
PharmacologyBIOAVAILABILITY : It is the fraction of the unchanged drug
reaching systemic circulation following administration by any route.
Intravenous route – bioavailability is ~ 1 or 100%.
For drugs administered orally, bioavailability may be less than 100% for two main reasons, incomplete absorption and first pass metabolism.

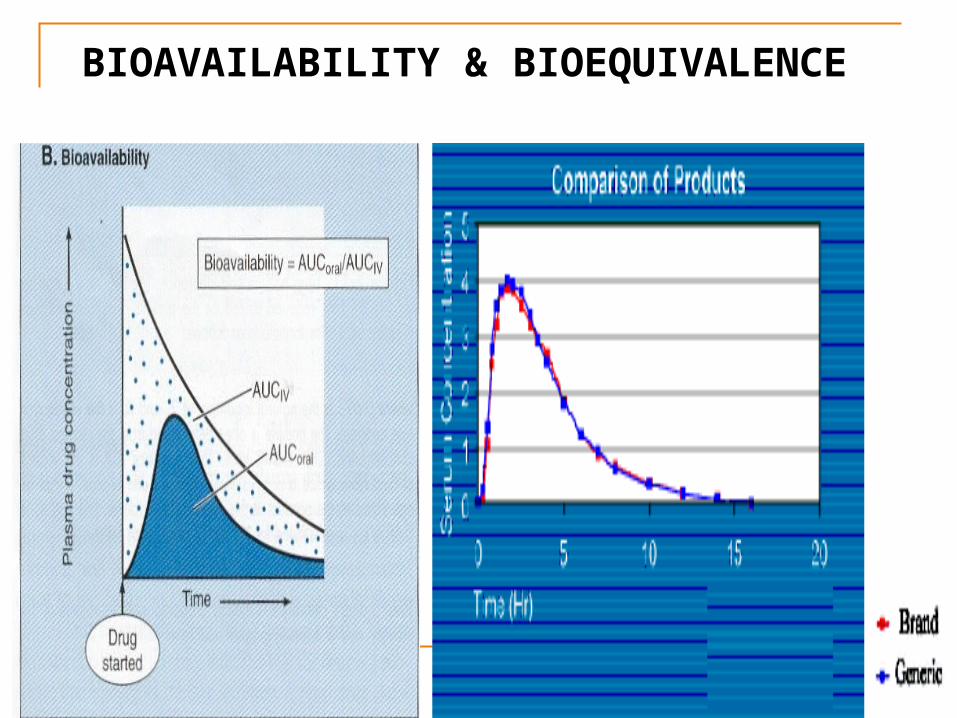
BIOAVAILABILITY & BIOEQUIVALENCE

Bioavailability of a drug
Oral dose = Intravenous dose / F
For example, the oral bioavailability (F) of digoxin (lanoxin) is 0.7 and i.v dose 175 ug.
Oral dose of digoxin = 175 ug / 0.7 = 250 ug
F

PharmacologyBIOEQUIVALENCE:
For two drugs to be bioequivalent, they must have the same bioavailability and the same plasma profile, i.e. the curve must have the same shape.
They must have the same Cmax and Tmax.
Cmax: The maximum plasma concentration attained by a drug-administration.
Tmax: The time at which maximum concentration is reached.

