Embed Size (px)
Citation preview

Acta Tropica, 59(1995)187-196 187 © 1995 Elsevier Science B.V. All rights reserved 0001-706X/95/$09.50
ACTROP 00462
A highly sensitive and rapid procedure for direct PCR detection of Leishmania infantum within
human peripheral blood mononuclear cells
Sophie RaveP'*, G6rard Cuny a, Jacques Reynes b, Francisco Veas a
aLaboratoire Rbtrovirus-Parasites, Institut Frangais de Recherche Scientifique pour le DOveloppement en Cooperation (ORSTOM), 911, avenue Agropolis, B.P.5045, 34032 Montpellier, France, bClinique des
Maladies Infectieuses A, H@ital Gui de Chauliac, Centre Hospitalier Universitaire, 34059 Montpellier, France
(Received 17 October 1994; revised 13 January 1995; accepted 16 January 1995)
We have developed a highly sensitive, simple and rapid procedure to detect Leishmania infantum within human macrophages. It only requires ficoll preparation of peripheral blood mononuclear cells from the patient, and their direct use for Leishmania kDNA amplification by polymerase chain reaction. Under these conditions, about one parasite can be detected in a one million human cell environment. Results, including those of a hybridization step to confirm the diagnosis specificity, are obtained within 24 h, a very short period as compared to current diagnostic methods. This procedure is of particular interest for early detection and early drug treatment of leishmaniasis, especially in the case of HIV coinfection. Furthermore, the method could be useful for monitoring the efficiency of new leishmaniasis treatments in infected patients.
Key words: Visceral Leishmaniasis; Leishmania; PBMCs; PCR; Diagnosis; Sensitivity
1. Introduction
Leishmaniasis affects more than 12 million people worldwide and is therefore a major health problem. It is caused by protozoan parasites that are transmitted to human and dog hosts through bites of infected sandflies. Leishmania is an obligate intramacrophage parasite: after phagocytosis, it is retained in the cell parasitopho- rous vacuoles. In these vesicles, the parasite is able to escape the host defense system and proliferate, ultimately causing lysis of the host cells and infection of the surround- ing macrophages.
Leishmania infantum species is responsible for human visceral leishmaniasis (VL). Moreover, in endemic areas visceral leishmaniasis is considered as an HIV-associated opportunistic infection (Montalban et al., 1989; Alt6s et al., 1991; Medrano et al., 1992). In these coinfection cases, classical leishmaniasis treatments with antimonium compounds are frequently ineffective (Peters et al., 1990).
Leishmaniasis is often difficult to diagnose, especially in asymptomatic patients
* Corresponding author: Tel: (33) 67 61 74 21, fax (33) 67 61 74 34.
SSDI 0001-706X(95)00079-8

188
harbouring only a few parasites and in HIV infection cases give a high percentage of negative serologies (Berenguer et al., 1989). Current diagnostic procedures are indirect or not sensitive enough. Antibody-based tests (indirect fluorescent antibody test, enzyme-linked immunoabsorbance assay), are thus not able to distinguish between past and present infections, cannot detect early infections and can crossreact with antibodies against other pathogens. Montenegro skin tests are negative in HIV- infected patients with CD4 + lymphocyte counts of less than 250/mm 3. Beside these methods, the most common diagnostic method, in vitro culture of bone-marrow aspirates, is laborious and time-consuming.
Faster and more sensitive diagnostic methods are therefore essential, especially in cases of HIV infection, for early detection (necessary for treatment) and also for monitoring the efficiency of new leishmaniasis treatments such as amphotericin B (Alvar, 1994).
In recent years, DNA-based techniques have shown high potential for diagnosis of various parasites. For Leishmania, different methods based on hybridization with nucleic acid probes have been reported, but these techniques often lack sensitivity for routine detection (Smith et al., 1989; Howard et al., 1991; Gramiccia et al., 1992). Sensitivity has been improved with the advent of the polymerase chain reaction (PCR) technique. However, the methods described so far required biopsy, which is stressful for patients, and purification of the target DNA before amplifica- tion, which is time-consuming (de Bruijn et al., 1992; Smyth et al., 1992; Lopez et al., 1993).
