Embed Size (px)
Citation preview

LymphomasLymphomasDr.CSBR.Prasad, M.D.,

Techniques to study the nature of lymphomas (1970 onwards)
• 1-Immunophenotyping• 2-Cytogenetics• 3-Molecular analysis

Immunophenotyping
• Helps to differentiate benign from malignant process
• Helps to differentiate B & T cell neoplasms• Helps to subcategorize B & T cell
lymphomas

Immunophenotyping
• Commonly three methods are used and they yeild similar results.
1-Immunohistochemistry (IHC) 2-Immunofluorescence (IF) 3-Flow cytometry

Immunophenotyping
Attached enzymes (usually horse radish peroxidase or alkaline phosphatase), or a fluorescent substance is tagged to the antibodies
.

Immunophenotyping - IHC
Can you tell them apart? One panel shows the small lymphoid cells from a case of small
lymphocytic lymphoma. The other shows benign small lymphocytes from a reactive lymph node. Which is which?

CD3, a pan-T-cell marker
Immunophenotyping - IHC
CD20 (L26), a pan-B-cell marker.

CD20 postivity in B-cell neoplasm
Immunophenotyping - IHC

• All lymphoid cells are reactive for CD45 (leukocyte common antigen, or LCA).
• B-cells: are reactive for CD19, CD20 and CD22. Certain low-grade B-cell lymphomas are reactive for two markers otherwise usually found on T-cells: CD5 and CD43. Follicular center cell lymphomas (as well as very different fish, lymphoblastic lymphomas) are frequently CD10(+).
• T-cells: Pan T-cell markers (present on almost all T-cells) include CD2, CD3, CD5, and CD7 (the early childhood markers). Most T-cells mark with either CD4 (helper cells) or CD8 (suppressor cells or cytotoxic cells).
• Natural-killer cells: These are frequently associated with CD16, CD56, or CD57.
Immunophenotyping – IHCCluster Designation Numbers


Immunophenotyping – Flow cytometry

Techniques to study the nature of lymphomas (1970 onwards)
• 1-Immunophenotyping• 2-Cytogenetics• 3-Molecular analysis

Cytogenetics
• Helps to identify translocation, deletions etc.
• Examples: Burkitt’s t(8;14) Mantle cell lymphoma t(11;14) ALCL t(2;5) Follicular lymphoma t(14;18)

Techniques to study the nature of lymphomas
• Molecular analysis is used to find out --Ig gene rearrangement in B-cell
malignancies and --T-cell receptor gene rearrangement in
T-cell malignancies. These rearrangements are too subtle to be
detected by conventional cytogenetics.

Important points regarding lymphoid neoplasms
• There is no benign lymphoid neoplasm.• Lymphomas are diagnosed by histological examination of the
lymphnode or the involved tissue.• Some times it’s necessary to use markers to differentiate reactive
process from lymphomas.• B-cell lymphomas are the most common variety (80-85%).• In patients with lymphomas immune abnormalities are very
common. (infections, autoimmunity and second malignancy).• NHL from the start is a widely disseminated neoplasm.• Spread of NHL is unpredictable where as spread in HD is
predictable.• Clinical presentation: NHL: 2/3 - nodal, 1/3 - extranodal HD: ~100% nodal.

Classification of Lymphomas
Helps in :1-recognising a lymphoma by their features.2-grouping them to understand the biological
principle that underlie their apperance.3-assessing the prognosis and give guidance
in Tx.

Classification of Lymphomas--History1942 Gall & Melory Morphology
1966 Rappaport Morphology
1974 Lukes Immu + Morphology
1975 Kiel Immu+Mor+Grade
1982 Working formula Morphology +Grade
1992 Modified Kiel Mor+IHC+Gra
1994 REAL Mor+IHC+Mol+Clin.prof
1997-2000 WHO Mor+IHC+Mol+Clin.prof

Classification of NHL:1. Non Gall-Rappaport lymphoma2. Non Rappaport-Non-Gall-Lukes Lymphoma3. Non Lukes-Kiel lymphoma4. Non Rappaport-Non Kiel-Non Gall-Non
Lukes lymphoma
Classification of Lymphomas--History
Source: Letter published in ‘The Lancet.’ during late 70s.