PharmacologyVolume of distribution : It is defined
as the volume in which the amount of drug would need to be uniformly distributed to produce the observed blood concentration. The volume of distribution (Vd) of a drug is given by:
D Vd = --------- Loading Dose =
Vd X C C D = Total amount of the drug in the
body C = Drug concentration in plasma
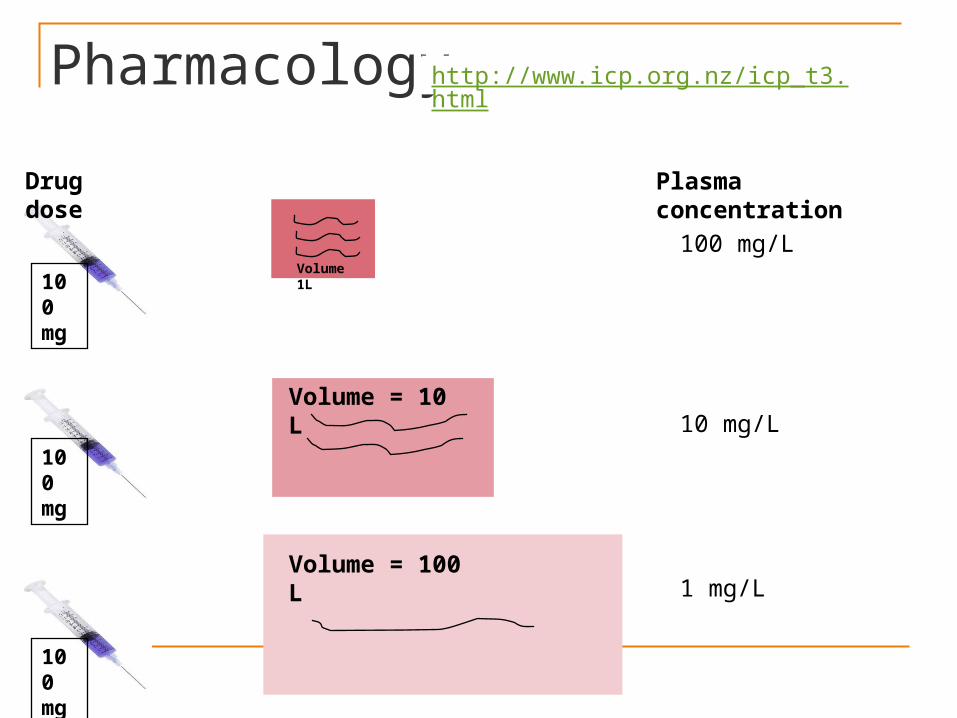
Pharmacology
100 mg
Drug dose
Plasma concentration
100 mg/L
100 mg
100 mg
10 mg/LVolume = 10 L
Volume = 100 L 1 mg/L
Volume 1L
http://www.icp.org.nz/icp_t3.html

Volume of distribution

PharmacologyVOLUME OF DISTRIBUTION
Vd of about 4 L: Present mainly in the vascular compartment. e.g., Heparin.
Vd of about 10 L: Present in extra cellular fluid, but are unable to penetrate cells. e.g., Mannitol.
Vd of about 42 L: Drugs are able to pass most biologic barriers and are distributed in total body water (extra and intracellular) eg., Alcohol.
Vd > 42 L: drugs are extensively stored within specific cells or tissues. e.g., Chloroquine
Intracellular fluid 28L
Plasma 4L
Extracellular fluid 10L

PharmacologyPLASMA PROTEIN BINDING : Acidic drugs bind to albumin. Basic drugs bind to acid glycoprotein. Plasma protein bound drugs are restricted
to the vascular compartment. Bound fraction (storage) is not available
for action Sulfonamides when administered to a
neonates can displace bilirubin from binding sites and can cause kernicterus.
Sulfonamides competes with protein binding of warfarin and can result in increased free plasma concentration of warfarin leading to bleeding.

