Embed Size (px)
Citation preview

Genetics of Cognitive Disorders in Elderly
Dr Ravi SoniDM-SR II
Dept. of Geriatric Mental HealthKGMU, LKO

Contents:
• Genetics of
1. Alzheimer’s disease
2. Fronto-temporal lobar degeneration
3. Lewy body disease
4. Vascular dementia [CADASIL]
5. Huntington’s disease

Alzheimer’s disease genetics• Classified into two subtypes depending on the age of onset
1. Early onset Alzheimer’s Disease [EOAD]: also called familial AD
– Starts before the age of 65 years, typically in late 40s and early 50s– Accounts for 1-5% AD patients– Most obvious family aggregation of AD patients– Mendelian autosomal dominant pattern of inheritance [<1% AD
patients
2. Late onset Alzheimer’s Disease [LOAD]: also called sporadic AD
– Starts after the age of 65 years– Accounts for >95% of cases

Diag = Age diagnosed
Diag. 57
Diag. 50 Diag. 58
Diag. 52= Female
= Male

This is suggestive of a type of inheritance called Autosomal Dominant

Early onset AD
• Caused by mutation into 3 genes• Which is highly penetrant• Genes encode protein involved in Amyloid Precursor
Protein [APP] and Aβ generation
• Genes:– APP: Chromosome 21– Presenilin 1 [PSEN1]: chromosome 14– Presenilin 2 [PSEN2]: chromosome 1
Zhangyu Zou et al. Clinical genetics of Alzheimer’s Disease. BioMed Research International. Volume 2014, Article ID 291862 http://dx.doi.org/10.1155/2014/291862

APP Gene• A𝛽 is generated from APP by two endoproteolytic cleavage events
catalyzed by 𝛽-secretase and 𝛽-secretase• The most common form of A𝛽 is 40 amino acids in length and is called
A𝛽40• The longer form, A𝛽42, is less abundant than A𝛽40 but is deposited first
and is more amyloidogenic.
Beta-secretase is also known as beta site APP cleaving enzyme 1 (BACE1) Gamma-secretase is a multiprotein complex consisting of presenilin (PS), nicastrin (NCT), anterior pharynx defective 1 (APH1), and presenilin enhancer protein 2 (PEN-2)

APP gene continued….• APP gene mutations are found in EOAD families
– 33 different APP mutations have been identified in AD patients to date, including 23 missense mutations, nine duplications, and one deletion.
– Most of the mutations are dominantly inherited.
• Most of these mutations are positioned in the vicinity of the
𝛽- and 𝛽-secretase cleavage sites and therefore they influence APP proteolytic processing and/or aggregation.
• Alpha-secretase is a third protease that prevents the formation of toxic Aβ peptides.
• Dysfunctional activity of these three proteases, results in Aβ accumulation, which stimulates diverse cell signaling pathways, and lastly resulting in synaptic degeneration, neuronal loss, and cognitive decline


PSEN 1 and PSEN 2 gene
• Mutations in the PSEN1 and PSEN2 genes have been identified in EOAD families.
• These genes encode for presenilin 1 and presenilin 2 proteins respectively, required for 𝛽-secretase to produce A𝛽 from APP.
• To date, approximately 200 different AD-related PSEN1 mutations and 22 AD-related PSEN2 mutations have been detected.
• The presenilins act as aspartyl proteases that carry out 𝛽-secretase cleavage of APP to produce A𝛽. Therefore, mutations in the PSEN1 and PSEN2 genes increase the ratio of A𝛽42 to A𝛽40.
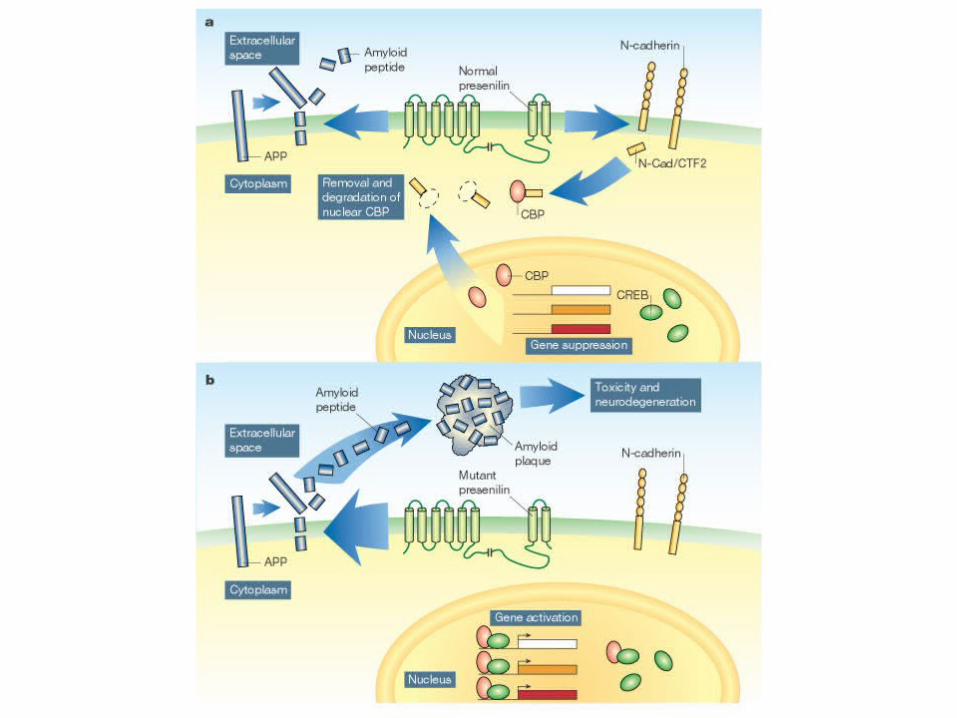


