Embed Size (px)
Citation preview

Proof of concept of WGS based surveillance: meningococcal disease
Martin MaidenDepartment of Zoology

http://zoo-maidenlab.zoo.ox.ac.uk/@MaidenLab

… and our sponsors & collaborators

Population genomics: the gene-by-gene approach
Complete
Sequence
Annotation
Bacterial Isolate
Genome Sequence
Database
(BIGSDB)
Contigs
Gene sequencesProvenance/phenotyp
e information
Jolley, K. A. & Maiden, M. C. (2010). BIGSdb: Scalable analysis of bacterial
genome variation at the population level. BMC Bioinformatics 11, 595.

Data submitters:
currently >1300;
Data curators:
currently >90 MLST
schemes
Sequence
definitionsMLST, rMLST,
antigen genes, core
genome, pan-
genome
Gene A
Gene B
Gene C
Gene D
Allele1: TTTGATACTGTTGCCGAAGGTTT
Allele2: TTTGATACCGTTGCCGAAGGTTT
Allele3: TTTGATTCCGTTGCCGAAGGTTT
>750 citations
Isolate datasets
• provenance
• phenotype
• gene content
• allelic variation
• genomes
Linked to:
Population
annotation
• locus classification
• description
• biochemical
pathway
• Core + accessory
genome analysis
• Association studies
Comparative
genomics
PubMLST
1998*, 2003
Gene-by-gene
analysis using
reference genome or
defined loci
Molecular typing
Species identification
Epidemiology
Vaccine coverage/
impact
Linking genotype
to phenotype
Outbreak investigation
Population structure
>8000 unique visitors/month*http://mlst.zoo.ox.ac.uk

PubMLST RESTful API facilitates data exchange
• All data accessible
via JSON API
• Authenticated
(OAuth) access to
protected resources
• Data submission
available soon
http://rest.pubmlst.org
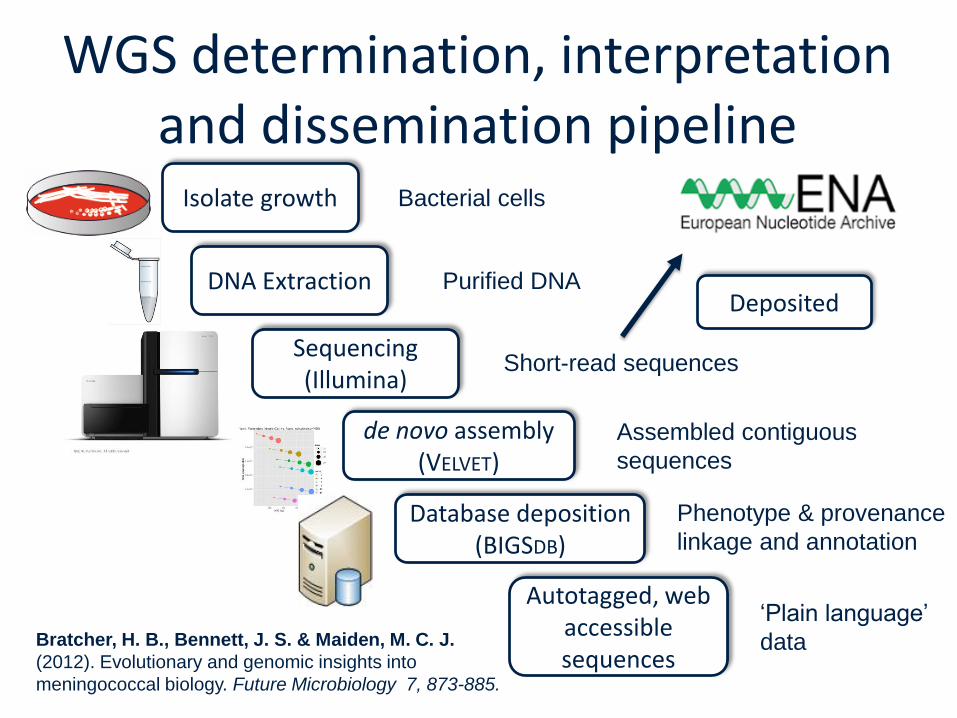
WGS determination, interpretation and dissemination pipeline
Isolate growth
DNA Extraction
Sequencing (Illumina)
de novo assembly(VELVET)
Database deposition (BIGSDB)
Autotagged, web accessible sequences
Bacterial cells
Purified DNA
Short-read sequences
Assembled contiguous
sequences
Phenotype & provenance
linkage and annotation
‘Plain language’
data Bratcher, H. B., Bennett, J. S. & Maiden, M. C. J.
(2012). Evolutionary and genomic insights into
meningococcal biology. Future Microbiology 7, 873-885.
Deposited

MLST
(7 loci)
16S rRNA
sequences
(1 locus)
Ribosomal MLST
(53 loci)
Strain
Lineage/
Clonal Complex
Species
Family
Order
Class
Phylum
Genus
Whole genome
MLST
(>500 loci)- Core genome
MLST
- Accessory
genome MLST
Hierarchical genome analysis
Clone
Meroclone
Maiden Maiden, M. C., van Rensburg, M. J., Bray, J. E., Earle, S. G., Ford, S. A., Jolley, K. A. & McCarthy, N. D. M.C.J. et al. 2013. MLST revisited: the gene-by-gene approach to bacterial genomics. Nat Rev Microbiol. 2013 Sep 2. doi: 10.1038/nrmicro3093.PMCID: PMC3980634

Neisseria structure and characterisation
Jolley, K. A., Brehony, C. & Maiden, M. C. (2007). Molecular typing of
meningococci: recommendations for target choice and nomenclature. FEMS
Microbiol Rev 31, 89-96.
Component Phenotypic Genotypic
Capsule Serogroup cps region
OMPS Serotype,Subtype, etc.
porA, porB, fetA, etc.
Housekeepinggenes
MLEE MLST
Ribosomes MALDITOF 16s rRNA,rMLST
Neisseria meningitidis B: P1.7,16: F3-3: ST-32 (cc32)

Validation of WGS pipeline• 108 diverse meningococcal isolates,
sequenced with 54bp Illuminareads.
• Assembled with VELVET and uploaded into BIGSDB.
• Comparison of 24 typing loci (total of 2592 loci) previously characterised by Sanger sequencing in all isolates.
• There were 34 (1.3%) allelic differences found in 20 of the de novo assembled genomes.
• 30 discrepancies (1.15%) attributable to Sanger sequence errors (mislabelling, editing errors).
• 4 discrepancies (0.15%) attributable to Velvet assembly. These were all in the same porA allele (a repeat sequence).
Bratcher, H. B., Corton, C., Jolley, K. A., Parkhill, J. & Maiden, M. C. (2014). A gene-by-
gene population genomics platform: de novo assembly, annotation and genealogical analysis of
108 representative Neisseria meningitidis genomes. BMC Genomics 15, 1138.