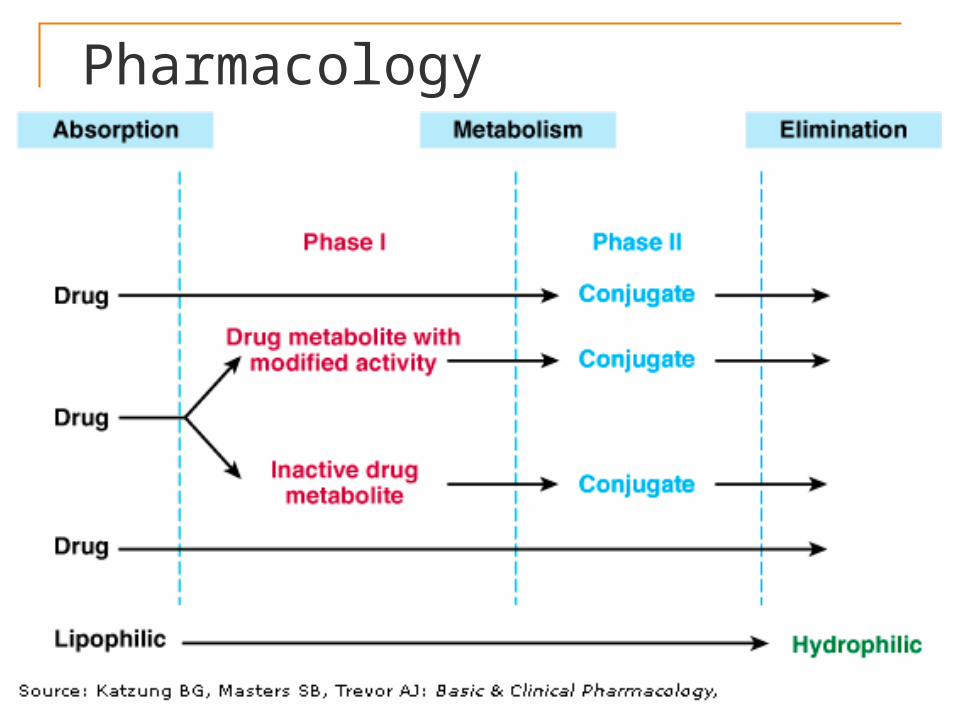
PharmacologyMETABOLISM It renders the lipid soluble drug to water
soluble. The primary sites for metabolism is liver,
others include --- kidney, intestine, lungs and plasma
It can be of two types: • Phase I reactions –
Microsomal - cytochrome P 450 Non-microsomal metabolism• Phase II reactions – conjugation
reactions.
Pharmacology

PharmacologyBiotransformation can leads to --- Inactivation Active metabolite from an
inactive drug – Prodrug e.g. Levodopa dopamine
Enalapril enalaprilat Active metabolite from an active
drug e.g. Diazepam oxazepam
Imipramine desipramine

Phase I reactions : Cytochrome P 450 enzyme system:1. Oxidative dealkylations – Morphine, codeine2. Deamination – Amphetamine, diazepam3. Desulfuration - Thiopental Cytochrome P450-independent
oxidations1. Flavin mono-oxygenase – Chlorpromazine,
amitriptyline2. Dehydrogenases – Alcohol Reductions1. Carbonyl reductions – Methadone, naloxone2. Nitroreductions – Clonazepam Hydrolyses1. Ester – Procaine, succinylcholine 2. Amides – Lidocaine, indomethacin

Drug metabolism

Drug metabolismo INHIBITION OF CYP 450 DRUG
METABOLISM : Eg : cimetidine, erythromycin,
clarithromycin, grapefruit juice, SSRI o CYP 450 ENZYME INDUCTION : Eg : Chronic alcohol, carbamazepine,
phenobarbital, phenytoin, rifampin, St. John’s wort.
Lopinavir is co-formulated with Ritonavir in order to take advantage of P450 inhibition by Ritonavir which increases the beneficial effect of Lopinavir.

CYTOCHROME P 450 2 D6Absent in about 7% CaucasiansHyperactive in about 30% East AfricansCatalyzes the metabolism of codeine, many TCAs and beta blockersInhibited by fluoxetine, paroxetine, quinidine
Eg., Codeine is actually converted to morphine, a better analgesic. Codeine itself is much less active as an analgesic, but causes nausea and other adverse effects. The absence of cytochrome P450 2D6 in 7% of Caucasians means that these individuals cannot metabolize codeine to the active metabolite, morphine, and therefore will get little, if any, pain relief from codeine.

