Embed Size (px)
Citation preview

Pre-leukemiaDr. Akshay Agarwal
Moderator : Dr. Sangeeta Sharma

Introduction
Pre-leukemia are a group of disorders that most
commonly present with cytopenia(s) and has a
tendency to progress into acute leukemias unless
attended to clinically.
These include Myeloproliferative syndromes,
Marrow failure syndromes and congenital defects.

Disorders implicated in the
Pathogenesis of AML
Marrow Failure Syndromes Congenital Defects
Myelodysplastic Syndrome Down’s Syndrome
Fanconi’s Anemia Bloom Syndrome
Shwachman-Diamond Syndrome Monosomy 7 syndrome
Amegakaryocytic Thrombocytopenia Klinefelter Syndrome
Blackfan-Diamond Syndrome Turner Syndrome
Kostmann Agranulocytosis Neurofibromatosis
Familial Aplastic Anemia Congenital Dysmorphic syndromes
Dyskeratosis Congenita

Myelodysplastic Syndrome
• Derived from 2 words:
• Myelo – the Myeloid series
• Dysplastic – dysplasia meaning disordered
growth

Dysplastic features of
Erythroid series• Megaloblastosis
• Karyorrhexis
• Nuclear irregularity
• Fragmentation or multinucleation
• Ring Sideroblasts
• Cytoplasmic Vacuoles
• PAS positivity

