Embed Size (px)
Citation preview

FLASHPATHH A Z E M A L I

CYSTIC FIBROSIS
H A Z E M A L I

CLINICAL• Autosomal recessive disorder of ion transport that affects fluid
secretion in exocrine glands and in the epithelial lining of the respiratory, gastrointestinal, and reproductive tracts.
• Common disease in Caucasian– 1 in 2500 live births have disease– 1 in 20 are carriers
• Much lower incidence in other ethnic groups
• Usually manifests in Children and Adolescents

CLINICALClinical picture:• Respiratory:
– Recurrent infections– COPD “Bronchiectasis”
• GIT, Liver & Pancreas:– Pancreatic insufficiency “Steatorrhea, malnutrition”– Hepatobiliary disease and cirrhosis– Meconium ileus
• Genitourinary:– Congenital bilateral absence of the vas deferens “Infertility”
Note that the severity of clinical picture “phenotype”depends on the type of the mutation
(See Later)

CLINICALCommon associated respiratory infections:• Allergic bronchopulmonary aspergillosis• Pseudomonas aeruginosa• Hemophilus influenza• Mycobacteria (atypical)• Staphylococcus aureus
Two unique organisms for cystic fibrosis:• Burkholderia cepacia• Stenotrophomonus maltophilia
• Both are aggressive• Both are treated with trimethoprim-
sulfamethoxazole

CLINICAL• Treatment:
– Antibiotics for respiratory infections
– Postural drainage and chest physiotherapy for airway clearance
– Lung transplant for end-stage lung disease
– Pancreatic enzyme replacement therapy and supplemental feedings• Remember “Fibrosing Colonopathy”
– Colonic strictures due to submucosal fibrosis seen in children with CF receiving high-strength pancreatic supplements
– Oral ursodiol for biliary buildup/obstruction
– Assisted reproductive technologies for infertility

PATHOPHYSIOLOGYCFTR:• Cystic Fibrosis Transmembrane conductanceRegulator• Gene location: chromosome 7 (at 7q31)• Function:
– Regulate epithelial Chloride channel– Also regulate other ion channels
• E.g., Sodium, Potassium, and Bicarbonate ions
• Components: Five domains– Two trans-membrane domains– Two cytoplasmic nucleotide-binding domains (NBDs)– One regulatory (R) domain
• Activation:Agonists (e.g., acetylcholine) Increase cAMP Activates protein kinase A Phosphorylating the CFTR at the R domain (using ATP bound to NBDs) CFTR Activation
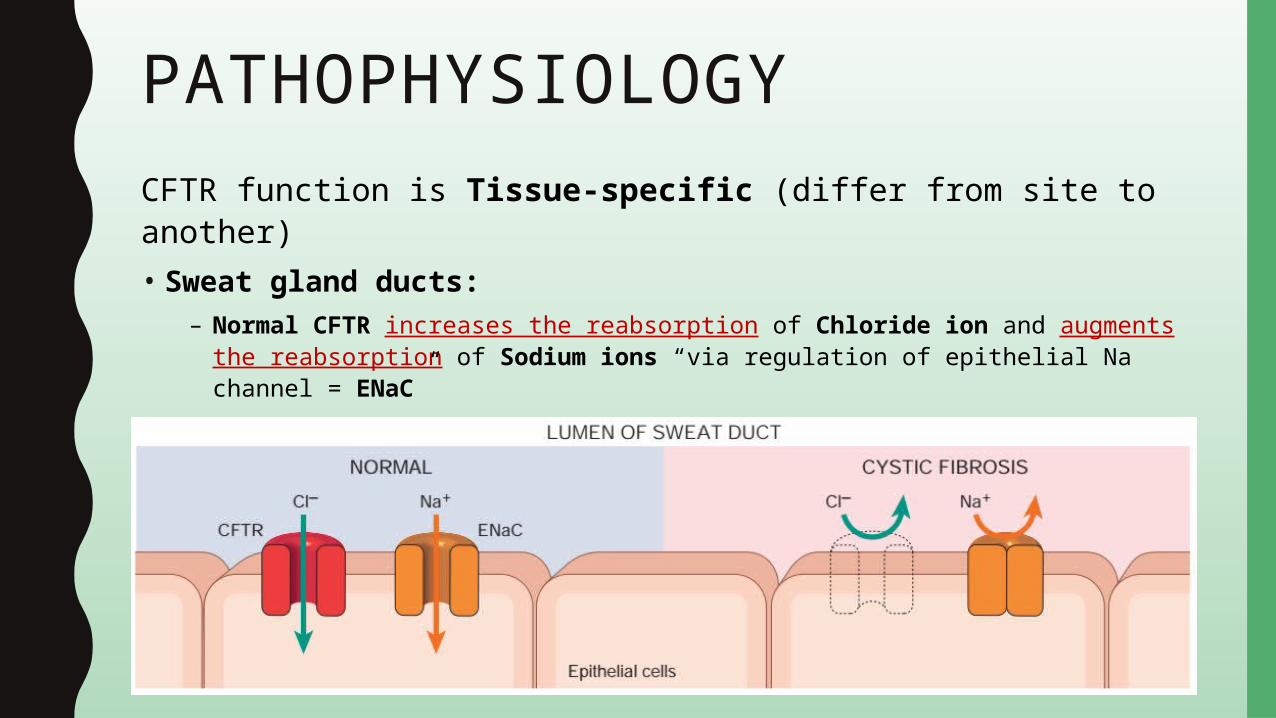
PATHOPHYSIOLOGYCFTR function is Tissue-specific (differ from site to another)• Sweat gland ducts:
– Normal CFTR increases the reabsorption of Chloride ion and augments the reabsorption of Sodium ions “via regulation of epithelial Na channel = ENaC”
– Mutated CFTR decreases the reabsorption of Sodium chloride• Hypertonic “Salty” sweat