• A large study at the NCI looked at 1175 cases of non-Hodgkin's lymphoma with respect to different types of classification.
• “… and concluded that each of the classifications had clinical value but none was clearly superior.” !!!!!!
Classification of Lymphomas--History

Classification of Lymphomas--History1942 Gall & Melory Morphology
1966 Rappaport Morphology
1974 Lukes Immu + Morphology
1975 Kiel Immu+Mor+Grade
1982 Working formula Morphology +Grade
1992 Modified Kiel Mor+IHC+Gra
1994 REAL Mor+IHC+Mol+Clin.prof
1997-2000 WHO Mor+IHC+Mol+Clin.prof

Working Formulation:• based solely on the morphology of H&E
stained sections• intended to translate among the previous
classifications, not to replace them. • categories do have clinical validity
(therapeutic and prognostic)and are based on relatively simple, reproducible morphologic features.
Classification of Lymphomas--History

• The criteria are both architectural (low magnification) and cytological (high magnification):
1. Architectural diffuse proliferation follicular proliferation 1. Cytological Nuclear outline cleaved (indented) non-cleaved Cell size small large mixed small and large
Classification of Lymphomas--History
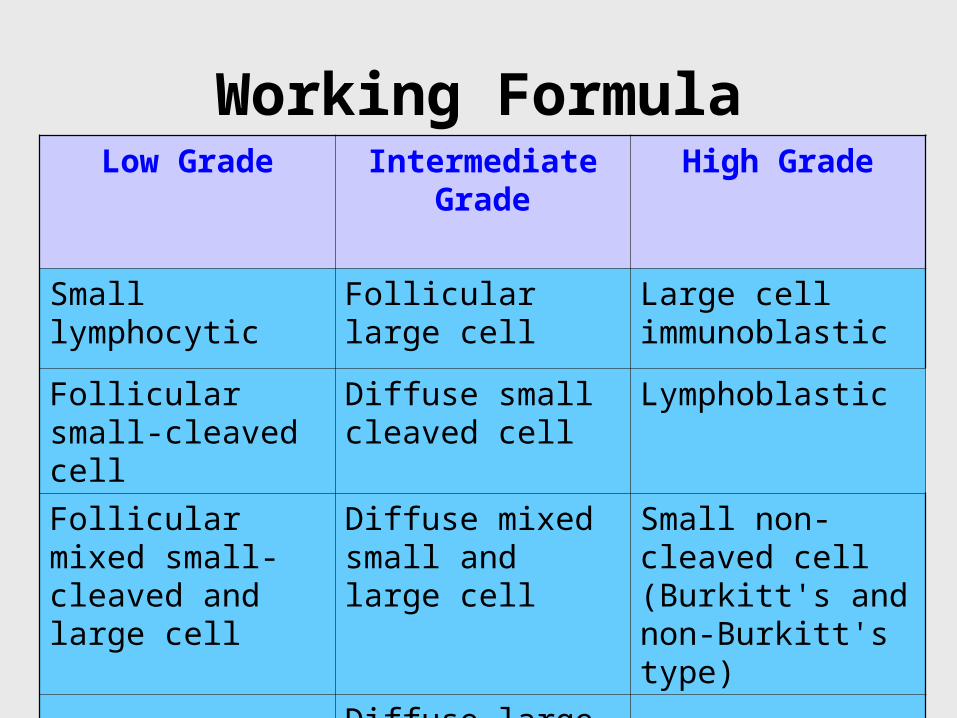
Working FormulaLow Grade Intermediate
GradeHigh Grade
Small lymphocytic Follicular large cell Large cell immunoblastic
Follicular small-cleaved cell
Diffuse small cleaved cell
Lymphoblastic
Follicular mixed small-cleaved and large cell
Diffuse mixed small and large cell
Small non-cleaved cell (Burkitt's and non-Burkitt's type)
Diffuse large cell

• As the years have rolled by, many lymphomas have been distinguished as individual entities with the help of immunologic, cytogenetic and molecular techniques.
• These lymphomas appear to be unique diseases.

