Embed Size (px)
Citation preview

Aliphatic peptides show similar self-assembly toamyloid core sequences, challenging the importanceof aromatic interactions in amyloidosisAnupama Lakshmanana,1, Daniel W. Cheongb,1, Angelo Accardoc, Enzo Di Fabrizioc, Christian Riekeld,and Charlotte A. E. Hausera,2
aInstitute of Bioengineering and Nanotechnology, Singapore 138669; bInstitute of High Performance Computing, Singapore 138632; cIstituto Italianodi Tecnologia, 16163 Genoa, Italy; and dEuropean Synchrotron Radiation Facility, 38043 Grenoble Cedex 09, France
Edited* by Alexander Rich, Massachusetts Institute of Technology, Cambridge, MA, and approved November 21, 2012 (received for review October 12, 2012)
The self-assembly of abnormally folded proteins into amyloid fibrilsis a hallmark of many debilitating diseases, from Alzheimer’s andParkinson diseases to prion-related disorders and diabetes type II.However, the fundamental mechanism of amyloid aggregationremains poorly understood. Core sequences of four to seven aminoacids within natural amyloid proteins that form toxic fibrils havebeen used to study amyloidogenesis. We recently reported a classof systematically designed ultrasmall peptides that self-assemble inwater into cross-β–type fibers. Here we compare the self-assemblyof these peptides with natural core sequences. These include coresegments from Alzheimer’s amyloid-β, human amylin, and calcito-nin. We analyzed the self-assembly process using circular dichro-ism, electron microscopy, X-ray diffraction, rheology, and moleculardynamics simulations. We found that the designed aliphatic pep-tides exhibited a similar self-assembly mechanism to several natu-ral sequences, with formation of α-helical intermediates being acommon feature. Interestingly, the self-assembly of a second core se-quence from amyloid-β, containing the diphenylalanine motif, wasdistinctly different from all other examined sequences. The diphe-nylalanine-containing sequence formed β-sheet aggregates withoutgoing through the α-helical intermediate step, giving a unique fiber-diffraction pattern and simulation structure. Based on these results,we propose a simplified aliphatic model system to study amyloid-osis. Our results provide vital insight into the nature of early inter-mediates formed and suggest that aromatic interactions are not asimportant in amyloid formation as previously postulated. This in-formation is necessary for developing therapeutic drugs that in-hibit and control amyloid formation.
Amyloid fibril formation is implicated in a wide range ofchronic degenerative diseases such as Alzheimer’s, Parkin-
son, and prion-related diseases, as well as others, such as diabetestype II (1). Although amyloidogenic proteins from unrelateddiseases do not share any sequence homology, the structuralproperties of the amyloid fibrils are similar (2). Non–disease-associated proteins can also form amyloids in vitro, suggestingthat the process is ubiquitous (3). Despite the clinical signifi-cance of amyloidogenesis and its widespread occurrence, theexact mechanism of amyloid formation remains unclear. Severalstudies have indicated that amyloids, commonly characterized bya highly ordered cross-β structure (4), are associated with protein-specific amyloidogenic core sequences of a few amino acids (2, 5,6). These core sequences, usually four to seven amino acids long,are able to self-assemble into fibrils physically, morphologically,and tinctorally similar to those observed with the entire protein(2, 6–9). Thus, they have the potential to serve as model systems,simplifying the study of amyloidosis. Furthermore, there isgrowing evidence that early oligomers during the self-assemblyprocess are more pathogenic and toxic compared with maturefibrils (10, 11). Clearly, there is an urgent need to identify thestructures and properties of the early oligomers to arrest molec-ular recognition and self-assembly in the earlier stages.Recently, we reported a class of rationally designed ultrasmall
peptides that self-assemble in water into amyloid-β−type fibers
(12). Based on this, we proposed a mechanism for amyloid for-mation involving a conformational transition of the structurallyunorganized monomers into metastable α-helical intermediatesthat terminate in cross-β structures (12). The current studycompares the self-assembly process of these peptides with natu-rally occurring amyloidogenic core sequences from three amy-loid-associated diseases, namely Alzheimer’s, diabetes type II,and thyroid medullary carcinoma. We aim to extend the self-as-sembly mechanism observed for rationally designed peptides tonatural amyloidogenic sequences. We studied the self-assembly ofdesigned and natural peptide sequences using circular dichroism,electron microscopy, X-ray fiber diffraction, rheology, and mo-lecular dynamics simulations.An increased occurrence of aromatic residues in natural core
sequences has led to widespread conclusions about the crucialrole played by these residues in molecular recognition and self-assembly (13–15). Comparing the self-assembly of our fully ali-phatic designed peptides with natural core sequences would alsohelp to determine the significance and effect of π–π interactionson amyloid formation.In addition, we wanted to identify and characterize key early
intermediates that drive self-assembly and can thus serve asa common target across diseases for therapeutic targeting andintervention.
ResultsThe investigated rationally designed peptides (LD6 and ID3),natural amyloidogenic core sequences (NL6, DF5, GA6, andKE7), and RADA are listed in Table 1. Their origins and specificsecondary structural transformations during self-assembly arealso given. LD6 (LIVAGD) and ID3 (IVD) are two extensivelycharacterized ultrasmall peptides (12, 16), namely the best-performing 6-mer with respect to propensity of gelation and gelstrength, and a 3-mer. The natural core sequences include defined“hotspots” within amyloid proteins of high amyloidogenic po-tential, without and with one or two aromatic residues at differentpositions in the sequence (2, 5–8, 14, 17). RADA is an ionic self-complementary 16-mer, known for forming stacks of flat β-sheets(18). All peptides were acetylated at the N terminus, with RADAadditionally amidated at the C terminus.
Amyloidogenic Core Sequences Self-Assemble into Similar Structuresas LD6 and ID3. NL6, DF5, GA6, and KE7 as well as LD6 and ID3self-assembled into hydrogels (Fig. S1 A–F). GA6 (GGVVIA) is
Author contributions: C.A.E.H. designed research; A.L., D.W.C., A.A., C.R., and C.A.E.H.performed research; E.D.F. contributed new reagents/analytic tools; A.L., D.W.C., C.R., andC.A.E.H. analyzed data; and A.L., D.W.C., C.R., and C.A.E.H. wrote the paper.