Genome and phenotype
• Whole genome MLST (wgMLST).
• Autotagger – runs regularly – tags all loci with known alleles (>2200 in Neisseria database.
• Each unique sequence given new allele number.
• Loci grouped into schemes.
• Linkage to phenotype & other information.
Jolley, K. A. & Maiden, M. C. (2013). Automated extraction of typing information for bacterial pathogens
from whole genome sequence data: Neisseria meningitidis as an exemplar. Euro Surveill 18 (4): 20379.

Meningitis Research Foundation Meningococcus Genome Library
• Charity funded.
• Open access
• All available England and Wales (& soon Scotland) meningococcal isolates.
• Assembled & annotated contiguous sequence data.
http://www.meningitis.org/current-projects/genome

Isolates in the MRF Genome Library –England and Wales
0
100
200
300
400
500
600
Z
Y
X
W/Y
W
NG
E
C
B
A

National Surveillance: MRF-MGL 2010-2012
Hill, D.M.C., Lucidarme, J., Gray S.J., Newbold , L.S., Ure, R., Brehony, C., Harrison,
O.B., Bray, J.E., Jolley, K.A., Bratcher H.B.,, Parkhill, J., Tang, C.M., Borrow, R., and
Maiden, M.C.J. Genomic epidemiology of age-associated meningococcal lineages in national
surveillance: an observational cohort study. Lancet Infectious diseases, DOI:
http://dx.doi.org/10.1016/S1473-3099(15)00267-4
.
• A total of 923 isolates from England, Wales and Northern Ireland.
• 899 from England and Wales:
• Scanned at >2000 loci;
• 2-313 alleles/locus;
• 219 STs, 22 clonalcomplexes;
• 496 rSTs (ribosomal sequence types);
• Most isolates (78%) belonged to 6 clonalcomplexes.

0
500
1000
1500
2000
2500
3000
1975 ~ 1985 ~ 1995 ~ 1999 2000 2001 ~ 2005 2006 2007 2008 2009 2010 2011 2012
41/44 269 11 32 8 213 23 167 174 22 Other UA NT
Retrospective epidemiology
Hill, D.M.C., Lucidarme, J., Gray S.J., Newbold , L.S., Ure, R., Brehony, C., Harrison,
O.B., Bray, J.E., Jolley, K.A., Bratcher H.B.,, Parkhill, J., Tang, C.M., Borrow, R., and
Maiden, M.C.J. Genomic epidemiology of age-associated meningococcal lineages in national
surveillance: an observational cohort study. Lancet Infectious diseases, DOI:
http://dx.doi.org/10.1016/S1473-3099(15)00267-4
.

Outbreak investigation
Mulhall, RM, Brehony, C, O’Connor, L, Bennett, D, Jolley, KA, Bray, J, Maiden, MCJ,
Cunney, R. Resolution of a protracted serogroup B meningococcal outbreak in a large extended
indigenous Irish Traveller Family in the Republic of Ireland during 2010 to 2013 using non-culture
PCR, WGS and publically accessible web-based tools. In preparation.

High resolution international epidemiology (W:cc11)
0
10
20
30
40
50
60
70
2005 2006 2007 2008 2009 2010 2011 2012 2013
n
year
W:cc11 England and Wales 2005 to 2013
Current UK
UK Hajj
UK1996 (n=3)1997 (n=2)1998 (n=2)
UK1975 (n=6)1987 (n=1)1989 (n=1)1990 (n=1)
UK1996 (n=2)1998 (n=1)
Argentina 2008-2012
Brazil 2008-2011
Current South Africa
Lucidarme, J., Hill, D.M., Bratcher, H.B., Gray S.J, du Plessis, M., Tsang, R.S.W., Vazquez, J.A., Taha, M.-K., Mehmet Ceyhan, Jamie Findlow J., Jolley, K.A., Maiden M.C.J., Borrow, R. (2015) Journal of Infection
0
10
20
30
40
50
60
70
80
90
2002 2003 2004 2005 2006 2007 2008 2009 2010 2011 2012
Nu
mb
er o
f C
ase
s
Year
N. meningitidis cases per year among inpatients in Bamako, Mali (2002-2012)
Group Ameningococcalcases
Group W135meningococcalcases

Protein vaccine antigens
0
10
20
30
40
50
60
70
80
90
100
0
100
200
300
400
500
600
700
800
900
1000
1100
1200
1300
all g
en
ogr
ou
ps
gen
ogr
ou
p B
on
ly
all g
en
ogr
ou
ps
gen
ogr
ou
p B
on
ly
all g
en
ogr
ou
ps
gen
ogr
ou
p B
on
ly
all g
en
ogr
ou
ps
gen
ogr
ou
p B
on
ly
all g
en
ogr
ou
ps
gen
ogr
ou
p B
on
ly
all g
en
ogr
ou
ps
gen
ogr
ou
p B
on
ly
Bexsero® MenBvac® MeNZB™ NonaMen rLP2086 VA-MENGOC-BC®
pe
rce
nta
ge
fre
qu
en
cy
Other
ST-60cc
ST-162cc
ST-11cc
ST-213cc
ST-32cc
ST-23cc
ST-269cc
ST-41/44cc


invasive isolate survey: proof of concept for WGS based surveillance
Epidemiological year 2011/2012
Dominique Caugant, Holly Bratcher, Carina Brehony, Martin Maiden, IBD-LabNet

799 representative IMD cases,15 countries
0
10
20
30
40
50
60
70
80
90
100
110
120
130
nu
mb
er
of
iso
late
s se
qu
en
ced
2011/12 2011 2012

Serogroup by country
0
25
50
75
100
125
150
175
200
225
250
nu
mb
er
of
iso
late
s
NovalueNG
Y
W
W/Y
X
E
C
B
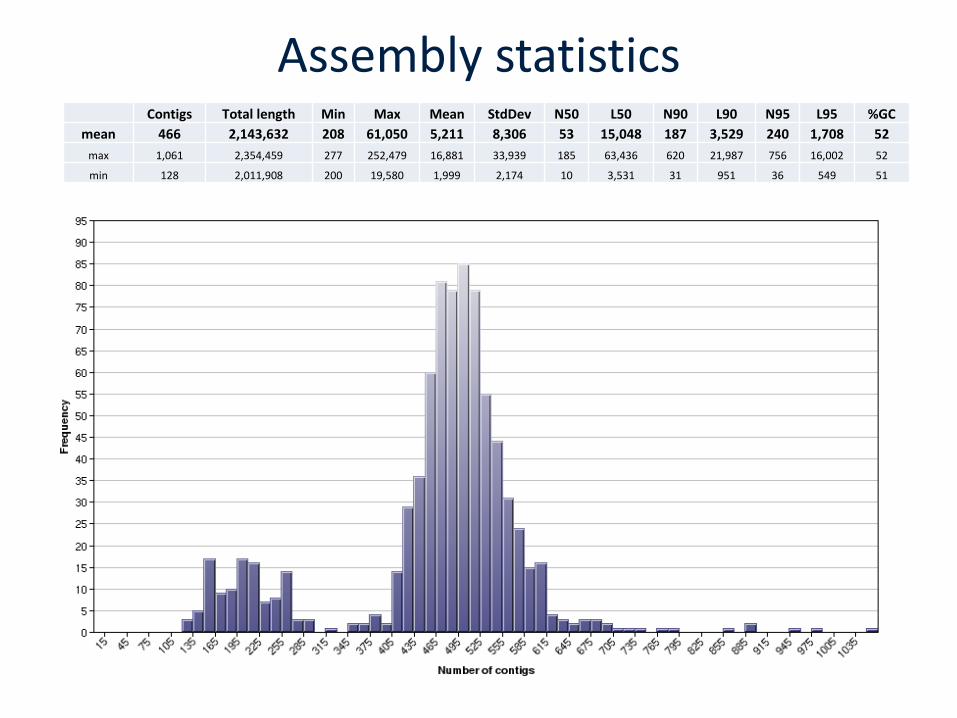
Assembly statisticsContigs Total length Min Max Mean StdDev N50 L50 N90 L90 N95 L95 %GC
mean 466 2,143,632 208 61,050 5,211 8,306 53 15,048 187 3,529 240 1,708 52
max 1,061 2,354,459 277 252,479 16,881 33,939 185 63,436 620 21,987 756 16,002 52
min 128 2,011,908 200 19,580 1,999 2,174 10 3,531 31 951 36 549 51