To overcome these drawbacks, we have developed a highly sensitive, simple and rapid procedure. It allows direct detection of the parasite in peripheral blood mono- nuclear cells (PBMCs) of the patient, with no target DNA purification, using a PCR technique. The highly variable regions of the kinetoplast DNA (kDNA) minicircles, previously used to design specific probes for another Kinetoplastida, Trypanosoma cruzi (Veas et al., 1991), were amplified using a pair of oligonucleotides designed from the conserved minicircle region. This multicopy target sequence (there are about 10000 minicircle copies per parasite) combined with a simplified sample preparation procedure allowed detection of about one parasite per 10 6 PBMCs (equivalent to 1 ml of blood), with the results obtained within 24 h.
2. Materials and methods
Cultures Leishmania infantum LEM 75 MON-1 was kindly provided by Professor Rioux
(Laboratoire d'Ecologie et de Pathologie Parasitaire, Montpellier, France). This strain was maintained at promastigote stage at 25°C in RPMI 1640 supplemented with 10% heat-inactivated fetal calf serum (FCS) (Biowhittaker France, Fontenay- sous-Bois).
Experimental infection of PBMCs with L. infantum PBMCs were obtained from heparinized blood of an healthy volunteer by centrifu-
gation (400 x g, 30 min) on Ficoll-Hypaque (Pharmacia, Uppsala, Sweden). After extensive washes in phosphate-buffered saline (PBS), the PBMCs were resuspended in RPMI 1640 supplemented with 10% heat-inactivated FCS, and plated into

189
60 x 15 mm culture dishes for 3 h at 37°C in a 5% CO2 atmosphere to ensure the monocyte adherence. L. infantum promastigotes were added at a ratio of 2 parasites per macrophage, considering that macrophages represent on average 20% of the total PBMCs, followed by 5 h incubation. All cells (adherent and non-adherent) were then harvested and washed in PBS. Infection was checked by microscopic examination of PBMCs that displayed amastigotes within the macrophage vacuoles. These infected PBMCs were aliquoted as 106 cells pellet and frozen at -20°C. At the same time, aliquots of 106 control uninfected PBMCs were prepared and stored at - 20°C.
Preparation of samples for PCR amplification Aliquoted PBMCs (infected or not), parasites (alone or mixed with 106 PBMCs),
were lysed by incubation in 25 ~tl of 50 mM KC1, 1.5 mM MgC12, 10 mM Tris-HC1 pH 9, 0.1% Triton X-100 and 60 I~g/ml proteinase K at 60°C for 1 h, followed by 15 min at 95°C to inactivate the proteinase. Amplification was then carried out directly in the tubes containing the sample lysates.
Polymerase chain reaction The 22-mer Lm5 (TGGTGTAAAATAGGCCAGGTGG) and Lm3G
(CCTACCCGCGGGACCAGAAAAG) primers were designed from the conserved region of L. infantum kDNA minicircles.
Purified kDNA from L. infantum LEM 75 MON-1 (called kDNA throughout this paper) in lysis buffer was used as positive control and lysis buffer alone was used as negative control. Amplification of the prepared samples was done in 1.5 mM MgC12, 50 mM KC1, 10 mM Tris-HC1 pH 9, 0.1% Triton X-100, in the presence of 0.2 mM of each deoxyribonucleotide, 15 pmol of each primer and 2.5 units of Taq DNA polymerase (Promega Corporation, Madison, USA) in a 50 ~tl final volume. Samples were overlaid with mineral oil and initially denatured at 92°C for 5 min. Cycles consisted of annealing at 62°C for 30 sec, extension at 70°C for 2 min and denatur- ation at 92°C for 30 sec. These programmes were run for 45 cycles on a PHC-2 thermocycler (Techne, UK). Amplification products were visualised by electrophore- sis in 1% agarose gel with 0.5 lxg/ml ethidium bromide.
Hybridization of the PCR products The PCR products were transferred from agarose gels onto Hybond N + nylon
membranes (Amersham, UK) by pocket-blotting according to Cuny et al. (1991), or directly dot blotted on membranes. The 700 bp fragment generated by PCR amplification of the kDNA was used as probe. It was radiolabeled with 0taZP-dCTP (Amersham, UK) or labeled with digoxigenin-ll-dUTP (DIG) (Boehringer Mannheim, Germany) using the random priming method (Feinberg et al., 1983). Membranes were prehybridized at 65°C in rapid hybridization buffer (Amersham, UK) for 1 h prior to addition of the denatured radioactive probe. Hybridization was carried out at 65°C for 3 h under rotary shaking. Membranes were then washed twice with 2 x standard saline phosphate EDTA (SSPE), 0.1% sodium dodecyl sulfate (SDS) at room temperature, once in 0.2 × SSPE, 0.1% SDS and twice in 0.1 × SSPE, 0.1% SDS at 65°C, air dried and autoradiographed at -70°C on Hyperfilm-MP (Amersham, UK) using intensifying screens. When necessary, filters were stripped by washing twice in 0.5N NaOH at 65°C for 30 min. Hybridization

190
was done with the DIG-labeled probe under the same temperature and stringency conditions as used with the radioactive probe. After washing, detection was per- formed using an alkaline phosphatase anti-digoxigenin conjugate according to the manufacturer's instructions (Boehringer Mannheim, Germany).