PATHOPHYSIOLOGY• Lung / GIT / Pancreas:
– Normal CFTR increases the secretion of Chloride ion and reduces the reabsorption of Sodium ions “via regulation of epithelial Na channel = ENaC”
– Mutated CFTR decreases the secretion of Chloride ion and augments the reabsorption of Sodium ion (with passive Water reabsorption)• Hyperconcentrated “dehydrated” viscid secretions

PATHOPHYSIOLOGY• As regards the Bicarbonate ion:
– Normal CFTR increases the concentration of Bicarbonate ion into the lumen “via regulation of anion exchangers = SLC26”• Normal Alkaline secretion
– Mutated CFTR decreases the concentration of Bicarbonate ion into the lumen• Acidic secretion increased mucin precipitation and plugging,
increase bacterial activity

GROSS• Nose:
– Nasal polyps• Single or Multiple polypoid masses• Soft and edematous
• Lung:– Bronchiectasis
• Enlarged, dilated bronchi– Diameter of the bronchus should exceed the diameter of the accompanying bronchial artery
• Dilated bronchi are extending to pleural surface• Filled with yellow-green mucopurulent secretions

GROSS• Pancreas:
– Cystic changes “2ry to ductal obstruction”• Multiple, small cysts (1-3 mm in diameter)• Filled with thick, tenacious secretions
• Hepatobiliary:– Bile duct obstruction
• Enlarged, dilated bile ducts• filled with thick, tenacious secretions
– End-stage: Cirrhosis

