
Univ.-Prof. Dr.-Ing. K.-Ch. Thienel
Frühjahrstrimester 2022
Bauchemie und Werkstoffe des Bauwesens
Chemie und Eigenschaften mineralischer
Baustoffe und Bindemittel

2
Inhaltsverzeichnis
1
1 Gesteine und Verwitterungsprodukte 4
1.1 Urgestein 4
1.2 Verwitterung 5
2 Silikate und Aluminate 7
2.1 Kieselsäure und Silikate 7
2.2 Aluminiumsalze und Aluminate 9
2.3 Technische Silikate 9
2.3.1 Alkalisilikate 9
2.3.2 Gläser 10
2.4 Ton 10
2.4.1 Kristallstruktur der Tone 10
3 Keramische Baustoffe 13
3.1 Ziegel 13
3.2 Blähton und Blähschiefer 14
4 Anorganische Bindemittel 16
4.1 Lehm 16
4.2 Gips und Anhydrit 17
4.2.1 Rohstoffe 17
4.2.2 Herstellung 18
4.2.3 Erhärtung 20
4.3 Magnesiabinder 21
4.4 Kalk 21
4.4.1 Rohstoffe 21
4.4.2 Luftkalk 21
4.4.3 Kalk mit hydraulischen Eigenschaften 23

3
4.4.4 Bezeichnung der Kalke und praktische Anwendung [6] 24
4.5 Latent hydraulische Stoffe und Puzzolane 26
4.6 Zement 28
4.6.1 Geschichtliche Entwicklung [8] 28
4.6.2 Zementherstellung 28
4.6.3 Chemische Zusammensetzung 33
4.6.4 Mineralische Zusammensetzung 35
4.6.5 Eigenschaften der Klinkermineralien 36
4.6.6 Nebenbestandteile 37
4.6.7 Erhärtung 37
4.6.8 Einflüsse auf die Festigkeitsentwicklung 46
4.6.9 Einfluss der Mahlfeinheit 46
4.6.10 Einflüsse von Zumahlstoffen 47
4.6.11 Genormte und bauaufsichtlich zugelassene Zemente 51
5 Literatur 56
Das vorliegende Skript basiert in weiten Teilen auf dem Skriptum „Bindemittel“ zur
Grundvorlesung in Baustoffkunde von Herrn Univ.-Prof. Dr.-Ing. Dr.-Ing. E.h. P. Schießl vom
Lehrstuhl für Baustoffkunde und Werkstoffkunde der Technischen Universität München [1].
Für dessen freundliche Genehmigung möchte ich mich ausdrücklich bedanken.
In der vorliegenden Fassung wurde bei vielen Abbildungen die Originalquelle ergänzt. Diese
Arbeit ist noch nicht vollständig abgeschlossen. Hinweise auf noch fehlende oder ggf.
unzutreffende Quellenangaben werden gerne in der nächsten Version aufgenommen.

4
1 Gesteine und Verwitterungsprodukte
1.1 Urgestein
Die wichtigsten Grundstoffe anorganischer Baustoffe sind Quarz, Kalkstein, Ton und
Gipsstein. Sie entstammen als Verwitterungsprodukte dem Urgestein. Das Urgestein ist ein
Erstarrungsgestein, das an der Erdoberfläche durch das Auskristallisieren sich abkühlender
Schmelzflüsse entstand. Diese Schicht aus Urgestein (Lithosphäre) bildet mit einer Dicke von
etwa 16 km die äußere Schale der Erde (Bild 1).
Bild 1: Aufbau des Erdinneren [2]
Das Urgestein bildet 95 % der Gesteine. Seine Bestandteile sind im Wesentlichen Siliziumoxid
und Aluminiumoxid, sowie Eisenoxid, Magnesiumoxid, Natriumoxid und Kaliumoxid in
unterschiedlichen Anteilen (siehe Bild 2). Der Sauerstoffgehalt des Urgesteins liegt bei etwa
50 %, da seine Bestandteile Oxide sind. Die durch Verwitterung und Lösungsvorgänge aus dem
Urgestein entstandenen Sedimentgesteine bilden die restlichen 5 % der Lithosphäre.
Nach ihrer Entstehungsgeschichte (Genese) unterscheidet man zwischen:
• Magmatiten
z. B.: Granit, Syenit, Diorit, Gabbro, Basalt, Porphyr, Diabas
• Sedimentiten
z. B.: Sandstein, Kalkstein
• Metamorphiten
z. B.: Gneis, Quarzit
Magmatite entstehen beim Erstarren silikatischer Schmelzen (Magmen) in der äußeren
Erdkruste (Tiefengestein) oder an der Erdoberfläche (Ergussgestein). Beim Abkühlen erfolgt
die Kristallisation in bestimmten Ausscheidungsreihenfolgen, die in Phasendiagrammen
nachvollzogen werden können. Je nach Gehalt an SiO2 wird chemisch unterschieden zwischen
basischen (52 % < SiO2) und sauren (65 % > SiO2) Magmatiten.

5
Bild 2: Zusammensetzung der Erdrinde
Sedimentgesteine entstehen durch chemisch physikalische oder biologische Verwitterung (vgl.
Kapitel 1.2). Metamorphite sind das Resultat einer Umkristallisation vorhandener Gesteine
durch teilweises Aufschmelzen in größeren Erdtiefen (Druck- und Temperaturerhöhung) im
Zuge von Gebirgsneubildungen (Metamorphose).
Ein typischer Vertreter der Tiefengesteine ist Granit. Er besteht aus Quarzkristallen, die durch
Feldspat verkittet sind. Feldspate sind alkali- bzw. erdalkalihaltige Aluminiumsilikate. Sie
werden nach ihren Alkalikomponenten in Kalifeldspat (Orthoklas), Natronfeldspat (Albit) und
Kalkfeldspat (Anorit) unterschieden.
1.2 Verwitterung
Das Urgestein besteht aus dem Anhydrid der Kieselsäure (SiO2, Quarz), dessen Verbindungen
mit Aluminiumoxid (Aluminiumsilikat) und dessen Verbindung mit Erdalkali und
Aluminiumoxiden. Diese Bestandteile des Urgesteins unterscheiden sich in ihren
Eigenschaften, insbesondere hinsichtlich ihrer Löslichkeit erheblich. Der Grad der Löslichkeit
der Salze kann anhand der Stellung im Periodensystem der Elemente festgestellt werden. Je
weiter links die Kationen im Periodensystem stehen, desto leichter sind sie löslich. SiO32--
Anionen bilden nahezu unlösliche Verbindungen. Dies gilt auch für die kieselsauren Salze des
Aluminiums (Aluminiumsilikate). Calciumsilikate sind bereits in geringem Umfang löslich und
Alkalisilikate relativ leicht löslich (Wasserglas).
Nachdem das Urgestein durch Temperaturwechsel in seinem Gefüge mechanisch aufgelockert
wurde, konnten die in der Uratmosphäre vorhandenen Säuren (Schwefelsäure, Salzsäure,
Kohlensäure) mit den Alkaliverbindungen wasserlösliche Salze bilden (Steinsalz, Kalisalz), die
dann ausgewaschen wurden und in Meeren oder Binnenwässern nach dem Eintrocknen die
heutigen Salzlagerstätten formten. Die schwerer löslichen Kalk- und Magnesiumverbindungen
wurden durch das kohlensaure Wasser in die entsprechenden Carbonate umgesetzt und als
Kalkstein (CaCO3), Magnesit (MgCO3) und Dolomit (CaCO3 + MgCO3) abgelagert. Die
Schwefelsäure der Uratmosphäre wandelte Kalk- und Magnesiumverbindungen der Feldspate
SiO2
60%
Al2O3
15%Fe2O3
7%
CaO4.00%
MgO3.00%
K2O3%
Na2O
3%
Tongestein4.00%
Sandstein0.75%
Kalkstein0.25%
Other5.00%

6
in Gips (CaSO4) und Magnesiumsulfat (MgSO4) um. Die durch die chemischen Reaktionen
entstandenen unlöslichen bzw. schwer löslichen Gesteine werden als chemische
Sedimentgesteine bezeichnet.
Die Verwitterung löst die Alkali- und Erdalkaliverbindungen aus den Feldspaten und lässt diese
zu einer fein pulvrigen Masse zerfallen, die vorwiegend aus Ton (Aluminiumsilikate) besteht.
Eine Systematik, der durch die verschiedenen Schritte der Verwitterung entstehenden
Baustoffe, zeigt Bild 3.
Bild 3: Gesteinsverwitterung [3]

7
2 Silikate und Aluminate Silizium und Aluminium sind nach Sauerstoff die am häufigsten vorkommenden Elemente der
Erdrinde. Ihre Verbindungen bilden mehr als 90 % der festen Erdkruste. Sie sind die Grundlage
für Zement, Keramik und Glas.
2.1 Kieselsäure und Silikate
Als Kieselsäuren werden die Sauerstoffsäuren des Siliziums (SiO2 · n H2O) bezeichnet.
Orthokieselsäure (Monokieselsäure) H4SiO4 = (Si(OH)4) = 2 (SiO2 · H2O) ist eine sehr
schwache Säure. Sie entsteht durch Zersetzung von Siliziumtetrahalogeniden (SiF4, SiCl4,
SiBr4, SiI4) mit Wasser. Zwischen den Si-Atomen besteht bei der Orthokieselsäure keine
Bindung. An die vier Sauerstoffatome sind Wasserstoffionen angelagert. Die Orthokieselsäure
ist sehr unbeständig und wird unter Wasserabspaltung über die Zwischenstufe
Orthodikieselsäure H6Si2O7:
2 Si(OH)4 → H6Si2O7 + H2O
zur Metakieselsäure (H2SiO3)n überführt. Bei dieser Reaktion werden Si – O – Si-Bindungen
ausgebildet:
Bild 4: Übergang von der Orthokieselsäure zur Metakieselsäure
Wird auch das letzte Wasser entfernt, entsteht das Kieselsäureanhydrid SiO2 (Quarz). Hier ist
die Verknüpfung so vollständig, dass jedes Sauerstoffatom gleichzeitig zwei Tetraedern
angehört. Es resultiert daraus eine räumliche Struktur (Gerüst- oder Tektosilikate), wie sie auch
bei Feldspaten, Zeolithen und weitgehend in Gläsern vorliegt.
Silikate sind Salze der Kieselsäure (vgl. Skript Bauchemie). Sie entstehen durch den Austausch
der Wasserstoffionen gegen Metallionen. Allen Silikatmineralen ist ein gemeinsames
Bauprinzip eigen, deshalb lassen sie sich relativ einfach in eine systematische Ordnung bringen.
Die Grundbausteine aller Silikate sind SiO4-Tetraeder (Bild 5). Ein Siliziumatom ist dabei von
vier Sauerstoffatomen umgeben. Die Sauerstoffatome berühren sich wegen ihrer Größe (rIon =
1,45 Å) [4] und dazwischen bleibt Platz für das deutlich kleinere Siliziumatom (rIon = 0,39 Å)
[4] (der freie Raum heißt Tetraederlücke).
Bild 5: SiO44--Tetraeder

8
Eine weitere Eigenschaft der Silikate besteht in der Fähigkeit der Sauerstoffatome, gleichzeitig
an verschiedenen SiO4-Komplexen teilzuhaben. Daraus ergeben sich neben isolierten SiO4-
Tetraedern weitere zusammengesetzte Bauelemente:
• isolierte Tetraeder,
• Doppeltetraeder,
• Ringstrukturen,
• Einfach- und Doppelketten,
• Schichtstrukturen und
• Gerüststrukturen.
[SiO4]4- [Si2O7]6-
Bild 6: Einzel-, Doppel- oder Gruppensilikate (z. B.: Akermanit (Ca2MgSi2O7) [5]
Silikate mit Ketten- oder Bandstruktur sind in der Regel parallel zu den Ketten bzw. Bändern
leicht zu spalten, da die Verbindungen innerhalb der Ketten bzw. Bänder wesentlich fester ist
als die Ionenbindung (vgl. Skript Bauchemie), die sie zu größeren Gebilden zusammenhalten.
Die resultierende Faserstruktur tritt besonders markant beim Asbest auf.
Die Silikate mit einer Schichtstruktur sind ebenfalls sehr leicht parallel zur Schichtebene zu
spalten. Besonders typisch dafür ist der Glimmer. Die weiche Beschaffenheit von Talk beruht
auf den sehr gut gegeneinander verschieblichen Schichten.
[Si4O10]4-
Bild 7: Schichtstruktur (z. B. im Kaolinit (Al4(OH)8Si4O10) (vgl. auch Bild 10)) [5]

9
2.2 Aluminiumsalze und Aluminate
Aluminium kann aufgrund seines amphoteren Verhaltens sowohl als Anion als auch als Kation
von Salzen auftreten. Die wässrige Lösung von Aluminiumsalzen reagiert durch die eintretende
Hydrolyse sauer.
Die Aluminiumionen sind klein (rIon = 0,50 Å) [4]. Im Vergleich zu den Sauerstoffionen (rIon =
1,45 Å) [4] und können daher sowohl tetraedrisch als auch oktaedrisch von Sauerstoffionen
umgeben sein. Die tetraedrische Anordnung stellt sich ein, wenn SiO4- Tetraeder durch AlO4-
Tetraeder ersetzt sind. Dies ist der Fall bei Alumosilikaten. Beispiele für die Gruppe der
Alumosilikate sind einige wichtige Feldspate wie:
• Albit Na[AlSi3O8],
• Orthoklas K[AlSi3O8] und
• Anorit Ca[Al2Si2O8].
Da das AlO4-Tetraeder verglichen mit dem SiO4-Tetraeder eine zusätzliche negative Ladung
aufweist, muss ein Ladungsausgleich durch Kationen erfolgen.
Aluminiumoktaeder AlO6 liegt bei den Aluminiumsilikaten vor. Sie ist in vielen
Tonmineralien anzutreffen (siehe 2.4).
Neben den reinen Formen gibt es Verbindungen, in denen sowohl AlO4-Tetraeder und AlO6-
Oktaeder nebeneinander auftreten. Typische Vertreter sind:
• Mullit Al3[6][Al3[4]Si2O13] = 3 Al2O3 2 SiO2 und
• Sillimanit Al[6][Al[4]SiO5] = Al2O3 SiO2.
2.3 Technische Silikate
Als technische Silikate können die Alkalisilikate, Gläser und die Tonwaren bezeichnet werden.
2.3.1 Alkalisilikate
Alkalisilikate werden durch das Verschmelzen von reinem Quarzsand (SiO2) mit
Alkalicarbonaten (Soda (Na2CO3) bzw. Pottasche (K2CO3)) bei etwa 1300 °C mit
unterschiedlichen Molverhältnissen hergestellt.
SiO2 + 2 M2CO3 → M4SiO4 + 2 CO2, 2 SiO2 + M2CO3 → M2Si2O5 + CO2,
SiO2 + M2CO3 → M2SiO3 + CO2 und 4 SiO2 + M2CO3 → M2Si4O9 + CO2.
Mit
M: Natrium bzw. Kalium
Es entstehen stückige, glasige Brocken. Wegen ihrer Wasserlöslichkeit werden sie als
„Wassergläser“ bezeichnet. Flüssiges Wasserglas wird durch das Auslösen von stückigem Glas
in heißem Wasser in Druckkesseln (150 °C bei 5 bar) hergestellt.
Kaliwasserglas wird als Bindemittel für Fassadenfarben (Silikatfarben, Mineralfarben) genutzt.
Auf neuem Putz bildet sich mit Calciumhydroxid Calciumsilikat:
K2SiO3 + Ca(OH)2 + CO2 → CaSiO3 + K2CO3 + H2O
Auf altem Putz entsteht bindendes SiO2-Gel:
K2SiO3 + CO2 → SiO2 + K2CO3

10
Wasserglas ist ein wetterfestes Bindemittel für Farbpigmente. Da das aus Natriumwasserglas
entstehende Soda Kristalle bildet, die als Ausblühungen stören, wird für diese Zwecke
Kaliwasserglas genutzt. Natriumwasserglas wird unter anderem als Flammschutzmittel für
Holz eingesetzt. Weitere Einsatzgebiete im Bauwesen sind z. B. Säurekitte.
2.3.2 Gläser
Unter einem Glas im weiteren Sinne versteht man eine amorphe, d.h. ohne Kristallisation
erstarrte (metastabile), beim Erwärmen nur allmählich erweichende unterkühlte Schmelze,
deren Atome eine Nahordnung, aber keine gerichtete Fernordnung besitzen (s. Skript Glas).
Normales Glas ist ein Gemenge aus Natrium- und Calciumsilikat. Rohstoffe sind Quarzsand,
Kalkstein und Soda, die vereinfacht wie folgt reagieren:
CaCO3 + SiO2 → CaSiO3 + CO2,
Na2CO3 + SiO2 → Na2SiO3 + CO2.
Im Normalglas liegt ein unregelmäßiges Netz von SiO4-Tetraedern vor, die durch mehrwertige
Metalle zusammengehalten werden und zwischen denen die einwertigen Alkaliionen
willkürlich eingelagert sind (siehe Skript Bauchemie). Die übliche Zusammensetzung von Glas
ist ca. 73 % SiO2, 15 % Na2O und 12 % CaO. Die Zusammensetzung wird verändert, um
bestimmte Eigenschaften zu erreichen. Für schwer schmelzbare Gläser (Thüringer Glas) wird
Pottasche (K2CO3) anstelle von Soda eingesetzt, für stark lichtbrechende Gläser (Flintglas)
ersetzt Blei ganz oder teilweise das Calcium (Bleiglas).
2.4 Ton
Tone sind Verwitterungsprodukte der Feldspate. Sie bestehen nach dem Herauslösen der
Alkali- und Erdalkalianteile aus dem verbliebenen Aluminiumsilikat. Dieses kommt in
verschiedenen Formen mit unterschiedlichen SiO2-Gehalten vor. Die wichtigsten
Verbindungen sind:
Kaolinit: Al2O3 SiO2 2 H2O und
Montmorillonit: Al2O3 2 SiO2 H2O + n H2O.
Tone können in ihrer Kristallstruktur in unterschiedlichem Maße Wasser aufnehmen. Kaolinit
hat die geringste Wasseraffinität, Montmorillonit kann hingegen bis maximal die 7-fache
Wassermenge binden. Die meisten in der Natur vorkommenden Tone bestehen aus
unterschiedlichen Anteilen von Kaolinit und Montmorillonit, was zu unterschiedlichen
Wasseraufnahmen führt.
2.4.1 Kristallstruktur der Tone
Tonminerale sind zumeist kristallin und bestehen aus dünnen, sechseckigen Blättchen (: 0,2
– 1 µm, d: 0,001 µm). In der Idealvorstellung liegen Schichten aus AlO6-Oktaedern vor, die im
Fall des Montmorillonit beiderseits in Schichten aus SiO4 eingelagert sind. In den
Kaolinitischen Tonen wechseln sich Tetraederschichten (SiO4-Tetraeder) (Bild 8) mit
Oktaederschichten (Al- und Mg-Hydroxid) ab (Bild 9). Die Verknüpfung beider Schichten
erfolgt über den Einbau von Sauerstoffatomen der Tetraederschichten an Hydroxidpositionen
der Oktaederschichten. Die verschiedenen Tone unterscheiden sich in der Schichtenfolge.
Kaolinit ist ein Vertreter der Zweischichtmineralien, in denen je eine Tetraederschicht mit einer
Oktaederschicht verbunden ist (Bild 10 links). Montmorillonit ist ein Vertreter der
Dreischichtmineralien, in denen eine Oktaederschicht mit jeweils zwei Tetraederschichten
verbunden ist (Bild 10 rechts).