3. Results
Specificity and sensitivity of PCR primers Lm5 and Lm3G The choice of the two primers Lm5 and Lm3G was based on kDNA minicircle
sequence analysis of different Leishmania strains (Durand et al., manuscript in preparation). This pair of oligonucleotides, designed from the conserved minicircle region, allowed amplification of purified L. infantum kDNA by PCR, resulting in a single 700 bp band (lane 1, Fig. 1A), the expected size corresponding to the entire high variable regions of kDNA minicircles. These primers were also assayed using other purified kDNA species: whereas Leishmania donovani, the species equally responsible for human VL, resulted in amplification of the 700 bp band, Leishmania major gave a less intense band at about 600 bp, and another Kinetoplastida, Trypanosoma cruzi, was not amplified (lanes 2-4, Fig. 1A).
The ability of these primers to amplify kDNA from intact L. infantum parasites was checked using promastigotes maintained in culture. The amplification product from 1000 L. infantum parasites appeared as a single 700 bp band (lane 2, Fig. 2), as from the kDNA positive control.
Serial dilutions of L. infantum promastigotes (10-fold dilution steps from 1000 to
A M 1 2 3 4 5 B I 2 3 4 5
1353 1078 872
603 700
Fig. 1. Specificity of PCR primers Lm5 and Lm3G. (A) Amplification products from various Kinetoplastida. (B) Hybridization of amplification products with radiolabeled probe. Lanes are: Markers(M), d~X174 DNA/HaelII; 1, 1 ng L. infantum kDNA (kDNA positive control); 2, 2 ng L.
donovani kDNA; 3, 2 ng L. major kDNA; 4, 2 ng T. cruzi kDNA; 5, lysis buffer negative control.

M 1 2 3 4 5 6
191
1353
1078
872
603
Fig. 2. Sensitivity of PCR primers Lm5 and Lm3G. Lanes are: Markers(M) as in Fig. 1; 1, 1 ng kDNA positive control; 2, 1000 L. infantum; 3, 100 L. infantum; 4, 10 L. infantum; 5, 1 L. infantum; 6, lysis
buffer negative control.
1) were then amplified to establish the detection limit. The intensity of the unique amplified 700 bp band was clearly correlated with the number of Leishmania assayed and as few as 10 parasites could be detected (lane 4, Fig. 2). The 700 bp band was not observed with the lowest parasite dilution. This did not necessarily indicate amplification failure because it was not possible to ensure that a single parasite was really present in the PCR reaction. However, when only one parasite was assayed a smaller artefactual 600 bp band often appeared (lane 5, Fig. 2), but this PCR product did not hybridize with a specific probe (lane b5, Fig. 5).
Detection o f intra-macrophage L. infantum Experimental infection of human PBMCs was performed as described in the
Materials and Methods to determine whether it was possible to detect parasites within the macrophage vacuoles of patients' PBMCs. Direct observation of infected cells, after eosin/methylene blue staining, revealed 1 parasite per monocyte on average i.e. approximately 2 × 105 L. infantum in the 10 6 PBMCs infected sample. Direct amplification of 106 infected PBMCs resulted in the same single 700 bp band (lane 2, Fig. 3A) as obtained with the kDNA positive control and parasite alone, indicating that the presence of cellular DNA did not hinder detection of Leishmania kDNA. No 700 bp band was detected after amplification of an equivalent number of uninfected PBMCs (lane 3, Fig. 3A).
No amplification was observed when the same experiment was carried out using 2 x 10 6 infected PBMCs (data not shown), suggesting that the PCR reaction was hindered with very high loads of cellular material.