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.1A.L. and D.W.C. contributed equally to this work.2To whom correspondence should be addressed. E-mail: [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1217742110/-/DCSupplemental.
www.pnas.org/cgi/doi/10.1073/pnas.1217742110 PNAS | January 8, 2013 | vol. 110 | no. 2 | 519–524
BIOPH
YSICSAND
COMPU
TATIONALBIOLO
GY

an aliphatic core sequence from the transmembrane domain ofamyloid-β (Aβ) with a high propensity for amyloid aggregation(17). Another core sequence is KE7 (KLVFFAE), containing thesequence LVFFA (19) with its diphenylalanine (FF) motif,considered essential for aggregation of Aβ (15, 20). There is aminimum critical concentration for each peptide below which nogelation is observed. Above the critical value, the speed of hy-drogel formation followed the order LD6 ≅DF5 > NL6 >GA6 >KE7 > ID3, with the order LD6 > ID3 as previously observed(12, 16). Formation of a solid gel was determined by visual in-spection (turning the vial upside down). At 12 mM, LD6 formedthe most shape-compact and homogeneous gel, whereas NL6,GA6, and DF5 showed slight phase separation of the fibrousaggregates from the aqueous phase (Fig. S1 A–F). KE7 formedhydrogels and aggregates with different properties that will bedescribed below.The morphology of fiber networks constituting the hydrogels
was evaluated by field emission scanning electron microscopy(FESEM) (Fig. 1 A, B, and D–F). Interestingly, the naturalsequences NL6, DF5, and GA6 formed fiber networks with sim-ilar morphology to the aliphatic peptides, with helical twists andturns observed for individual fibers as well as formation of fiberbundles (Fig. 1 A, B, and D–F). The SEM image of the NL6residue attached to a glass capillary showed a corrugated mor-phology (Fig. S2A), whereas the higher-resolution SEM imagerevealed fibers with periodic helical turns (Fig. 1C).
X-Ray Fiber Diffraction of NL6. Raster diffraction of the NL6 resi-due dried on a superhydrophobic substrate revealed fibrousaggregates (Fig. 2A), which were also visible in the electronmicroscope images (Fig. 1 C and D and Fig. S2A). The convec-tive flow-induced nanofibrillar alignment in the drying residueresulted in diffraction patterns with fibrous texture (21) revealinga cross-β pattern (Fig. 2B) (22–24). The azimuthally averagedintensity profiles of the meridian and equator are shown in Fig. 2C and D. We identified on the meridian a 0.46-nm peak assecond-order β-strand periodicity for a 0.92-nm period (a axis)(Fig. 2C). The weak 100 reflection was assumed to be part of theasymmetric 1.02-nm peak, which also contains the tails of anequatorial peak. A broad 0.40-nm peak was attributed to short-range ordered β-sheet material. The equatorial 0.97-nm re-flection (Fig. 2D) corresponded to the correlation found alongthe chain direction (b axis) in amyloid structures. The >0.97-nmspacings observed on the equator were attributed to macrolatticeordering of β-crystallites along the chain-axis (b) direction, forexample due to a helical twist (25). We attributed the reflectionsto 0k0 orders with 2.07 nm (k = 1), 0.97 nm (k = 2), 0.66 nm (k =3), 0.34 nm (k = 6) for a 1.96(2) nm b axis (average of orders 2/3)(Fig. 2 C and D). The b-axis dimension corresponds nearly toa fully extended 6-mer peptide chain of 0.35 × 6 = 2.1 nm. Theequatorial 0.66-nm peak was attributed to the c-axis 001 re-flection (intersheet direction) (Fig. 2C). This suggests an ortho-rhombic unit cell with a = 0.92 nm, b = 1.96 nm (chain direction),c = 0.66 nm, α = β = γ = 90°.
Rheology. Characterization of the different peptide hydrogels wasdone by oscillatory rheology. ID3 was not included, due to itssmaller size compared with the other peptides. To ensure com-parability and standard sample preparation for all the peptides,the ring-cast method was used (SI Materials and Methods). Sur-prisingly, LD6 formed the stiffest, most shape-compact, andhomogeneous gel compared with all examined peptides (Fig. 3 Aand B and Fig. S1). This is reflected by the high elastic modulus(G′ value) of over 104 Pa and the visual appearance of the gel,which assumes the well-defined shape of the mold (Fig. 3A). Inaddition, no separation of the fibrous aggregates from the aque-ous phase was observed. The G′ value for LD6 is higher than thatof RADA, an ionic self-complementary 16-mer peptide, at thesame molar concentration (18). In contrast to LD6, KE7 formeda weak viscous gel with the lowest G′ value (Fig. 3A). In all cases,the trend of G′ values supported our empirical observation on thestiffness of the hydrogels. The linear viscoelastic range (LVR) wassimilar for LD6, NL6, DF5, and GA6 (Fig. 3B). KE7, however,showed a higher LVR, with a yield point similar to that of RADAand different from the other peptides (Fig. 3B). The G′′ values ofthe peptides also showed the same trend as the G′ values, withLD6 being the highest and KE7 the lowest (Fig. S1G). The Tan(δ) value for all peptide hydrogels was lower than 1, showingdominance of the G′ (solid-like) component over the G′′ (fluid-like) component.
Table 1. Sequences of investigated peptides
Peptide sequence Origin (associated amyloid disease) (ref.)α-Helical
intermediatesHelical
fibers/β-turns
LIVAGD (LD6) Designed synthetic peptide (12) Yes YesIVD (ID3) Designed synthetic peptide (12) Yes YesNFGAIL (NL6) Human amylin (diabetes type II) (8) Yes YesKLVFFAE (KE7) Human amyloid-β (1–42) (Alzheimer’s) (7) No NoGGVVIA (GA6) Human amyloid-β (1–42) (Alzheimer’s) (5, 17) Yes YesDFNKF (DF5) Human calcitonin (thyroid medullary carcinoma) (2) Yes YesRADARADA RADARADA (RADA) Designed synthetic peptide (18) No No
This table includes rationally designed ultrasmall peptides and core sequences of five to seven amino acids from naturally occurring amyloid proteins.All peptides were acetylated at the N terminus.
Fig. 1. Morphological characterization of the peptide hydrogels using FESEM.(A) ID3 (12 mM) at a magnification of 30,000×. (B) LD6 (12 mM) at 50,000×. (C)High-resolution SEM image of NL6 attached to a glass capillary. (D) NL6 (12 mM)at 25,000×. (E) GA6 (12 mM) at 50,000×. (F) DF5 (12 mM) at 50,000×.
520 | www.pnas.org/cgi/doi/10.1073/pnas.1217742110 Lakshmanan et al.

