
Carcinoma polmonare non-
microcitoma (CPNM)
Giulio Metro
S.C. Oncologia Medica – Ospedale Santa Maria della
Misericordia, Azienda Ospedaliera di Perugia
“Corso di laurea in tecniche di laboratorio biomedico”
15 Marzo 2016, Perugia

erb-b1EGFRHER1
neu Eb-b2HER2
Erb-b3HER3
Erb-b4HER4
= Dominio tirosin-chinasico
Dominio che legail ligando
Dominio trans-membrana
La famiglia dell’EGFR come bersaglio terapeutico
Nessun ligandospecifico. Partner
preferito di dimerizzazione Hereguline
NRG2NRG3
Heregulineβ-cellulina
EGF, TGFa,Amphiregulin
Dominio tirosin-chinasico

• La famiglia dei recettori HER/ErbB (Human Epidermal growth factor
Receptor) è un gruppo di proteine di membrana ad attività tirosin-chinasica
composto da quattro membri: EGFR (ErbB1), HER2 (ErbB2), ErbB3 ed
ErbB4.
• Per ciascuno di questi 4 recettori si identifica un dominio extracellulare che
interagisce con uno specifico ligando, una porzione trans-membrana e un
dominio intracellulare.
• Tutti questi recettori svolgono un’importante azione di trasduzione dei
segnali tra ambiente extra- e intra-cellulare.
Sono espressi in numerosi tipi di tessuti, epiteliali, mesenchimali, nervosi,
ove rivestono un ruolo fondamentale nella proliferazione, crescita,
sopravvivenza, differenziazione e migrazione della cellula, e sono coinvolti
nello sviluppo e nella progressione di numerosi tipi di tumori.
Tutti i recettori della famiglia ErbB promuovono la
crescita e la progressione di numerosi tipi di tumori

• L’attivazione aberrante dei 4 recettori
della famiglia ErbB con conseguente
deregolazione del segnale di trasdu-
zione può avvenire con diversi
meccanismi:
1. iperespressione dei recettori sulla
membrana delle cellule tumorali;
2. mutazioni attivanti i geni dei recettori ErbB;
3. aumentata espressione genica
• La tabella a lato riporta il tipo di
deregolazione presente in alcuni dei
più frequenti tipi di tumore.
Tutti i recettori della famiglia ErbB promuovono la
crescita e la progressione di numerosi tipi di tumori
Alterazioni molecolari a carico dei recettori della famiglia
ErbB osservati nei tumori umani

• Il legame tra i recettori della famiglia ErbB
e i rispettivi ligandi induce la omo o la
eterodimerizzazione dei recettori stessi.2
I recettori, combinandosi tra di loro, possono
formare 4 omo-dimeri (per accoppiamento di
due recettori dello stesso tipo) o 6
eterodimeri (per accoppiamento di 2
recettori di tipo diverso).
• In seguito alla omo-/etero-dimerizzazione
del recettore, vengono attivate numerose
proteine citoplasmatiche, tra cui Ras/MAPK,
e PI(3)chinasi/Akt che scatenano una serie
di reazioni a cascata lungo le vie di
trasduzione del segnale che regolano i
processi di proliferazione, differenziazione e
sopravvivenza cellulare.
La omo- e la etero-dimerizzazione dei recettori della famiglia
ErbB attivano meccanismi che contribuiscono alla crescita
tumorale
Omo- ed etero-dimerizzazione dei recettori
della famiglia ErbB
Omodimeri Eterodimeri Omodimeri

Anatomia del sistema respiratorio che mostra la trachea ed entrambi I polmoni con i lobi e le vie respiratorie.
L’ossigeno viene inalato nei polmoni e passa tramite le sottili membrane degli alveoli nel flusso sanguigno (vedi
ingrandimento)

Fattori di rischio
• Fumo attivo di sigaretta (responsabile di ca. l’85%
dei tumori al polmone) (durata>>intensità)
• Fumo passivo
• Radon ambientale (gas radioattivo prodotto dal
decadimento dell’uranio)
• Asbesto (minerale non più in utilizzo)
• Altri fattori (suscettibilità genetica, inquinamento
ambientale, fattori virali?, della dieta?)

