necrosis isseldom observed. However, in spite of the presence ofnumerous TUNEL-positive nuclei in some patients, thetypical morphologic features of mononucleated cellsundergoing apoptosis are scarce in muscle cells.
We have hypothesized that myofiber functionality
GSHPxactivityc
TUNELd
(%)Fasd
(%)Caspase 1d
(%)Caspase 3d
(%)
COX–neg.fiberse
(%)
Percentage ofCOX–neg.
fibersf
positive forSOD
— — — — 2 50
53 — — — — — —48 — — 2 — — —
45.5 — — — — — —2 — — 2 — —
42 — — — — — —
60 3 2 — — 3 33
— — — — — —61 2 — 2 — — —35 — — — — — —28 — — — — — —
I, complex I (39.62 6 9.44); II, complex II (1.73 6 0.43); complex I 1 IIIV, complex V; residual activity as percentage of normal: values ofered normal.
, and Fas.
yb
II,
II,
II,
eins):0.9);nsid
ase 3
and viability can be compromised only when multiple

Em
700 Di Giovanni et al.
apoptotic nuclei within the single fiber are present(Mirabella et al., 2000). Accordingly, we have recentlydemonstrated that massive muscle atrophy and myosinloss in acute quadriplegic myopathy are accompaniedby the presence of 70–100% of apoptotic nuclei (DiGiovanni et al., 2000). In the same way, in our patientswith severe COX deficiency, atrophy of fibers is presentwhen almost all nuclei are apoptotic (Figs. 1 and 3).
Interestingly, our data suggest that OXPHOS impair-ment per se is not sufficient in determining apoptoticfeatures and overexpression of ROS detoxifying en-
FIG. 1. TUNEL and COX histochemistry on the same section (A, B,D are serial sections). (A) The vast majority of muscle fibers are COX-TUNEL-positive nuclei are present within COX-negative fibers (patiincreased Mn SOD (C) and Cu/Zn SOD (D) immunoreactivity (sCOX-negative fibers in the first muscle biopsy of a patient with revestaining with a parallel reduction of TUNEL-positive nuclei is eviden
zymes. In fact, muscle biopsies with defects of complex p
I and complex II showed sporadic myofibers with ap-optotic nuclei and SOD-positive staining, while GSHPxactivity was not significantly different from nonmito-chondrial control muscles. Other authors have sug-gested that the experimental block of respiratory chaindownstream complex II may cause electrons to accu-mulate around complex I and ubiquinone, from wherethey can be directly donated to oxygen to give anionsuperoxide (Turrens et al., 1985; Kwong et al., 1998;
sposito et al., 1999). Similarly, in our patients, impair-ent of COX activity along with preservation of com-
Mn SOD (C) and Cu/Zn SOD (D) immunohistochemistry (B, C, andive and show TUNEL-positive nuclei (patient 6, table 1). (B) Multiple5, Table 1). On serial sections, the same muscle fibers have stronglyrrowheads). (E) Numerous TUNEL-positive nuclei are present in
COX deficiency (patient 12 a, Table 1). (F) Diffuse increase of COXthe second biopsy (patient 12b). Original magnification: 3250 (A–F).
E, F);negatent 1ee arsiblet in
lex I or II may stall electron flow through the electron

1
pcB(nang
701Apoptosis and Oxidative Stress in Mitochondrial Diseases
transport chain determining increased ROS production.By contrast, a deficiency of the first two complexes ofthe respiratory chain may reduce the electrons forma-tion and the consequent anion superoxide generation:this could explain the low levels of ROS detoxifyingenzymes in our complex I- and II-deficient muscles. Adefect of COX with diminished translocation of protonsout of the mitochondria and reduction of the protonmotor force would also cause a partial block down-stream complex IV, reducing, but not impeding ATPsynthesis by ATP synthase (complex V). Importantly,reduction of ATP cellular level is a proapoptotic factor,while its absolute lack induces necrosis (Richter et al.,995).Previously published reports demonstrated that com-
lex I deficiency in cell lines is associated with in-reased ROS production (Pitaken and Robinson, 1996;arrientos and Moraes, 1999). Barrientos and Moraes
1999) conducted their experiments in human–ape xe-omitochondrial cybrids with partial complex I defectnd osteosarcoma-derived cell lines treated with rote-one showing impairment of cell respiration, cell
FIG. 2. Caspase-1, caspase-3, and Fas immunocytochemistry. Diffusein several muscle fibers (patient 9, Table 1). Fas immunoreactivity is pin the form of granular staining in several muscle fibers (C) (patientOriginal magnification: 3250 (A, B), 3125 (C, D).