Molecular Dynamics Simulations. Self-assembly of the peptidesNL6, ID3, and LD6 was individually investigated from simu-lations of 64 molecules in a cubic box of water. Cube edge di-mension was 6.8 nm for ID3, 8 nm for LD6, and 8.2 nm for NL6.(Movies S1, S2, and S3). Because the peptides are very short, itwas possible to observe self-assembly from our all-atom simu-lations. Furthermore, the final concentrations of the peptidesolutions (Table S1) are much higher than that encounteredunder experimental conditions, as we assumed that a higherconcentration would speed up the dynamics of the aggregation.It is important to note that amyloidosis occurs over a much longerperiod of many years within the human body and, therefore, ahigher starting concentration would be required to form theaggregates within the simulation timescale. The simulations wereperformed for 50 ns, except for NL6. NL6 revealed a highly dis-ordered aggregate at the end of 50 ns, and thus the simulationwas extended for an additional 50 ns to a total of 100 ns. In-terestingly, all three peptides self-assembled from monomers intoa partially organized but stable fiber (Fig. 4 A–C). Accounting forperiodic boundary conditions, the peptides effectively formed aninfinitely long fiber along a single axis (Fig. 4 A–C).Fig. S5 A–C shows the density maps along the cross-section of
the fiber, giving an estimate of the fiber dimensions. The trimerID3 formed a relatively compact fiber that is more cylindrical inshape, with a fiber diameter of around 2.7 nm. LD6 and NL6 alsoformed clear fibers, albeit less ordered and compact. These pep-tides exhibited a more ellipsoidal cross-section, with diametersconsistent with the relative length of the peptides. We performedthree independent simulations for each peptide to ensure thatdifferent simulation trajectories result in similar aggregate mor-phology. All simulations resulted in partially organized but stablefibers. The cluster size distribution of the peptides over the courseof the 50-ns simulations was calculated to gain insights into thedynamics of the self-assembly (Fig. S3 A–C). All three peptidesformed a single, stable aggregate fairly rapidly, with LD6 showingthe fastest dynamics where a single stable fiber was formed at 12–13 ns. NL6 formed a single cluster at around 20 ns, whereas theID3 peptides formed a single cluster at around 28 ns. This is asexpected, because NL6 and LD6 have longer hydrophobic tails,
which increase the kinetics of aggregation. The cluster size dis-tribution also shows relatively few clusters of sizes between 10and 64. This suggests that a minimum seed size of around 10peptides is necessary for oligomeric cluster formation, afterwhich rapid growth to the single large cluster occurs. Furtherstudies are needed for confirmation.
Structural Transition via α-Helical Intermediates Points to a CommonFeature of Amyloidosis. Based on our published hypothesis of self-assembly, the early initiation steps involve antiparallel pairing oftwo peptide monomers and structural transition to an α-helicaldimer. The helical dimers then assemble into β-type fibers beforefurther condensation to cross-β aggregates (12). Surprisingly,natural core sequences NL6, DF5, and GA6 showed similar ran-dom coil to α-helix to β-turn transitions with increasing peptideconcentration (Fig. 5). The α-helical intermediates were charac-terized by a negative π–π* transition near 222 nm, a negative splitπ–π* transition near 208 nm, and a positive peak near 192 nmcorresponding to a π–π* transition (12). DF5 in particular formedvery stable α-helical intermediates, which appeared withinminutes at concentrations below 100 μM (Fig. 5C).
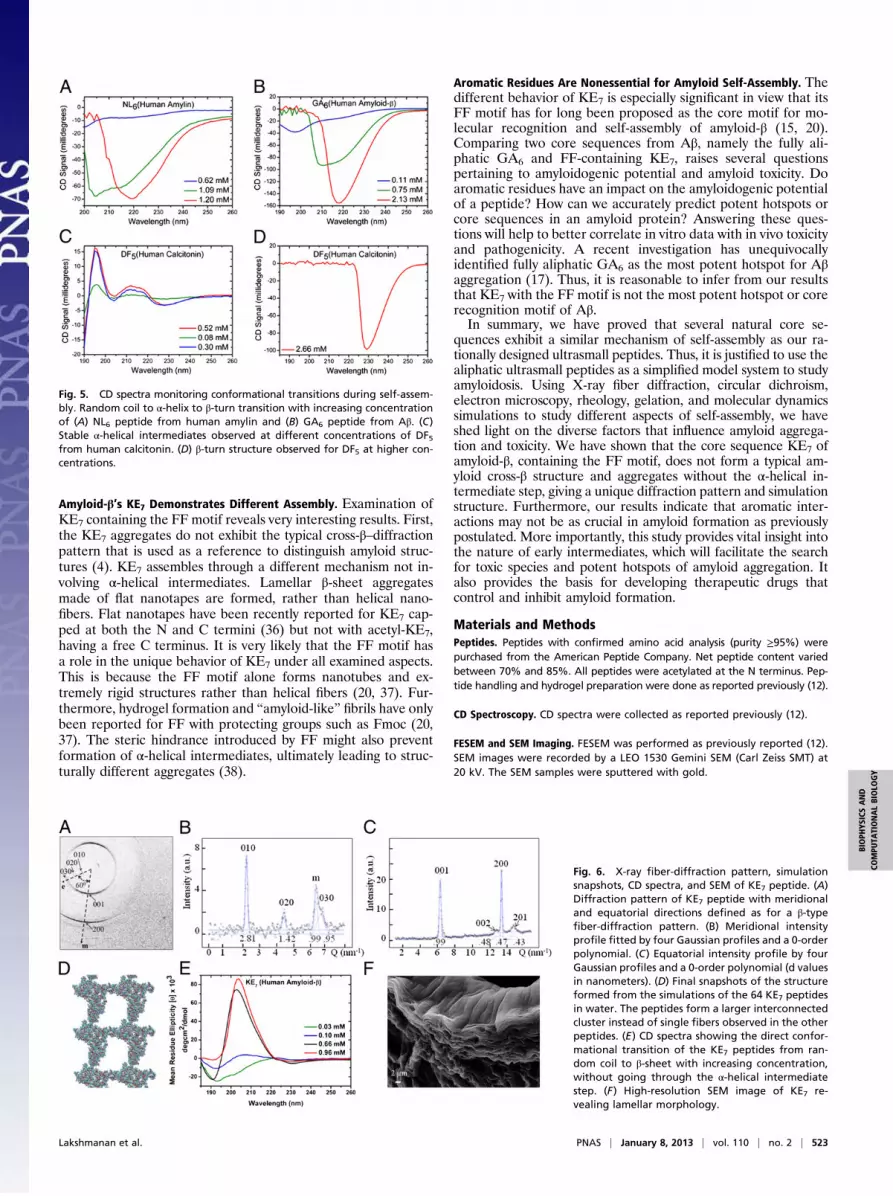
KE7 Shows a Different Fiber-Diffraction Pattern, Morphology, andSelf-Assembly Mechanism. Interestingly, KE7 (KLVFFAE) withits FF motif not only formed aggregates and hydrogels withdistinctly different properties but also showed a unique X-ray–diffraction pattern related to the convective flow alignment ofthe evaporating droplet on the superhydrophobic substrate (Fig.6 A–C). The gelation capacity and mechanical stiffness of theformed hydrogel were notably lower than that of LD6 and the
Fig. 2. X-ray fiber diffraction of NL6. (A) Composite diffraction image of NL6residue based on a raster-diffraction scan with 1-μm step increment. (B) Asingle “pixel” at the position marked by an arrow in A corresponds to a fiber-diffraction pattern. The meridional (m) and equatorial (e) directions are in-dicated. (C) Meridional intensity profile fitted by three Gaussian profiles anda 0-order polynomial. The d = 1.02 nm peak contains both meridional andequatorial components. (D) Equatorial intensity profile fitted by five Gaussianprofiles and a 0-order polynomial. The experimental data in red were not usedfor the fit (d values in nanometers). The fit of the 2.07 nm peak is less precisedue to the low-angle intensity tail, and the 0.34 nm peak due to severaloverlapping peaks.
