Embed Size (px)
Citation preview

B IO
TE
HC N OL
O GAI
CHEMIAORGANICZNA
Budowa
Fluorowcopochodne zawierające atom fluorowca związany z atomem węgla sp3 mają budowę
przestrzenną zbliżoną do budowy węglowodorów. Wiązanie węgiel - fluorowiec utworzone przez
orbital 2sp3 atomu węgla oraz orbital nsp3 atomu fluorowca (n = 2,3,4,5 odpowiednio dla
poszczególnych fluorowców poczynając od fluoru) różni się jednak dość istotnie od wiązania węgiel
- wodór. Parametry tych wiązań podaje poniższe zestawienie:
Wiązanie C - H C - F C - Cl C - Br C - I
Długość [pm] 110 137 177 191 212
Energia [kJ/mol] 435 453 352 293 235
Wiązania węgiel - fluorowiec są spolaryzowane, co powoduje pojawienie się na atomie
fluorowca cząstkowego ładunku ujemnego. Ani ten fakt, ani znacznie większy (w porównaniu z
atomem wodoru) promień atomowy fluorowca nie wpływają jednak znacząco na deformację kąta
tetraedrycznego H - C - X. Natomiast w miarę wzrostu ilości atomów fluorowca przy tym samym
atomie węgla, wzrastają kąty między wiązaniami C-X, maleją zaś kąty H-C-X.
C
H
Cl
H
H
108,28 o
110,64 o177 pm
110 pm
C
H
Cl
H
Cl
116,36o
106,35 o
Fluorowcopochodne zawierające atom fluorowca związany z atomem węgla sp2 mają budowę
rezonansową na skutek sprzężenia p - Sprzężenie to można zapisać następującymi strukturami
granicznymi:
C C X C C X
X - atom fluorowca
Konsekwencją tego sprzężenia w halogenku winylu jest: a) skrócenie długości wiązania węgiel -
fluorowiec (rys. 2), co powoduje, że wiązanie to staje się mocniejsze, b) mniejsza polaryzacja
wiązania węgiel - fluorowiec w stosunku do odpowiedniego halogenku etylu (patrz: następny rozdział
- MOMENT DIPOLOWY), c) mała reaktywność (np. cząsteczka halogenku winylu nie ulega
reakcjom typowym dla fluorowcopochodnych alkilowych). Podobne własności mają
fluorowcopochodne aromatyczne.
CH2 CH Cl
173 pmCl
169 pm
chlorek winylu chlorobenzen
B IO
TE
HC N OL
O GAI
CHEMIAORGANICZNA
Własności fizyczne
TEMPERATURA WRZENIA: W temperaturze 25C fluorowcopochodne są na ogół cieczami.
Gazami są jedynie fluoropochodne zawierające do 3 atomów węgla, monochloropochodne zawierające
do 2 atomów węgla oraz bromometan. Ciałami stałymi są np. tri- lub tetrajodopochodne. Obserwuje
się tu następujące typowe prawidłowości:
a. wzrost temperatury wrzenia wraz ze wzrostem masy molowej w obrębie szeregów homologicznych
Tabela 1
X CH3X C2H5X CH3CH2CH2X CH3CH2CH2CH2X
F -78oC -38oC -3oC 32oC
Cl -24oC 12oC 47oC 78oC
Br 4oC 38oC 71oC 102oC
I 42oC 72oC 102oC 130oC
b. wzrost temperatury wrzenia ze wzrostem masy atomowej fluorowca w halogenkach zawierających
tę samą grupę alkilową lub arylową (patrz - Tabela 1 i Tabela 2)
c. zmniejszanie się temperatury wrzenia izomerycznych fluorowcopochodnych wraz ze wzrostem
stopnia rozgałęzienia łańcucha węglowego
Tabela 2
X n-butyl - X izo-butyl - X sec-butyl - X t-butyl - X
F 32oC 23oC 24oC 12oC
Cl 78oC 69oC 68oC 51oC
Br 102oC 91oC 91oC 73oC
I 130oC 120oC 119oC 100oC (rozkł.)
Ciekawym zjawiskiem jest fakt, że większość perfluoropochodnych ma niższą temperaturę wrzenia
od odpowiadających im węglowodorów (mimo, że ich masa molowa jest ok. 4-krotnie większa),
a także od temperatury wrzenia monofluoropochodnej o tym samym łańcuchu węglowym np.
butan - t.wrz. -1C ( M.cz.=58 g/mol )
perfluorobutan - t.wrz. -5C, ( M.cz.=238 g/mol )
(podczas gdy t.wrz. 1-fluorobutanu wynosi 32C)
Przyczyną tego zjawiska mogą być znaczne siły odpychania elektrostatycznego między
cząsteczkami perfluoropochodnych na skutek występowania dużych cząstkowych ładunków ujemnych
na równomiernie rozmieszczonych w cząsteczce atomach fluoru. Podobne zjawisko nietypowej
zmiany temperatury wrzenia ze wzrostem masy molowej podobnych chemicznie cząsteczek obserwuje
się dla niektórych fluorozwiązków nieorganicznych (np. JF5 - t.wrz = 97oC, JF7 - t.wrz.= 4 oC ). Dla
perchloropochodnych nie obserwuje się już tego zjawiska (polaryzacja wiązań węgiel-fluor i węgiel-
chlor jest co prawda podobna, jednakże promień kowalencyjny chloru jest znacznie większy od
promienia fluoru i stąd w perchlorowcopochodnych mniejsza jest koncentracja cząstkowego ładunku
ujemnego).
