
1
Rapid apoptosis induction by IGFBP-3 involves an IGF -independent nucleo-mitochondrial translocation of RXRα/Nur77
Kuk-Wha Lee1, Liqun Ma1, Xinmin Yan1, Bingrong Liu1, Xiao-kun Zhang2 &Pinchas Cohen1*
1Division of Pediatric Endocrinology, Mattel Children's Hospital at UCLA, DavidGeffen School of Medicine, Los Angeles, CA, USA2Cancer Center, The Burnham Institute, La Jolla, CA, USA
Running title: IGFBP-3 translocates RXRα/Nur77
*To whom correspondence may be addressed. Tel: (310) 206-5844. Fax: (310)206-5843. E-mail: [email protected]
SUMMARY
Insulin-like growth factor binding protein-3 (IGFBP-3) induces apoptosis by its
ability to bind IGFs as well as its IGF-independent effects involving binding to
other molecules including the retinoid X receptor-α (RXRα). Here we describe
that in response to IGFBP-3, the RXRα binding partner nuclear receptor Nur77,
rapidly undergoes translocation from the nucleus to the mitochondria, initiating
an apoptotic cascade resulting in caspase activation within 6 hours. This
translocation is a type 1 IGF receptor signalling-independent event as IGFBP-3
induces Nur77 translocation in R- cells. IGFBP-3 and Nur77 are additive in
inducing apoptosis. GFP-Nur77 transfection into RXRα WT and KO MEFs and
subsequent treatment with IGFBP-3 shows that RXRα is required for IGFBP-3
induced Nur77 translocation and apoptosis. Addition of IGFBP-3 to 22RV1 cell
lysates enhanced the ability of GST- RXRα to “pull-down” Nur77, and
overexpression of IGFBP-3 enhanced the accumulation of mitochondrial RXRα.
This unique non-genotropic nuclear pathway supports an emerging role for
IGFBP-3 as a novel, multi-compartmental signaling molecule involved in
induction of apoptosis in malignant cells.
JBC Papers in Press. Published on February 24, 2005 as Manuscript M412757200
Copyright 2005 by The American Society for Biochemistry and Molecular Biology, Inc.
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

2
INTRODUCTION
Over the past decade, multiple lines of investigation have validated Insulin-
like growth factor binding protein-3 (IGFBP-3) as an inducer of cellular apoptosis,
effects that can be unrelated to its IGF-binding (1). Importantly, several groups
have now reported successful in vivo treatment of cancer models with IGFBP-3,
either as a single-agent or in combination with chemotherapeutic agents (2-4).
However, the molecular mechanisms by which IGFBP-3 induces apoptosis
remain largely unknown at present.
Several novel IGFBP-3 binding partners have been recently identified that
may participate in its IGF-independent pro-apoptotic effects (1). We and others
demonstrated that retinoid X receptor-α (RXRα) is a binding partner for IGFBP-3
(5,6), and that RXRα is required for IGFBP-3 apoptotic effects (5). Indeed, IGFBP-
3 potentiates RXRE-mediated signalling while inhibiting signalling via other
RXRα heterodimeric partners (5-7). Our discovery of IGFBP-3 binding to RXRα
suggested that its apoptotic effects might involve a RXRα-dependent
transcriptional mechanism. Most published reports have evaluated IGFBP-3
induced apoptosis at 24 to 72 hours (8-11), consistent with a transcriptional
mechanism. However, we have recently described apoptosis activation by
IGFBP-3 (as evidenced by caspase activation and histone associated DNA
fragmentation ELISA) as early as 1-6 hours after IGFBP-3 exposure, suggesting
a mechanism that does not require de novo gene transcription (12,13).
The orphan nuclear receptor Nur77 (also known as NGFI-B (14) and TR3
(15)) is a nuclear receptor transcription factor and is an important regulator of
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

3
apoptosis in different cells (16). It is a member of the orphan steroid receptor
family, which also includes Nor1 and Nurr1. This family is essential for
apoptosis of self-reactive immature thymocytes following stimulation of the T-cell
receptor (17,18). In response to synthetic apoptotic stimuli, Nur77 translocates
from the nucleus to the mitochondria to induce cytochrome c release and
apoptosis in leukemia (19), lung (20), ovary (21), stomach (22), colon (23), and
prostate cancer cells (24). Subcellular localization of Nur77 is important for its
biologic function. In the nucleus, it functions as a transcription factor to mediate
cell proliferation events. Targeted to the mitochondria, it takes on a novel role as
a mediator of apoptosis, not unlike the role played at the mitochondria by another
transcription factor, p53 (25). Importantly, Nur77 can also heterodimerize with
RXRα (26) and participate in its transcriptional activities (26-28). The mitogenic
effect of Nur77 requires its DNA binding and transactivation functions in the
nucleus whereas both are dispensable for the apoptotic effects of Nur77 at the
mitochondria (29).
Because the nuclear receptor RXRα is an intracellular binding partner for
IGFBP-3, we hypothesized that IGFBP-3 would modify RXRα/Nur77 heterodimeric
DNA binding, shifting this heterodimer from a DNA binding state to one that
targets mitochondria. Mitochondrial translocation of RXRα/Nur77 would then
result in the release of cytoplasmic cytochrome c, activation of intracellular
caspases, and induction of apoptosis.
Herein we report evidence that IGFBP-3 is a rapid biological signal
molecule for RXRα/Nur77 translocation. Our results reveal a new interaction
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

4
between the nuclear receptor and IGFBP superfamilies and identify IGFBP-3 as a
unique signal modulator of both traditional and novel nuclear receptor roles at
the junction of cellular proliferation and apoptosis.
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

5
EXPERIMENTAL PROCEDURES
Materials--Celtrix (Mountain View, California) provided recombinant human
IGFBP-3. IGF-1 was a generous gift from Pharmacia Corporation (Stockholm,
Sweden). Commercial antibodies included: anti-human IGFBP-3 from DSL
(Webster, Texas), anti-Nur77 from Geneka Biotechnology (Montreal, Canada),
anti-RXRα from Santa Cruz Biotechnologies (Santa Cruz, California), anti-
cytochrome c from Pharmingen (BD Biosciences, Palo Alto, California), and anti-
β-actin from Sigma-Aldrich (St. Louis, Missouri). PMP70 Antibody and cathepsin
S antibodies were from Zymed (South San Francisco, CA) and R & D systems
(Minneapolis, MN) respectively. For the western immunoblot utilizing the R-
MEFs, polyclonal rabbit anti-Nur77 antibody (Harlan Biosciences, Indianapolis,
Indiana) was generated against two specific N-terminal peptides (Genemed
Synthesis, San Francisco, California) derived from human Nur77 peptide
sequence. Sera was purified on protein A/G column (Amersham/Pharmacia,
Sunnyvale, California) and verified by Western blotting. Nur77 banding pattern
was confirmed using CCRF-CEM nuclear extract (Active Motif, Carlsbad,
California). SDS-polyacrylamide gel electrophoresis (PAGE) reagents, Tween,
and fat-free milk were purchased from Bio-Rad (Hercules, California). ECL
reagents were from Amersham (Sunnyvale, California). Full length IGFBP-3 and
Nur77 cDNAs was cloned into pLP-IRESneo mammalian expression vector via
pDNR-mediated CreatorTM technology (Clontech, Palo Alto, California). The
cloning of GFP-Nur77 has been described (24). LipofectAMINE and PLUS
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