Classification of Lymphomas--History1942 Gall & Melory Morphology
1966 Rappaport Morphology
1974 Lukes Immu + Morphology
1975 Kiel Immu+Mor+Grade
1982 Working formula Morphology +Grade
1992 Modified Kiel Mor+IHC+Gra
1994 REAL Mor+IHC+Mol+Clin.prof
1997-2000 WHO Mor+IHC+Mol+Clin.prof

WHO classification of lymphomas:
1-The World Health Organization (WHO) classification is a modification of the Revised European-American Lymphoma (REAL) classification.
2-Lymphomas are classified based on Morphology, IHC, Molecular abnormality and Clinical profile.
3-This classification recognizes 3 major categories of lymphoid malignancies based on morphology and cell lineage (B-cell, T/NK cell & Hodgkin’s Lymphoma).
4-Both lymphomas and lymphoid leukemias are included in this classification.
ex: B cell CLL & SLL
Lymphoblastic lymphoma & T-cell acute lymphocytic leukemia

WHO classification of lymphomas
•B-cell neoplasms •Precursor B-cell neoplasms
•Precursor B-cell acute lymphoblastic leukemia (B ALL) •Lymphoblastic lymphoma
•Peripheral B-cell neoplasms •B-cell CLL/small lymphocytic lymphoma •B-cell prolymphocytic leukemia •Lymphoplasmacytic lymphoma/immunocytoma •Mantle cell lymphoma •Follicular lymphoma •Extranodal marginal zone B-cell lymphoma of mucosa-associated lymphoid tissue (MALT) type •Nodal marginal zone lymphoma (with or without monocytoid B-cells) •Splenic marginal zone lymphoma (with or without villous lymphocytes) •Hairy cell leukemia •Plasmacytoma/plasma cell myeloma •Diffuse large B-cell lymphoma •Burkitt lymphoma

•T-cell and putative NK-cell neoplasms
•Precursor T-cell neoplasms
•Precursor T-cell acute lymphoblastic leukemia (T-ALL)
•Lymphoblastic lymphoma
•Peripheral T-cell and NK-cell neoplasms
•T-cell CLL/prolymphocytic lymphoma
•T-cell granular lymphocytic leukemia
•Mycosis fungoides/Sézary syndrome
•Peripheral T-cell lymphoma, not otherwise characterized
•Hepatosplenic gamma/delta T-cell lymphoma
•Subcutaneous panniculitislike T-cell lymphoma
•Angioimmunoblastic T-cell lymphoma
•Extranodal T-cell/NK-cell lymphoma, nasal type
•Enteropathy-type intestinal T-cell lymphoma
•Adult T-cell lymphoma/leukemia (with human T-cell leukemia virus type 1 [HTLV-1])
•Anaplastic large cell lymphoma, primary systemic type
•Anaplastic large cell lymphoma, primary cutaneous type
•Aggressive NK-cell leukemia
WHO classification of lymphomas

Acute lymphoblastic leukemia--Mostly ALLs with lymphomatous presentation are of
pre T-cell type.--They present as mediatinal masses
(Lymphadenopathy, thymic involvement) and splenomegaly.
--Tdt is positive in 95% of the cases (present in both B & T)
--To differentiate B from T immunotyping is required.--It’s also important to differentiate ALL from myeloid
leukemia.

Cytogenetic defect:90% of ALL have numeric / structural defect.>59% have hyperploidy and polyploidyT(12;21), t(9;22), t(4;11).
Many of these chromosomal aberrations dysregulate the expression of transcription factors essential for normal hemopoietic cell development resulting in arreasted development and accumulation of immature progenitor cells.
Acute lymphoblastic leukemia

Clinical features: Abrupt onset, anemia, fever, bleeding, Bone pain, lymphadenopathy, hepatosplenomegaly, testicular involvement is common in ALL, CNS manifestations.
Prognosis: In ALL is good. 90% achieve CR and 2/3 may be cured.Worse prognosis: -age <2years -Presentation in adolescent and young adults -peripheral blast count >1,00,000 (high tumor burden) -presence of t(9;22)Favourable prognosis: -age between 2 & 10 -Low tumor burden -early pre B-ALL phenotype -t(12;21)
Acute lymphoblastic leukemia