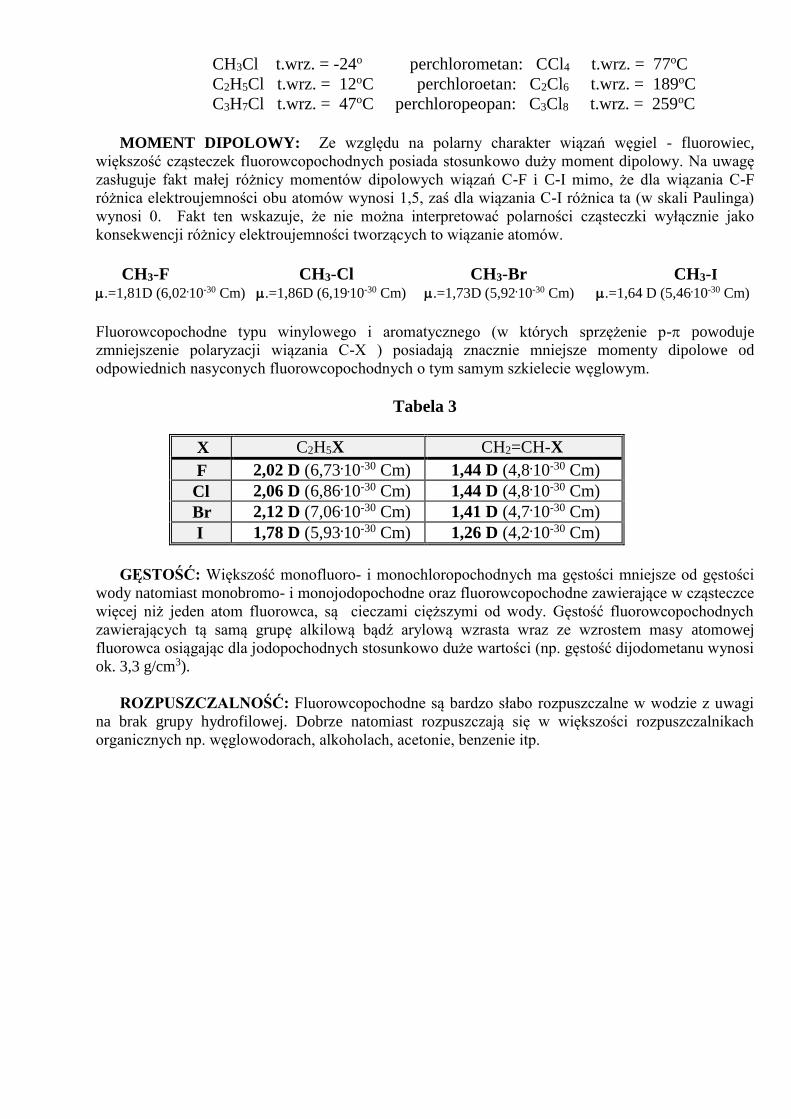
CH3Cl t.wrz. = -24o perchlorometan: CCl4 t.wrz. = 77oC
C2H5Cl t.wrz. = 12oC perchloroetan: C2Cl6 t.wrz. = 189oC
C3H7Cl t.wrz. = 47oC perchloropeopan: C3Cl8 t.wrz. = 259oC
MOMENT DIPOLOWY: Ze względu na polarny charakter wiązań węgiel - fluorowiec,
większość cząsteczek fluorowcopochodnych posiada stosunkowo duży moment dipolowy. Na uwagę
zasługuje fakt małej różnicy momentów dipolowych wiązań C-F i C-I mimo, że dla wiązania C-F
różnica elektroujemności obu atomów wynosi 1,5, zaś dla wiązania C-I różnica ta (w skali Paulinga)
wynosi 0. Fakt ten wskazuje, że nie można interpretować polarności cząsteczki wyłącznie jako
konsekwencji różnicy elektroujemności tworzących to wiązanie atomów.
CH3-F CH3-Cl CH3-Br CH3-I
.=1,81D (6,02.10-30 Cm) .=1,86D (6,19.10-30 Cm) .=1,73D (5,92.10-30 Cm) .=1,64 D (5,46.10-30 Cm)
Fluorowcopochodne typu winylowego i aromatycznego (w których sprzężenie p- powoduje
zmniejszenie polaryzacji wiązania C-X ) posiadają znacznie mniejsze momenty dipolowe od
odpowiednich nasyconych fluorowcopochodnych o tym samym szkielecie węglowym.
Tabela 3
X C2H5X CH2=CH-X
F 2,02 D (6,73.10-30 Cm) 1,44 D (4,8.10-30 Cm)
Cl 2,06 D (6,86.10-30 Cm) 1,44 D (4,8.10-30 Cm)
Br 2,12 D (7,06.10-30 Cm) 1,41 D (4,7.10-30 Cm)
I 1,78 D (5,93.10-30 Cm) 1,26 D (4,2.10-30 Cm)
GĘSTOŚĆ: Większość monofluoro- i monochloropochodnych ma gęstości mniejsze od gęstości
wody natomiast monobromo- i monojodopochodne oraz fluorowcopochodne zawierające w cząsteczce
więcej niż jeden atom fluorowca, są cieczami cięższymi od wody. Gęstość fluorowcopochodnych
zawierających tą samą grupę alkilową bądź arylową wzrasta wraz ze wzrostem masy atomowej
fluorowca osiągając dla jodopochodnych stosunkowo duże wartości (np. gęstość dijodometanu wynosi
ok. 3,3 g/cm3).
