Embed Size (px)
Citation preview

ZOa-Hydroxysteroid Dehydrogenase of Porcine Testes
PURIFICATION AND PROPERTIES
(Received for publication, August 2, 1971)
FUMIKO SATO, YOSHINARI TAKAGI, AND MIKIO SHIKITA
Frona the National Institute of Radiological Sciences, Chiba-shi, Japan
SUMMARY
A soluble ZOa+hydroxysteroid dehydrogenase was found in porcine testes. The enzyme content was roughly propor- tional to the wet weight of the gland, when glands weighing 160 to 440 g were examined. The enzyme has been purified to near homogeneity by a six-step procedure. The molecular weight of the enzyme was estimated to be about 35,000 by gel filtration methods. The mobility of the enzyme protein in sodium dodecyl sulfate polyacrylamide gel electrophoresis also corresponded to this molecular weight, suggesting that the enzyme has no apparent subunit structure. The purified enzyme sedimented as a symmetrical boundary with a sedimentation coefficient of 2.9. Sucrose gradient centrifu- gation gave a similar sedimentation coefficient for the en- zyme.
The enzyme catalyzes the reduction of 17a-hydroxyproges- terone to 17cr,ZOar-dihydroxypregn-4-en-3-one with oxida- tion of an equimolar amount of NADPH. Apparently the enzyme has a dual pyridine nucleotide specificity although the Michaelis constant for NADH is 14 to 50 times larger than that for NADPH. The enzyme exhibits pH optimum between 5.5 and 7.5 both for the NADH- and NADPH- linked reactions, and its isoelectric point is pH 5.2.
Progesterone, corticosterone, and 17oc-hydroxycorticos- terone were much less reactive than 17ol-hydroxyproges- terone as hydrogen acceptors in the ZOa-hydroxysteroid dehydrogenase reaction. The enzyme had no 17P-hydroxy- steroid dehydrogenase activity toward estrone. The maxi- mum rate of the enzymatic reduction of 17a-hydroxyproges- terone was observed at 50”, while the enzyme was rapidly inactivated at temperatures higher than 55”. The enzyme was stable at -ZOO, but unstable at 25”. Partial restoration of the original activity could be achieved by addition of cysteine and other su.lfhydryl compounds. p-Chloromer- curibenzoate, heavy metal ions, and pyridoxal 5’-phosphate were strong inhibitors, while albumin and EDTA had no influence upon the enzymatic activity.
Previous reports from this laboratory (I, 2) have described the properties of the 20a-hydroxysteroid dehydrogenase of rat testes. Similar 20a-hydroxysteroid dehydrogenases seems to be present in the testes of other mammals (I). This paper concerns the puri-
fication and characterization of t’he 20a-hydroxysteroid dehydro- genase in porcine testes. The purified porcine testicular 20oL-hy- droxysteroid dehydrogenase exhibits a selective substrate speci- ficity toward 17or-hydroxyprogesterone and a dual specificity for NADPH and NBDH, being quite different from the rat ovarian 20a-hydroxysteroid dehydrogenase which specifically cat,alyzes the reversible reduction of progesterone only in the presence of NrlDPH (3, 4). A preliminary account of the porcine testicular 20oc-hydroxyst’eroid dehydrogenase and identification of the re- action product, 17a(, 20a-dihydroxypregn -4. en- 3 -one, has been reported (5).
EXPERIMENTAL PROCEDURE
Materials-[4J4C]Progesterone and 17a-hydroxy[4-14C]proges- terone were purchased from the Radiochemical Centre, Amersham. These radioactive steroids were diluted with respective cold ste- roids and purified by recrystallizations. The purity of the ste- roids was confirmed by thin layer chromatography and aubo- radiography of the chromatograms. The steroids were dissolved in absolute methanol to a concentration of 151 nmoles and about 10,000 cpm per 20 ~1. p-Chloromercuribenzoate was purified by repeated precipitation with 1 N HCl. P-Mercaptoethyl[14Cj- guanidine was prepared by the intramolecular rearrangement re- action of 2-aminoethyl[14C]isothiuronium bromide hydrobromide which was synthesized from [14C]thiourea and 2-bromoethylamine HBr. Pyridine nucleotide cofactors, NADPII and NADIT, were purchased from Sigma Chemical Co. DEhE-cellulose (Whatman DE-32) was washed successively with 0.5 N NaOH, H20, 0.5 N HCl, 0.5 N NaOH, and Hz0 in the conventional wag, and kept suspended in 5 m&T phosphate buffer (pH 7.0) containing 1 rnM EDTA. Hydroxylapatite was purchased from Serva (Heidel- berg) and washed with II,0 and 5 rnlnf phosphate buffer (pH 7.0). A saturated solution of ammonium sulfate was neutralized with NH40H and stored at 0”; in the experiments described below the degree of saturation was based upon the value at 0”. Organic solvents were dried and distilled prior to use. Water was de- ionized and then redistilled. The reference proteins used in the gel filtration, sodium dodecyl sulfate polyacrylamide gel electro- phoresis, and in the sucrose density gradient centrifugation experiments were all obtained commercially and employed with- out purification. Porcine testes were obtained from a slaughter house and kept in a deep freezer (-20’) for several months be- fore use.
Assay of WOa-Hydroxysteroid Dehydrogenase-Unless otherwise specified, t,he enzyme was incubated with 151 nmoles (50 pg) of 170c-hydroxy[4m14C]progesterone in a total volume of 1 ml of 0.1
815
by guest on September 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

816 %Ooc-Hydrox ysteroid Dehydrogenase of Testes Vol. 247, No. 3
- SOLVENT FRONT
+S 40mm
+-P
7mm - STARTING LI NE
(t20 m’m FIG. 1. Separation of the reaction product 17or,20cu-dihydroxy-
[4-1*C]pregn-4-en-3-orle (P) from the substrate 17a-hydroxy[4-ldC]- progesterone (S) by chromatography on a small silica gel thin laver Dlate. Right, photograph of the spots of the steroids nnder ultra;iolet light; Zefl, the autoradiogram of the same thin layer chromatograph pl>Lte. The amount of radioactivity determined by the liquid scint.illation spectrometer was 3910 cpm and 2770 cpm for S and P, respectively.
M phosphate buffer (1111 7.0) containing 10 IYIM cysteine and 0.5 lllM NADPH. The enzymatic reaction was initiated by the addi- tion of 20 ~1 of a methnnolic solution of the steroid, allowed to pro- ceed for 30 min at 37”, and then terminated by extraction of the steroid with an esccss of CH&&. Recovery of the radioactivity was around SOO,:,
The CH2C12 extract was concentrated to dryness under vacuum and an aliquot was chromatographed on silica gel thin layer (Silica f, plastic basis, Tokyo-Kasei Co.). The size of ea,ch chromato- gram plate was 20 x 50 mm. The chromatograms were run twice in the same direction with a mixture of CHCls-ethyl ace- tate-ethanol (25:2: I, v/v). Fig. 1 shows the separation of the reaction product 170(, 20a-dihydroxypregn-4-en-3-one from the substrate 17cu-hytlroxyprogesterone by this chromatography. Spot.s of these steroids were tlet’ected under ultraviolet light and separated from each other by cutting the chromatogram with scissors.
