Embed Size (px)
Citation preview

This article appeared in a journal published by Elsevier. The attachedcopy is furnished to the author for internal non-commercial researchand education use, including for instruction at the authors institution
and sharing with colleagues.
Other uses, including reproduction and distribution, or selling orlicensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of thearticle (e.g. in Word or Tex form) to their personal website orinstitutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies areencouraged to visit:
http://www.elsevier.com/authorsrights

Author's personal copy
A first-principles study of electron attachment to the fully hydratedbromonucleobases
Miłosz Wieczór a,1, Paweł Wityk a,1, Jacek Czub a,⇑, Lidia Chomicz b, Janusz Rak b,⇑a Faculty of Chemistry, Gdansk University of Technology, Narutowicza 11/12, 80-233 Gdansk, Polandb Faculty of Chemistry, University of Gdansk, Wita Stwosza 63, 80-308 Gdansk, Poland
a r t i c l e i n f o
Article history:Received 20 December 2013In final form 29 January 2014Available online 6 February 2014
a b s t r a c t
Degradation mechanism of four brominated nucleobases (BrX), potential DNA radiosensitizers, is studiedin explicit water solution, using ab initio molecular dynamics. Several fs long dynamics is needed tolocalize an electron on the nucleobase. Produced by electron attachment BrX anion radical degradesthrough the bromide anion abstraction, barrier-free (purines) or with low barrier (pyrimidines), to areactive nucleobase radical. Such differences in dissociative behavior of purines and pyrimidines suggestthat brominated purine anions should dissociate more effectively than their pyrimidine counterparts.
� 2014 Elsevier B.V. All rights reserved.
1. Introduction
The fundamental mode of action of radiotherapy in cancertreatment [1] lies in interactions between the products of waterradiolysis and cellular DNA [2]. As indicated by radiation chemistryexperiments employing radical scavengers, hydroxyl radicals (OH�)constitute a major factor responsible for the type of damageinduced by ionizing radiation (IR) [3]. Although a given dose ofIR produces comparable amounts of solvated electrons and hydro-xyl radicals in water [4], the latter species seems to be exclusivelyeffective in strand break formation [3]. Indeed, as was shown bythe Sanche group [5], a 25-bp DNA duplex labeled with radioactivephosphorus and irradiated with as much as 700 Gy of 1.18 and1.33 MeV photons in a neutral aqueous solution containing ahydroxyl radical scavenger demonstrated no strand breaks withdenaturing PAGE electrophoresis.
Therefore, a rational mean of DNA sensitization to IR should‘activate’ hydrated electrons otherwise inactive against the nativebiopolymer. Given that the excess electrons are able to triggerstrand breaks in DNA, as was proven via the ultra-high vacuumbombardment of plasmid DNA [6], an IR-induced signal for apopto-sis could then be initiated with a lesser dose and even underhypoxic conditions – a common state for cancer cells – that triggertheir radio-resistance [7]. Modified nucelobases incorporated dur-ing DNA biosynthesis could play a role as such hydrated electron‘activators’. In order to sensitize the biopolymer, these derivativeshave to be better electron acceptors than the natural nucleobases.
The latter requirement results from the fact that electrons are sta-bilized in water by more than 3 eV [8], while the electron affinity ofnative nucleobases bound in DNA amounts to c. 2 eV [9]. Finally, asshown by our recent studies on modified uracils [10], the chemicalbond connecting a substituent to a nucleobase residue has to berelatively weak to assure efficient dissociation of an anion thatleaves behind a reactive nucleobase radical capable of inducing aDNA strand break via secondary chemical reactions [11].