• PSEN1 mutations account for the majority (78%) of the EOFAD mutations identified, followed by APP (18%), with PSEN2 mutations found in only a few families (4%).
• Together, the mutations in these three genes are thought to be responsible for 30–50% of autosomal dominant AD cases and about 0.5% of AD cases in general.

Late onset AD• For most cases of LOAD, the risk of developing AD is assumed to be
determined by genetic variants combined with lifestyle and environmental exposure factors.
• The 𝜀4-allele of the apolipoprotein E gene (APOE) on chromosome 19q13.2 is the only well-established genetic risk factor for LOAD.
• Apolipoprotein E (ApoE) gene is polymorphic and it has three isoforms: Apo 2, Apo 3 and Apo 4. The 4 allele has been linked to both late ɛ ɛ ɛ ɛonset family forms and sporadic forms of AD, whereas 2ɛ allele was found to offer protection.

ApoE gene continued….• In central nervous system, ApoE is produced mainly by astrocytes and
take part in transport of cholesterol to neurons through ApoE receptors, and is a crucial cholesterol carrier in brain and has possible role in neuroplasticity-related phenomena because it requires changes in membrane lipids.
• ApoE has beneficial effect for neuronal protection but it is isoforms dependent, like Apo 4 isoform is less effective than Apo 2 and Apo 3ɛ ɛ ɛ
• There is possibility that Apo 4 may not be toxic but it simply has less ɛprotective effects than 2 and 3. ɛ ɛ
• ApoE has protective role against free radicals and has significant antioxidant property but it is also isoforms dependent like, effect is clearly perceptible with isoform 2 but less so with 3 and even less so with 4ɛ ɛ ɛ