ROZPUSZCZALNOŚĆ: Fluorowcopochodne są bardzo słabo rozpuszczalne w wodzie z uwagi
na brak grupy hydrofilowej. Dobrze natomiast rozpuszczają się w większości rozpuszczalnikach
organicznych np. węglowodorach, alkoholach, acetonie, benzenie itp.

B IO
TE
HC N OL
O GAI
CHEMIAORGANICZNA
Synteza
Metody syntezy fluorowcopochodnych alkilowych
1. HALOGENOWANIE ALKANÓW ( reakcja substytucji rodnikowej, SR)
Metoda ta pozwala na otrzymywanie bromo- i chloropochodnych w inicjowanej światłem
wolnorodnikowej reakcji alkanów z bromem lub chlorem. W procesach tych powstaje na ogół
mieszanina halogenopochodnych i stąd metoda ta ma ograniczone zastosowanie.
Przykład:
CH3CHCH3
CH3
CH3CCH3
CH3
Br
CH3CHCH2Br
CH3Br2 , hv
+
99% 1% 2. ADDYCJA ELEKTROFILOWA FLUOROWCÓW DO ALKENÓW I ALKINÓW
W stereospecyficznej addycji (addycja „trans”) chloru bromu lub jodu do alkenów tworzą się
odpowiednie dihalogenopochodne wicynalne, zaś w przypadku addycji do alkinów -
tetrahalogenopochodne zawierające po dwa atomy fluorowca przy sąsiednich atomach węgla.
Przykłady:
a) w reakcji cykloheksenu z chlorem tworzy się racemiczny trans - 1,2-dichlorocykloheksan
R A C E M A T
H
Cl H
Cl H
ClH
Cl
+Cl2
b) w reakcji (E)-2-butenu z bromem tworzy się mezo-2,3-dibromobutan
(E)-2-buten
(2R,3S)-2,3-dibromobutan
mezo -2,3-dibromobutan
H Br
H Br
CH3
CH3
Br2
CH CH
CH3
CH3
c) w reakcji (Z)-2-butenu z bromem tworzy się racemiczny 2,3-dibromobutan
R A C E M A T
(2S,3S)-2,3-dibromobutan
HBr
BrH
CH3
CH3
+
(Z)-2-buten
CH CH
CH3 CH3
Br2
CH3
CH3
Br H
H Br
(2R,3R)-2,3-dibromobutan

d) w reakcji 2-butynu z chlorem tworzy się 2,2,3,3-tetrachlorobutan
Cl2CH3CCl2CCl2CH3C C
CH3
CH3Cl
Cl
Cl2C CCH3 CH3
3. ADDYCJA ELEKTROFILOWA FLUOROWCOWODORÓW DO ALKENÓ I ALKINÓW
W tej niestereospecyficznej reakcji addycji tworzą się monofluorowcopochodne (z alkenów) lub
difluorowcopochodne (z alkinów). Addycję tę cechuje wysoka regioselektywność (określana tzw.
Regułą Markownikowa) determinowana trwałością tworzącego się przejściowo karbokationu.
przykłady:
a) addycja elektrofilowachlorowodoru do 1-metylocykloheksenu daje achiralną monochloropochodną
(reg. Markownikowa)
achiralny
Cl
CH3
Cl
CH3
H
CH3
b) addycja bromowodoru do achiralnego 4-metylocyklopentenu daje mieszaninę czterech
stereoizomerów
trans - R A C E M A TBr
H
CH3
H
Br
H
CH3
H+
+
H
Br
CH3
H
+
CH3
H
CH3
HH
CH3
H
H
Br
CH3
H
Brcis - R A C E M A T
c) addycja jodowodoru do 1-butenu daje racemiczny jodek sec-butylu (reg. Markownikowa)
R A C E M A T
C
C2H5
CH3
HI C
C2H5
CH3
HI+
ICH3CH2CHCH3
HCH3CH2CH CH2
d) addycja chlorowodoru zarówno do cis- jak i trans-2-pentenu prowadzi do mieszaniny tych samych
trzech produktów: achiralnego 3-chloropentanu i racemicznego 2-chloropentanu
+
achiralny
+ C
C3H7
CH3
HClC
C3H7
CH3
HCl
C2H5CHCH2CH3
Cl
Cl
Cl
C2H5CH2CHCH3
C2H5CHCH2CH3
+
H
HCH CH
C2H5
CH3
CH CH
C2H5 CH3
(E)-2-penten
(Z)-2-penten R A C E M A T

e) addycja chlorowodoru do propynu prowadzi do produktu podstawionego geminalnie (reg.
Markownikowa)
HClCH3CCl2CH3CH3C CH2
ClHCl
C CCH3 H
4. ADDYCJA RODNIKOWA BROMOWODORU DO ALKENÓW (reakcja Karasha)
Alkeny ulegają addycji bromowodoru w obecności nadtlenków dając monobromopochodne.
Reakcja ma charakter rodnikowy i nie jest stereospecyficzna. Cechuje ją natomiast regioselektywność
(warunkowana trwałością tworzącego się przejściowo rodnika) przeciwna niż w przypadku addycji
elektrofilowej (tworzy się w przewadze produkt niezgodny z reguła Markownikowa)
a) w reakcji HBr z izobutenem w przewadze tworzy się 1-bromo-2-metylopropan
CH3C CH2
CH3HBr
ROORCH3C CH2
CH3
H Br
b) w reakcji HBr z 1-metylocykloheksenem tworzy się mieszanina 4 stereoizomerów
HBr
ROORCH3 CH3
Br
CH3
Br
CH3BrCH3 Br
RACEMAT trans- RACEMAT cis-
+ + +
5. REAKCJA ALKOHOLI Z KWASAMI FLUOROWCOWODOROWYMI
Alkohole reagują ze wszystkimi kwasami fluorowcowodorowymi dając odpowiednie halogenki.