5-bromouracil (5BrU) belongs to the class of sensitizersdescribed above. When administered to cells as a deoxynucleoside,it substitutes thymine during DNA biosynthesis [12], possessessubstantially larger electron affinity than native nucleobases andits anion undergoes efficient decomposition leading to the geno-toxic uracil-5-yl radical [13–15]. These very promising propertiesof 5BrU resulted in clinical trials terminated in the late nineteennineties [16]. However, in the last 10–15 years one can note arenewed interest in halogenated uridine analogs (5BrdU and5IdU) as IR-radiosensitizing agents [17]. The remaining bromi-nated nucleobases (BrX): 5-bromocytosine (5BrC), 8-bromoade-nine (8BrA) and 8-bromoguanine (8BrG) have not been tested onpatients so far, although they seem to have properties similar to5BrU. For instance, Sevilla et al. [18] demonstrated, using low tem-perature EPR, that 5BrC incorporated in DNA was approximatelythree-fold more efficient as an electron scavenger than cytosineitself. Similarly, reactions between hydrated electrons and8-bromo-20-deoxyadenosine [19–21] or 8-bromo-20-deoxyguano-sine [22,23] lead to a rapid release of bromide ion followed by fasthydrogen atom abstraction from the C50 position of the 20-deoxyri-bose moiety.
An important prerequisite for radiodamage to occur is anefficient dissociative electron attachment. Very recently, we have
http://dx.doi.org/10.1016/j.cplett.2014.01.0520009-2614/� 2014 Elsevier B.V. All rights reserved.
⇑ Corresponding authors.E-mail addresses: [email protected] (J. Czub), [email protected] (J. Rak).
1 These authors contributed equally.
Chemical Physics Letters 595–596 (2014) 133–137
Contents lists available at ScienceDirect
Chemical Physics Letters
journal homepage: www.elsevier .com/ locate /cplet t

Author's personal copy
reported the results of density functional theory (DFT) studies de-voted to electron attachment-induced degradation of four BrXmolecules (Figure 1) both in the gas phase and in an aqueous envi-ronment [15]. In the above-mentioned study, water was accountedfor using the PCM method [24], i.e. with a continuum model ofsolvent. It is worth noting, however, that the PCM approach,although generally useful, is unable to properly account for suchspecific interactions as e.g. hydrogen bonds between water mole-cules and the solute. In fact, Smyth and Kohanoff [25] observedthat the surrounding water orbitals participate in stabilizing theexcess electrons on nucleobases and nucleosides. Therefore, onecan conclude that a substantial overestimation of the kineticbarrier for electron-induced dissociation of the phosphodiesterbond in the cytidine nucleotide – which was predicted in a hybridquantum mechanical/molecular mechanical (QM/MM) moleculardynamics (MD) simulation [26] – probably resulted from the factthat the water molecules were treated classically and thereforethe excess electron was not allowed to enter the MM region.
In this Letter we report on the results of ab initio MD simula-tions of dissociative electron attachment to four methylatedbromonucleobases (BrX, see Figure 1). The first-principlesapproach within the DFT framework applied in the current Letterallowed us to model the surrounding water molecules at the samelevel of theory as used for the nucleobases. Consequently, our sim-ulations provide the first complete picture of the bromonucleobasedissociation process in which the stabilizing effect of water mole-cules and specific solute–solvent interactions are representedexplicitly. By modeling the process in a thermally fluctuating envi-ronment and using enhanced sampling methods, our approachallowed us to capture underlying enthalpic and entropic drivingforces for the dissociation process. The resulting free energyprofiles give an accurate description of electron-induced decompo-sition of bromonucleobases in an aqueous solution, and may serveas benchmark data for further studies of the electron-induceddissociation in BrX-labeled DNA.