Radioact’ivity was c’ctermined with a liquid scintillation spec- trometer (Nuclear Chicago Corp., Mark I) by placing the chro- matogram strip on the bottom ol a vial containing 10 ml of toluene-2,5 - diphenyloxazole (0.4vi)-1,4-bis[2-(5-phenyloxazo- lyl)]benzene (0.01 o/). The apparent radioactivity g;l,adually increased by 20 to 3094, reaching a plateau in 2 hours. If the chromatogram strip was placed upside down in the counting vial, the apparent radioactivity decreased by 5 to 6%. The efficiency of the radioactivity determination was approximately 65yk. Sufficient counts were accumulated to determine the radioac- tivity with an error of less than 50/;. Background counts from the vial containing the chromatogram strip of the size similar to that of the steroid spots were 20 to 50 cpm. The recovery of the steroid was calculated from the percentage distribution of radio- activity among the chromatogram spots and the amount of the substrate steroid added initially.
The same procedure as described above for the NADPH-linked 20cr-hydroxysteroid dehydrogenase was employed also for the assay of the NADH-linked 20ol-hydroxysteroid dehydrogenase. In the routine assay, the final concentrat,ion of NADH was 1 .O mu.
Enzyme Units-The amount of the enzyme reducing 1. nmole of 17a-hydroxyprogesterone per min under t’he conditions of assay described above was convent,ionally defined as 1 unit of 2Oa-hy- droxysteroid dehydrogenase activity. The specific activity was expressed as units per mg of protein.
Determinations-‘l’roteii~s were estimated by t’he method of
Lowry et ul. (6) with crystalline bovine serum albumin as a stand- ard. In monitoring column chromatography effluents, the light absorbance at 280 nm was taken as a measure of the protein con- centration. Phosphorous concentrations were measured by the method of Allen (7). A molar absorbance of 6220 at 340 nm was used to calculate NADPH and NADH concentrations. The pH of solutions was determined with a glass electrode (Toa Denpa, model HM-5A). Lactate dehydrogenase activity was assayed by the rate of oxidation of NADH (66 MM) in the presence of pyruvic acid (330 pM) in phosphate buffer (30 mM, pH 7.4). Alcohol de- hydrogenase activity was assayed by the rate of reduction of NAIY (7.5 mM) with ethanol (400 rn$ in pyrophosphate buffer (16 mM, pH 8.8). The reverse reaction of alcohol dehydrogenase was assayed by incubation of NADH (110 PM) with acetaldehyde (830 PM) in Tris-chloride buffer (0.1 M, pH 8.0). Peroxidase ac- tivity was assayed by the method described in the manual supplied by Worthington Biochemical Corp. (8). For these spectrophoto- metric enzyme assays, the absorbance of each sample was recorded at 25” in a Cary model 14 recording spectrophotometer.
Chromatography-A peristaltic pump (Perpex, LKB-Produker AR) was used for the control of the flow rate of column chromatog- raphy. Sephadex G-150 gel filtration was run by reverse flow. The void volume of the Sephades and Bio-Gel columns was meas- ured with Blue Dextran 2000 (Pharmacia Fine Chemicals).
EEectrophoresis-Discontinuous polyacrylamide gel electropho- resis was performed by the method of Davis (9). The gels were prepared with 7.50/, monomer concentration in the Tris-chloride buffer (pH 8.9), but the arnount of persulfate catalyst used was reduced to 35 mg per ml which was one-half the concentration rec- ommended by the original author (9). The enzyme solution was mixed with the same volume of a 50% sucrose solution containing 50 IIIM P-mercaptoethanol and 50 ~1 of this was placed on top of the gels. The proteins were electrophoretically treated at 5-10” at a current of 2 ma per tube and with the Tris-glycine buffer (pH 8.3) containing 50 mM P-mercaptoethanol. Electrophoresis was stopped when the tracking dye (bromphenol blue) traveled a dis- tance of 55 mm in the running gel. The gels were stored wrapped in Saran Wrap at 4” for about 22 hours, while one of the gels was stained for proteins and then quickly destained. The unstained gels were cut into slices 3 to 5 mm thick so that the protein bands of the corresponding stained gel were not cut in two. Each slice was homogenized in I .O ml of 0.1 111 phosphate buffer (pH 7.0) with a glass-Teflon homogenizer and assayed for 20ol-hydroxy- steroid dehydrogenase activity. The enzyme protein was also electrophoretically treated by the standard method in the absence of fi-mercaptoethanol. Sodium dodecyl sulfate polyacrylamide gel electrophoresis was done as described by Weber and &born (10). The glass gel tubes used for this electrophoresis were 10 cm long according to the instructions by the original authors. All of the gels were stained for protein with 1 y. Amido schwarz in 7.5% acetic acid. Isoelectric fractionation was carried out with the LKB 8101 Ampholine electrofocusing equipment by the method of Vesterberg and Svensson (11). In a separate experiment, it was shown that carrier ampholyte did not interfere with the 20a-hy- drosysteroid dehydrogenase assay.
Ceentrij~ation-A Tominaga model 90 centrifuge (rotor 9) and an International Preparative Ultracentrifuge (model B-60; rotor A-170) were used for the preparation of the enzyme. These pre- parative centrifuges were operated at 0”.
The sedimentation velocity of the purified enzyme was deter- mined with a Beckman-Spinco model E analytical centrifuge
by guest on September 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Issue of February 10, 1972 F. Sato, Y. Takagi, and M. Xhilcita s17
equipped with schlieren optics. Centrifugation was done at 59,780 rpm in the analyt.ical rotor An-D at 20” and pictures were taken at 0, 32, 64, 96, and 128 min after the maximal velocity was attained in the centrifuge. The sedimentation coefficient for each protein concentration was obtained from an average of four deter- minations at the 32-min intervals.
Sucrose density gradient centrifugation was performed accord- ing to Martin and Ames (12). Linear gradients with sucrose con- centrations from 5 to 20%) in 0.1 M phosphate buffer (pH 7.0) were established in 5-ml cellulose acetate butyrate tubes. The gra- dients were centrifuged for 19.3 hours at 4” and at 37,000 rpm in an SW 40 rotor in a Hitachi model 55 1’ ultracentrifuge.
Other Procedures-The enzyme solution was concentrated by ultrafiltration with Sartorius collodion bags (Gmbh). Auto- radiograms were taken of the thin layer chromatograms by placing the x-ray film and the chromatograms between two sheets of glass and storing these plates in the dark for 1 or 2 weeks. A digital computer (Tosbac 3400 model 31) was used for the least squares analysis of a linear relationship between two parameters. In all cases reported below, the criteria for the linearity depended on a correlation coefficient of 0.95 or above.
RESULTS
Assay o.f Enzyme
Reaction Product--When the initial homogenate of the tissue (7000 x g supcrnatant) was incubated with 17a-hydroxy[4-14C]- progesterone, several radioactive products other than 17~) 2001- dihydrosyprcgn-4-en-3-one vvere produced in minor amounts. However, after the ammonium sulfate fractionation stage of the purification, 17~~) 20a-dihydrosylpregn-4-en-3-one leas the role product of the enzyme reaction. The purity and identification of this reaction product was established as reported previously (5), by its melting point (207-alOo), color reactions, chemical modifica- tion reactions, absorption spect,ra in methauol and in concentrated sulfuric acid, chromatographic mobilities, and recrystallizations with authentic steroid.
Kinetics and Xtoichiometry-Under the conditions of the assay as described above, reduction of the substrate steroid was linear with time and enzyme concentration, n-hen the enzyme was added in amounts sufficient to permit 5 to 405-i conversion of t.he steroid in the incubation of 30 min.
The stoichiometry of the 20a-hydroxysteroid dchydrogenase reaction between NADPH and steroid a-as determined by the spectrophotometric recording of the XADPH oxidation and by radioactive measurement of 17a, 20cu-dihydroxypregn-4en-3-one obtained at the end of the reaction. In the beginning, the mix- tures contained 151 mpmoles of 17cu-hydroxy[4-Wlprogesterone and 209 mpmoles of NADPH in each 3.0 ml of 0.1 M phosphate buffer (pI-I 7.0) containing 10 mM P-mercaptoethylguanidine. In the reaction of 42 min with 2 units of the enzyme, 38 and 37 nmoles of NADPH and steroid were reacted, respectively. With 1 unit of the enzyme, 28 nmoles of each of them was reacted in 65 min.