11
Bild 8: SiO4-Tetraeder als Schichtsilikatstruktur
Bild 9: AlO6-Oktaeder als Schichtsilikatstruktur
Bild 10: Schematische Darstellung der Schichtstruktur von Kaolinit (links) und
Montmorillonit (rechts)
Da die Silikatschichten beim Montmorillonit nicht fest miteinander verbunden sind, können sie
durch das Einlagern von Wasser ihren Abstand untereinander vergrößern (Quellen) oder bei
Wasserabgabe verringern (Schwinden). Das eingelagerte Zwischenschichtwasser ist an den
festen Oberflächen recht stark gebunden. Die Bindung wird mit zunehmender Dicke des
Wasserfilms geringer. Dies ist die Ursache für die sehr unterschiedlichen mechanischen
Eigenschaften von trockenem und feuchtem Ton.

12
Das Einbauen von Wasser in die Zwischenschichten und das damit verbundene Quellen sorgt
dafür, dass Ton bei Wasserzufuhr die vorhandenen Hohlräume in der Haufwerksstruktur
schließt. Diese abdichtende Eigenschaft ist in der Natur bei den Quellhorizonten zu finden.
Technisch wird die abdichtende Fähigkeit beim Einsatz als Dichtungsstoff
(Deponieabdichtung, Bentonit-Abdichtungen von Schlitzwänden) genutzt.
Beim Schwinden wird ein Teil des verdunstenden Wasservolumens als äußere
Schwindverformung sichtbar. Ein anderer Teil verursacht ein inneres Schwinden und lässt
zwischen den Tonmineralien Hohlräume entstehen.

13
3 Keramische Baustoffe Grundlage für die Herstellung keramischer Baustoffe sind tonhaltige Massen, die neben dem
Hauptbestandteil Ton noch Feldspat- und Quarzanteile enthalten. Durch den Brennvorgang
wird die Struktur des Tons verändert. Dies wird hier am Beispiel des Kaolinits erläutert. Nach
einer ersten Entwässerungsphase werden bei ca. 400 °C die zwischen den Si- und Al-Atomen
sitzenden Hydroxylgruppen abgespalten. Da das Wasser über die Hydroxylgruppen
aufgenommen wurde, führt dieser Vorgang zu einer geringeren Wasseraufnahme. Oberhalb von
etwa 500 °C wird der Ton zersetzt und bildet das so genannte Metakaolinit. Diese behält
andeutungsweise die ursprüngliche Kristallstruktur, setzt sich jedoch aus amorphem SiO2 und
Al2O3 zusammen. Das Metakaolinit wird oberhalb von 800 °C wieder zersetzt. Die entstehende
Oxidmischung ist sehr reaktiv und bildet poröse Strukturen. Bei etwa 1200 °C beginnen die
Kristalle zu verschmelzen und sintern schließlich zusammen. Das entstehende Mullit nimmt
kein Wasser auf. Tabelle 1 gibt am Beispiel von Kaolinit einen Überblick über die chemischen
Reaktionen bei der thermischen Behandlung.
Tabelle 1: Chemische Umsetzung von Kaolinit bei der thermischen Behandlung
Temperatur
[°C]
Vorgang Chemische Umsetzung
20 - 200 Abgabe von freiem Wasser
(Trocknen des Tons)
200 – 450 Abgabe von adsorbiertem
Wasser
450 – 600 Abgabe von Kristallwasser,
Bildung von Metakaolinit Al4(OH)8Si4O10 → 2 (Al2O3 2 SiO2) + 4 H2O
600 – 950 Bildung eines reaktiven
Oxidgemisches Al2O3 2 SiO2 → Al2O3 + 2 SiO2
950 - 1500 Bildung von Mullit 3 Al2O3 + 2 SiO2 → 3 Al2O3 2 SiO2 (3/2-Mullit)
3.1 Ziegel
Ziegeleiprodukte entstehen durch das Brennen tonhaltiger Massen. Da reiner Ton beim
Brennen zu sehr schwindet, wird er entweder durch die Zugabe anderer Mineralien wie z. B.
Quarzsand gemagert oder gleich Lehm eingesetzt. Der Tongehalt der Rohmassen liegt je nach
Anforderung zwischen 20 und 60 %. Ziegeltone können in feiner Verteilung (< 1 mm) bis zu
25 % Kalkstein (CaCO3) enthalten. Größere Kalkteile führen bei späterem Ablöschen des
entstandenen Brandkalks zu Schäden. Die mitunter braune Farbe von Ton rührt her aus den
unterschiedlichen Gehalten an Eisenhydroxid (Fe(OH)3). Beim Brennen wandelt es sich in rotes
Eisenoxid (Fe3O3) und sorgt für die charakteristische rote Farbe vieler Ziegeleierzeugnisse.
Daneben wirken Eisenverbindungen als Flussmittel und erniedrigen den Schmelzpunkt des
Tons.
Über die Zusammensetzung und die Temperaturkurve (Temperatur über Zeit) können
Eigenschaften der fertigen Ziegeleiprodukte gezielt eingestellt werden (Bild 11 und Bild 12).
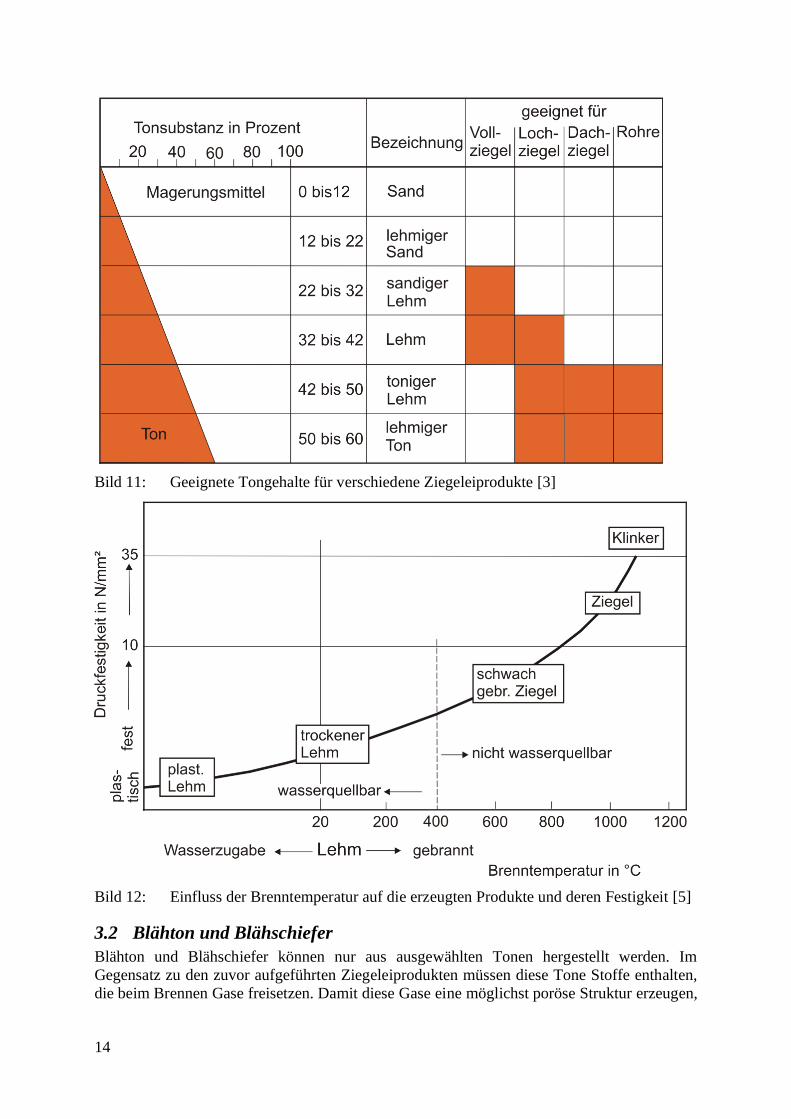
14
Bild 11: Geeignete Tongehalte für verschiedene Ziegeleiprodukte [3]
Bild 12: Einfluss der Brenntemperatur auf die erzeugten Produkte und deren Festigkeit [5]
3.2 Blähton und Blähschiefer
Blähton und Blähschiefer können nur aus ausgewählten Tonen hergestellt werden. Im
Gegensatz zu den zuvor aufgeführten Ziegeleiprodukten müssen diese Tone Stoffe enthalten,
die beim Brennen Gase freisetzen. Damit diese Gase eine möglichst poröse Struktur erzeugen,

15
muss sich beim Brennen eine äußere Sinterschicht bilden. Die Gase können aus
unterschiedlichen anorganischen Quellen stammen:
• Fe2O3 → FeO + O2,
• CaCO3 → CaO + CO2,
• MgCO3 → MgO + CO2,
• CaSO4 → CaO + SO3 und
• 4 FeS2 + 7 O2→ 2 Fe2O3 + 4 SO2.
Ebenso ist das thermische Zersetzen organischer Bestandteile (z. B.: Zugabe von Kohlenstaub,
Sulfitablauge oder Schweröl) eine mögliche Gasquelle.
Zum Ausbilden der äußeren Sinterschicht muss der Ton in ausreichendem Maße basische Stoffe
(Na2O, K2O, CaO, MgO) und Eisenoxid enthalten. Sie bilden mit der Kieselsäure (H2SiO3) des
Tons niedrig schmelzende Silikate. Der Quarzgehalt des Tons sollte möglichst niedrig sein, da
er nicht an der Expansion beteiligt ist.
Bild 13: Blähton (links) und Blähschiefer (rechts)

16
4 Anorganische Bindemittel Den anorganischen Bindemitteln gemeinsam ist ihre (zumeist) natürliche Rohstoffquelle,
Abbau, Transport, Aufbereitung und das Brennen (Ausnahme: Anhydrit). In den meisten Fällen
ist eine Feinmahlung erforderlich. Die einzelnen Verfahrensstufen und die eingesetzten
technischen Einrichtungen sind teilweise ähnlich, lediglich Gips weicht erheblich von den
anderen Baustoffen ab. Bei der Herstellung durchlaufen die anorganischer Bindemittel
endotherme chemische Prozesse. Umgekehrt laufen später auf der Baustelle zum Teil
ausgeprägt exotherme chemische Reaktionen ab.
Die anorganischen Bindemittel werden in zwei Hauptgruppen unterteilt:
Luftbindemittel, die an Luft erhärten und im erhärteten Zustand bedingt wasserlöslich sind
(Lehm, Gips, Luftkalk und Magnesiabinder), und
Hydraulische Bindemittel, die an Luft erhärten und unter Wasser erhärten (hydraulischer
Kalk, Zement).
4.1 Lehm
Lehm ist vielleicht das älteste Bindemittel, allerdings erlebt der Lehm bei der Suche ökologisch
orientierter Bauherren nach einem natürlichen Bindemittel derzeit wieder an Bedeutung. Lehm
besteht aus Ton mit Beimengungen von Sand, Eisenverbindungen und anderem.
Lehm besteht aus sehr feinkörnigem Material ( 2 µm), die ihn im lufttrockenen Zustand
recht hart werden lassen. Die Tonbestandteile des Lehms liegen dann als Aluminiumsilikat-
hydrat vor. Zusätzliches Wasser wird vom Ton als Hydratwasser gebunden (vgl. Kapitel 2.4)
und macht ihn bildsam:
Al2O3 SiO2 2 H2O + n H2O ⥬ Al2O3 SiO2 (H2O)n+2
fester Ton bildsamer Ton
Der Vorgang ist reversibel. Lehmputze und -mörtel, die fest bleiben sollen, sind daher
unbedingt vor Feuchtigkeit zu schützen.
Bild 14: Bauwerke aus Lehmziegeln (Sanaa, Jemen) (Quelle: I. Zumurud, 2005)

17
4.2 Gips und Anhydrit
4.2.1 Rohstoffe
4.2.1.1 Natürliche Gipssteinvorkommen
Gips ist Calciumsulfat, das in verschiedenen Hydratstufen in Bindung mit oder auch ohne
Kristallwasser vorliegen kann. Das in der Natur vorkommende Gipsgestein ist Calciumsulfat-
Dihydrat (CaSO4 2 H2O); die in der Natur anstehende kristallwasserfreie Form des
Calciumsulfats wird als Anhydrit (CaSO4) bezeichnet. Natürliche Gipssteinvorkommen
(CaSO4 2 H2O) finden sich oft in den Sedimenten ausgetrockneter Meere in der Nähe von
Salzlagerstätten. Ursache ist ein Gipsgehalt des Meerwassersalzes von etwa 4,6 %. Beim
Austrocknen werden zunächst die schwer löslichen Carbonate, danach die besser löslichen
Sulfate und schließlich die Chloride ausgeschieden. Wo geologische Veränderungen für diese
Bedingungen gesorgt haben, entstand kristallwasserfreier Gips, so genannter Anhydrit. [6]
Gips und Anhydrit sind in den geologischen Formationen des Perm – wozu der Zechstein gehört
– ferner des Trias – im Muschelkalk und im Keuper – sowie des Tertiärs anzutreffen. Die
ältesten Vorkommen sind die des Zechsteins mit einem Alter von rund 255 Millionen Jahren.
Die Gipse des Muschelkalkes sind durchschnittlich 239 Millionen Jahre alt, während die
Keupergipse etwa weitere 7 Millionen Jahre jünger sind. In Deutschland finden sich
Gipsgesteine des Zechsteins vor allem im Norden, während Muschelkalk- und Keupergipse
weitgehend auf Süddeutschland beschränkt sind. [6]
Der auskristallisierte Gips enthält zunächst 21 % Kristallwasser. Dieses Kristallwasser kann bei
hohem Druck und höheren Temperaturen entweichen.
4.2.1.2 Gips aus technischen Prozessen
In zahlreichen technischen Prozessen entsteht Calciumsulfat als Nebenprodukt. Es bildet sich
meist durch Umsetzung von Calciumverbindungen – im allgemeinen Calciumcarbonat oder
Calciumhydroxid – mit Schwefelsäure oder – wie bei der Rauchgasentschwefelung – mit dem
Schwefeldioxid der Rauchgase. [6]
Rauchgas-Entschwefelungs-Anlagen-Gips (REA-Gips)
REA-Gips entsteht bei der Entschwefelung der Rauchgase von Kraftwerken, die mit fossilen
Brennstoffen befeuert werden (vgl. Skript Bauchemie). Er wird bei der nassen
Rauchgasentschwefelung mit Kalk(stein)waschverfahren nach Oxidation mit Luft, Abtrennung
der Gipskristalle, Waschen und Filtrieren gezielt gewonnen. Gips aus
Rauchgasentschwefelungsanlagen (REA-Gips) ist das feuchte, feinteilige kristalline
Calciumsulfat-Dihydrat – CaSO4 2 H2O – mit hoher Reinheit. Dieser REA-Gips ist ein direkt
verwertbarer Rohstoff. [6]
In Folge der politischen Entscheidung, die Kohleverstromung in Deutschland zu beenden, wird
diese Quelle in den nächsten Jahren versiegen [7, 8]. Dies wird sich massiv auf den vermehrten
Abbau natürlicher Gipsvorkommen nach sich ziehen.
Phosphogips
Phosphogips entsteht bei der Phosphorsäure-Herstellung im Nassverfahren durch Reaktion der
Phosphaterze (Calciumphosphat) mit Schwefelsäure. Er ist aus technischen und
wirtschaftlichen Gründen als Rohstoff für die Gipsindustrie ohne Bedeutung:
Ca5(PO4)3F + 5 H2SO4 + 10 H2O ⥬ 5 CaSO4 2 H2O + 3 H3PO4 + HF.

18
Fluoroanhydrit
Fluoroanhydrit entsteht bei der Flusssäure-Herstellung durch Reaktion von Flussspat mit
konzentrierter Schwefelsäure. Fluoroanhydrit wird auch synthetischer Anhydrit genannt [6]:
CaF2 + H2SO4 ⥬ CaSO4 + 2 HF.
Sonstige technisch entstandene Gipse
Bei einer Reihe weiterer chemischer Prozesse, wie z. B. bei der Herstellung von Caprolactam
(Vorprodukt für hoch polymeres Gusspolyamid), Weinsäure, Zitronensäure und Oxalsäure oder
bei der Aufbereitung von Dünnsäure aus der Titandioxid-Herstellung, fallen gewisse Mengen
von Gips an.
4.2.2 Herstellung
Gips und Anhydrit für den technischen Einsatz werden durch Brennen aus den oben genannten
Rohstoffen hergestellt. Das Ausgangsmaterial durchläuft dabei verschiedene
temperaturabhängige Veränderungen, die sich auf die Eigenschaften der hergestellten Produkte
auswirken. In Tabelle 2 sind die Phasen des Systems CaSO4 – H2O und deren Eigenschaften
aufgeführt. Außer dem Calciumsulfat-Dihydrat, dem Calciumsulfat-Halbhydrat, dem Anhydrit
III und dem Anhydrit II gibt es als fünfte Phase noch den Anhydrit I, der jedoch nur bei
Temperaturen über etwa 1180 °C existiert.
Tabelle 2: Phasen im System CaSO4-H2O und ihre Eigenschaften [6]
Chemische Formel der
Phase CaSO4 2 H2O CaSO4 ½ H2O CaSO4 III CaSO4 II
Bezeichnung Calciumsulfat-
Dihydrat
Calciumsulfat-
Halbhydrat
Anhydrit III Anhydrit II
Weitere Bezeichnung Naturgips,
Rohgips,
Gipsstein
-Halbhydrat,
-Gips,
Stuckgips,
-Halbhydrat,
-Gips,
Autoklavgips
löslicher
Anhydrit
Naturanhydrit,
Rohanhydrit,
Anhydritstein,
synthetischer
Anhydrit, erbrannter
Anhydrit
Formen -Form
-Form
-A III
-A III
A II-s (schwer
löslich)
A II-u (unlöslich)
A II -E (Estrichgips)
Kristallwasser [M.-%] 20,92 6,21 0 0
Löslichkeit in Wasser
bei 20 °C [g (CaSO4)/l]
2,06 8,8 6,7
8,8 6,7
2,7
Stabilität < 40 °C metastabil metastabil 40 – 1180 °C
Bildungstemperatur
im Labor
: 45 – 200 °C in
trockener Luft
: > 45 °C in
Wasser
50 °C im
Vakuum,
100 °C an
Luft
200 – 1180 °C
Bildungstemperatur
im technischen Prozess
: 120 – 180 °C
trocken
: 290 °C
trocken
300 – 900 °C
A II-s: 300 – 500 °C