6
Reagent were from Invitrogen (Carlsbad, California). All other chemicals were
from Sigma-Aldrich.
Cell Culture--22RV1 cells, A172 cells, CCRF-CEM, and F9 embryonal carcinoma
cells from ATCC (Manassas, VA), and F9 RXRα -/- cells (kind gift of Dr. P.
Chambon) were maintained in Dulbecco’s modified Eagle’s medium containing
10% fetal calf serum (Life Technologies, Carlsbad, California), 100 units of
penicillin/ml, and 100 units of streptomycin/ml in a humidified environment with
5% CO2.
Mouse Embryonic Fibroblast (MEF) Generation--Fibroblasts from an IGF-I
receptor knockout and corresponding wild type mouse were generated from 18-
day embryos as described previously (30) and were designated R- and WT MEFs
respectively. The R- cells were maintained in Dulbecco's modified Eagle's
medium containing 10% fetal bovine serum and Geneticin (G418). All cells were
used before passage 6.
Apoptosis ELISA--Cells (2500 cells/cm2) were seeded on 96-well plates.
Following overnight attachment, cells were washed with PBS and serum starved
overnight, before incubation with the indicated conditions in a total of 100 µl
volume. Roche Photometric Cell Death ELISA (Indianapolis, IN) was performed
according to the manufacturer’s instructions to quantify histone-associated DNA
fragments (mono- and oligo-nucleosomes) generated by apoptotic cells. This
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

7
immunoassay is based on the sandwich-enzyme principle and used separate
mouse monoclonal antibodies directed against DNA fragments and histones.
Briefly, cell lysates were placed into a 96-well plate coated with streptavidin-
linked, anti-histone antibody. Peroxidase-labeled mouse monoclonal DNA
antibodies were used to localize and detect the bound, fragmented DNA by
photometry of 2,2'-azino-bis-[3-ethylbenzathiazoline sulfonate] as the substrate.
Calcium ionophore treatment served as the positive control, and serum-free
medium (SFM) as the negative control. Each experimental condition was
performed in triplicate. Reaction products in each 96-well plate were read using
a Bio-Rad microplate reader. Mean absorbance data at 405 nm (±SD) were
plotted. Studies to provide evidence that the absorbance values generated by the
assay are linearly related to growth were performed and confirmed in a
published paper from our lab (31).
Caspase assays--The caspase assay was done using Apo-ONETM homogenous
caspase -3/-7 assay (Promega) and performed according to manufacturer’s
instructions. rhIGFBP-3 was used at a final concentration of 1 µg/ml.
Immunofluorescence Confocal Microscopy--Ten thousand cells were plated on
coverglasses in serum containing media for 2 days. Cells were then incubated
in serum-free media with or without IGFBP-3 before staining for
immunofluorescence. After three washes in phosphate-buffered saline (PBS),
fixation and permeabilization of the cells were performed with 1%
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

8
paraformaldehyde in PBS for 15 min at room temperature and 0.2% Triton X-100
in PBS for 15 min on ice, and cells were washed twice with PBS. Nur77 or
IGFBP-3 protein localization was detected using hIGFBP-3/ Nur77 polyclonal
antibodies (diluted 1:1,000) followed by fluorescein/Texas Red antibody from
Vector Laboratories (Burlingame, California). Specimens were incubated with
primary antibodies in PBS for 1 h at room temperature, with secondary
antibodies in PBS for 40 min at room temperature, and then incubated with
Hoechst (Electron Microscopy Sciences, Ft. Washington, Pennsylvania) for 2
minutes. Samples were analyzed using inverted confocal microscopy (Leica,
Inc., Germany), equipped by digital camera Himamatsu (Japan), and operated by
QED-image software.
Subcellular fractionation Procedures--Nu-CLEAR Protein Extraction KitTM was
from Sigma-Aldrich. Subcellular fractions were isolated according to the
manufacturer’s protocol. The ApoalertTM Cell Fractionation Kit (BD Clontech) was
used to isolate a mitochondrial fraction from the cytoplasm of cells. Purity of the
fraction was assessed by immunoblot for PMP70 (peroxisomal) and cathepsin S
(lysosomal) contamination.
Transient Transfections--Cells (2 x 104) were seeded in 96-well culture plates.
Reagents were appropriately scaled up to 6-well plates for transfections that
were followed by mitochondrial isolation and subsequent western
immunoblotting. Transfections were done with LipofectAMINE:PLUS Reagent as
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

9
directed by manufacturer (Invitrogen). Typically, 50 ng of β-galactosidase
expression vector (pSV-β-Gal, Promega, Madison, Wisconsin), and 50 ng
expression vector containing IGFBP-3 and/or Nur77 were mixed with carrier DNA
to give 0.2 mg total DNA per well. After 24-48 hours transfection, caspase activity
was quantitated and normalized for transfection efficiency to measurements of
aliquots of co-transfected β-galactosidase gene activity (β-galactosidase enzyme
assay system, Promega).
GST Pull-down--The GST-RXRα fusion vector encoding the full-length RXRα
molecule and was the generous gift of Dr. D. J. Mangelsdorf and has been
previously described (32). GST-RXRα fusion protein was produced in GST-
RXRα-transformed Escherichia coli DH5, which were lysed and loaded on
glutathione-sepharose 4B beads (Sigma). Ten µg of purified GST-RXRα bound
to beads was incubated with 500 µg of cell lysate, with or without 200 ng of
recombinant IGFBP-3 protein and then separated by centrifugation. The bound
proteins were analyzed by nonreducing SDS-PAGE followed by Western blotting
using anti- Nur77 antibody. Experiments were repeated three times.
Densitometric and statistical analysis--Densitometric measurement of
autoradiographs was performed using computer scanned densitometry. All
experiments were repeated at least three times. Means +/- SD are shown.
Statistical analyses were performed using ANOVA utilizing InStat (GraphPad, San
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

10
Diego, California). Differences were considered statistically significant when p<
0.005, denoted by **.
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

11
RESULTS
IGFBP-3 Induces Rapid Induction of Apoptosis—We have previously observed
that in A172 glioblastoma cells, IGFBP-3-induced caspase activation reaches its
maximum at one to six hours, after which it decreases (12). Likewise, in human
macrovascular umbilical vein endothelial cells, VEGF-induced survival of HUVEC
is inhibited by IGFBP-3, via the induction of apoptosis in a type 1 IGF receptor-
independent manner utilizing the neutralizing antibody αIR3 (13). We therefore
confirmed these rapid effects in a variety of cell lines. Mouse Embryonic
Fibroblasts (MEF) exhibited a 70% increase in apoptosis as evidenced by
fluorometric assessment of caspase 3/7 activation (Fig. 1A) as early as 2 hours
post treatment. This induction was maximal to nearly 2.5 fold over baseline at 6
hours. Similarly, in the human glioblastoma cell line A172, an almost 2-fold
increase in apoptosis was detected as early as one hour (Fig 1B). In the 22RV1
prostate cancer cell line (Fig. 1C), a significant 32% increase in caspase
activation was induced by the addition of IGFBP-3. This subsequently rose to a
40 and 51% increase over serum free levels at 6 and 24 hours respectively.
These results confirm that IGFBP-3 induction of apoptosis, assessed in multiple
cell lines, is a rapid event. Experiments were repeated three times.
IGFBP-3 Induces Rapid Nucleo-Mitochondrial Translocation of Nur77--To
investigate whether Nur77 translocation could mediate the pro-apoptotic effects
of IGFBP-3 in CaP cells, we first established that IGFBP-3 leads to nucleo-
mitochondrial translocation of Nur77. We performed a time course of IGFBP-3
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