Surveillance data coverage: 7 MLST loci
795 assigned MLST profiles
0
50
100
150
200
250
300
350
400
450
500
550
600
650
700
750
800
100 99.0-99.9 ≤99.9
nu
mb
er
of
iso
late
s
percent loci defined
4 missing ST profiles (0.5%)165-270 contigs / genome7 loci identified / isolate6 loci assigned / isolate

0102030405060708090
100110120130140150160170180190200210220230240250 unassigned
ST-92
ST-8
ST-750
ST-53
ST-226
ST-198
ST-116
ST-1117
ST-1
ST-364
ST-334
ST-254
ST-1157
ST-865
ST-461
ST-174
ST-162
ST-167
ST-35
ST-60
ST-22
ST-18
ST-103
ST-213
ST-23
ST-269
ST-11
ST-32
ST-41/44
Clonal complexes by country

Surveillance data coverage: PorA & FetA
0
50
100
150
200
250
300
350
400
450
500
550
600
650
700
750
800
100 66.7 33.3
nu
mb
er
of
iso
late
s
percent loci assigned (n=3)
21 partial antigen profiles (2.6%)216-677 contigs / genome1-2 loci assigned / isolate
14 no PorA VR1 allele12 no PorA VR2 allele
6 no FetA VR allele

Surveillance data coverage: 5 BAST loci
0
50
100
150
200
250
300
350
400
450
500
550
600
650
100 80-90 60-70 40-50
nu
mb
er
of
iso
late
s
percent loci defined
Over all 597 with partial profile (74.8%)
14 no PorA VR1 allele12 no PorA VR2 allele
130 no NadA peptide allele*3 no fHbp peptide allele
19 no NHBA peptide allele
44 only 2-3 loci identified (5.5%)average 495 contigs / genome
3 no PorA VR1 allele11 no PorA VR1, VR2 alleles
3 no fHbp/NadA peptide alleles19 no NHBA/NadA peptide alleles

Top 37 BAST profiles
0
1
2
3
4
5
6
7
8
9
10
11
No
val
ue 4
22
3 3
28
8
34
9
84
22
8 8
23
7
22
2
10
71
25
7
94
26
7
10
14
10
72 5
38
22
7
57
8
96
2
13
13
10
22
0
10
15
10
16
13
57
21
9
22
1
24
7
24
8
38
4
89
8
10
74
11
04
11
73
12
26
12
71
pe
rce
nt
of
iso
late
s
BAST profile (present in at least 3 isolates)
2011/12
2011
2012
BAST vaccine profile 1
fHbp_peptide: 1 | NHBA_peptide: 2 | NadA_peptide: 8 | PorA_VR1: 7-2 |
PorA_VR2: 4

Ribosomal (rMLST) data coverage798 assigned rMLST profiles
0
50
100
150
200
250
300
350
400
450
500
550
600
650
700
750
800
100 95.0-99.9 ≤99.9
nu
mb
er
of
iso
late
s
percent loci defined (n=51)
1 missing rST profile
(0.1%)
1061 contigs
50 loci identified
52 loci assigned

0
25
50
75
100
125
150
175
200
225
250
100 99.0-99.9 98.0-98.9 97.0-97.9 96.0-96.9 95.0-95.9 90.0-94.9 ≤89.9
nu
mb
er
of
gen
om
es
percent of cgMLST tagged (n=1605)
Core genome (cgMLST) locus coverage
177 (22.2%)
genomes

0
25
50
75
100
125
150
100 99.0-99.9 98.0-98.9 97.0-97.9 96.0-96.9 95.0-95.9 90.0-94.9 ≤89.9
nu
mb
er
of
iso
late
s
percent of tagged loci (n=1605)
(n=3)
cgMLST coverage: MRF-MGL 2013/2014

Scalable genomic epidemiology
Centuries+ decades years months weeks days hours
Evolution emergence epidemiology diagnosis
COLOMBIA 2004
(n=37)
Y
32%
B
51%
W-135
3%
C
14%
AFRICAN
MENINGITIS BELT
2003-2004
(n=501)
Other
1,2%
A
79%
W-135
20%
AUSTRALIA 2004
(n=361)
Other
7,2%
C
20%
A
0,3%
B
68%
W-135
3,3%
Y
2,2%
WESTERN
EUROPE 2002
(n=3,982)
A
0,1%
C
29%
Other
1,0%
B
64%
W-135
3,6%
Y
2,3%
RUSSIA 2002-2004
(n=1,899)
B
32%
A
36%C
22%
Other
10%
CHILE 2003
(n=193)
Other
5%
C
14%B
78%
W-135
1%
Y
2%
CANADA 2003*
(n=148)
W-135
7%
C
24%
B
43%
Other
1%
Y
25%
UNITED STATES 2003
(n=200)
Y
27%
C
21% B
44%
Other
6%W-135
2%
TAIWAN 2001
(n=43)
Y
19%
A
4,7%
W-135
41%
B
33%
C
2,3%
THAILAND 2001
(n=36)
Other
2%
B
81%
W-135
17%
SAUDI ARABIA
2002
(n=21)
B
10%
W-135
76%
A
14%
BRAZIL 2004
Sao Paulo state
(n=520)
B
36%
C
58%
Other
6%
NEW ZEALAND 2004
(n=252)
C
8%
Other
0,8%
B
87%
W-135
3,6%
Y
0,4%
SOUTH AFRICA 2003
(n=264)
Other
1%W-135
9%
B
29%
A
34%
C
11%
Y
16%
URUGUAY 2001
(n=53)
C
11%
B
83%
Other
6%
COLOMBIA 2004
(n=37)
Y
32%
B
51%
W-135
3%
C
14%
AFRICAN
MENINGITIS BELT
2003-2004
(n=501)
Other
1,2%
A
79%
W-135
20%
AUSTRALIA 2004
(n=361)
Other
7,2%
C
20%
A
0,3%
B
68%
W-135
3,3%
Y
2,2%
WESTERN
EUROPE 2002
(n=3,982)
A
0,1%
C
29%
Other
1,0%
B
64%
W-135
3,6%
Y
2,3%
RUSSIA 2002-2004
(n=1,899)
B
32%
A
36%C
22%
Other
10%
CHILE 2003
(n=193)
Other
5%
C
14%B
78%
W-135
1%
Y
2%
CANADA 2003*
(n=148)
W-135
7%
C
24%
B
43%
Other
1%
Y
25%
UNITED STATES 2003
(n=200)
Y
27%
C
21% B
44%
Other
6%W-135
2%
TAIWAN 2001
(n=43)
Y
19%
A
4,7%
W-135
41%
B
33%
C
2,3%
THAILAND 2001
(n=36)
Other
2%
B
81%
W-135
17%
SAUDI ARABIA
2002
(n=21)
B
10%
W-135
76%
A
14%
BRAZIL 2004
Sao Paulo state
(n=520)
B
36%
C
58%
Other
6%
NEW ZEALAND 2004
(n=252)
C
8%
Other
0,8%
B
87%
W-135
3,6%
Y
0,4%
SOUTH AFRICA 2003
(n=264)
Other
1%W-135
9%
B
29%
A
34%
C
11%
Y
16%
URUGUAY 2001
(n=53)
C
11%
B
83%
Other
6%
0.1
UK 1993
Case 1
Carrier 1
FAM18
USA
1983
Carrier 2
Carrier 3
Cases 3 & 6
Remote
cases 1 & 2
Carrier 4
Carrier 5


