Embed Size (px)
Citation preview

HDC Webinar Series
Introduced by Dwayne Spradlin, CEO, Health Data Consortium
Clinical Trial Data Transparency:Explaining Governance for Public Data Sharing
Chris Boone, Avalere Health LLC

Clinical Trial Data TransparencyExplaining Governance Considerations for PublicData SharingChris Boone

Objectives and Outputs for Today’s Webinar
3
PROJECT OVERVIEW
Avalere recognizes the current landscape and the drive of data transparency.This session provides an overview of the clinical trial data transparencydiscussion in the EU and discusses some of the key barriers to effectivelygoverning the process for collecting, storing, securing, authorizing access, andauditing the data from a regulatory perspective.
MEETING OBJECTIVES
● Review the rationales for and benefits/risks of clinical trial transparency
● Discuss responsible use of public data sharing
● Analyze key barriers to overcome with data collection and standardization
● Describe legal and policy implications allowing public access to data
● Identify and describe reasonable data sharing models

Overall Approach To The Project
4
AVALERE FOLLOWED A FOUR-STEP PROCESS TO COMPLETE THE RESEARCH
DevelopResearchStrategy
Summarizeand Report
Key FindingsConsultInternalAvalereExperts
Conduct aLiteratureand Policy
Review
Define: Criteria for
structuredsearch ofwhite andgreyliterature
Developoutline
Consult residentAvalere experts
Use expertinsights to guidetargetedliterature review
Identifycrediblesources thatdiscuss theEU regulatoryframeworkand its history
Address datainfrastructureneeds
Summarize keyfindings acrosseach domain infinal report
“White paper”provideshighlights inconsolidatedformat

Key Definitions for Today’s Discussion
5
● IPD: Individual participant data
● CSR: clinical study reports
● Data sharing: “the responsible entity making the data available via open or restricted
access, or exchanged among parties”
● Data generator: “may include industry sponsors, data repositories, and researchers
conducting clinical trials”
● Data holder: “to mean the entity or entities that have access to data, including
regulatory agencies, journals to which manuscripts are submitted, and other
repositories such as ClinicalTrials.gov in the US”
● Intervention: “a process or action that is the focus of a clinical trial. This can include
giving participants drugs, medical devices, procedures, vaccines, and other products
that are either investigational or already available.”

Background

Context of the Study
7
● Clinical trials are essential to determining the safety of medical interventions and
their ability to achieve particular health outcomes
● Clinical trials are required by regulatory authorities around the world before a new
medical product can be brought to market, or before a new indication, formulation,
or target population can be approved for an intervention already on the market
● Vast amounts of data are generated over the course of a clinical trial – held by
sponsors
● Data from clinical trials, including participant-level data, are being shared by
sponsors and investigators more widely than ever before
● Some sponsors have voluntarily offered data to researchers, some journals now
require authors to agree to share the data underlying the studies they publish
● For more than a decade, policymakers have sought to expand public access to
information about planned, ongoing, and completed clinical trials
● The EMA has proposed the expansion of access to data submitted in
regulatory applications

8
“Science is built on data: its collection,analysis, publication, reanalysis,critique, and reuse. However, thecurrent system of scientific publishingworks against maximum disseminationof the scientific data underlyingpublications. Barriers include inability toaccess data, restrictions on usageapplied by publishers or data providers,and publication that is difficult toreuse…”
“Science is built on data: its collection,analysis, publication, reanalysis,critique, and reuse. However, thecurrent system of scientific publishingworks against maximum disseminationof the scientific data underlyingpublications. Barriers include inability toaccess data, restrictions on usageapplied by publishers or data providers,and publication that is difficult toreuse…”