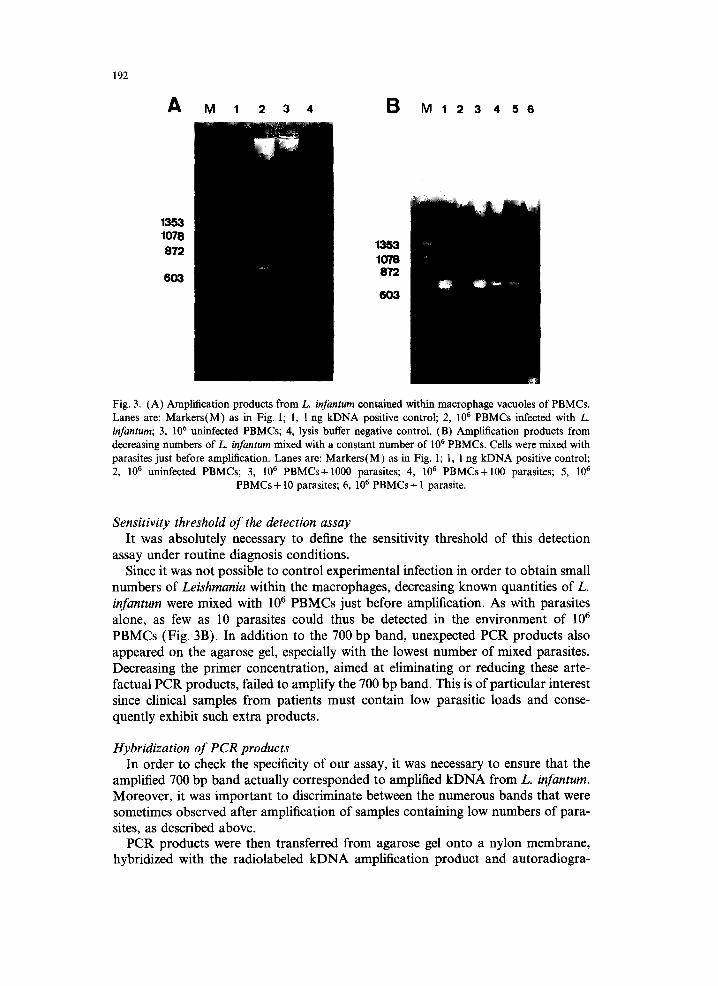
192
A M 1 2 3 4 B M 1 2 3 4 5 6
1353 1078 872
603
1353 1078 872
603 6 O ~ .......
Fig. 3. (A) Amplification products from L. infantum contained within macrophage vacuoles of PBMCs. Lanes are: Markers(M) as in Fig. 1; 1, 1 ng kDNA positive control; 2, 10 6 PBMCs infected with L. infantum; 3, 106 uninfected PBMCs; 4, lysis buffer negative control. (B) Amplification products from decreasing numbers of L. infantum mixed with a constant number of 106 PBMCs. Cells were mixed with parasites just before amplification. Lanes are: Markers(M) as in Fig. 1; 1, 1 ng kDNA positive control; 2, 10 6 uninfected PBMCs; 3, 106 PBMCs+1000 parasites; 4, 106 PBMCs+100 parasites; 5, 10 6
PBMCs+ 10 parasites; 6, 106 PBMCs+ 1 parasite.
Sensitivity threshold of the detection assay It was absolutely necessary to define the sensitivity threshold of this detection
assay under routine diagnosis conditions. Since it was not possible to control experimental infection in order to obtain small
numbers of Leishmania within the macrophages, decreasing known quantities of L. infantum were mixed with 106 PBMCs just before amplification. As with parasites alone, as few as 10 parasites could thus be detected in the environment of l06 PBMCs (Fig. 3B). In addition to the 700 bp band, unexpected PCR products also appeared on the agarose gel, especially with the lowest number of mixed parasites. Decreasing the primer concentration, aimed at eliminating or reducing these arte- factual PCR products, failed to amplify the 700 bp band. This is of particular interest since clinical samples from patients must contain low parasitic loads and conse- quently exhibit such extra products.
Hybridization of PCR products In order to check the specificity of our assay, it was necessary to ensure that the
amplified 700 bp band actually corresponded to amplified kDNA from L. infantum. Moreover, it was important to discriminate between the numerous bands that were sometimes observed after amplification of samples containing low numbers of para- sites, as described above.