Fig. 3. Rheological characterization of peptide hydrogels at a concentrationof 12 mM. (A) Frequency sweep studies (storage modulus G′ as a function ofangular frequency ω) show that LD6 forms a shape-compact gel with thehighest mechanical stiffness and KE7 forms a weaker gel with lowest me-chanical stiffness. (Inset) LD6 (Left) and KE7 (Right) gels made by the ring-castmethod. (B) Amplitude sweep studies (storage modulus G′ as a function ofstrain γ) show a similar LVR profile for peptide hydrogels with helical fibernetworks and a broader LVR region for KE7 and RADA. At least five in-dependent measurements (n = 5) were taken for each peptide.
Lakshmanan et al. PNAS | January 8, 2013 | vol. 110 | no. 2 | 521
BIOPH
YSICSAND
COMPU
TATIONALBIOLO
GY

other natural core sequences (Fig. 3A and Fig. S1). This could beattributed to the formation of apparently short and flat nanotapesrather than nanofibers (Fig. S2C). In addition, the high-resolutionSEM image showed no fibrillar morphology like NL6 (Fig. 1 Cand D) but rather a lamellar organization (Fig. 6F and Fig. S2B).In contrast to other investigated core sequences, the circular
dichroism (CD) spectra of KE7 showed no α-helical interme-diates (Fig. 6E). With increasing concentration, a direct randomcoil to β-sheet transition was observed, but no β-turn. Theβ-sheet structure and molecular packing as inferred from the flatnanotapes and lamellar organization could explicate the lowmechanical stiffness of KE7 hydrogels (Fig. 3A and Fig. S1G).The lamellar layers slide over each other, unlike dense helicalfiber networks of other peptides that exert higher resistance toapplied shear. Furthermore, KE7’s LVR was very similar to thatof RADA and different from that of the other peptides (Fig. 3Band Fig. S1H). RADA also forms a β-sheet structure and showsa layer-like morphology by FESEM (Fig. S2D).The composite diffraction image obtained by raster diffraction of
the KE7 residue showed mainly domains defined by weakly textured∼0.47-nm β-sheet reflections (Fig. S4A). Based on themodel of slab-like β-crystallites for KE7 (26), we tentatively assume nonperiodicstacking of slabs with a preferred slab orientation in each domain.We observed strongly textured diffraction patterns in a zone wherethe drying drop was arrested on the substrate before forming a res-idue. Amyloid cross-β–structure formation has already been shownfor this zone for residues obtained by drying lysozyme solution dropson a superhydrophobic substrate (27). A diffraction pattern from theinterface zone, however, did not show the characteristic cross-β–fiber pattern of ID3, LD6, and NL6, with the meridional andequatorial directions at 90° but rather at ∼60° (Fig. 6A). We thusexclude a structure based on an assembly of nanotubes as observedfor KE7 in acid solution (28) on the grounds of the lamellar mor-phology (Fig. 6F) and suggest the model of lamellar stacking ofβ-sheet slabs along the (b) chain axis (26). For the correspondingunit-cell choice (26), we identified the strong meridional 200 re-flection (0.94 nm; H-bonding direction), 001 reflection (0.99 nm;interchain packing), as well as the weaker 002 (0.48 nm) and 201(0.43 nm) reflections (Fig. 6C). The equatorial reflections suggesta period of 2.81 nm along the stacking direction (b axis) (Fig. 6B).Based on the coincidence of the [100] and [001] directions, wetentatively assume an intergrowth of slabs that are rotationally dis-ordered around the stacking direction. In view of a 3.03-nm b axis forwild-type KE7 (26), the 2.81 nm value observed for the current KE7with lamellar morphology suggests tilted 7-mer chains. For a pos-sible monoclinic structure with a unique c axis, the chains would betilted in the a/c plane by ∼30° from a normal to the a/b plane (Fig.S4B). We cannot, however, exclude at present a triclinic unit cell.The simulations of KE7 were carried out as before. All three
independent simulations of 50 ns resulted in similar structures.Due to the high concentration and longer peptide length, thepeptides rapidly aggregated, forming a single cluster around 10ns (Fig. S3D). The cluster was highly disorganized at this stage,but remained stable throughout the simulations. At the end, KE7
formed a single large interconnected structure spanning thelength of the simulation box, contrary to the other peptides thatformed isolated fibers. Fig. 6D shows a snapshot of the finalstructure from one KE7 simulation. The density maps of the finalstructure were taken along the other two orthogonal planes (Fig.S5 D and E). The density maps clearly show that KE7 formsstructures that are less dense and compact compared with theother peptides. KE7 formed disordered aggregates that wereconnected along all three axes, unlike the other peptides, whichassembled into isolated fiber-like structures along a single axis.
DiscussionThe aim of this study was to compare the proposed self-assemblymechanism for amyloid-like ultrasmall peptides with naturallyoccurring amyloidogenic peptides. Interestingly, the self-assemblyof several natural core sequences proceeded via discrete stages sim-ilar to the designed ultrasmall peptides, particularly with α-helicalstructures as intermediates. This observation supports growing evi-dence that early intermediates in amyloidosis are the toxic andpathogenic species (10, 11). The toxicity of amyloidogenic peptides/proteins is increasingly linked to their membrane permeabilizationand disruption capability (11, 29–31). The α-helical intermediatesobserved in our study could play a key role, as α-helical structures areknown to facilitate membrane–peptide interaction and subsequentmembrane disruption (32). In addition, intermediates can promoteaggregation by increasing the local concentration of aggregatingspecies (31). This could occur by helix formation and helix–helixassociation, or by creation of an anchor point via membrane–peptideinteractions for fiber assembly and elongation (3, 31, 33). In a diversecellular environment, conformational change of the amyloidogenicspecies to an α-helical state could well be the trigger for self-as-sembly. In fact, the work of Kelly and coworkers on transthyretinsuggests that induced conformational changes are sufficient to pro-mote spontaneous assembly of a functional protein into amyloidfibrils (34). Previously, α-helical structures have been reported as keykinetic intermediates for amyloid formation (31, 33). Here, we extendthis observation to small natural core sequences of four to sevenamino acids. These intermediates are observed by simply dissolvingthe peptides in water, without the presence of helix-inducing com-pounds such as hexafluoroisopropanol, membranes, or a hydropho-bic environment. Among the natural core sequences, DF5, with itsvery stable α-helical intermediates, is the fastest to self-assemble intofibrous aggregates. It is entirely conceivable that the conformationaltransition from a random coil to an α-helical structure drives thespeed of self-assembly.