Clinical Trial DataTransparency
Less Flexible More Flexible
Clinical Trial DataTransparency
Less Flexible More Flexible
There is an Ongoing Push for Greater Transparency of Clinical TrialData in Europe and the United States
9
IncreasingSharing
Requirements
IncreasingSharing
Requirements
LimitedSharing
Initiativesand Offers
LimitedSharing
Initiativesand Offers
REGULATORS MUST WEIGH THE RISKS AND BENEFITS OF SHARING CLINICAL TRIAL DATA
EMAEMA FDAFDAUK House
ofCommons
UK Houseof
Commons
PhRMA
PhRMA
EFPIAEFPIA
EuropeanAlliance forPersonalize
Medicine
EuropeanAlliance forPersonalize
Medicine
IOMIOM
IndividualCompanies/Institutions
IndividualCompanies/Institutions
Regional National
EFPIA: European Federation of Pharmaceutical Industries and AssociationsEMA: European Medicines AgencyIOM: Institute of MedicinePhRMA: Pharmaceutical Research and Manufacturers of America

Growing pressure from academic groups and patient-consumeradvocates to release clinical trial data led to EMA decision
10
EMA TAKES THE FOLLOWING VIEWS AND POSITIONS:
● Enabling public scrutiny and secondaryanalysis of CTs
● Protection of personal data (PPD)
● Respect for the boundaries of patients’informed consent
● Protection of commercially confidentialinformation (CCI)
● Ensuring future investment in bio-pharmaceutical research anddevelopment (R&D)
● Addressing the consequences ofinappropriate secondary data analysis
● Protecting the Agency’s and the EuropeanCommission’s deliberations and decision-making process
● Ensuring that transparency is a two-waystreet

Timeline* of Developments Related to Clinical Data Transparencyin Europe