Szybkość tych reakcji zależy od rodzaju kwasu fluorowcowodorowego (szybkość wzrasta wraz z
liczbą masową fluorowca) oraz od rzędowości alkoholu (szybkość reakcji wzrasta ze wzrostem
rzędowośći)
a) Alkohole I-rzędowe reagują łatwo jedynie z kwasami: bromowodorowym i jodowodorowym.
Reakcja z kwasem chlorowodorowym jest powolna i wymaga katalizatora (stosuje się chlorek
cynku).
b) Alkohole II i III-rzędowe reagują z kwasem chlorowodorowym bez katalizatora. (Przykładowo:
chlorek t-butylu otrzymuje się z bardzo dobrą wydajnością poprzez kilkunastominutowe
wytrząsanie t-butanolu z kwasem chlorowodorowym w rozdzielaczu.)
Reakcje te są dokładnie omówione w rozdziale dotyczącym alkoholi. Biegną one wg mechanizmu
jednocząsteczkowego SN1 w przypadku alkoholi III-rzędowych, allilowych i benzylowych. Akohole I
i II-rzędowe reagują wg mechanizmu SN2 lub SN1 (w zależności od warunków reakcji).
R-OH + HX R-X + H2O
W reakcjach tych nie można otrzymać czystego optycznie halogenku z optycznie czynnego alkoholu
(z uwagi na całkowity lub częściowy przebieg tych reakcji wg mechanizmu SN1). Pozwalają na to
reakcje wymienione w punktach: 5 i 8.
6. REAKCJA ALKOHOLI Z CHLORKIEM TIONYLU
Alkohole reagują łatwo z chlorkiem tionylu w stereospecyficznej reakcji substytucji
wewnątrzcząsteczkowej (połączonej z eliminacją dwutlenku siarki) w tworzącym się przejściowo
estrze kwasu chlorosiarkowego(IV) (reakcja SNi). Reakcja przebiega z retencją konfiguracji.
Etap I R-OH + SOCl2 R-OSOCl + HCl

Etap II
C
R1
R2
H
O
SCl
O
C
R1
R2
H
Cl + SO2
sumarycznie: R-OH + SOCl2 R-Cl + SO2 + HCl
Reakcja ta przebiega szybko i jest bardzo dogodna z praktycznego punktu widzenia, gdyż oprócz
halogenku tworzą się w niej jedynie produkty gazowe. Jej modyfikacją jest reakcja z dodatkiem
pirydyny, która przebiega z inwersją konfiguracji (reakcja SN2 ). Jej mechanizm jest następujący:
Etap I
HCl+ C
R1
R2
H
OH
SOCl2C
R1
R2
H
OSOCl
Etap II C5H5N + HCl C5H5NH+ + Cl-
Etap III
C
R1
R2
H
O
SCl
O
C
R
R2
H
Cl + SO2Cl Cl+
7. REAKCJA ALKOHOLI Z HALOGENKAMI FOSFORU (PCl3 i PBr3)
Alkohole reagują z trichlorkiem i tribromkiem fosforu dając odpowiedni halogenek i kwas
fosforowy. Jakkolwiek reakcja przebiega równie szybko jak reakcja z chlorkiem tionylu to jednak jest
mniej dogodna z praktycznego punktu widzenia ze wzgledu na konieczność oddzielania tworzącego
się w toku reakcji kwasu fosforowego.
3 R-OH + PX3 3 R-X + H3PO3
8. REAKCJA ALKOHOLI Z JODEM LUB BROMEM W OBECNOŚCI FOSFORU
Modyfikacją powyższej reakcji jest zastosowanie - zamiast halogenku fosforu - jodu (lub bromu) i
czerwonego fosforu. Przyjmuje się, że halogenek fosforu tworzy się in situ i reaguje z alkoholem wg.
podanego w punkcie 6 równania reakcji. Reakcja z zastosowaniem jodu pozwala na otrzymywanie
jodków alkilowych z bardzo wysokimi wydajnościami.
6 R-OH + 2P + 3I2 6 R-I + 2H3PO3
9. SUBSTYTUCJA NUKLEOFILOWA W SULFONIANACH I SILILOKSYALKANACH
Sulfoniany alkilowe reagują bardzo wydajnie z halogenkami nieorganicznymi dając - w reakcji
substytucji - odpowiednie halogenki alkilowe. Jest to typowa reakcja SN2 (rozdział 5.2) i przebiega z
inwersją konfiguracji
Cl C6H5SO3+ C
R1
R2
H
ClC
R1
R2
H
O S C6H5
O
O

Podobnie reagują sililoksyalkany:
Br CH3SiO
CH3
CH3
+ C
R1
R2
H
BrC
R1
R2
H
O Si CH3
CH3
CH3
10. REAKCJA ZWIĄZKÓW KARBONYLOWYCH Z PENTACHLORKIEM FOSFORU
Reakcja aldehydów lub ketonów z pentachlorkiem fosforu pozwala na otrzymanie gem -
dichloropochodnych:
R1COR2 + PCl5 R1CCl2R2 + POCl3 (Ri - H, grupa alkilowa lub arylowa)
Metody syntezy fluorowcopochodnych allilowych
1) Chlorek allilu otrzymuje się w reakcji chloru z propynem w temp. 350oC.