Il “peso” del tumore al polmone
Parkin D, et al. CA Cancer J Clin 2005;55:74–108; Ferlay J, et al. Ann Oncol 2007;18:581–592
Tumore del polmone in Europa:
292.000 nuovi casi
253.300 morti
Tumore del polmone nel mondo:
1.5 millioni di nuovi casi
1.18 millioni di morti
CPNM rappresenta >80% dei tumori al polmone

Carbone. Semin Oncol 1997
Charloux, et al. Int J Epidemiol 1997
Adenocarcinoma
classico
(75–90%)
Carcinoma non-
squamoso
(70–75%)
Carcinoma
squamoso
(25–30%)
CPNM (ca. 80%)
Suddivisione del CPNM
Fumo fattore di rischio
preponderante
Fumo importante fattore
di rischio ma si osserva
spesso anche in chi non
ha mai fumato

Risultati per terapie a base di cisplatino (11 studi)
100
80
60
40
20
0
Sopra
vviv
enza (
%)
0 6 12 18 24Tempo dalla randomizzazione (mesi)
Terapia di supporto + chemio
Terapia di supporto
NSCLC Collaborative Group, BMJ 1995
La chemioterapia a base di platino migliora la
sopravvivenza nel CPNM metastatico (non operabile)

CPNM metastatico: stiamo raggiungendo
sopravvivenze più lunghe?
Supporto:2–5 months
Platino agente singolo:6–8 months
Doppiette con platino:8–10 mesi
Sopravvivenza mediana (mesi)
Le combinazioni di chemioterapia hanno fallito
nell’incrementare in maniera significativa la sopravvivenza mediana oltre gli 8-10 mesi
Un “plateau” terapeutico si èraggiunto. Le nuove combinazioni di chemioterapia non miglioreranno la sopravvivenza
Schiller, et al. NEJM 2002; Sandler, et al. NEJM 2006

Verso la terapia personalizzata
• Ogni paziente è unico e con peculiari
caratteristiche cliniche e molecolari.
• Il trattamento che funziona in un paziente potrebbe
non funzionare o funzionare meno in un altro
paziente
• “Ricamare” la terapia più appropriata
focalizzandosi su determinati bersagli è l’unico
modo per migliorare i risultati del trattamento.

Angiogenesi e cancro

Bevacizumab: anticorpo contro il fattore di
crescita vascolare endoteliale (secreto)

So
pra
vviv
en
za
me
dia
na
(m
esi)
15
10
5
0
12.3
13.4 13.6
14.6
SAiL4AVAiL3
(7.5mg/kg)E45991 AVAiL2
(15mg/kg)
1Sandler, et al. N Engl J Med 2006
2,3Reck, et al. Ann Oncol 2010
4Crinò, et al. Lancet Oncol 2010
5Wozniak et al. ASCO 2010
13.3
ARIES5
(7.5 or
15mg/kg)
Sopravvivenza > 12 mesi con l’aggiunta del bevacizumab
alla chemioterapia nel trattamento del CPNM metastatico

• Solo marcatori clinici; farmaco tossico nel tumore
squamoso dove non trova impiego (solo
adenocarcinoma)
• Massima efficacia nell’istologia adenocarcinoma
• Nessun marcatore biologico che predica la
sensibilità identificato sin’ora
Ci sono biomarcatori che predicano la
sensibilità al bevacizumab?

Evolution of NSCLC subtyping
Li, et al. J Clin Oncol 2013

Sottogruppi molecolari
nell’adenocarcinoma
Pao & Hutchinson Nat Med 2012

CPNM con mutazione EGFR o
riarrangiamento genico ALK o ROS1
• Le cellule tumorali contengono multiple anomalie
genetiche ed epigenetiche
• Nonostante tale complessità, la loro crescita può
spesso essere bloccata dall’inattivazione di un
singolo oncogene
• Questo fenomeno, chiamato “oncogene-
addiction”, fornisce un forte razionale per la
terapia molecolare a bersaglio
• In tal senso il CPNM dei mai o “deboli” fumatori è
spesso una malattia “oncogene-addicted”

Da: Sun, et al. Nat Rev Cancer 2007
CPNM nei mai fumatori: incidenza

CPNM nei mai fumatori e istologia
Da: Sun, et al. Nat Rev Cancer 2007
Nettamente prevalente l’adenocarcinoma

CPNM nei mai fumatori e biologia
Da: Sun, et al. Nat Rev Cancer 2007
La mutazione di EGFR identifica un CPNM non
correlato al fumo, mentre la mutazione di KRAS
identifica spesso un tipo di CPNM fumo-relato