Contigs Total length Min Max Mean StdDev N50 L50 N90 L90 N95 L95 %GC
mean 306 2,133,479 209 88,174 7,847 12,456 33 22,789 117 5,194 151 2,688 52
max 612 2,278,600 273 258,183 19,478 32,854 80 64,227 289 16,887 370 9,336 52
min 109 2,026,649 200 30,309 3,499 4,877 12 7,569 35 1,670 44 942 51
MRF 2013/2014 assembly statistics

0
25
50
75
100
125
150
175
200
225
250
275
100 99.0-99.9 98.0-98.9 97.0-97.9 96.0-96.9 95.0-95.9 90.0-94.9 ≤89.9
nu
mb
ero
f is
ola
tes
percent of cgMLST tagged (n=1605)
Core genome data coverage: MRF 2014/2015
188 (24.7%) genomes

MRF 2014/2015 assembly statistics
H Bratcher, C Brehony, M Maiden, D Caugant . IDB-LabNet 2015
mean 323 2,126,265 202 80,153 6,900 10,841 35 19,614 126 4,366 162 2,215
max 516 2,278,600 273 219,677 18,381 28,710 67 58,245 236 15,605 305 9,336
min 116 2,037,538 200 34,143 4,184 5,990 13 8,869 43 1,992 53 1,065
Contigs Total length Min Max Mean StdDev N50 L50 N90 L90 N95 L95

Age association of meningococcal genotypes (MRF-MGL 2010-2012)
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
<1 1-4 5-9 10-14 15-19 20-24 25-29 30-39 40-49 50-69 >70
Pro
po
rtio
n o
f ca
ses
Age category
Minor clonal complexes
ND
ST-174 complex
ST-461 complex
ST-162 complex
ST-22 complex
ST-23 complex/Cluster A3
ST-213 complex
ST-60 complex
ST-41/44 complex/Lineage 3
ST-269 complex
ST-32 complex/ET-5 complex
ST-11 complex/ET-37 complex
Hill, D.M.C., Lucidarme, J., Gray S.J., Newbold , L.S., Ure, R., Brehony, C., Harrison,
O.B., Bray, J.E., Jolley, K.A., Bratcher H.B.,, Parkhill, J., Tang, C.M., Borrow, R., and
Maiden, M.C.J. Genomic epidemiology of age-associated meningococcal lineages in national
surveillance: an observational cohort study. Lancet Infectious diseases, DOI:
http://dx.doi.org/10.1016/S1473-3099(15)00267-4
.

Population annotation
Harrison, O.B., Bray, J.A., Maiden, M.C., and Caugant, D.A. (2015) Genomic Analysis of the Evolution and Global Spread of Hyper-invasive Meningococcal Lineage 5. Ebiomedicine, 2(3), 234-243doi:10.1016/j.ebiom.2015.01.004.

Validation against four reference genomes
Isolate Loci present in draft genome
Identical loci Discrepantloci
Incomplete loci
Discrepantbases in annotated loci
Z2491 1872/1867 (99.8%)
1801 (96.2%) 19 (1%) 51 (2.7%) 32 (0.002%)
FAM18 1905/1914 (93.2%)
1775 (93.2%) 23 (1.2%) 107 (5.6%) 24 (0.001%)
G2136* 1897/1904 (99.6%)
1757 (92.6%) 47 (2.5%) 93 (4.9%) 90 (0.005%)
H44/76* 1967/1975 (99.2%)
1821 (92.6%) 49 (2.55) 97 (4.9%) 76 (0.004%)
Draft genomes generated by VELVET assembly of Illumina reads and deposited
in BIGSDB without further curation.
Annotations compared with GENOMECOMPARATOR.
* Finished genomes primarily generated with Roche 454 technology.

Phenotypic serogroup by country
0
25
50
75
100
125
150
175
200
225
250
nu
mb
er
of
iso
late
s
No value
NG
Y
W
W/Y
X
E
C
B
A
H Bratcher, C Brehony, M Maiden, D Caugant . IDB-LabNet 2015

Indexing the genome: Neiss loci
gene 122540..122974
/gene="rplK"
/locus_tag="NMC0119"
/db_xref="GeneID:4676186"
CDS 122540..122974
/gene="rplK"
/locus_tag="NMC0119"
/note="binds directly to 23S ribosomal RNA"
/codon_start=1
/transl_table=11
/product="50S ribosomal protein L11"
/protein_id="YP_974250.1"
/db_xref="GI:121634005"
/db_xref="GeneID:4676186"
/translation="MAKKIIGYIKLQIPAGKANPSPPVGPA
LGQRGLNIMEFCKAFNAATQGMEPGLPIPVVITAF
ADKSFTFVMKTPPASILLKKAAGLQKGSSNPLTNK
VGKLTRAQLEEIAKTKEPDLTAADLDAAVRTIAGS
ARSMGLDVEGVV“
Database: RefSeq
Entry: NC_008767
LinkDB: NC_008767
LOCUS NC_008767 2194961 bp DNA circular CON 10-
JUN-2013
DEFINITION Neisseria meningitidis FAM18 chromosome, complete
genome.
pubMLST.org/Neisseria
Sequence definition database
“LOCUS TAG IDENTIFIER”
NMC0119 (FAM18)
NMA0146 (020-06)
NGO1855 (FA 1090)
LOCUS “ALIASES” for
‘seed
sequences
’