MICROSCOPY• Nose:
– Nasal polyps• Covering:
– Respiratory epithelium “pseudostratified columnar ciliated”– Delicate basement membrane (not thickened)
• Core:– Loose, Edematous– Large cystic mucous glands, with inspissated secretion in their lumina
• Contain acid mucin (not neutral mucin)– Mixed inflammatory cells
• May contain some eosinophils (not extensive)– Chronic cases fibrotic cores
Poly
ps in
CF
Glan
ds c
onta
in a
cid m
ucin
Stai
ns b
lue
with
Alci
an b
lue/
PAS
DD fr
omIn
flam
mat
ory/
alle
rgic
poly
ps
Infla
mm
ator
y po
lyps
Glan
ds c
onta
in n
eutra
l m
ucin
Stai
ns p
urpl
e re
d wi
th
AB/P
AS
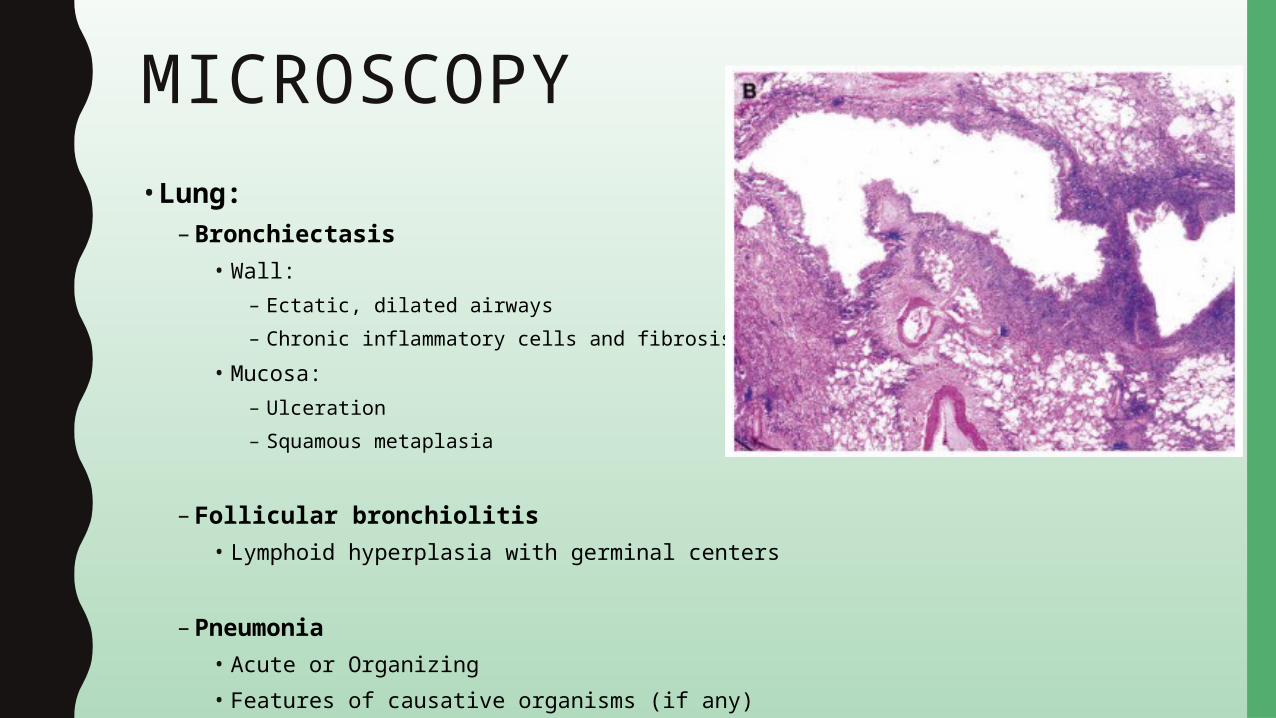
MICROSCOPY• Lung:
– Bronchiectasis• Wall:
– Ectatic, dilated airways– Chronic inflammatory cells and fibrosis
• Mucosa:– Ulceration– Squamous metaplasia
– Follicular bronchiolitis• Lymphoid hyperplasia with germinal centers
– Pneumonia• Acute or Organizing• Features of causative organisms (if any)

MICROSCOPY• Pancreas:
– Cystic changes• Ectatic, dilated ducts filled with eosinophilic material• Obstruction-related changes:
– Exocrine acinar atrophy– Replacement of atrophic lobules by interstitial fibrosis– Scattered islets of Langerhans could be seen
– Grading:• Grade I: accumulation of secretion• Grade II: exocrine atrophy• Grade III: atrophy with lipomatosis• Grade IV: fibrosis with total obliteration of theexocrine glands and ducts with scattered islets