ATP
Ras-Raf-MAPK
Proliferazione
Pi3K-AKT
Sopravvivenza
Ligando
Dominio extra-cellulare
Dominio trans-membrana
Dominio tirosin-chinasico
Fosforilazione della tirosina
Wild-type EGFR Mutant EGFR
Arteaga 2006; Gadzar et al 2004; Hendricks et al 2006; Sordella et al 2004
La mutazione dell’EGFR causa un cambiamento
conformazionale del recettore

• Le delezioni nell’esone 19 e la mutazione punti-
forme L858R nell’esone 21 sono le mutazioni
attivanti più comuni del dominio tirosino chinasico
di EGFR (oltre il 90% di tutte le mutazioni).
• Nei pazienti con NSCLC, la presenza di queste
mutazioni di EGFR è predittiva di una significativa
risposta clinica agli inibitori tirosino chinasici di
EGFR (EGFR TKI). Si pensa che queste
mutazioni, aumentando l’affinità dell’inibitore per il
recettore, rappresentino il meccanismo molecolare
alla base dell’efficacia clinica dei TKI.
• Il rimanente 10% circa delle mutazioni consistono
in rare mutazioni missense riscontrabili
principalmente nell’esone 18 ma anche negli esoni
20 e 21.
Deregolazione del recettore EGFR (ErbB1)
Le delezioni nell'esone 19 e la
mutazione puntiforme L858R
nell'esone 21 rappresentano il 90%
delle mutazioni di EGFR13

Mutazioni nel gene dell’EGFR
Riely, et al. Clin Cancer Res 2006
Exons 1–16
Exons 18–24
Exons 25–28
EGFR transcript
Exon 17
Extracellulardomain
Trans-membrane
domain
Tyrosine-kinase domain
Regulatory domain
Confer sensitivity/resistance to EGFR TKIs
Unclear effect on sensitivity to EGFR TKIs
18
18
19
20
21
Deletions
L858R
G719A/S
L861X
P694X
V700D
E709X
G735S
V738F V742A
T751IS752Y
D761N
A763V
N765A
S768I
T783A
L792P
L798F
G810SN826S
L838VT847I
I853TA859T
E866K
L833VH835L
H850NV851X
G863DA864T
L730F P733L
E746K
D761Y
D770_N771 insNPG
T790M
EGFR
TKI = tyrosine-kinase inhibitor

Distribuzione della mutazione dell’EGFR
in base all’esone
Sequist, et al. J Clin Oncol 2007

Mutazione dell’EGFR: più frequente nei
mai fumatori e nel sesso femminile
D’Angelo, et al. J Clin Oncol 2011

L’avvento degli inibitori dell’EGFR a partire dai
primi anni 2000
EGFR Inhibitors
EGFR Inhibitors:
Monoclonal antibody – cetuximab, etc.
Small molecule TK inhibitors – gefitinib, erlotinib
From Howard West,
M.D., OncTalk.com

Presenza mutazione Assenza mutazione
Test di interazione, p<0.0001
HR (95% CI) = 0.48 (0.36, 0.64)
p<0.0001
No. eventi gefitinib, 97 (73.5%)
No. eventi C / P, 111 (86.0%)
Gefitinib (n=132)
Carboplatin / paclitaxel (n=129)
HR (95% CI) = 2.85 (2.05, 3.98)
p<0.0001
No. eventi gefitinib , 88 (96.7%)
No. eventi C / P, 70 (82.4%)
132 71 31 11 3 0129 37 7 2 1 0
108103
0 4 8 12 16 20 24
GefitinibC / P
0.0
0.2
0.4
0.6
0.8
1.0
Pro
bab
ilit
y o
f p
rog
ressio
n-f
ree s
urv
ival
A rischio :91 4 2 1 0 085 14 1 0 0 0
2158
0 4 8 12 16 20 24
0.0
0.2
0.4
0.6
0.8
1.0
Pro
bab
ilit
y o
f p
rog
ressio
n-f
ree s
urv
ival
Gefitinib (n=91)
Carboplatin / paclitaxel (n=85)
Months Months
Sopravvivenza libera da malattia con gefitinib vs.
chemioterapia in base alla presenza/assenza della
mutazione dell’ EGFR