● Prior to 2010, EMA released clinical trial reports upon request. It is now working to finalize its policy onpublishing clinical trial data proactively once the decision-making process on an application for a EU-widemarket authorization is complete
● As a separate but concurrent initiative, EMA is working to make summaries of the results of clinical trialspublicly available through the EU Clinical Trials Register
● In December 2013, Council of the European Union released the revised Proposal for Regulation on ClinicalTrials (aimed at repealing Directive 2001/20/EC). If approved by the European Parliament in 2014, each EUmember state will automatically be subject to the regulation and new requirements for public transparency ofclinical trials’ results
● There have also been national debates about data transparency:
● For example - U.K.: House of Commons ordered a report Access to clinical trial information and thestockpiling of Tamiflu (Dec 18 203)
● Europe is more pro-active than the U.S., and works within a different legal construct, but biopharma data isoften based on global development activities
Mar 19-20 2014
Discussion aboutEMA Policy at
EMA ManagementBoard Meeting
Dec 12 2013Jun 24 2013
11*Timeline not to scale.
Jul 17 2012
EuropeanCommission
adopts proposal toreplace Clinical
Trials Directive of2001
EMA releasesdraft Policy 70 fora 3-month public
consultation
EMA releases keyprinciples
Council of EUreleases proposal
on sharing ofclinical trial data
Dec 17 2013 May 22 2014
EU Elections
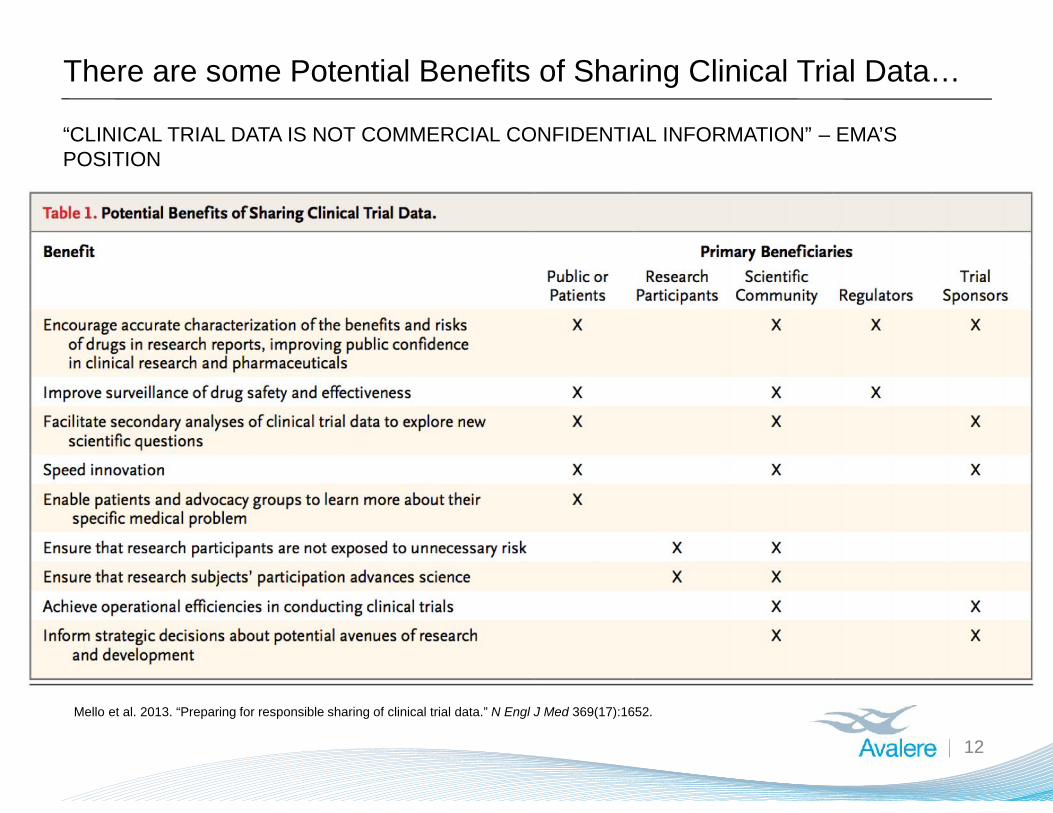
There are some Potential Benefits of Sharing Clinical Trial Data…
12
“CLINICAL TRIAL DATA IS NOT COMMERCIAL CONFIDENTIAL INFORMATION” – EMA’SPOSITION
Mello et al. 2013. “Preparing for responsible sharing of clinical trial data.” N Engl J Med 369(17):1652.