Solvent jar Steroid--‘t%e final concentration of methanol in the reaction mixture was 2$; in the routine assay system. Change of the methanol concentration from 1 to 45; had no effect on the rate of the enzyme reaction. When propylene plycol, having a final concentration of 2.5O4, was used to dissolve the substrate steroid (2), the rate of enzyme reaction was 60 to 70% louver than the rate of reaction when methanol was used as the solvent for the st,eroid. No effect was observed on the 20a-hydrosysteroid dehydrogenase activit,y at various stages of 1)urificwtioii from Sicl’s 1 to 6, when
TABLE I Purijication of porcine testicuh 2Ool-hyrlroxysteroitl dehydroyenase
step
1. 7,000 X g supernatant.. 105,000 X g supernatant pH 5.5 supernatant.
2. 45 to 67% (NH,),SO, precipitate.
3. DEAF-cellulose effluent, Fraction 51 to 61.
4. Sephadex G-150 filtrate, Fraction 31 to 36.
5. TTydroxylapatite runoff fraction.. 6. Bio-Gel P-100 filtrate, Fraction 23
to25...
Protein
nz!z
7,400 :I, 100 4,100 4,540
222
35.7 19.c
7.1
Total e”Zyllle activity
uni1s miis/mg
3,000 0.063 4,960 0.24
4,310 0.31
2,410 0.53
1,370
936
6.14
26.2
8G6 43.6
584 81.9
bovine serum albumin was added to the assay system in concen- trations of 0.3 and 1.0 mg per ml.
Enzyme Content
Porcine testes obtained from the slaughter house varied in
weight from 160 to 440 g (after decapsulation). The 20a-hy- droxysteroid dehydrogenase content of the soluble fraction of the testicular homogenates (105,000 x g supernatant) was roughly proportional to the wet weight of the gland, although there was a small tendency for larger glands to contain a greater amount of the enzyme per unit weight of tissue. The removal of some of the particulate material by ultracentrifugation significantly in- creased the 20a-hydroxysteroid dehydrogenase activity, despite the inevitable loss of a portion of the enzyme in the fluffy ,sedi- ments.
PurQkation Procedures
All of the procedures were carried out in the cold room (5-7”) with solutions cooled in an ice xvater bath. A typical purification is described below and summarized in Table I. The figures in the table are values corrected for the aliquots which were kept stored for the protein determination, etc., and were not carried through to the next step.
Step 1. Extraction of Enzyme-Two testes were thawed in the cold room overnight and decapsulated. The tissue (782 g) was dissected into small pieces with scissors and homogenized in 100- to 150-g portions with the same volume of 0.02 M phosphate buffer (pH 7.0) containing 5 mnr EDTA. The homogenization was per- formed for 2 min with a Waring Hlendor. The homogenates were combined and centrifuged at 7,000 x g for 30 min. The resulting supernatant fluid (900 ml) was then centrifuged at 105,000 x g for 90 min. The supernatant solution was retained and the resi- due was discarded.
The 105,000 x g supernatant (600 ml) was brought to pH 5.5 by the addition of 130 ml of 0.1 s HCl. Following centrifugation (7,000 x g, 10 min), the gummy sediment was discarded. Kith- out delay, the supernatant solution was neutralized to pH 7.0 with 0.1 N NaOH to yield a red transparent solution (760 ml), which contained most of the 20a-hydroxysteroid dehydrogenase sctiv- ity.
Step 1. Ammonium Sulfate Fractionation-To the pH 5.5 supernatant solution, saturated ammonium sulfate solution was added to achieve 45% saturation. The mixture was stirred in an
by guest on September 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from
SlS $?Oa-Hydroxysteroid Dehydrogenase of Testes Vol. 247, No. 3
L c
I I I I I, T I 17.5 7 .*
15.0 - 6
s T I I
:12.5 - 5 I
y 2.5-g,
& 8
2OF
8
s o-8 0: 0 10 20 30 40 50 60 70
FRACTION NUMBER
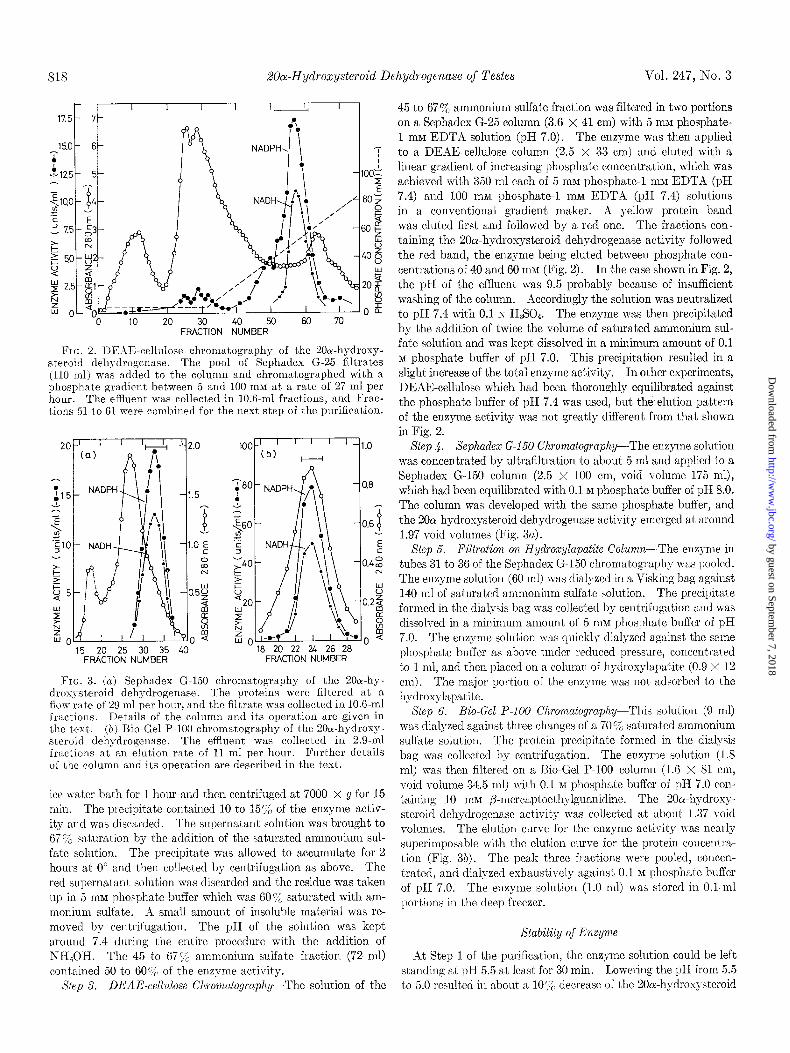
FIG. 2. DEAE-cellulose chromatography of the 20ol-hydroxy- steroid dehydrogcnase. The pool of Sephadex G-25 filtrates (110 ml) was added to the column and chromatographcd with a phosphate gradient between 5 and 100 mM at a rate of 27 ml per hour. The efflllent was collected in 10.6.ml fractions, and Frac- tions 51 to 61 were combined for the next step of the purification.
loo-’ ’ ’ ’ ’ ’ - 1.0 (b) H
18 20 22 24 26 28 FRACTION NUMBER
FIG. 3. inj Sephadex G-150 chromatography of the 20cr-hy- droxysteroid dehydrogenase. The proteins wcrc filtered at a flow rate of 29 ml per hollr, and the filtrate was collected in 10.6.ml fractious. Details of the column and its operation are given in the text. (b) Bio-Gel P-100 chromatography of the 20a-hydroxy- steroid dehydrogenasc. The effluent was collected in 2.9.ml fractions at an elution rate of I1 ml per hour. Further details of the column and its operation are described in the text.
ice \T-atcr bat,h for 1 hour and then centrifuged at 7000 x g for 15 min. The precipitate contained 10 to 15y0 of the enzyme activ-
ity and was discarded. The supernatant solution was brought to 67’,;, saturation by the addition of the saturated ammonium sul- fate solution. The precipitate was allowed to accumulate for 2
hours at 0” and then collected by centrifugation as above. The red supernatant solution was discarded and the residue was taken up in 5 mM phosphate buffer which was 6096 saturated with am- monium sulfat,e. il small amount of insoluble material was re- moved by centrifugation. The pH of the solution was kept around 7.4 during the entire procedure with the addition of NII,OH. The 45 to 6751 ammonium sulfate fraction (72 ml) contained 50 to 609; of the enzyme activity.