Gefitinib vs. chemioterapia nei pazienti con
mutazione dell’EGFR
Mok et al 2009; Kobayashi et al 2009; Mitsudomi et al 2009
NEJ002
HR (95% CI) = 0.36 (0.25, 0.51)
p<0.001
Days
Gefitinib (n=98)
C/P (n=100)
0.0
0.2
0.4
0.6
0.8
1.0
0 100 200 300 400 500
p<0.001
HR (95% CI) = 0.49 (0.34, 0.71)
p<0.0001
Months
0
20
40
60
80
100
0 10 20 30 40
WJTOG 3405
Gefitinib (n=58)
C/D (n=59)
p<0.0001
62.1
32.2
0
20
40
60
80
100

Erlotinib vs. chemioterapia in pazienti EGFR-
mutati: sopravvivenza libera da progressioneP
FS
pro
bab
ilit
y
1.0
0.8
0.6
0.4
0.2
0
Erlotinib (n=82)
Gem/carbo (n=72)
HR=0.16 (0.10–0.26)Log-rank p<0.0001
Time (months)
0 5 10 15 20 25
Patients at risk
Erlotinib 82 70 51 20 2 0
GC 72 26 4 0 0 0
13.14.6

Parametro Erlotinib Chemioterapia
Tossicità grado 3 & 4 45% 81%
Modifiche/interruzione di
dose per tossicità23% 47%
Sospensione del
trattamento per tossicità5% 14%
Rosell, et al. ASCO 2011
Differenza in tossicità

N EGFR RR (%) PFS (mesi) OS (mesi)
278 Classiche esone 19-21 74.1 8.5 19.6
272 Wild-type 16.5 2.0 10.4
11 Inserzione esone 20 0 1.4 4.8
15 G719 53.3 8.1 16.4
15 L861 60.0 6.0 15.2
15 Mutazioni rare 20.0 1.6 11.1
Wu J et al. Clin Cancer Res 2011;17:3812-3821
Sensibilità differente a gefitinib ed erlotinib a seconda
del tipo di mutazione dell’EGFR

Nei pazienti con mutazione dell’EGFR,
gefitinib ed erlotinib rispetto alla
chemioterapia:
• Producono un tasso di risposte tumorali più
elevato
• Producono una migliore sopravvivenza libera
da progressione tumorale
• Sono associati ad una migliore qualità di vita
• Inducono una minore tossicità

Study First line Median OS Ref
IPASS Gefitinib 21.6M Yang CH et. al. Ann Oncol
2010:21;LBA2
Pac/Car 21.9M
NEJ002 Gefitinib 27.7M Inoue A. et al. JCO 2011
Pac/Car 26.6M
WJOG3405 Gefitinib 30.9M Lancet Oncology;
2010:11;121-8
Doc/cis Not reached
First-SIGNAL Gefitinib 30.6M Lee JS. et.al. JTO;2009;4:
PRS 4
Gem/cis 26.5M
CAMP Gefitinib 27.7M Morita S. et. al. CCR
2009;15:4493-98
Chemotherapy 25.7M
Spanish Erlotinib 27.0M Rosell R. et al. NEJM
2009;361;958-67
Sopravvivenza del paziente con CPNM EGFR-mutato trattato
con EGFR-TKIs

Inibitori reversibili: Afatinib and Dacomitinib
Afatinib1
• Orally bioavailable, small molecule TKI
• Designed to irreversibly bind to the ATP binding pocket of EGFR and HER2
• Highly specific for EGFR and HER2– EGFR IC50: 0.50nM– HER2 IC50: 14nM
Dacomitinib2
•Irreversible inhibitor of the tyrosine kinases of EGFR (HER1), HER2, HER4
– ‘Pan-HER’ inhibitor
•Preclinical activity against
– EGFR sensitising mutations
– EGFR T790M
– wild-type HER2
– mutant HER2NCI-H1975
1. Li, et al. Oncogene 2008
2. Engleman, et al. Cancer Res 2008

Endpoint primario: sopravvivenza libera da progressione
Review indipendente‒ tutti i pazienti randomizzatiS
op
ravviv
en
za
libe
rada p
rog
ressio
ne
(pro
ba
bili
tà)
1.0
0.8
0.6
0.4
0.2
0.0
Pz a rischio
Afatinib 230 180 151 120 77 50 31 10 3 0
Cis/Pem 115 72 41 21 11 7 3 2 0 0
Sopravvivenza libera da progressione (mesi)0 3 6 9 12 15 18 21 24 27
Afatinib
n=230
Cis/pem
n=115
SLP evento, n (%) 152 (66) 69 (60)
SLP (mesi) 11.1 6.9
Hazard ratio
(95% intervallo di
confidenza)
0.58 (0.43–0.78)
p=0.0004
47%
22%
Yang JC, et al.