2. Methods
Each of the four models employed in our simulations consisted ofa single BrX molecule surrounded by 67 molecules of water in a cu-bic PBC box with a fixed box vector length of 13.4 Å. The initial geom-etries were obtained by equilibration in the NPT ensemble usingforce field based molecular dynamics. The Born–Oppenheimer MDsimulations were performed using the Quickstep module [27] inthe cp2k software package and the gaussian and augmented-plane-wave (GAPW) density functional method [28]. We used thePBE exchange–correlation functional [29] in combination withGoedecker–Teter–Hutter (GTH) [30] pseudopotentials and thedouble-zeta split valence augmented with polarization functions(DZVP) basis set. The plane-wave representation was truncatedat 400 Ry. All calculations were performed using the unrestrictedKohn–Sham (UKS) [31] scheme and a typical timestep of 0.5 fs.To estimate and compare the stability of BrX molecules uponelectron attachment, we computed free energy profiles for C–Br
bond dissociation in the BrX anion radicals. These were obtainedby means of constrained ab initio molecular dynamics (AIMD)simulations, with the C–Br bond length employed as the reactioncoordinate. To this end, 10 sampling windows were used forsuccessive C–Br distances, varying from 1.7 to 3.5 Å with an incre-ment of 0.2 Å. Each window was simulated for 20 ps. Theconstraint forces necessary to keep the C–Br distance fixed,obtained through the Shake algorithm [32], were averaged overthe last 15 ps of the generated trajectories. These averages werethen integrated with respect to distance to produce the potentialof mean force (PMF), an equivalent of the free energy profile[33]. The RC-dependence of free energy is defined to within anadditive constant by an integral (see Eq. (1)). The integrand dA
dn0,corresponding to the mean force acting along the reaction coordi-nate, can be replaced with mean value of a respective Lagrangemultiplier k(n) [33] to yield the Eq. (1) in form of Eq. (2).
AðnÞ ¼Z n
n0
dAdn
dn ð1Þ
AðnÞ ¼ �Z n
n0< kðnÞ > dn ð2Þ
The latter integral was approximated by the Simpson rule andinterpolated to produce the free energy profile. For standard errorestimation, correlations in the Lagrange multiplier time serieswere taken into account.
The standard free energies for C–Br dissociation were obtainedby integrating the PMF-derived probability distribution, assuminga standard concentration of 1 M and setting the dissociationboundary at 2.0 Å (we consider BrX� to be dissociated for a C–Brdistance greater than 2.0 Å). In order to validate our methodology,analogous free energy calculations were applied to neutral BrXmolecules as well.
3. Results
3.1. Electron attachment process
Since the previous studies on the subject essentially neglectedthe effect of explicit solvent molecules on electron affinity due tomethodology limitations, it is worthwhile to try to assess the roleof this explicit environment on the dynamics of the excess chargeattachment. To tackle this problem, we simulated the process ofelectron attachment to a neutral solute molecule in an equilibratedsystem and computed the time-dependence of the Mulliken spinpopulation. Prior to being completely attached by the respectivebrominated base, the excess electron partially (ca. 25–35%) scattersthroughout the solvent molecules and it takes 6–12 fs for it to fullylocalize on the solute (Figures 2 and 3).
Such short timescales imply that BrX molecules are stronglyfavored as electron acceptors, provided that the electron itself isaccessible (e.g. that a hydrated electron can be readily extractedfrom the bulk solvent). The dynamic accommodation of theattached electron is associated with structural changes in the sys-tem, i.e. reorientation of the solvent molecules and perturbation ofthe planarity of the purine/pyrimidine ring. This 6–12 fs delay inlocalization is not observed when the starting geometry comesfrom an equilibrated anion-radical system, which additionally con-firms that the geometry perturbation is associated with the processof electron attachment. Interestingly, the electron localizes fasterin the case of pyrimidines, possibly owing to their simpler ringgeometries and larger electron affinities (EAs). Moreover, bromi-nated bases are characterized by general shortening of electronlocalization time. Indeed, for non-substituted bases, this time,
Figure 1. Structures of methylated bromonucleobases (BrX), along with theirabbreviated names.
134 M. Wieczór et al. / Chemical Physics Letters 595–596 (2014) 133–137

Author's personal copy
calculated with the same methodology, amounts to as much as15–25 fs [25].