Bacterial Isolate Genome Sequence Database (BIGSDB)
CCATCCCGTTGTCGAACAGCAGGTACGCCA
CTTCACCGCCAACCACACCGACCTTGACCAC
AAACACCGCCTCATGCTGCTCACCGGCCCC
AATATGGGCGGCAAATCCACCTACATGCGCAGGAACCCTCAAAGCCGTTTTCCCGGAAAACC
TATCCACAGCCGAACAGCTCCGCCAAGCCA
TTTTGCCCGAACCTTCCGTCTGGCTGAAAGA
CGGCAATGTCATCAACCACGGTTTTCATCCC
GAACTGGACGAATTGCGCCGCATTCAAAACC
ATGGCGACGAATTTTTGCTGGATTTGGAAGC
CAAGGAACGCGAACGTACCGGTTTGTCCAC
ACTTAAAGTCGAGTTCAACCGCGTTCACGGC
TTTTACATTGAATTGTCCAAAACCCAAGCCG
AACAAGCACCTGCCGACTACCAACGCCGGC
AAACCCTTAAAAACGCCGAACGCTTCATCAC
GCCGGAACTGAAAGCCTTTGAAGACAAAGT
GCTGACTGCTCAAGAGCAAGCCCTCGCCTT
AGAAAAACAACTCTTTGACGGCGTATTGAAA
AACCTTCAGACGGCATTGCCGCAGCTTCAAA
AAGCCGCCAAAGCCGCCGCCGCGCTGGAC
GTGTTGTCCACATTTTCAGCCTTGGCAAAAG
AGCGGAACTTCGTCCGCCCCGAGTTTGCCG
ACAAGTCGCGCTGATTGTTT
AACCTTCAGACGGCATTGCCGCAGCTTCAAA
AAGCCGCCAAAGCCGCCGCCGCGCTGGAC
GTGTTGTCCACATTTTCAGCCTTGGCAAAAG
AGCGGAACTTCGTCCGCCCCGAGTTTGCCG
ACTATCCGGTTATCCACATCGAAAACGGCCG
CCATCCCGTTGTCGAACAGCAGGTACGCCA
CTTCACCGCCAACCACACCGACCTTGACCAC
AGGAACCCTCAAAGCCGTTTTCCCGGAAAAC
CTATCCACAGCCGAACAGCTCCGCCAAGCC
ATTTTGCCCGAACCTTCCGTCTGGCTGAAAG
ACGGCAATGTCATCAACCACGGTTTTCATCC
CGAACTGGACGAATTGCGCCGCATTCAAAAC
CATGGCGACGAATTTTTGCTGGATTTGGAAG
CCAAGGAACGCGAACGTACCGGTTTGTCCA
CACTTAAAGTCGAGTTCAACCGCGTTCACGG
CTTTTACATTGAATTGTCCAAAACCCAAGCC
GCCCCGAGTTTGCCGACTATCCGGTTATCCA
CATCGAAAACGGCCGCCATCCCGTTGTCGA
ACAGCAGGTACGCCACTTCACCGCCAACCA
CACCGACCTTGACCACAAACACCGCCTCATG
CTGCTCACCGGCCCCAATATGGGCGGCAAA
TCCACCTACATGCGCCAAGTCGCGCTGATTGTTT
AGGAACCCTCAAAGCCGTTTTCCCGGAAAAC
CTATCCACAGCCGAACAGCTCCGCCAAGCC
ATTTTGCCCGAACCTTCCGTCTGGCTGAAAGACGGCAATGTCATCAACCACGGTTTTCATCC
CGAACTGGACGAATTGCGCCGCATTCAAAAC
CATGGCGACGAATTTTTGCTGGATTTGGAAG
CCAAGGAACGCGAACGTACCGGTTTGTCCA
CACTTAAAGTCGAGTTCAACCGCGTTCACGGCTTTTACATTGAATTGTCCAAAACCCAAGCC
GAACAAGCACCTGCCGACTACCAACGCCGG
CAAACCCTTAAAAACGCCGAACGCTTCATCA
CGCCGGAACTGAAAGCCTTTGAAGACAAAGT
GCTGACTGCTCAAGAGCAAGCCCTCGCCTTAGAAAAACAACTCTTTGACGGCGTATTGAAA
AACCTTCAGACGGCATTGCCGCAGCTTCAAA
AAGCCGCCAAAGCCGCCGCCGCGCTGGAC
GTGTTGTCCACATTTTCAGCCTTGGCAAAAGAGCGGAACTTCGTCCGCCCCGAGTTTGCCG
ACTATCCGGTTATCCACATCGAAAACGGCCG
CCATCCCGTTGTCGAACAGCAGGTACGCCA
CTTCACCGCCAACCACACCGACCTTGACCAC
AAACACCGCCTCATGCTGCTCACCGGCCCCAATATGGGCGGCAAATCCACCTACATGCGC
CAAGTCGCGCTGATTGTTT
abcZ
adk
aroE
fumC
gdh
pdhC
pgm
porA
porB
fetA
penA
rpoB
16S
Locus X
Locus Y
Sequence
bin
Jolley, K. A. & Maiden, M. C. (2010). BIGSdb:
Scalable analysis of bacterial genome variation at
the population level. BMC Bioinformatics 11, 595.
Locus
definitions
tables:
annotation
source Locus Allele Provenance
abcZ 2 Country UK
adk 3 Year 2013
aroE 4 serogroup B
gdh 8 Disease carrier
pdhC 4 Age 23
pgm 6 Source Swab
... etc... ... etc ...

Acknowledgements
Julia BennettWT
Carly Bliss
Holly BratcherWT
James BrayWT
Carina BrehonyWT
Marianne Clemence
Ali Cody
Fran Colles
Kanny DialloWTF
Sarah Earle
Suzanne Ford
Odile HarrisonWT
Sofia Hauck
Dorothea Hill
Lisa Rebbets
Melissa Jansen van Rensburg
Keith JolleyWT
Jasna Kovac
Jenny MacLennanWT
Noel McCarthyWTF
Maddi Pearce
Samuel SheppardWTF
Helen Strain
Eleanor Watkins
Helen Wimalarathna

Population genomics: the gene-by-gene approach
Complete Sequence
Annotation
Bacterial Isolate Genome Sequence Database (BIGSDB)
Contigs
Gene sequencesProvenance/phenotype information
Jolley, K. A. & Maiden, M. C. (2010). BIGSdb: Scalable analysis of bacterial genome variation at
the population level. BMC Bioinformatics 11, 595.

Bacterial typing requirements
1. Universal, in that they are applicable to all bacteria.
2. Natural, reflecting genealogical relationships while retaining the capacity to describe closely related organisms with distinct properties.
3. Understandable, so that the output and the process by which the system has been arrived at are transparent, easily interpreted and reproducible, and where possible the system should be backwards compatible with previous approaches.
4. Expandable, to account for the incompleteness of our knowledge of diversity, and flexible enough to accommodate changes in this knowledge.

Bacterial typing requirements
5. Portable, because methods need to be easily carried out in any laboratory and the data need to be freely exchanged by the use of generic methodologies, reagents and bioinformatics pipelines
6. Technology independent, so that the data used are independent of the means of their collection (this means that schemes adopted now need to retain their validity as data improve)
7. Readily available to the entire community

Bacterial typing requirements
8. Scalable, so that methods are sufficiently fast and inexpensive to be useable in real time for large or small numbers of isolates (this scalability is especially important for clinical applications and large-scale bacterial population analyses)
9. Accommodate a wide range of variation so that they can encompass both close and distant genealogical relationships
10. Broadly accepted by those who use them and open to contributions from members of the community.