Step 3. DEAE-cellulose Chromatography-The solution of the
45 to 67Y0 ammonium sulfate fraction was filtered in two portions on a Sephadex G-25 column (3.6 x 41 cm) with 5 rnM phosphate- 1 InM EDTA solution (pH 7.0). The enzyme was then applied to a DEAE-cellulose column (2.5 x 33 cm) and &ted Jvith a linear gradient of increasing phosphate concentration, which was achieved with 350 ml each of 5 rnM phosphate-l mM EDTA (pH 7.4) and 100 mM phosphate-l mM EDTA (pH 7.4) solutions in a conventional gradient maker. A yellow protein band was eluted first and followed by a red one. The fractions con- taining the 20a-hydroxysteroid dehydrogenase activity followed the red band, the enzyme being eluted between phosphate con- centrations of 40 and 60 mar (Fig. 2). In the case shown in Fig. 2, the pH of the effluent was 9.5 probably because of insufficient washing of the column. Accordingly the solution was neutralized to pH 7.4 with 0.1 N 1180~. The enzyme was then precipitated by the addition of twice the volume of saturated ammonium sul- fate solution and was kept dissolved in a minimum amount of 0.1 M phosphate buffer of pH 7.0. This precipitation resulted in a slight increase of the total enzyme activity. In other experiments, DEAE-cellulose which had been thoroughly equilibrated against the phosphate buffer of pH 7.4 was used, but the elution pattern of the enzyme activity was not greatly different from that shown in Fig. 2.
Step 4. Xephadex G-150 Chromatography-The enzyme solution was concentrated by ultrafiltration to about 5 ml and applied to a Sephadex G-150 column (2.5 x 100 cm, void volume 175 ml), which had been equilibrated with 0.1 M phosphate buffer of pH 8.0. The column was developed with the same phosphate buffer, and the 20or-hydroxysteroid dehydrogenase activity emerged at around 1.97 void volumes (Fig. 3~).
Step 5. Filtration on EIydroxylapatite Column- The enzyme in tubes 31 to 36 of the Sephadex G-150 chromatography was pooled. The enzyme solution (60 ml) was dialyzed in a Visking bag against 140 ml of saturated ammonium sulfate solution. The precipitate formed in the dialysis bag was collected by centrifugation and was dissolved in a minimum amount of 5 mM phosphate buffer of pH 7.0. The enzyme solut’ion was quickly dialyzed against the same phosphate buffer as above under reduced pressure, conccntrat’ed to 1 ml, and then placed on a column of hydroxylapatite (0.9 x 12 cm). The major portion of the enzyme was not adsorbed to the hydroxglapatite.
Step 6. Bio-Gel P-100 Chromatography-This solution (9 ml) was dialyzed against three changes of a 70cA saturated ammonium sulfate solution. The protein precipit’ate formed in the dialysis bag \vas collected by centrifugation. The enzyme solution (1.8 ml) was then filtered on a &o-Gel P-100 column (1.6 X 81 cm, void volume 34.5 ml) with 0.1 RI phosphate buffer of pI-I 7.0 con- taining 10 ICM fl-mercaptoethylguanidine. The 20a-hydroxy- steroid dehydrogenase activity was collected at about 1.37 void volumes. The elution curve for the enzyme activity was nearly superimposable wit,h the elution curve for the protein concentra-
tion (Fig. 3b). The peak three fractions were pooled, concen-
trated, and dialyzed exhaustively against 0.1 M phosphate buffer
of pH 7.0. The enzyme solution (1.0 ml) was stored in O.l-ml portions in the deep freezer.
Stability of Enzyme
At Step 1 of the purification, the enzyme solution could be left
standing at pH 5.5 at least for 30 min. Lowering the pH from 5.5
to 5.0 resulted in about R 107; decrease of the 20ar-hydrosysteroid
by guest on September 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from
Issue of February 10, 1972 F. Xato, Y. Takagi, and M. Xhilcita 819
dehydrogenase activity. Most of the activity was destroyed when the pH of the solution was lowered below 4.2. The en- zyme was considerably more stable at an alkaline pH, even as high as 9 to 10. At the Step 2, the 45 to 67% ammonium sulfate fraction could be stored at 4” overnight or at -20” for months without a large loss of the enzyme activity. There was approxi- mately a 30% decrease of the enzyme activity in 5 hours when a portion of the DEAE-cellulose chromatography effluent of Fig. 2 was left standing at 4” without neutralization. Twenty per cent of the activity applied to the Sephadex and Bio-Gel columns could not be accounted for. When 50 mM Tris-chloride-l mM EDTA solution (pII 7.4) was used instead of the phosphate buffer, the loss of the enzyme activity during the gel filtration amounted to 607,.
At all stages of these purification procedures, the enzyme was more stable at 4’ than at 25“. The purified enzyme remained stable for several days, when it was stored at 4” in concentrations of about 2 mg of protein per ml in 0.1 M phosphate buffer of pH 7.0. Under these conditions of storage, changes of pH from 6.0 to 9.0 had no significant effect on the stability of the enzyme. No activity was lost when the same enzyme solution was stored at -20” for months, while more than half of the activity was lost in 3 days when it was left standing at 25”. Repeated thawing and freezing of the enzyme solution resulted in a significant decrease of the enzyme activity. This decrease was even more prominent if the enzyme solution contained sulfhydryl compounds such as cysteine or /?-mercaptoethylguanidine.
When the pH 5.5 supernatant solution from Step 1 of the pu- rification was heated to 60” for 10 min, the enzyme activity was completely destroyed. A similar heat inactivation of the enzyme occurred with the 45 to 67V0 ammonium sulfate fraction.
Properties of Puri&d Enzyme
The physical and catalytic properties of the 20oc-hydroxysteroid dehydrogenase were studied with enzyme from Step 6.