Similar Assembly for Amyloid Core Sequences and Aliphatic Peptides.We have shown that naturally occurring NL6 (NFGAIL) andDF5 (DFNKF), containing one or two aromatic residues, self-assemble via the same mechanism as aliphatic peptides LD6 andID3. The final self-assembled structures formed by NL6, GA6, andDF5 are morphologically and structurally similar to the designedpeptides. Our multimodal analysis of the amyloid peptide aggre-gates clearly indicates that aromatic residues are not essential forself-assembly. Nevertheless, they alter the kinetics of aggregationas seen previously with the whole amyloid protein (35) and in-crease the proportion of disordered aggregates. Molecular dy-namics simulations show that NL6 and KE7 form more disorderedaggregates compared with LD6 and ID3 in the initial stages. Inaddition, NL6 takes longer to organize into an ordered fiber similarto that of LD6 and ID3. KE7 shows the fastest dynamics, with a singlecluster formed in 10 ns; however, the cluster is highly disordered andloosely aggregated and a compact fiber is not seen within the sim-ulated time frame. Thus, aromatic and hydrophobic interactionsdue to the phenylalanine residue shorten the lag phase for creationof a nucleus, but do not lead to long-range fiber networks requiredfor hydrogelation. This is supported by our observation that KE7 isthe slowest to form a hydrogel among the 5- to 7-mers, whereas NL6and DF5 form hydrogels with slight phase separation due to rapidclumping of fibrous aggregates. Aliphatic LD6, on the other hand,forms the stiffest and most homogeneous hydrogel.
Fig. 4. Molecular dynamics simulations in water and density maps of fibers.(A−C) Final snapshots of the fibers from simulations of (A) 64 NL6, (B) 64 ID3,and (C) 64 LD6 in water. Surprisingly, all three peptides form partially or-ganized fibers during the simulation.
522 | www.pnas.org/cgi/doi/10.1073/pnas.1217742110 Lakshmanan et al.
Amyloid-β’s KE7 Demonstrates Different Assembly. Examination ofKE7 containing the FF motif reveals very interesting results. First,the KE7 aggregates do not exhibit the typical cross-β–diffractionpattern that is used as a reference to distinguish amyloid struc-tures (4). KE7 assembles through a different mechanism not in-volving α-helical intermediates. Lamellar β-sheet aggregatesmade of flat nanotapes are formed, rather than helical nano-fibers. Flat nanotapes have been recently reported for KE7 cap-ped at both the N and C termini (36) but not with acetyl-KE7,having a free C terminus. It is very likely that the FF motif hasa role in the unique behavior of KE7 under all examined aspects.This is because the FF motif alone forms nanotubes and ex-tremely rigid structures rather than helical fibers (20, 37). Fur-thermore, hydrogel formation and “amyloid-like” fibrils have onlybeen reported for FF with protecting groups such as Fmoc (20,37). The steric hindrance introduced by FF might also preventformation of α-helical intermediates, ultimately leading to struc-turally different aggregates (38).
Aromatic Residues Are Nonessential for Amyloid Self-Assembly. Thedifferent behavior of KE7 is especially significant in view that itsFF motif has for long been proposed as the core motif for mo-lecular recognition and self-assembly of amyloid-β (15, 20).Comparing two core sequences from Aβ, namely the fully ali-phatic GA6 and FF-containing KE7, raises several questionspertaining to amyloidogenic potential and amyloid toxicity. Doaromatic residues have an impact on the amyloidogenic potentialof a peptide? How can we accurately predict potent hotspots orcore sequences in an amyloid protein? Answering these ques-tions will help to better correlate in vitro data with in vivo toxicityand pathogenicity. A recent investigation has unequivocallyidentified fully aliphatic GA6 as the most potent hotspot for Aβaggregation (17). Thus, it is reasonable to infer from our resultsthat KE7 with the FF motif is not the most potent hotspot or corerecognition motif of Aβ.In summary, we have proved that several natural core se-
quences exhibit a similar mechanism of self-assembly as our ra-tionally designed ultrasmall peptides. Thus, it is justified to use thealiphatic ultrasmall peptides as a simplified model system to studyamyloidosis. Using X-ray fiber diffraction, circular dichroism,electron microscopy, rheology, gelation, and molecular dynamicssimulations to study different aspects of self-assembly, we haveshed light on the diverse factors that influence amyloid aggrega-tion and toxicity. We have shown that the core sequence KE7 ofamyloid-β, containing the FF motif, does not form a typical am-yloid cross-β structure and aggregates without the α-helical in-termediate step, giving a unique diffraction pattern and simulationstructure. Furthermore, our results indicate that aromatic inter-actions may not be as crucial in amyloid formation as previouslypostulated. More importantly, this study provides vital insight intothe nature of early intermediates, which will facilitate the searchfor toxic species and potent hotspots of amyloid aggregation. Italso provides the basis for developing therapeutic drugs thatcontrol and inhibit amyloid formation.
Materials and MethodsPeptides. Peptides with confirmed amino acid analysis (purity ≥95%) werepurchased from the American Peptide Company. Net peptide content variedbetween 70% and 85%. All peptides were acetylated at the N terminus. Pep-tide handling and hydrogel preparation were done as reported previously (12).
CD Spectroscopy. CD spectra were collected as reported previously (12).
FESEM and SEM Imaging. FESEM was performed as previously reported (12).SEM images were recorded by a LEO 1530 Gemini SEM (Carl Zeiss SMT) at20 kV. The SEM samples were sputtered with gold.
Fig. 5. CD spectra monitoring conformational transitions during self-assem-bly. Random coil to α-helix to β-turn transition with increasing concentrationof (A) NL6 peptide from human amylin and (B) GA6 peptide from Aβ. (C)Stable α-helical intermediates observed at different concentrations of DF5from human calcitonin. (D) β-turn structure observed for DF5 at higher con-centrations.
Fig. 6. X-ray fiber-diffraction pattern, simulationsnapshots, CD spectra, and SEM of KE7 peptide. (A)Diffraction pattern of KE7 peptide with meridionaland equatorial directions defined as for a β-typefiber-diffraction pattern. (B) Meridional intensityprofile fitted by four Gaussian profiles and a 0-orderpolynomial. (C) Equatorial intensity profile by fourGaussian profiles and a 0-order polynomial (d valuesin nanometers). (D) Final snapshots of the structureformed from the simulations of the 64 KE7 peptidesin water. The peptides form a larger interconnectedcluster instead of single fibers observed in the otherpeptides. (E) CD spectra showing the direct confor-mational transition of the KE7 peptides from ran-dom coil to β-sheet with increasing concentration,without going through the α-helical intermediatestep. (F) High-resolution SEM image of KE7 re-vealing lamellar morphology.
Lakshmanan et al. PNAS | January 8, 2013 | vol. 110 | no. 2 | 523
BIOPH
YSICSAND
COMPU
TATIONALBIOLO
GY

Rheology. Viscoelasticity of peptide hydrogels was measured using an ARES-G2 rheometer (TA Instruments). A serrated stainless steel, parallel-plategeometry of 8-mm diameter was used and the gap distance was maintainedbetween 0.8 and 2 mm. Oscillatory frequency sweep studies were performedfor a range of 0.1–100 rad/s, using either a 0.1% or 1% strain. Oscillatoryamplitude sweep studies were conducted from 0.1 to 100% strain with anangular frequency of 1 rad/s. All measurements were done at 25 °C. Thering-cast method used for hydrogel preparation is described in SI Materialsand Methods.
