Embed Size (px)
Citation preview

Chemical mechanism and kinetic study on the ocimene ozonolysis
reaction in atmosphere
Xiamin Sun[a,b], Jing Bai[a], Yuyang Zhao[a], Chenxi Zhang[a], Yudong Wang[a], jingtian Hu[a]
a. Environment Research Institute, Shandong University, Jinan 250100, PR China
b. State Key Laboratory of Solid Lubrication, Lanzhou Institute of Chemical Physics, Chinese Academy of Science, Lanzhou 730000, PR China
Atmospheric Environment. 45, (2011), 6197-6203

Main objective of the paper.
Obtain theoretical kinetic constants of ocimene ozonolysis and its reaction pathway, in order to estimate atmospheric lifetimes of the reaction species.

Background information:
Ocimene is a monoterpene released to the atmosphere by the vegetation. Reactions of biogenic monoterpenes with ozone are responsible for a significant amount of aerosol formation in the atmosphere†.
†Hoffman et. al. Journal of Atmospheric Chemistry, 12, (1997), 181-194.

Atkinson† describes ozonolysis in the following schemes:
†R. Atkinson. Atmospheric Environment, 34, (2000), 2063-2101.

R. Atkinson. Atmospheric Environment, 34, (2000), 2063-2101.
It is important to understand the elementary reactions that form the mechanism for ozonolysis because some intermediates react with (or produce) OH radicals, NOx, SOx, carbon dioxide, and oxygen in the atmosphere. E.g.

Computational methods:
The paper is purely computational, therefore, no experiments are made. The computational methods can be divided in two parts:
- Geometry optimization of intermediates and transition states using a hybrid DFT model (MPWB1K) using Gaussian 03. Used to get the potential energy profile.
- Kinetic calculations using Rice-Ramsperher-Kassel-Marcus Theory, and Canonical Variational TST (with the computer program PLYRATE v9.7)
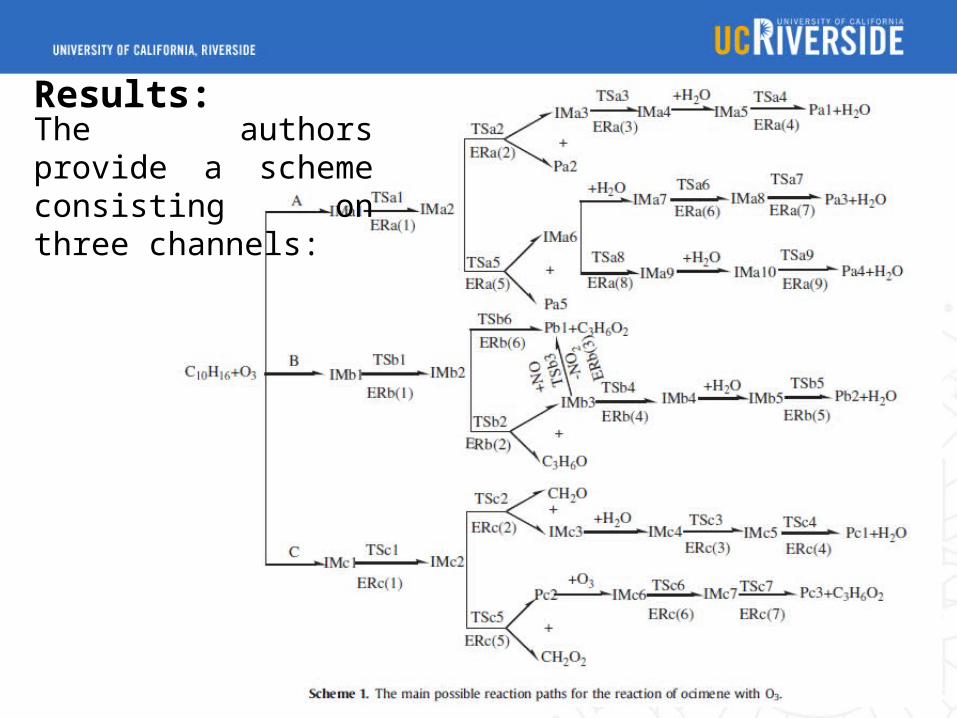
The authors provide a scheme consisting on three channels:
Results:

Let’s focus on channel A:
ERa(1)
ERa(2)
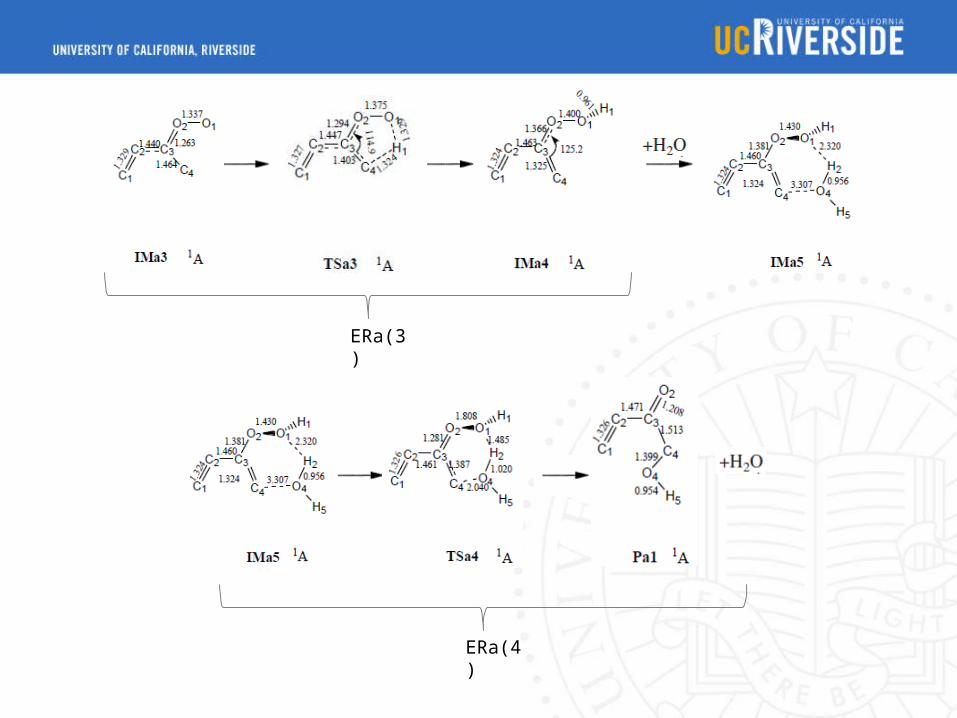
ERa(3)
ERa(4)
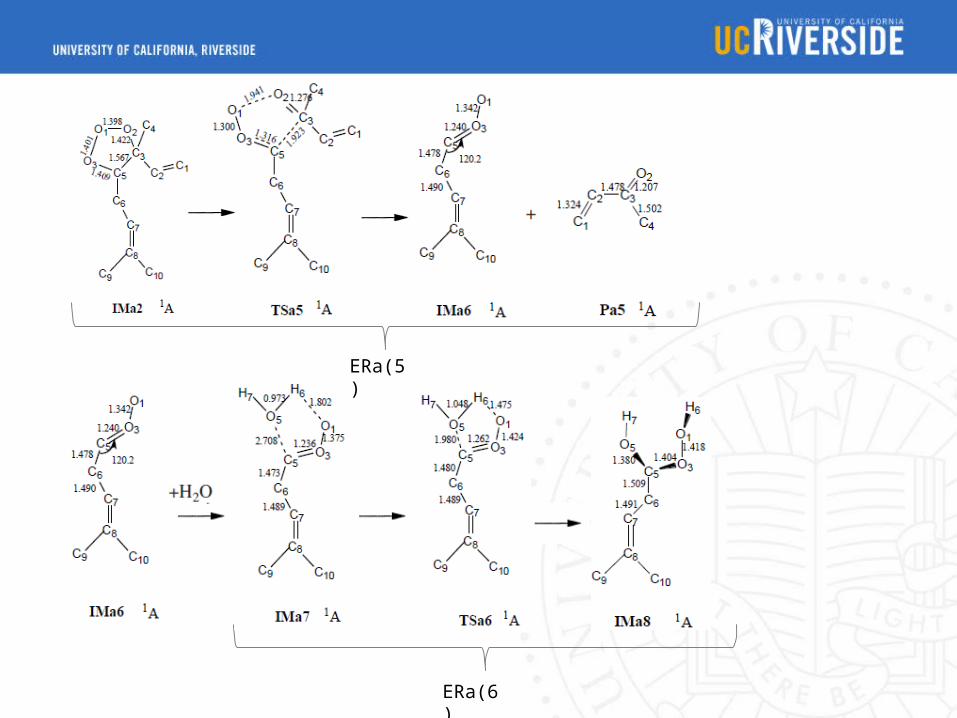
ERa(5)
ERa(6)
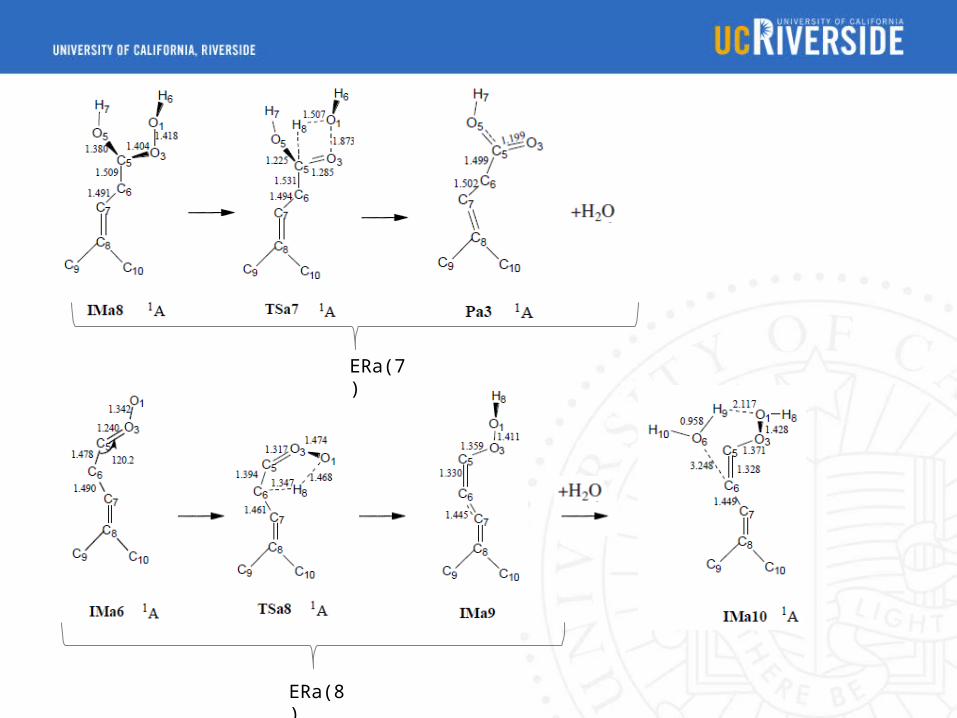
ERa(7)
ERa(8)