12
treatment and observed the subcellular localization of Nur77 by indirect
immunofluorescence confocal microscopy in 22RV1 CaP cells. Within 15
minutes there was strong cytoplasmic appearance of red-staining Nur77
compared to minimal cytoplasmic labeling at time 0 (Fig. 2A). The faint nuclear
staining seen at time 0 may reflect inaccessibility of anti-Nur77 antibody to the
protein secondary to RXR heterodimerization or Nur77 homodimerization. The
cytoplasmic Nur77 presence was maintained throughout this 2-hour time course
although some Nur77 reappeared in a nuclear location by the end of 2 hours.
To confirm that IGFBP-3 is a biologic Nur77 translocation signal, we also
assessed relative Nur77 concentrations in nuclear and cytoplasmic fractions of
IGFBP-3-treated 22RV1 CaP cells by Western immunoblot at 0, 0.5, 1, and 3
hours (Fig. 2B). Cells treated with IGFBP-3 showed the gradual disappearance
of nuclear Nur77 over the course of 3 hours associated with some increase in
the amount of cytoplasmic Nur77. Furthermore, a dramatic increase was shown
to be via mitochondrial targeting as subdivision of the cytoplasmic fraction into a
mitochondrial enriched fraction revealed the appearance of a prominent Nur77
band as detected by Western immunoblot as early as 1 hour (Fig. 2C).
Membranes were probed with PMP70 and cathepsin S to show that the isolated
mitochondrial fraction was free of peroxisomal / lysosomal contamination.
Similar results were obtained for the LAPC-4 prostate cancer cell line and A172
glioblastoma cell line (data not shown). A representative of three separate
experiments is shown.
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

13
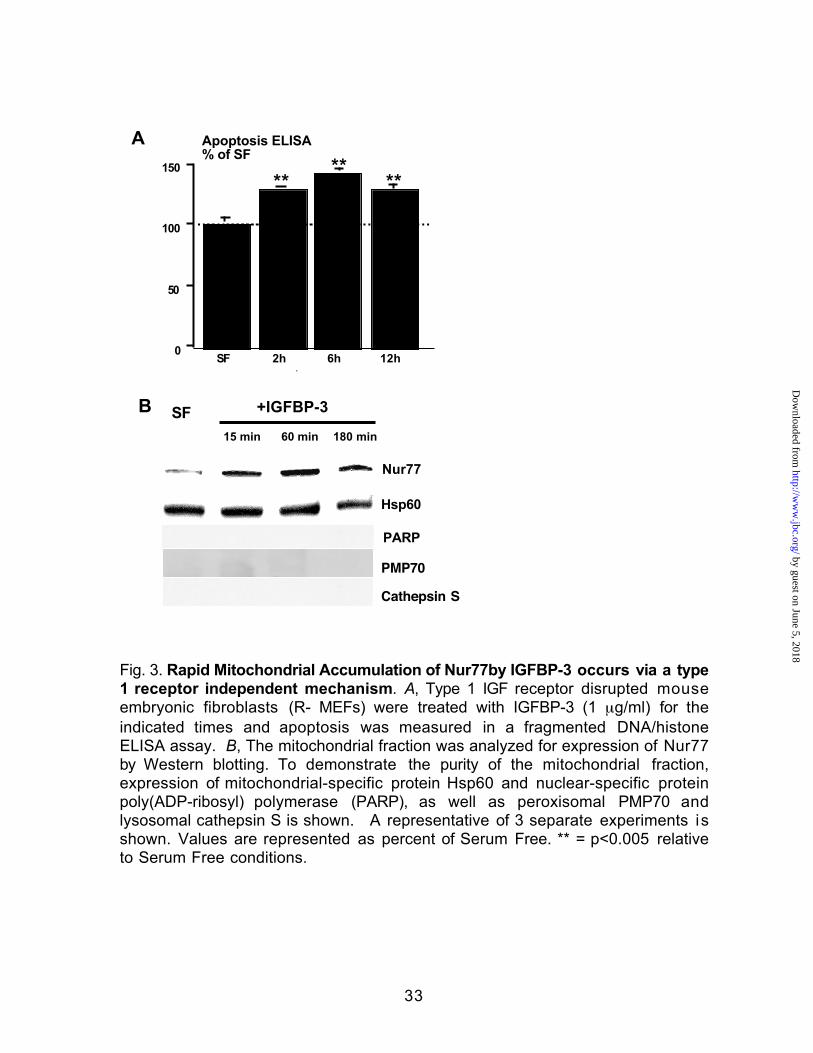
Rapid Mitochondrial Translocation of Nur77by IGFBP-3 occurs via a type 1
receptor independent mechanism--The possibility that IGFBP-3 acts to induce
apoptosis independently of IGFs and IGF receptors was investigated by testing
the ability of IGFBP-3 to induce apoptosis in the IGF receptor-negative (R-)
embryonic fibroblast cells derived from an IGF-1R knockout mouse (30). This
effect is mediated in part by a type 1 IGF receptor independent mechanism as
IGFBP-3 was still able to induce apoptosis, with a 32% increase over baseline at
2 hours, that was maximal at 6 hours in type 1 IGF receptor disrupted MEFs in a
fragmented DNA/histone ELISA (Fig. 3A). These cells have been shown
previously to neither bind nor respond to IGFs. IGFBP-3 induced mitochondrial
translocation of Nur77 in this unique system was further demonstrated by
immunoblotting analysis, which showed accumulation of Nur77 in the
mitochondrial fraction (Fig. 3B). To demonstrate the purity of the mitochondrial
fraction, expression of mitochondrial-specific protein Hsp60 and nuclear-specific
protein poly (ADP-ribosyl) polymerase (PARP) is shown as well as immunoblots
to PMP70 and cathepsin S (peroxisome/lysosome markers respectively). This
data suggests that Nur77 mitochondrial translocation by IGFBP-3 occurs
independent of signalling via the type 1 receptor. This experiment was repeated
three times.
Additive apoptotic effects of overexpression of IGFBP-3 and Nur77--To determine
whether mitochondrial targeting of Nur77 by IGFBP-3 plays a role in regulating
the release of cytochrome c from mitochondria into cytosol, the location of
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

