Embed Size (px)
Citation preview

BASICS OF PHARMACOKINETICS
Mohammed Asadullah Jahangir, M.Pharm, (PhD)

The duration of drug therapy ranges from a single dose of drug for acute conditions to drugs taken life long for chronic conditions.
The frequency of administration of drug in a particular dose is called as dosage regimen.
The kinetics of drug absorption, distribution, metabolism and excretion (KADME) and their relationship with the pharmacological, therapeutic or toxicological response in man and animals is called Pharmacokinetics.

Other important definitions in Pharmacokinetics- Clinical Pharmacokinetics – It is application of
pharmacokinetic principles in the safe and effective management of individual patient.
Population Pharmacokinetics – It is the study of pharmacokinetic differences of drugs in various population groups.
Toxicokinetics – It is the application of pharmacokinetic principles to the design, conduct and interpretation of drug safety evaluation studies.

Plasma Drug Concentration-Time Profile after oral administration of single dose of drug

Two categories can be evaluated from a plasma concentration time profile-
1. Pharmacokinetic Parameters
2. Pharmacodynamic Parameters
1. Pharmacokinetic Parameters- Peak plasma concentration (Cmax ) – It is expressed in mcg/ml. It gives the
rate of drug absorption. It depends upon-
-dose administered
-rate of absorption
-rate of elimination Time of peak concentration (tmax ) - It is expressed in hours. It gives the rate
of drug absorption. Onset time and onset of action is dependent on tmax. Area under the curve (AUC) – It is expressed in mcg/ml*hours. It gives the
extent of absorption. It is the important parameter in evaluating the bioavailability of drug from its dosage form.

2. Pharmacodynamic Parameters-
Minimum effective concentration (MEC) Maximum safe concentration (MSC) Onset of action Onset time Duration of action Intensity of action Therapeutic range Therapeutic index - The ratio of maximum safe
concentration to minimum effective concentration of the drug is called as the therapeutic index i.e. MSC/MEC.

Order of a reaction - The manner in which the concentration of drug influences the rate of reaction or process is called as the order of reaction.
For a general reaction,
dC/dt = -KCn
K= rate constant
n = order of reaction
n = 0, zero order kinetics
n = 1, first order kinetics
Basically three types of kinetics are encountered- Zero order kinetics First order kinetics Mixed order kinetics

Zero order kinetics (constant rate process)
dC/dt = -K0C0 = -K0
K0=zero order rate constant in mg/min
After rearrangement and integration the comes out to be,
C = C0-K0t
Zero order half life
T1/2=C0/2K0
The half life of zero order is proportional to the initial concentration of drug C0 and inversely proportional to K0
Examples of zero order- Controlled drug delivery such as that from i.m. implants or osmotic pumps Administration of a drug as a constant rate i.v. infusion

First order Kinetics (Linear kinetics)
If n=1,
dC/dt = -KC
K=first order rate constant, in time-1 or per hour
The rate of a first order reaction is directly proportional to the concentration of drug undergoing reaction i.e. greater the concentration, faster the reaction.

After rearrangement and integration the equation will give
C = C0e-Kt or, logC = logC0 - Kt/2.303
First order process is also called as mono-exponential rate process. A semilogarithmic plot of above equation gives a straight line with
slope = -K/2.303 and y-intercept = logC0

First order half life
t1/2 = 0.693/K
The half life of first order process is a constant and independent of initial drug concentration.
Most pharmacokinetic processes like absorption, distribution and elimination follow first order kinetics.

Mixed order kinetics (Nonlinear Kinetics)
Sometimes the kinetics of the pharmacokinetic processes fluctuates from predominantly first order to predominantly zero order, therefore such kind of kinetics is called mixed order kinetics. Mixed order kinetics is called dose dependent kinetics or nonlinear kinetics.
Example-
Drug absorption of vitamin C
Drug distribution of naproxen
Drug elimination of riboflavin
Nonlinearity is usually seen when the pharmacokinetic processes involves carriers or enzymes which are substrate specific, which gets saturated at high drug concentration i.e. capacity limited. Such capacity limited processes is described by Michaelis-Menten kinetics.

Methods for
Analysis of Pharmacokinetic Data
Model ApproachC
ompartment Model
Physiological Model
Distributed Parameter Model
Model-
Independen
t Approach
Noncompar-tmental Analysis
Mammillary
ModelPerfusion
Limited ModelCatenary Model