ERa(9)
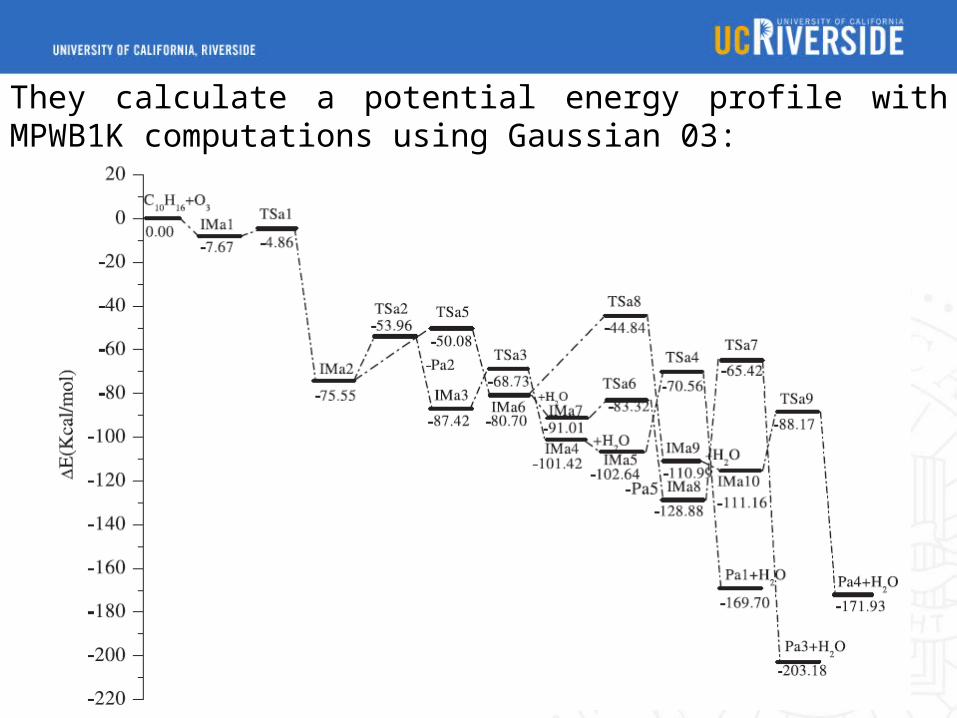
They calculate a potential energy profile with MPWB1K computations using Gaussian 03:

RRKM Theory is used for the following elementary reactions:
CVTST is used for the following isomerizations:

RRKM Theory:
RRKM Theory traces its origins to Lindemann-Christensen approach to unimolecular reactions:
Derivation of kinetic equations:

Assuming steady state:
Hinshelwood modified this treatments by considering collision theory:

Rice-Ramsperger-Kassel (RRK) consider the energy dependence:
- In order for A* to react, the critical energy must concentrate in one part of the molecule
- Free energy transfer between oscillators is assumed (loosely coupled and separated).
- The event that an oscillator has the required energy depends on statistical factors

The probability of an oscillator to have enough energy is calculated accordingly:
The rate constant for the conversion of the energized molecule into the activated complex will be proportional to this probability

The ratio between activation and de-activation rates of A now follows a distribution function:
The “first-order” rate is now calculated by integration:
With a high pressure limit, it reduces to:

www.chem.purdue.edu/mcluckey/.../2011_Prentice_SL_RRKM.pdf
Energy diagram of the different energies that are involved in RRKM theory

Marcus follows the same mechanism but considers the energy to be distributed in active (e.g vibrations) and inactive states (e.g. translations) and merges TST with the RRK theory.
The first step is calculated as an equilibrium:

Assume that the activated complex to be in steady-state.
The ½ factor is accounted to lack of reversibility in the last step.
The rate constant of the energized molecule depends on the energy of translation along the reaction coordinate:

Assume that the activated complex to be in steady-state.
The ½ factor is accounted to lack of reversibility in the last step.
The rate constant of the energized molecule depends on the energy of translation along the reaction coordinate:

The active vibrational-rotational modes of the activated complex cannot be assumed to be a continuous distribution.
Using a PIB approximation for the movement along delta:
After including a statistical factor l and accounting for some changes in rotational states

After integration, the cannonical “first-order” rate is obtained.
CVTST:
Using CTST we obtain the following expression:
CVTST obtains this canonical rate by integrating over all energies:
The minimum k(T) obtained when the dividing surface is varied is then chosen as the “closer” rate to the real rate constant.