3.2. C–Br bond dissociation
As can be seen from the free energy profiles shown in Figure 4A,the computed C–Br bond dissociation energies for the neutral BrXmolecules fall within the range of 59–67 kcal/mol. These valuesagree well with the experimental C–Br bond energy (below69 kcal/mol) [34] and thus strongly support the validity of ourmodel and methodology. Figure 4B shows that the chemicalstability of brominated nucleobases decreases dramatically uponelectron attachment. In each case, a high thermodynamic drivingforce for C–Br bond dissociation is observed, which convincinglydemonstrates that brominated analogs exhibit a strong tendency
to produce reactive base radicals. Furthermore, the profiles revealsignificant differences between brominated purines and pyrimi-dines. After electron attachment, the former break down readilyin a barrier-free process (see smooth purine curves in Figure 4B),while for brominated cytosine and brominated uridine a plateauand a small free energy barrier for dissociation is seen, respectively(the plateau observed for BrC seems to remain in agreement withthe experimental work for this species in DNA [18] that foundthe BrC anion radical to be a r⁄ radical). These findings suggest thatthe specific mechanism of the process may differ between bromi-nated purines and pyrimidines. Also, comparison of the calculatedfree energies for C–Br dissociation shows a considerable variationin the thermodynamic driving force for the dissociation of thestudied anion radicals, with values of �14.0, �19.3, �18.6 and�10.0 kcal/mol for BrA, BrG, BrC and BrU, respectively.
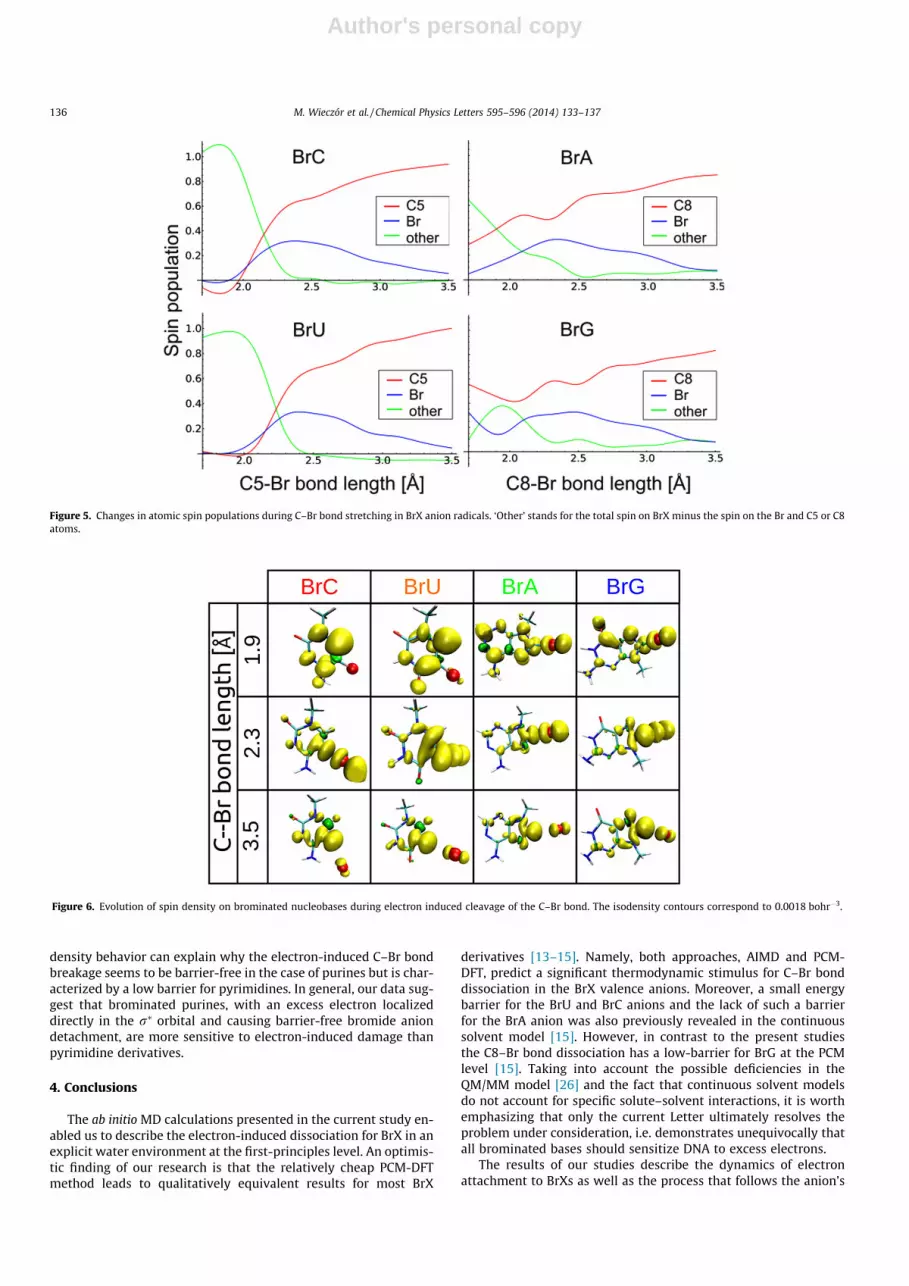
The C–Br bond cleavage occurs due to the presence of an excesselectron. Therefore, a quantitative analysis of spin population dis-tribution and its dependence on C–Br bond length is expected toreveal the underlying mechanism of the dissociation process andindicate possible differences between the BrX molecules. We foundthat when the excess electron is attached to the neutral purinebases at their equilibrium C–Br bond length (1.9 Å, Figure 4A), asignificant fraction of the spin density is localized from the begin-ning on the C8 and Br atoms, occupying the r⁄ orbital (Figures 5and 6) and destabilizing the C–Br bond. For pyrimidines, the evolu-tion of spin density is different: initially, the excess electron islocalized on the p⁄ orbital (barely any spin density on the C5 andBr atoms at 1.9 Å; see Figures 5 and 6). Only upon stretching ofthe C5–Br bond can one see a significant increase in spin popula-tion in the vicinity of the