X-Ray–Diffraction Experiments. Aqueous solutions of 2.5 mg/mL for KE7 and15 mg/mL for NL6 at pH 7 were prepared for X-ray fiber diffraction. Ap-proximately 4-μL solution drops were deposited by pipette onto a super-hydrophobic poly(methyl methacrylate) surface and evaporated withinabout 1 h under quasi–contact-free conditions into solid residues (21). Fur-ther details are given in SI Materials and Methods.
Computer Simulations. Simulations of four peptides, namely LD6, ID3, NL6, andKE7, were performed, using the Optimized Potentials for Liquid Simulations(OPLS) force field. The extended simple point charge model (SPC/E) modelwas used for water molecules and GROMACS version 4.0.5 was used formolecular dynamics simulations, as reported (12). Further details are given inSI Materials and Methods.
ACKNOWLEDGMENTS. We thank Ulrich Hauser (Institute of Physics I,University of Cologne) and Norma Greenfield (Robert Wood Johnson MedicalSchool) for helpful discussions, and Yihua Eva Loo and Michael Reithofer forproofreading. X-ray–diffraction experiments were performed with the sup-port of staff handling the ID13 beamline, and SEM images were recorded byI. Snigereva at the European Synchrotron Radiation Facility. Computing facil-ities were provided by the A*STAR Computing Resource Center. This work isfunded by the Institute of Bioengineering and Nanotechnology (BiomedicalResearch Council, Agency for Science, Technology and Research, Singapore)and the Institute of High Performance Computing (Science and EngineeringResearch Council, Agency for Science, Technology and Research, Singapore).
1. Chiti F, Dobson CM (2006) Protein misfolding, functional amyloid, and human disease.Annu Rev Biochem 75:333–366.
2. Reches M, Porat Y, Gazit E (2002) Amyloid fibril formation by pentapeptide andtetrapeptide fragments of human calcitonin. J Biol Chem 277(38):35475–35480.
3. Abedini A, Raleigh DP (2009) A role for helical intermediates in amyloid formation bynatively unfolded polypeptides? Phys Biol 6(1):015005.
4. Geddes AJ, Parker KD, Atkins EDT, Beighton E (1968) “Cross-β” conformation inproteins. J Mol Biol 32(2):343–358.
5. Sawaya MR, et al. (2007) Atomic structures of amyloid cross-beta spines reveal variedsteric zippers. Nature 447(7143):453–457.
6. Nelson R, et al. (2005) Structure of the cross-beta spine of amyloid-like fibrils. Nature435(7043):773–778.
7. Balbach JJ, et al. (2000) Amyloid fibril formation by A β 16–22, a seven-residuefragment of the Alzheimer’s β-amyloid peptide, and structural characterization bysolid state NMR. Biochemistry 39(45):13748–13759.
8. Tenidis K, et al. (2000) Identification of a penta- and hexapeptide of islet amyloidpolypeptide (IAPP) with amyloidogenic and cytotoxic properties. J Mol Biol 295(4):1055–1071.
9. Rambaran RN, Serpell LC (2008) Amyloid fibrils: Abnormal protein assembly. Prion 2(3):112–117.
10. Haass C, Selkoe DJ (2007) Soluble protein oligomers in neurodegeneration: Lessonsfrom the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol 8(2):101–112.
11. Glabe CG, Kayed R (2006) Common structure and toxic function of amyloid oligomersimplies a common mechanism of pathogenesis. Neurology 66(2 Suppl 1):S74–S78.
12. Hauser CAE, et al. (2011) Natural tri- to hexapeptides self-assemble in water to am-yloid β-type fiber aggregates by unexpected α-helical intermediate structures. ProcNatl Acad Sci USA 108(4):1361–1366.
13. Gazit E (2002) A possible role for π-stacking in the self-assembly of amyloid fibrils.FASEB J 16(1):77–83.
14. Azriel R, Gazit E (2001) Analysis of the minimal amyloid-forming fragment of the isletamyloid polypeptide. An experimental support for the key role of the phenylalanineresidue in amyloid formation. J Biol Chem 276(36):34156–34161.
15. Görbitz CH (2006) The structure of nanotubes formed by diphenylalanine, the corerecognition motif of Alzheimer’s β-amyloid polypeptide. Chem Commun (Camb) (22):2332–2334.
16. Mishra A, et al. (2011) Ultrasmall natural peptides self-assemble to strong tempera-ture-resistant helical fibers in scaffolds suitable for tissue engineering. Nano Today 6(3):232–239.
17. Colletier JP, et al. (2011) Molecular basis for amyloid-beta polymorphism. Proc NatlAcad Sci USA 108(41):16938–16943.
18. Zhang S, et al. (1995) Self-complementary oligopeptide matrices support mammaliancell attachment. Biomaterials 16(18):1385–1393.
19. Lynn DG, Meredith SC (2000) Review: Model peptides and the physicochemical ap-proach to beta-amyloids. J Struct Biol 130(2-3):153–173.
20. Gazit E (2007) Self-assembled peptide nanostructures: The design of molecularbuilding blocks and their technological utilization. Chem Soc Rev 36(8):1263–1269.