Pharmacology
Phase II reactions : It is the conjugation of a drug to form a polar (ionized) drug which can be easily excreted.
Glucuronide conjugation – diazepam, digoxin, morphine
Glutathione conjugation - acetaminophen
Sulfation conjugation – estrogens
Acetylation – sulfonamides, INH Methylation – epinephrine, histamine
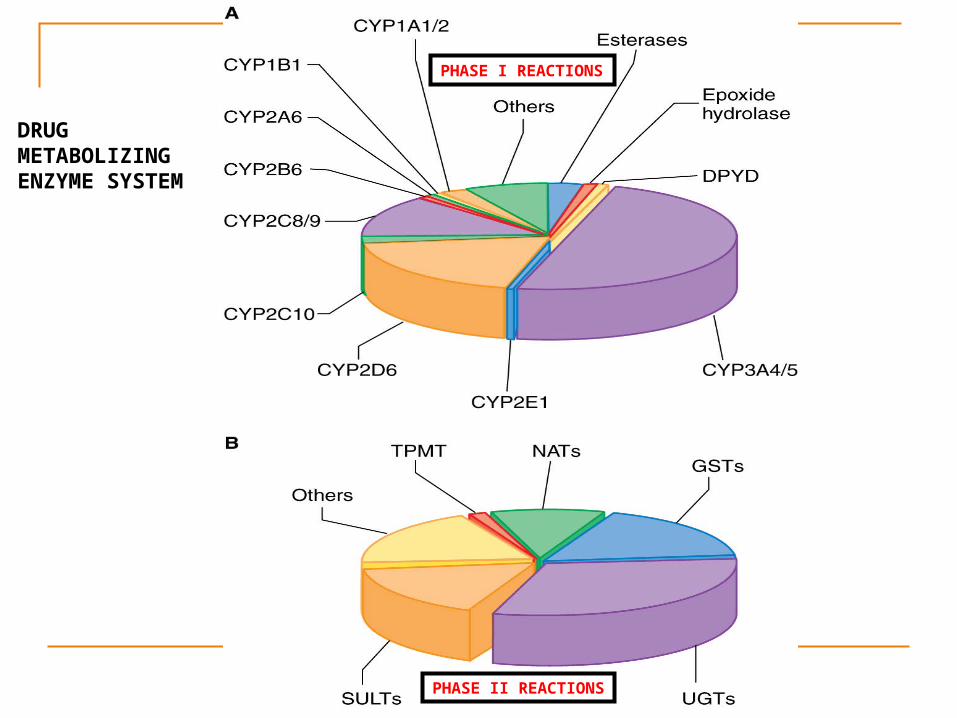
PHASE I REACTIONS
PHASE II REACTIONS
DRUG METABOLIZING ENZYME SYSTEM

PharmacologyEXCRETION Kidney : excretes all water soluble
drugs. Lipid soluble drugs are reabsorbed. Changes in urinary pH affects the
excretion of drugs acidic drugs are excreted in alkaline
urine and urine made basic by agents like sodium bicarbonate (NaHCO3), potassium citrate and acetazolamide.
basic drugs are excreted in acidic urine and urine made acidic by ammonium chloride (NH4Cl), Vitamin-C, Cranberry juice.

PharmacologyP-GLYCOPROTEIN is a protein that play a
protective role by preventing entry into body and promoting removal of foreign substances (drugs) from the body.
P-glycoprotein is expressed in intestinal mucosa, renal proximal tubules, blood-brain-barrier and in tumor cells (where it functions as a multi-drug resistance mechanism).
Induction of P-glycoprotein can result in decreased intestinal absorption, reduced entrance into the CNS and increased secretion of drugs into renal tubules.
Inducers of P-glycoprotein : Rifampin & St. John's wort
Inhibitors: Cimetidine & Grapefruit Juice.