rowth, free radical productions, and occurrence of
apoptosis; cell death was associated with ROS produc-tion more than respiratory chain dysfunction. Pitakenand Robinson (1996) studied fibroblasts from patientswith complex I deficiency and various clinical pheno-types. They observed Mn SOD induction that correlatedwith redox state of the cell rather than with complex Idefect per se. Moreover, they found a marked inductionof MnSOD even in fibroblasts from two patients withsevere Leigh disease and isolated COX deficiency. Thelack or scarcity of SOD induction in muscle biopsies ofour patients with complex I deficiency is in agreementwith the presence of none or mild muscle weaknesseven though the enzyme defect was also expressed inmuscle. It is, however, possible that clinically affectedCNS or fibroblasts of these patients would show ROSoverproduction and signs of apoptosis. In summary,both these papers and our data demonstrate that oxi-dative stress and apoptosis are selectively expressed inthe clinically affected target tissues, supporting theirpathogenic role. The complex interaction that leads toROS overproduction from a biochemical defect may bedifferent in different tissues and has still to be better
creased caspase-1 (A) and caspase-3 (B) immunoreactivity is detectedt both over the surface membrane and partially within the cytoplasmable 1), while no Fas staining is detectable in a control muscle (D).
ly inresen15, T
elucidated.

702 Di Giovanni et al.
Dismutation of anion superoxide to hydrogen perox-ide by SOD is a well-known defensive mechanism fromoxidative stress at the cellular level. On the other hand,reduction of hydrogen peroxide by the Fenton reactionto the highly toxic peroxidating hydroxyl radical can be,in some circumstances, even more harmful to the cell.GSHPx activity has instead the clear-cut role to metab-olize and eliminate hydrogen peroxide, partially avoid-ing conversion to hydroxyl radical. In our COX-defi-cient muscles, upregulation of these ROS detoxifyingenzymes do occur, indirectly indicating ROS overpro-duction. SOD and GSHPx, however, seem not to besufficient to prevent the apoptotic cascade. This maysuggest that SOD itself exerts a toxic action on the cellthrough peroxidation and that GSHPx is not able tofully antagonize this negative effect.
In patient 12 (Table 1) the partial reversibility of thedefect of COX, but not of the associated complex V, wasaccompanied by an increase of the already elevatedGSHPx activity and by a decrease of SOD immunola-beling and of apoptotic features. This evidence stronglysuggests that COX deficiency is a key event in thedetermination of oxidative stress and apoptosis. Thespontaneous reversibility of COX activity in a few pa-tients with mitochondrial myopathy is a well known,but still not understood phenomenon (Di Mauro andBonilla, 1997). Interestingly, in our patient, improve-
FIG. 3. Electron micrograph showing a hypotrophic muscle fiber whigher magnification it is visible extensive loss of myofilaments accomand other cellular debris (B). Original magnification: 33200 (A); 363
ment of COX defect and of muscle weakness was ac-
companied by an important reduction of apoptotic fea-tures and a doubling of GSHPx activity raising thepossibility of an effective antiapoptotic action by thisdetoxifying enzyme. Increase of GSHPx in the secondbiopsy is likely to be the delayed response to a longlasting oxidative stress, while COX activity was justrestored. Reduction of apoptotic features may also rep-resent a sign of plasticity of the apoptotic process inmuscle depending on the balance between pro- andantiapoptotic stimuli. If this were true, it would bepossible to rescue a multinucleated muscle cell if pro-apoptotic factors are removed when only a minority ofapoptotic nuclei is present. At the same time, the powerof regeneration of muscle could compensate for the lossof those cells irreversibly committed to death. In ourpatient, in second biopsy, signs of regeneration werepresent and may have contributed to the improvementof muscle pathology.
Apoptotic features and antioxidant enzymes expres-sion did not associate in our COX deficient patientswith a particular phenotype, but they roughly corre-lated with muscle weakness (Table 1). Moreover, inpatient 12, with a reversible defect of COX, the reduc-tion of apoptotic features paralleled the improvementof myopathy. Furthermore, in patients 6 and 13, whodied of untreatable cardiac failure, all nuclei of hearttissue were TUNEL-positive. To reinforce the concept,
ultiple nuclei with condensed chromatin (patient 7, Table 1) (A). Atd by abnormal mitochondrial profiles and accumulation of lipofuscin).
ith mpanie00 (B
in mitochondrial encephalomyopathies with complex I

o2g(
703Apoptosis and Oxidative Stress in Mitochondrial Diseases
and II deficiencies the absence of apoptotic featurescorrelated with the absence of weakness in most pa-tients (Table 2).