19
: 80 – 180 °C
nass
: 110 °C
nass
A II-u: 500 – 700 °C
AI I-E: > 700 °C
Vom Calciumsulfat-Halbhydrat sind zwei verschiedene Formen bekannt, die man als -
Halbhydrat und als -Halbhydrat bezeichnet. Sie entstehen bei unterschiedlichen
Brennbedingungen und unterscheiden sich in ihren physikalischen Eigenschaften.
• -Halbhydrat entsteht im geschlossenen Autoklaven. Das ausgetriebene Wasser wird
dabei teilweise zurückgehalten und kann die zunächst porige Kristallstruktur in eine
kristalline Struktur umwandeln. Unter dem Mikroskop sind bei -Halbhydrat gut
ausgebildete Kristalle zu erkennen. Das entstandene fein kristalline Gefüge ist dicht und
glänzt seidig (Bild 15 links).
• -Halbhydrat wird durch rasches Erhitzen in offenen Anlagen erzeugt. Das Wasser
entweicht schlagartig und lässt ein poröses äußeres Kristallgerüst entstehen. Unter dem
Mikroskop zeigt -Halbhydrat z. B. zerklüftete Teilchen der ehemaligen Dihydrat-
Körner oder –Kristalle. Die poröse Struktur verleiht dem -Halbhydrat ein stumpfes,
kreidiges Aussehen (Bild 15 rechts).
Die unterschiedlichen Strukturen von - und -Halbhydrat kommt in den unterschiedlichen
Materialeigenschaften zum Ausdruck. -Halbhydrat braucht wegen seines dichteren Gefüges
(größere Dichte und höheres Schüttgewicht) weniger Wasser und liefert dadurch etwa die
dreifache Festigkeit wie -Halbhydrat, seine baupraktische Bedeutung ist allerdings gering.
Bild 15: -Halbhydrat (links) und -Halbhydrat (rechts)
Anhydrit III, auch löslicher Anhydrit genannt, existiert ebenfalls in zwei Formen, die als -
und als -Anhydrit III bezeichnet werden. Anhydrit II entspricht in seiner chemischen
Zusammensetzung dem natürlich vorkommenden Anhydrit; er entsteht bei der vollständigen
Entwässerung von natürlichem oder technisch entstandenem Dihydrat, Halbhydrat oder
Anhydrit III.
Die Entwässerung (Calcinierung oder Calcination) des Gipses erfolgt im technischen Betrieb
bei höheren Temperaturen als im Laboratorium, damit die Verweilzeit in den Brennaggregaten
möglichst kurz sein kann. Dabei muss man jedoch in Kauf nehmen, dass technisch erzeugte
Gipse aufgrund der kurzen Zeit zum Einstellen der chemischen Gleichgewichte keine reinen
Phasen, sondern Mischungen verschiedener Phasen sind. Beim erbrannten Anhydrit II – dem
so genannten Hochbrandgips – unterscheidet man drei Varianten. Die Unterschiede dieser drei
Varianten liegen in der verschiedenen Reaktionsfreudigkeit mit Wasser. Der Anhydrit II-u
reagiert verhältnismäßig träge, während der Anhydrit II-s und der Anhydrit II-E etwas schneller
hydratisieren. Für die Entstehung von Anhydrit II-s (schwer löslicher Anhydrit) gilt als

20
Faustregel ein Entstehungsbereich unter 500 °C, während der Anhydrit II-u (unlöslicher
Anhydrit) zwischen 500 °C und 700 °C entsteht. Der Anhydrit II-E (Estrichgips) bildet sich
oberhalb 700 °C. [6]
4.2.3 Erhärtung
Die Erhärtungsreaktion ist grundsätzlich lediglich eine Umkehrung des Herstellprozesses. Bei
Wasserzugabe reagieren die Halbhydrate und die Anhydrite II und III zu Calciumsulfatdihy-
drat:
CaSO4 1/2 H2O + 3/2 H2O ⥬ CaSO4 2 H2O
CaSO4 + 2 H2O ⥬ CaSO4 2 H2O
Im Zuge dieser Hydratation entstehen zunächst die nadelförmigen Kristalle des CaSO4 2 H2O,
die sich gegenseitig verfilzen und verwachsen. Danach bildet CaSO4 1/2 H2O zunächst eine
gesättigte Lösung von 8 g/l, die aber in Bezug auf CaSO4 2 H2O übersättigt ist (Löslichkeit
2,06 g/l, vgl. Tabelle 2). Aus der übersättigten Lösung scheiden sich CaSO4 2 H2O-Kristalle
aus. Bild 16 zeigt den geschlossenen Gipskreislauf.
Bild 16: Gipskreislauf [1]
Das gemeinsame Volumen der Mischungsbestandteile Halbhydrat und Wasser ist größer als
das Volumen des entstehenden Gipses. Die dadurch verursachte anfängliche
Volumenkontraktion von 7 – 9 % im noch nicht festen Zustand wird durch eine bleibende
Expansion von etwa 0,7 % überlagert, die die nach allen Seiten wachsenden Kristallnadeln
verursachen.
Die Kristallisationsvorgänge können durch Kristallisationskeime (z. B. Spachtel mit
Gipsresten) oder geeignete Zusätze beschleunigt werden. Beschleuniger sind z. B. Glaubersalz
(Na2SO4), Schwefelsäure und Sulfate.
Eine Verzögerung der Kristallisation kann durch Leim, Dextrin, Zucker, Alaun, Wasserglas
und Kalkmilch erreicht werden.
Naturvorkommen
Erhärtung
= Hydratation
→ Brennen
Im Drehrohrofen
130 - 180°C
(Austreiben von
Kristallwasser)
Mahlen +
Anmachen mit
Wasser
Gipsstein
CaSO4 · 2H2O
Gipsteig
2CaSO4 · 1/2H2O + 1,5H2O
„Halbhydrat“
CaSO4 · 1/2H2O

21
4.3 Magnesiabinder
Magnesiabinder ist ein nichthydraulisches Bindemittel und wird nach seinem Erfinder auch in
irreführender Weise „Sorelzement“ genannt. Magnesiabinder wird durch das Mischen von
kaustisch (ätzend) gebranntem Magnesit (Magnesia, MgO) mit in Wasser gelöstem
Magnesiumchlorid (MgCl2) hergestellt. Das Magnesiumchlorid ist dabei nicht an der Reaktion
beteiligt, sondern dient lediglich als Katalysator:
MgO + H2O + MgCl2 ⥬ Mg(OH)2 + MgCl2.
Die Erhärtung erfolgt durch die sich gegenseitig durchdringenden und verfilzenden feinen
Kristallnadeln des entstehenden Magnesiumhydroxids. Die Erhärtung ist in wenigen Stunden
abgeschlossen.
Der Vorteil des Magnesiabinders liegt in seiner Fähigkeit, große Mengen Füller, insbesondere
Holzmehl, Sägemehl und –späne zu so genanntem Steinholz zu binden, das z. B. für Estriche
eingesetzt werden kann. Nachteilig ist, dass das MgCl2 nicht chemisch eingebunden ist und das
Chlorid beim Eindringen in Betonkonstruktionen zur Korrosion des Bewehrungsstahls und
anderer Baumetalle (Blei, Kupfer, Zink und Aluminium) führen kann. Ein weiterer Nachteil ist
die fehlende Wasserbeständigkeit. Magnesiabinder haben heute keine baupraktische Bedeutung
mehr.
4.4 Kalk
Der Einsatz von Kalk als Bindemittel reicht bis in die Jungsteinzeit zurück. Bekannte
Bauwerke, für die Kalkmörtel verwendet wurde, sind u. a. die Pyramiden von Gizeh, der Palast
von Pergamon und die minoischen Paläste.
4.4.1 Rohstoffe
Als Rohstoffe für die Herstellung aller Baukalke kommen Kalkstein (CaCO3), mit Ton
verunreinigter Kalkstein als Kalkmergel und Dolomit (CaMg(CO3)2) zur Anwendung. Sie sind
als Sedimentgesteine vor allem durch Ausfällen von CaCO3 zu feinem Kalkschlamm
(chemische Sedimente) oder aus den kalkhaltigen Schalen und Skeletten abgestorbener
Meerestiere (Muschelkalk) entstanden.
4.4.2 Luftkalk
4.4.2.1 Herstellung
Luftkalk (Weißkalk) wird aus möglichst reinem Kalkstein mit einem CaCO3–Gehalt von
mindestens 95 % (besser 97 %) genutzt. Beim Kalkbrennen spaltet sich bei 900 °C aus
Kalkstein (CaCO3) Kohlenstoffdioxid ab und es entsteht Calciumoxid:
CaCO3 ⥬ CaO + CO2
Der gebrannte Kalk wird mit Wasser gelöscht.
CaO + H2O ⥬ Ca(OH)2 + 1150 J/g
Es wird zwischen Trocken- und Nasslöschen unterschieden. Beim Nasslöschen wird ein
Kalkbrei hergestellt. Beim heute industriell üblichen Trockenlöschen wird nur so viel Wasser
zugegeben, dass als Endprodukt ein trockenes Pulver mit geringer Restfeuchte entsteht. Dieses
Kalkhydrat kommt als Weißkalkhydrat in den Handel. In beiden Fällen muss der Löschvorgang
vor der weiteren Verarbeitung abgeschlossen sein. Anderenfalls kann es beim Erhärten zu
Treiberscheinungen kommen, da Calciumhydroxid unter einer Volumenvergrößerung von ca.
70 % entsteht.
22
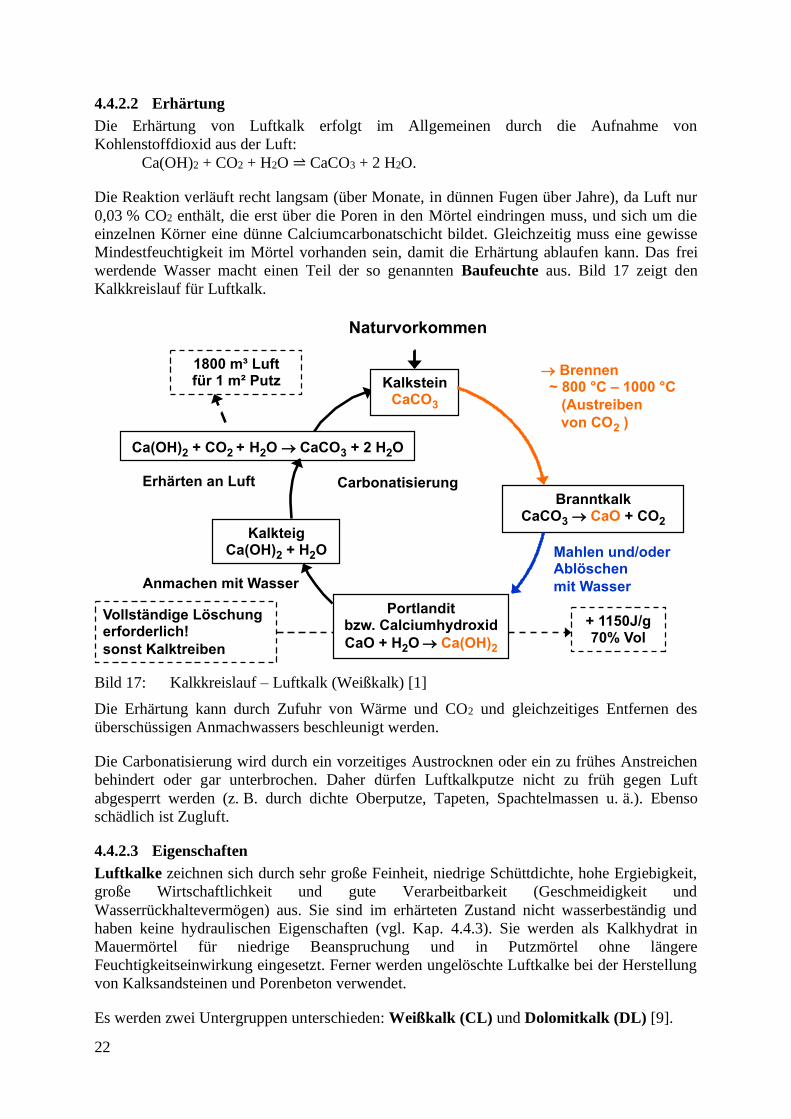
4.4.2.2 Erhärtung
Die Erhärtung von Luftkalk erfolgt im Allgemeinen durch die Aufnahme von
Kohlenstoffdioxid aus der Luft:
Ca(OH)2 + CO2 + H2O ⥬ CaCO3 + 2 H2O.
Die Reaktion verläuft recht langsam (über Monate, in dünnen Fugen über Jahre), da Luft nur
0,03 % CO2 enthält, die erst über die Poren in den Mörtel eindringen muss, und sich um die
einzelnen Körner eine dünne Calciumcarbonatschicht bildet. Gleichzeitig muss eine gewisse
Mindestfeuchtigkeit im Mörtel vorhanden sein, damit die Erhärtung ablaufen kann. Das frei
werdende Wasser macht einen Teil der so genannten Baufeuchte aus. Bild 17 zeigt den
Kalkkreislauf für Luftkalk.
Bild 17: Kalkkreislauf – Luftkalk (Weißkalk) [1]
Die Erhärtung kann durch Zufuhr von Wärme und CO2 und gleichzeitiges Entfernen des
überschüssigen Anmachwassers beschleunigt werden.
Die Carbonatisierung wird durch ein vorzeitiges Austrocknen oder ein zu frühes Anstreichen
behindert oder gar unterbrochen. Daher dürfen Luftkalkputze nicht zu früh gegen Luft
abgesperrt werden (z. B. durch dichte Oberputze, Tapeten, Spachtelmassen u. ä.). Ebenso
schädlich ist Zugluft.
4.4.2.3 Eigenschaften
Luftkalke zeichnen sich durch sehr große Feinheit, niedrige Schüttdichte, hohe Ergiebigkeit,
große Wirtschaftlichkeit und gute Verarbeitbarkeit (Geschmeidigkeit und
Wasserrückhaltevermögen) aus. Sie sind im erhärteten Zustand nicht wasserbeständig und
haben keine hydraulischen Eigenschaften (vgl. Kap. 4.4.3). Sie werden als Kalkhydrat in
Mauermörtel für niedrige Beanspruchung und in Putzmörtel ohne längere
Feuchtigkeitseinwirkung eingesetzt. Ferner werden ungelöschte Luftkalke bei der Herstellung
von Kalksandsteinen und Porenbeton verwendet.
Es werden zwei Untergruppen unterschieden: Weißkalk (CL) und Dolomitkalk (DL) [9].
Kalkteig Ca(OH)2 + H2O
Branntkalk CaCO3 ® CaO + CO2
Kalkstein CaCO3
Ca(OH)2 + CO2 + H2O ® CaCO3 + 2 H2O
Vollständige Löschung erforderlich!
sonst Kalktreiben
Portlandit bzw. Calciumhydroxid
CaO + H2O ® Ca(OH)2
Carbonatisierung Erhärten an Luft
Anmachen mit Wasser
® Brennen ~ 800 °C – 1000 °C
(Austreiben
von CO2 )
+ 1150J/g 70% Vol
Mahlen und/oder Ablöschen
mit Wasser
1800 m³ Luft für 1 m² Putz
Naturvorkommen

23
4.4.3 Kalk mit hydraulischen Eigenschaften
4.4.3.1 Natürlicher Hydraulischer Kalk
Natürlicher Hydraulischer Kalk (NHL) wird aus einem Gemisch aus Kalk mit mehr oder
weniger tonhaltigen oder kieselsäurehaltigen Kalksteinen hergestellt, das bei Temperaturen von
1000 °C bis 1200 °C, also noch unterhalb der Sintergrenze von 1400 °C gebrannt wird. Das
Kalk-Ton-Gemisch muss nicht gesondert produziert werden, sondern kommt in der Natur in
großer Menge in Form von Mergel vor und enthält keine weiteren Zusätze. Beim Brennen
finden Oberflächenreaktionen (Festkörperreaktionen) zwischen den Reaktionspartnern
(CaCO3 bzw. CaO, SiO2 und Al2O3) statt, weshalb die Rohmischung fein gemahlen sein muss:
CaCO3 ⥬ CaO + CO2,
2 CaO + SiO2 ⥬ 2 CaO SiO2 (Dicalciumsilikat) und
3 CaO + 2 Al2O3 ⥬ 3 CaO 2 Al2O3 (Tricalciumaluminat).
Calciumsilikat und –aluminat sind kalkübersättigte Mineralien, die bei Zugabe von Wasser in
andere Verbindungen unter Einbau von Kristallwasser (Hydrate) übergehen. Diese Hydrate
erhärten ohne Luft mit und in Wasser und werden deshalb „hydraulische Komponenten“
genannt. Der bei den Festkörperreaktionen nicht gebundene CaO-Anteil reagiert wie Luftkalk
durch die Aufnahme von Kohlendioxid.
Die Eigenschaften Natürlicher Hydraulischer Kalke werden durch den Anteil an
hydraulischen Komponenten (Tonanteil) bestimmt. Natürlicher Hydraulischer Kalk wird mit
einem Gehalt an Ton oder reaktionsfähigen kieselsauren Bestandteilen von etwa 10 % bis 30 %
hergestellt. Beim Brennen stellen sich die in Tabelle 3 aufgeführten Verhältnisse zwischen den
Tonanteilen und dem Calciumoxid ein. Das Reaktionsverhältnis zwischen Kalk und Ton liegt
bei den ungesinterten hydraulischen Bindemitteln bei Kalk : Ton = 60 : 40. Die daraus
resultierenden hydraulischen Anteile sind ebenfalls in Tabelle 3 enthalten.
Bei einem Tongehalt von etwa 10 % besitzt NHL ein begrenztes hydraulisches
Erhärtungsvermögen. Es überwiegen jedoch die charakteristischen Kalkeigenschaften. Dieser
NHL löscht aufgrund seines hohen CaO-Gehalts mit Wasser. Die Reaktion ist weniger intensiv
als bei Luftkalk. Bei einem Tongehalt von etwa 20 % löscht NHL nur sehr träge. Bis zu einem
Gehalt an Ton oder reaktionsfähigen kieselsauren Bestandteilen von etwa 20 % dient NHL als
Mauer- und Putzmörtel, bei denen höhere, aber begrenzte Festigkeit sowie eine gewisse
Widerstandsfähigkeit gegen Feuchtigkeitseinwirkung gefordert werden.
Durch einen höheren Gehalt an reaktionsfähigen kieselsauren Bestandteilen von etwa 30 %
(vgl. Tabelle 3) erhält der Natürliche Hydraulische Kalk neben der Carbonaterhärtung ein
ausgeprägtes hydraulisches Erhärtungsvermögen. Er ist nach Luftlagerung von bis zu 3 Tagen
auch unter Wasser beständig. Es findet keine Löschreaktion mehr statt, weil der Kalkanteil an
die Hydraulefaktoren gebunden ist. Die Verarbeitungseigenschaften liegen zwischen denen der
Luftkalke und der Zemente. Trasskalk hat durch die Feinheit des verwendeten Trasses (Trass
siehe Tabelle 6) und den LP-Zusatz (LP = Luftporenbildner) ein hohes
Wasserrückhaltevermögen, ein großes Verformungsvermögen und damit eine geringe
Rissempfindlichkeit sowie eine erhöhte Widerstandsfähigkeit gegen das Auslaugen von CaO
und gegen Frost-Tauwechsel. Diese Natürlichen Hydraulischen Kalk werden als Mauer- und
Putzmörtel eingesetzt, bei denen die Festigkeit der Luftkalke und der anderen hydraulisch
erhärtenden Kalke nicht ausreicht oder eine schnellere Erhärtung gefordert wird. Sie sind in
gleicher Weise wie Kalkzementmörtel zu verarbeiten. Sie dienen als Mauermörtel für den
Schornsteinbau und als Bindemittel für Außenputze bei ungünstigen Witterungsverhältnissen.
Trasskalk wird u. a. für Mauer- und Fugenmörtel bei schlagregenbeanspruchtem Mauerwerk,