21. Accardo A, et al. (2010) In situ X-ray scattering studies of protein solution droplets
drying on micro- and nanopatterned superhydrophobic PMMA surfaces. Langmuir 26
(18):15057–15064.22. Inouye H, et al. (2002) Molecular organization of amyloid protofilament-like assembly
of betabellin 15D: Helical array of beta-sandwiches. Biophys J 83(3):1716–1727.23. Serpell LC, Benson M, Liepnieks JJ, Fraser PE (2007) Structural analyses of fibrinogen
amyloid fibrils. Amyloid 14(3):199–203.24. Sumner Makin O, Serpell LC (2004) Structural characterisation of islet amyloid poly-
peptide fibrils. J Mol Biol 335(5):1279–1288.25. Blake C, Serpell L (1996) Synchrotron X-ray studies suggest that the core of the
transthyretin amyloid fibril is a continuous β-sheet helix. Structure 4(8):989–998.26. Inouye H, Gleason KA, Zhang D, Decatur SM, Kirschner DA (2010) Differential effects
of Phe19 and Phe20 on fibril formation by amyloidogenic peptide A beta 16–22 (Ac-
KLVFFAE-NH2). Proteins 78(10):2306–2321.27. Accardo A, et al. (2011) Lysozyme fibrillation induced by convective flow under quasi
contact-free conditions. Soft Matter 7(15):6792–6796.28. Mehta AK, et al. (2008) Facial symmetry in protein self-assembly. J Am Chem Soc 130
(30):9829–9835.29. Last NB, Rhoades E, Miranker AD (2011) Islet amyloid polypeptide demonstrates
a persistent capacity to disrupt membrane integrity. Proc Natl Acad Sci USA 108(23):
9460–9465.30. Brender JR, Salamekh S, Ramamoorthy A (2012) Membrane disruption and early
events in the aggregation of the diabetes related peptide IAPP from a molecular
perspective. Acc Chem Res 45(3):454–462.31. Butterfield SM, Lashuel HA (2010) Amyloidogenic protein-membrane interactions:
Mechanistic insight from model systems. Angew Chem Int Ed Engl 49(33):5628–5654.32. Knight JD, Hebda JA, Miranker AD (2006) Conserved and cooperative assembly of
membrane-bound α-helical states of islet amyloid polypeptide. Biochemistry 45(31):
9496–9508.33. Abedini A, Raleigh DP (2009) A critical assessment of the role of helical intermediates
in amyloid formation by natively unfolded proteins and polypeptides. Protein Eng
Des Sel 22(8):453–459.34. Colon W, Kelly JW (1992) Partial denaturation of transthyretin is sufficient for amy-
loid fibril formation in vitro. Biochemistry 31(36):8654–8660.35. Marek P, et al. (2007) Aromatic interactions are not required for amyloid fibril for-
mation by islet amyloid polypeptide but do influence the rate of fibril formation and
fibril morphology. Biochemistry 46(11):3255–3261.36. Tao K, et al. (2011) Self-assembly of short A beta(16–22) peptides: Effect of terminal
capping and the role of electrostatic interaction. Langmuir 27(6):2723–2730.37. Lakshmanan A, Zhang S, Hauser CAE (2012) Short self-assembling peptides as building
blocks for modern nanodevices. Trends Biotechnol 30(3):155–165.38. Lakshmanan A, Hauser CAE (2011) Ultrasmall peptides self-assemble into diverse
nanostructures: Morphological evaluation and potential implications. Int J Mol Sci 12
(9):5736–5746.
524 | www.pnas.org/cgi/doi/10.1073/pnas.1217742110 Lakshmanan et al.

Supporting InformationLakshmanan et al. 10.1073/pnas.1217742110SI Materials and MethodsRing-Cast Method. For the rheology experiments, a ring-castmethod was used for hydrogel preparation to ensure uniform gelvolume and facilitate easy transfer of the intact gel onto the stagefor measurements. For comparison, a standard concentration of12 mMwas used for all peptides and measurements were taken 1–2 d post gelation. After vortexing, 200-μL aliquots of the peptidesolution were immediately transferred into plastic ring casts of 9-mm diameter and placed on clean 35-mm tissue-culture dishes.The top and bottom of each ring cast was covered with a layer ofparafilm, and the tissue-culture dish was tightly sealed withparafilm to prevent evaporation.
X-Ray Diffraction.Aqueous solutions of 2.5 mg/mL for KE7 and 15mg/mL for NL6 at pH 7 were prepared for X-ray fiber diffraction.Approximately 4-μL solution drops were deposited by pipetteonto a superhydrophobic poly(methyl methacrylate) surface andevaporated within about 1 h under quasi–contact-free conditionsinto solid residues (1). Raster-diffraction experiments (2) wereperformed on the residues using a monochromatic beam of λ =0.08122 nm, focused to a 200 (h) × 150 (v) nm2 spot with about 2 ×109 photons/s flux using Si-refractive lenses in a crossed geometry(3). A Maxipix detector (4), provided by the European SynchrotronRadiation facility, with 512 × 512 pixels of 50 × 50 μm2 each wasused for data collection with ≤5 s per pattern.
Molecular Dynamics Simulation. All of the investigated peptides(LD6, ID3, NL6, and KE7) have been described using the all-atom OPLS force field (5), and the water molecules were con-sidered explicitly using the SPC/E model (6). This force-field andwater-model combination has been found to give good results for
the hydration properties of amino acids (7). Molecular dynamicssimulations in the isothermal–isobaric ensemble were performedusing GROMACS version 4.0.5 (8) at time steps of 2 fs. Periodicboundary conditions were applied in all three directions. Cutoffradii were set at 0.9 nm for electrostatic interactions and 1.4 nmfor Lennard-Jones interactions. Long-range electrostatic inter-actions were treated using the particle-mesh Ewald method (9).Temperature coupling was achieved using the Berendsen ther-mostat, but with an additional stochastic term to ensure a correctkinetic energy distribution and produce a correct canonical en-semble (10). Pressure coupling was achieved with the Berendsenbarostat (11). Relaxation times of 1 and 2 ps were used for thethermostat and barostat, respectively.The initial conformations of the peptides were obtained from
the final structure of a 50-ns molecular dynamics simulation ofa single molecule of each peptide in water at a temperature andpressure of 298 K and 1 bar, respectively. This was to ensurea relaxed starting conformation for the monomer before theassembly process. Subsequently, 64 peptides were initially spacedevenly within the simulation box and solvated with water. A highconcentration range was chosen so as to be able to observe fiberformation within our simulation time. The concentrations of thepeptide solutions considered are given in Table S1. The box wasthen subjected to an initial energy minimization to remove anyspurious overlaps before the molecular dynamics simulationswere performed on each system with the temperature coupled to298 K and the pressure isotropically coupled to 1 bar. Three setsof simulations were performed for each peptide and concentra-tion to establish the general validity of the results.
1. Accardo A, et al. (2010) In situ X-ray scattering studies of protein solution dropletsdrying on micro- and nanopatterned superhydrophobic PMMA surfaces. Langmuir26(18):15057–15064.
2. Riekel C, Burghammer M, Davies R, Gebhardt R, Popov D (2009) Fundaments of softcondensed matter scattering and diffraction with microfocus techniques. Applicationsof Synchrotron Light to Scattering and Diffraction in Materials, Lecture Notes inPhysics, eds Ezquerra TA, Garcia-Gutierrez M, Nogales A, Gomez M (Springer,Heidelberg), Vol 776, pp 91–104.
3. Schroer CG, et al. (2005) Hard X-ray nanoprobe based on refractive X-ray lenses. ApplPhys Lett 87(12):124103-1–124103-3.
4. Ponchut C, et al. (2002) Evaluation of a photon-counting hybrid pixel detector arraywith a synchrotron X-ray source. Nucl Instrum Methods Phys Res A 484(1-3):396–406.
5. Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL (2001) Evaluation andreparametrization of the OPLS-AA force field for proteins via comparison withaccurate quantum chemical calculations on peptides. J Phys Chem B 105(28):6474–6487.
6. Berendsen HJC, Grigera JR, Straatsma TP (1987) The missing term in effective pairpotentials. J Phys Chem 91(24):6269–6271.
7. Hess B, van der Vegt NF (2006) Hydration thermodynamic properties of amino acidanalogues: A systematic comparison of biomolecular force fields and water models. JPhys Chem B 110(35):17616–17626.
8. Hess B, Kutzner C, van der Spoel D, Lindahl E (2008) GROMACS 4: Algorithms forhighly efficient, load-balanced, and scalable molecular simulation. J Chem TheoryComput 4(3):435–447.