Bacterial typing methods
• Universal, in that they are applicable to all bacteria
• Natural, reflecting genealogical relationships while retaining the capacity to describe closely related organisms with distinct properties
• Understandable, so that the output and the process by which the system has been arrived at are transparent, easily interpreted and reproducible, and where possible the system should be backwards compatible with previous approaches
• Expandable, to account for the incompleteness of our knowledge of diversity, and flexible enough to accommodate changes in this knowledge
• Portable, because methods need to be easily carried out in any laboratory and the data need to be freely exchanged by the use of generic methodologies, reagents and bioinformatics pipelines
• Technology independent, so that the data used are independent of the means of their collection (this means that schemes adopted now need to retain their validity as data improve)
• Readily available to the entire community
• Scalable, so that methods are sufficiently fast and inexpensive to be useable in real time for large or small numbers of isolates (this scalability is especially important for clinical applications and large-scale bacterial population analyses)
• Able to accommodate a wide range of variation so that they can encompass both close and distant genealogical relationships
• Broadly accepted by those who use them and open to contributions from members of the community.

cnl meningococci & other species
Claus, H., Maiden, M. C., Maag, R., Frosch, M. & Vogel, U. (2002). Many carried meningococci lack the genes required for capsule synthesis and transport. Microbiology 148, 1813-1819.Harrison, O. B., Claus, H., Jiang, Y., Bennett, J. S., Bratcher, H. B., Jolley, K. A., Corton, C., Care, R., Poolman, J. T., Zollinger, W. D., Frasch, C. E., Stephens, D. S., Feavers, I., Frosch, M., Parkhill, J., Vogel, U., Quail, M. A., Bentley, S. D. & Maiden, M. C. J. (2013). Description and Nomenclature of Neisseria meningitidis Capsule Locus. Emerging Infectious Diseases 19, 566-573.

First generation genomics:single locus typing and MLST
aroE
gdh
pgm
adkpdhC
fumC
porA
fetA
abcZ
Maiden, MCJ, Bygraves, JA, Feil, E, Morelli, G, Russell, JE, Urwin, R, Zhang, Q, Zhou, J, Zurth, K,
Caugant, DA, Feavers, IM, Achtman, M & Spratt, BG. 1998. Multilocus sequence typing: a portable
approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad
Sci USA 95, 3140-3145.
Maiden, MC. 2006. Multilocus Sequence Typing of Bacteria. Annu Rev Microbiol 60, 561-588.
Jolley KA, Brehony C, Maiden MC. 2007. Molecular typing of meningococci: recommendations for target
choice and nomenclature. FEMS Microbiol Rev 31, 89-96.
• Neisseria seven-locus ST summarises 3284bp.
• That is 0.15% of the 2.18Mbp genome.
• 11,001 STs in PubMLSTdatabase (September 2014).
• 469-750 alleles per locus.
• Many polymorphisms per locus.

GENOMECOMPARATOR: gene-by-gene analysis

GENOMECOMPARATOR: rapid comparative genomics
Jolley, K. A., Hill, D. M., Bratcher, H. B., Harrison, O. B., Feavers, I. M., Parkhill, J. &
Maiden, M. C. (2012). Resolution of a meningococcal disease outbreak from whole genome
sequence data with rapid web-based analysis methods. J Clin Microbiol. 50(9):3046-53.
SPLITSTREE 4.0
NEIGHBORNET

Ribosomal multi-locus sequence typing, rMLST
Jolley, K. A., Bliss, C. M., Bennett, J. S., Bratcher, H. B., Brehony, C. M., Colles, F. M., Wimalarathna, H. M., Harrison, O. B., Sheppard, S. K., Cody, A. J. & Maiden, M. C. (2012). Ribosomal Multi-Locus Sequence Typing: universal characterisation of bacteria from domain to strain. Microbiology 158, 1005-1015.
• Isolate characterisation from ‘domain to strain.
• Indexes the 53 ribosomal genes.• PubMLST.org/rMLST, provides a look-up table
available on the web.• Ribosomal sequence types, rSTs related to
appropriate nomenclatures, October 2014:• 99,996 genome sequences;• 977 genera;• 2,531 unique species ;• rSTs defined for 6 groups, Neisseria and
Campylobacter to clonal complex level.

Meningococcal genealogies: cgMLST, rMLST, & MLST
Bratcher, H.B., Maiden, M.C. Unpublished.

Lineage 5: 40 years of global disease and reverse vaccinology
1,886 (95%) core loci
52 (3%) accessory
Harrison, O. B., Bray, J. E., Maiden, M. C. J. & Caugant, D. A. Genomic Analysis of the Evolution and
Global Spread of Hyper-invasive Meningococcal Lineage 5. EBioMedicine.
Harrison, O.B., Hill, D.M., Maiden, M.C.J. unpublished.

Variability across the lineage 5 (ST-32 complex) genome
229 loci identical
1,600 loci p-distance values below
0.002
Harrison, O.B., Bray, J.A., Maiden, M.C., and Caugant, D.A. (2015)
Genomic Analysis of the Evolution and Global Spread of Hyper-
invasive Meningococcal Lineage 5. Ebiomedicine, 2(3), 234-243
doi:10.1016/j.ebiom.2015.01.004.

Meningitis Research Foundation Meningococcus Genome Library
• Charity funded.
• Open access
• All available England and Wales (& soon Scotland) meningococcal isolates.
• Assembled & annotated contiguous sequence data.
http://www.meningitis.org/current-projects/genome

MRF-MGL isolates 2010-2012• A total of 923 isolates from
England, Wales and Northern Ireland.
• 899 from England and Wales:
• Scanned at >1600 loci;
• 2-313 alleles/locus;
• 219 STs, 22 clonalcomplexes;
• 496 rSTs (ribosomal sequence types);
• Most isolates (78%) belonged to 6 clonalcomplexes.
ST-41/44 complex
237 isolates
ST-269 complex
171 isolatesST-11 complex, 59 isolates
ST-213 complex
75 isolates
ST-23 complex
120 isolates
ST-32 complex
42 isolates

0
500
1000
1500
2000
2500
3000
1975 ~ 1985 ~ 1995 ~ 1999 2000 2001 ~ 2005 2006 2007 2008 2009 2010 2011 2012
41/44 269 11 32 8 213 23 167 174 22 Other UA NT
Meningococcal clonal complexes and disease: England and Wales
Hill, D.M.C., Lucidarme, J., Gray S.J., Newbold , L.S., Ure, R., Brehony, C., Harrison,
O.B., Bray, J.E., Jolley, K.A., Bratcher H.B.,, Parkhill, J., Tang, C.M.,, Borrow, R., and
Maiden, M.C.J. Genomic epidemiology of age-associated meningococcal lineages in national
surveillance: an observational cohort study. Submitted.