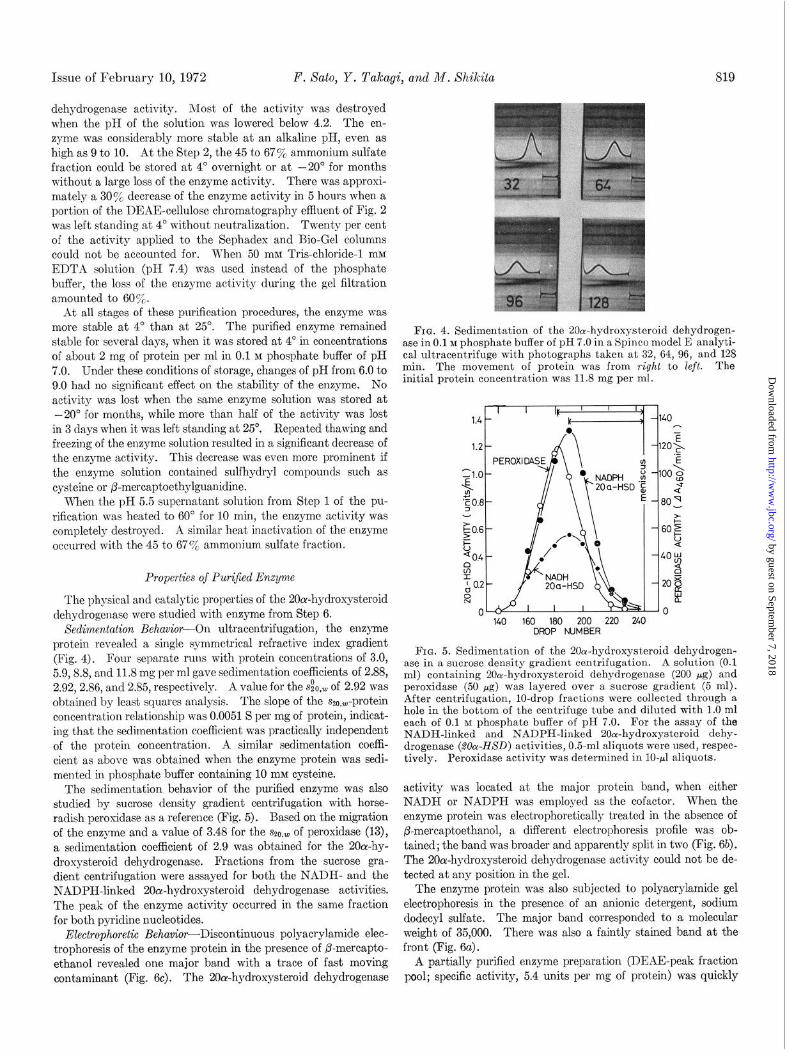
Sedimentation Behavior-On ultracentrifugation, the enzyme protein revealed a single symmetrical refractive index gradient (Fig. 4). Four separate runs with protein concentrations of 3.0, 5.9,8.8, and 11.8 mg per ml gave sedimentation coefficients of 2.88, 2.92,2.86, and 2.85, respectively. A value for the s&,,,, of 2.92 was obtained by least squares analysis. The slope of the szo,W-prot,ein concentration relationship was 0.0051 S per mg of protein, indicat- ing that the sedimentation coefficient was practically independent of the protein concentration. A similar sedimentation coeffi- cient as above was obtained when the enzyme protein was sedi- mented in phosphate buffer containing 10 mM cyst’eine.
The sedimentation behavior of the purified enzyme was also studied by sucrose density gradient centrifugation with horse- radish peroxidase as a reference (Fig. 5). Based on the migrat,ion of the enzyme and a value of 3.48 for the SZ~,,,, of peroxidase (13), a sedimentation coefficient of 2.9 was obtained for the 2Oo1-hy- droxysteroid dehydrogenase. Fractions from the sucrose gra- dient centrifugation were assayed for both the NADH- and the NADPH-linked 20oc-hydroxysteroid dehydrogenase activities. The peak of the enzyme activity occurred in the same fraction for both pyridine nucleotides.
Electrophoretic Behavior-Discontinuous polyacrylamide elec- trophoresis of the enzyme protein in the presence of a-mercapto- ethanol revealed one major band with a trace of fast moving contaminant (Fig. 6~). The 2Oor-hydroxysteroid dehydrogenase
FIG. 4. Sedimentation of the 20a-hydroxysteroid dehydrogen- ase in 0.1 M phosphate buffer of pH 7.0 in a Spinco model E analyti- cal ultracentrifuge with photographs taken at 32, 64,96, and 1‘28 min. The movement of protein was from right to left. The initial protein concentration was 11.8 mg per ml.
$0.6 F 2 0.4
z & 0.2
w
0 140 160 180 200 220 240
DROP NUMBER
FIG. 5. Sedimentation of the 2(lor-hydroxysteroid dehydrogen- ase in a sucrose density gradient centrifugation. A solution (0.1 ml) containing 20a-hydroxysteroid dehydrogenase (200 pg) and peroxidase (50 pg) was layered over a sucrose gradient (5 ml). After centrifugation, lo-drop fractions were collected through a hole in the bottom of the centrifuge tube and diluted with 1.0 ml each of 0.1 M phosphate buffer of pH 7.0. For the assay of the NADH-linked and NADPH-linked 2Oor-hydroxysteroid dehy- drogenase (Z&Y-HSD) activities, 0.5-ml aliquots were used, respec- tively. Peroxidase activity was determined in IO-r1 aliquots.
activity was located at the major protein band, when either NADH or NADPH was employed as the cofactor. When the enzyme protein was electrophoretically treated in the absence of @mercaptoethanol, a different electrophoresis profile was ob- tamed; the band was broader and apparently split in two (Fig. 6b). The 20Lu-hydroxysteroid dehydrogenase activity could not be de- tected at any position in the gel.
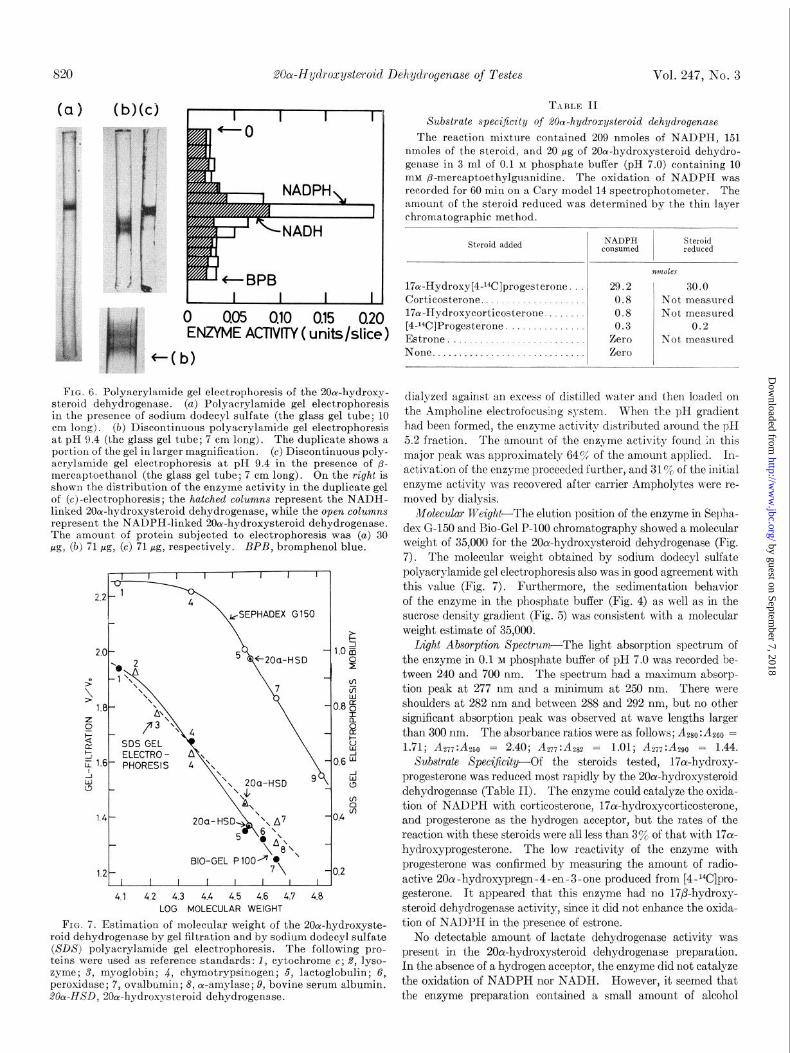
The enzyme protein was also subjected to polyacrylamide gel electrophoresis in the presence of an anionic detergent, sodium dodecyl sulfate. The major band corresponded to a molecular weight of 35,000. There was also a faintly stained band at the front (Fig. 6~).