9. Essmann U, et al. (1995) A smooth particle mesh Ewald method. J Chem Phys 103(19):8577–8593.
10. Bussi G, Donadio D, Parrinello M (2007) Canonical sampling through velocityrescaling. J Chem Phys 126(1):014101.
11. Berendsen HJC, Postma JPM, van Gunsteren WF, Di Nola A, Haak JR (1984) Moleculardynamics with coupling to an external bath. J Chem Phys 81(8):3684–3690.
Lakshmanan et al. www.pnas.org/cgi/content/short/1217742110 1 of 6

Fig. S1. Peptide hydrogels prepared by the ring-cast method and their rheological characterization. The molar concentration used for all peptides was 12 mM.(A) LD6. (B) NL6. (C) KE7. (D) GA6. (E) DF5. (F) RADA. Images were taken 24 h after gel preparation, just before rheology measurements. (G) Frequency sweepstudies measuring loss modulus G′′ as a function of angular frequency ω. The trends are similar to those observed for the storage modulus (G′), with LD6 havingthe highest and KE7 the lowest value. The G′′:G′ ratio (Tanδ) for all of the measured samples was much lower than 1, indicating dominance of the elastic solid-like component over the viscous fluid-like component. (H) Amplitude sweep (G′′ vs. strain percent γ) shows a similar strain response for peptide hydrogels withhelical fiber networks and a different strain response for KE7 and RADA, which form lamellar β-sheets. KE7 and RADA alone show a temporary increase in theloss modulus at a strain corresponding to the end of their linear viscoelastic range. The lamellar morphology and the viscous nature of the KE7 and RADAhydrogels most likely lead to greater energy loss, especially around the yield point of the material.
A B
200 nm 200 nm
C D
Fig. S2. Morphological characterization of the peptide hydrogels using electron microscopy. (A) The scanning electron microscopy (SEM) image of NL6 at-tached to a glass capillary showing a corrugated morphology. (B) SEM image of KE7 attached to a glass capillary showing a hollow shape. Lamellar layers can bediscerned even at this low magnification. (C) KE7 (12 mM) at a magnification of 60,000× showing flat nanotapes. (D) RADA (12 mM) at a magnification of40,000× showing a flatlayer-like morphology.
Lakshmanan et al. www.pnas.org/cgi/content/short/1217742110 2 of 6

Fig. S3. Cluster size distribution as a function of time for the (A) 64 ID3, (B) 64 LD6, (C) 64 NL6, and (D) 64 KE7 peptides. All peptides form a single cluster within30 ns, including the tripeptide ID3. KE7 showed the fastest dynamics, where a single aggregate was formed at around 10 ns. However, the aggregate was moredisordered and loosely connected compared with the other simulated peptides. LD6 formed a single stable cluster at around 12–13 ns, whereas NL6 formeda single cluster at around 20 ns. ID3 was slowest to form a single cluster, at around 30 ns. This is as expected, because KE7 has the longest hydrophobic tail,which increases the kinetics of aggregation, whereas ID3 has the shortest hydrophobic tail.
Lakshmanan et al. www.pnas.org/cgi/content/short/1217742110 3 of 6

Fig. S4. X-ray fiber diffraction and proposed structural model for KE7. (A) Composite diffraction image of bulk KE7 residue revealing two domains withhomogeneous β-reflection orientation. A fully extended 7-mer peptide chain at an angle of 30° to the fiber axis has a length of cos30°*(0.35 × 7) = 2.1 nm,which is smaller than b = 2.81 nm [010]. This would agree with the KE7 raster diffraction. We performed raster diffraction on the bulk KE7 residue with 1-μmstep resolution. The composite diffraction image shown in Fig. S4A is based on the prominent 0.47-nm β-sheet reflection. The image reveals two domains withpreferred β-reflection texture. (B) Model of monoclinic slab with inclined 7-mer chains forming antiparallel β-sheets. The slabs are assumed to be rotationallydisordered along the stacking direction (c*). The structural model is based on β-sheet slabs stacked along the fiber axis (1). The diffraction pattern (Fig. 6A) doesnot show the common orthogonal meridional and equatorial directions of a cross-β pattern but rather an angle of ∼60°, although the same types of reflectionsare observed as for wild-type KE7 (1). The correlation length along the fiber axis is calculated as ∼3 nm from the width of the 010 reflection by Scherrer’sequation (2), which corresponds roughly to the dimension of a single slab (1). We note that [100] and [001] directions coincide in the pattern and that the angleof both directions with the [010] direction is ∼60° (Fig. 6A), suggesting tilted 7-mer chains. The tilting of the chains from a normal to the plane of the slabs isdeduced roughly from sin−1(2.81/3.03)∼70°. For a monoclinic lattice with unique c axis, the chains could be tilted in the a/c plane as shown schematically. Atriclinic space group can, however, not be excluded at present.
1. Inouye H, Gleason KA, Zhang D, Decatur SM, Kirschner DA (2010) Differential effects of Phe19 and Phe20 on fibril formation by amyloidogenic peptide A beta 16–22 (Ac-KLVFFAE-NH2).Proteins 78(10):2306–2321.
2. Klug HP, Alexander LE (1974) X-Ray Diffraction Procedures for Polycrystalline and Amorphous Materials (Wiley Interscience, New York), 2nd Ed.
Fig. S5. Molecular dynamics simulations of 64 peptide molecules in water. (A–C) Density maps of the fibers taken along the plane perpendicular to fiber axisfor NL6, ID3, and LD6, respectively. This can give a rough estimate of the fiber dimensions. (D and E) Density maps of the final structure of KE7 taken along the yand z axes, respectively. The cluster is seen to span the entire box length in all three directions.
Lakshmanan et al. www.pnas.org/cgi/content/short/1217742110 4 of 6

Movie S1. Simulation of 64 ID3 peptides in water. The formation of a fiber can be clearly observed.
Movie S1
Table S1. Concentrations of peptide solutions
Peptide Npep Nwat xpep mM
NL6 1 4,106 0.000243 13.2864 15,748 0.00405 192.75
ID3 1 4,123 0.000242 13.2864 9,074 0.007 338.0
LD6 1 4,108 0.000243 13.2864 14,833 0.004296 207.57
KE7 1 4,095 0.000244 13.2864 21,152 0.00302 145.78
Npep and Nwat are the number of peptide and water molecules in thesimulation box, respectively. xpep is the mole fraction of peptides. The con-centrations in mM are given in the last column.
Lakshmanan et al. www.pnas.org/cgi/content/short/1217742110 5 of 6

Movie S2. Simulation of 64 LD6 peptides in water. The formation of a fiber can be clearly observed.
Movie S2
Movie S3. Simulation of 64 NL6 peptides in water. The formation of a fiber can be clearly observed.
Movie S3
Lakshmanan et al. www.pnas.org/cgi/content/short/1217742110 6 of 6





