14
cytochrome c was examined during the course of IGFBP-3 treatment.
Immunoblotting of a cytoplasmic fraction that was depleted of mitochondria
showed that the addition of 1 µg/ml of IGFBP-3 caused a greater than 3-fold
increase in the appearance of cytoplasmic cytochrome c at 30 minutes, which
increased to almost 6-fold at 60 minutes that was sustained at 180 minutes (Fig.
4A).
The release of cytoplasmic cytochrome c is directly upstream of caspase
activation in the mitochondrial pathway of apoptosis (33). Overexpression of
Nur77 in thymocytes induces massive apoptosis (34). We co-expressed by
transient transfection both IGFBP-3 and Nur77 in mammalian expression vectors
to assess effects on caspase activation in 22RV1 cells. Expression of both
IGFBP-3 and Nurr7 in combination resulted in additive effects on caspase
activation (Fig. 4B), indicating additivity between IGFBP-3 and Nur77 in their
apoptotic effects. Protein expression was confirmed with immunoblots of whole
cell lysates from transfected cells (Fig. 4C).
IGFBP-3 induced Nur77 Translocation is RXRα-dependent, and involves co-
migration of RXRα/Nur77 heterodimers to mitochondria--We previously reported
that the nuclear receptor RXRα is a nuclear binding partner for IGFBP-3 and is
required for IGFBP-3 induced apoptosis (5). Also, in response to nerve growth
factor (NGF), nuclear RXRα/Nur77 heterodimeric complexes translocate to the
cytoplasm in PC12 pheochromocytoma cells (35). A recent paper also describes
the carrier role of RXRα to assist Nur77 translocation in the 9-cis retinoic acid-
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

15
dependent apoptosis of gastric cancer cells (36). We hypothesized that pro-
apoptotic IGFBP-3 would translocate RXRα/Nur77 heterodimers to the
mitochondria. To investigate the role of RXRα in IGFBP-3-induced Nur77
translocation, F9 RXRα+/+ (wild-type, WT) embryonal carcinoma cells and F9
RXRα-/- cells were treated with 1 µg/ml IGFBP-3 overnight, nuclear fractions were
isolated, resolved via SDS-PAGE, and immunoblotted for the presence of Nur77.
Bands were quantitated by densitometric analysis and normalized to nuclear
PARP. As expected, IGFBP-3 induced a >50% reduction of nuclear Nur77 in the
wild-type cells, consistent with an RXRα/Nur77 translocation event (Fig. 5A). In
contrast, treatment of the sister RXRα-/- line with IGFBP-3 resulted in an increase,
rather than decrease, in nuclear Nur77. To confirm this observation, we
transfected green fluorescent protein (GFP)-Nur77 in the same cell lines, treated
with IGFBP-3, and visualized these cells utilizing confocal microscopy.
Expression of GFP-Nur77 in the RXRα WT line showed a predominantly nuclear
distribution consistent with its function as a transcription factor at basal
conditions (Fig. 5B). Treatment with 1 µg/ml of IGFBP-3 resulted in the rapid
appearance of extranuclear GFP-Nur77 observed within 15 minutes. This effect
was not seen in the sister RXRα-/- line, as IGFBP-3 treatment has no effect on the
translocation of nuclear GFP-Nur77. In addition, transfection of the Nur77
overexpression vector into F9 RXRα+/+ cells induced a marked increase in
caspase activation while transfection into the sister F9 RXRα-/- line failed to
induce any significant increase in caspase activity (Fig. 5C). Indeed, addition of
200 ng of IGFBP-3 to 500 µg of 22RV1 cell lysate demonstrated that IGFBP-3
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

16
enhanced the ability of GST-RXRα to “pull-down” Nur77 (Fig. 5D), indicating that
IGFBP-3 augments the ability of RXRα/Nur77 to physically associate. In fact,
isolation of mitochondria from cells transfected with IGFBP-3 revealed a 3-fold
increase in mitochondrial RXRα (Fig. 5E) that was not seen in cells transfected
with control expression vector, demonstrating that IGFBP-3 leads to the co-export
of RXRα and Nur77. Together, our results demonstrate that the mechanism of
IGFBP-3 induced Nur77 translocation, like IGFBP-3-induced apoptosis, requires
RXRα, and involves co-migration of RXRα/Nur77 heterodimers to mitochondria.
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

17
DISCUSSION
Multiple lines of in vitro, in vivo, and clinical evidence point to IGFBP-3 as
an anti-cancer molecule (37). In vitro, IGFBP-3 has been shown to induce
apoptosis in a variety of cancer models via both IGF-dependent and
–independent mechanisms (1). In vivo, recent publications report efficacy either
as single agent or in chemotherapy combinations in non-small cell-lung cancer
(2) and colon cancer (3). IGFBP-3 gene expression is commonly lost in human
cancer cell lines and xenografts as detected in DNA microarray analysis of
cancerous compared with non-cancerous cells (38). Decreased IGFBP-3
expression is associated with prostate cancer progression, demonstrating more
frequent loss of expression in advanced disease, in both human (39) and mouse
(40) models. In addition, recent evidence demonstrates that methylation of the
IGFBP-3 promoter is one mechanism by which the silencing of IGFBP-3
expression in cancer cells is achieved (41).
Despite promising pre-clinical evidence using IGFBP-3 as a cancer
therapeutic (2-4), controversy remains as to the complex role of IGFBP-3 in
various tumors. IGFBP-3 modulates cellular proliferation with dual actions that
either enhance IGFs or inhibit their actions as well as actions that are idependent
of its binding to IGFs (1). Evidence for this duality has been reported in renal cell
(42,43), lung (44,45), and breast and other cancers (46). Interestingly, an
outcome prediction model for prostate cancer was established utilizing HoxC6
and IGFBP-3 expression, as IGFBP-3 was positively associated with Gleason
score (47). However, recent expression profiling of HoxC6 siRNA transfections
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

18
and HoxC6 overexpression identified IGFBP-3 as a potential proapoptotic
repression target of HoxC6 in prostate cancer (48). Clearly, more work needs to
be done to examine the role of IGFBP-3 in cellular proliferation and apoptosis.
We have recently demonstrated in a prostate cancer model, the
requirement for IGFBP-3 secretion and re-uptake by endocytic pathways
(specifically caveolin- and transferrin receptor-mediated) for apoptosis induced
by transforming growth factor (TGF)β (49). After internalization, IGFBP-3 rapidly
localizes to the nucleus where it interacts with RXRα and other factors (1).
Nuclear import is a nuclear localization signal-dependent process and mediated
by importin-β factor (50).
Our observation that IGFBP-3 translocates Nur77 has several important
implications. First, because IGFBP-3 is a biological signal versus the previously
described chemical apoptosis inducers (i.e. calcium ionophores, etoposide,
rexinoids) (24); this implies that the normal prostate epithelial cell has an
endogenous signal (IGFBP-3), which can induce a programmed cell death
cascade upon cancer surveillance. In fact, we have recently reported that
EWS/FLI-1, an abnormal transcription factor resulting from oncogenic fusion in
Ewing’s tumor, binds the IGFBP-3 promoter in vitro and in vivo and represses its
activity. Moreover, IGFBP-3 silencing can partially rescue the apoptotic phenotype
caused by EWS/FLI-1 inactivation. IGFBP-3-induced Ewing cell apoptosis relies
on both IGF-1-dependent and -independent pathways. These findings therefore
identify the repression of IGFBP-3 as a key event in the development of Ewing's
sarcoma (51).
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