C–Br bond. This is coupled to the mixingof the r⁄ type orbital with the initial p⁄ state. Electron transferfrom the p⁄ to r⁄ state occurs only if these two states are equalin energy. In order to reach this point on the potential energy sur-face, the C5–Br bond has to elongate (up to a certain point), whichis associated with an increase in the total energy and manifests asan energy barrier in the free energy profile. This difference in spin
Figure 2. Initial evolution of the Mulliken spin populations on brominatednucleobases following attachment of an excess electron. Initial configurations wereobtained from 20 ps equilibrium simulations of neutral BrX molecules.
Figure 3. Dynamics of electron attachment to neutral BrU in water. The isodensity contours correspond to 0.0003 bohr�3.
Figure 4. Free energy profiles for dissociation of the C–Br bond in neutral BrXs (A) and their radical anions (B).
M. Wieczór et al. / Chemical Physics Letters 595–596 (2014) 133–137 135
Author's personal copy
density behavior can explain why the electron-induced C–Br bondbreakage seems to be barrier-free in the case of purines but is char-acterized by a low barrier for pyrimidines. In general, our data sug-gest that brominated purines, with an excess electron localizeddirectly in the r⁄ orbital and causing barrier-free bromide aniondetachment, are more sensitive to electron-induced damage thanpyrimidine derivatives.
4. Conclusions
The ab initio MD calculations presented in the current study en-abled us to describe the electron-induced dissociation for BrX in anexplicit water environment at the first-principles level. An optimis-tic finding of our research is that the relatively cheap PCM-DFTmethod leads to qualitatively equivalent results for most BrX
derivatives [13–15]. Namely, both approaches, AIMD and PCM-DFT, predict a significant thermodynamic stimulus for C–Br bonddissociation in the BrX valence anions. Moreover, a small energybarrier for the BrU and BrC anions and the lack of such a barrierfor the BrA anion was also previously revealed in the continuoussolvent model [15]. However, in contrast to the present studiesthe C8–Br bond dissociation has a low-barrier for BrG at the PCMlevel [15]. Taking into account the possible deficiencies in theQM/MM model [26] and the fact that continuous solvent modelsdo not account for specific solute–solvent interactions, it is worthemphasizing that only the current Letter ultimately resolves theproblem under consideration, i.e. demonstrates unequivocally thatall brominated bases should sensitize DNA to excess electrons.