CLL / SLLBoth are identical in morphologically, phenotypically and
genotypically.But differ only in the degree of peripheral blood lymphocytosis (CLL
ab.lym.count >4000/cumm)Histology: 1-Diffuse effacement of architecture of LN 2-populated by small lymphocytes 3-Proliferation centers (by prolymphocytes)PBS & BM: 1-Lymphocytosis 2-Smudge cells 3-Non-paratrabacular aggregates of small lymphocytes.
Peripheral B-cell neoplasms

CLL / SLL

Proliferation center - prolymphocytes
CLL / SLL

Non-paratrabacular infiltration of lymphoma cells
CLL / SLL

Immunophenotyping: B-cell markers CD19, CD20. dim sIg. characteristic CD23Chrmosomal defects: del 13q, 11q, 17q Trisomy 12.CF: >50y, often asymptomatic, Lymphadenopathy, white count >2lakhs. Richter syndromeLab: Hypogammaglobulinemia > infections Autoantibodies > hemolytic anemia, & thrombocytopenia
CLL / SLL

A variant of SLL called "atypical SLL" fails to express CD23; and like mantle cell lymphoma it expresses bright CD20 and surface light chain and FMC7. Often these are the cases with trisomy12.
It may be mistaken for Mantle cell lymphoma (cyclinD1+)
CLL / SLL

CLL/SLL
This lymphoma is very indolent but relentless, with median survivals of almost a decade.
Incurable

Follicular lymphoma
Most common form of NHLMiddle aged malesHistology: Nodular & nodular diffuse growth patterns Two cell types 1-Centrocyte, 2-centroblast BM-Paratrabacular infiltration Liver-portal triad infiltration

A follicular origin may also be inferred from a combination of soft signs: immunophenotype (CD10+), cytogenetic t(14;18), and morphologic (the presence of small cleaved B-cells).
Immunoprofiles: CD19, 20, 10 + sIg+ No CD5 (Differentiates from Mantle cell lymphoma)
BCL-2 over expression (differentiates it from reactive follicle)
Follicular lymphoma

Cytogenetics: Typical t(14;18) Here H Ig gene of chr#14 is translocated to BCL-2 gene on chr#18.
This results in over expression of BCL2 and there by preventing apoptosis.
CF: Indolent lymphoma and is incurable. Survival 7-9yrs Transformation to DLBL may occur (activation of c-Myc)
Follicular lymphoma

Follicular lymphoma

Follicles dispersed throughout the lymphnode with loss of architecture
Follicular lymphoma

Follicular lymphoma

Berard’s grading of FLGrades 1, 2 & 3
Follicular lymphoma

Paratrabacular infiltration of lymphoma cells
Follicular lymphoma

DLBCL
Males >60yrsHistopath: Large cells 4-5x the small lymphocyte Diffuse growth pattern Nucleus is round to oval vesicular with 2-3
nucleoli adjacent to the nuclear membrane. There may be multinulceated giant cells

DLBCL

DLBCL
Several of these cells are excellent examples of the typical cell of large cell lymphoma, the centroblast. It has open (clear) chromatin and several moderately large nucleoli that cling to the nuclear membrane

DLBCL
The WHO classification of DLBCLs takes note of several morphological variants:
• Centroblastic • Immunoblastic • T-cell/histiocyte-rich • Lymphomatoid granulomatosis type • Anaplastic B-cell • Plasmablastic as well as 3 specific subtypes: • Mediastinal (thymic) large B-cell lymphoma • Primary effusion lymphoma • Intravascular large B-cell lymphoma

DLBCL-Intravascular lymphoma
Small blood vessels in the dermis plugged with tumor cells