CH2 CH CH3
Cl2
350 Co
CH2 CH CH2 Cl
2) Ogólną metodą wprowadzania bromu w pozycję allilową (w pozycję sąsiadującą z podwójnym
wiązaniem) jest reakcja alkenów z N-bromoimidem kwasu bursztynowego (N-
bromosuccinimidem - NBS)
CH2 CH2
C C
O OH HO O
Kwas bursztynowy (Succinic acid )
Imid kwasu bursztynowego (Succinimide )
CH2 CH2
C C
O N O
H
CH2 CH2
C C
O N O
Br
N-bromoimid kwasu bursztynowego (N-Bromsuccinimide )
Reakcję tę prowadzi się najczęściej w czterochlorku węgla w temperaturze wrzenia.
CH2 CH2
C C
O N O
H
CH2 CH2
C C
O N O
Br
CH3 CH2 CH CH2+ +CH3 CH CH CH2
Br
CCl4

Metody syntezy fluorowcopochodnych winylowych
Fluorowcopochodne tej grupy otrzymuje się zasadniczo dwoma metodami:
1) Częściowa addycja halogenowodoru do alkinów
CHCCH3
HBrCH2CCH3
Br
2) Częściowa eliminacja halogenowodoru w geminalnych lub wicynalnych
difluorowcopochodnych:
CH3 CH2 CH2 CHCl2KOH
alk.CH3 CH2 CH CH Cl
Metody syntezy fluorowcopochodnych arylowych
1. SUBSTYTUCJA ELEKTROFILOWA W ARENACH
W reakcji substytucji elektrofilowej otrzymuje się jedynie chloro- lub bromopochodne
aromatyczne:
Ar-H + X2 + katalityczna ilość Fe Ar-X + HX ( X= Cl, Br)
Reakcjom tym ulega większość związków aromatycznych niezależnie od charakteru posiadanych grup
funkcyjnych.
2. SUBSTYTUCJA W SOLACH DIAZONIOWYCH
Ważną metodą syntezy fluorowcopochodnych aromatycznych, której zaletą jest wysoka
wydajność oraz łatwość oddzielenia pożądanego produktu od pozostałych reagentów jest metoda
wykorzystująca jako substraty aminy aromatyczne., które łatwo otrzymuje się z odpowiednich
nitrozwiązków. Aminy aromatyczne przeprowadza się najpierw w stosunkowo trwałe w niskiej
temperaturze sole diazoniowe. Substytucja grupy diazoniowej atomem fluorowca pozwala na
otrzymanie dowolnego rodzaju halogenku aromatycznego:
Ar FBF4Ar N2
Ar I
Ar Br
Ar Cl
KI
CuBr
CuCl
NaBF4
ClAr N NAr N NAr NH20 C
oNaNO2,HCl,
Sól diazoniowa
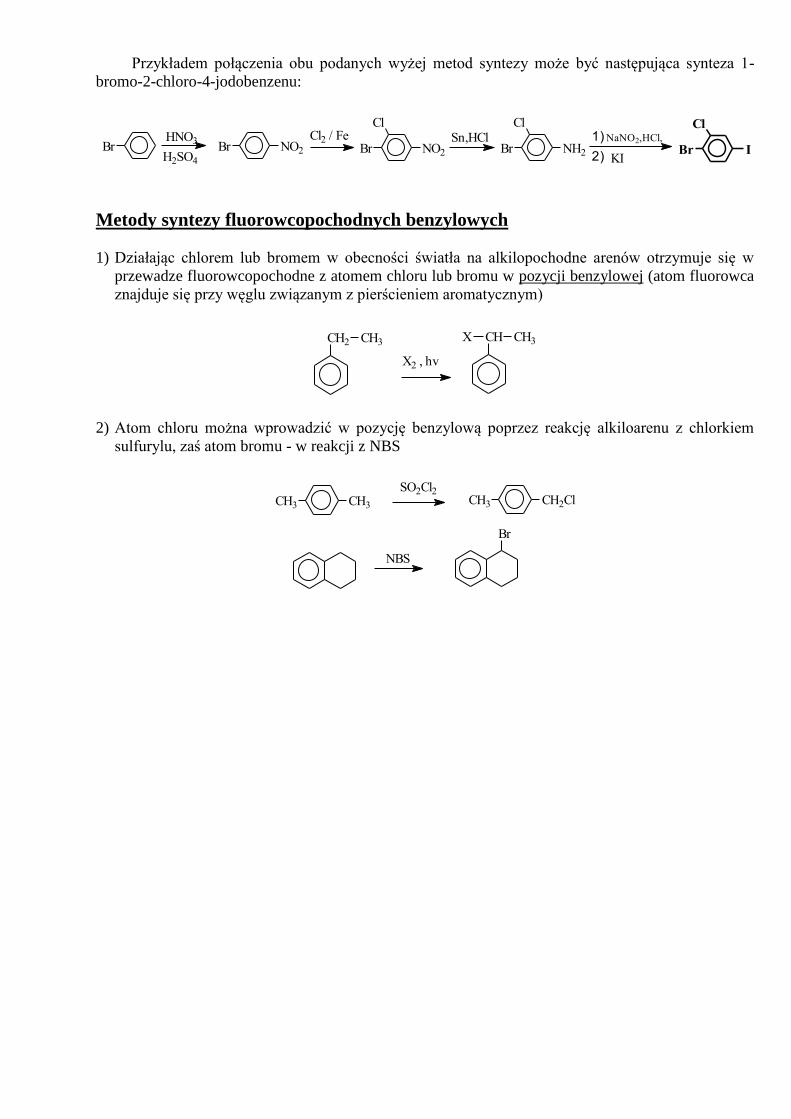
Przykładem połączenia obu podanych wyżej metod syntezy może być następująca synteza 1-
bromo-2-chloro-4-jodobenzenu:
BrH2SO4
HNO3NO2Br
Cl2 / FeNO2Br
ClSn,HCl
2)
1) NaNO2,HCl,
KINH2Br
Cl
IBr
Cl
Metody syntezy fluorowcopochodnych benzylowych
1) Działając chlorem lub bromem w obecności światła na alkilopochodne arenów otrzymuje się w
przewadze fluorowcopochodne z atomem chloru lub bromu w pozycji benzylowej (atom fluorowca
znajduje się przy węglu związanym z pierścieniem aromatycznym)
CH2 CH3 CH CH3X
X2 , hv
2) Atom chloru można wprowadzić w pozycję benzylową poprzez reakcję alkiloarenu z chlorkiem
sulfurylu, zaś atom bromu - w reakcji z NBS
NBS
Br
CH3 CH3
SO2Cl2CH3 CH2Cl