The RRKM microcanonical k(3) rate is obtained from the following.
The collision deactivation is calculated by the following:
The Beyer-Swinehart algorithm is used to calculate the number of states and the density of states†. The POLYRATE program is used for CVTST calculations‡.
†Moon et al. J Am Soc Mass Spectrom, 18, (2007), 1063-1069.‡Corchado et al., 2007. POLYRATE Version 9.7. University of Minnesota Minneapolis.
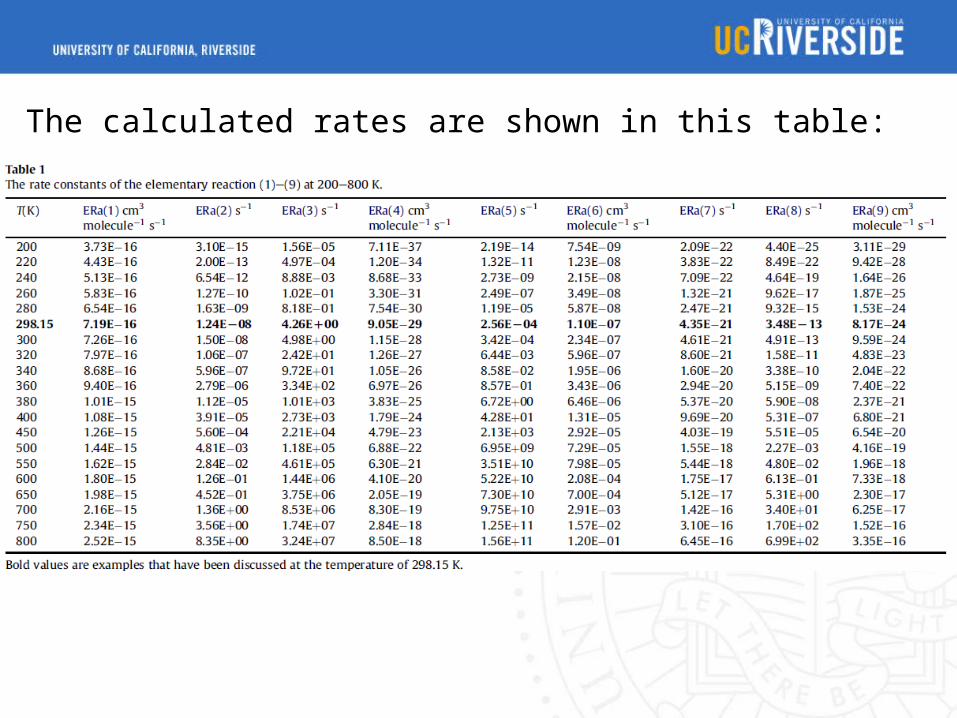
The calculated rates are shown in this table:

The lifetimes are obtained:

Arrhenius formulas were fitted to the following T dependence:

Conclusions:
› The authors that the lifetime for ocimene was 86 min (short lifetime). IMa2 and IMa9 belong to middle lifetime species (2.56 and 2.60 d). IMa4 and IMa8 belong to long lifetime species.
› The authors conclude that the kinetic information obtained is useful to understand the formation of secondary organic aerosols.
› Water acts as an activator for OH transfer and promotes further reaction.
› Oxidation occurs spontaneously once it is initiated by ozone.









![Hiroshi FUJISAKI,1,2, Yong ZHANG, and John E. STRAUB · tative description of chemical reactions [2]. In the celebrated RRKM theory of an absolute reaction rate for isolated molecules,](https://img.dokumen.tips/doc/110x75/5f0f35b87e708231d44307e2/hiroshi-fujisaki12-yong-zhang-and-john-e-straub-tative-description-of-chemical.jpg)