2015 vaccine introductions
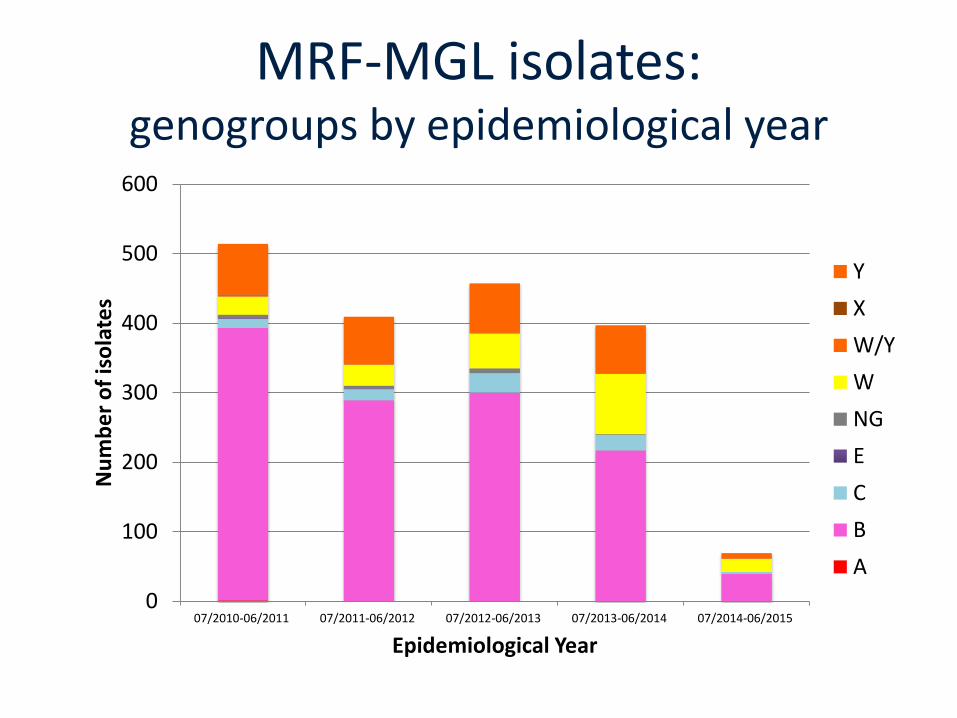
MRF-MGL isolates:genogroups by epidemiological year
0
100
200
300
400
500
600
07/2010-06/2011 07/2011-06/2012 07/2012-06/2013 07/2013-06/2014 07/2014-06/2015
Nu
mb
er
of
iso
late
s
Epidemiological Year
Y
X
W/Y
W
NG
E
C
B
A
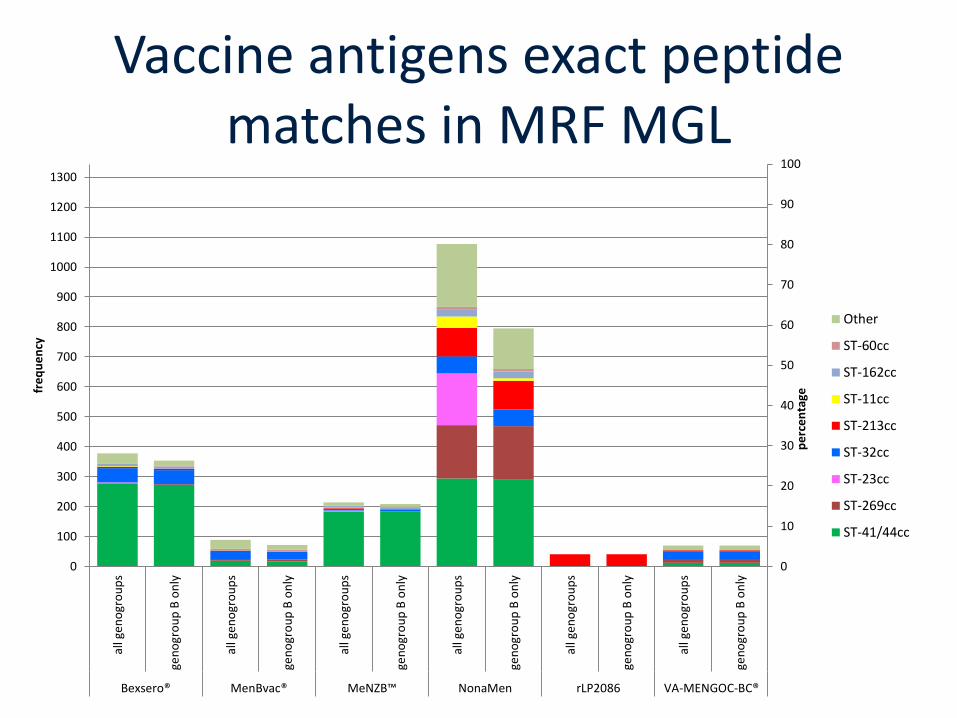
Vaccine antigens exact peptide matches in MRF MGL
0
10
20
30
40
50
60
70
80
90
100
0
100
200
300
400
500
600
700
800
900
1000
1100
1200
1300
all g
en
ogr
ou
ps
gen
ogr
ou
p B
on
ly
all g
en
ogr
ou
ps
gen
ogr
ou
p B
on
ly
all g
en
ogr
ou
ps
gen
ogr
ou
p B
on
ly
all g
en
ogr
ou
ps
gen
ogr
ou
p B
on
ly
all g
en
ogr
ou
ps
gen
ogr
ou
p B
on
ly
all g
en
ogr
ou
ps
gen
ogr
ou
p B
on
ly
Bexsero® MenBvac® MeNZB™ NonaMen rLP2086 VA-MENGOC-BC®
pe
rce
nta
ge
fre
qu
en
cy
Other
ST-60cc
ST-162cc
ST-11cc
ST-213cc
ST-32cc
ST-23cc
ST-269cc
ST-41/44cc

0
5
10
15
20
25
<1 1 2 3
4-6
7-9
10
-12
13
-15
16
-18
19
-21
22
-24
25
-27
28
-30
31
-33
34
-36
37
-39
40
-43
44
-46
47
-49
50
-52
53
-55
56
-58
59
-61
62
-64
65
-67
68
-70
71
-73
74
-76
77
-79
80
-82
83
-85
86
-88
89
-91
92
-94
95
-97
>9
7
NK
Pro
po
rtio
n o
f IM
D C
ase
s Ep
ide
mio
logi
cal
Ye
ar (
%)
Patient Age (Years)
2010/11
2011/12
0
1
2
3
4
5
6
7
8
9
<1 1-3 4-6 7-9 10-11P
rop
ort
ion
of
Cas
es
Epid
em
iolo
gica
l Ye
ar (
%)
Patient Age (Months)
Age distribution of isolates in meningococcal genome library