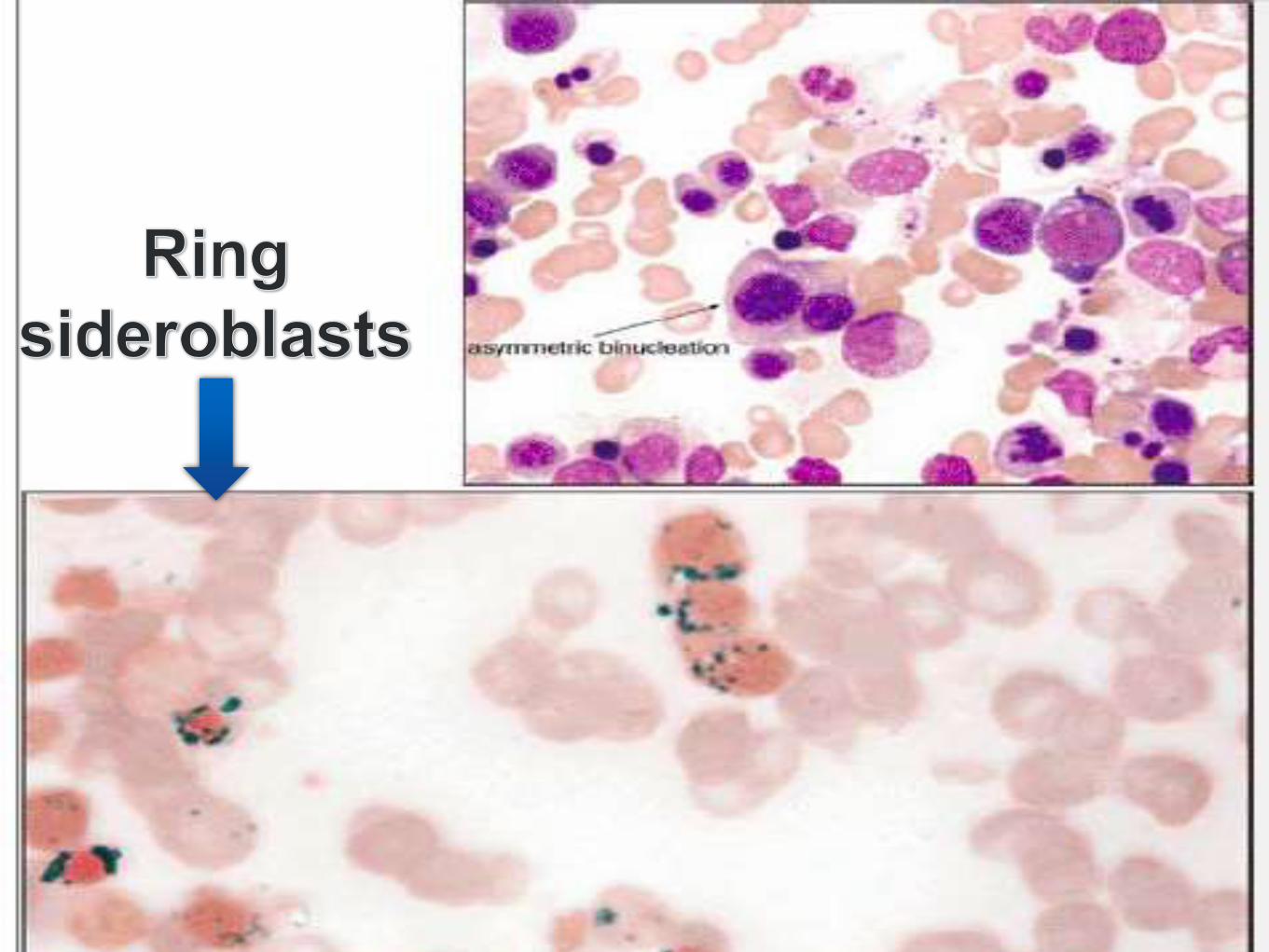
Iron stain

Dysplastic features of
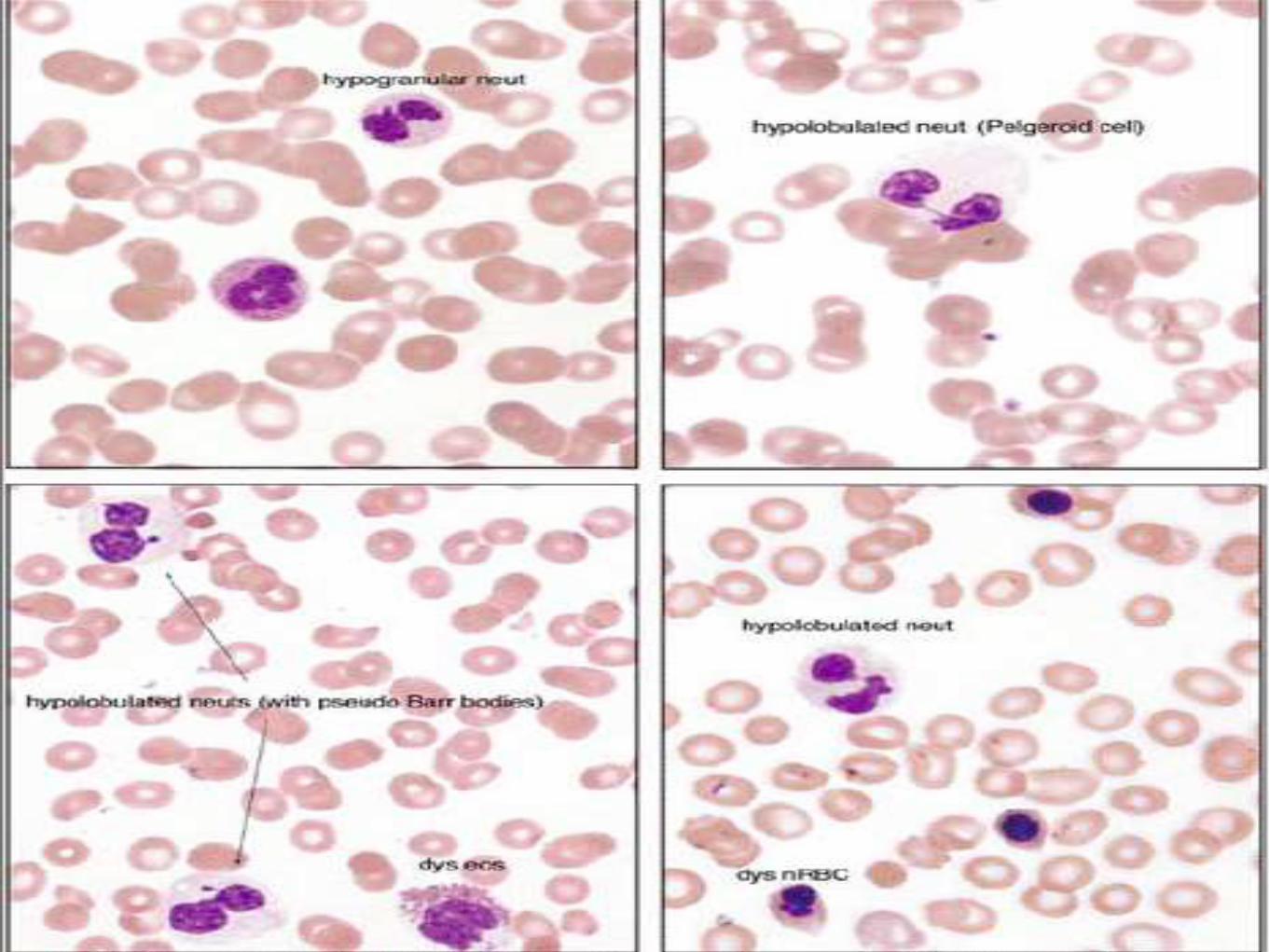
Granulocytic series
• Hypogranular neutrophil
• Pseudo-Pelger Huet anomaly
• Bizzare segmented neutrophils