B IO
TE
HC N OL
O GAI
CHEMIAORGANICZNA
Reaktywność
Zastąpienie atomu wodoru w cząsteczce alkanu atomem fluorowca pociąga za sobą duże zmiany
we własnościach chemicznych cząsteczki. Z biernej chemicznie staje się ona bardzo reaktywna, łatwo
ulegając przemianom pod wpływem wielu czynników - szczególnie cząsteczek o charakterze
nukleofilowym. Fluorowcopochodne mogą reagować z nukleofilami w dwojaki sposób:
1) Nukleofil wchodzi na miejsce fluorowca: mamy wówczas do czynienia z REAKCJĄ
SUBSTYTUCJI (podstawienia), którą - w zależności od charakteru czynnika nukleofilowego - można
przedstawić jednym z poniższych równań:
C C
H
X
(nuklefil może być cząsteczką obojętną - np. lub anionem np. OH )
C C
H
Nu
C C
H
Nu
+
+
XNu
NuX
NH3 X - atom fluorowca
2) Atakujący nukleofil odrywa atom wodoru od węgla sąsiadującego z węglem związanym z
fluorowcem (tzw. węgla ). W wyniku odejścia protonu oraz anionu halogenkowego, z wyjściowej
cząsteczki tworzy się cząsteczka alkenu. Proces ten nazywamy REAKCJĄ ELIMINACJI.
Nu H
Nu H
+
+
C C
H
X C C
C C
+
+
XNu
NuX
Są dwie zasadnicze przyczyny wspomnianej wyżej dużej reaktywności fluorowcopochodnych:
1. PIERWSZĄ PRZYCZYNĄ jest względna łatwość oderwania anionów halogenkowych (X-) od
atomu węgla. Przykładowo: powyższe reakcje (substytucji i eliminacji) nie zachodzą dla amin
(zawierających w grupę NH2 zamiast halogenu) lub alkoholi (zawierających grupę OH) gdyż aniony:
NH2- i OH- trudno jest oderwać od atomu węgla mimo, że np. wiązanie węgiel-tlen jest także
spolaryzowane. Mówi się, że aniony halogenkowe są grupami łatwo odchodzącymi w przeciwieństwie
do anionów OH- i NH2-. Brak jest przekonywującego wyjaśnienia tego zjawiska. Własność tę można
uważać za jakościowo analogiczną do własności silnej kwasowości halogenowodorów (aniony
halogenkowe łatwo jest oderwać od protonu) w zestawieniu z bardzo słabą kwasowością amoniaku i
wody (aniony: OH- i NH2- trudno jest oderwać od protonu). W oparciu o analogiczne spostrzeżenia dla
szeregu innych związków organicznych przyjmuje się, że opisane wyżej reakcje substytucji i eliminacji
zachodzą tym łatwiej im niższa jest zasadowość grupy odchodzącej (a tym samym: im wyższa jest
kwasowość sprzężonego z tą grupą kwasu). Niską zatem zasadowość anionów halogenkowych można
uważać za ważny powód determinujący różnorodność przemian, jakim ulegają fluorowcopochodne.

Łatwość odchodzenia anionów halogenkowych (w porównaniu z innymi grupami) wiąże się m.in. z
możliwością HETEROLITYCZNEJ dysocjacji wiązania węgiel-fluorowiec w środowisku polarnym, z
wytworzeniem wysoce reaktywnego i nietrwałego KARBOKATIONU.