19
IGFBP-3 mediates the effects of multiple anti-proliferative and pro-
apoptotic biological agents including TGFβ (52), tumor necrosis factor-α (53),
retinoids (54), p53 (55), and 1,25 dihydroxyvitamin D3 (56). In addition, IGFBP-3
gene expression is commonly lost in human prostate cancer cell lines and
xenografts, and was detected in DNA microarray analysis of normal compared
with cancerous cells (38). Decreased IGFBP-3 expression is associated with
prostate cancer progression, demonstrating more frequent loss of expression in
advanced disease, in both human and mouse models (37-39). Low IGFBP-3
levels in prostate cancer imply impairment of RXRα/Nur77 translocation and
subsequent apoptosis of cancerous cells.
We have shown that expression of IGFBP-3 and Nur77 together are
additive in their pro-apoptotic effects. The importance of both these genes that
are inactivated on the cellular path to immortalization is supported by the fact that
IGFBP-3 binding and proteolysis is the target of the E7 protein encoded by
human papillomavirus type 15, one of the few viral genes that can immortalize
primary human cells and thereby override cellular senescence (57), and that
Nur77 is inactivated by the Epstein-Barr virus transactivator EBNA2, essential for
the immortalization of B-cells (58). These small, lean, viral genomes would
presumably selectively inactivate critical apoptosis-inducing host proteins that
would hinder the viral program of self-propagation. Additionally, two recent
papers have now described a non-genotropic carrier function of RXRα to
transport Nur77 to the mitochondria to initiate a mitochondria-dependent
apoptotic pathway (36,59).
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

20
Finally, we have described a novel interface between the nuclear receptor
superfamily and the growth and survival-regulating IGF-IGFBP axis. Beyond its
initial description as a serum carrier for the growth-promoting IGFs, IGFBP-3 has
emerged as a multi-functional, intrinsic, IGF-independent, signaling protein that
mediates important autocrine and paracrine regulation of growth and
homeostasis in a variety of tissues (1). Whereas we have previously described
IGFBP-3 binding to the nuclear receptor RXRα and supershifting RXR:RXRE
complexes in EMSA assays, modulating traditional nuclear receptor roles as
transcription factors via modulation of signaling via the RXRE and presumably
taking on a co-activator/co-repressor role in the nucleus (5-7), we currently
describe IGFBP-3 as a modulator of novel nuclear receptor roles as extra-nuclear
mediators of cellular processes. The fact that both IGFBP-3 and Nur77 are
dramatically suppressed by androgens and are upregulated during apoptosis
induced by castration in the ventral rat prostate affords another unique in vivo
model to study IGFBP-3-induced Nur77 translocation (60,61). This phenomenon
also suggests that uncontrolled androgen receptor signalling implicated in
androgen-independent prostate cancer involves the loss of the IGFBP-3 / Nurr77
apoptotic pathway.
It is now well recognized that Nur77 mediates apoptosis (62) through both
transactivation-dependent (63,64) and –independent (17,19,20,22,24) pathways.
The movement of transcription factors (such as RXRα and Nur77), kinases, and
DNA replication factors between the nucleus and cytoplasm is important in
regulating their activity (65). Nur77 interacts with Bcl-2 at the mitochondria,
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

21
inducing a conformational change that exposes its BH3 domain, resulting in Bcl-
2 conversion from an anti- to pro-apoptotic molecule (66). Our results regarding
RXRα/Nur77 suggest a model that may explain how RXRα/Nur77 activity is
regulated, at least partially, by the presence of IGFBP-3. Abnormal Nur77
transcriptional activity may have oncogenic potential because a Nur77 fusion
protein that is 270 times as active as the native receptor in activating gene
expression is produced through chromosomal translocation in extraskeletal
myxoid chondrosarcoma (67). Recent X-ray crystallographic analysis of the
Nur77 Drosophila homologue revealed the absence of both a classic ligand
binding pocket and coactivator binding site (68). Nur77 is often overexpressed in
cancer cells, due to the uncontrolled expression of growth factors that induce its
synthesis and subsequent transactivation (28,61). Agents, such as IGFBP-3, that
induce Nur77 translocation, may inhibit growth and promote apoptosis of cancer
cells.
In conclusion, the elucidation of the signaling pathways involved in the
anti-proliferative, pro-apoptotic effects of IGFBP-3 is important both for our basic
understanding of the mechanism of action of IGFBP-3 on a cellular level and to
devise new therapeutic approaches to treat prostate cancer constituting IGFBP-3
or related derivatives alone or in combination with synergistic agents. Our
present findings indicating that IGFBP-3 induces a rapid RXRα/Nur77
translocation event along with previous findings of modulation of slower DNA
transcriptional events may herald an “amplification” loop in prostate cancer
apoptosis signaling. Mutagenesis and identification of the specific peptide
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

22
regions of each molecule involved in apoptosis signaling will prove to be a
beneficial adjunct to the delivery of these molecules in a broad range of cancer
therapeutics.
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

23
ACKNOWLEDGEMENTS
The authors wish to thank Dr. P. Chambon for use of the F9 RXRα-/- and WT line,
and Dr. D. Mangelsdorf for the GST-RXRα fusion vector. We also thank David
Hwang, John F. Garcia, and Sarah T. Kerfoot for expert technical assistance.
Supported in part, by a Prostate Cancer Foundation award and NIH grants,
RO1AG20954, P50CA92131, RO1CA100938 (PC); as well as a fellowship award
from the Giannini Foundation (KL), a grant from the Stein-Oppenheimer
Foundation (KL), a grant from the Lawson Wilkins Pediatric Endocrinology
Society (KL), and NIH grant 2K12HD34610 (KL, PI ERB McCabe). The authors
declare that they have no competing financial interests.
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