24
in der Denkmalpflege, für Bodenverfestigungen und im Fahrbahnunterbau verwendet.
Tabelle 3: Anhaltswerte für die Zusammensetzung Natürlicher Hydraulischer Kalke
Zusammensetzung in Massenanteilen
Tonbestandteil 13 20 28
Calciumoxid 87 80 72
Hydraulischer
Anteil (≈ 2,5 Ton) 33 50 70
Freies Calciumoxid 67 50 30
4.4.3.2 Formulierter Kalk (FL)
Formulierter Kalk besteht hauptsächlich aus Luftkalk (CL) und/oder Natürlichem
Hydraulischem Kalk (NHL) mit Zusätzen aus anderem hydraulischem und/oder puzzolanem
Material. Er erstarrt und erhärtet nach dem Mischen mit Wasser (hydraulisch). Die Erhärtung
erfolgt zusätzlich durch die Reaktion mit atmosphärischen Kohlenstoffdioxid
(Carbonatisierung). Als Zusätze kommen Zement (vgl. Kap. 4.6), Puzzolane (vgl. Kap. 4.5),
Kalksteinmehl und Hüttensand (vgl. Kap. 4.5) in Frage.
4.4.3.3 Hydraulischer Kalk (HL) Chemische Anforderungen an Weißkalk und
Dolomitkalk
Hydraulischer Kalk ist ein Bindemittel, das aus Kalk und anderen Materialien wie Zement (vgl.
Kap. 4.6), Hüttensand, Flugasche (vgl. Kap. 4.5), Kalksteinmehl und anderen Materialien
besteht. Er erstarrt und erhärtet unter Wasser (hydraulisch). Atmosphärisches
Kohlenstoffdioxid trägt zum Erhärtungsprozess bei.
4.4.4 Bezeichnung der Kalke und praktische Anwendung [1]
Die Bezeichnung der Luftkalke richtet sich nach ihrem (CaO + MgO)-Gehalten (Tabelle 4).
Tabelle 4: Anforderungen an die chemische Zusammensetzung von Weißkalk und
Dolomitkalk [9]
Art Bezeichnu
ng
Chemische Zusammensetzung als Massenanteil in Prozent
CaO +
MgO
MgO CO2 SO3 Verfügbare
r Kalk
Wei
ßk
alk
CL 90 ≥ 90 ≤ 5 ≤ 4 ≤ 2 ≥ 80
CL 80 ≥ 80 ≤ 5 ≤ 7 ≤ 2 ≥ 65
CL 70 ≥ 70 ≤ 5 12 ≤ 2 ≥ 55
Dolo
mit
kalk
DL 90-30 ≥ 90 ≥ 30 ≤ 6 ≤ 2 k. A.
DL 90-5 ≥ 90 > 5 ≤ 6 ≤ 2 k. A.
DL 85-30 ≥ 85 ≥ 30 ≤ 9 ≤ 2 k. A.
DL 85-5 ≥ 85 > 5 ≤ 9 ≤ 2 k. A.
Die Bezeichnung der Kalke mit hydraulischen Eigenschaften richtet sich nach ihrer
Mindestdruckfestigkeit nach 28 Tagen und ihrem Ca(OH)2 - Gehalt (Tabelle 5).

25
Um die negativen Folgen einer spät und langsam ablaufenden Löschreaktion zu vermeiden,
werden hydraulische Kalke in der Regel bei der Herstellung im Werk trocken gelöscht. Kalke
mit hydraulischen Eigenschaften stellen den Übergang zwischen Luft erhärtenden und Wasser
härtenden Bindemitteln dar.
Kalke sind mit anderen Bindemitteln und untereinander mischbar, jedoch nicht mit
Tonerdezement oder Sulfathüttenzement. Hydraulisch erhärtende Kalke dürfen nicht mit Gips
vermischt werden, da es hier zum Gipstreiben kommt.
Tabelle 5: Arten und charakteristische Festigkeitswerte der Kalke mit hydraulischen
Eigenschaften [9]
Art Bezeichnung Verfügbarer Kalk
als Ca(OH)2 [M.-%]
Druckfestigkeit [MPa]
7 Tage 28 Tage
Natü
rlic
her
Hyd
rau
lisc
her
Kalk
NHL 2 ≥ 35 - ≥ 2 bis ≤ 7
NHL 3,5 ≥ 25 - ≥ 3,5 bis ≤ 10
NHL 5 ≥ 15 ≥ 2 ≥ 5 bis ≤ 15
Fom
uli
erte
r K
alk
FL A 2
≥ 40 bis ≤ 80
- ≥ 2 bis ≤ 7
FL A 3,5 - ≥ 3,5 bis ≤ 10
FL A 5 ≥ 2 ≥ 5 bis ≤ 15
FL B 2
≥ 25 bis ≤ 80
- ≥ 2 bis ≤ 7
FL B 3,5 - ≥ 3,5 bis ≤ 10
FL B 5 ≥ 2 ≥ 5 bis ≤ 15
FL C 2
≥ 15 bis ≤ 80
- ≥ 2 bis ≤ 7
FL C 3,5 - ≥ 3,5 bis ≤ 10
FL C 5 ≥ 2 ≥ 5 bis ≤ 15
Hyd
rau
lisc
her
Kalk
HL 2 ≥ 10 - ≥ 2 bis ≤ 7
HL 3,5 ≥ 8 - ≥ 3,5 bis ≤ 10
HL 5 ≥ 4 ≥ 2 ≥ 5 bis ≤ 15
Baukalke sind Bindemittel, die DIN EN 459-1 entsprechen [9]. Baukalk wird vorwiegend als
Kalkhydrat oder als Feinkalk geliefert. Kalkteig und Stückkalk besitzen nur noch
untergeordnete Bedeutung. Bild 18 zeigt eine schematische Zuordnung der Kalkarten und der
Anwendungsgebiete.

26
Bild 18: Schema für Kalkarten und Anwendungsbereiche [9]
4.5 Latent hydraulische Stoffe und Puzzolane
Latent hydraulische Stoffe besitzen die Fähigkeit, bei einer entsprechenden Anregung
hydraulisch erhärten zu können. Durch die Anregung kann sich die latente (im Sinne von
verborgene oder ruhende) hydraulische Eigenschaft entfalten. Dies geschieht jedoch in einem
weitaus geringeren Maßstab, als bei einem hydraulischen Bindemittel sodass keine technisch
nutzbaren Festigkeiten erreicht werden. Die Hydraulizität eines latent hydraulischen Stoffes
wird bestimmt durch seine physikalisch-mineralogischen Eigenschaften. Die wichtigsten
Kennwerte sind Glasgehalt und Mahlfeinheit. Als Anreger dienen Calciumhydroxid, Luftkalke
und Portlandzement, in einigen Fällen genügt auch Gips.
Das meistverwendete latent hydraulische Material ist Hüttensand. Er wird durch Granulieren
(Mahlfeinheit!) von Hochofenschlacken hergestellt, wie sie bei der Stahlherstellung als
Nebenprodukte anfallen.
Puzzolane Stoffe reagieren in Verbindung mit Ca(OH)2 (aus Portlandzement oder Kalk) in
wässriger Lösung unter Bildung von CSH-Phasen. Puzzolane Stoffe erhärten nicht selbst.
Entscheidend ist das Vorhandensein reaktionsfähiger Kieselsäure.
Bild 19: Beteiligung an der hydraulischen Erhärtung
Der wesentliche Unterschied zwischen latent hydraulischen und puzzolanen Stoffen liegt in
dem jeweils enthaltenen Kalkanteil. Er liegt bei latent hydraulischen Stoffen zwischen 30 %
und etwa 55 %, bei Puzzolanen im Allgemeinen unter 10 %. Puzzolane können auf natürlichem
oder künstlichem Wege entstehen. Ein gängiger Weg zum Erhalt eines Stoffes mit einem hohen
Gehalt reaktionsfähiger Kieselsäure ist das plötzliche Abkühlen von SiO2-reichen Schmelzen

27
von hohen Temperaturen (hoher Glasgehalt).
Der Begriff Puzzolane geht auf den Namen des Ortes Putuoli (Pozzuoli) am Vesuv zurück, in
dessen Nähe eine Erde gefunden wurde, die schon von den Römern in Verbindung mit Kalk als
hydraulisches Bindemittel genutzt wurde. Ähnliche Vorkommen wurden in der Nähe anderer
aktiver oder erloschener Vulkane gefunden (Santorinerde, Trass und andere). Tabelle 6 gibt
einen Überblick über natürliche Puzzolane, Tabelle 7 enthält industriell erzeugte latent
hydraulische Stoffe und Puzzolane. Die Puzzolane haben je nach ihrer Entstehung
unterschiedliche Zusammensetzungen. Allen gemeinsam ist ein hoher Gehalt an Kieselsäure
(SiO2), der im Allgemeinen zwischen 50 und 80 % liegt. Im Gegensatz zum unter normalen
Bedingungen inerten Quarzmehl mit einem SiO2-Gehalt von über 95 % liegt bei den
Puzzolanen das SiO2 in einer anderen Struktur vor, die sie zur Reaktion mit Kalk befähigt.
Tabelle 6: Puzzolane natürlichen Ursprungs
Name Vorkommen Eigenschaft/Herkunft
Puzzolanerde Nahe Neapel, Italien Umwandlungsprodukte
vulkanischer Auswurfmassen
bzw. Lavaströme
Santorinerde Santorin, Griechenland
Trass Eifel, Bayern, Steiermark
Diatomeenerde, Kieselgur Lüneburger Heide Meeressediment,
Panzer der Kieselalge Molererde Dänemark
Gaize Frankreich Verwitterungsprodukt kieseliger
Gesteine Tripel Ukraine
Tabelle 7: Industriell erzeugte latent hydraulische Stoffe und Puzzolane
Name Eigenschaft Herstellung/Gewinnung
Hüttensand Latent hydraulisch abgeschreckte Hochofenschlacke
aus dem Hochofenprozess
Ziegelmehl Puzzolan aufgemahlener Bruch von
schwach gebrannten Ziegeln
Ölschieferrückstände Puzzolan Nicht brennbarere Rückstände aus
der Ölschieferverbrennung
Flugasche Puzzolan
Asche aus den Elektrofiltern von
Kraftwerken und
Müllverbrennungsanlagen
Silikastaub Puzzolan
Filterasche aus der
Bauxitverhüttung und der
Silizium- sowie
Ferrosiliziumgewinnung
Die in den Puzzolanen enthaltene Kieselsäure ist amorph und dem Glaszustand ähnlich. Die
einzelnen SiO2-Moleküle sind dadurch in der Lage, mit Säuren und Laugen zu reagieren.
Calciumhydroxid reagiert mit der amorphen, fein verteilten Kieselsäure wie folgt:
3 Ca(OH)2 + 2 SiO2 + H2O ⥬ 3 CaO 2 SiO2 H2O + 3 H2.

28
Das Reaktionsprodukt Calciumsilikathydrat ist dieselbe Verbindung, die bei der Hydratation
von Zement entsteht. Die Mischungen von Puzzolanen mit dem CaO-Spender Kalk nennt man
Puzzolankalke, solche mit Zement Puzzolanzemente.
4.6 Zement
4.6.1 Geschichtliche Entwicklung [10]
Der Begriff Zement tauchte, wenn auch mit völlig anderer Bedeutung, erstmals bei den alten
Römern auf. Sie bezeichneten ein betonartiges Mauerwerk aus Bruchsteinen mit gebranntem
Kalk als Bindemittel und Puzzolanen als Gesteinskörnung als Opus Caementitum. Der
entscheidende Schritt hin zum heutigen Zement ist wohl dem Engländer J. SMEATON 1756
gelungen. Er fand heraus, dass der Tongehalt im Bindemittel ausschlaggebend für die
Hydraulizität des „Zements“ ist. 1796 stellt J. PARKER erstmals industriell hydraulische Kalke
her (Romancement).
Das für die Zementherstellung optimale Verhältnis von 3 Teilen Kalkstein zu einem Teil Ton
(d. h. 25 – 30 % Ton) wurde bereits 1820 von VICAT und J. F. JOHN gefunden.
Als Geburtsjahr des Portlandzements gilt 1824. Maurermeister Joseph ASPDIN meldete ein
Patent zur Herstellung eines Zements an, den er aus einer Schlämme aus Kalk und Ton brannte.
Das gebrannte Gemisch wurde nachfolgend zu einem feinen Pulver zermahlen und als
Portlandcement verkauft. Der Name geht auf die Farbe der mit Portlandcement hergestellten
Bauwerke zurück, die dem Naturstein auf der Insel Portland ähnelt. Bei diesem
„Portlandcement“ handelte es sich noch nicht um Zement im heutigen Sinne, weil das Produkt
nicht bis zu Sinterung gebrannt war. Dieser Schritt erfolgte erst 1843 mit der Weiterentwicklung
des Verfahrens durch den Sohn William ASPDIN.
4.6.2 Zementherstellung
Zement wird aus Kalkstein, Ton und insbesondere ihrem natürlichen Gemisch Mergel
hergestellt. Aus wirtschaftlichen Gründen (Transportkosten) erfolgt die Produktion in der Regel
in der unmittelbaren Nähe der Rohstoffvorkommen (Bild 20).
Die Rohstoffe werden im Steinbruch abgebaut und in Hammer- bzw. Walzenbrechern grob
zerkleinert. In dieser Form werden einzelnen Rohstoffkomponenten im Mischbett
vorhomogenisiert, um größerer Schwankungen in der chemischen Zusammensetzung der
Lagerstätten auszugleichen, und anschließend zwischengelagert. Die nach Kalk- und Tongehalt
getrennten Rohstoffkomponenten werden über geregelte Dosiereinrichtungen in genau
vorgegebenen Verhältnissen in die mit Windsichtern kombinierten Rohmühlen (Walzen- bzw.
Kugelmühlen) aufgegeben und fein gemahlen. Gegebenenfalls werden zum Einstellen der
gewünschten Rohmehlzusammensetzung Korrekturstoffe wie Bauxit, Quarzsand und/oder
Eisenoxid zugesetzt. Der Spielraum für die optimale Zusammensetzung des Rohmehls ist klein,
wie aus dem Dreistoff-Diagramm CaO – Al2O3 – SiO2 (Rankin-Diagramm (Bild 21)) abzulesen
ist. [10-12]

29
Bild 20: Zementwerke in Deutschland – Geologische Verbreitung der Rohstoffe [11]
Bild 21: Rankin-Diagramm [12, 13]

30
Während des Mahlens wird das Mahlgut mit heißem Ofenabgas getrocknet, das durch die
Mahlanlage gesaugt wird. Die Trocknung sehr feuchter Rohstoffe erfolgt wegen der benötigten
erheblich größeren Heißgasvolumenströme nicht in der Rohmühle selbst, sondern in speziell
dafür installierten Trommeltrocknern. Durch eine Verstellung der Austragseinrichtungen an
den Vorratsbehältern der einzelnen Rohmehlkomponenten ist eine Änderung der
Rohmehlrezeptur möglich. Verbleibende Inhomogenitäten werden in eigenen Siloanlagen
mittels Fluidisierung oder mechanischer Umwälzvorrichtungen vereinheitlicht. Bild 22 zeigt
das Ablaufschema der Zementherstellung vom Abbau der Rohstoffe bis zum Verladen des
fertigen Zements. [1, 11, 12]
Bild 22: Ablaufschema der Zementherstellung
4.6.2.1 Vorwärmer
Beim heute üblichen Trockenverfahren durchläuft das fertige Rohmehl anschließend den
Vorwärmer. Er besteht aus einem System von 4 bis 6 Zyklonen (Bild 23), durch die das
Rohmehl im Gegenstrom zu den Ofengasen geführt wird.
Das Rohmehl entzieht in den einzelnen Zyklonen dem aufsteigenden Ofengas Wärme. Beim
Eintritt in den Ofen hat das Rohmehl eine Temperatur von etwa 750 °C – 850 °C. In den
Zyklonstufen beginnt bereits die stoffliche Umwandlung. Tonmineralien zersetzen sich ab etwa
400 °C - 500 °C und geben dabei ihr Kristallwasser ab, während die Entsäuerung (Calcinierung)
der Carbonate bei ca. 800 °C - 1000 °C von statten geht. In der untersten Zyklonstufe kommt
es nur zu einer partiellen Calcinierung, bei der etwa 20 – 40 % der Carbonate entsäuert werden.
Eine weitere Verbesserung für Zementofenanlagen bedeutet der Einsatz von Vorcalcinatoren.
Dabei wird die gesamte Brennstoffenergie, die zum Brennen des Zementklinkers erforderlich
ist, zwischen der Hauptfeuerung im Drehrohr und der Zweitfeuerung am Vorcalcinator
aufgeteilt. Mit dieser Zweitfeuerung zwischen Drehrohr und Wärmetauscher wird so viel

31
Energie dem System zugeführt, dass der im Brenngut enthaltene Carbonatanteil bei Verlassen
des Vorcalcinators fast vollständig dissoziiert ist. Der wesentliche Vorteil des Vorcalcinators
besteht darin, dass die für die Decarbonatisierung des Rohmehls erforderliche Energie durch
die Zweitfeuerung schon bei 900 °C - meist durch flammenlose Verbrennung - freigesetzt
werden kann und unmittelbar für die Dissoziation zur Verfügung steht. Somit ist auch der
Einsatz von minderwertigen Brennstoffen möglich. Daneben entlastet der Vorcalcinator die
Sinterzone des Drehrohrofens bei gleichzeitiger Erhöhung der Klinkerkapazität bei
unveränderten Ofenabmessungen.
Bild 23: Schema eines 4-stufigen Zyklonvorwärmers mit Rohmehl- und Gasführung [14]
4.6.2.2 Brennen des Rohmehls zu Zementklinker [10, 15-17]
Das Brennen des Brenngutes zu Zementklinker erfolgt bei Temperaturen bis 1450 °C im
Drehrohrofen. Drehrohröfen sind unter 3 ° bis 4 ° geneigt liegende, feuerfest ausgekleidete
Rohre mit einem Durchmesser von ca. 3,5 m bis 5,0 m und einer durchschnittlichen Länge von
40 m bis 70 m (Trocken- bzw. Halbtrockenverfahren). Infolge der Neigung und Drehung (0,5
bis 4,5 Umdrehungen pro Minute) des Ofens läuft das am oberen Ofenende zugeführte
Brenngut einer Kohlenstaub-, Öl- oder Gasflamme entgegen, die als Primärfeuerung bezeichnet
wird. Daneben können Ersatzbrennstoffe wie Altreifen über die Sekundärfeuerung am oberen
Ofenende direkt dem Drehrohr aufgegeben werden.
Das Rohmehl durchläuft im Ofen mehrere Temperaturzonen: Nach dem Eintritt in den Ofen
gelangt es zuerst in die Calcinierungszone. Hier wird das Calciumcarbonat im Rohmehl mit
steigender Temperatur zunehmend entsäuert. Während der Verweildauer von etwa 20 – 30
Minuten heizt sich das Brenngut auf etwa 1000 °C auf.
In der anschließenden Übergangszone reagieren die vorliegenden Bestandteile (gebrannter
Kalk und entwässerter Ton) bei steigender Temperatur miteinander (Leichtbrand). Bei ca.
1250 °C erweicht das Rohmehl, wobei aus dem Calciumoxid und den Bestandteilen des Tons
Calciumsilikate, Calciumaluminate und Calciumaluminatferrite entstehen. Das
Reaktionsprodukt entspricht bei dieser Temperatur im Wesentlichen einem hydraulischen Kalk,
bei dem das Calciumoxid hauptsächlich als Dicalciumsilikat (2 CaO SiO2) vorliegt.