The results of our studies describe the dynamics of electronattachment to BrXs as well as the process that follows the anion’s
Figure 5. Changes in atomic spin populations during C–Br bond stretching in BrX anion radicals. ‘Other’ stands for the total spin on BrX minus the spin on the Br and C5 or C8atoms.
Figure 6. Evolution of spin density on brominated nucleobases during electron induced cleavage of the C–Br bond. The isodensity contours correspond to 0.0018 bohr�3.
136 M. Wieczór et al. / Chemical Physics Letters 595–596 (2014) 133–137

Author's personal copy
formation. Our calculations reveal that electron attachment isabout two-fold faster for brominated nucleobases than for their na-tive counterparts [25]. Furthermore, the current findings lead to aconsistent picture, in which purine bromoderivative anionsdissociate in a barrier-free manner, while small barriers are pre-dicted for pyrimidine anion breakage. The latter result may suggestthat brominated purines, if incorporated into cellular DNA, mightbe even more efficient radiosensitizers than BrU itself. However,the fact that bromopurine anions dissociate easier than theirbromopyrimidine counterparts does not mean that bromopurinesare better DNA sensitizers. The formation of bromonucleobaseanion is an indispensable condition for sensitization and thereare other sites in DNA of comparable or even larger EA than thoseof purine bromoderivatives which can compete for an electron.Indeed, the B3LYP EAs of 1.87 and 1.36 eV for BrA and BrG, respec-tively [15], are comparable or smaller than the EAs of 1.85 and2.01 eV for T and C, respectively [35]. Nevertheless, the actual EAof a given base in DNA strongly depends on its local sequence[36,37]. Thus, bromopurines do sensitize DNA to electrons[38–40] although less effectively than bromouracil [41,42] proba-bly because of their lower electron affinities.
Acknowledgments
This work was supported by the Polish National Science Center(NCN) under Grant No. N N204 156040 (J.R.) This research was alsosupported in part by PL-Grid Infrastructure and ACK Cyfronet AGHresources.
References
[1] M. Joiner, A. van der Kogel, Basic Clinical Radiobiology, fourth ed., HodderArnold, London, UK, 2009.
[2] R.L. Warters, K.G. Hofer, Radiat. Res. Q 12 (1977) 389.[3] B.D. Michael, P. O’Neill, Science 287 (2000) 1603.[4] C. von Sonntag, The Chemical Basis of Radiation Biology, Taylor and Francis,
London, UK, 1987.[5] S. Cecchini, S. Girouard, M.A. Huels, L. Sanche, D.J. Hunting, Biochemistry 2005
(1932) 44.[6] B. Boudaïffa, P. Cloutier, D. Hunting, M.A. Huels, L. Sanche, Science 287 (2000)
1658.[7] B.T. Oronsky, S.J. Knox, J. Scicinski, Transl. Oncol. 4 (2011) 189.
[8] B. Abel, U. Buck, A.L. Sobolewski, W. Domcke, Phys. Chem. Chem. Phys. 14(2012) 22.
[9] J. Gu, Y. Xie, H.F. Schaefer III, Nucleic Acids Res. 35 (2007) 5165.[10] L. Chomicz, M. Zdrowowicz, F. Kasprzykowski, J. Rak, A. Buonaugurio, Y. Wang,
K.H. Bowen, J. Phys. Chem. Lett. 4 (2013) 2853.[11] C.J. Burrows, J.G. Muller, Chem. Rev. 98 (1998) 1109.[12] T.J. Kinsella, P.P. Dobson, J.B. Mitchell, A.J. Fornace Jr., Int. J. Radiat. Oncol. Biol.