Recent research
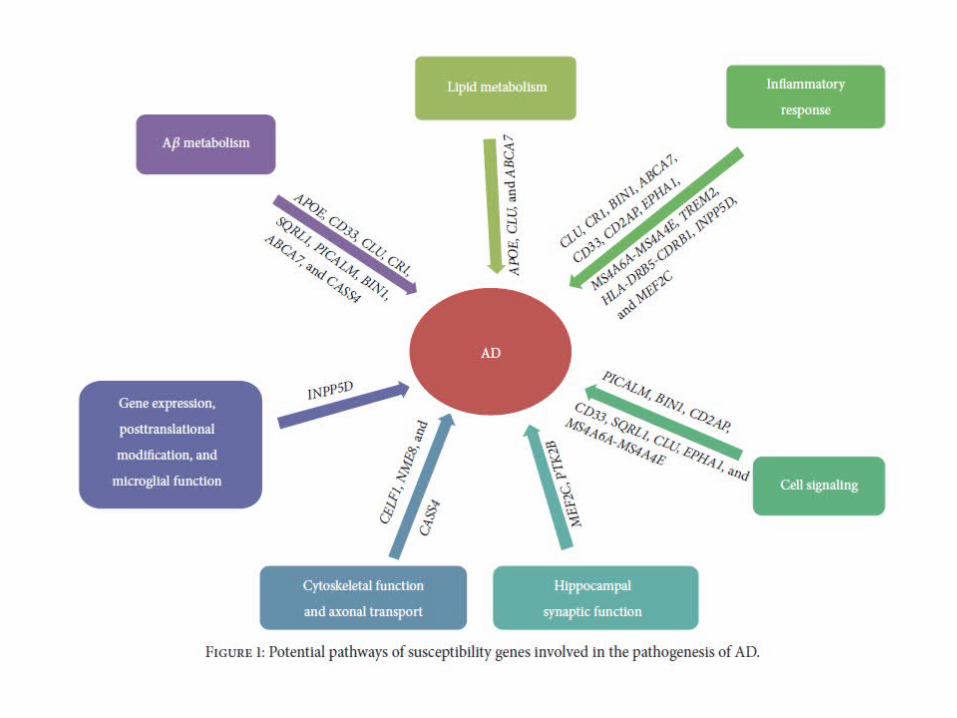
• GWAS [Genome wise association studies] have identified candidate genes associated with LOAD:
• Clusterin (CLU)• Complement component receptor 1 (CR1)• Phosphatidylinositol binding clathrin assembly protein (PICALM)• Bridging integrator 1 (BIN1)• Sialic acid binding Ig-like lectin (CD33)• CD2-associated protein (CD2AP)• Membrane spanning 4A gene cluster (MS4A6A/MS4A4E)• Ephrin receptor A1 (EPHA1)• ATP-binding cassette transporter (ABCA7)• Triggering receptor expressed on myeloid cells 2 (TREM2)

Risk genes for LOAD
• These AD risk genes affect various pathways, roughly falling into four pathways:
1. A𝜀 metabolism,
2. Lipid metabolism,
3. Immune and complement system/inflammatory response, and
4. Cell signaling

Genetics of FTLD• Genetically complex disorder• Inherited as an autosomal dominant trait with high penetrance in
majority of cases• Genetic linkage studies have revealed FTLD loci on chromosome 3p,
9, 9p, and 17q.• The most prevalent genes are PGRN (progranulin) and MAPT
(microtubule-associated protein tau), both located on chromosome 17q21.
• Other potential genes are – VCP (Valosin-containing protein) gene, – Chromosome 9 open reading frame 72 (C9ORF72), – TAR DNA binding protein (TARDBP) and – Charged multi vesicular body protein (CHMP2B) — are linked to
frontotemporal dementia.• Presenilin-1 gene (PSEN1) and Lrrk2 (leucine-rich repeat kinase) are
being studied for their possible role in frontotemporal dementia • The autosomal dominant form of FTLD linked to chromosome
17q21 is termed FTDP-17.

Genetics
Autosomal dominant pattern of inheritance• About 10% of people with frontotemporal dementia inherited in an
autosomal dominant pattern
Familial pattern of inheritance• A large group of families (20-40%) have an inheritance pattern that is
termed familial
Sporadic pattern of inheritance• The majority of frontotemporal dementia (50-70%) is sporadic. • This means the disorder does NOT appear to be inherited, and the risk to
family members is almost the same as that of the general population

PGRN Gene• Encodes for PGRN protein• PGRN, expressed in neurons and activated microglia• involved in tissue remodeling by activating signaling cascades that control cell cycle progression and cell motility• PGRN mutations occur in 26% of familial FTD cases.• PGRN mutation is associated with the expression of truncated and hyperphosphorylated isoforms of TDP-43 (TAR DNA binding protein 43). • Under pathologic conditions, TDP-43 relocates from the neuronal nucleus to the cytoplasm resulting in loss of TDP-43 nuclear functions
(Presence of this ubiquinated protein outside the nucleus, in cytoplasm suggest that it has some important regulatory functions in cell, and loss of which results in death of affected neurons, same mutation is seen in patients with ALS)

MAPT Gene• MAPT having 37 mutations within the microtubule-binding region or exon
10, produce tau isoforms with either three microtubule-binding repeats (3R-tau) or four repeats (4R-tau).
• In Pick’s disease 3R-tau accumulates.• Missense and deletion mutations disrupts the binding of tau to
microtubules resulting in accumulation of unbound tau.• MAPT mutations on exons 1, 9, and 11 to 13 account for the dementia-
dominant phenotype. • The parkinsonism-plus–predominant phenotype is associated with
mutations within intron and exon 10, leading to the overproduction of 4R-tau isoforms.