Dysplastic features of
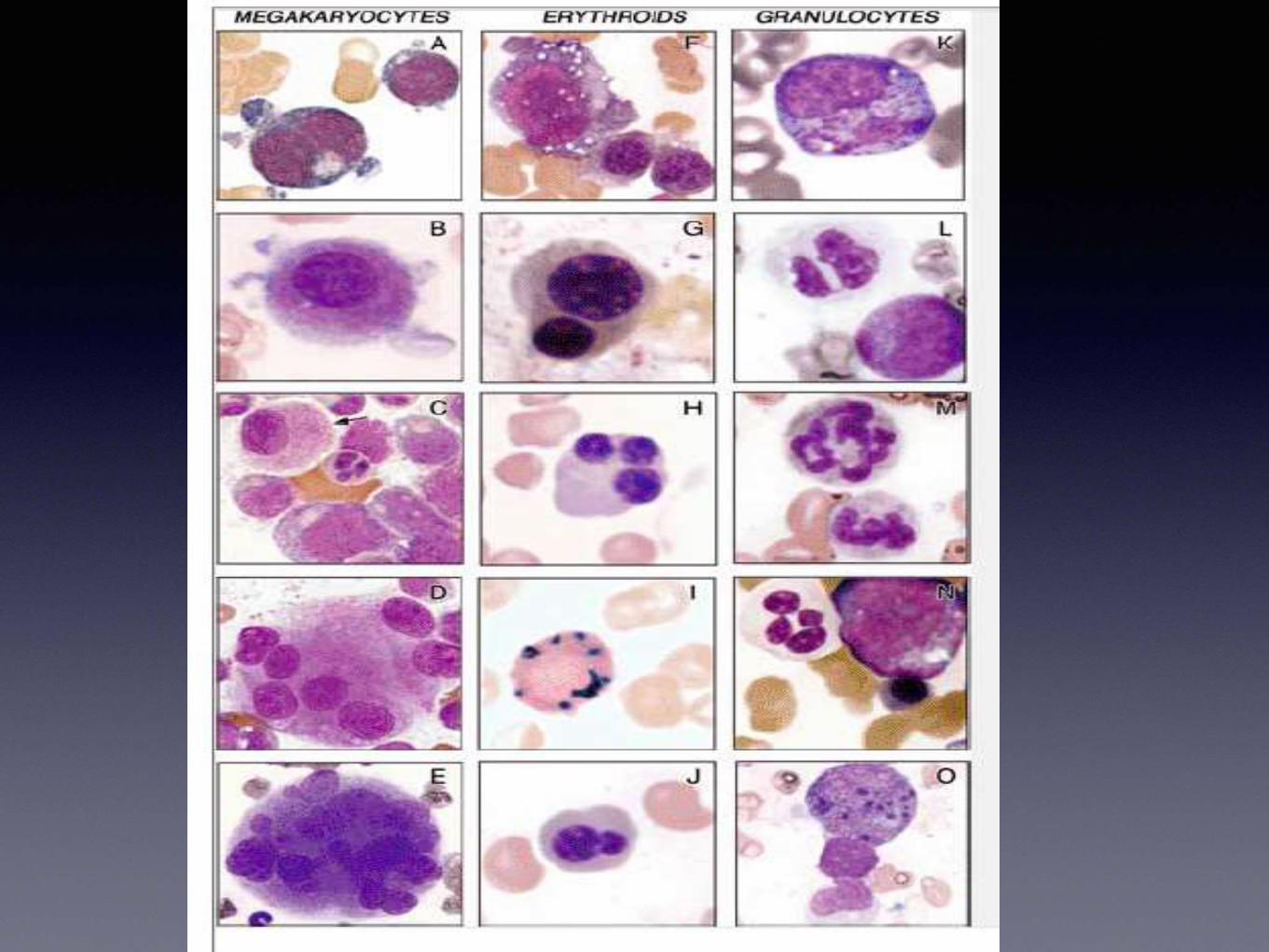
Megakaryocytic series
• Micromegakaryocytes
• Non lobated or multiple nuclei