Taken together, all these data strengthen the patho-genic role of oxidative stress and apoptosis on skeletaland heart muscle dysfunction in mitochondrial dis-eases.
On the other side human COX deficiency may repre-sent a “natural” model to investigate the importance ofmitochondria and OXPHOS dysfunction in the apopto-tic pathways. This may be useful also for a better un-derstanding of degenerative diseases. In the last years,in fact, the role of mitochondria in oxidative stress andapoptotic cell death has become particularly relevant tothe pathogenesis of various neurodegenerative disor-ders (Schapira, 1999; Leonard and Schapira, 2000).
Further studies both in cellular systems and in vivowill help to define the chain of events leading to apo-ptosis and to design new approaches to therapy actingon the modulation of proapoptotic factors and the in-
FIG. 4. Glutathione peroxidase activity in muscle homogenates. Thef patients: COX deficient muscles (a), complex I and II (b) and nonmit5th and 75th percentiles, and whiskers represent 5th and 95th percenroup (b) and by 43.2% compared with group (c). Statistical significanP 5 0.011). Number in each bar indicates the number of muscles h
hibition of oxidative stress mechanisms.
EXPERIMENTAL METHODS
We studied skeletal muscle biopsies, obtained withinformed consent, from 17 patients with mitochondrialencephalomyopathies and COX deficiency, 11 patientswith mitochondrial respiratory complex I and II defects,and 8 patients who had proven to be free of muscledisease. In one patient with a defect of complex Vassociated in addition to spontaneously reversible COXdeficiency we performed our experiments in two con-secutive muscle biopsies (pt.12, Table 1). In this child,affected by myopathy and hypertrophic cardiomyopa-thy, muscle weakness improved with the increase ofCOX activity. In patients 6 and 13, affected by myop-athy and cardiomyopathy, who died respectively at age2 and 8 days of severe untreatable heart failure, we alsostudied postmortem cardiac muscle specimens.
Among the group of COX deficiency, six patients hadan identified nuclear or mitochondrial gene defect:
-and-whisker” plot of glutathione enzymatic activity for each groupndrial controls (c). The small box indicates the median, box representsGroup (a) has increased enzymatic activity by 51.5% compared with
as reached between (a) and (b) (P 5 0.0072) and between (a) and (c)genates for group.
“boxochotiles.ce womo
three children with Leigh syndrome had a mutation in

BbicwU
704 Di Giovanni et al.
the SURF 1 gene, one patient with Kearns-Sayre syn-drome (KSS) had the “common deletion” of mitochon-drial DNA (mtDNA), one patient with progressive ex-ternal ophtalmoplegia (PEO) and myopathy had aG5521A mutation in the mitochondrial gene of tRNAtrp
and the sixth patient, with PEO and cardiomyopathy,had a A3243G transversion in the tRNAleu(UUR)gene.Clinical, genetic, biochemical, and morphological dataare summarized in Tables 1 and 2.
TUNEL and Immunohistochemistry
The TUNEL (terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling) technique was ap-plied for detection of nuclear DNA fragmentation insitu. Frozen muscle sections from patients and controlswere incubated under the same coverslip with TUNELreaction mixture and incorporated fluorescein-dUTPwas detected by using alkaline phosphatase-conjugatedanti-fluorescein antibodies according to the manufac-turer’s instruction (In Situ Cell Death Detection kit,
oehringer). Negative experimental controls were incu-ated with label solution without terminal transferase
nstead of TUNEL reaction mixture, while a positiveontrol was set up by preincubating muscle sectionsith DNase I for 10 min before the TUNEL procedure.nfixed 10-mm muscle sections adjacent to those ana-
lyzed by means of TUNEL were processed for immu-nocytochemistry as follows: sections were dried atroom temperature, fixed in cold acetone, and pretreatedwith 0.3% H 2O2 in PBS to quench the endogenousperoxidase activity, rinsed in PBS, and incubated with10% normal serum (goat or rabbit depending on thesecondary antibodies used) for 60 min to mask nonspe-cific absorption sites. Sections were then incubated for1 h at room temperature with one of the followingantibodies: rabbit polyclonal against human Fas (C-20Santa Cruz, diluted 1:200), rabbit polyclonal anti-hu-man caspase 1 (Santa Cruz, diluted 1:100), goat poly-clonal anti-human Caspase 3 (Santa Cruz, diluted1:200), mouse monoclonal anti-human Cu/Zn SOD(RBI, diluted 1:300), sheep polyclonal anti-human MnSOD (Calbiochem, diluted 1:300). Omission of the pri-mary antibodies or their replacement by preimmunesera was used for control experiments. After severalrinses in PBS, the sections were incubated with theappropriate biotinylated secondary antibodies (goat an-ti-mouse, goat anti-rabbit, or rabbit anti-goat IgG)washed in PBS and then incubated with the Avidin–biotin–peroxidase complex according to the manufac-turer’s instructions (Vectastain ABC, Vector Laborato-
ries, Burlingame, CA). Peroxidase staining wasobtained by incubating the sections in 0.075% DAB and0.002% H2O2 in 50 mM Tris buffer, pH 7.6, for 10 min.In order to evaluate the relative amount of TUNEL-positive nuclei within individual fibers and their corre-lation with COX-negative fibers and SOD-positive fi-bers, TUNEL was also performed on the same musclesections after histochemistry for COX, and compared toserial sections processed by Mn SOD and Cu/Zn SODimmunohistochemistry. The count of fibers withTUNEL-positive nuclei and of fibers positive by immu-nocytochemistry was performed in at least a hundredmuscle fibers per section and expressed as percentages.The number of TUNEL-positive nuclei and of apoptosisrelated-proteins was correlated with the age at time ofbiopsy, phenotype, degree of muscle weakness, severityof biochemical defect, and SOD-positive fibers. SOD-positive myofibers were also compared to COX-nega-tive fibers (tables).