R
C
H H
CC
H H
H
X
H
+ R
C
H H
CC
H H
H
H
KARBOKATION
+ X
Jest to proces bardzo powolny a szybkość jego zależy między innymi od rzędowości atomu węgla
związanego z fluorowcem. Wytworzony karbokation ulega szybkiej reakcji następczej, która może być
jednym z trzech poniższych procesów:
a) addycji innego nukleofila obecnego w ukłądzie reakcyjnym. Mówimy wówczas, że wyjściowa
fluorowcopochodna ulega reakcji SUBSTYTUCJI (SN)
b) odszczepieniu protonu od sąsiedniego atomu węgla przez czynnik zasadowy i utworzeniu podwójnego
wiązania. Mamy wówczas do czynienia z reakcją ELIMINACJI (E)
c) przegrupowaniu do bardziej trwałego karbokationu, który następnie ulega reakcji a) lub b)
2) DRUGĄ PRZYCZYNĄ reaktywności fluorowcopochodnych jest polarny charakter wiązania:
węgiel – fluorowiec (Rys. 1). Fakt ten implikuje ważną konsekwencję: pojawienie się w cząsteczce
fluorowcopochodnej centrum elektrofilowego podatnego na atak czynników nukleofilowych
R
C
H H
CC
H H
H
X
H
+
X - atom fluorowca
Rys. 1
Centra z cząstkowym ładunkiem dodatnim wykazują powinowactwo do czynników
nukleofilowych. Centrum takie (zwane centrum elektrofilowym) może tworzyć nowe wiązanie z
nukleofilem pozbywając się przy tym innej grupy zwanej grupą odchodzącą (patrz Rys. 2) - reakcja taka
jest reakcją SUBSTYTUCJI. W cząsteczkach fluorowcopochodnych może ona przebiegać na atomie
węgla związanym z fluorowcem z uwagi na występujący na nim cząstkowy ładunek dodatni oraz na
fakt, że pierwotnie związana z węglem grupa (atom fluorowca) może ulec oderwaniu (jest grupą łatwo
odchodzącą). Atakujący cząsteczkę nukleofil ma jednak własności zasadowe i może w związku z tym
atakować także protony w łańcuchu alkilowym, co może prowadzić do ich oderwania od cząsteczki.
(inaczej: procesu kwasowo-zasadowej wymiany protonu). W wyniku oderwania protonu od łańcucha
węglowego tworzy się jednak na ogół wysokoenergetyczny i bardzo nietrwały karboanion (Rys. 3),
który może się stabilizować jedynie w procesie odwrotnym tzn. addycji protonu. Atak nukleofila na
większość atomów wodoru jest zatem nieskuteczny i na ogół nie powoduje zmian w cząsteczce (z tego
właśnie powodu węglowodory nasycone nie reagują z czynnikami nukleofilowymi).
Rys. 2
A YNu + Nu A + Y
(gr. odchodząca)(nukleofil)

C
C
H H
CC
H H
H
X
H
C
H H
H
HH
+
Nu
+
C
C
H H
CC
H H
H
X
H
C
H
H
HH
.. + NuH
Rys. 3
Wyjątek stanowią tu atomy wodoru przy węglu . Na tych atomach węgla, reakcja oderwania
protonu może przebiegać łatwiej niż na innych atomach węgla obecnych w cząsteczce. Jest to
spowodowane specyficzną możliwością połączenia procesu oderwania protonu z eliminacją anionu
fluorowca oraz jednoczesnego utworzenia wiązania między atomami węgla i . W zależności od
budowy cząsteczki (przede wszystkim od rzędowości atomu węgla związanego z atomem fluorowca),
oraz własności atakującego czynnika nukleofilowego, atom węgla związany z fluorowcem (atom
) oraz atomy wodoru przy węglach KONKURUJĄ ZE SOBĄ w procesie wychwytu
czynnika nukleofilowego prowadząc w efekcie do - wspomnianych wyżej - dwu ważnych w chemii
organicznej procesów: REAKCJI SUBSTYTUCJI (SN) oraz REAKCJI ELIMINACJI (E):
C
C
H H
CC
H H
H
X
H
C
H H
H
HH
+C
C
H H
CC
H H
H
Nu
H
C
H
H
HH
H
+
NReakcja SNu
X
C
C
H H
CC
H H
H
X
H
C
H H
H
HH
+
+
Nu
X
Reakcja E +
C
C
H
CC
H H
HC
H H
H
HH
H
NuH
Podsumowanie rozważań:
Reakcja SN jest reakcją substytucji na atomie węgla związanym z grupą odchodzącą (w przypadku
fluorowcopochodnych jest to atom węgla związany z fluorowcem). Reakcja E przebiega z
odszczepieniem protonu przy węglu połączonym z eliminacją anionu fluorowca. W zależności od
warunków reakcji, procesy: SN oraz E mogą przebiegać:
1) poprzez stadium karbokationu, który stabilizuje się następnie bądź przez addycję nukleofila (SN) lub
eliminację protonu (E). Reakcje te są zatem DWUETAPOWE.
2) bez wstępnej jonowej dysocjacji wiązania C-X, lecz poprzez atak nukleofila na jeden z dwu atomów:
- atom węgla związany z fluorowcem (reakcja SN) lub atom wodoru przy węglu (reakcja E). Reakcje
te są JEDNOETAPOWE.

Reakcje SN i E nie dotyczą wyłącznie fluorowcopochodnych. Ulegają im również inne grupy
związków organicznych jak: alkohole, etery, estry - szczególnie estry kwasów sulfonowych,
sililoksyalkany itp. Przebieg tych reakcji, ich mechanizmy, kinetyka, stereochemia oraz wzajemna
konkurencja były bardzo szczegółowo badane w pierwszej połowie obecnego stulecia. Ustalono szereg
praw rządzących ich przebiegiem pozwalających ze stosunkowo dużym prawdopodobieństwem
przewidywać kierunek reakcji oraz warunki jej przebiegu.