Pharmacokinetic Model Approach Compartment Models: The body is divided into hypothetical compartments
arranged either in series or parallel to each other, communicating with each other. These compartments are virtual and is considered as tissue or group of tissues that have similar drug distribution characteristics i.e. similar blood flow and affinity. Rate of drug movement between compartments follows first order kinetics.
Depending upon whether the compartments are in series or parallel to each other they are divided in two categories-
-Mammillary Model
-Catenary Model
Mammillary Model - It is the most common model. In this model the central compartment or the compartment 1is connected parallel to the peripheral compartment. The central compartment has high vascularity and high perfusion like lungs, liver, kidneys. Elimination also occurs through these compartments in most cases. The peripheral compartments or tissue compartment or compartment denoted by numbers 2,3,4...etc have low vascularity and poor perfusion. Movements of drugs between the compartment follows first order kinetics.
[Note-K12 denotes the drug movement from compartment one to compartment two or from central compartment to one of the peripheral compartment.]
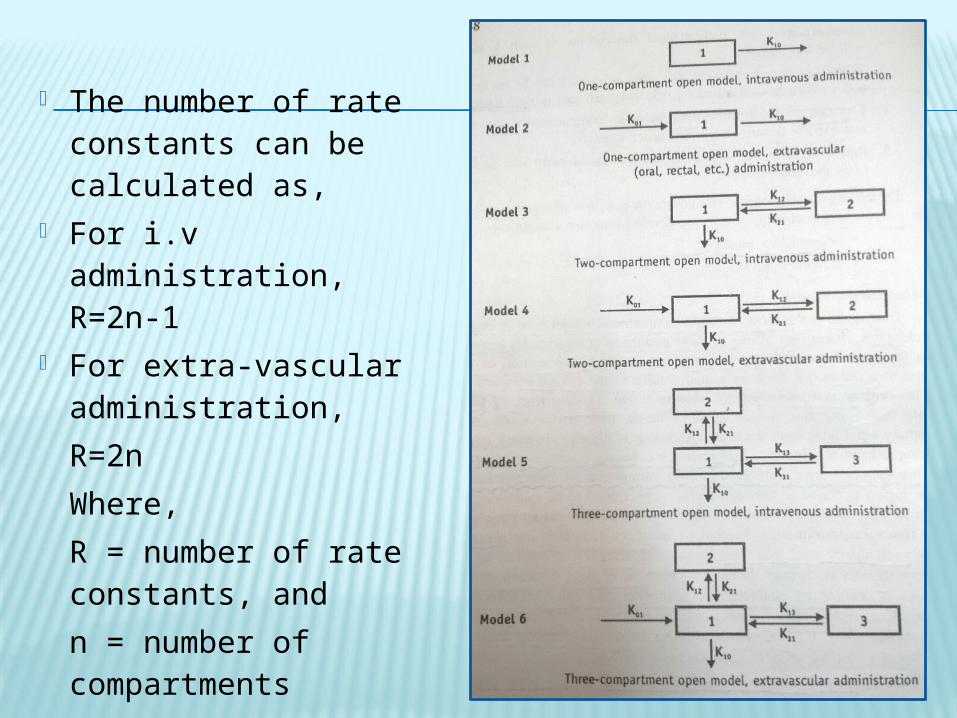
The number of rate constants can be calculated as,
For i.v administration, R=2n-1
For extra-vascular administration, R=2nWhere, R = number of rate constants, and n = number of compartments

Catenary Model – In catenary model the compartments are joined in series.
This model is rarely used.
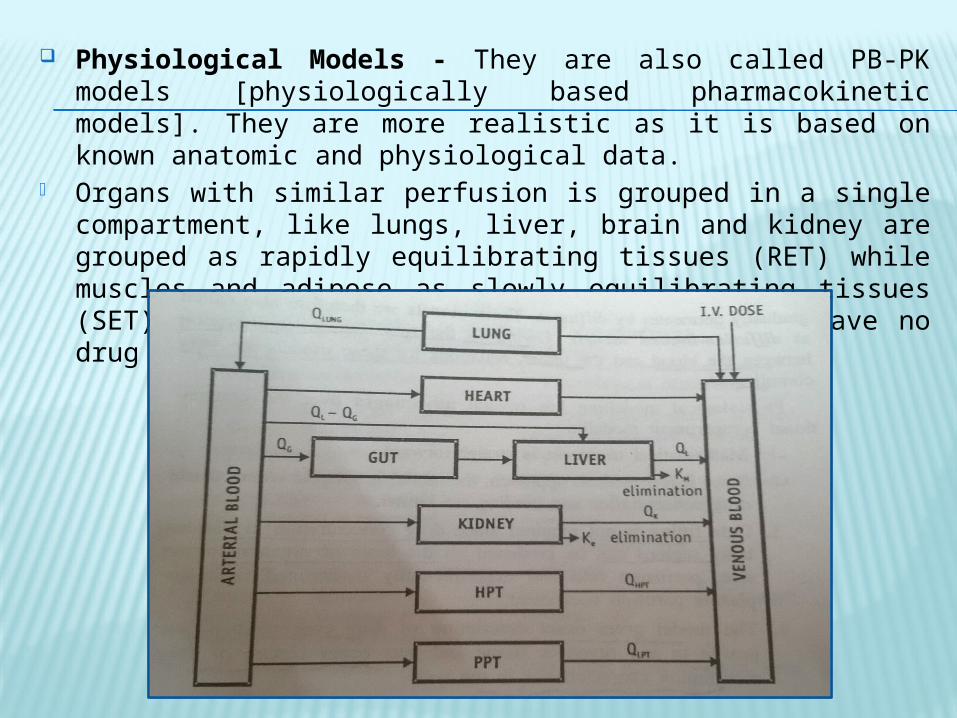
Physiological Models - They are also called PB-PK models [physiologically based pharmacokinetic models]. They are more realistic as it is based on known anatomic and physiological data.
Organs with similar perfusion is grouped in a single compartment, like lungs, liver, brain and kidney are grouped as rapidly equilibrating tissues (RET) while muscles and adipose as slowly equilibrating tissues (SET). Organs or tissues such as bones that have no drug penetration is excluded.

