Embed Size (px)
Citation preview

A Case for Cystic Fibrosis
Presentation by: Chris Lim

History CC: “Our daughter always seems to be catching any respiratory
illness that comes around.” HPI: 3 year old Caucasian female presents with a severe upper
respiratory infection PMHx: Thought to have had a “meconium ileus” at birth
• Issue was said to have resolved itself and there were no further complications
Meds: None Allergies: None Hospitalizations: Unknown PSH: Unknown SocHx:
• Parents state they do not smoke in home• Mother states that pt has bad eating habits• Patient lives at home with parents & is only child
FmHx:• Mother stated she has two deceased brothers, both lost to
different types of pneumonia (one brother at age 10, one at age 21)
• Maternal grandmother had a sibling who died from pneumonia (unknown age)

Review of Systems
General:
+ Cannot gain weight
Respiratory:
+ Shortness of breath
+ Productive cough
GI:
+ Greasy, foul-smelling feces

Physical Examination
BP: 90/60 Respiratory Rate: 26
Temperature: 100.6°F Pulse: 102 bpm
Patient appears to be moderately distressed
Only in 20th percentile for height & weight based on age
TM’s are intact w/no erythema
Oropharynx is clear w/no erythema or tonsillar exudate
Supple neck
Cardiovascular tachycardia w/no evidence of murmur
Bilateral expiratory wheezing/rhonci in lungs bilaterally
Digital clubbing of the extremities

Differential Diagnosis
DDx
DD
Ddx
D/Dx

The Differential Diagnosis / Differential Diagnostic Procedure
Systematic method to identify the presence of a disease or condition, when there are many alternative entities possible
• Based on symptoms, patient history
Hypothetico- Deductive method, (process of elimination)
• Utilizing medical knowledge• Flowcharts or algorithms. • “Working Down the List”
Potential qualifiers or disease conditions can be viewed as the hypotheses.
DDx will guide which tests/panels to order (if necessary).
• “Work-up”
DDx will influence the flow towards Assessment & Plan. • Conducted to provide a list to confirm or R/O
conditions. • Re-checking the patient condition and follow-up may
require additional differential diagnosis, to verify the beneficial or harmful effects of the primary diagnosis.

DDx:
I. Symptoms List
II. Possible Causes
III.
a) Most Dangerous/High Stakes
b) Most Likely
c) Low Stakes
d) Unlikely
IV. R/O or treat possible causes

Central Problem Statement:
Upper Reparatory Tract Infection, SOB and productive cough in a 3 y/o Caucasian girl with a history of URI, anorexia, steatorrhea, tachycardia, bilateral wheezing/rhonchi & digital clubbing

?

POSSIBLE PATHOLOGICAL PROCESSES:
V- Vascular
I – Inflammatory/ Infectious
N - Neoplastic
D – Degenerative/ Deficiency/ Drugs
I – Idiopathic/ Intoxication / Iatrogenic
C – Congenital
A – Autoimmune / Allergic / Anatomic
T - Traumatic
E – Endocrine/ Environmental
M - Metabolic
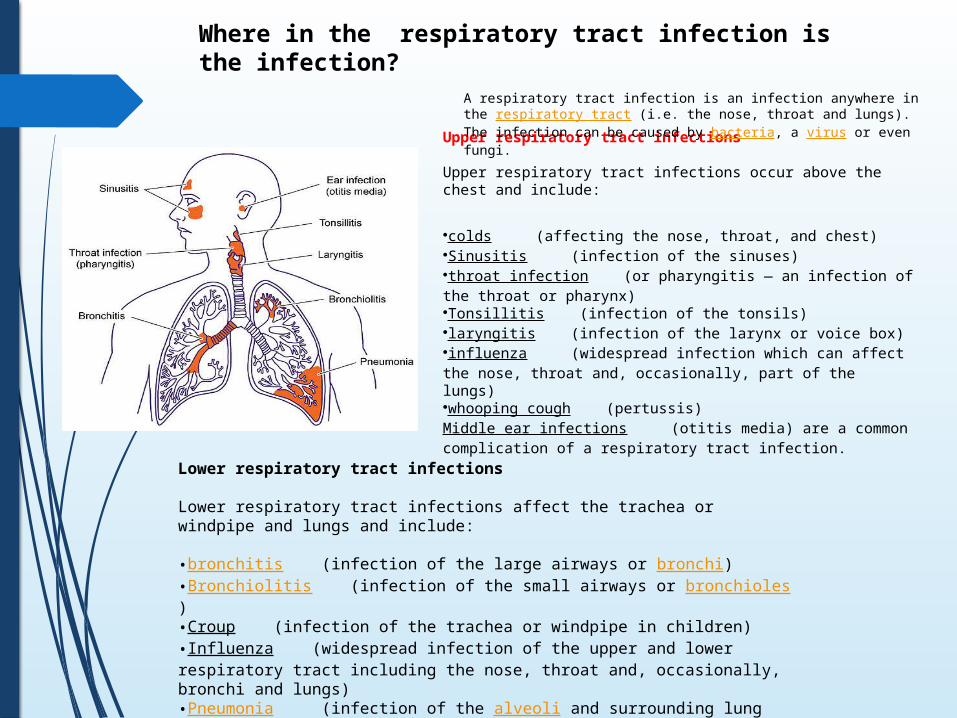
Upper respiratory tract infections
Upper respiratory tract infections occur above the chest and include:
•colds (affecting the nose, throat, and chest) •Sinusitis (infection of the sinuses) •throat infection (or pharyngitis — an infection of the throat or pharynx) •Tonsillitis (infection of the tonsils) •laryngitis (infection of the larynx or voice box) •influenza (widespread infection which can affect the nose, throat and, occasionally, part of the lungs) •whooping cough (pertussis) Middle ear infections (otitis media) are a common complication of a respiratory tract infection.
Where in the respiratory tract infection is the infection?
A respiratory tract infection is an infection anywhere in the respiratory tract (i.e. the nose, throat and lungs). The infection can be caused by bacteria, a virus or even fungi.
Lower respiratory tract infections
Lower respiratory tract infections affect the trachea or windpipe and lungs and include:
•bronchitis (infection of the large airways or bronchi)•Bronchiolitis (infection of the small airways or bronchioles)•Croup (infection of the trachea or windpipe in children)•Influenza (widespread infection of the upper and lower respiratory tract including the nose, throat and, occasionally, bronchi and lungs)•Pneumonia (infection of the alveoli and surrounding lung tissue)