Electron Microscopy
For electron microscopic study, muscle samples werefixed with 2.5% glutaraldehyde in 0.1 M phosphatebuffer, pH 7.4, postfixed in 1% osmium tetraoxide, de-hydrated in a graded ethanol series, and embedded inEpon 812. Ultrathin sections were cut and stained withuranyl acetate and lead citrate, and examined with a208S Philips electron microscope.
Glutathione Peroxidase Activity
To evaluate the activity of the antioxidant enzymeGSHPx frozen muscle samples from the same biopsiesprocessed for TUNEL and immunohistochemistry wereweighted and homogenized in six volumes of 50 mMTris–HCL, pH 7.5, containing 5 mM EDTA and 1 mMdithiothreitol. Samples were then centrifuged at 8000gfor 15 min at 6°C. Noncollagen protein concentrationwas determined from the clarified homogenate. GSHPxactivity was assayed, in the supernatant, according toPaglia and Valentine, with Bioxytech GPx-340 kit (Col-orimetric assay for cellular glutathione peroxidase, Oxisinternational). Briefly, GSHPx activity was obtained bymeasuring spectrophotometrically at 340 nm at 25°C for3 min NADPH oxidation in presence of reduced gluta-thione and ter-butyl hydroperoxide.
Statistical analysis was performed by the t Studenttest, and GSHPx activity was compared in COX defi-cient muscles (a), complex I and II (b), and nonmito-chondrial controls (c). Moreover, the analysis of vari-
ance for GSHPx and COX activities was evaluated
R
R
R
S
S
W
705Apoptosis and Oxidative Stress in Mitochondrial Diseases
through the ANOVA test in the patients reported inTable 1. A value of P , 0.05 was considered significant.
REFERENCES
Anderson, K. M., Seed, A., Ou, D., and Harris, J. E. (1999). Free radicaland reactive oxygen species in programmed cell death. Med. Hy-potheses 52: 451–463.
Barrientos, A., and Moraes, C. T. (1999). Titrating the effects of mito-chondrial complex I impairment in the cell physiology. J. Biol. Chem.274: 16188–16197.
Carrier, H., Flocard, F., Tagliati, V., Arrigo, A. P., and Godinot, C.(2000). Immunolabeling of mitochondrial superoxide dismutaseand of Hsp60 in muscle harbouring a respiratory chain deficiency.Neuromusc. Disord. 10: 144–149.
Crompton, M. (1999). The mitochondrial permeability transition poreand its role in cell death. Biochem. J. 341: 233–249.
Di Giovanni, S., Mirabella, M., D’Amico, A., Tonali, P., and Servidei,S. (2000). Apoptotic features accompany acute quadriplegic myop-athy. Neurology 55: 854–858.
Di Mauro, S., and Bonilla, E. (1997). Mitochondrial encephalomyopa-thies. In The Molecular and Genetic Basis of Neurological Diseases (R. N.Rosenberg, S. B. Prusiner, S. Di Mauro, and R. L. Barchi Eds.), pp.201–235. Butterworth-Heinemann, Boston.
Duranteau, J., Chandel, N. S., Kulioz, A., Shao, Z., and Schumacher,P. T. (1998). Intracellular signaling by reactive oxygen species dur-ing hypoxia in cardiomyocytes. J. Biol. Chem. 273: 11619–11624.