32
Während des ab 1200 °C beginnenden und bis auf eine Temperatur von rund 1450 °C
ansteigenden Garbrands wird weiteres Calciumoxid gebunden. Das Material durchläuft diese
Zone in etwa 10 Minuten. Aus dem Dicalciumsilikat bildet sich das für Zement
charakteristische Tricalciumsilikat:
2 CaO SiO2 + CaO ⥬ 3 CaO SiO2.
Die Reaktion erfolgt unterhalb des Schmelzpunktes durch Diffusion der Teilchen und wird als
Sintern bezeichnet. Die kurze Durchlaufzeit durch diese heiße Zone reicht nicht aus, um alle
chemischen Umwandlungen vollständig ablaufen zu lassen. Das chemische Gleichgewicht wird
unter technischen Brennbedingungen nicht erreicht. Aus dem Rohmehl entsteht eine harte
Masse, meist in Form von Kugeln. Der Name „Zementklinker“ entstammt der Ähnlichkeit mit
dem ebenfalls bis zum Sintern gebrannten keramischen Klinker. Am Ende des Drehrohrofens
sinkt die Temperatur wieder auf 1350 °C – 1200 °C. In dieser Kühlzone kristallisieren
Aluminat- und Ferritphasen. Bild 24 zeigt den Zusammenhang zwischen den sich bildenden
Phasen und den Temperaturen im Drehrohrofen.
Bild 24: Phasenbildung des Zementklinkers im Drehrohrofen
Der Zementklinker wird nach Verlassen des Drehrohrofens möglichst rasch in Rost-, Trommel-
oder Satellitenkühlern abgekühlt, um zu verhindern, dass das Tricalciumsilikat wieder zerfällt.
Die beim Kühlen des Klinkers auf 800 °C bis 900 °C erhitzte Luft strömt als Sekundärluft in
das Drehrohr und dient dort als Verbrennungsluft. Das Einblasen des Brennstoffes erfolgt
mittels Primärluft. Die zur Klinkerkühlung bevorzugt eingesetzten Rostkühler benötigen mehr
Luft zu Kühlzwecken, als für das Brennen des Klinkers erforderlich ist. Die überschüssige
Luftmenge kann gegebenenfalls als Wärmebilanz verbessernde Tertiärluft dem Vorcalcinator
zugeführt werden, oder wird nach der Nutzung der Abwärme und entsprechender Entstaubung
in die Atmosphäre freigesetzt.
Die Tabelle 8 fasst die bei der Zementherstellung in den verschiedenen Temperaturbereichen
ablaufenden Vorgänge und die zugehörigen Wesentlichen chemischen Umsetzungsreaktionen
nochmals zusammen.

33
Tabelle 8: Chemische Umsetzungen bei der Zementherstellung (vereinfacht)
Temperatur
in °C Vorgang Chemische Umsetzung
20 – 200 Abgabe von freiem Wasser
(Trocknung)
200 - 400 Abgabe von adsorbiertem
Wasser
450 - 600 Tonzersetzung, Bildung von
Metakaolinit Al4(OH)8Si4O10 ⥬ 2 (Al2O3 2 SiO2) + 4 H2O
600 - 950
Zersetzung der Metakaolinit,
Ausbilden reaktiver
Oxidmischungen, Beginn der
Kalksteinentsäuerung in
Gegenwart von Al2O3 + SiO2
Al2O3 2 SiO2 ⥬ Al2O3 + 2 SiO2
CaCO3 ⥬ CaO + CO2
800 - 1000
Kalksteinzersetzung,
gleichzeitiges Bilden von
Calciumsilikat und
Calciumaluminat
CaCO3 ⥬ CaO + CO2
3 CaO + Al2O3 + 2 SiO2 ⥬ 2 (CaO 2 SiO2) +
CaO Al2O3
1000 - 1300
Schmelzen des Rohmehls,
Kalkaufnahme durch
Dicalciumsilikat,
Tricalciumaluminat, Bilden
von
Tetracalciumaluminatferrit
CaO + SiO2 ⥬ CaO 2 SiO2
2 CaO + SiO2 ⥬ 2 CaO SiO2
2 CaO + CaO Al2O3 ⥬ 3 CaO Al2O3
3 CaO + CaO Al2O3 + Fe2O3 ⥬
4 CaO Al2O3 Fe2O3
1200 - 1450 Weitere Kalkaufnahme durch
Tricalciumsilikat CaO + 2 CaO SiO2 ⥬ 3 CaO SiO2
4.6.2.3 Vermahlen des Klinkers mit diversen Zumahlstoffen zu Zement
Der abgekühlte Klinker wird noch im Kühler durch einen Brecher grob zerkleinert und ebenso
wie die zementeigenen Zumahlstoffe in Großraumsilos oder geschlossenen Hallen
zwischengelagert. Anschließend werden je nach Zementrezeptur Klinker sowie diverse
Zumahlstoffe (Natur-, REA-Gips, Hochofenschlacke, ...) miteinander gemischt und in den
Zementmühlen gemahlen. Für das Feinmahlen des Zements (Mahlfeinheit mindestens 85 % <
70 m) werden überwiegend Kugelmühlen - in selteneren Fällen Wälzmühlen - verwendet. Die
meisten Zementmühlen arbeiten in Kombination mit einstellbaren Windsichtern, die den
Zement mit der geforderten Feinheit aus dem Mahlgut aussondern.
4.6.3 Chemische Zusammensetzung
Ausgangsbasis für die Herstellung des Zements ist ein ungefähres Mischungsverhältnis
Kalkstein : Ton = 3 : 1. Durch den Verlust des Kristallwassers verringert sich der Anteil des
Tons auf etwa 22 % der ursprünglichen Einwaage. Nach der Entsäuerung des Kalksteins
verbleiben 43 % CaO, sodass sich für die Zement bildenden Reaktionen Calcium und Ton im
Verhältnis 2 : 1 gegenüberstehen.

34
Die Eigenschaften des Zements werden sehr stark von seiner Zusammensetzung beeinflusst.
Um diese Feinheiten erfassen zu können, wurden verschiedene Kennziffern eingeführt, die als
Moduln bezeichnet werden. Die verschiedenen Kennwerte sind in Tabelle 9 zusammengefasst.
Tabelle 9: Kennwerte zur chemischen Beurteilung des PZ-Klinkers [17]
Bezeichnung Abkürzung Formel Üblicher Bereich
für Portlandzement
Kalkstandard I KSt I 32322 7,01,18,2
100
OFeOAlSiO
CaO
++ 92 – 102
Kalkstandard III
(für MgO 2,0
%)
KSt III ( )
32322 65,018,18,2
75,0100
OFeOAlSiO
MgOCaO
++
+ 90 – 102
Kalkstandard III
(für MgO 2,0
%)
KSt III ( )
32322 65,018,18,2
50,1100
OFeOAlSiO
MgOCaO
++
+ 90 – 102
Hydraulemodul HM 32322 OFeOAlSiO
CaO
++ 2,0 – 2,4
Silikatmodul SM 3232
2
OFeOAl
SiO
+ 1,8 – 3,0
Tonerdemodul TM 32
32
OFe
OAl 1,5 – 2,5
Für die chemische Zusammensetzung des Portlandzementklinkers lassen sich auf Basis dieser
Formeln folgende Richtwerte berechnen [17]:
KSt 90 - 97 → Klinker für normalen Portlandzement
KSt 97 - 102 → Klinker für schnell erhärtenden, hochfesten Portlandzement
SM < 1,8 → Klinker für SiO2 reichen Portlandzement
SM = 2 → Klinker für normalen Portlandzement
SM > 3 → Klinker für SiO2 armen Portlandzement
TM < 1,5 → Klinker für sulfatbeständigen Portlandzement
TM = 2 → Klinker für normalen Portlandzement
TM > 4 → Klinker für Aluminatzement
Für die Berechnung der Zusammensetzung ist es wichtig, dass der Zement
1. nicht mehr Kalk enthält, als durch die Hydraulefaktoren gebunden werden kann, weil
ansonsten die Gefahr des Kalktreibens besteht.
2. nicht wesentlich weniger Kalk enthalten ist, weil sonst die (Früh-) Festigkeit zu gering
ausfällt.

35
Die durchschnittliche Zusammensetzung eines Zementklinkers gibt Bild 25 wider. Nicht
dargestellt sind die Nebenbestandteile (Gips, Alkalien, Feuchtigkeit ...), die mit etwa 6 bis 8 %
an der Zusammensetzung beteiligt sind.
Bild 25: Durchschnittliche Zusammensetzung eines Zementklinkers
4.6.4 Mineralische Zusammensetzung
In der Zementchemie ist es wegen der geringen Anzahl der maßgeblichen Ausgangsbestandteile
üblich, den einzelnen Oxide Kurzbezeichnungen zu geben. Es bedeuten:
C = CaO CH = Ca(OH)2
S = SiO2 H = H2O
A = Al2O3 S = SO3
F = Fe2O3 M = MgO
Aus den oxidischen Hauptbestandteilen setzen sich die in Tabelle 10 aufgeführten
Hauptklinkerphasen zusammen. Für die ersten drei Klinkerphasen kann die Zusammensetzung
eindeutig angegeben werden. Beim Tetracalciumaluminatferrit ist dies nicht in jedem Fall
möglich, da die Verbindung aus unterschiedlichen Mischkristallen besteht und die sich in
Abhängigkeit vom Verhältnis Al2O3 : Fe2O3 im Rohmehl bilden. C4AF ist die mittlere
Zusammensetzung, die im technischen Klinker vorgefunden wird.
Tabelle 10: Hauptbestandteile des Zementklinkers, chemische Formel und Kurzbezeichnung
[17]
Bezeichnung Formel Kurzbezeichnung Durchschnittlicher
Gehalt im PZ [%]
Tricalciumsilicat oder Alit 3 CaO·SiO2 C3S 40 - 80
Dicalciumsilicat oder Belit 2 CaO·SiO2 C2S 2 - 30
Tricalciumaluminat 3 CaO·Al2O3 C3A 3 - 15
Tetracalciumaluminatferrit 4
CaO·Al2O3·Fe2O3
C4AF 4 - 15
67
22
85
65
20
63
63
18
41
0
10
20
30
40
50
60
70
80
CaO SiO2 Al2O3 Fe2O3
Chem
ische Z
usam
mensetz
ung i
n %
Maximal
Mittel
Minimal
SiO2 Al2O3 Fe2O3

36
4.6.5 Eigenschaften der Klinkermineralien
In praktisch jedem PZ-Klinker sind die vier Klinkerphasen enthalten. Ihr jeweiliger Anteil
richtet sich nach der Zusammensetzung des Rohmehls. Geringfügige Veränderungen in der
chemischen Zusammensetzung können dabei deutliche Verschiebungen in der mineralischen
Zusammensetzung bewirken Tabelle 11.
Tabelle 11: Auswirkung von Variationen der chemischen Zusammensetzung des Rohmehls auf
die potenzielle Bildung der Klinkermineralien in M.-%
Oxide 1 2 3
CaO 66,0 63,0 66,0
SiO2 20,0 22,0 20,0
Al2O3 7,0 7,7 5,5
Fe2O3 3,0 3,3 4,5
Rest 4,0 4,0 4,0
Mineralien
C3S 60 27 68
C2S 13 43 7
C3A 14 15 7
C4AF 9 10 14
C S 3 3 3
Tricalciumsilicat (C3S) Alit
Mit einem Anteil von etwa 2/3 ist C3S der Hauptbestandteil des Portlandzementklinkers. Auf
seinen Eigenschaften beruht der Unterschied zu hydraulischem Kalk. Eine schnelle Erhärtung
und hohe Hydratationswärme gehen auf das C3S zurück. Es ist unempfindlich gegen das
Einwirken von Sulfaten und weist bei der Hydratation eine mittlere Schwindneigung auf.
Dicalciumsilicat (C2S) Belit
Das C2S zeichnet sich im Gegensatz zum C3S durch eine langsamere, jedoch stetige Erhärtung
aus, die mit einer niedrigeren Hydratationswärme einhergeht. C2S hat die gleiche
Schwindneigung und ist gegen Sulfateinwirkung beständig. Vom C2S existieren zwischen
20 °C und 1500 °C fünf Modifikationen. Im Klinker liegt es zumeist als β-C2S vor.
Tricalciumaluminat (C3A)
C3A erstarrt sehr schnell in Kontakt mit Wasser und entwickelt dabei eine sehr große
Hydratationswärme. C3A begünstigt das Schwinden und ist gegen Sulfateinwirkungen
empfindlich. Das mit den Sulfaten gebildete voluminöse Salz ist die Ursache für das so
genannte Sulfattreiben.
Tetracalciumaluminatferrit (C4AF) oder C2(A,F)
C4AF erhärtet langsam und erreicht keine hohe Festigkeit. Die entstehende Hydratationswärme
ist entsprechend gering, ebenso die Schwindneigung. Gegen Sulfateinwirkung ist C4AF
unempfindlich.

37
4.6.6 Nebenbestandteile
Neben den vier Hauptbestandteilen können im Klinker noch
• Freikalk (CaO),
• Periklas (MgO) und
• Alkalien
als Nebenbestandteile enthalten sein.
4.6.6.1 Freikalk
Der Freikalkgehalt liegt in der Regel bei etwa 1 %. Er wird verursacht durch
• einen zu hohen Kalkgehalt im Rohmehl,
• zu grobes Rohmehl,
• ungenügend homogenisiertes Rohmehl,
• zu niedrige Brenntemperatur,
• zu langsame Abkühlung (Zerfall des C3S),
• ungleichmäßige Aufnahme der Brennstoffasche und
• zu hohen MgO-Gehalt.
Im Klinker ist der Freikalkgehalt auf < 2 % zu begrenzen, damit der daraus hergestellte Zement
raumbeständig ist und um das schädliche Kalktreiben (1,9-fache Volumenvergrößerung) zu
verhindern.
4.6.6.2 Periklas
Periklas kann ebenso wie Freikalk zu Treiberscheinungen führen. Bei der Hydratation
MgO + H2O ⥬ Mg(OH)2
nimmt das entstehende Mg(OH)2 (Brucit) das 2,2-fache des Ausgangsvolumens ein. Diese
Reaktion läuft sehr langsam und kann sich über Jahre bis Jahrzehnte hinziehen. Der MgO-
Gehalt im Klinker ist auf 5 % zu begrenzen.
4.6.6.3 Alkalien
Im Klinker kommen Alkalien in folgenden Anteilen vor:
• K2O 0,1 bis 0,5 % und
• Na2O 0,1 bis 0,8 %.
Die Alkalien entstammen den Rohstoffen und zum Teil den Brennstoffen und liegen primär als
Alkalisulfate vor.
4.6.7 Erhärtung
Infolge der chemischen Reaktion zwischen Zementkorn und Wasser erstarrt und erhärtet der
Zement. Die Reaktion von Zement mit Wasser setzt sich aus zwei unterschiedlichen
chemischen Reaktionen zusammen:
Die Hydrolyse setzt unmittelbar nach dem Kontakt des Zements mit dem Anmachwasser ein.
Dabei wird Kalk aus den Klinkerphasen in geringem Umfang freigesetzt und reagiert mit dem
Wasser unter Bildung von OH--Ionen:
CaO + H2O ⥬ Ca2+ + 2 OH-
Der pH-Wert steigt umgehend auf pH = 12,5 und sorgt in der Folge für den Korrosionsschutz
des im Beton eingebetteten Bewehrungsstahls. Der Festigkeitsbeitrag des Calciumhydroxids ist
belanglos.

38
Die eigentliche Erhärtungsreaktion ist die Hydratation. Sie verläuft wesentlich langsamer als
die Hydrolyse und beginnt in der Regel nicht vor einer Stunde nach dem Anmachen. Bei der
Hydratation werden die Klinkerphasen in wasserhaltige Hydratphasen umgewandelt.
4.6.7.1 Hydratation der Hauptklinkerphasen [18, 19]
4.6.7.1.1 Hydratation des C3S
Bei der Hydratation des C3S werden Calciumsilicathydrate (C-S-H-Phasen) variabler
chemischer Zusammensetzung sowie Portlandit (CH) gebildet. Daher kann nur eine allgemeine
Formel angegeben werden
C3S + (y + z) H ⥬ CxSHy + z CH
wie in folgendem Beispiel:
2 C3S + 7 H ⥬ C3S2H4 + 3 CH.
oder in Oxidschreibweise:
2 (3 CaO SiO2) + 7 H2O ⥬ [3 CaO 2 SiO2 4 H2O] + 3 Ca(OH)2
Bei den C-S-H-Phasen unterscheidet man je nach Zahlenwert von x und y in C-S-H(I) und C-
S-H(II). C3S2H4 gilt als durchschnittliche Zusammensetzung der C-S-H-Phasen bei normaler
Erhärtung. Anhand der Wärmeentwicklung (Bild 26) wird die Hydratation in 5 Stadien
eingeteilt (Tabelle 12)
Bild 26: Hydratationswärmeentwicklung von C3S [18, 19]

39
Tabelle 12: Stadien der C3S-Hydratation [18, 19]
Stadium Zeit Reaktionskinetik Chemische
Prozesse
Phasenbeschreibung
1 Anfangshydrolyse
(Bild 27)
erste
Minuten
nach dem
Anmachen
chemisch
kontrollierte,
schnelle Reaktion
Beginn der
Hydrolyse, in
Lösung gehen
von Ionen
Ausbildung einer eng
anliegenden
Reaktionsschicht von ca.
20 – 30 nm
2 Ruheperiode ca. 20 min –
2 h
kernkontrollierte,
langsame Reaktion
kontinuierliche
Lösung von
Ionen
erste heterogene
Keimbildung
3 Beschleunigungs-
periode (Bild 28)
2 h - 11 h chemisch
kontrollierte,
schnelle Reaktion
Beginn der
Bildung von
Hydratations-
produkten
große, dünnplattige
Portlanditkristalle (CH),
kurze, stumpfnadelige
C-S-H-Faserbündel
4 Verzögerungsperi
ode
11 h - 26 h chemisch und
diffusionskontrollier
te Reaktion
kontinuierliche
Bildung von
Hydratations-
produkten
eindimensionales
Wachstum der C-S-H-
Fasern, Zusammen-
lagerung dieser Fasern,
stärkere Verzahnung der
Partikel, Verwachsungen
der plattigen CH-
Kristalle
5 Stetige Periode
(auch
Finalperiode)
(Bild 29)
ab ca. 26 h Diffusions-
kontrollierte
Reaktion
langsame
Bildung von
Hydratationspro
dukten
weiteres
Längenwachstum der C-
S-H-Fasern, Verzahnung
Bild 27: 5 min hydratisiertes C3S, gelförmige Hüllen um die C3S-Körner [18, 19]
6
Bild 6:
C-S-H in Kalksandstein: auf
ein Quarzkorn gewachsene
schwertförmige
Tobermoritkristalle,
1,13 nm Tobermorit [C5S6H5],
Kristalldicke: 20 nm,
Spitzen: <10 nm.
P = PortlanditC = CalcitCS = Calciumsilicat (C 3S)
P
P
P
C
C
CC
CS
CS
CS
CS
CS
Abb. 7:
Röntgenogramm von
hydratisierenden C3S nach 28 d:
eine Identifikation der C-S-H-
Phasen kann nicht vorgenommen
werden.
Auch die Ausbildung der C-S-H-Phasen als zunächst langfaserig und später kurzfaserig
kann nicht bestätigt werden. Vielmehr sind die C-S-H-Phasen am Anfang eher
kurzfaserig und wachsen im weiteren Hydratationsverlauf bis auf eine Länge von
1,5 µm (siehe Bild 3). Die ESEM-Bilder 8 – 13 zeigen die Gefügeausbildung während
des Hydratationsverlaufs bei Raumtemperatur zu ausgewählten Zeiten (siehe Tabelle 1).
Bild 8: 5 min hydratisiertes C3S, gelförmige Hüllen um
die C3S-Körner.
Bild 9: 140 min hydratisiertes C3S, Ausbildung einer
voluminösen C-S-H-Schicht mit Wabenstruktur.