PCR products were then transferred from agarose gel onto a nylon membrane, hybridized with the radiolabeled kDNA amplification product and autoradiogra-

! 2 3 4 5 6
193
700
Fig. 4. Hybridization of amplification products, transferred from agarose gels onto nylon membranes, with radiolabeled probe. Lanes are : 1, 1 ng kDNA positive control; 2, 10 6 uninfected PBMCs; 3, 10 6
PBMCs+1000 L. infantum; 4, 106 PBMCs+100 L. infantum; 5, 106 PBMCs+10 L. infantum; 6, 10 6
PBMCs + 1 L. infantum.
phed. A single 700 bp band was revealed in the samples containing parasites (lanes 3-5, Fig. 4), it was the same size as in the kDNA positive control (lane I, Fig. 4). The fact that no hybridization was observed using PCR products from L. donovanL L. major and Trypanosoma cruzi (lanes 2-4, Fig. 1B), indicates that this is a specific and sensitive diagnostic technique for Leishmania infantum.
Since there was hybridization only with the 700 bp band, we considered a much faster technique (in view of routine analysis): direct dot blotting of the PCR products and hybridization with the radiolabeled probe. This experiment was carried out using various amplification products previously analysed on 1% agarose gels (for details on these products see legend of Fig. 5). Presence and intensity of the signals (Fig. 5A) were completely correlated with the previously observed electrophoretic results (Figs. 2, 3A and 3B).
Hybridization with the DIG labeled probe was assayed on the same filter to avoid the use of radioactivity, which has short storage life and requires safety conditions. Detection, performed using an alkaline phosphatase anti-DIG conjugate resulted in the same sensitivity threshold as with the radiolabeled probe (Fig. 5B).
4. Discussion
A highly sensitive test is required to diagnose leishmaniasis at a very early parasite infection stage. The PCR technique can provide this high sensitivity, especially when the target is a multicopy sequence. In this manner, as few as 10 fg of purified kDNA from Leishmania has been successfully amplified by several laboratories (de Bruijn

194
A 1 2 3 4 5 6 7 8 9 ~
c
B 1 2 3 4 5 6 7 8 9 10
Fig. 5. Hybridization of PCR amplification products dot blotted onto nylon membranes, using (A) radiolabeled or (B) non radioactive probe. Lane a: 1, 1 ng kDNA positive control; 2, 1000 L. infantum; 3, lysis buffer negative control; 7, 1 ng kDNA positive control; 8, 106 PBMCS infected with L. infantum; 9, 106 uninfected PBMCs; 10, lysis buffer negative control. Lane b: 1, 1 ng kDNA positive control; 2, 1000 L. infantum; 3, 100 L. infantum; 4, 10 L. infantum; 5, 1 L. infantum; 6, lysis buffer negative control. Lane c: 1, 1 ng kDNA positive control; 2, 106 PBMCs+1000 L. infantum; 3, 106 PBMCs+100 L. infantum; 4, 106 PBMCs+ 10 L. infantum; 5, 106 PBMCs+ 1 L. infantum; 6, 106 uninfected PBMCs; 7,
lysis buffer negative control.
et al., 1992; Smyth et al., 1992). Since each kinetoplast contains about 10 fg kDNA (Borst et al., 1976), it would theoretically be possible to detect a single parasite in a biological sample. However, the sensitivity may be dramatically decreased, particu- larly with human biopsy material because of inaccessibility of the kDNA and/or PCR inhibition by tissue components (Rodgers et al., 1990).
In our study we chose to not purify the DNA, which can result in loss of material (de Bruijn et al., 1993), especially with low parasite loads. Therefore, in a first step we amplified L. infantum kDNA without any purification step. Using serial parasite dilutions, we showed that specific amplification products could be obtained with as few as 10 Leishmania, a similar detection limit as obtained with purified kDNA.
Besides the sensitivity, it is important to develop a diagnostic method that causes little stress for the patient and with minimal sample processing. Moreover, the results must be provided very quickly. The new procedure that we propose, involving blood sampling, preparation of PBMCs by Ficoll and their direct use for Leishmania kDNA amplification, with results available in 24 h, fulfils all of these requirements.

195
The potential offered by using blood samples to detect the parasite is supported by the following facts: (i) Leishmania have been observed in peripheral blood leukocytes from visceral leishmaniasis patients coinfected with HIV (Fillola et al., 1992; Martinez et al., 1993), (ii) amplification of Leishmania kDNA from purified DNA of peripheral blood of kala-azar patients has provided promising results (Smyth et al., 1992).