Large vessel filled with lymphoma cells
DLBCL-Intravascular lymphoma

Angiotropic lymphoma, Large B-Cell
DLBCL-Intravascular lymphoma

DLBCL
Centroblastic type

Burkitt’s lymphomaBurkitt's lymphomas come in at least 3 sorts, all of which are more prevalent in
males:
Endemic Burkitt's lymphoma (a WHO classification subtype): a childhood lymphoma prevalent in equatorial Africa and intimately associated with both Epstein-Barr virus infection and a characteristic translocation of the MYC gene.
Sporadic Burkitt's lymphoma (a WHO classification subtype): a world-wide lymphoma affecting slightly older patients, also associated with MYC changes but less so with EBV infection.
Burkitt's-like lymphoma (a WHO classification morphologic variant): a rather different disease affecting an older population and not notably associated with the MYC gene or EBV infection.
All 3 kinds have also been called "small non-cleaved cell lymphoma”

Molecular abnormalities:All forms are associated with translocation of c-myc gene on chr#8. The
partner is usually the IgH locus on chr#14. t(8;14) others t(8;22), t(2;8)Immunophenotyping: CD19, 20 and 10 + BCL-6+
Clinical features: Children and young adults Extranodal sites are most commonly involved Very aggressive But, responds to chemotherapy well.
Burkitt’s lymphoma

Endemic burkitt’s most commonly affects Children and young adults
& Extranodal sites are
most commonly involved
Burkitt’s lymphoma

Small non-cleaved cell, high magnification. Most nuclei have 1 or 2 prominent nucleoli. Note the "tingible body" macrophage at the left with debris in its cytoplasm
Burkitt’s lymphoma

Burkitt’s lymphoma
Starry sky pattern

BURKITT'S CELL
Burkitt’s lymphoma

Mycosis fungoides / Sezary syndrome
NHL involving Helper T-cells (CD4+).Has predilection to involve SKIN.It’s an indolent lymphoma.
Stages: Premycotic phase, Plaque phase & Tumor phase.

Mycosis fungoides
Histopathology:• Infiltration of epidermis and upper dermis
by neoplastic T-cells.• These cells have cerebriform nucleus (Sezry
cells)• Extracutaneous spread occurs most
commonly to LN and BM.

Mycosis fungoides
Erythematous patches

Mycosis fungoides
Multiple confluent purple 0.5-2 cm nodules some with overlying crust and scale

Asymptomatic red plaques on chest, abdomen, back and proximal extremities that waxed and waned for several years before they were clinically evaluated and biopsied.
Mycosis fungoides

Diffuse red scaly rash with scattered indurated plaques studded with comedones and follicular pustules.
Mycosis fungoides

There is a dense mononuclear cell infiltrate throughout the dermis with focal epidermal erosion.
Mycosis fungoides

Exocytosis is present (arrow).
Mycosis fungoides

Numerous eosinophils and plasma cells are present in the infiltrate. .
Mycosis fungoides

Another high power view showing prominent lymphocytic exocytosis without spongiosis or keratinocyte hole formation. Also note the coarse collagen fibers in the papillary dermis
Mycosis fungoides

Immunohistochemical staining shows increased numbers of CD4 positive lymphocytes.
Mycosis fungoides

Sezary syndrome
Features:• Generalized exfoliative erythroderma• No tumefactions• Associated with Leukemia of Sezary cells• Indolent lymphoma with 8-9yrs survival• Transformation to large cell lymphoma can
occur as a terminal event.

Sezary syndrome
Exfoliative erythroderma

Sezary syndrome
This is a cerebriform lymphocyte from a patient with Sezary's syndrome. It is an abnormal lymphocyte. It has super-clumped chromatin with large aggregates or blocks. The cytoplasm is blue and scanty.

Sezary syndrome
Sezary cells are atypical lymphocytes with a grooved or cerebriform nucleus seen both in tissue and blood. The Sézary cell is named after the French dermatologist Sézary A (1880-1956).