Respiratory Tract Infection
1) Nasopharynx
2) Larynx
3) Pulmonarya) Lung Parenchyma
4) Chronic Obstructive Pulmonaryb) Emphysemac) Chronic Bronchitisd) Bronchial Asthma


Common
Allergies
Asthma or reactive airway disease
Gastroesophageal reflux disease
Infections• Bronchiolitis
• Bronchitis
• Pneumonia
• Upper respiratory infection
Obstructive sleep apnea
Uncommon
Bronchopulmonary dysplasia
Foreign body aspiration
Causes of Wheezing in Children and Infants
Rare
Bronchiolitis obliterans
Congenital vascular abnormalities
Congestive heart failure
Cystic fibrosis
Immunodeficiency diseases
Mediastinal masses
Primary ciliary dyskinesia
Tracheobronchial anomalies
Tumor or malignancy
Vocal cord dysfunction

Question Indications
How old was the patient when the wheezing started?
Distinguishes congenital from noncongenital causes
Did the wheezing start suddenly?
Foreign body aspiration
Is there a pattern to the wheezing?
Episodic: asthma
Persistent: congenital or genetic cause
Is the wheezing associated with a cough?
GERD, sleep apnea, asthma, allergies
Is the wheezing associated with feeding?
GERD
Is the wheezing associated with multiple respiratory illnesses?
Cystic fibrosis, immunodeficiency
Is the wheezing associated with a specific season?
Allergies: fall and spring
Croup: fall to winter
Human bocavirus*
Human metapneumovirus: December through April
RSV: fall to spring
Does the wheezing get better or worse when the patient changes position?
Tracheomalacia, anomalies of the great vessels
Is there a family history of wheezing?
Infections, allergic triad
Questions to Distinguish the Etiology of Wheezing in Children
GERD = gastroesophageal reflux disease RSV = respiratory syncytial virus.

Signs and symptoms Presumptive diagnosis Further evaluation
Associated with feeding, cough, and vomiting
Gastroesophageal reflux disease 24-hour pH monitoring Barium swallow
Associated with positional changes Tracheomalacia; anomalies of the great vessels
AngiographyBronchoscopyCT Chest radiographyor MRI
EchocardiographyAuscultatory crackles, fever Pneumonia Chest radiography
Episodic pattern, cough; patient responds to bronchodilators
Asthma Allergy testingPulmonary function testing
Trial of albuterol (Proventil)Exacerbated by neck flexion; relieved by neck hyperextension
Vascular ring AngiographyBarium swallowBronchoscopyChest radiographyCT or MRI
Heart murmurs or cardiomegaly, cyanosis without respiratory distress
Cardiac disease AngiographyChest radiographyEchocardiography
History of multiple respiratory illnesses; failure to thrive
Cystic fibrosis or immunodeficiency Ciliary function testing
Immunoglobulin levels
Sweat chloride testingSeasonal pattern, nasal flaring, intercostal retractions
Bronchiolitis (RSV), croup, allergies Chest radiography
Stridor with drooling Epiglottitis Neck radiography
Sudden onset of wheezing and choking
Foreign body aspiration Bronchoscopy
Differential Diagnosis of Wheezing According to Characteristic Signs and Symptoms

Pediatric Aspergillosis is a severe complication of Cystic Fibrosis



Bronchiectasis can cause Cystic Fibrosis

?