We evaluated our method using experimentally infected human PBMCs. This approach offers the advantage of mimicking the actual biological sample with parasites inside macrophage vacuoles. Detection of Leishmania infantum was success- fully obtained under these well-defined conditions. One million PBMCs (equivalent to 1 ml of blood) is the present maximal cellular environment before PCR inhibition. This is not a major drawback since larger clinical blood samples could be processed by dividing them into l06 PBMCs aliquots, each one subsequently submitted to PCR. Our lowest detection limit in such an environment is about 1 parasite. Moreover, this diagnosis can be supplied within 24 h, i.e. much faster than the two- week period required for the bone-marrow culture method.
The specificity of our assay was clearly demonstrated by hybridization of the radiolabeled kDNA positive control amplification product probe only with the 700 bp band of the transferred electrophoretic PCR products. Likewise, we were able to use a simple dot blot of the PCR products and hybridization with the non- radioactive DIG-probe in routine diagnosis. This is much faster and allows process- ing of a large number of samples.
The procedure presented here should provide a means for early and rapid diagnosis and consequently early and rapid drug treatment of leishmaniasis. This is of particu- lar interest for AIDS patients who require rapid treatment. It could also help in monitoring the response to and efficiency of new leishmaniasis treatments in HIV patients. Indeed, although they initially respond to treatment with antimonium compounds, these patients frequently follow a chronic-relapsing course (Montalban et al., 1990), and new drugs are needed for treatment and prophylaxis of leishmania- sis in such patients (WHO/TDR news, 199l; Lazanas et al., 1993).
Lastly, it could be especially useful for epidemiological studies of visceral leishma- niasis in these HIV infected patients. Such cases could presently be underestimated because of the difficulties in diagnosing leishmaniasis with current methods in AIDS patients.
Our method is now being assessed with clinical blood samples by comparison with the standard diagnostic procedure, bone-marrow culture.
Acknowledgements
We would like to thank Patrick Durand, C6cile Brengues and Pierre Kengne for sequence data, Nicole Vidal for oligonucleotide synthesis and Eric Delaporte for critical reading of the manuscript.
References
Alt6s, J., Salas, A., Riera, M., Udina, M., Galm6s, A., Balanzat, J., Ballesteros, A., Buades, J., Salva, F. and Villalonga, C. (1991) Visceral leishmaniasis: another HIV-associated opportunistic infection? Report of eight cases and review of the literature. AIDS 5, 201-207.

196
Alvar, J. (1994) Leishmaniasis and AIDS co-infection: the Spanish example. Parasitol. Today 10, 160-163. Berenguer, J., Moreno, S., Cercenado, E., Bernaldo de Quiros, J.C.L., Garcia de la Fuente, A. and
Bouza, E. ( 1989 ) Visceral leishmaniasis in patients infected with human immunodeficiency virus (HIV). Ann. Intern. Med. 111, 129-132.
Borst, P. and Fairlamb, A.H. (1976) DNA of parasites, with special reference to kinetoplast DNA. In Biochemistry of Parasites and Host Parasite Relationships (ed. H. Van den Bossche), pp. 169-92. Amsterdam: North Holland Publishing Co.
de Bruijn, M.H.L. and Barker, D.C. (1992) Diagnosis of new world leishmaniasis: specific detection of species of the Leishmania braziliensis complex by amplification of kinetoplast DNA. Acta Trop. 52, 45-58.
de Bruijn, M.H.L., Labrada, A., Smyth, A.J., Santrich C. and Barker, D,C. (1993) A comparative study of diagnosis by the polymerase chain reaction and by current clinical methods using biopsies from Colombian patients with suspected leishmaniasis. Trop. Med. Parasitol. 44, 201-207.
Cuny, G., Veas, F. and Roiz6s, G. (1991) "Pocket blotting": a method for transferring nucleic acids onto nylon membranes. Anal. Biochem. 193, 4548.
Feinberg, A.P. and Vogelstein, B. (1983) A technique for radiolabelling DNA restriction endonuclease fragments to high specific activity. Anal. Biochem. 132, 6-13.
Fillola, G., Corberand, J.X., Laharrague, P.F., Levenes, H., Massip, P. and Recco P. (1992) Peripheral intramonocytic leishmanias in an AIDS patient. J. Clin. Microbiol. 30, 3284-3285.