• The myelodysplastic syndromes (MDSs) aredisorders caused by a clonal expansion ofhematopoietic stem cells in which maturation isabnormal (dysplastic) and production ineffective,resulting in many cells being destroyed before theyreach the systemic circulation. Anemia typicallyoccurs, often accompanied by thrombocytopeniaand/or neutropenia. Some cases develop fromexposure to ionizing radiation or cytotoxicchemotherapy, usually with alkylating agents, and inthese circumstances the bone marrow ischaracteristically hypocellular and partially fibrotic

Classification Sr.
No.
Disease Blood findings Bone marrow findings
1 Refractory cytopenias with unilineage dysplasia
(RCUD)
Uni / bi-cytopenias
No or rare blasts (<1%)
Unilineage dysplasia: >
10% of the cells in one
myeloid lineage
<5% blasts
<15% of erythroid
precursors are ring
sideroblasts
2 Refractory anemia with ring siderblasts (RARS) Anemia
No blasts
>15% of erythroid
precursors are ring
sinderblasts
Erythroid dysplasia only
<5% blasts
3 Refractory cytopenia with multilineage
dysplasia
(RCDM)
Cytopenia(s)
No or rare blasts
No Auer rods
<1 x 109/L monocytes
Dysplasia in >10% of the
cells in >2 myeloid
lineages
<5% blasts
No auer rods
+/- 15% ring sideroblasts

Sr.
No.
Disease Blood findings Bone marrow findings
4 Refractory Anemia with excess blasts -1
(RAEB-1)
Cytopenia
<5% blasts
No auer rods
<1 x 109/L monocytes
Unilineage or
multilineage dysplasia
5-9% blasts
No auer rods
5 Refractory Anemia with excess blasts -2
(RAEB -2)
Cytopenia
5-19% blasts
+/- auer rods
<1 x 109/L monocytes
Unilineage or
multilineage dysplasia
10-19% blasts
+/- auer rods
6. Myelodysplastic syndrome – unclassified
(MDS-U)
Cytopenia
<1% blasts
Unequivocal dysplasis
in ,10% cells
<5% blasts
7. MDS associated with isolated del (5q) Anemia
Normal to increased
platelet count
No or rare blasts
Normal to increased
megakaryocytes with
hypolobated nuclei
<5% blasts
Isolated del(5q)
No auer rods

Fanconi’s Anemia
• Autosomal recessive
• FA is the result of a genetic defect in a cluster of
proteins responsible for a DNA repair.

• As a result, the majority of FA patients develop
cancer, most often acute myelogenous leukemia
in 33%, and 90% develop bone marrow failure by
the age of 40.
• Most commonly M4 (Myelomonocytic) and M5
(monocytic)

• Significant risk of solid tumors like
• Hepatic tumors
• Squamous cell carcinoma
• 28% calculated risk by the age 40

Cell and Molecular Biology
• FA cells characteristically display a high frequency of spontaneous chromosomal breakage and hypersensitivity to DNA cross-linking agents.
• A subset of patients also present with biallelicmutations in BRCA2 gene
• BRCA2 protein is important in the repair of DNA damage.
• Thus, cells lacking BRCA2 inaccurately repair damaged DNA and are hypersensitive to DNA crosslinking agents.

Signs and Symptoms
• During childhood, short stature and skin pigmentation,
including café au lait spots, may become apparent.
• The first sign of a hematologic problem is usually
petechiae and bruises, with later onset of pale
appearance, feeling tired, and infections.
• Somatic abnormalities such as Skeletal (absent
thumbs, radial hypoplasia, scoliosis), genitourinary (
inferitility, horseshoe kidneys) , gastrointestinal,
cardiac and neurological may be present

Bilateral thumb hypoplasia

Treatment
• Administration of oxymetholone, corticosteroids
can show response in upto 70% patients but
many will become refractory after variable time
• Hematopoietic stem cell transplant is the
treatment of choice.