Esposito, L. A., Melov, S., Panov, A., Cottrell, B. A., and Wallace, D. C.(1999). Mitochondrial disease in mouse results in increased oxida-tive stress. Proc. Natl. Acad. Sci. USA 96: 4820–4825.
Leonard, J. V., and Schapira, A. H (2000). Mitochondrial respiratorychain disorders II: Neurodegenerative disorders and nuclear genedefects. Lancet 355: 389–394.
Kroemer, G., and Reed, J. C. (2000). Mitochondrial control of celldeath. Nature Med. 6: 513–519.
Kwong, L. K., and Sohal, R. S. (1998). Substrate and site specificity ofhydrogen peroxide generation in mouse mitochondria. Arch. Bio-chem. Biophys. 350: 118–126.
Mirabella, M., Di Giovanni, S., Silvestri, G., Tonali, P., and Servidei, S.(2000). Apoptosis in mitochondrial encephalomyopathies with mi-tochondrial DNA mutations: A potential pathogenic mechanism.Brain 123: 93–104.
Mitsui, T., Kawai, H., Nagasawa, M., Kunishige, M., Akaike, M.,
Kimure, Y., and Saito, S. (1996). Oxidative damage to skeletalmuscle DNA from patients with mitochondrial encephalomyopa-thies. J. Neurol. Sci. 139: 111–116.
Ohkoshi, N., Mizusawa, H., Shiraiwa, N., Shoji, S., Harada, K., andYoshizawa, K. (1995). Superoxide dismutase of muscle in mitochon-drial encephalomyopathies. Muscle and Nerve 18: 1265–1271.
Pitkanen, S., and Robinson, B. H. (1996). Mitochondrial complex Ideficiency leads to increased production of superoxide radicals andinduction of superoxide dismutase. J. Clin. Invest. 98: 345–351.
ichter, C. (1992). Reactive oxygen and DNA damage in mitochon-dria. Mutat. Res. 275: 249–255.
ichter, C., Gogvadze, V., Laffranchi, R., Sclapbach, R., Schweizer, M.,Suter, M., Walter, P., and Yaffee, M. (1995). Oxidants in mitochon-dria: From physiology to diseases. Biochem. Biophys. Acta 1271:67–74.
ichter, C., Schweizer, M., Cossarizza, A., and Franceschi, C. (1996).Control of apoptosis by the cellular ATP level. FEBS Lett. 378:107–110.
chapira, A. H. (1999). Mitochondrial involvement in Parkinson’sdisease, Huntington disease, hereditary spastic paraparesis andFriedreich ataxia. Biochim. Biophys. Acta 1410: 159–170.
later, A. F. G., Nobel, C. S. I., and Orrenius, S. (1995). The role ofintracellular oxidants in apoptosis. Biochem. Biophys. Acta 1271:59–62.
Stangel, M., Zettl, U. K., Mix, E., Zielasek, J., Toyka, K. V., Hartung,H. P., and Gold, R. (1996). H2O2 and Nitric Oxide-mediated oxi-dative stress induce apoptosis in rat skeletal muscle myoblasts.J. Neuropathol. Exp. Neurol. 55: 36–43.
Tan, S., Sagara, Y., Liu, Y., Pamela, M., and Scubert, D. (1998). Theregulation of reactive oxygen species production during pro-grammed cell death. J. Cell. Biol. 141: 1423–1432.
Turrens, J. F., Alexandre, A., and Lehninger, A. L. (1985). Ubisemiqui-none is the electron donor for superoxide formation by complex IIIof heart mitochondria. Arch. Biochem. Biophys. 237: 408–414.
Turner, N. A., Xia, F., Azhar, G., Zhang, X., Liu, L., and Wei, J. Y.(1998). Oxidative stress induces DNA fragmentation and caspaseactivation via the c-Jun NH2- terminal kinase pathway in H9c2cardiac muscle cells. J. Mol. Cell. Cardiol. 30: 1789–1801.
Von Harsdorf, R., Li, P. F., and Dietz, R. (1999). Signaling pathways inreactive oxygen species-induced cardiomyocytes apoptosis. Circu-lation 99: 2934–2941.olvetang, E. J., Johnson, K. L., Krauer, K., Ralph, S. J., and Linnane,A. W. (1994). Mitochondrial respiratory chain inhibitors induce
apoptosis. FEBS Lett. 339: 40–44.Received December 14, 2000Revised January 25, 2001
Accepted February 2, 2001





