… but there are some Risks or Unintended Consequences as well.
13
o Patient Privacy: It is difficult to effectively de-identify certain data –especially data from trials concerning rare diseases or other small trials
o Poorly Conducted Analyses: Could also lead to unskilled analysts,market competitors, or others with strong private agendas to publicizepoorly conducted analyses
o Dis-incentivizes future research: Could affect incentives to invest inresearch to develop new medical products.
o Operational Costs: Could potentially administratively burdensome tooperate since regulatory agencies are already overwhelmed
1
2
3
4

Clinical Data Management

What is Clinical Data Management (CDM)
15
DEFINED AS “THE DEVELOPMENT, EXECUTION AND SUPERVISION OF PLANS,POLICIES, PROGRAMS AND PRACTICES THAT CONTROL, PROTECT, DELIVER ANDENHANCE THE VALUE OF DATA AND INFORMATION ASSETS” IN THE CLINICALTRIAL ARENA
CDM Domains
DataGovernance
DataArchitecture
DatabaseManagement
Data SecurityManagement
Data QualityManagement
Reference andMaster DataManagement
DataWarehousingand BusinessIntelligence
MetadataManagement
Lu et al. 2010. “Clinical data management: Current status, challenges, and future directions from industry perspectives.” OpenAccess Journal of Clinical Trials 2:93-105.
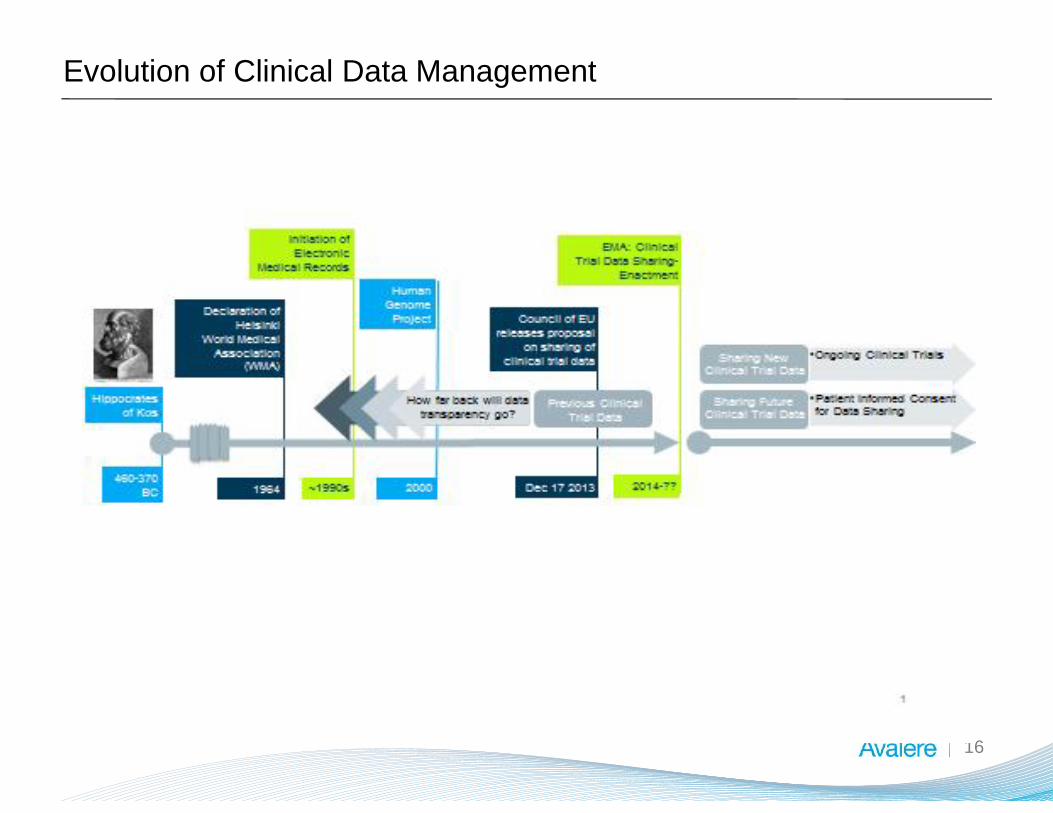
Evolution of Clinical Data Management
16

What Data is Shared?
17
● Depending on the regulatory jurisdiction, data might or might not be shared or made available
to the public for secondary uses
● Shared data might include both summary data and individual patient data
● In the US, if a sponsor is seeking regulatory approval, data are shared in confidence with
regulators
● In the EU, the EMA has undertaken regulatory action to share anonymized clinical trial data
with external requestors.
● Select study data might also be made available to individual researchers on a case by case
basis upon request, or could be made publicly available, usually at the summary level, for
example, through publication in a peer-reviewed journal or through publicly accessible clinical
trial registration sites (e.g. ClinicalTrials.gov)
● Data not shared includes:o Analyzable data setso Clinical study reports (CSRs)o Individual participant data (IPD)

Current Practices in Data Disclosure
18
● Publication in peer-reviewed scientific journal is currently the primary
methodo Scientific journal articles generally contain a brief summary of the trial
background, research question(s), methodology, results, figures andtables, and discussion
● Clinical trial sponsors seeking regulatory approval from authorities such as
the FDA and the EMA must submit detailed CSRs and IPD as required,
which forms the basis of the marketing application for a product
● Case-by-case basis upon request
● Discussions have not been specific regarding which datasets might be
shared

Providers and Recipients of Shared Data
19
PROVIDERS
● Individual participants in a clinical trial
● Clinical trial funders (e.g. govnerment,
industry, foundations, or advocacy
organizations)
● Contract research organizations
● Principal investigators or their institutions
● Site principal investigators of a multisite
trial
● Data coordinating center
● Regulatory agencies
● Systematic reviewers and guideline
developers
RECIPIENTS
● Individuals participating in the trial
● Secondary researchers
● Institutions supporting the researchers
● Funding agencies
● IRBs or scientific peer review committees
● The DSMB or DMC for another clinical
trial
● Educators
● A disease advocacy group seeking to
advance research
● Prospective plaintiffs or attorneys
● Competitors of the industry sponsor
● Members of the media
● Interested members of the public