MICROSCOPY• Hepatobiliary:
– Ductular reaction “2ry to Bile duct obstruction”• Portal tracts expansion by inflammation and increased numbers of bile
ductules• Bile ductules are dilated and contain plugs of a light eosinophilic material
– Portal fibrosis, bridging fibrosis and cirrhosis

SPECIAL STUDIESLaboratory tests:• Elevated sweat chloride (>60 mEql/L)
– Remember that sweat glands are morphologically unaffected.
• Abnormal nasal trans-epithelial potential difference– Useful in cases with low sweat chloride
“Milder CFTR mutations“
• Azoospermia on semen analysis– Obstructive type (due to structural abnormalities of the vas
deferens)

SPECIAL STUDIESMolecular studies: CFTR gene mutation
More than 1800 mutations, classified into Six groups:Clas
sDefect Result
I Defective protein synthesis No CFTR protein synthesis from the start
IIDefective protein folding and glycosylation(in Golgi/endoplasmic reticulum)
CFTR protein is synthesized, but it is degraded before reached the cell surface
IIIDefective ATP binding(to NBDs)
CFTR protein reaches the cell surface in normal amount, but it is Nonfunctioning
IVDefective chloride ion transport/conductance(through transmembrane domains)
CFTR protein reaches the cell surface in normal amount, but with reduced function
V Defective introns’ splicing(within the gene)
CFTR protein reaches the cell surface in reduced amount
VIDefective regulatory role on other ion channels(e.g., Na, K, HCO3)
CFTR protein reaches the cell surface in normal amount, but with altered function

SPECIAL STUDIESClass I, II, and III Total loss of function (Severe clinical picture “phenotype”)
– I.e., Bronchiectasis, pancreatic insufficiency, male infertility, hepatic cirrhosisExamples • ΔF508 deletion
– Class II “Defective protein folding”– The most common mutation in Caucasian
• W1282X nonsense mutation– Class I “Defective protein synthesis”– The most common mutation in Ashkenazi Jews
Class IV, V, and VI Reduced function (Milder clinical picture “phenotype”)– I.e., Congenital bilateral absence of vas deference and infertility
Example R117H missense mutation (in trans with 5T allele)– Class V “Defective chloride ion transport/conductance”
• 5T allele modifies Poly-T tract in intron 8 and reduces its splicing efficacy

SPECIAL STUDIES• Remember that cystic fibrosis is an autosomal recessive disease
– Mutation of one allele carrier– Mutation of both alleles disease
• Also the type of mutation plays a role in the overall phenotype
– If two “severe” mutations (e.g. class I, II, III) severe clinical picture– If one “severe” + one “mild” mutation (e.g. class IV, V, VI) less
severe– If two “mild” mutations very mild

DIFFERENTIAL DIAGNOSISL u n g :“ O t h e r c o n g e n i t a l / c y s t i c l u n g d i s e a s e s ”• Congenital:
– Bronchogenic cysts– Congenital pulmonary cysts– Congenital pulmonary airway malformation– Congenital lobar emphysema– Pulmonary sequestration
• Acquired:– Emphysema– Healed abscess– Honeycombing

DIFFERENTIAL DIAGNOSIS
Also:
“ Other causes o f pancreat ic insufficiency, chron ic pancreat i t is “
“ Other causes o f b i le ductu lar react ion , c i r rhos is “
“ Other causes o f nasa l po lyps ”

DIFFERENTIAL DIAGNOSISPrimary cilia dyskinesis:• Immotile cilia, Kartagener syndrome, Young syndrome,
secondary cilia dyskinesis
• Ultrastructural abnormalities affect virtually all cilia and are characterized by:
– Loss of dynein arms– Absence of radial spokes– Transposition or absence of microtubules– Compound or disorientated cilia

WWW.
DO NOT FORGET TO SEARCH FOR MORE PICS AND VIRTUAL SLIDES

THANK YOUH A Z E M A L I