Do Specific Mutations Cause Specific Forms of Frontotemporal Dementia?
• PGRN mutations: bvFTD, PNFA, CBS, PSP but not to ALS
• C9ORF72 mutations: FTD and ALS
• VCP mutations: FTD and paget disease of bone
• CHMP2B mutations: FTD, FTD-ALS and ALS
• TARDBP mutations: both sporadic and familial ALS


Dementia of Lewy bodies
• Dementia with Lewy bodies is a complex brain disorder and a key member of the Lewy body disease spectrum.
• Its genetic etiology is unclear, and information is scattered.
• Results of molecular genetic studies imply a genetic and mechanistic overlap with Alzheimer disease, Parkinson disease with dementia, and Parkinson disease.
• Clinical and pathological evidence supports the notion that Alzheimer disease (AD), dementia with Lewy bodies (DLB), and Parkinson disease (PD) with dementia are different members of the same disease continuum
Bram Meeus, Jessie Theuns, Christine Van Broeckhoven. The Genetics of Dementia With Lewy BodiesWhat Are We MissingArch Neurol. 2012;69(9):1113-1118. doi:10.1001/archneurol.2011.3678.


Continued….
• Information regarding genetic susceptibility to DLB is very less
– Considered as sporadic disorder because of late onset of the disease with no familial predisposition and the result of twin investigation did not support a major genetic etiology of DLB.
– Clustering of dementias in families of patients with DLB– Overlapping symptoms with AD and PD with dementia
• Molecular genetic investigations in cohorts of unrelated and largely sporadic patients with DLB have revealed genetic variations in genes that are involved in the pathogenic pathways leading to related neurodegenerative disorders.
• These include PD (α-synuclein gene [SNCA ]) and AD (amyloid precursor protein gene [APP ], presenilin 1 gene [PSEN1 ], and presenilin 2 gene [PSEN2 ]).

Vascular Dementia-CADASIL
• CEREBRAL AUTOSOMAL DOMINANT ARTERIOPATHY WITH SUBCORTICAL INFARCTS AND LEUKOENCEPHALOPATHY
• Inherited small vessel disease causing stroke and subcortical vascular dementia that starts in early adulthood and progresses over time.
• This is a nonatherosclerotic, nonamyloid angiopathy involving small arteries and capillaries of the brain and other organs.
• Caused by missense mutations in the Notch3 gene on chromosome 19p13

CADASIL
• 1977: family w/hereditary, multi-infarct dementia syndrome
• Presents in mid-20s to age 45• Stroke, dementia, migraine with aura, mood
disorders• Shortened life span
– Most die by age 65• Unknown prevalence
– 400 families world-wide– 2/100,000– Largely undiagnosed

What leads to CADASIL?
• Mutations in notch3 gene– Odd number of cysteine
residues in Notch3 receptor extracellular domain
– Impaired clearance of cleavage product
• Alterations of vascular smooth muscle
• Presence of granular osmiophilic deposits

Genetics

Notch3 gene mutation• Mature vascular smooth muscle cells require
continued function of the Notch 3 pathway– Continued survival– Usually missense mutation– More than 50 have been found– Spontaneous mutations have been described
• Leads to accumulation of protein in membranes of smooth muscles and, ultimately, fibrosis and luminal narrowing– Blood vessels are narrowed and weak and do not
react to fluctuations of CO2 and BP– Capillaries, veins are involved
• Generalized vasculopathy

Huntington Disease
• Autosomal Dominant Progressive Chorea and Dementia• Defective huntingtin protein [chromosome 4]• Degeneration of cholinergic and GABA-ergic cell in BG• Relative excess of Dopamine

Huntington Disease• IT15/Huntingtin gene on 4p16.3 cloned in 1993• Disease mutation - CAG expansion in exon 1
Repeat number Outcome– 10-28: normal, no transmission of HD– 29-35: normal, paternal meiotic instability– 36-39: reduced penetrance (25%: 36 repeats, 90%: 39
repeats)– 40-100+: will develop HD if person lives long enough
• Increased meiotic instability in males - Paternal transmission of expanded allele associated with over 3/4 of juvenile disease
• Encodes 348 kD huntingtin protein which is a target for caspase 3, a protease associated with neuronal apoptosis



Anticipation

King George’s Medical University UP, Lucknow, INDIA King George’s Medical University UP, Lucknow, INDIA





