Phys. 13 (1987) 733.[13] S.D. Wetmore, R.J. Boyd, L.A. Eriksson, Chem. Phys. Lett. 343 (2001) 151.[14] X. Li, L. Sanche, M.D. Sevilla, J. Phys. Chem. A 106 (2002) 11248.[15] L. Chomicz, J. Rak, P. Storoniak, J. Phys. Chem. B 116 (2012) 5612.[16] M.D. Prados et al., Int. J. Radiat. Oncol. Biol. Phys. 45 (1999) 1109.[17] T.J. Kinsella, Cancer Biol. Ther. 7 (2008) 1567 (and references therein).[18] Y. Razskazovskii, S.G. Swarts, J.M. Falcone, C. Taylor, M.D. Sevilla, J. Phys. Chem.
B 101 (1997) 1460.[19] R. Flyunt, R. Bazzanini, C. Chatgilialoglu, Q.G. Mulazzani, J. Am. Chem. Soc. 122
(2000) 4225.[20] C. Chatgilialoglu, M. Guerra, Q.G. Mulazzani, J. Am. Chem. Soc. 125 (2003)
3839.[21] F. Boussicault, P. Kaloudis, C. Caminal, Q.G. Mulazzani, C. Chatgilialoglu, J. Am.
Chem. Soc. 130 (2008) 8377.[22] C. Chatgilialoglu, C. Caminal, M. Guerra, Q.G. Mulazzani, Angew. Chem. Int. Ed.
44 (2005) 6030.[23] C. Chatgilialoglu, C. Caminal, A. Altieri, G.C. Vougioukalakis, Q.G. Mulazzani, T.
Gimisis, M. Guerra, J. Am. Chem. Soc. 128 (2006) 13796.[24] M. Cossi, V. Barone, R. Cammi, J. Tomasi, Chem. Phys. Lett. 255 (1996) 327.[25] M. Smyth, J. Kohanoff, Phys. Rev. Lett. 106 (2011) 238108.[26] P. Schyman, A. Laaksonen, H.W. Hogosson, Chem. Phys. Lett. 462 (2008) 289.[27] J. Vandevondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing, T.J. Hutter,
Comput. Phys. Commun. 167 (2005) 103.[28] G. Lippert, J. Hutter, M. Parrinello, Theor. Chem. Acc. 103 (1999) 124.[29] C. Adamo, V. Barone, J. Chem. Phys. 110 (1999) 6158.[30] S. Goedecker, M. Teter, J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys. 54
(1996) 1703.[31] J.B. Krieger, Y. Li, G.J. Iafrate, Phys. Rev. A 45 (1992) 101.[32] J.P. Ryckaert, G. Ciccotti, H.J.C. Berendsen, J. Comput. Phys. 23 (1977) 327.[33] M. Sprik, G. Ciccotti, J. Chem. Phys. 109 (1998) 7737.[34] J.-L. Lin, A.V. Teplyakov, S. Bent, J. Phys. Chem. 100 (1996) 10721.[35] J. Gu, J. Leszczynski, H.F. Schaefer III, Chem. Rev. 112 (2012) 5603.[36] M. Kobyłecka, J. Leszczynski, J. Rak, J. Am. Chem. Soc. 130 (2008) 15683.[37] M. Kobyłecka, J. Leszczynski, J. Rak, J. Chem. Phys. 131 (2009) 085103.[38] T. Kimura, K. Kawai, S. Tojo, T. Majima, J. Org. Chem. 69 (2004) 1169.[39] K. Polska, J. Rak, A.D. Bass, P. Cloutier, L. Sanche, J. Chem. Phys. 136 (2012)
075101.[40] Y. Park, K. Polska, J. Rak, J.R. Wagner, L. Sanche, J. Phys. Chem. B 116 (2012)
9676.[41] A. Manetto, S. Breeger, C. Chatgilialoglu, T. Carell, Angew. Chem. Int. Ed. 45
(2006) 318.[42] D. Fazio, C. Trindler, K. Heil, C. Chatgilialoglu, T. Carell, Chem. Eur. J. 17 (2011)
206.
M. Wieczór et al. / Chemical Physics Letters 595–596 (2014) 133–137 137





