A partially purified enzyme preparation (DEAE-peak fraction pool; specific activity, 5.4 units per mg of protein) was quickly
by guest on September 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from
20a-Hyd~oxysteroid Dehydrogenase of Testes Vol. 247, No. 3
+(b)
0 005 OJO OJ5 0.20 ENZYME ACTIVITY ( units/slice)
FIG. 6. Polyacrylamide gel electrophoresis of the 20rw-hydroxy- steroid dehydrogenase. (a) Polyacrylamide gel electrophoresis in the presence of sodium dodecyl sulfate (the glass gel tube; 10 cm long). (b) Discontinuous polyacrylamide gel electrophoresis at pH 9.4 (the glass gel tube; 7 cm long). The duplicate shows a portion of the gel in larger magnification. (c) Discontinuous poly- acrylamide gel electrophoresis at pH 9.4 in the presence of p- mercaptoethanol (the glass gel tube; 7 cm long). On the right is shown the distribution of the enzyme activity in the duplicate gel of (c)-electrophoresis; the hatched columns represent the NADH- linked 2Ocu-hydroxysteroid dehydrogenase, while the open columns represent the NADPH-linked 200c-hydroxysteroid dehydrogenase. The amount of protein subjected to electrophoresis was (a) 30 pg, (b) 71 pg, (c) 71 pg, respectively. BPB, bromphenol blue.
17,- .,.- I I I I I I I I I
4.1 4.2 4.3 4.4 4.5 4.6 4.7 4.8 LOG MOLECULAR WEIGHT
FIG. 7. Estimation of molecular weight of the 2Oa-hydroxyste- roid dehydrogenase by gel filtration and by sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis. The following pro- teins were used as reference standards: 1, cytochrome c; 2, lyso- zyme; S, myoglobin; 4, chymotrypsinogen; 5, lactoglobulin; 6, peroxidase; 7, ovalbumin; 8, or-amylase; 9, bovine serum albumin. 20~~HSD, 20cu-hydroxysteroid dehydrogenase.
TABLE II
Substrate specificity of dOa-hydroxysteroid dehydrogenase
The reaction mixture contained 209 nmoles of NADPH, 1~51 nmoles of the steroid, and 20 pg of 2Oa-hydroxysteroid dehydro- genase in 3 ml of 0.1 M phosphate buffer (pH 7.0) containing 10 mM p-mercaptoethylguanidine. The oxidation of NADPH was recorded for 60 min on a Cary model 14 spectrophotometer. The amount of the steroid reduced was determined by the thin layer chromatographic method.
Steroid added NADPH consumed
Steroid reduced
17cr-Hydroxy[4-‘*C]progesterone. Corticosterone.................... 17ol-Hydroxycorticosterone. [4J4C]Progesterone.
Estrone None.............................
nnuJles
29.2 30.0 0.8 Not measured 0.8 Not measured 0.3 0.2
Zero Not measured Zero
dialyzed against an excess of distilled water and then loaded on the Ampholine electrofocusing system. When the pH gradient had been formed, the enzyme activity distributed around the pH 5.2 fraction. The amount of the enzyme activity found in this major peak was approximately 645r, of the amount applied. In- activation of the enzyme proceeded further, and 31 Y0 of the initial enzyme activity was recovered after carrier Ampholytes were re- moved by dialysis.
Molecular lVeight--The elution position of the enzyme in Sepha- dex G-150 and Bio-Gel P-100 chromatography showed a molecular weight of 35,000 for the 20cr-hydroxysteroid dehydrogenase (Fig. 7). The molecular weight obtained by sodium dodecyl sulfate polyacrylamide gel electrophoresis also was in good agreement with this value (Fig. 7). Furthermore, the sedimentation behavior of the enzyme in the phosphate buffer (Fig. 4) as well as in the sucrose density gradient (Fig. 5) was consistent with a molecular weight estimate of 35,000.
Light Absorption Spectrzlm-The light absorption spectrum of the enzyme in 0.1 M phosphate buffer of pH 7.0 was recorded be- tween 240 and 700 nm. The spectrum had a maximum absorp- tion peak at 277 nm and a minimum at 250 nm. There were shoulders at 282 nm and between 288 and 292 nm, but no other significant absorption peak was observed at wave lengths larger than 300 nm. The absorbance ratios were as follows; Azso: As60 = 1.71; As~~:A~S,, = 2.40; A*T,:Azsz = 1.01; A2,,:-4~~0 = 1.44.
Substrate Xpeci$city-Of the steroids tested, 17a-hydroxy- progesterone was reduced most rapidly by the 2Otu-hydroxysteroid dehydrogenase (Table II). The enzyme could catalyze the osida- tion of NADPH with corticosterone, 17c+hydroxycorticosterone, and progesterone as the hydrogen acceptor, but the rates of the reaction with these steroids were all less than 3% of that with 17a- hydroxyprogesterone. The low reactivity of the enzyme with progesterone was confirmed by measuring the amount of radio- active 200~ - hydroxypregn - 4 -en - 3 -one produced from [4 -r4C]pro- gesterone. It appeared that this enzyme had no 17/3-hydroxy- steroid dehydrogenase activity, since it did not enhance the oxida- tion of NADPH in the presence of estrone.
No detectable amount of lactate dehydrogenase activity was present in the 20ahydroxysteroid dehydrogenase preparation. In the absence of a hydrogen acceptor, the enzyme did not catalyze the oxidation of NADPH nor NADH. However, it seemed that the enzyme preparation contained a small amount of alcohol
by guest on September 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Issue of February 10, 1972 F. Sate, Y. Talcayi, and M. Xhilcita 521
dehydrogenase activity. Thus, 2.6 units of the 20a-hydroxy- steroid dehydrogenase, which oridized 2.7 nmoles of NADPH per min in the presence of 170c-hydroxyprogesterone, reduced less than 0.06 nmole per min of NAD+ in the presence of ethanol under the coiiditions of assay optimal for alcohol dehydrogenase. The presence of a small amount of alcohol dehydrogenase activity was al.;0 shown by the reverse reaction. The same amount of the 200(- hydroxysteroid dehydrogcnase as above oxidized 0.08 nmole of N&\l)H per min in the presence of acetaldehyde in the assay de- scribed under “Esperimental Procedure,” whereas the enzyme cau~l no increaqe of NADPH oxidation in the presence of acetaldchyde mlder the assay conditions optimal for the steroid rciluction.
Cofurlors--~~-ithout addition of NADPH or NADH, l’ia-hy- rll,o\;!-I)t’ogesterolle \vas not reduced by the 200c-hydroxysteroid tlch>-drogenasc. I?oth N,111I-‘H and NADH could serve as the (sofactor of the enzyme. In various chromatographies (Figs. 2 and 3) as well as in t,hc sucrose density gradient centrifugation (Fig. 5), the N;\DI-I- and NhDPHliuked enzyme activities moved together. Furthermore, when the solution of the purified enzyme at a concentration of 1.1 mg of protein per ml was left standing at 4” and the enzyme activity was determined in the absence of cys- teine, the NXDR- and NADPH-linked enzyme activities de- creased hi parallel. I f cysteine (10 m&f) was added to the partially inactivated enzyme solution, however, the NADH-linked activity nas restored to a greater extent than the NADPH-linked activity.