24
REFERENCES
1. Firth, S. M., and Baxter, R. C. (2002) Endocr Rev 23, 824-8542. Lee, H. Y., Moon, H., Chun, K. H., Chang, Y. S., Hassan, K., Ji, L., Lotan, R.,
Khuri, F. R., and Hong, W. K. (2004) J Natl Cancer Inst 96, 1536-15483. Kirman, I., Poltoratskaia, N., Sylla, P., and Whelan, R. L. (2004) Surgery
136, 205-2094. Qingnan, Y., Banerjee, K., Paterson, J., Alami, N., Shiry, L., and Leland-
Jones, B. (2003) in American Association for Cancer Research July 11-14,Washington, DC
5. Liu, B., Lee, H. Y., Weinzimer, S. A., Powell, D. R., Clifford, J. L., Kurie, J. M.,and Cohen, P. (2000) J Biol Chem 275, 33607-33613.
6. Schedlich, L. J., O'Han, M. K., Leong, G. M., and Baxter, R. C. (2004)Biochem Biophys Res Commun 314, 83-88
7. Ikezoe, T., Tanosaki, S., Krug, U., Liu, B., Cohen, P., Taguchi, H., andKoeffler, H. P. (2004) Blood 104, 237-242
8. Granata, R., Trovato, L., Garbarino, G., Taliano, M., Ponti, R., Sala, G.,Ghidoni, R., and Ghigo, E. (2004) Faseb J 18, 1456-1458
9. Kim, H. S., Ingermann, A. R., Tsubaki, J., Twigg, S. M., Walker, G. E., andOh, Y. (2004) Cancer Res 64, 2229-2237
10. Perks, C. M., McCaig, C., Clarke, J. B., Clemmons, D. R., and Holly, J. M.(2002) Biochem Biophys Res Commun 294, 988-994
11. Butt, A. J., Fraley, K. A., Firth, S. M., and Baxter, R. C. (2002) Endocrinology143, 2693-2699
12. Ikonen, M., Liu, B., Hashimoto, Y., Ma, L., Lee, K.-W., Niikura, T., Nishimoto,I., and Cohen, P. (2003) Proc Natl Acad Sci U S A 100, 13042-13047
13. Franklin, S. F., Ferry, R. J., and Cohen, P. (2003) J Clin Endocrinol Metab88, 900-907
14. Milbrandt, J. (1988) Neuron 1, 183-18815. Chang, C., and Kokontis, J. (1988) Biochem Biophys Res Commun 155,
971-97716. Hazel, T. G., Nathans, D., and Lau, L. F. (1988) Proc Natl Acad Sci U S A
85, 8444-8448.17. Liu, Z. G., Smith, S. W., McLaughlin, K. A., Schwartz, L. M., and Osborne, B.
A. (1994) Nature 367, 281-284.18. Rajpal, A., Cho, Y. A., Yelent, B., Koza-Taylor, P. H., Li, D., Chen, E., Whang,
M., Kang, C., Turi, t. G., and Winoto, A. (2003) EMBO J. 22, 6526-653619. Zhang, Y., Dawson, M. I., Mohammad, R., Rishi, A. K., Farhana, L., Feng, K.
C., Leid, M., Peterson, V., Zhang, X. K., Edelstein, M., Eilander, D., Biggar,S., Wall, N., Reichert, U., and Fontana, J. A. (2002) Blood 100, 2917-2925
20. Dawson, M. I., Hobbs, P. D., Peterson, V. J., Leid, M., Lange, C. W., Feng,K. C., Chen, G., Gu, J., Li, H., Kolluri, S. K., Zhang, X.-k., Zhang, Y., andFontana, J. A. (2001) Cancer Res 61, 4723-4730
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

25
21. Holmes, W. F., Soprano, D. R., and Soprano, K. J. (2003) J Cell Biochem89, 262-278
22. Wu, Q., Liu, S., Ye, X. F., Huang, Z. W., and Su, W. J. (2002) Carcinogenesis23, 1583-1592
23. Wilson, A. J., Arango, D., Mariadason, J. M., Heerdt, B. G., and Augenlicht,L. H. (2003) Cancer Res 63, 5401-5407
24. Li, H., Kolluri, S. K., Gu, J., Dawson, M. I., Cao, X., Hobbs, P. D., Lin, B.,Chen, G., Lu, J., Lin, F., Xie, Z., Fontana, J. A., Reed, J. C., and Zhang, X.(2000) Science 289, 1159-1164.
25. Mihara, M., Erster, S., Zaika, A., Petrenko, O., Chittenden, T., Pancoska, P.,and Moll, U. M. (2003) Mol Cell 11, 577-590
26. Forman, B. M., Umesono, K., Chen, J., and Evans, R. M. (1995) Cell 81,541-550.
27. Perlmann, T., and Jansson, L. (1995) Genes Dev 9, 769-782.28. Wu, Q., Dawson, M. I., Zheng, Y., Hobbs, P. D., Agadir, A., Jong, L., Li, Y.,
Liu, R., Lin, B., and Zhang, X. K. (1997) Mol Cell Biol 17, 6598-6608.29. Kolluri, S. K., Bruey-Sedano, N., Cao, X., Lin, B., Lin, F., Han, Y.-H.,
Dawson, M. I., and Zhang, X.-k. (2003) Mol Cell Biol 23, 8651-866730. Sell, C., Rubini, M., Rubin, R., Liu, J. P., Efstratiadis, A., and Baserga, R.
(1993) Proc Natl Acad Sci U S A 90, 11217-1122131. Rajah, R., Valentinis, B., and Cohen, P. (1997) J Biol Chem 272, 12181-
12188.32. Janowski, B. A., Willy, P. J., Devi, T. R., Falck, J. R., and Mangelsdorf, D. J.
(1996) Nature 383, 728-731.33. Reed, J. C. (1997) Cell 91, 559-56234. Weih, F., Ryseck, R. P., Chen, L., and Bravo, R. (1996) Proc Natl Acad Sci
U S A 93, 5533-5538.35. Katagiri, Y., Takeda, K., Yu, Z. X., Ferrans, V. J., Ozato, K., and Guroff, G.
(2000) Nat Cell Biol 2, 435-440.36. Lin, X. F., Zhao, B. X., Chen, H. Z., Ye, X. F., Yang, C. Y., Zhou, H. Y., Zhang,
M. Q., Lin, S. C., and Wu, Q. (2004) J Cell Sci37. Ali, O., Cohen, P., and Lee, K. W. (2003) Horm Metab Res 35, 726-73338. Schwarze, S. R., DePrimo, S. E., Grabert, L. M., Fu, V. X., Brooks, J. D., and
Jarrard, D. F. (2002) J Biol Chem 8, 839. Hampel, O. Z., Kattan, M. W., Yang, G., Haidacher, S. J., Saleh, G. Y.,
Thompson, T. C., Wheeler, T. M., and Marcelli, M. (1998) J Urol 159, 2220-2225.
40. Kaplan, P. J., Mohan, S., Cohen, P., Foster, B. A., and Greenberg, N. M.(1999) Cancer Res 59, 2203-2209.
41. Chang, Y. S., Wang, L., Suh, Y. A., Mao, L., Karpen, S. J., Khuri, F. R., Hong,W. K., and Lee, H. Y. (2004) Oncogene 23, 6569-6580
42. Hintz, R. L., Bock, S., Thorsson, A. V., Bovens, J., Powell, D. R., Jakse, G.,and Petrides, P. E. (1991) J Urol 146, 1160-1163
43. Cheung, C. W., Vesey, D. A., Nicol, D. L., and Johnson, D. W. (2004)Kidney Int 65, 1272-1279
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