DDx Summary:I. Symptoms List
• CC & HPI
II. Possible Causes
• (Slides 9-12)
III.
a) Most Dangerous/High Stakes
• Bronchiectasis*
b) Most Possible
• Cystic fibrosis
c) Low Stakes
• Pediatric aspergillosis
d) Unlikely
• Pediatric gastroesophageal reflux
• Pediatric hypersensitivity pneumonitis
IV. R/O or treat possible causes
• Workup of DDx→Dx
*Bronchiectasis—permanent dilation of thebronchioles, loss of airway, air is trapped, may be concomitant with tumor or necrotizing infection.

Working Diagnosis:
Cystic fibrosis

Cystic fibrosis: clinical presentation
Cystic fibrosis is caused by a mutation in an ion channel, this affects exocrine secretions in multiple organ systems.
• Respiratory manifestations include persistent, productive cough; hyperinflation of the lung fields on chest radiographs; and pulmonary function tests consistent with obstructive airway disease. Sinus disease is present in a majority of patients.
• Pancreatitis and insufficient exocrine function of the pancreas (leading to CF-related diabetes) are common symptoms, along with oily stools and malabsorption of fats and proteins,.
• Reduced bone mineral content and hypertrophic osteoarthropathy (abnormal proliferation of skin and osseous tissue at the distal parts of the extremities, leading to digital clubbing) may also be observed in patients with CF.
• Neonates frequently present with meconium ileus and demonstrate poor weight gain.

Cystic fibrosis: diagnostic criteria
A diagnosis of cystic fibrosis requires clinical symptoms consistent with CF in at least one organ system as well as evidence of a dysfunctional CFTR (cystic fibrosis transmembrane conductance receptor).
• Evidence of a dysfunctional CFTR can come in the form of elevated sweat chloride (≥60 mmol/L in a sufficient quantity [75 mg/15μL in 30 minutes] of collected sweat), abnormal nasal potential differences (NPD), or presence of two disease-causing mutations in CFTR in each parent allele.
The patient presented with a severe upper respiratory infection as well as a history of recurring respiratory illness. She has frequent greasy stools and a history remarkable for meconium ileus—an obstruction of the distal small intestine in neonates.
Physical exam finding reveal that she is only in the 20th percentile for height and weight. Her extremities revealed digital clubbing.


Epidemiology Most common life-limiting autosomal recessive disease in
Caucasions.
Point mutation in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) protein also known as an ATP-binding cassette (ABC) transporter.
Most common mutation is ΔF508-CFTR, occurs in >90% U.S. patients and 66% of all CF worldwide.
CFTR gene is located on chromosome 7q31.2
230,000 base pairs and is 1,480 amino acids long
CFTR protein contains two ATP-hydrolyzing domains composed of 6 α-helices each.

Requires the presence of two copies to manifest symptoms of disease.

Epidemiology cont.
Chloride ion channels are used in creating digestive juices, mucus, and sweat.
Around 1,500 other known mutations.
- Point mutation causes misfold in the protein.
- Protein remains trapped in the ER.- Disrupts chloride transport.
Mutated CFTR proteins don’t allow chloride, iodine, or thiocyanate to cross the cell membrane.

PathophysiologyLungs
Normal lung epithelium secretes chloride into the airway lumen via CFTR.
Impaired chloride transport leads to:
• Increased sodium and water absorption from airways into the blood.
• Dehydration of mucociliary blanket coating epithelial cells.
• Accumulation of thick mucus in the airways • Nutrient rich environment for reoccurring pulmonary
infections.• Burkholderia cepacia, Haemophilus influenza,
Pseudomonas aeruginosa, and Staphyloccus aureus.• Excessive neutrophilic inflammation

Pathophysiology cont.Gastrointestinal
80-90% of CF individuals have abnormal pancreatic function.
Gastrointestinal problems with cystic fibrosis due to inability of the pancreas to supply digestive enzymes to the intestine.
Volume of pancreatic enzymes decreases—pancreas secretes thick mucus
• Causes malabsorption of proteins and influences absorption of vitamin A, D, E, K
Abdominal pain, diarrhea, and steatorrhea is common.
20% of individuals with CF present with meconium ileus.
Meconium ileus is not related to the severity of CF.