Blood criteria to define Sézary syndrome as recently proposed by ISCL are the following:
• An absolute Sézary cell count of 1000 cells/cumm or more • An increase in CD3 or CD4 positive cells resulting in a CD4/CD8
ratio of 10 or more • Aberrant expression of pan T cell markers by flow cytometry,
deficient CD7 expression • Increased relative or absolute lymphocyte counts with evidence of
a T cell clone in the blood by Southern blot or PCR technique
Sezary syndrome
ISCL-International Society for Cutaneous Lymphomas

Diagnosis of Sézary syndrome requires the presence of the triad of
1. Erythroderma, 2. Lymphadenopathy and 3. 10% or more of atypical mononuclear cells in
the peripheral smear. Many experts now consider a circulating Sézary cell
count which exceeds 1000 cells/cumm as characteristic of Sézary syndrome.
Sezary syndrome

Hodgkin’s disease
• B-cell neoplasm.• Characterised by the presence of RS-giant cells• RS-giant cells make only minor fraction of the
tumor.• Major fraction is formed by the cytokine recruited
inflammatory cells.• Age: Bimodel age distribution (2nd and 6th decade).

Patients usually present with a slowly expanding, non-tender lymph node, mostly cervical, axillary, or inguinal. Only rarely are axial abdominal and pelvic lymph nodes or Waldeyer's ring involved initially.
Hodgkin’s disease

Classification ( 5 types):1. Classic HD --Nodular sclerosing --Lymphocyte rich --Lymphocyte depletion --Mixed cellularity2. Nodular Lymphocyte predominant variety
Hodgkin’s disease

Nodular Lymphocyte predominant variety
In this variety RS cells have a characteristic B-cell immunophenotype distinct from that of the classical HD subtypes.
Hodgkin’s disease

RS-giant cell:-15-45µm in diameter.-Single or multiple nuclei-Each nucleus will have large inclusion, the
size of a lymphocyte / RBC.-Perinuclear halo / clearing.-Cytoplasm is abundant.-Positive for CD 15 and CD 30 and negative
for CD 45 and B&T markers.
Hodgkin’s disease

A patient of Dorothy Reed.
Hodgkin’s disease

Binucleate diagnostic Reed-Sternberg cells
Hodgkin’s disease

Size of the nucleolus is that of a lymphocyte (arrow) / RBC (circle)
Hodgkin’s disease
RS-giant cell

Diagnostic Reed-Sternberg cells
Hodgkin’s disease

RS-giant cell
Hodgkin’s disease

In this case CD30 (left) stains the cytoplasm of the Reed-Sternberg cells diffusely and also the perinuclear Golgi apparatus. CD15 (right) shows crisp membrane staining as well as Golgi staining.
Hodgkin’s disease IHC of Reed-Sternberg cell

Cytokines /chemokines and their possible main clinical and biological effects
Hodgkin’s disease
IL-1/TNF-alfa Systemic symptoms
IL-5 Eosinophilia
IL-6 Plasma cell response
IL-8 Neutrophil recruitment
TGF-ß Fibrosis & immunosupression
IL-13 & IL-9 Autocrine pathways
IL-7 Inflammatory infiltrate
Source: Hematol Oncol 2001; 19: 1-17

In fact to make the diagnosis of HL microscopically, two findings are needed:
1) Reed-Sternberg cells and variants and 2) an appropriate inflammatory background.
The inflammatory response is ultimately the result of cytokines produced by the tumor cells.
Hodgkin’s disease

LYMPHOCYTE PREDOMINANCE HODGKIN'S DISEASE
It is likely that this category of Hodgkin's disease contains two discrete entities:
1-Classic Hodgkin's disease, representing the end of the spectrum of mixed cellularity in which Hodgkin cells are relatively infrequent.
2-Nodular lymphocyte predominance in which nodularity is minimal or absent

There are two critical features for the diagnosis of this subtype:
1-the rarity of Hodgkin cells (RS-cells) and 2-the absence of fibrous bands (diagnostic of
nodular sclerosis)
LYMPHOCYTE PREDOMINANCE HODGKIN'S DISEASE

Small lymphocytes generally dominate the reactive element, although, contrary to what the name of the subtype might imply, the predominance of lymphocytes is not the defining feature. Histiocytes are variable in number; eosinophils are generally few in number.
LYMPHOCYTE PREDOMINANCE HODGKIN'S DISEASE

Mixed Cellularity Hodgkin Disease
1- Variety of different cell types in the background inflammatory component.
2- It lacks the fibrous bands (of the nodular sclerosis subtype)
3- Has more numerous Reed-Sternberg cells, and
4- Has a slightly worse prognosis.