Gramiccia, M., Smith, D.F., Angelici, M.C., Ready, P.D. and Gradoni, L. (1992) A kinetoplast DNA probe diagnostic for Leishmania infantum. Parasitology 105, 29-34.
Howard, M.K., Kelly, J.M., Lane, R.P. and Miles, M.A. (1991) A sensitive repetitive DNA probe that is specific to the Leishmania donovani complex and its use as an epidemiological and diagnostic reagent. Mol. Biochem. Parasitol. 44, 63-72.
Lazanas, M.C., Tsekes, G.A., Papandreou, S., Harhalakis, N., Scandali, A., Nikiforakis, E. and Saroglou, G. (1993) Liposomal amphotericin B for leishmaniasis treatment of AIDS patients unresponsive to antimonium compounds. AIDS 7, 1018-1019.
Lopez, M., Inga, R., Cangalaya, M., Echevarria, J., Llanos-Cuentas, A., Orrego, C. and Arevalo, J. (1993) Diagnosis of Leishmania using the polymerase chain reaction: a simplified procedure for field work. Am. J. Trop. Med. Hyg. 49, 348-356.
Martinez, P., de la Vega, E., Laguna, F., Soriano, V., Puente, S., Moreno, V., Sentchordi, M.J., Garcia- Aguado, C. and Gonzalez-Lahoz, J. (1993) Diagnosis of visceral leishmaniasis in HIV-infected individ- uals using peripheral blood smears. AIDS 7, 227-230.
Medrano, F.J., Hernandez-Quero, J., Jim6nez, E., Pineda, J.A., Rivero, A., Sanchez-Quijano, A., Velez, I.D., Viciana, P., Castillo, R., Reyes, M.J., Carvajal, F., Leal, M. and Lissen, E (1992) Visceral leishmaniasis in HIV-l-infected individuals: a common opportunistic infection in Spain? AIDS 6, 1499-1503.
Montalban, C., Martinez-Fernandez, R., Calleja, JL., Garcia-Diaz, J.D., Rubio, R., Dronda, F., Moreno, S., Yebra, M., Barros, C., Cobo, J., Martinez, M.C., Ruiz, F. and Costa, J.R. (1989) Visceral leishmaniasis (kala-azar) as an opportunistic infection in patients infected with the human immuno- deficiency virus in Spain. Rev. Infect. Dis. 1 I, 655-660.
Montalban, C., Calleja, J.L., Erice, A., Laguna, F., Clotet, B., Podzamczer, D., Cobo, J., Mallolas, J., Yebra, M., Gallego, A. and the Co-operative Group for the Study of Leishmaniasis in AIDS (1990) Visceral leishmaniasis in patients infected with human immunodeficiency virus. J. Infect. 21,261-270.
Peters, B.S., Fish, D., Golden, R., Evans, D.A., Bryceson, A.D.M. and Pinching, A.J. (1990) Visceral leishmaniasis in HIV infection and AIDS: clinical features and response to therapy. Q. J. Med. 283, 1101-1111.
Rodgers, M.R., Popper, S.J. and Wirth, D.F. (1990) Amplification of kinetoplast DNA as a tool in the detection and diagnosis of Leishmania. Experimental Parasitology 71,267-275.
Smith, F.D., Searle, S., Ready, P.D., Gramiccia, M. and Ben-Ismail, R. (1989) A kinetoplast DNA probe diagnostic for Leishmania major: sequence homologies between regions of Leishmania minicircles. Mol. Biochem. Parasitol. 37, 213-224.
Smyth, A.J., Ghosh, A., Hassan, Md.Q., Basu, D., de Brnijn, M.H.L., Adhya, S., Mallik, K.K. and Barker, D.C. (1992) Rapid and sensitive detection of Leishmania kinetoplast DNA from spleen and blood samples of kala-azar patients. Parasitology 105, 183-192.
Veas, F., Breniere, S.F., Cuny, G., Brengues, C., Solari, A. and Tibayrenc, M. (1991) General procedure to construct highly specific kDNA for clones of Trypanosoma cruzi for sensitive detection by polymerase chain reaction. Cell. Mol. Biol.37, 73-84.
WHO "AIDS, Leishmaniasis danger of clash highlighted".( 1991 ) TDR news 36, 1-10.





