Pharmacology
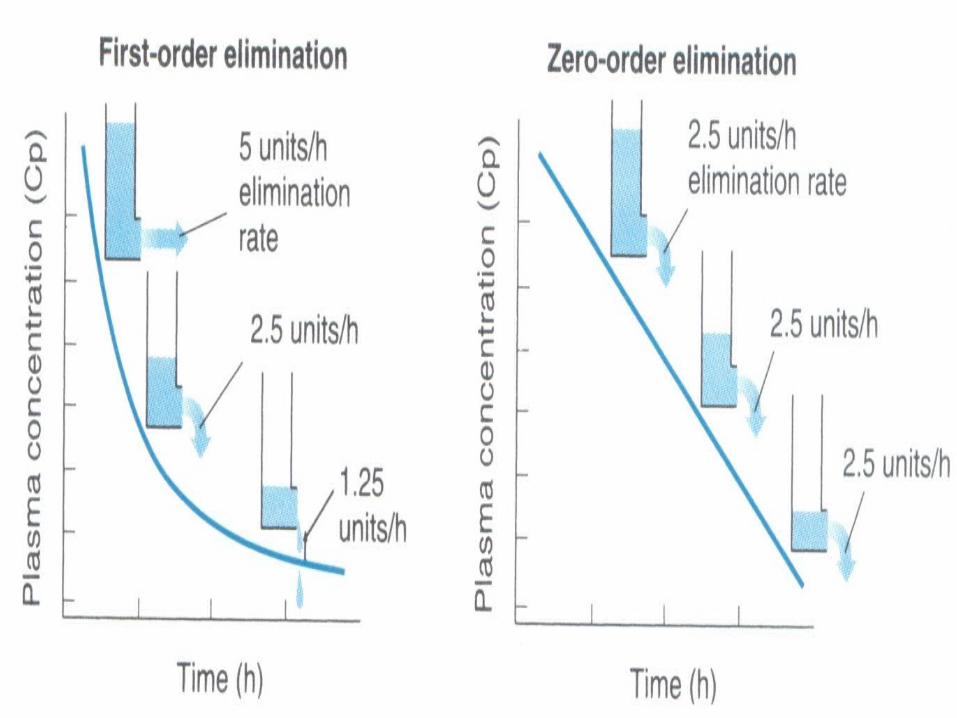
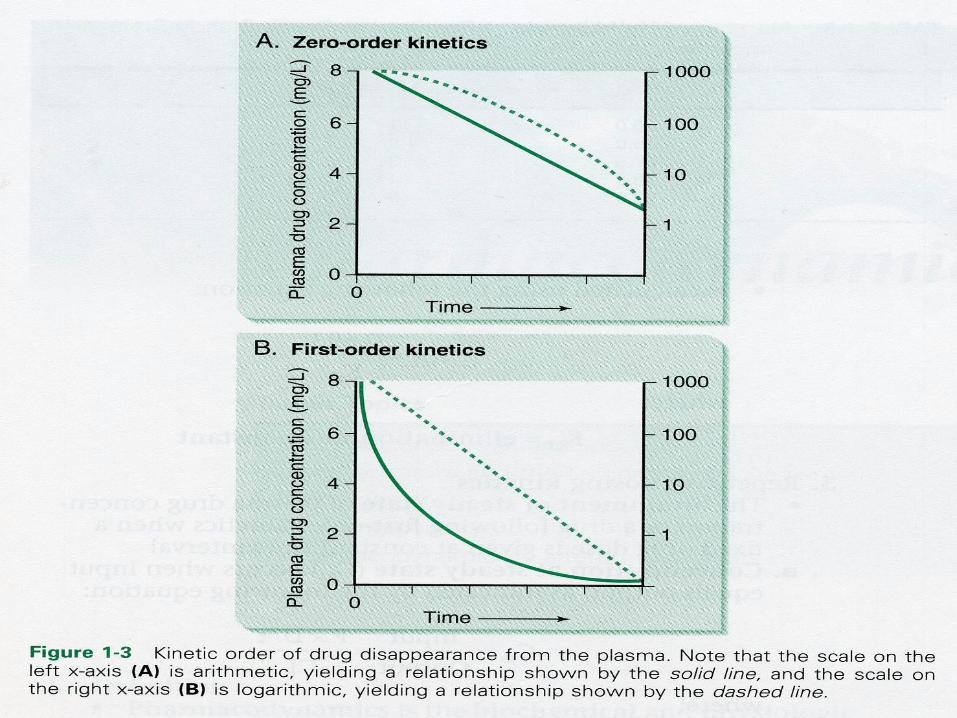
FIRST ORDER KINETICS : The rate of elimination of the
drugs is proportional to the plasma concentration.