The inflammatory infiltrate includes lymphocytes, eosinophils, neutrophils, and histiocytes. Though not seen here, plasma cells might participate also.
Mixed Cellularity Hodgkin Disease

Nodular sclerosis is a bit of a specialty item.
1-More common in women than men, and it 2-Very often presents with a mediastinal mass. 3-Named for the dense fibrous bands that subdivide
involved lymph nodes, 4-It has a prognosis slightly better than mixed
cellularity HL. 5-A morphologically variant Reed-Sternberg cell called
the "lacunar cell" is found (In formalin-fixed tissue)
Nodular sclerosis variety of Hodgkin Disease

Nodular sclerosis variety of Hodgkin Disease

Lacunar cells are a feature of nodular sclerosis Hodgkin disease and are not found in other subtypes. In formalin-fixed tissue, the cytoplasm around Reed-Sternberg cell nuclei retracts, leaving a cleared space possibly
spanned by a few shreds of cytoplasm. The nuclei are also contracted and have diminished nucleoli.
Nodular sclerosis variety of Hodgkin Disease

A diagnostic (multi-nucleated) Reed-Sternberg cell lies dead-center. Just below it is a non-diagnostic, uninuclear cell that has been called a "Reed-Sternberg variant" or a "Hodgkin" cell. Although this cell is characteristic of Hodgkin disease, the pathologist
who plays by the rules will scrutinize a node suspected of Hodgkin disease involvement until a diagnostic cell is found.
Nodular sclerosis variety of Hodgkin Disease
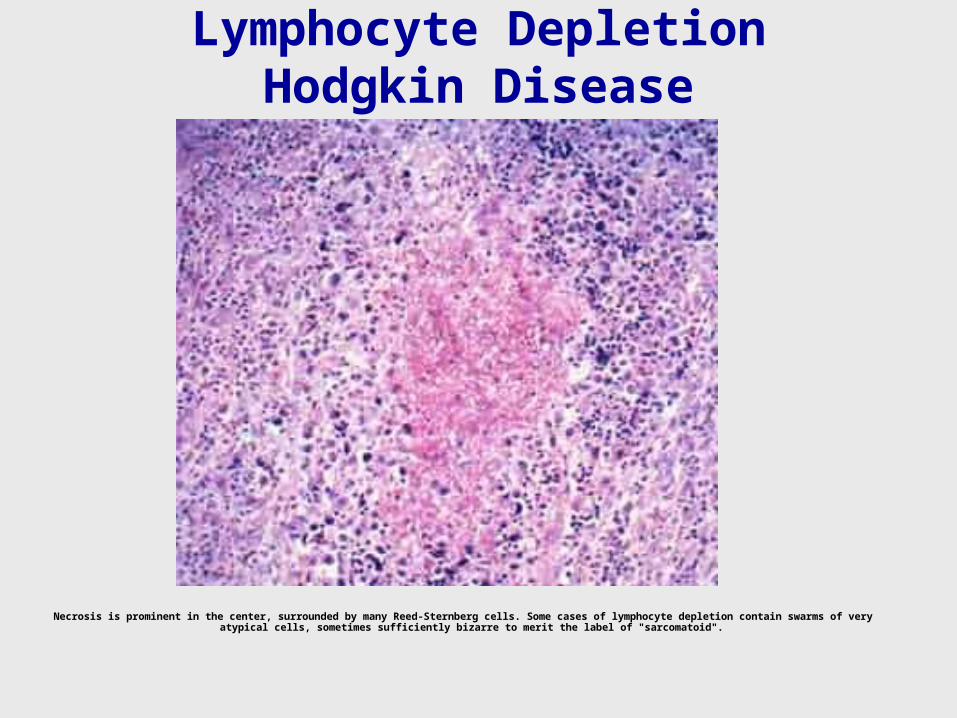
Lymphocyte Depletion Hodgkin Disease
Necrosis is prominent in the center, surrounded by many Reed-Sternberg cells. Some cases of lymphocyte depletion contain swarms of very atypical cells, sometimes sufficiently bizarre to merit the label of "sarcomatoid".