Toxicity: indirect comparison of reversible
versus irreversible EGFR-TKIs
Gefitinib
NEJSG 002
n=114
IPASS
n=607
First-SIGNAL
n=159
WJTOG3405
n=87
Rash 71.0 (5.3) 66.2 (3.1) 72.3 (1.3) 74 (2)
Diarrhoea 34.2 (0.9) 46.6 (3.8) NR 47(1)
Fatigue 10.5 (2.6) NR 28.3 (0.6) 34 (2)
Anorexia NR 21.9 (1.5) 44.7 (0) NR
Stomatitis 9.6 (0) 17.0 (0.2) NR 19 (0)
Paronychia NR 13.5 (0.3) NR 28 (1)
Vomiting 6.1 (0.9) 12.9 (0.2) NR NR
Erlotinib
OPTIMAL
n=83
CALGB30406
n=81
73.5 (2.4) NR (7.4)
25.3 (1.2) NR (4.9)
4.8 (0) NR (1.2)
NR NR
13.3 (1.2) NR
3.6 (0) NR
NR NR
Afatinib
LUX-3
n=229
37 (16.2)
33 (14.4)
3 (1.3)
7 (3.1)
20 (8.7)
26 (11.4)
7 (3.1)

Basale Regressione tumorale Progressione(mediana 9 mesi)
L’insorgenza di resistenza inevitabile

Le cellule resistenti sono selezionate dal
trattamento
Sensibili Resistenti
Prima di
gefitinib/erlotinib
Alla progressione di
malattia in corso di
gefitinib/erlotinib
Nel momento
della massima
risposta tumorale

• Sostituzione di una metionina con treonina alla posizione 790 (T790M) nel dominio chinasico nell’esone 20 aumenta l’affinit dell’EGFR per l’ATP
• ~ 50% dei pazienti con resistenza acquisita a gefitinib/erlotinib
• Rara ma possibile causa di resistenza primaria qualora sia presente dall’inizio
Pao PLos Med 2005; Kobayashi NEJM 2005; Yun PNAS 2008
Mutazione secondaria in pazienti con CPNM EGFR mutato
resistente a gefitinib/erlotinib: T790M

La mutazione T790M nel CPNM
Godin-Heyman Cancer Res 2007


AZD9291 e Rociletinib in pazienti EGFRT790M+
Rociletinib
AZD9291
RR:53%
RR:61%
Sequist L, ASCO 2015
Janne PA, NEJM 2015

• Il DNA libero circolante è estratto direttamente
dal plasma o siero
da Oxnard, IASLC 2013
Biopsia liquida

Studio di Gefitinib fase IV (N= 106)
Douillard, et al. EMCTO 2013

Douillard et al., Br J Cancer 2014; Douillard et al., J Thorac Oncol 2014
Frequenza di mutazione di EGFR tra tumore e
plasma
• La concordanza nella mutazione EGFR è risultataconcorde in 615 di 652 pazienti valutati con entrambi i campioni
*Per i pz valutabili sia per plasma che tessuto
*

Douillard, et al. J Thorac Oncol 2014
Efficacia del trattamento in base alla presenza di
mutazione nel tumore vs. plasma

April 2014: l’indicazione EMA per gefitinib è
cambiata

Identificazione della fusione di EML4-ALK Fusion
nel CPNM
Soda M, et al. Nature. 2007;448:561–67.
~3.6 kb
EML4
EML4–ALK variante 1
HELP1 496 981
WDBasic
1 496 1059
1 1058 1620
TM
EML4-ALK variant 1
Exon 13
ALK EML4
297 bpExon 21
KinaseALK
EML, echinoderm microtubule-associated protein-like 4;
HELP, hydrophobic echinoderm mizroxbule-associated protein-like protein