They are of two categories- Perfusion rate limited/blood flow rate limited - It is based on the
assumption that the drug movement within a body region is much more rapid that its rate of delivery to that region by the perfusing blood. However it is only applicable to the highly membrane permeable drugs i.e. low molecular weight, poorly ionised and highly lipophilic drugs example thiopental, lidocaine etc.
Diffusion rate limited/membrane permeation rate limited models - It is more complex and is applicable to drugs in which the cell membrane acts as a barrier like for highly polar, ionised and charged drugs.
Distributed parameter model - This model is analogous to physiological model. This model is specifically useful for assessing regional differences in drug concentrations in tumours or necrotic tissues.

Applications of pharmacokinetic models It helps in understanding the behaviour of drugs in
patients. It helps in predicting the concentration of drug in various
body fluids It helps in optimizing the dosage regimen for individual
patients. It helps in evaluating the risk of toxicity. It helps in correlating plasma drug concentration with
pharmacological response. It helps in determining the influence of altered
physiology on drug ADME. It helps in explaining the drug interactions.

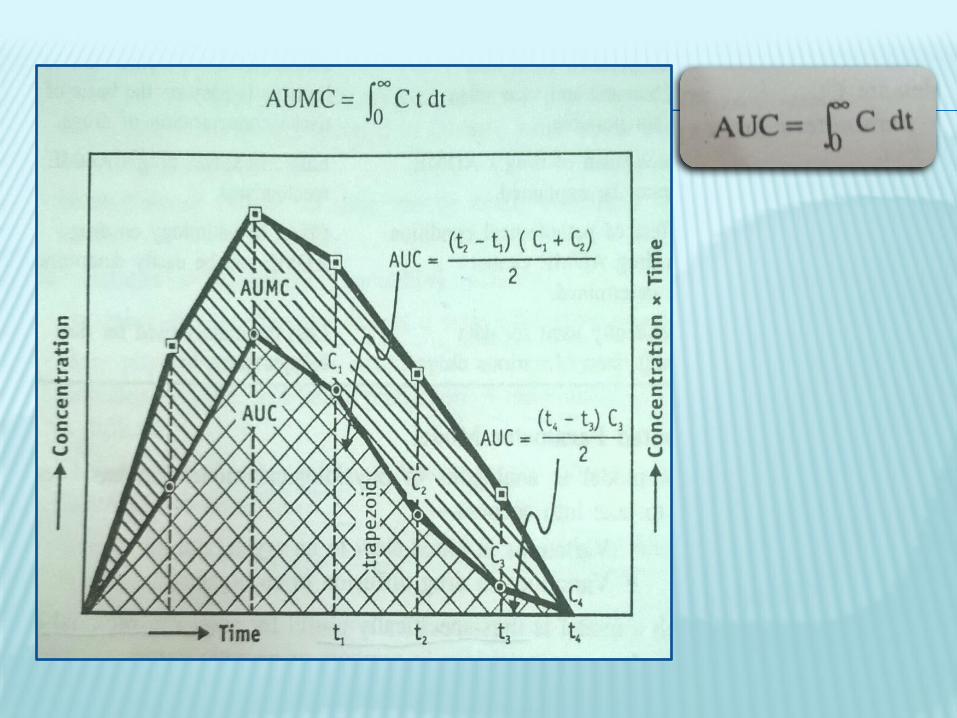
Non-compartmental Analysis or Model-Independent method- It follows linear kinetics so it can be applied to any compartment model. It is based on the statistical moments theory, which involves collection of experimental data following a single dose of drug.
MRT=AUMC/AUC MRT= mean residence time of drug
AUMC= area under the first moment curve
AUC= area under curve/zero moment curve
AUMC is obtained by the plot of product of plasma drug concentration and time vs time i.e. C.t vs t
AUC is obtained by plotting C vs t.

Application of Model Independent/Non-compartmental Model-
It is used to estimate the important pharmacokinetic parameters like bioavailability, clearance and apparent volume of distribution.
It is used in determining half life, rate of absorption and first order absorption rate constant of the drug.

THANK YOU





