Nodular Lymphocyte-Predominance Hodgkin's Lymphoma
1-Have a B-cell immunophenotype (i.e. CD-20+)2-Negative for CD15 and CD30 3-Typical Reed-Sternberg cells are absent4-Polylobated variants called L & H cells
(Lymphocytic and/or histiocytic; colloquially "popcorn cells” are seen.

Additional distinctive clinical features setting apart nodular lymphocyte predominance HL include:
• an indolent though relapsing course with an excellent prognosis;
• occasional cases relapsing as high-grade B-cell non-Hodgkin's lymphoma;
• a peak incidence in males in their 30's and 40's, without the bimodal age pattern of classic HL;
• a greater tendency to be restricted to cervical lymph nodes.
Nodular Lymphocyte-Predominance Hodgkin's Lymphoma

L & H cells OR“Pop corn” cells
Nodular Lymphocyte-Predominance Hodgkin's Lymphoma

L & H cells
Nodular Lymphocyte-Predominance Hodgkin's Lymphoma

L & H cells
Nodular Lymphocyte-Predominance Hodgkin's Lymphoma

ImmunophenotypeThe immunophenotypes of the Reed-Sternberg cells and variants in the two subgroups of HL are mirror
images:
CD15 CD30 LCA(all
leukocytes)
CD20(B-
cells)
EMA
Classic Hodgkin + + – – –Lymphocyte Predominance
– – + + +

Hodgkin's Lymphoma (HL) Non-Hodgkin's Lymphoma (NHL)
Stage / Grade Because HL begins as a localized process that spreads slowly from one nodal region to another, stage (degree of spread) is important. In some cases grade may be significant, but it is mostly unremarked.
Because NHL is usually systemic from the get-go, grade influences prognosis and therapy more than stage.
Source of Mass Neoplastic cells are usually <1% of the mass, which is mostly benign inflammatory cells
Almost all the mass is neoplastic lymphoid cells
Immune Deficiency Usually cell-mediated (T-cell): mycobacterial, fungal, viral, & protozoal infections.
Usually humoral (antibodies from B-cells): bacterial infections.
GI / Waldeyer's Involvement Rare. Common.
Marrow Involvement Significant. In many cases not important.
Extra-Nodal Involvement 10% of cases 40% of cases.
Treatment HL is always treated, but milder, localized cases may receive only radiotherapy.
Indolent NHLs may remain untreated for years. Almost all treatment, however, is for systemic disease and thus utilizes chemotherapy. Radiotherapy may be an adjunct.

Ann Arbor Staging ClassificationStage I
Involvement of a single lymph node or extra-lymphatic site (IE) Stage II
Involvement of 2 or more lymph node regions on the same side of the diaphragm or localized involvement of an extra-lymphatic site (IIE)
Stage III Involvement of lymph node regions on both sides of the diaphragm or
localized involvement of an extra-lymphatic organ or site (IIIE) or spleen (IIIS) or both (IIISE)
Stage IV Diffuse or disseminated involvement of one or more extra-lymphatic
organs with or without associated lymph node involvement
The stage can also have a designation of "A" for asymptomatic or "B" for constitutional symptoms.
Hodgkin’s disease - staging

To make a patient "B", the patient must have one or more of the following:
1-Unexplained fever >38° C 2-Night sweats at least of moderate severity 3-Weight loss must be at least 10% of initial body weight
in the 6 months preceeding evaluation.
Other potentially important symptoms of HL such as fatigue, alcohol related pain or pruritus are not considered in the staging system.
----Hoffbrand's PG-Hematology 3rd Ed, 507p.
Hodgkin’s disease - staging

Prognosis is dictated by:
1-Bulk of the tumor2-Stage of the disease
Histological variety has very little role to play in determining the prognosis.
Hodgkin’s disease - staging

Which is a better term?
Hodgkin’s Lymphoma or Hodgkin’s Disease ?
Hodgkin’s disease





