Inversion Translocation
Or
ALK
Partner gene product
ALK fusion protein*
Tumour cell
proliferation
Cell
survival
PI3K
BAD
AKT
STAT3/5
mTOR
S6K
RAS
MEK
ErK
PLC-Y
PIP2
IP3
1Inamura K, et al. J Thorac Oncol. 2008;3:13–17. 2Soda M, et al. Proc Natl Acad Sci. U S A. 2008;105:19893–97.
Figure based on: Chiarle R, et al. Nat Rev Cancer. 2008;8(1):11–23. Mossé YP, et al. Clin Cancer Res. 2009;15(18):5609–14; and Pfizer Inc, data on file.
*La localizzazione sub-cellulare del gene di fusione ALK, sebbene sia probabilmente nel citoplasma, non è cinfermata.1,2
BAD, BCL2-associated agonist of death; STAT3, signal transducer and activator of transcription 3; S6K, ribosome protein S6 kinase;
ERK, extracellular signal-regulated kinase.
Vie di traduzione tradotte da EML4-ALK


EML4/ALKEtà
mediana Maschi Donne
Mai
fumatoriFumatori Adenoca.
Squamo-
si
129/3933
(3.3%)
59
(29-79)
56/1451
(3.9%)
51/1017
(5.0%)
83/762
(10.8%)
36/1534
(2.3%)
118/2168
(5.4%)
8/870
(0.9%)
Caratteristiche cliniche dei pazienti con
CPNM ALK+
• Bassa età mediana
• Più comune nei pazienti che non hanno mai
fumato o hanno fumato poco
• Quasi esclusivamente adenocarcinomi
Da una revisione di 14 studi della letteratura

Crizotinib
– Inibitore competitivo potente e selettivo per l’ATP delle tirosin-
chinasi di ALK e MET e delle loro varianti oncogeniche1,2
– Crizotinib somministrato in capsule da 250 mg due volte al
giorno3
1. Zou, et al. Cancer Res 2007; 2. Christensen et al. Mol Cancer Ther 2007; 3. Kwak, et al. N Engl J Med 2010
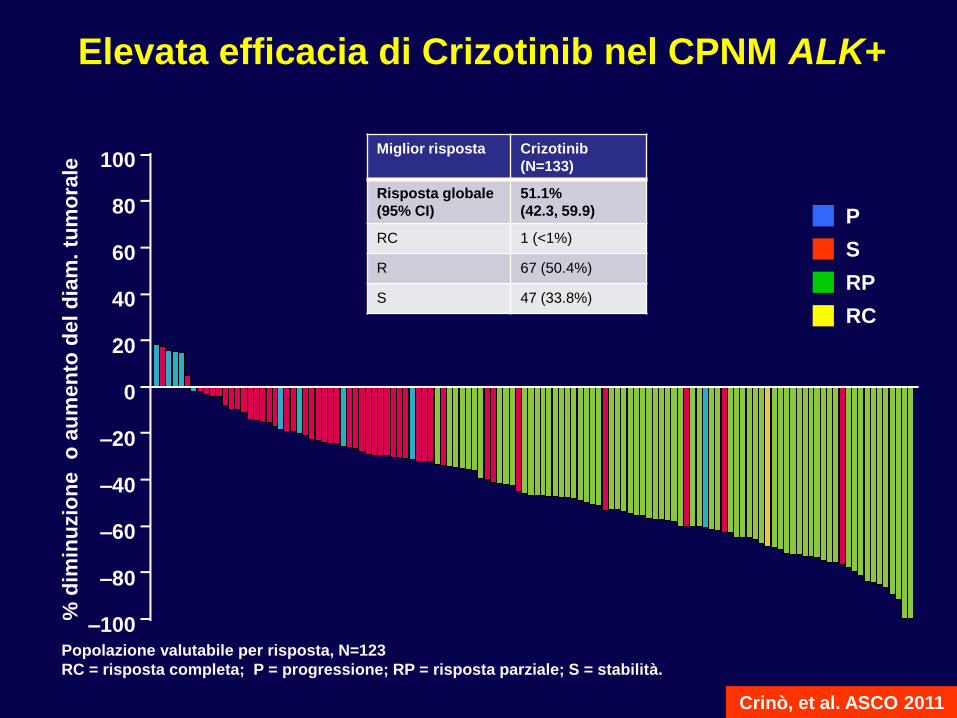
Popolazione valutabile per risposta, N=123
RC = risposta completa; P = progressione; RP = risposta parziale; S = stabilità.
100
80
60
40
20
0
–20
–40
–60
–80
–100
% d
imin
uzio
ne
o a
um
en
to d
el
dia
m.
tum
ora
le
P
S
RP
RC
Crinò, et al. ASCO 2011
Elevata efficacia di Crizotinib nel CPNM ALK+
Miglior risposta Crizotinib
(N=133)
Risposta globale
(95% CI)
51.1%
(42.3, 59.9)
RC 1 (<1%)
R 67 (50.4%)
S 47 (33.8%)