With freshly thawed enzyme and in the presence of 10 mM cys- teine, 1Iichaelis constants of 750 and 52 PM were determined for N:lDH and NADPII, respectively, from double reciprocal plots. When a portion of the same enzyme solution had been left stand- ing at 4” until the activity had decreased to half of the initial value, the I<, values for NAIlI and NADPH were determined to be 1.1 m&r and 22 q, respectively.
pH Opfimum-The enzyme showed a broad pH optimum be- tween pH 5.5 to 7.5, when NADPH was used as the coenzyme. Addition of cysteine (10 mM) to the assay system did not cause a large change in this pH optimum profile. When NADH was used as the coenzy me, the maximal rate of the enzyme reaction occurred at pH 6.8 to 7.2. For both NADH- and NADPH-linked reac- tions, the enzyme activity was completely inhibited in solutions of the pH values above 10 or below 4, regardless of whether cysteine was present or absent. In this study, four kinds of buffer systems were used; glycine-HCl (pH 2.0 to 3.5), sodium phosphate (pH 5.5 to 7.0), Tris-chloride (pH 7.5 to 9.0), and glycine-NaOH (pH 9.0 to 13.0). The use of buffer solutions of different kinds had no in- fluence upon the rate of the enzyme reaction, if the pH of the solutions were the same.
Inhibitors-The 20a-hydrosysteroid dehydrogenase activity Teas strongly inhibited by low concentrations of sulfhydryl re- agents such as pmchloromcrcuribenzoate, 5, %dithiobis-@nitro- benzoic acid), and iodoacetamide, while EDTA had no effect on the enzyme activity (Table HI). Heavy metal ions, such as Ag+ and Cu++, were also powerful inhibitors. The inhibition by 0.2 rnM p-chloromercuribenzoate could be reversed by filtration of the mixture on a small Sephadex G-25 column. The addition of cys- teamine to the assay in a concentration of 10 mM also reversed the inhibition by 0.2 InM p-chloromercuribenzoate. The inhibi- tion by 0.1 mM cupric ions could be partially reversed by subse- quent addition of EDTA in a concentration of 25 mM to the assay. Divalent metal ions, such as Ca++, Fe++, Rig++, Mn++, Cd++, and Zn++, did not seem to have any significant effect on the enzyme reaction. Another t.ype of compound which also
TABLE III Inhibitors of 20a-hydroxysteroid dehydrogenase
Compound
_____ ~
p-Chloromercuribenzonte
Monoiodoacetamide
Dithiobisnitrobenzoate
AgNOz cuso 4
CdSO,
Pyridoxal 5’.phosphate
EDTA
CaC1.2 FeS04 MgClz
Mn(CH&OO)t ZnSOc
Concentration Enzyme activity,
percentage of control
M
1 x 10-K
5 x 10-E
1 x 10-h
2 x 10-d
1 x 10-4
1 x 10-z
1 x 10-z
9 x IO-”
1 x 10-d
%
57 22 14
2
105 88 58
11
8.9
1 x 10-d 8.6 1 x 10-4 7.1 5 x 10-d 6.9 1 x 10-d 95 5 x 10-d 87
1 x 10-d
1 x 10-S
1 x 10-Z
2.5 X 1O-2
96 40
8.9
94
1 x 10-S 96 5 x lo-” 97 1 x 10-Z 117 1 x 10-S 104 1 x 10-S 98 1 x 10-a 99
inhibited the 20ar-hydrosysteroid dehydrogenase was pyridoxal 5’-phosphate. The inhibition by 10 rnM pyridoxal 5’-phosphate exceeded 90 %.
Eflect of Xulfhydryl Compounds-As mentioned above, the 2001- hydroxysteroid dehydrogenase activity was gradually destroyed by leaving the enzyme solution at room temperature. The en- zyme activity could be partially restored by the addition of sulf- hydrgl compounds, although high concentrations of these com- pounds inhibited the enzyme. Cysteine was most effective among several sulfhydryl compounds examined (Fig. 8). For the enzyme preparation used in this specified experiment, ,&mcrcaptoethanol and glutathione were both entirely ineffective in restoring the en- zyme activity. However, the efficacy of the sulfhydryl com- pounds depended upon the extent of the inactivation of the en- zyme. Thus, if the enzyme activity had decreased to convert 8.9 nmoles of 17a-hydroxyprogesterone per min per mg of protein in the assay without cysteine, 1 m&I P-mercaptoethanol increased the activity by 2309,; if the activity had been further decreased to convert 5.4 nmoles of the steroid per min per mg of protein, @- mercaptoethanol was no longer effective. Cysteine (10 m&l) in- creased the activity of these same preparations by 510 and 667%, respectively. It should be noted that cysteine had no significant influence on the enzyme preparation of a specific activity of 82 units per mg.
Reactivation of the enzyme by the sulfhydryl compounds was complet,ed almost instantaneously. No significant difference was observed in the extent of reactivation, when the enzyme was pre- viously incubated with B-mercaptoethylguanidine for various
by guest on September 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

s22 2Oa-Hydroxysteroicl Dehydrogenase of Testes Vol. 247, Xo. 3
-2501 /
p;/, , y
10-4 10-3 10-Z 10-l CONCENTRATION OF SH COMPOUNDS (M)
FIG. 8. Partial reactivation of the 200c-hydroxysteroid dehydro- genase by various sulfhydryl compounds. ,4n enzyme prepara- tion, which had lost 92yc of the initial activity (80 unit,s per mg), was previously incubated for 5 min at 25” in 0.1 M phosphate buffer (pH 7.0) containing NADPH (0.5 mM) and the sulfhydryl com- pound specified in the figure. Then the sltbstrate 17a-hydroxy- [4-14C]progesterone was added to start the enzymic reaction. Other conditions of the assay were the same as described under “Experimental Procedure.” The results are expressed in terms of a percentage of the control assay (the assay without addition of any sulfhydryl compound). DTT, dithiothreitol; MEG, p-mer- captoethylgltanidine; MEA, p-mercaptoethylamine (cysteamine); GSH, glutathione; HOCHL’HDS’H, p-mercaptoethanol.
periods of time between 1 and 60 min. A study with P-mercnpto-
eth\l[14C]Ruallidille showed that the enzyme remained activated
although the sulfhydryl compound was removed from the enzyme
solution by subsequent dialysis.
lZ$ect of Temperature of Assay-The rat,e of the 20oc-hydroxy- steroid dehydrogenase reaction was determined between 0 and 70”. The reaction rate increased exponentially with the increase of the assay temperature, while it rapidly decreased to zero when the temperature exceeded 55”. The maximal rate was achieved at about 50” and the rate at 0” was about one-tenth of the rate at 50”. The acth-ation energy for the 20ol-hydroxysteroid dehydrogenase reaction was estimated to be 7250 cal by Arrhenius plots over the range of O-55”.
DISCUSSION
When the crude tissue extract from porcine testes was centri- fuged at 105,000 x g, its apparent 20a-hydroxysteroid dehydro- genase activity was increased significantly. It seems likely that adsorption of the substrate steroid to particulate matter was re- sponsible for the low 20cr-hydroxysteroid dehydrogenase activity in the initial crude tissue extract. Another explanation for this result would be the removal of a particulate pyridine nucleotide oxidase. According to Billiar and Little (14), fatty acids inhibit the placental 2Oa-hydroxysteroid dehydrogenase, and the addition of albumin to the assay system reverses this inhibition. There was no effect of albumin on the testicular 20cr-hydroxysteroid dehydrogenase at any stage of its purification.