Diagnostic Tests: Sweat Chloride
Chloride Concentration for Children and Adults
Result
≤ 39 mmol/L Normal
40 - 59 mmol/L Intermediate
≥ 60 mmol/L Indicative of CF

Further Diagnostic Testing
Genetic Testing—identify F508del mutation
Chest CT—detect bronchiectasis and clogged airways
Sinus CT—mucus/pus/polyps/damage
Sputum Cultures—detects bacteria that is characteristic to CF—Pseudomonas aeruginosa
Fecal Elastase—measure pancreatic elastase-1

Testing after Diagnosis
Imaging Tests
Lung Function Tests
Sputum Culture
Organ function Tests
• Regular diabetes testing after the age of 10

Screening
Prenatal Screening
• Genetic testing for parents
• Amniocentesis or chorionic villus sampling
Newborn Screening
• Elevated IRT levels in blood

Treatment Options
No cure, goal is to minimize symptoms to prevent complications
Accomplish this by:
• Preventing & controlling lung infections
• Loosening & removing mucus from lungs
• Preventing & treating intestinal blockage
• Providing adequate nutrition

Short-Term Treatment Options
Antibiotics to treat respiratory infection
NSAIDs to treat fever
Mucus-thinning drugs and bronchodilators to treat cough, SOB
Oral pancreatic enzymes to aid digestion

Long-Term Treatment Options
Chest Physical Therapy
• Loosens chest mucus, performed 1-4x/day
• Can be performed by someone clapping on patient’s chest or with a chest clapper, vibrating vest, or other breathing devices
Pulmonary Rehabilitation
• Long-term program including nutritional counseling, exercise training, energy conserving strategies, breathing techniques, and psychological counseling

Patient & Family Education
Screen future newborn children
High-calorie, high-fiber, high-salt diet
Fat-soluble vitamins
Drink lots of fluids
Ensure up-to-date immunizations
Hand washing
Exercise

Patient & Family Education“The beneficial role of aerobic exercise in maintaining
health in CF has been extensively reported in previous studies. A recent Cochrane review concluded that physical training has a positive effect on exercise capacity, strength and lung function. Exercise improves transepithelial potential difference in the CF airway, improves mucus clearance, increases recruitment in lung ventilation and enhances airway clearance. Aerobic capacity has been related to improved survival, quality of life and professional achievement in patients with CF. Children with CF who regularly exercise, enjoy improved quality of life while there are also cost-benefit implications since they require fewer antibiotics. Exercise recommendations include various aerobic activities 3-5 times per week for 20-45 min at intensity levels of 60-85% of maximum heart rate depending on the severity of CF lung disease.”
-“Aerobic exercise and respiratory muscle strength in patients with cystic fibrosis”, Theodore Dassios, Anna Katelari, Stavros Doudounakis, Gabriel Dimitriou

Questions & Comments

ReferencesBastien N, Chui N, Robinson JL, et al. Detection of human bocavirus in Canadian children in a 1-year study. J Clin Microbiol. 2007;45(2):610–613.
Dassios, Theodore, Anna Katelari, Stavros Doudounakis, and Gabriel Dimitriou. "Aerobic Exercise and Respiratory Muscle Strength in Patients With Cystic Fibrosis." Respiratory Medicine 107.5 (2013): 684-90. Web.
De Boeck, Kristiane, et al. "Cystic fibrosis: terminology and diagnostic algorithms." Thorax 61.7 (2006): 627- 635.
"Diagnosis: Testing." Johns Hopkins Cystic Fibrosis Center Diagnosis Testing EmSweat Testem Comments. Web. 5 Dec 2014. http://www.hopkinscf.org.
Finder JD. Understanding airway disease in infants. Curr Probl Pediatr. 1999;29(3):65–81.
Goldjian, Edward F. Rapid Review Pathology, p. 273-314. 2011.
Katkin JP. Cystic Fibrosis: Clinical Manifestations and Diagnosis. UpToDate. 2014. http://www.uptodate.com.
Martinati LC, Boner AL. Clinical diagnosis of wheezing in early childhood. Allergy. 1995;50(9):701–710.
Mayo Clinic Staff. "Cystic Fibrosis." Diseases and Conditions. Mayo Foundation for Medical Education and Research, 13 June 2012. Web. 06 Dec. 2014.
Porth, Carol M. Essentials of Pathophysiology. 3rd ed. N.P.: Lippincott Williams, 2011.
University of Miami Miller School of Medicine. Genetics Awareness Project. 2014. http://geneticsawareness.org.
US Department of Health and Human Services. Expert Panel Report 3: Guidelines for the Diagnosis and Management of Asthma. National Institute of Health: National Heart, Lung, and Blood
Institute. 2007.
Virant FS, Shapiro GG. Evaluation of the child with chronic cough and/or wheezing. In: Tinkelman DG, Naspitz CK. Childhood Asthma: Pathophysiology and Treatment. 2nd ed. New York, NY: Dekker; 1993:303–327.