Giorno 7 Giorno 14
Ou, et al. J Thorac Oncol. 2010;5(12):2044–46.
Sintomi all’inizio Miglioramento dei sintomi
TosseMiglioramento significativo dal giorno 3, completamente risolto entro 2
settimane
Febbricola serotina Risoltasi entro il giorno 3
Anoressia Incremento ponderale di 1.5 kg in 2 settimane
Dolore al collo per infiltrazione tumorale Risoltosi entro il giorno 3
Esempio di rapida risposta in paziente
ALK+ in trattamento con Crizotinib

Dicembre 2011: Inizio crizotinib Gennaio 2012 Aprile 2012
Riduzione della lesione principale
E scomparsa dei noduli controlateraliRisposta completa
In Novembre 2011, FISH positiva per la traslocazione di EML4-ALK

Modificata da Gerber and Minna, Cancer Cell 2010
Inibizione di ALK: dalla scoperta alla terapia a
bersaglio in tempi record
Bersaglio
EGFR
Anno di
identificazione
1978
ALK 2007
Anno di correlazione tra
efficacia trattamento ed
aberrazione genetica
2004
2010

Riarrangiamento genico scoperto nel 1987
Produce un recettore di membrana ad attività tirosin-chinasica
Tale recettore è orfano (non identificato ligando) vedi HER2
Presente nel CPNM, glioblastoma multiforme (tumore primitivo
cerebrale) e colangiocarcinoma (tumore delle vie biliari)
49% di identità con ALK nelle sequenze amino-acidiche del
dominio tirosin-chinasico e 77% in quelle del sito di legame per
l’ATP
CPNM riarrangiato per ROS1: un nuovo
sottogruppo genomico di CPNM
reviewed in: Ou, et al. Expert Anticancer Ther 2012

from: Ou, et al. Expert Anticancer Ther 2012
Recettore prodotto dal
riarrangiamento di ROS1

Caratteristiche cliniche del CPNM
riarrangiato per ROS1
Circa l’1% di tutti gli adenocarcinomi
Incidenza percentuale sovrapponibile tra razza asiatica e
razza caucasica
L’età mediana di insorgenza del tumore con
riarrangiamento di ROS1 è circa 50 anni
Circa ¾ dei pazienti con riarrangiamento di ROS1 sono mai
fumatori
reviewed in Ou, et al. Expert Anticancer Ther 2012

ParametroROS1-positivi
N=14
Miglior risposta
Risposta completa
Risposta parziale
Malattia Stabile
Malattia progressiva
1
7
4
2
Risposta tumorale globale 57.1%
Durata mediana del
trattamento (settimane)25.7
% controllo di malattia a 8
settimane79%
Attività del Crizotinib nel CPNM con
riarrangiamento di ROS1
Shaw, et al. ASCO 2012

Basale dopo 4 settimande di Crizotinib
Riarrangiamento di ROS1 e
sensibilità al Crizotinib (1)
Image courtesy of Ignatius Ou

Riarrangiamento di ROS1 e
sensibilità al Crizotinib (2)
Shaw, et al. ASCO 2012

FISH IHC
26.09.2012 26.10.2012 27.12.2012

Conclusioni• Il CPNM, in particolare se adenocarcinoma e legato ad una
bassa esposizione al fumo, è spesso definito dalla
presenza di una specifica alterazione genetica
• La mutazione dell’EGFR e il riarrangiamento genico di ALK
o ROS1 identificano ciascuno una malattia oncogene-
addicted
• L’importanza della malattia oncogene-addicted consiste nel
poter utilizzare terapie molto efficaci dirette contro il
bersaglio in questione
• La biopsia liquida è una valida alternativa al tessuto per
conoscere la presenza di mutazione “sensibile” dell’EGFR
sia prima del trattamento che quella di resistenza T790M in
corso di trattamento con inibitori dell’EGFR gefitinib,
erlotinib, afatinib
Recommended

