Current Model for Clinical Trials Results Dissemination
20
Van Valkenhoef et al. “Deficiencies in the transfer and availibility of clinical trials evidence: a review of existing systems andstandards.” BMC Medical Informatics and Decision Making 2012, 12:95.
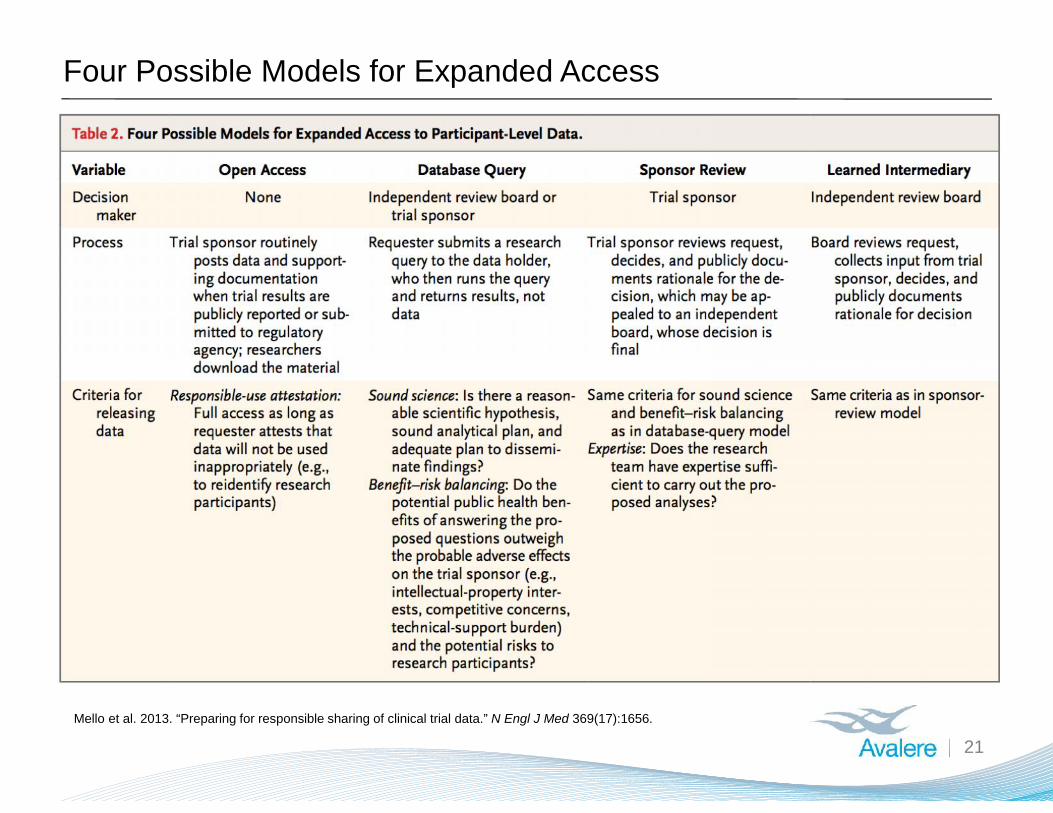
Four Possible Models for Expanded Access
21Mello et al. 2013. “Preparing for responsible sharing of clinical trial data.” N Engl J Med 369(17):1656.

Alternative Model for Clinical Trials Results Dissemination
22
Van Valkenhoef et al. “Deficiencies in the transfer and availibility of clinical trials evidence: a review of existing systems andstandards.” BMC Medical Informatics and Decision Making 2012, 12:95.