26
44. Yu, H., Spitz, M. R., Mistry, J., Gu, J., Hong, W. K., and Wu, X. (1999) J NatlCancer Inst 91, 151-156
45. London, S. J., Yuan, J. M., Travlos, G. S., Gao, Y. T., Wilson, R. E., Ross, R.K., and Yu, M. C. (2002) J Natl Cancer Inst 94, 749-754
46. Renehan, A. G., Zwahlen, M., Minder, C., O'Dwyer, S. T., Shalet, S. M., andEgger, M. (2004) Lancet 363, 1346-1353
47. Singh, D., Febbo, P. G., Ross, K., Jackson, D. G., Manola, J., Ladd, C.,Tamayo, P., Renshaw, A. A., D'Amico, A. V., Richie, J. P., Lander, E. S.,Loda, M., Kantoff, P. W., Golub, T. R., and Sellers, W. R. (2002) Cancer Cell1, 203-209
48. Ramachandran, S., Liu, P., Young, A. N., Yin-Goen, Q., Lim, S. D., Laycock,N., Amin, M. B., Carney, J. K., Marshall, F. F., Petros, J. A., and Moreno, C.S. (2005) Oncogene 24, 188-198
49. Lee, K. W., Liu, B., Ma, L., Li, H., Bang, P., Koeffler, H. P., and Cohen, P.(2004) J Biol Chem 279, 469-476
50. Schedlich, L. J., Le Page, S. L., Firth, S. M., Briggs, L. J., Jans, D. A., andBaxter, R. C. (2000) J Biol Chem 275, 23462-23470.
51. Prieur, A., Tirode, F., Cohen, P., and Delattre, O. (2004) Mol Cell Biol 24,7275-7283
52. Hwa, V., Oh, Y., and Rosenfeld, R. G. (1997) Endocrine 6, 235-242.53. Rajah, R., Lee, K. W., and Cohen, P. (2002) Cell Growth Differ 13, 163-17154. Gucev, Z. S., Oh, Y., Kelley, K. M., and Rosenfeld, R. G. (1996) Cancer Res
56, 1545-1550.55. Buckbinder, L., Talbott, R., Velasco-Miguel, S., Takenaka, I., Faha, B.,
Seizinger, B. R., and Kley, N. (1995) Nature 377, 646-64956. Boyle, B. J., Zhao, X. Y., Cohen, P., and Feldman, D. (2001) J Urol 165,
1319-132457. Mannhardt, B., Weinzimer, S. A., Wagner, M., Fiedler, M., Cohen, P.,
Jansen-Durr, P., and Zwerschke, W. (2000) Mol Cell Biol 20, 6483-6495.58. Lee, J. M., Lee, K.-H., Weidner, M., Osborne, B. A., and Hayward, S. D.
(2002) Proc Natl Acad Sci U S A 99, 11878-1188359. Cao, X., Liu, W., Lin, F., Li, H., Kolluri, S. K., Lin, B., Han, Y. H., Dawson, M.
I., and Zhang, X. K. (2004) Mol Cell Biol 24, 9705-972560. Nickerson, T., Pollak, M., and Huynh, H. (1998) Endocrinology 139, 807-
81061. Uemura, H., and Chang, C. (1998) Endocrinology 139, 2329-2334.62. Mu, X., and Chang, C. (2003) J Biol Chem 278, 42840-4284563. Philips, A., Lesage, S., Gingras, R., Maira, M. H., Gauthier, Y., Hugo, P., and
Drouin, J. (1997) Mol Cell Biol 17, 5946-5951.64. Wilson, T., Fahrner, T., Johnston, M., and Milbrandt, J. (1991) Science 252,
1296-130065. Nigg, E. A. (1997) Nature 386, 779-78766. Lin, B., Kolluri, S. K., Lin, F., Liu, W., Han, Y. H., Cao, X., Dawson, M. I.,
Reed, J. C., and Zhang, X. K. (2004) Cell 116, 527-54067. Labelle, Y., Bussieres, J., Courjal, F., and Goldring, M. B. (1999) Oncogene
18, 3303-3308
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

27
68. Baker, K. D., Shewchuk, L. M., Kozlova, T., Makishima, M., Hassell, A.,Wisely, B., Caravella, J. A., Lambert, M. H., Reinking, J. L., Krause, H.,Thummel, C. S., Willson, T. M., and Mangelsdorf, D. J. (2003) Cell 113,731-742
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

28
Figure Legends
Fig. 1. Rapid Activation of Apoptosis by IGFBP-3. A, Time course apoptosis
induction of Mouse Embryonic Fibroblasts (MEFs) after treatment with 1 µg/ml of
IGFBP-3. Apoptosis induction was quantitated by fluorometric measurement of
activated caspase 3/7. Values are represented as percent of Serum Free. B,
Time course of human glioblastoma line A172 apoptosis induction after
treatment with 1 µg/ml of IGFBP-3. Apoptosis induction was quantitated by
fluorometric measurement of activated caspase 3/7. Values are represented as
percent of Serum Free. C, Time course of human prostate cancer line 22RV1
apoptosis induction after treatment with 1 µg/ml of IGFBP-3. Apoptosis induction
was quantitated by fluorometric measurement of activated caspase 3/7. Values
are represented as percent of Serum Free. ** = p<0.005 relative to Serum Free
conditions.
Fig. 2. IGFBP-3 induces nucleo-mitochondrial translocation of Nur77. A,
Indirect immunofluorescent confocal microscopy of 22RV1 cells after treatment
with IGFBP-3. Nur77 is labeled in red. Nuclei are labeled in blue. Note rapid
appearance of cytoplasmic Nur77. SFM, Serum Free Medium B, Western
immunoblot of subcellular fractions of 22RV1 prostate cancer cells after
treatment with 1mcg/ml IGFBP-3, probed for Nur77. PARP, and Hsp60 are
loading controls and show purity of the nuclear fraction. C, 22RV1 mitochondrial
fraction isolated after treatment with 1mcg/ml IGFBP-3. Membrane was probed
with PARP, PMP70, and Cathepsin S to show purity of the mitochondrial fraction.
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

29
Fig. 3. Rapid Mitochondrial Accumulation of Nur77by IGFBP-3 occurs via a type
1 receptor independent mechanism. A, Type 1 IGF receptor disrupted mouse
embryonic fibroblasts (R- MEFs) were treated with IGFBP-3 (1 µg/ml) for the
indicated times and apoptosis was measured in a fragmented DNA/histone
ELISA assay. B, The mitochondrial fraction was analyzed for expression of Nur77
by Western blotting. To demonstrate the purity of the mitochondrial fraction,
expression of mitochondrial-specific protein Hsp60 and nuclear-specific protein
poly(ADP-ribosyl) polymerase (PARP), as well as peroxisomal PMP70 and
lysosomal cathepsin S is shown. A representative of 3 separate experiments is
shown. Values are represented as percent of Serum Free. ** = p<0.005 relative
to Serum Free conditions.
Fig. 4. Additive effects on apoptosis by IGFBP-3 and Nur77. A, Release of
cytoplasmic cytochrome c by IGFBP-3. Values are expressed as fold increase
from baseline derived from densitometric analysis of western immunoblots of
mitochondria-depleted cytoplasmic fractions probed with cytochrome c. Values
were normalized for loading with β-actin. **P<0.005 relative to time 0. B, Caspase
activation post transient transfection of IGFBP-3, Nur77; alone and in
combination. Values are nomalized to β-galactosidase expression to adjust for
transfection efficiency. **P<0.005 relative to vector alone, and also for
combination compared to Nur77 alone. C, Western immunoblot of
overexpressing transiently transfected cells whole cell lysates.
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

30
Fig. 5. RXRα is required for IGFBP-3-induced Nur77 translocation. A,
Differential subcellular localization of Nur77 to IGFBP-3 in RXRα+/+ and RXRα-/-
cells. Values are expressed as fold increase from baseline derived from Nur77
densitometric analysis of western immunoblots of nuclear fractions of F9 cells
after treatment with IGFBP-3. Values are normalized to nuclear PARP to adjust
for loading. **P<0.005 relative to no treatment. B, Confocal microscopy of F9 cells
transfected with GFP-Nur77. Note extranuclear appearance of GFP-Nur77 after
treatment with IGFBP-3. C, Caspase activation post transfection of Nur77
expression vector. **P<0.005 relative to control vector alone. D, GST-RXRα pull-
down of 22RV1 cell lysates treated with IGFBP-3. IGFBP-3 enhances the ability of
RXRα and Nur77 to physically associate. CCRF-CEM nuclei was used as a
positive control for Nur77 protein expression E, Overexpression of IGFBP-3
enhances mitochondrial RXRα accumulation. Mitochondrial fraction of 22RV1
cells transiently transfected with IGFBP-3 expression vector. Fraction is
immunoblotted with anti- RXRα.
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