40
Bild 28: 4h hydratisiertes C3S, erste stumpfnadelige C-S-H-Bündel (Länge bis 300 nm) und
Portlandit [18, 19]
Bild 29: 21 d hydratisiertes C3S, spitznadelige C-S-H-Phasen (Länge bis 900 nm) [18, 19]
4.6.7.1.2 Hydratation des -C2S
Während der Hydratation des -C2S bildeten sich die gleichen faserförmigen
Hydratationsprodukte wie während der C3S-Hydratation. Allerdings verläuft diese Hydratation
wie oft beschrieben deutlich langsamer als die des C3S. Die Reaktionsprodukte unterscheiden
sich nur im Hinblick auf die geringere Menge an abgespaltenem Portlandit. Sie können
ebenfalls mit der allgemeinen Formel
2 C2S + (2 - x + y) H ⥬ CxSHy + (2 – x) CH
wie in folgendem Beispiel gebildet werden:
2 C2S + 5 H ⥬ C3S2H4 + CH.
oder in Oxidschreibweise:
2 (2 CaO SiO2) + 4 H2O ⥬ [3 CaO 2 SiO2 3 H2O] + 1 Ca(OH)2
Der Festigkeitsbeitrag der im Vergleich zu den C-S-H-Phasen riesengroßen Portlanditkristalle
dürfte gering sein. Die großen tafelförmigen Kristalle könnten im Gegenteil sogar als
Gleitflächen bei Belastung wirken.
7
Bild 10: 4h hydratisiertes C3S, erste stumpfnadelige C-S-
H-Bündel (Länge bis 300 nm) und Portlandit.
Bild 11: 16 h hydratisiertes C3S, spitznadelige C-S-H-
Phasen (Länge bis 400 nm).
Bild 12: 21 d hydratisiertes C3S, spitznadelige C-S-H-
Phasen (Länge bis 900 nm).
Bild 13: 360 d hydratisiertes C3S, Verzahnung der bis
1200 nm langen C-S-H-Phasen und Portlandit.
2. 2 Hydratation des bbbb-C2S
Während der Hydratation des b-C2S bildeten sich die gleichen faserförmigen Hydrata-
tionsprodukte wie während der C3S-Hydratation. Allerdings verläuft diese Hydratation
wie oft beschrieben deutlich langsamer als die des C3S. Nach ca. einem Jahr Hydrata-
tionszeit konnten die gleichen Faserproportionen wie beim hydratisierenden C3S beob-
achtet werden. Bei einer Untersuchung nach 3 Jahren an hydratisiertem b-C2S wurden
extrem lange C-S-H-Fasern registriert (Bild 14). Dies könnte ein weiterer Beitrag zur
Klärung der guten Nacherhärtung infolge der b-C2S-Hydratation sein.
7
Bild 10: 4h hydratisiertes C3S, erste stumpfnadelige C-S-
H-Bündel (Länge bis 300 nm) und Portlandit.
Bild 11: 16 h hydratisiertes C3S, spitznadelige C-S-H-
Phasen (Länge bis 400 nm).
Bild 12: 21 d hydratisiertes C3S, spitznadelige C-S-H-
Phasen (Länge bis 900 nm).
Bild 13: 360 d hydratisiertes C3S, Verzahnung der bis
1200 nm langen C-S-H-Phasen und Portlandit.
2. 2 Hydratation des bbbb-C2S
Während der Hydratation des b-C2S bildeten sich die gleichen faserförmigen Hydrata-
tionsprodukte wie während der C3S-Hydratation. Allerdings verläuft diese Hydratation
wie oft beschrieben deutlich langsamer als die des C3S. Nach ca. einem Jahr Hydrata-
tionszeit konnten die gleichen Faserproportionen wie beim hydratisierenden C3S beob-
achtet werden. Bei einer Untersuchung nach 3 Jahren an hydratisiertem b-C2S wurden
extrem lange C-S-H-Fasern registriert (Bild 14). Dies könnte ein weiterer Beitrag zur
Klärung der guten Nacherhärtung infolge der b-C2S-Hydratation sein.

41
4.6.7.1.3 Hydratation des C3A
C3A ist im Vergleich zu den anderen Phasen sehr reaktiv und reagiert deshalb am schnellsten
mit dem Anmachwasser. Bei Abwesenheit eines Sulfatträgers bilden sich sofort große
hexagonale, dünne Calciumaluminathydrat-Kristalle (C-A-H), die leicht die Porenräume
überbrücken können und daher für eine schnelle Verfestigung des Frischbetons verantwortlich
sind (Bild 30). Ausgenutzt wird dieser Effekt zum Beispiel bei der Hydratation von
Schnellzement.
2 C3A + 21 H ⥬ C4AH13 + C2AH8.
C4AH13 und C2AH8 sind instabil und wandelt sich in stabiles C3AH6 um:
C4AH13 + C2AH8 ⥬ 2 (C3AH6) + 9 H.
Bild 30: C3A ohne Sulfatzusatz: auf ein C3A-Korn aufgewachsene dünntafelige
Calciumaluminatkristalle (d 50 nm) nach 20 min Hydratation (rechts Detail) [18,
19]
Da ein schnelles Ansteifen in den meisten Fällen unerwünscht ist, wird dem aufgemahlenen
Klinker ca. 5% Gips bzw. Anhydrit zugegeben. Es entstehen dabei in den ersten Minuten nach
Wasserzugabe kleine hexagonale Ettringitkristalle (Tricalciumaluminatsulfathydrat) von
100 nm bis 400 nm Größe, die auf den Klinkeroberflächen aufwachsen, siehe Bild 31. Die
Bildung von C-A-H wird dabei zugunsten des frühen Ettringitwachstums unterdrückt.
Bild 31: Sofortige Bildung von Ettringit in einer C3A- Halbhydrat-Mischung nach 2 min
Hydratation [18, 19]
9
Bild 15: Hydratation eines C3A ohne Sulfatzusatz: auf
ein C3A-Korn aufgewachsene dünntafelige Calcium-
aluminatkristalle (d » 50 nm).
Bild 16: C3A ohne Sulfatzusatz nach 20 min
Hydratation (Detailvergrößerung von Bild 15).
Ettringit ist als primäres Hydratationsprodukt nur so lange stabil, wie ausreichend Sulfat
zur Verfügung steht. Da das Sulfatangebot im Zement für eine vollständige
Trisulfatbildung niemals ausreicht (bei 10% C3A wären dafür 9% SO3, bzw. 19% Gips
erforderlich), werden in Abhängigkeit von internen (gelöstes Sulfat, pH-Wert,
Feuchtigkeitsangebot, Carbonatgehalt) und externen Bedingungen (Temperatur)
Monosulfat und sulfatfreie Calciumaluminate gebildet. Während dieser
Sekundärreaktion werden die primär gebildeten Ettringithüllen aufgebrochen, so daß das
C3A mit einer verminderten Sulfatmenge zu Monosulfat weiterreagieren kann (Bilder 17
und 18).
Bild 17:
C3A- Halbhydrat-Mischung nach
2 min Hydratation: nach dem
Anmachen bildet sich sofort
Ettringit.
9
Bild 15: Hydratation eines C3A ohne Sulfatzusatz: auf
ein C3A-Korn aufgewachsene dünntafelige Calcium-
aluminatkristalle (d » 50 nm).
Bild 16: C3A ohne Sulfatzusatz nach 20 min
Hydratation (Detailvergrößerung von Bild 15).
Ettringit ist als primäres Hydratationsprodukt nur so lange stabil, wie ausreichend Sulfat
zur Verfügung steht. Da das Sulfatangebot im Zement für eine vollständige
Trisulfatbildung niemals ausreicht (bei 10% C3A wären dafür 9% SO3, bzw. 19% Gips
erforderlich), werden in Abhängigkeit von internen (gelöstes Sulfat, pH-Wert,
Feuchtigkeitsangebot, Carbonatgehalt) und externen Bedingungen (Temperatur)
Monosulfat und sulfatfreie Calciumaluminate gebildet. Während dieser
Sekundärreaktion werden die primär gebildeten Ettringithüllen aufgebrochen, so daß das
C3A mit einer verminderten Sulfatmenge zu Monosulfat weiterreagieren kann (Bilder 17
und 18).
Bild 17:
C3A- Halbhydrat-Mischung nach
2 min Hydratation: nach dem
Anmachen bildet sich sofort
Ettringit.

42
Ettringit ist als primäres Hydratationsprodukt nur so lange stabil, wie ausreichend Sulfat zur
Verfügung steht. Da das Sulfatangebot im Zement für eine vollständige Trisulfatbildung
niemals ausreicht (bei 10 % C3A wären dafür 9 % SO3, bzw. 19 % Gips erforderlich), werden
in Abhängigkeit von internen (gelöstes Sulfat, pH-Wert, Feuchtigkeitsangebot, Carbonatgehalt)
und externen Bedingungen (Temperatur) Monosulfat und sulfatfreie Calciumaluminate
gebildet. Während dieser Sekundärreaktion werden die primär gebildeten Ettringithüllen
aufgebrochen, sodass das C3A mit einer verminderten Sulfatmenge zu Monosulfat
weiterreagieren kann (Bild 32).
Bildung von Ettringit: 2 C3A + 3 CS H2 + 26 H ⥬ 2 C3A 3 CS H32.
Bildung von Monosulfat: 2 C3A 3 CS H32 + 2 C3A + 4 H ⥬ 3 C3A CS H12.
Bild 32: Deutlich ausgebildete hexagonale Monosulfatkristalle in einer C3A- Halbhydrat-
Mischung nach 24 h Hydratation [18, 19]
Die Hydratation von C3A verläuft in Abhängigkeit vom Sulfatgehalt sehr unterschiedlich.
Allgemein können in Abhängigkeit vom Gips/C3A-Verhältnis die folgenden
Hydratationsprodukte erwartet werden:
CS H2 / C3A stabile Hydratationsprodukte
> 3,0 Trisulfat und Gips (führt zu Sulfattreiben)
3,0 Trisulfat
1,0 ... 3,0 Trisulfat und Monosulfat
1,0 Monosulfat
< 1,0 Monosulfat und C4AH13, C2AH8 bzw. C3A(CS , CH)H12
0 C3AH6
10
Bild 18:
C3A- Halbhydrat-Mischung nach
24h Hydratation: deutlich
ausgebildete hexagonale
Monosulfatkristalle.
2.4 Hydratation des C2(A,F)
Die Hydratation des C2(A,F) verläuft in Abhängigkeit vom Eisengehalt deutlich träger
als die des C3A. Als Hydratationsprodukte werden meist in Abhängigkeit vom
Sulfatgehalt adäquat zum C3A Calciumaluminatferritmonosulfat (kurz AFm) und
Calciumaluminatferrittrisulfat (kurz AFt) angegeben.
Für die Untersuchungen wurde C2(A,F) durch Brennen im Muffelofen synthetisiert.
Eine Untersuchung der Klinkeranschliffe zeigte, daß das Material trotz einer homogenen
Rohstoffmischung ein sehr inhomogenes Brennprodukt ergab. Mittels
Elektronenstrahlmikroanalyse (ESMA) mit dem Energiedispersiven Spektrometer
(EDX) konnten sehr starke Schwankungen im Aluminat- und Ferritgehalt bestimmt
werden (Bilder 19 und 20).
Langjährige Erfahrungen im Bereich der Bauschadensermittlung zeigen, daß praktisch
nie ein Eisenenttringit (AFt) bzw. ein Eisenmonosulfat (AFm) nachgewiesen werden
konnte. Die Hydratation von reinem C2F führte dagegen eindeutig zur Bildung von
Eisenettringit. Die Frage, ob bei der normalen Portlandzementhydratation überhaupt AFt
bzw. AFm entsteht, sollte in Zukunft genauer untersucht werden. Ebenfalls ist in diesem
Zusammenhang die Begriffsbildung von AFt und AFm zu diskutieren.
Während der Hydratation von Portlandzement konnte für C2(A,F) folgende
Beobachtung gemacht werden: Als Hydratationsprodukt dieser Phase stellte sich reiner
Aluminatettringit dar. Die Körner des Ausgangsmaterials zeigten sich als
eisenangereicherte und aluminiumarme (ausgelaugte) Reste im Gefüge (Bilder 21
und 22).

43
4.6.7.1.4 Hydratation des C4AF
Die Hydratation des C4AF verläuft in Abhängigkeit vom Eisengehalt ähnlich aber deutlich
träger als die des C3A. In der Literatur werden als Hydratationsprodukte meist in Abhängigkeit
vom Sulfatgehalt adäquat zum C3A Calciumaluminatferritmonosulfat (kurz AFm) und
Calciumaluminatferrittrisulfat (kurz AFt) angegeben. Neuere Untersuchungen deuten darauf
hin, dass sich das Hydratationsprodukt dieser Phase als reiner Aluminatettringit darstellt. Die
Körner des Ausgangsmaterials zeigten sich als eisenangereicherte und aluminiumarme
(ausgelaugte) Reste im Gefüge.
Ohne Sulfatträger kommt es zu folgenden Reaktionen:
2 C4AF + 32 H ⥬ C4(A,F)H13 + 2 C2(A,F)H8 + (A,F)H3.
C4(A,F)H13 und C2(A,F)H8 sind instabil und wandelt sich in stabiles C3(A,F)H6 um:
C4(A,F)H13 + 2 C2(A,F)H8 ⥬ 2 C3(A,F)H6 + 9 H.
In Gegenwart eines Sulfatträgers bilden sich bei der Hydratation die AFt-
(Calciumaluminatferrittrisulfat) und die AFm-Phase (Calciumaluminatferritmonosulfat) aus.
AFt wird zuerst gebildet und wandelt sich später zumindest teilweise in AFm um oder bleibt
bei hohen Sulfatgehalten bestehen:
( ) ( ) .HFA,2HSC3FA,C4H110HSC12AFC3 332424 +→+
AFt-Phase
( ) ( ) ( ) .HFA,2HSCFA,C6H12HSC3FA,C4AFC3 31233244 +→++
AFm-Phase
Bild 33: "Ausgelaugtes" C4AF-Korn in einer C2(A,F)-Gips-CH-Mischung [18, 19]
4.6.7.2 Hydratation des Portlandzements [10, 16, 18, 19]
Das bei der Hydratation entstehende Zementgel besteht aus feinsten, submikroskopischen,
kolloidalen Reaktionsprodukten. Der anfangs plastische Zementleim geht mit fortschreitender
Zeit in den erhärteten Zementstein über. Zunächst wird Calciumhydroxid Ca(OH)2 in Form
hexagonaler Kristalle gebildet, die praktisch keine Verfestigung verursachen. Nach ein bis
mehreren Stunden kristallisieren infolge der schnellen Hydratation von Tricalciumsilikat (C3S)
12
Bild 22:
„Ausgelaugtes“ C4AF-Korn in
einer C4AF-Gips-CH-Mischung,
(markierter Bereich wurde mittels
EDX untersucht).
3. Hydratation von Portlandzement
Die Hydratation eines technischen Portlandzementes verläuft teilweise deutlich
verschieden zu der Hydratation einzelner Klinkerphasen. Infolge des Zusammenspiels
der Hydratationsreaktionen mehrerer nebeneinander vorliegender Phasen stellt sich ein
anderes chemisches Gleichgewicht in der wässrigen Phase ein, was sich wiederum
deutlich auf die Bildung der Hydratationsprodukte auswirkt.
Der zeitliche Verlauf der PZ-Hydratation läßt sich analog zu der C3S-Hydratation in
5 Perioden einteilen. Unmittelbar nach dem Anmachen (Induktionsperiode) setzt eine
erste Reaktion des C3A unter Mitwirkung des als Abbinderegler eingesetzten Sul-
fatträgers zu Ettringit und Syngenit ein (Bild 23 und 24). Diese primär entstandenen
prismatischen Ettringitkristalle besitzen eine Länge von ca. 300 – 500 nm und eine
Dicke von ca. 50 – 250 nm. Ein weiteres deutliches Kristallwachstum in Längsrichtung
setzt erst nach mehreren Stunden wieder ein, wobei die Hauptwachstumsphase zwischen
12 und 24 h beobachtet wird. Die Länge der Kristalle beträgt jetzt ca. 2,5 µm (s.a. 31
und 32).
Bei alkalireichen Portlandzementen wird neben Ettringit zu Reaktionsbeginn zusätzlich
Syngenit (K2SO4 . CaSO4
. H2O) gebildet. Es wurde auch bei einem Zement mit einem
sehr niedrigen Alkaligehalt etwas verzögert eine Syngenitbildung beobachtet (Bild 25
und 26). Die Bedeutung der temporären Syngenitbildung für das Erstarrungsverhalten
bedarf weiterer Untersuchungen.
Nach etwa 5 Stunden Hydratationsdauer zersetzen sich die leistenförmigen
Syngenitkristalle unter Bildung von sekundärem Gips und Kaliumsulfat (Bild 28 und
29). Dieser Vorgang setzt etwa zeitgleich mit der Bildung erster nadelförmiger C-S-H-
Phasen ein und ist am Ende der Beschleunigungsphase abgeschlossen.
Die C-S-H-Phasen bilden sich zu Beginn der Beschleunigungsperiode auf der C3S-
Klinkerphase (Bild 27). Es sind vereinzelt kleine (< 200 nm Länge) stark inhomogen
verteilte C-S-H-Büschel zu beobachten. Im weiteren Hydratationsverlauf (bis zu 24
Stunden) wachsen die C-S-H-Phasen kontinuierlich bis auf eine Länge von 600 nm,
wobei die Nadelspitzen einen Durchmesser von ca. 5 nm aufweisen (Bild 31 und 32).