After it was purified to near homogeneity, the 20ar-hydroxy- steroid dehydrogenase exhibited a dual specificity for NADPH and NADH. Several pieces of information suggest the presence of a single enzyme with dual pyridine nucleotide specificity. (a)
Similar rates of migration of the two enzyme activities in the DEAF, aud gel filtration chromatographies as well as in the su- crose density gradient centrifugation. (b) Concomitant location of the enzyme activities at a single protein band in discontinuous polyacrylamide gel electrophoresis. (c) Parallel decrease of the enzyme activity during storage at 4”. (d) Similarity of the pH optimum for the enzyme activity. These findings suggest that the porcine testicular 20&ydroxysteroid dehydrogenase and the rat ovarian 20a-hydroxysteroid dehydrogenase are different enzymes, since the latter specifically utilizes NADPH and does not react with 17oc-hydrosyprogesterone (4).
The molecular weight of the 20cu-hydroxysteroid dehydrogenase was estimated to be 35,000 by gel filtration methods. Sodium dodecyl sulfate polyacrylamide gel elcctrophoresis of the enzyme protein suggested a similar molecular weight for the enzyme. The enzyme had the same sedimentation coefficient whether it was sedimented in 0.1 M phosphate buffer or in a sucrose density gra- dient. Thus, the enzyme does not dissociate iu the presence of an anionic detergent and it does not aggregate or dissociate in the presence of high concentrations of sucrose. These results suggest that the 20cr-hydroxysteroid dehydrogenase has no subunit struc- ture, unlike other dehydrogenases such as lactate (15), alcohol (16), malate (17), glyceraldehyde 3-phosphate (18) dehydro- genases.
Several other hydroxysteroid dehydrogenascs, such as 17@hy- droxysteroid dehydrogenases (19-21) and 16a-hydroxysteroid de- hydrogenase (22), are quite unstable and a high concentration of glycerol is needed for their stabilization. Among these dehydro- genases, the placental 17i0-hydroxysteroid dehydrogenase is unique for its extreme sensitivity to the cold (23). On the other hand, the porcine testicular 20~.hydrorystcroid dehydrogenase is rela- tively stable; it can be left standing at 4” for a week without a large loss of the activity. It is noted that the 20c+hydroxysteroid dehydrogenase which has been isolated from the rat ovary b) TViest (24) is also stable at 4”.
All of the hydrosysteroid tlehydrogcnases, except the 170.hy- droxysteroid dehydrogennse of rabbit liver (20), possess a sulf- hydryl group or groups related to their catalytic activity. This was also shown for the 20a-hydroxysteroid dehydrogenase of por- cine testes. On the other hand, a powerful metal chelator, EDT.& does not inhibit the activities of most of the hydroxysteroid de- hydrogcnases including the 20oc-hydroxysteroid dehydrogenase reported here. In this regard, the 16cr-hydroxysteroid dehydro- genase of rat kidney (22) is an exception; EDTA effectively in- hibits this enzyme activity. There are many dehydrogenases which are inhibited by pyridoxal 5’-phosphate; e.g. glutamic (25), 6-phosphogluconic (26), glyceraldehyde 3-phosphate (27)) malate (28) dehydrogenases. The 20ol-hydroxysteroid dehydrogenase of porcine testes was also inhibited by pyridoxal 5’-phosphate.
1. 2.
3. 4. 5.
G.
7. 8.
REFERENCES
SHIKITA, M., dND T2~vnoa~, B. (1965) Biochemistry, 4, 1189. SHIKITA, M., IKASO, I-I., AXD TMMOKI, B. (1967) Biochemistry,
6, 1760. WIEST, W. G., (1959) J. Biol. Chem., 234, 3115. WILCOX, R. B., AND WIEST, W. G., (1966) Steroids, ‘7, 395. SHIICITA, M., OGISO, T., .GD T~MAOKI, B. (1969) Endocrinol.
Jap., 16, 11. LOTVRY, 0. H., ROSEBROUGH, ?J. J., FaRR, A. L., AND RAND~LJ,,
R. J. (1951) J. Biol. Chem., 193, 265. ALLEN, R. J. L. (1940) Biochem. J., 34, 858. Worthington Manual 1.11.1.7. (1967) Worthington Biochemical
Corp., Freehold, N. J.
by guest on September 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Issue of F‘cbrunry lo, 1972 F. Xato, Y. Talcagi, and If. Xhikita 523
9. I).\vIs, 13. <J. (19Gi) 11 nn. zv. Y. /ic.ad. Sci. 121, 404. 10. WEHIX, Ki.. .\SD Osrw~r, M. (1969) J. Biol. Chum. 244, 440G. ll.q\~I~:s~~I~I~Bl:I~~;. o., .lSI) s\~lx%3lN, II. (1966) dcla Chenr. &and.,
20, 820. 12. .\l.wns, Ii. Ct., .\ND AMBS, B. N”;. (1961) J. Viol. Ch,em. 236,
13i2. 13. CLTIL, I:.. .\SD OGSToS: A. G. (1951) Biochenl. J., 49, 105. 14. %l,LI.iR, I:. B., .INL) I~IT’I’LI:, I~. (1071) &rlocrinoloyy, 88, 263. 15. APPICLL.\, I<., .IXD >I.u<I<ERT, C. I,. (1981) Biochem. Biophys.
Res. Cwmw~., 6, 171. 1G. I>I~uM, 1). I,:., T~.\RRISOS, J. H., IV, T,I, T-K., I~IWHUNE, J. L.,
ASD 1..\LLI.I.:, B. I,. (1907) P,OC. .\.O,t. lt Cd &i. c. 8. .t ., 57, 1434.
18. &RRINGTOX, W. F., rich K.~RR, G. M. (19G5) J. Mol. Biol., 13, 885.
19. JARNMK, J., ADAMS, J. A., WILLMMS-ASHMSN, H. G., AND TBL~LAY, I’. (1962) J. Biol. Chem.. 237, 345.
20. BILL, P., .IND BREUER, II. (1970) Hoppe-Seyler’s Z. Physiol. Chem., 351, 1011.
21. KAUTSKY, M. I’., .\ND H.\GI~RMAN, n. 11. (1970) J. Biol. Chem., 246, 1978.
22. MICIGS, R. A., .\SD RY.~, K. J. (1960) .I. Biol. Chem., 241,401l. 23. J.UUBM<, J., SI:I;DS, A. I5., JR., -ISI) TM,.~L~Y, P. (1966) Uio-
chemistry, 6, 12G9. 24. WIES~~, W. G. (1969) Methods Enzymol., 15, 638. 25. ANDERSON, B. hf., ANDERSOX, C. I)., MD CEIURCHICH, J. E.
(196G) Biochemistry, 5, 2893. 26. RIPP~\, AI., SPANIO, L., ,\xD POSTRNOLI, S. (1967) Arch. Bio-
them. Biophys., 118, 48. 27. RONCHI, S., ZAPPONI, M. C., .UVD FEI~RI, G. (1969) Eur. J. Bio-
them., 8, 325. 28. YOST, F. J., JR., ASD HARRISON, J. H. (1971) Biochem. Biophys.
Res. Commun., 42, 516.
by guest on September 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Fumiko Sato, Yoshinari Takagi and Mikio ShikitaPROPERTIES
-Hydroxysteroid Dehydrogenase of Porcine Testes: PURIFICATION ANDα20
1972, 247:815-823.J. Biol. Chem.
http://www.jbc.org/content/247/3/815Access the most updated version of this article at
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/247/3/815.full.html#ref-list-1
This article cites 0 references, 0 of which can be accessed free at
by guest on September 7, 2018
http://ww
w.jbc.org/
Dow
nloaded from





