Constant fraction of the drug from the plasma is eliminated in unit time.
80mg 40mg 20mg 10mg 5mg
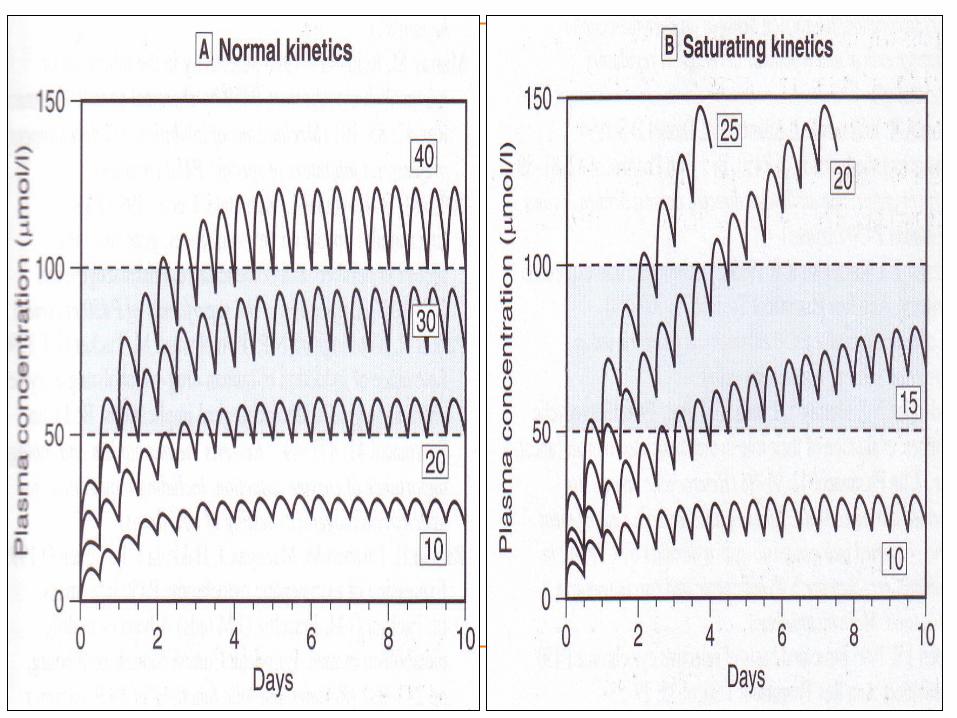
Most of the drugs follow first order kinetics (within the therapeutic ranges).
Pharmacokinetics