44
und der langsameren Reaktion des Dicalciumsilikats (C2S) Calciumsilikathydrate (CSH) in
Form von Nadeln oder Leisten. Die sehr feinen Kristalle verwachsen miteinander und bilden
ein Netzwerk. Hierauf beruht die Festigkeitsentwicklung des Zements.
Der Zement bindet etwa 25 M.-% Wasser chemisch, d. h., es wird in das Kristallgitter der
Minerale fest eingebaut. Außer diesem chemisch gebundenen Wasser bindet der Zement noch
etwa 10 bis 15 M.-% Wasser durch Adsorption. Dieses physikalisch gebundene Wasser reagiert
nicht mit unhydratisierten Zementteilchen, sondern bleibt physikalisch an die Oberfläche der
feinen Kristalle gebunden (spezifische Oberfläche etwa 1000mal so groß wie die des Zements!).
Das Wasser in den so genannten Gelporen wird "Gelwasser" genannt. Unter dem Begriff
Zementgel werden die feinen Kristalle und das Gelwasser zusammengefasst. Sie bilden eine
Einheit, weil ein Austreiben des Gelwassers nur durch eine erhöhte Temperatur erfolgen kann
und dies eine irreversible Zerstörung des Zementsteingefüges bedeutet. Die Summe aus
chemisch gebundenem Wasser und Gelwasser, die dem Zement zur vollständigen Hydratation
zur Verfügung stehen muss, beträgt also etwa 35 bis 40 M.-% (entspricht einem
Wasserzement-Wert von 0,35 bis 0,40).
Die Hydratation des Zements ist, wie alle chemischen Reaktionen, von der Temperatur
abhängig. Sie verläuft i. A. in der Wärme schneller und in der Kälte langsamer. Tiefe
Temperaturen können die Reaktion völlig unterbrechen. Wasserentzug (Austrocknung)
unterbricht die Hydratation ebenfalls. Die Hydratation des Zements verläuft exotherm, d. h.
beim Erstarren und Erhärten wird entsprechend dem Reaktionsfortschritt Wärme frei, die so
genannte „Hydratationswärme“.
Wenn fein gemahlener reiner Portlandzementklinker mit Wasser gemischt wird, reagiert das
Tricalciumaluminat unverzüglich mit Wasser und bewirkt dabei ein unerwünschtes zu frühes
Erstarren des Zementleims ("Löffelbinder"). Aus diesem Grund wird vor oder während dem
Mahlen des Zementklinkers Gips oder Anhydrit als Abbinderegler zugesetzt. Wenn Wasser
zugegeben wird, reagieren diese Sulfatträger mit dem Tricalciumaluminat unter Bildung von
Ettringit (= Trisulfat). Mit dieser Reaktion wird das Erstarren des Zementleims verhindert.
Später wandelt sich Ettringit ohne schädliche Nebenwirkungen in Monosulfat um (vgl. Kapitel
4.6.7.1.3).
Bezogen auf die Gefügeentwicklung während des Verfestigungsprozesses unterscheidet man
drei Hydratationsstufen (Bild 34). Nach dem Mischen mit Wasser tritt mit der Bildung von
CSH-Phasen eine erste merkliche Verfestigung auf, die Erstarrungsbeginn genannt wird. Mit
dem Erstarrungsbeginn endet die erste Hydratationsstufe. Während der weiteren Reaktion
bilden sich die Calciumsilikathydrate zunächst langfaserig (2. Hydratationsstufe) und dann
kurzfaserig (3. Hydratationsstufe) aus.
Am Erstarrungsende sind bereits 15 % der Klinkerminerale hydratisiert. Das Erstarren geht in
das Erhärten über, wenn die faserförmigen Kristalle (langfaserige CSH-Phasen) die
wassergefüllten Zwischenräume der Zementkörner überbrücken und sich somit ein stabiles
Grundgefüge ausbildet. Später wachsen die kurzfaserigen CSH-Phasen in die noch
vorhandenen Poren hinein und verdichten so das Grundgefüge.
Die verschiedenen Klinkerminerale reagieren unterschiedlich schnell mit Wasser und bilden
darüber hinaus unterschiedliche Hydratationsprodukte (vgl. Kapitel 4.6.7.1). Daraus folgen
gravierende Unterschiede in der zeitlichen Entwicklung der Druckfestigkeit (Bild 35).

45
Bild 34: Schematische Darstellung der Bildung der Hydratphasen ([1] nach [20-22])
Bild 35: Druckfestigkeitsentwicklung der Klinkerminerale [23]

46
4.6.7.3 Messung der Zementerhärtung
Die fortschreitende Verfestigung des Zements nach dem Anmachen mit Wasser geht über ein
anfängliches Ansteifen und das Erstarren bis zum Erhärten. Aus betontechnologischen Gründen
ist es sehr wichtig, den Verlauf der Verfestigung definiert erfassen zu können. Ein Zement muss
ausreichend lange verarbeitbar sein, muss sich aber auch ausreichend schnell verfestigen
(Entschalen, Belastbarkeit).
Mit Erstarren wird der Übergang des Zementleims vom plastischen in den festen Zustand
bezeichnet. Die Erstarrungszeiten (Beginn und Ende) werden nach DIN EN 196-3 mit dem
Eindringversuch (Vicat-Nadel) bestimmt (Bild 36).
Bild 36: Bestimmung der Erstarrungszeiten mit dem Nadelgerät nach DIN EN 196-3 [24]
4.6.8 Einflüsse auf die Festigkeitsentwicklung
Durch die chemische Zusammensetzung des Zements (die bis auf Zumahlstoffe, der des
Rohmehls entspricht) ist näherungsweise auch die mineralogische Zusammensetzung definiert
(vgl. Kapitel 4.6.4). Dabei wird hier stillschweigend unterstellt, dass der Brennprozess optimal
eingestellt ist und alle Phasen bis zum Gleichgewichtszustand reagieren. Die mineralogische
Zusammensetzung wiederum bestimmt die physikalischen und chemischen Eigenschaften des
Zements. Dazu gehören zum Beispiel die Geschwindigkeit der Reaktion mit Wasser und die
Wärme, die dabei freigesetzt wird. Hinzu kommt, dass durch die Hydratation verschiedener
Minerale natürlich unterschiedliche Reaktionsprodukte und damit auch unterschiedliche
Gefüge entstehen. Die Folgen sind gut zu erkennen, wenn die Festigkeitsentwicklung durch
Hydratation der reinen Klinkerminerale verglichen wird (vgl. Bild 35). Ausschließlich die
Calciumsilikate liefern hohe Druckfestigkeiten. Der entscheidende Unterschied zwischen C3S
und C2S liegt in der Geschwindigkeit der Hydratation und Gefügeentwicklung.
4.6.9 Einfluss der Mahlfeinheit
Je feiner ein Zement gemahlen ist, desto größer ist seine Oberfläche und desto schneller ist
seine Reaktion mit dem Anmachwasser, was sich wiederum in der Festigkeitsentwicklung
ausdrückt.
Die Mahlfeinheit kann sich nachteilig auswirken. Ist sie zu klein, so ist keine völlige
Hydratation möglich. Das an den Außenflächen des Zementkorns gebildete Gel verhindert
nämlich den Zutritt des Wassers zu dem noch nicht hydratisierten Kern des Zementkorns.

47
Deshalb kann bei gröberen Zementkörnern durch den Mangel an Wasser im Korninneren die
Hydratation zum Erliegen kommen. Dagegen können sehr feine Zementkörner nahezu
vollständig hydratisieren. Wenn allerdings die Mahlfeinheit zu groß ist, so hat der Zement einen
größeren Bedarf an Anmachwasser, was die Festigkeit vermindert. Außerdem ist unter diesen
Umständen mit schnellerem und stärkerem Schwinden zu rechnen.
Folgende Anforderungen müssen erfüllt werden:
• Die spezifische Oberfläche nach Blaine muss mindestens 2200 cm²/g betragen (in
Sonderfällen 2000 cm2/g).
Erklärung: Spezifische Oberfläche ist die auf die Masse bezogene Oberfläche einer
verdichteten Zementprobe. Sie wird ermittelt mit dem Luftdurchlässigkeitsverfahren
nach Blaine.
• Der Rückstand auf dem Prüfsiebgewebe 0,2 (Maschenweite 0,2 mm) darf höchstens
3 M.-% betragen.
Bild 37: Einfluss der Mahlfeinheit auf die Festigkeitsentwicklung von Zementklinker [25]
4.6.10 Einflüsse von Zumahlstoffen
Latent hydraulische und puzzolanische Stoffe werden häufig als Zumahlstoffe verwendet.
Hierfür gibt es verschiedene Gründe wirtschaftlicher, technologischer und ökologischer Natur:
• Energieeinsparung (Zementklinkerproduktion erfordert hohe Brenntemperatur),
• weniger Schadgasemission (da weniger Zementklinker produziert werden muss),
• Rohstoffeinsparung (viele Zumahlstoffe sind industrielle Nebenprodukte),
• Deponieeinsparung (Verwertung industrieller Nebenprodukte) und
• mit Zumahlstoffen können bestimmte Zementeigenschaften für bestimmte
Anwendungsfälle gezielt verbessert werden.
Portlandhüttenzement (CEM II/A-S, CEM II/B-S) und Hochofenzement (CEM III) werden
zusammenfassend auch als Hüttenzemente bezeichnet. Sie sind ein Gemisch aus
Portlandzementklinker und granulierter Hochofenschlacke, miteinander vermahlen unter
Zusatz von wenigen Prozenten Gips oder Anhydrit zur Regelung der Erstarrungszeiten.
Portlandhüttenzement enthält 6 bis 35 M.-% Hüttensand, Hochofenzement 36 bis 80 M.-%. Der
Rest zu 100 M.-% ist Portlandzementklinker.

48
Der Hüttensand wird durch das bei der Hydratation der Calciumsilikate frei werdende
Calciumhydroxid angeregt. Die Erhärtung der Hüttenzemente erfolgt mit wachsendem
Hüttensandgehalt zunehmend träger als die von Portlandzement wegen der vergleichsweise
langsameren Reaktion des Hüttensandes. Daher enthalten Hüttenzemente nach 28 Tagen in der
Regel weniger gebundenes Wasser als Portlandzement, haben allerdings meist einen höheren
Anteil gebundenes Wasser im Endzustand. Ihre Endfestigkeit ist in der Regel höher als bei
einem Portlandzement unter vergleichbaren Bedingungen (Bild 38). Um die langsamere
Anfangserhärtung (Bild 39) zu beschleunigen, werden insbesondere Hochofenzemente im
Allgemeinen feiner gemahlen.
Da mit zunehmendem Hüttensandgehalt eine langsamere Erhärtung einhergeht, ist besondere
Sorgfalt bezüglich der Nachbehandlung geboten. Ein frühzeitiges Austrocknen bedeutet das
Ende der Nacherhärtungsphase und sollte verhindert werden. Die so genannte
Nachbehandlungsempfindlichkeit wächst mit dem Hüttensandgehalt.
Bild 38: Vergleich der Festigkeitsentwicklung von Portland- und Hochofenzement [1]
Bild 39: Einfluss von Hüttensand auf die Festigkeitsentwicklung [16]
Wie beim Portlandzement wirkt sich die chemische Zusammensetzung des Klinkers
(ausgedrückt z. B. über den Kalkstandard (vgl. Kapitel 4.6.3)) und der Gehalt an amorpher
Kieselsäure (Glasgehalt) im Hüttensand auf die Festigkeit aus (Bild 40).
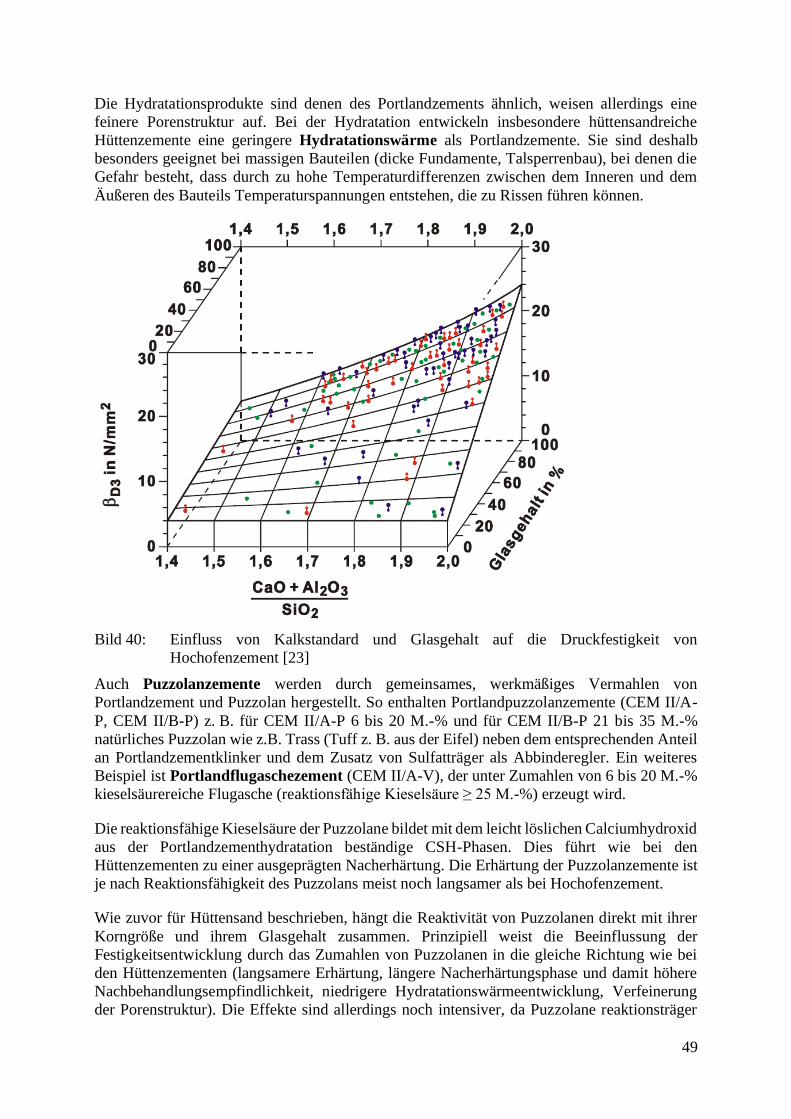
49
Die Hydratationsprodukte sind denen des Portlandzements ähnlich, weisen allerdings eine
feinere Porenstruktur auf. Bei der Hydratation entwickeln insbesondere hüttensandreiche
Hüttenzemente eine geringere Hydratationswärme als Portlandzemente. Sie sind deshalb
besonders geeignet bei massigen Bauteilen (dicke Fundamente, Talsperrenbau), bei denen die
Gefahr besteht, dass durch zu hohe Temperaturdifferenzen zwischen dem Inneren und dem
Äußeren des Bauteils Temperaturspannungen entstehen, die zu Rissen führen können.
Bild 40: Einfluss von Kalkstandard und Glasgehalt auf die Druckfestigkeit von
Hochofenzement [23]
Auch Puzzolanzemente werden durch gemeinsames, werkmäßiges Vermahlen von
Portlandzement und Puzzolan hergestellt. So enthalten Portlandpuzzolanzemente (CEM II/A-
P, CEM II/B-P) z. B. für CEM II/A-P 6 bis 20 M.-% und für CEM II/B-P 21 bis 35 M.-%
natürliches Puzzolan wie z.B. Trass (Tuff z. B. aus der Eifel) neben dem entsprechenden Anteil
an Portlandzementklinker und dem Zusatz von Sulfatträger als Abbinderegler. Ein weiteres
Beispiel ist Portlandflugaschezement (CEM II/A-V), der unter Zumahlen von 6 bis 20 M.-%
kieselsäurereiche Flugasche (reaktionsfähige Kieselsäure ≥ 25 M.-%) erzeugt wird.
Die reaktionsfähige Kieselsäure der Puzzolane bildet mit dem leicht löslichen Calciumhydroxid
aus der Portlandzementhydratation beständige CSH-Phasen. Dies führt wie bei den
Hüttenzementen zu einer ausgeprägten Nacherhärtung. Die Erhärtung der Puzzolanzemente ist
je nach Reaktionsfähigkeit des Puzzolans meist noch langsamer als bei Hochofenzement.
Wie zuvor für Hüttensand beschrieben, hängt die Reaktivität von Puzzolanen direkt mit ihrer
Korngröße und ihrem Glasgehalt zusammen. Prinzipiell weist die Beeinflussung der
Festigkeitsentwicklung durch das Zumahlen von Puzzolanen in die gleiche Richtung wie bei
den Hüttenzementen (langsamere Erhärtung, längere Nacherhärtungsphase und damit höhere
Nachbehandlungsempfindlichkeit, niedrigere Hydratationswärmeentwicklung, Verfeinerung
der Porenstruktur). Die Effekte sind allerdings noch intensiver, da Puzzolane reaktionsträger

50
sind als latent hydraulische Stoffe. Dies ist darauf zurückzuführen, dass Puzzolane aufgrund
ihres geringen CaO-Gehaltes keine eigene Hydraulizität besitzen.
Bild 41: Festigkeitsbeitrag von Flugasche [26]
Bild 42: Hydraulizität (D: Zumahlstoffe auf dem deutschen Markt) [27]

51
4.6.11 Genormte und bauaufsichtlich zugelassene Zemente
Die meisten Zemente sind in den Normen DIN 1164-10 bis -12 [28-30] und DIN EN 197-1 [31]
genormt. Hinzu kommen Sonderzement mit sehr niedriger Hydratationswärme in DIN EN
14216 [32] und bauaufsichtlich zugelassene Zemente. Die Zemente werden unterschieden nach
• Art,
• Festigkeitsklasse und
• durch Angabe besonderer Eigenschaften.
Die Unterscheidung der Zementart erfolgt anhand der Hauptbestandteile. Als
Hauptbestandteil werden Anteile mit mehr als 5 % der Gesamtsumme aller Haupt- und
Nebenbestandteile verstanden. Nebenbestandteile des Zements sind anorganische
mineralische Stoffe, die bei der Klinkerherstellung entstehen, oder hierfür als Ausgangsstoff
eingesetzt werden und nicht als Hauptbestandteil im Zement enthalten sind. Der Anteil aller
Nebenbestandteile darf nicht mehr als 5 % der Gesamtsumme aller Haupt- und
Nebenbestandteile betragen. Die genormten Zemente enthalten die folgenden
Hauptbestandteile:
• Portlandzementklinker (K),
• Hüttensand (granulierte Hochofenschlacke (granulated blast furnace slag)) (S),
• Natürliches Puzzolan (P) wie z. B. Trass,
• Natürliches getempertes Puzzolan (Q): Dies sind thermisch aktivierte Gesteine
vulkanischen Ursprungs, Tone, Schiefer oder andere Sedimentgesteine.
• Kieselsäurereiche Flugasche (cendre volnate) (V): Sie besteht im Wesentlichen aus
reaktionsfähigem Siliciumdioxid (SiO2) und Aluminiumoxid (Al2O3) und hat
puzzolanische Eigenschaften.
• Kalkreiche Flugasche (W): Sie besteht im Wesentlichen aus reaktionsfähigem
Calciumoxid (CaO), reaktionsfähigem Siliciumdioxid (SiO2) und Aluminiumoxid
(Al2O3) und hat latent hydraulische und/oder puzzolanische Eigenschaften.
• Gebrannter Schiefer (burnt shale) (T): Bei ca. 800 °C gebrannter Schiefer,
insbesondere gebrannter Ölschiefer, weist in fein gemahlenem Zustand ausgeprägte
hydraulische Eigenschaften wie Portlandzement und daneben puzzolanische
Eigenschaften auf.
• Kalkstein (limestone) (L, LL): Kalksteinmehl, das als Hauptbestandteil für Zement
eingesetzt wird, muss folgende Anforderungen erfüllen:
o CaCO3-Gehalt ≥ 75 M.-%
o Gehalt an organischem Kohlenstoff (TOC):
▪ LL: TOC ≤ 0,20 M.-%,
▪ L: TOC ≤ 0,50 M.-%.
• Silicastaub (D) entsteht bei der Reduktion von hoch reinem Quarz mit Kohle in
Lichtbogenöfen bei der Herstellung von Silicium- und Ferrosiliciumlegierungen und
besteht aus sehr feinen kugeligen Partikeln mit einem Gehalt an amorphem
Siliciumdioxid von mindestens 85 %.
Die Zementfestigkeit wird durch die Klassen (22,5), 32,5, 42,5 oder 52,5 gekennzeichnet. Sie
bezeichnet die Mindestdruckfestigkeit nach 28 Tagen. Als Hinweis auf die Anfangsfestigkeit
ist der Buchstabe N (normale Anfangsfestigkeit), der Buchstabe R (hohe Anfangsfestigkeit)
oder der Buchstabe L (langsame Anfangsfestigkeit) hinzuzufügen.
Die Bezeichnung von Normalzementen mit niedriger Hydratationswärme erfolgt durch
Anhängen von -LH. Die Hydratationswärme von Normalzementen LH mit niedriger