Guidance Considerations for the Future ofData Sharing

To minimize potential harms and maximize the public health withincreased sharing of clinical trial data, one must do the following:
24
● Respecting all promises made by trial investigators to study participants,
including individuals’ informed consent, as well as contracts and
agreements with institutions and research facilities;
● Safeguarding study participant privacy such that individual patients cannot
be identified through subsequent studies; and
● Ensuring the quality of data, including the appropriateness of the
availability, use and reliability of the data such that conclusions from
subsequent research are valid, and will not undermine current and future
therapies and their appropriate use in the interests of public health.
1
2
3

What is Data Quality?
25
“Data quality” has been broadly defined as “data strong enough to supportconclusions and interpretations equivalent to those derived from error-freedata
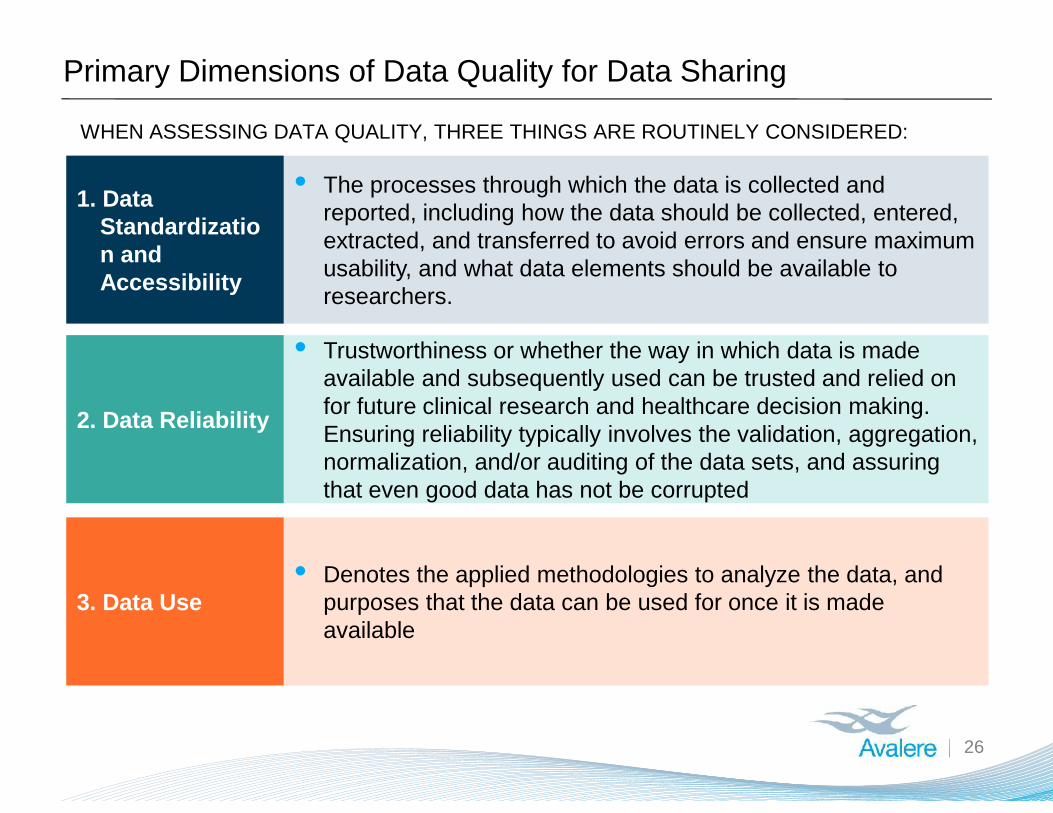
Primary Dimensions of Data Quality for Data Sharing
26
1. DataStandardization andAccessibility
2. Data Reliability
3. Data Use
• The processes through which the data is collected andreported, including how the data should be collected, entered,extracted, and transferred to avoid errors and ensure maximumusability, and what data elements should be available toresearchers.
• Trustworthiness or whether the way in which data is madeavailable and subsequently used can be trusted and relied onfor future clinical research and healthcare decision making.Ensuring reliability typically involves the validation, aggregation,normalization, and/or auditing of the data sets, and assuringthat even good data has not be corrupted
• Denotes the applied methodologies to analyze the data, andpurposes that the data can be used for once it is madeavailable
WHEN ASSESSING DATA QUALITY, THREE THINGS ARE ROUTINELY CONSIDERED:

Data Standardization and Accessibility
27
ISSUES WITH DATA STANDARDIZATION AND ACCESSIBLITY MAY LIMIT THE ABILITY OFSUBSEQUENT RESEARCH TO DRAW CONCLUSIONS
MAJOR FINDINGS
1.1: One component of achieving subsequently integrate-able data is defining whatconstitutes proper collection and reporting of data in the first place. Common terms andstandards for data entry, collection, and transfer can enable standardized reporting.
1.2: It is important to note which data elements are available from a single study andare the same data elements available from other studies (i.e., what is the level ofconsistency of the data available across multiple studies).
1.3: Appropriate cross-study analyses require consistent data element accessibilityacross initial data sets.
1.4: Another consideration is the current heterogeneity of clinical data management(CDM) systems, software systems that assist in the process of collecting and managingtrial data, which can compromise the quality of data by hindering its exchange. Someexpect this lack of interoperability between the various systems to hinder the exchangeof trial data in Europe

Data Reliability
28
POOR DATA RELIABILITY HINDERS MEANINGFUL USE OF ALL DATA
MAJOR FINDINGS
1.1: For the processes of validation, normalization, and auditing to yield high qualitydata that can be used in an interoperable manner among different European memberstates, or between other stakeholders, standardized information technology solutionsneed to be in place to facilitate them.
1.2: In response to this challenge, the European Clinical Research InfrastructureNetwork (ECRIN) Working group on Data Centers recently developed standardrequirements through expert consensus. These standards specify the criteria “for highquality [Good Clinical Practice] GCP-compliant data management in multinationalclinical trials.”
1.3: A good start to ensuring data integrity and the potential for appropriate publichealth conclusions to be reached in secondary studies may be requiring that recipientsof data to be shared under the EMA’s publication and access to clinical trial data draftpolicy certify that they will abide by these standards.

Data Use
29
LACK OF CLARITY IN RESEARCH PROTOCOLS MAY LIMIT EXPECTED GAINS FROMINCREASED CLINICAL TRIAL DATA SHARING
MAJOR FINDINGS
1.1: Clear standards for reporting on methodology - for both those submitting clinicaltrial data as well as those accessing it subsequently - help researchers reachappropriate conclusions.
1.2: Protocols for secondary studies and analyses will also need to be made public andbe subjected to the same standards of methodological rigor as the original research, orindeed, any other clinical research.
1.3: The worst case scenario is a flawed study impacting regulatory approvals – eitherby allowing a truly ineffective or unsafe drug on the market or preventing a trulyeffective or safe drug access to the market. For products already approved, flawedstudies can cause extreme confusion or unnecessary public anxiety, and even result indrugs being wrongfully suspended or withdrawn from the market.

Noteworthy Strategies and Tools to Address Challenges
30
● Accountability for the release of the data
● Educational campaigns to support consumers and the general public
● Training for researchers
● Tools to facilitate informed consent
● Organize research/consensus meetings around clinical data standards
● Mechanisms to validate secondary research
● Outcomes assessments

Special Thanks to Project Team
PhRMA Team• Jonathan Kimball, MA (Deputy Vice President, International Affairs)
Avalere Team
• Gillian Woollett, MA, DPhil (Senior Vice President, FDA Practice)
• Chris Boone, PhD, MSHA (Vice President, Evidence Translation & ImplementationPractice )
• Brenda Huneycutt, PhD, JD, MPH (Senior Manager, FDA Practice)
• Judit Illes, BCL/LLB, MS (Senior Associate, Evidence Translation & ImplementationPractice)
• Kathryn Jackson, PhD (FDA Fellow)
• Debbie Garner, MBA (Europe, Middle East and Africa Regional Director)
31





