PharmacologySATURATION / ZERO ORDER
KINETICS: The rate of the elimination of
drug is constant irrespective of the plasma concentration.
Constant amount of drug is eliminated in unit time. Eg : Phenytoin, Alcohol, Aspirin.
80mg 70mg 60mg 50mg 40mg

PharmacologyHALF LIFE : Plasma half life: It is the time
required for the drug to reduce to half its original plasma value.
Half life is a concept which is applied only to drugs following first order kinetics.
The half‑life of a drug is given by: 0.693
0.693 x Vd t½ = ------------ =
---------------- Ke
CLKe = Elimination constant, is calculated
by Ke = CL / Vd

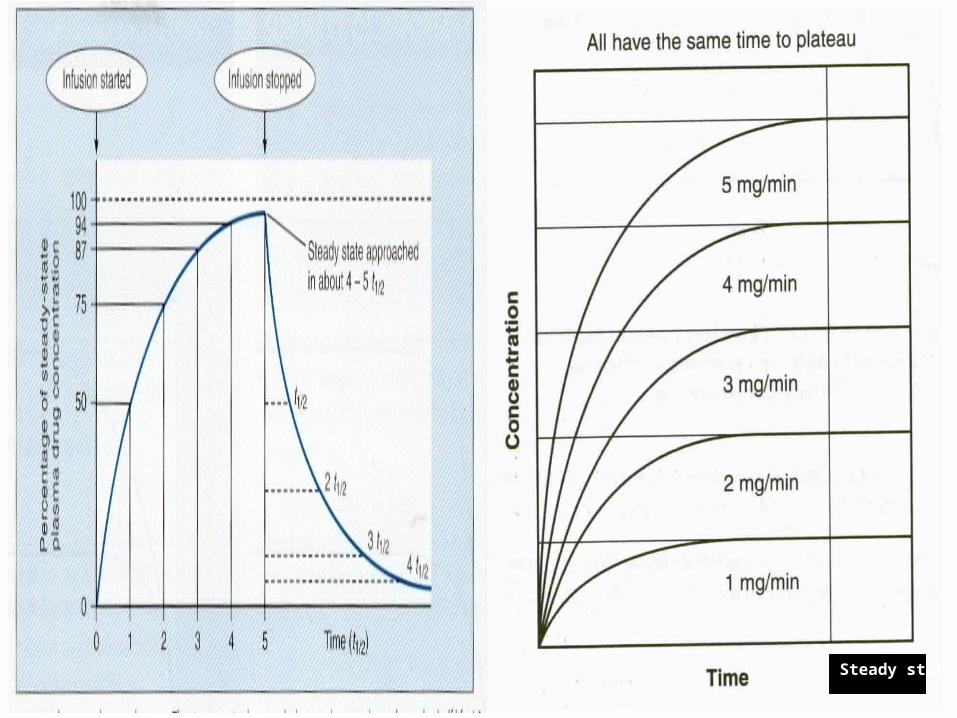
Pharmacology Steady state is the state when
the Rate in = Rate out. The time to reach the steady
state is dependent only on half life of a drug and is independent of the dose size and the frequency of administration.
Exception : different doses as in loading dose and then maintenance dose.


Half life and steady state
Half life 1 2 3 4 5 6 7 8
90% of steady state = 3.3 half life

Pharmacology
Rate of Infusion : Irrespective of the rate of
infusion, it takes the same amount of time to reach the steady state.
If the rate of infusion is doubled, then the plasma level of the drug at steady state is doubled.
Steady state

Pharmacology
CLEARANCE : It is the volume of the plasma from which the drug is completely removed in unit time.
Rate of elimination Clearance = --------------------------- Plasma concentration

Pharmacology Drugs which follow a first‑order
kinetics reach a steady state when the rate of input is equal to the rate of elimination.
At the steady state : Rate of infusion = Rate of
elimination Rate of elimination CL = ----------------------------- Drug concentration

Pharmacokinetics At steady state Rate of infusion = Css X
CL Dosing rate = Css X CL Maintenance dose (i.v) = Dosing
rate x Dosing interval τ (hours) Maintenance Dose (Oral) = Css X CL X τ / F
Dose X F
Css = ----------------
CL X τ

Steady state concentration
Dose X FCss = ------------------ CL X τ
What will be the new steady state concentration, if the dose interval is reduced from every 8 hrs to every 4 hrs considering the dose and route of administered used are same in that individual?
A. Increase 4 timesB. Increase 2 timesC. Remains sameD. Decrease by halfE. Decrease by 1/4

Pharmacology If a kinetic process is first-order, the
clearance and the half‑life of the drug are constant and independent of the dose.
If a kinetic process is zero-order, the clearance and the half‑life of the drug are not constant, but are dose-dependent.
Rate of elimination Clearance = ------------------------------- Plasma conc. Vd t½ = 0.693 -------- CL

PharmacologyCalculation of the loading dose When the desired plasma concentration is
known. Loading dose = Vd X desired conc in
plasma
Patients CL Corrected dose = Average dose/day X
-----------------------
Normal CL
Q. A patient was taking gentamicin for the treatment of urinary tract infection at the dose of 80 mg 12th hourly with the normal kidney function. What is the best and the appropriate dose of the gentamicin in the patients with kidney function reduced to 50 %?A. 80mg 6th hourlyB. 40mg every dayC. 60mg 12th hourlyD. 80mg every day E. 20 mg 8th hourly

Steady state plasma concentration

Recommended

