Dyskeratosis Congenita
• X-linked recessive, autosomal dominant and
autosomal recessive forms
• Bone marrow failure develops below the age of
20 years; 80-90% will develop bone marrow
abnormalities by age 30 yrs.

Cell and Molecular Biology
• Peripheral blood and bone marrow metaphases from patients show unbalanced chromosomal rearrangements in the absence of any clastogenicagents
• Chromosome/genomic instability
• Defect in DKC1 gene leading to defective ribosome production
• Hematopoietic progenitor studies have shown reduced numbers of all progenitors.

Signs and Symptoms
• Characterized by the mucocutaneous triad of
abnormal skin pigmentation, nail dystrophy and
mucosal leucoplakia
• Bone marrow failure is the principal cause of
early mortality with an early predisposition to AML
• Non-cutaneous lesions such as short stature,
pulmonary disease, esophageal strictures have
been documented.

Treatment
• Transient successful responses to
granulocyte/macrophage colony-stimulating
factor, Granulocyte colony-stimulating factor and
erythropoietin have been reported
• Steroid oxymetholone
• Hematopoietic Stem cell transplant

Shwachman-Diamond
Syndrome• Shwachman–Diamond syndrome is characterized
by an autosomal recessive mode of inheritance. The gene that is mutated in this syndrome lies on the long arm of chromosome 7 at cytogenetic position 7q11
• rare congenital disorder characterized by exocrine pancreatic insufficiency, bone marrow dysfunction, skeletal abnormalities, and short stature. After cystic fibrosis (CF), it is the second most common cause of exocrine pancreatic insufficiency in children.

Cell and Molecular biology
• The SDS gene is on 7q11.22 which is mutated in
90% patients.
• Its importance in indicated in RNA metabolism
and ribosome biogenesis.
Hematological
abnormalities• Neutropenia - Low neutrophil counts leave patients at
risk of developing severe recurrent infections that may be life-threatening.
• Anemia and thrombocytopenia may also occur. Bone marrow is typically hypocellular, with maturation arrest in the myeloid lineages that give rise to neutrophils, macrophages, platelets and red blood cells.
• Patients may also develop progressive marrow failure or transform to acute myelogenous leukemia.

Treatment
• G-CSF
• Incidence of myelodysplasia and transformation
to AML is seen in 15-25%
• Particularly AML with erythroid differentiation: M6

Blackfan-Diamond
Syndrome• Most pedigrees suggest an autosomal dominant
mode of inheritance with incomplete penetrance.
Approximately 10–25% of DBA occurs with a
family history of disease.
• Inherited pure red cell aplasia is a congenital
erythroid aplasia that usually presents in infancy.
• DBA causes anemia, without substantially
affecting the other blood

Diagnostic criteria• Normochromic macrocytic Anemia
• Reticulocytopenia
• Normocellular bone marrow with selective
deficiency of erythroid precursors
• Decreased leukocyte counts with normal to
increased platelet counts
• Elevated erythrocyte deaminase activity

Kostmann Agranulocytosis
• Kostmann disease is a form of severe congenital
neutropenia which is a rare autosomal recessive
condition in which severe chronic neutropenia is
detected soon after birth.

Diagnosis • An absolute neutrophil count chronically less than 500/mm3,
usually less than 200/mm3, is the main sign of Kostmann's.
• Other elements include the severity of neutropenia, and other normal findings (hemoglobin, platelets)
• Isolated neutropenia in infants can occur in viral infections, autoimmune neutropenia of infancy, bone marrow suppression from a drug or toxin, hypersplenism, and passive placental transfer of maternal IgG
• The bone marrow usually shows early granulocyte precursors, but myelopoietic development stops at the promyelocyte and/or myelocyte stage, so that few maturing forms are seen.

Bloom Syndrome
• are autosomal recessive disorder characterized
by short stature, caused by mutations in the BLM
gene leading to mutated DNA helicase protein
formation.

Signs and Symptoms
• short stature and a rash on the face
• moderate immune deficiency, characterized by deficiency in certain immunoglobulin classes and apparently leading to recurrent pneumonia and ear infections.
• low birth weight
• Hypogonadism is characterized by a failure to produce sperm, hence infertility in males, and premature cessation of menses (premature menopause)

• Thank you





