52
Hydratationswärme nach DIN EN 197-1 darf 270 J/g Zement nicht überschreiten. Die Prüfung
erfolgt entweder nach DIN EN 196-8 über 7 Tage oder nach DIN EN 196-9 über 41 h.
Durch Kombination der Hauptbestandteile entstehen die Produkte der Familie der
Normalzemente nach EN 197-1. Sie sind mit ihren Bezeichnungen in Tabelle 13 angegeben.
Es werden folgende fünf Hauptzementarten unterteilt:
• CEM I Portlandzement,
• CEM II Portlandkompositzement ,
• CEM III Hochofenzement,
• CEM IV Puzzolanzement sowie
• CEM V Kompositzement.
Tabelle 13: Zusammensetzung der 27 Normalzemente nach DIN EN 197-1 [31]
Diese Zemente werden aus ökologischen Gründen ergänzt durch Zemente nach DIN EN 197-5
[33], die einen weiter reduziertem Klinkergehalt aufweisen: Portlandkompositzement CEM
II/C-M und Kompositzement CEM VI (Tabelle 14).
Tabelle 14: Zusammensetzung der Portlandkompositzement CEM II/C-M und
Kompositzement CEM VI nach DIN EN 197-5 [33]
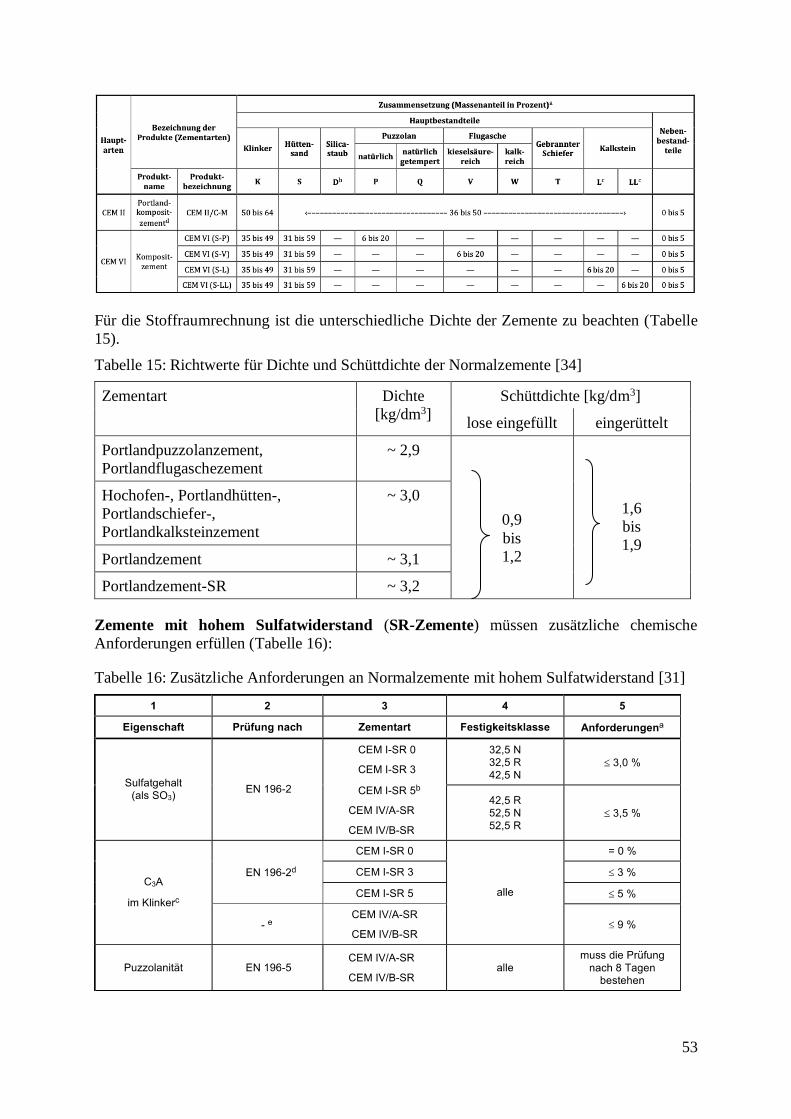
53
Für die Stoffraumrechnung ist die unterschiedliche Dichte der Zemente zu beachten (Tabelle
15).
Tabelle 15: Richtwerte für Dichte und Schüttdichte der Normalzemente [34]
Zementart Dichte
[kg/dm3]
Schüttdichte [kg/dm3]
lose eingefüllt eingerüttelt
Portlandpuzzolanzement,
Portlandflugaschezement
~ 2,9
0,9
bis
1,2
1,6
bis
1,9
Hochofen-, Portlandhütten-,
Portlandschiefer-,
Portlandkalksteinzement
~ 3,0
Portlandzement ~ 3,1
Portlandzement-SR ~ 3,2
Zemente mit hohem Sulfatwiderstand (SR-Zemente) müssen zusätzliche chemische
Anforderungen erfüllen (Tabelle 16):
Tabelle 16: Zusätzliche Anforderungen an Normalzemente mit hohem Sulfatwiderstand [31]

54
Neben den Normalzementen nach DIN EN 197-1 gibt es Zemente mit besonderen
Eigenschaften nach DIN 1164. Für sie gelten von den Anforderungen in DIN EN 197
abweichende oder zusätzliche Eigenschaften, die in der Bezeichnung des Zements angegeben
werden müssen. Zu diesen Zementen zählen:
• Zement mit niedrigem wirksamen Alkaligehalt – NA nach DIN 1164-10 [28],
• Zement mit verkürztem Erstarren – FE und – SE nach DIN 1164-11 [29] und
• Zement mit einem erhöhten Anteil an organischen Bestandteilen – HO nach DIN 1164-
12 [30].
Zum Vermeiden einer betonschädlichen Alkalireaktion kann es aufgrund der Eigenschaften
bestimmter Gesteinskörnungen in einigen Bundesländern erforderlich sein, in bestimmten
Anwendungsfällen für die Betonherstellung Zemente mit einem niedrigen wirksamen
Alkaligehalt (NA-Zemente) zu verwenden. Der Gesamtalkaligehalt wird als Na2O-Äquivalent
nach folgender Formel berechnet (Angaben [M.-%]):
Na2O-Äquivalent = Na2O + 0,658 · K2O
Tabelle 17: Zusätzliche Anforderungen an NA-Zemente [34]
Zementart Anforderungen an das Na2O Äquivalent [M.-%]
CEM I bis CEM V ≤ 0,60
Darüber hinaus gilt für folgende Zemente die Anforderung:
CEM II/B-S ≤ 0,70
CEM III/A 1) ≤ 0,95
CEM III/A 2) ≤ 1,10
CEM III/B ≤ 2,00
CEM III/C 1) Gilt bei Hüttensandgehalt ≤ 49 M.-%. 2) Gilt bei Hüttensandgehalt ≥ 50 M.-%.
Zemente mit frühem Erstarren (FE-Zemente) sind durch einen frühen Erstarrungsbeginn
gekennzeichnet. Sie ermöglichen bei entsprechend kurzen Misch-, Transport- und
Verarbeitungszeiten die Herstellung von Beton nach DIN EN 206-1 [35] in Verbindung mit
DIN 1045-2 [36] z. B. für Betonfertigteile [34].
Bei Zementen mit schnellem Erstarren (SE-Zemente)wird der Erstarrungsbeginn nach weniger
als 45 Minuten erreicht. Sie sind für die normale Betonherstellung nicht geeignet. Ihre
Anwendung beschränkt sich auf spezielle Herstellverfahren wie z. B. Trockenspritzbeton. Sie
sind zurzeit nur als Portlandzement (CEM I) zugelassen [34].
Zement mit erhöhtem Anteil organischer Bestandteile (HO-Zemente) dürfen abweichend von
der DIN EN 197-1 maximal 1 M.-% organische Bestandteile enthalten. Sie enthalten stark
verflüssigend wirkende Zusätze, die die Konsistenz des dazu hergestellten Zementleims
verändern [34].
Neben diesen Normalzementen gibt es Sonderzemente. So ist Zement mit sehr niedriger
Hydratationswärme (very low heat) nach DIN EN 14216 [32] in der Festigkeitsklasse 22,5
(Tabelle 18) genormt. Die Hydratationswärme dieser Zemente darf 220 J/g nicht überschreiten.
Die Prüfung erfolgt entweder nach DIN EN 196-8 über 7 Tage oder nach DIN EN 196-9 über
41 h.

55
Die Bezeichnung eines solchen Sonderzements zeigt das nachfolgende Beispiel:
Sonder-Puzzolanzement mit sehr niedriger Hydratationswärme
EN 14216 - VLH IV/B (P) 22,5
Tabelle 18: Sonderzemente mit sehr niedriger Hydratationswärme nach DIN EN 14216 [32]
Sulfathüttenzement ist ein hüttensandhaltiger Zement, dessen Reaktion auf einer sulfatischen
Anregung beruht. Früher waren solche Zemente in Deutschland genormt, zwischenzeitlich aber
seit den 70er Jahren für Konstruktionsbeton nicht mehr zugelassen. Für Sulfathütttenzement
gilt DIN EN 15743 [37].
Straßenbauzement wird zum Herstellen von Fahrbahndecken nach ZTV Beton StB
verwendet. Dieser Zement erfüllt die entsprechenden Anforderungen an Feinheit,
Wasseranspruch Erstarren, Frühfestigkeit und Alkaligehalt [34].
Weißzement ist ein Zement nach DIN EN 197-1 [31] der für helle bis weiße Betone eingesetzt
wird.
Hydrophobierter (wasserabstoßender) Zement ist genormt nach DIN EN 197-1 [31].

56
5 Literatur [1] Schießl, P., Bindemittel. Skriptum zur Grundvorlesung, Lehrstuhl für Baustoffkunde
und Werkstoffprüfung, Editor. 2003: München. p. 54.
[2] Bibliographisches Institut & F.A. Brockhaus AG, PC-Bibliothek - Der Brockhaus in
Text und Bild Version 3.0, in Brockhaus 2002.
[3] Krenkler, K., Chemie des Bauwesens - Band 1: Anorganische Chemie. 1980, Berlin,
Heidelberg: Springer-Verlag XX, 494 p.
[4] Wikibooks: Tabellensammlung Chemie/ Atom- und Ionenradien. 2020 28. März 2020
[cited 2020 2020/04/03]; Available from:
http://de.wikibooks.org/wiki/Tabellensammlung_Chemie/_Atom-_und_Ionenradien.
[5] Reininger, G.; Schubert, V., Allgemeine und Anorganische Chemie - Vorlesungsskript
Universität Paderborn. 2006: Paderborn.
[6] GIPS-Datenbuch, ed. Bundesverband der Gipsindustrie e. V. 2013, Berlin:
Bundesverband der Gipsindustrie e. V.,. 108 p.
[7] BMUB, Klimaschutzplan 2050 – Klimaschutzpolitische Grundsätze und Ziele der
Bundesregierung, ed. Bundesministerium für Umwelt Naturschutz
und nukleare Sicherheit. 2016. 92 p.
[8] Bundesministerium für Wirtschaft und Energie (BMWi), Kommission „Wachstum,
Strukturwandel und Beschäftigung“ - Abschlussbericht. 2019, Berlin:
Bundesministerium für Wirtschaft und Energie (BMWi). 278 p.
[9] DIN EN 459-1. Baukalk – Teil 1: Begriffe, Anforderungen und Konformitatskriterien
(Building lime – Part 1: Definitions, specifications and conformity criteria). 2015, p.
51.
[10] Locher, F.W., Zement: Grundlagen der Herstellung und Verwendung. 2000,
Düsseldorf: Verlag Bau und Technik. 522 p.
[11] Basten, M.; Bröker, H.; Eber, B.; Hilger, J.; Lütkehaus, M.; Reimer, T.; Rosemann, H.;
Schauer, M.; Sprung, S. and Wertel, C., Zementrohstoffe in Deutschland - Geologie,
Massenbilanz, Fallbeispiele, ed. Bundesverband der Deutschen Zementindustrie e. V.
2002, Düsseldorf: Verlag Bau + Technik GmbH. 64 p.
[12] VDZ, Zement Taschenbuch. 50 ed. 2002, Düsseldorf: Verlag Bau+Technik. 792 p.
[13] Keil, F., Zement. 1971: Springer Berlin Heidelberg p.
[14] Achternbosch, M.; Bräutigam, K.-R., Herstellung von Zementklinker
Verfahrensbeschreibung und Analysen zum Einsatz von Sekundärbrennstoffen.
Technik und Umwelt, ed. Karlsruhe, F., 2000, Karlsruhe, Germany: Institut für
Technikfolgenabschätzung und Systemanalyse p.
[15] Wolter, A., Einfluss des Ofensystems auf die Klinkereigenschaften. ZKG International,
1985. 38(10): pp. 612-614.
[16] Taylor, H.F.W., Cement Chemistry. 2. ed. 1997, London: Thomas Telford Ltd. 459 p.
[17] Stark, J.; Wicht, B., Zement und Kalk: Der Baustoff als Werkstoff. BauPraxis. 2000,
Weimar: Birkhäuser Verlag. XI, 376 p.
[18] Stark, J.; Wicht, B. and Eckart, A., Neue Ansätze zur Zementhydratation (1/2). ZKG
international, 2001(1): pp. 52-60.
[19] Stark, J.; Wicht, B. and Eckart, A., Neue Ansätze zur Zementhydratation (2/2). ZKG
international, 2001(2): pp. 114-119.
[20] Richartz, W., Über die Gefüge- und Festigkeitsentwicklung des Zementsteins (1/2).
Beton, 1969. 19(5): pp. 203-206.
[21] Richartz, W., Über die Gefüge- und Festigkeitsentwicklung des Zementsteins (2/2).
Beton, 1969. 19(6): pp. 245-248.
[22] Locher, F.W.; Richartz, W. and Sprung, S., Erstarren von Zement; Teil 1: Reaktion und
Gefügeentwicklung. ZKG International, 1976. 29(10): pp. 435-442.

57
[23] Lerch, W.; Bogue, R.H., Heat of hydration of portland cement pastes. Bureau of
Standards Journal of Research, 1934. 12: pp. 645-654.
[24] DIN EN 196-3. Prüfverfahren für Zement - Teil 3: Bestimmung der Erstarrungszeiten
und der Raumbeständigkeit (Methods of testing cement - Part 3: Determination of
setting times and soundness). 2009, p. 17.
[25] Locher, F.W.; Wuhrer, J. and Schweden, K., Einfluß der Mahlfeinheit und der
Kornverteilung auf die Eigenschaften von Portland- und Hüttenzementen sowie von
hydraulischen Kalken. Tonindustrie-Zeitung, 1966. 90(12): pp. 547-554.
[26] Schießl, P., Wirkung von Steinkohlenflugaschen im Beton. Beton, 1990. 40(12): pp. 519-
523.
[27] Wesche, K., Baustoffe für tragende Bauteile - Band 2: Beton, Mauerwerk
(Nichtmetallisch-anorganische Stoffe): Herstellung, Eigenschaften, Verwendung,
Dauerhaftigkeit. 3. ed. Vol. 2. 1993, Wiesbaden: Bauverlag. XXII, 502 p.
[28] DIN 1164-10. Zement mit besonderen Eigenschaften – Teil 10: Zusammensetzung,
Anforderungen und Ubereinstimmungsnachweis von Zement mit niedrigem wirksamen
Alkaligehalt (Special cement – Part 10: Composition, requirements and conformity
evaluation for cement with low effective alkali content). 2013, p. 10.
[29] DIN 1164-11. Zement mit besonderen Eigenschaften - Teil 11: Zusammensetzung,
Anforderungen und übereinstimmungsnachweis von Zement mit verkürztem Erstarren
(Special cement - Part 11: Composition, Specification and conformity evaluation for
cement with short setting time). 2003, p. 16.
[30] DIN 1164-12. Zement mit besonderen Eigenschaften – Teil 12: Zusammensetzung,
Anforderungen und Ubereinstimmungsnachweis von Zement mit einem erhohten Anteil
an organischen Bestandteilen (Special cement – Part 12: Composition, specification
and conformity evaluation for cement with higher quantity of organic constituents).
2005, p. 7.
[31] DIN EN 197-1. Zement - Teil 1: Zusammensetzung, Anforderungen und
Konformitätskriterien von Normalzement (Cement - Part 1: Composition,
specifications and conformity criteria for common cements). 2011, p. 8.
[32] DIN EN 14216. Zement – Zusammensetzung, Anforderungen und Konformitatskriterien
von Sonderzement mit sehr niedriger Hydratationswarme (Cement – Composition,
specifications and conformity criteria for very low heat special cements). 2019, p. 24.
[33] DIN EN 197-5. Zement - Teil 5: Portlandkompositzement CEM II/C-M und
Kompositzement CEM VI (Cement - Part 5: Portland-composite cement CEM II/C-M
and Composite cement CEM VI). 2021, p. 12.
[34] Bosold, D.; Pickhardt, R. Zemente und ihre Herstellung. 2017, p. 9.
[35] DIN EN 206-1. Beton - Teil 1: Festlegung, Eigenschaften, Herstellung und
Konformität. 2001.
[36] DIN 1045-2. Tragwerke aus Beton, Stahlbeton und Spannbeton – Teil 2: Beton –
Festlegung, Eigenschaften, Herstellung und Konformität – Anwendungsregeln zu DIN
EN 206-1 (Concrete, reinforced and prestressed concrete structures – Part 2: Concrete
– Specification, properties, production and conformity – Application rules for DIN EN
206-1). 2008, p. 62.
[37] DIN EN 15743. Sulfathuttenzement – Zusammensetzung, Anforderungen und
Konformitatskriterien (Supersulfated cement – Composition, specifications and
conformity criteria). 2015, p. 27.
Recommended

