31
Figures
0
50
100
150
Caspase 3/7 activation% of SF
C
0
50
100
150
200
250
SF 1h 6h
A17224h
** **
**
Caspase 3/7 activation% of SF
0
50
100
150
200
250
300
SF 2h 6h 24h 48h
Caspase 3/7 activation% of SF
*
**
**
MEF
A B
SF 2h 24h6h
200
** ** **
22RV1
Fig. 1. Rapid Activation of Apoptosis by IGFBP-3. A, Time course apoptosisinduction of Mouse Embryonic Fibroblasts (MEFs) after treatment with 1 µg/ml ofIGFBP-3. Apoptosis induction was quantitated by fluorometric measurement ofactivated caspase 3/7. Values are represented as percent of Serum Free. B,Time course of human glioblastoma line A172 apoptosis induction aftertreatment with 1 µg/ml of IGFBP-3. Apoptosis induction was quantitated byfluorometric measurement of activated caspase 3/7. Values are represented aspercent of Serum Free. C, Time course of human prostate cancer line 22RV1apoptosis induction after treatment with 1 µg/ml of IGFBP-3. Apoptosis inductionwas quantitated by fluorometric measurement of activated caspase 3/7. Valuesare represented as percent of Serum Free. ** = p<0.005 relative to Serum Freeconditions.
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

32
SFM 15 min
1 hr 2 hr
A Nuclear Cytoplasmic
Time (h)
Nur77
PARP
Mitochondrial
0 0.5 1 3
Hsp60
C
0 0.5 1 3 0 0.5 1 3
Time (h)
Nur77
PARPHsp60
PMP70Cathepsin S
B
Fig. 2. IGFBP-3 induces nucleo-mitochondrial translocation of Nur77. A,Indirect immunofluorescent confocal microscopy of 22RV1 cells after treatmentwith IGFBP-3. Nur77 is labeled in red. Nuclei are labeled in blue. Note rapidappearance of cytoplasmic Nur77. SFM, Serum Free Medium B, Westernimmunoblot of subcellular fractions of 22RV1 prostate cancer cells aftertreatment with 1mcg/ml IGFBP-3, probed for Nur77. PARP, and Hsp60 areloading controls and show purity of the nuclear fraction. C, 22RV1 mitochondrialfraction isolated after treatment with 1mcg/ml IGFBP-3. Membrane was probedwith PARP, PMP70, and Cathepsin S to show purity of the mitochondrial fraction.
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from
33
Nur77
Hsp60
PARP
+IGFBP-3SF
15 min 60 min 180 min
PMP70
Cathepsin S
Apoptosis ELISA% of SF
-
B
0
50
100
150
****
**
SF 2h 6h 12h
A
Fig. 3. Rapid Mitochondrial Accumulation of Nur77by IGFBP-3 occurs via a type1 receptor independent mechanism. A, Type 1 IGF receptor disrupted mouseembryonic fibroblasts (R- MEFs) were treated with IGFBP-3 (1 µg/ml) for theindicated times and apoptosis was measured in a fragmented DNA/histoneELISA assay. B, The mitochondrial fraction was analyzed for expression of Nur77by Western blotting. To demonstrate the purity of the mitochondrial fraction,expression of mitochondrial-specific protein Hsp60 and nuclear-specific proteinpoly(ADP-ribosyl) polymerase (PARP), as well as peroxisomal PMP70 andlysosomal cathepsin S is shown. A representative of 3 separate experiments isshown. Values are represented as percent of Serum Free. ** = p<0.005 relativeto Serum Free conditions.
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

34
2
4
6
Fold Increase from baselineCytoplasmic cytochrome c
**
** **
0 min 30 min 60 min 180 min
Fold Increase from baseline Caspase 3/7 Activation
0.5
1
1.5
2
2.5
3
vector BP3 Nur77 BP3
**
**
+Nur77
A B
**
IGFBP-3
Nur77
vector BP3Nur77 BP3+
Nur77
C
βactin
Fig. 4. Additive effects on apoptosis by IGFBP-3 and Nur77. A, Release ofcytoplasmic cytochrome c by IGFBP-3. Values are expressed as fold increasefrom baseline derived from densitometric analysis of western immunoblots ofmitochondria-depleted cytoplasmic fractions probed with cytochrome c. Valueswere normalized for loading with β-actin. **P<0.005 relative to time 0. B, Caspaseactivation post transient transfection of IGFBP-3, Nur77; alone and incombination. Values are nomalized to β-galactosidase expression to adjust fortransfection efficiency. **P<0.005 relative to vector alone, and also forcombination compared to Nur77 alone. C, Western immunoblot ofoverexpressing transiently transfected cells whole cell lysates.
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

35
- + IGFBP-3
IGFBP-3 - + - +
F9WT
F9RXR-/-
3000
4000
5000
6000
Cas
pas
e 3/
7 ac
tiva
tio
nA
rbit
rary
Un
its Control vector
Nur77 vector
F9WT F9RXR-/-
**
F9WT F9RXR-/-
100
200
300
% o
f B
asel
ine
Nu
clea
rN
ur7
7
**
**
Nur77
Cell lysate
IGFBP-3GST-RXRα
CCRF-CEM
MitochondrialRXRα
Control IGFBP-3
+
-
+
- -
+
-
++ +
-
+
-
+
-- - --
+
A B
CD
E
Fig. 5. RXRa is required for IGFBP-3-induced Nur77 translocation. A,Differential subcellular localization of Nur77 to IGFBP-3 in RXRα+/+ and RXRα-/-
cells. Values are expressed as fold increase from baseline derived from Nur77densitometric analysis of western immunoblots of nuclear fractions of F9 cellsafter treatment with IGFBP-3. Values are normalized to nuclear PARP to adjustfor loading. **P<0.005 relative to no treatment. B, Confocal microscopy of F9 cellstransfected with GFP-Nur77. Note extranuclear appearance of GFP-Nur77 aftertreatment with IGFBP-3. C, Caspase activation post transfection of Nur77expression vector. **P<0.005 relative to control vector alone. D, GST-RXRα pull-down of 22RV1 cell lysates treated with IGFBP-3. IGFBP-3 enhances the ability ofRXRα and Nur77 to physically associate. CCRF-CEM nuclei was used as apositive control for Nur77 protein expression E, Overexpression of IGFBP-3enhances mitochondrial RXRα accumulation. Mitochondrial fraction of 22RV1cells transiently transfected with IGFBP-3 expression vector. Fraction isimmunoblotted with anti-RXRα.
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

Kuk-Wha Lee, Liqun Ma, Xinmin Yan, Bingrong Liu, Xiao Kun Zhang and Pinchas Cohennucleo-mitochondrial translocation of RXRalpha/Nur77
Rapid apoptosis induction by IGFBP-3 involves an IGF -independent
published online February 24, 2005J. Biol. Chem.
10.1074/jbc.M412757200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from
Recommended

