Contiguous sequences (contigs.)
Data sources
First generation ‘Next generation’
Archival
Short-read
sequence
data
DNA
Sequence on
preferred platform
(e.g. Illumina)
Bacteria
l isolate
Complete, assembled closed
genomes with annotation, available
from public databases (e.g. IMGD)
Clinical
specimen
TGGAGCAGATCGAGGAGAGCGAGTTCGACGC
TGGAGCAGATCGAGGAGAGCGAGTTCGACGC
TGGAGCAGATCGAGGAGAGCGAGTTCGACGC
TGGAGCAGATCGAGGAGAGCGAGTTCGACGC
TGGAGCAGATCGAGGAGAGCGAGTTCGACGC
TGGAGCAGATCGAGGAGAGCGAGTTCGACGC
TGGAGCAGATCGAGGAGAGCGAGTTCGACGC
TGGAGCAGATCGAGGAGAGCGAGTTCGACGCTGGAGCAGATCGAGGAGAGCGAGTTCGACGC
Assemble with
preferred software
(e.g. VELVET)

wgMLST ST-32 complex isolates
2063 CDS, 1,894 present in all isolates
Harrison, O.B. Maiden M.C., Caugant, D.A. Unpublished,

Rapid automated genome assembly
506 IsolatesIllumina Genome Analyzer GAIIxRead Lengths: 100 NucleotidesAverage Input FASTQ Filesize: 586MB
(258 million nucleotides)Average Number of Reads: 2.58 millionK-mer Range: 21-99
Median Final K-mer: 81Median N50: 37,503Average Number of Contigs: 209Average Program Time: 22 mins 31 secsTotal Program Time: 58 hours
Filesize (MB)
Pro
gram
Tim
e (h
h:m
m:s
s) Total AutoAssembler.pl Program Time Using 10 Threads Per Assembly
James Bray, unpublished

BIGSDB automated annotation
MLST definitions CCATCCCGTTGTCGAACAGCAGGTACGCCA
CTTCACCGCCAACCACACCGACCTTGACCAC
AAACACCGCCTCATGCTGCTCACCGGCCCC
AATATGGGCGGCAAATCCACCTACATGCGCAGGAACCCTCAAAGCCGTTTTCCCGGAAAACC
TATCCACAGCCGAACAGCTCCGCCAAGCCA
TTTTGCCCGAACCTTCCGTCTGGCTGAAAGA
CGGCAATGTCATCAACCACGGTTTTCATCCC
GAACTGGACGAATTGCGCCGCATTCAAAACC
ATGGCGACGAATTTTTGCTGGATTTGGAAGC
CAAGGAACGCGAACGTACCGGTTTGTCCAC
ACTTAAAGTCGAGTTCAACCGCGTTCACGGC
TTTTACATTGAATTGTCCAAAACCCAAGCCG
AACAAGCACCTGCCGACTACCAACGCCGGC
AAACCCTTAAAAACGCCGAACGCTTCATCAC
GCCGGAACTGAAAGCCTTTGAAGACAAAGT
GCTGACTGCTCAAGAGCAAGCCCTCGCCTT
AGAAAAACAACTCTTTGACGGCGTATTGAAA
AACCTTCAGACGGCATTGCCGCAGCTTCAAA
AAGCCGCCAAAGCCGCCGCCGCGCTGGAC
GTGTTGTCCACATTTTCAGCCTTGGCAAAAG
AGCGGAACTTCGTCCGCCCCGAGTTTGCCG
ACAAGTCGCGCTGATTGTTT
AACCTTCAGACGGCATTGCCGCAGCTTCAAA
AAGCCGCCAAAGCCGCCGCCGCGCTGGAC
GTGTTGTCCACATTTTCAGCCTTGGCAAAAG
AGCGGAACTTCGTCCGCCCCGAGTTTGCCG
ACTATCCGGTTATCCACATCGAAAACGGCCG
CCATCCCGTTGTCGAACAGCAGGTACGCCA
CTTCACCGCCAACCACACCGACCTTGACCAC
AGGAACCCTCAAAGCCGTTTTCCCGGAAAAC
CTATCCACAGCCGAACAGCTCCGCCAAGCC
ATTTTGCCCGAACCTTCCGTCTGGCTGAAAG
ACGGCAATGTCATCAACCACGGTTTTCATCC
CGAACTGGACGAATTGCGCCGCATTCAAAAC
CATGGCGACGAATTTTTGCTGGATTTGGAAG
CCAAGGAACGCGAACGTACCGGTTTGTCCA
CACTTAAAGTCGAGTTCAACCGCGTTCACGG
CTTTTACATTGAATTGTCCAAAACCCAAGCC
GCCCCGAGTTTGCCGACTATCCGGTTATCCA
CATCGAAAACGGCCGCCATCCCGTTGTCGA
ACAGCAGGTACGCCACTTCACCGCCAACCA
CACCGACCTTGACCACAAACACCGCCTCATG
CTGCTCACCGGCCCCAATATGGGCGGCAAA
TCCACCTACATGCGCCAAGTCGCGCTGATTGTTT
AGGAACCCTCAAAGCCGTTTTCCCGGAAAAC
CTATCCACAGCCGAACAGCTCCGCCAAGCC
ATTTTGCCCGAACCTTCCGTCTGGCTGAAAGACGGCAATGTCATCAACCACGGTTTTCATCC
CGAACTGGACGAATTGCGCCGCATTCAAAAC
CATGGCGACGAATTTTTGCTGGATTTGGAAG
CCAAGGAACGCGAACGTACCGGTTTGTCCA
CACTTAAAGTCGAGTTCAACCGCGTTCACGGCTTTTACATTGAATTGTCCAAAACCCAAGCC
GAACAAGCACCTGCCGACTACCAACGCCGG
CAAACCCTTAAAAACGCCGAACGCTTCATCA
CGCCGGAACTGAAAGCCTTTGAAGACAAAGT
GCTGACTGCTCAAGAGCAAGCCCTCGCCTTAGAAAAACAACTCTTTGACGGCGTATTGAAA
AACCTTCAGACGGCATTGCCGCAGCTTCAAA
AAGCCGCCAAAGCCGCCGCCGCGCTGGAC
GTGTTGTCCACATTTTCAGCCTTGGCAAAAGAGCGGAACTTCGTCCGCCCCGAGTTTGCCG
ACTATCCGGTTATCCACATCGAAAACGGCCG
CCATCCCGTTGTCGAACAGCAGGTACGCCA
CTTCACCGCCAACCACACCGACCTTGACCAC
AAACACCGCCTCATGCTGCTCACCGGCCCCAATATGGGCGGCAAATCCACCTACATGCGC
CAAGTCGCGCTGATTGTTT
abcZ
adk
aroE
fumC
gdh
pdhC
pgm
porA
porB
fetA
penA
rpoB
16S
Locus X
Locus Y
MLST definitions
database
External
definitions
databases
Sequence
bin
Jolley, K. A. & Maiden, M. C. (2010). BIGSdb:
Scalable analysis of bacterial genome variation at
the population level. BMC Bioinformatics 11, 595.












![[Type text] Meningococcal vaccinesncirs.org.au/sites/default/files/2020-02/Meningococcal... · 2020-02-10 · Meningococcal vaccines . f. or Australians | NCIRS Fact sheet: April](https://img.dokumen.tips/doc/110x75/5f3b89378aca2557ce785a5e/type-text-meningococcal-2020-02-10-meningococcal-vaccines-f-or-australians.jpg)
