Reakcje te można w sposób uproszczony zapisać następująco:
SN : R-Y + Nu R-Nu + Y
E : R-Y + Nu Alken + HNu + Y
gdzie: Y - grupa odchodząca (w substracie może być elektrycznie obojętna lub naładowana
dodatnio, zaś po odejściu odpowiednio: anionem lub cząsteczką obojętną)
Nu - nukleofil (może być cząsteczką obojętną lub anionem zaś po związaniu z atomem
węgla lub protonem odpowiednio: grupą naładowaną dodatnio lub elektrycznie obojętną)
Przykłady:
E
S N
NS
Br+CH3OH(CH3)2CH Br CH3O+ CH3CH CH2+
Cl+ CH3CH2 Cl +CH3CH2 SO3C6H5
H2O+(CH3)2CH I+ I(CH3)2CH OH2
C6H5SO3
Jak wspomniano wyżej, zarówno reakcje SN jak i reakcje E mogą być procesami jedno- lub
dwuetapowymi. W oparciu o badania kinetyczne stwierdzono, że procesy te różnią się rzędem reakcji
oraz jej cząsteczkowością. Ich charakterystykę przedstawia poniższa tabela:
Cząsteczkowość Rzędowość Wyrażenie na szybkość
reakcji
Proces JEDNOETAPOWY
reakcja dwucząsteczkowa
II rz
v = k1 . [R-X] . [Nu]
Proces DWUETAPOWY
Etap I - tworzenie karbokationu -
etap wolny
Etap II - reakcja karbokationu z
czynnikiem nukleofilowym
- etap szybki
reakcja jednocząsteczkowa
(w etapie decydującym o
szybkości reakcji czyli w
etapie powolnym bierze udział
jedna cząsteczka)
I rz
v = k2 . [R-X]
(szybkość reakcji nie zależy od
stężenia nukleofila)

B IO
TE
HC N OL
O GAI
CHEMIAORGANICZNA
Definicje i klasyfikacja
Reakcja SUBSTYTUCJI ( SN ) na atomie węgla sp3
Grupa nukleofilowa (Nu1) związana z atomem węgla sp3 ulega wymianie (substytucji) na inną grupę
nukleofilową (Nu2). CENTRUM ATAKU nukleofila Nu2 jest ATOM WĘGLA sp3 związany z
grupą odchodzącą ( atom
grupa ATAKUJĄCA
grupa ODCHODZĄCA
1 + Nu12
Nu+ 2
1 + Nu12
Nu+ 2
2+ Nu2 1
Nu+1C Nu
)(
)(
C Nu
C NuC Nu
C Nu C Nu
GRUPA ATAKUJĄCA (nukleofil Nu2) tworzy wiązanie z atomem kosztem swojej pary
elektronowej.
GRUPA ODCHODZĄCA (Nu1) odchodzi razem z parą elektronów, które wiązały ją z atomem
węgla.
PRZYKŁADY:
++
grupa ODCHODZĄCA
grupa ATAKUJĄCA
CH3CH2 F OHH2O
CH3CH2 OH F1)
2)
3)
CH3 OH
H+ Br CH3 Br + O
H
H(woda)
CH3CHCH2
CH3
Br + +NH3 CH3CHCH2
CH3
NH3 Br

Dwie możliwości przebiegu reakcji:
Reakcja: JEDNOCZĄSTECZKOWA, DWUETAPOWA, I rzędu
1C Nu
- prod. pośr. reakcji
JEDNOCZĄSTECZKOWY - decydujący o szybkości reakcji
1Nu+C
KARBOKATION
ETAP I - powolny
DWUCZĄSTECZKOWY - nie wpływa na szybkość reakcji
ETAP II - szybki
+
KARBOKATION
2Nu C Nu
2C
v = k C Nu1
SN1
S1
Reakcja: DWUCZĄSTECZKOWA, JEDNOETAPOWA, II rzędu
- nie jest to produkt pośredni reakcji STAN PRZEJŚCIOWY
("pięciowiązalny" atom węgla - wiązania narysowane linią przerywaną są wiązaniami słabymi)
2 1Nu C Nu
C NuC Nu1 + Nu12
Nu+ 2
v = k C Nu1 Nu2
SN2
S2
Reakcja ELIMINACJI ( E ) na sąsiednich atomach węgla
Związana z atomem węgla grupa nukleofilowa (Nu1) oraz proton przy atomie węgla ulegają
eliminacji pod wpływem czynnika zasadowego (Nu2). CENTRUM ATAKU nukleofila Nu2 jest ATOM
WODORU przy atomie węgla
+
H NuC NuC
H1
+ Nu12
Nu+2
CC +
+CC
grupa ATAKUJĄCA
grupa ODCHODZĄCA
2+ Nu
2 1Nu+
1C NuC
H
H Nu
+CC
2+ Nu
2 1Nu+
1C NuC
H
H Nu
GRUPA ATAKUJĄCA (nukleofil Nu2) odrywa proton przy węglu i tworzy z nim wiązanie
kosztem pary elektronowe. GRUPA ODCHODZĄCA (Nu1) odchodzi razem z parą elektronów,
które wiązały ją z atomem węgla. PATRA ELEKTRONÓW zaangażowana wcześniej w wiązanie z
protonem tworzy wiązanie między atomami węgla i .

PRZYKŁADY:
BrH NH3NH3 ++CH3C Br
CH3
CH2 H
(woda) OH
H++CH2CH2 O
H
HH
3)
2)
1) BrH ORORCH2CH Br
CH3
H
grupa ATAKUJĄCA
grupa ODCHODZĄCA
+ +CH2 CH
CH3
+
CH3C
CH3
CH2
+
HO S O
O
O
CH2 CH2 HO S O
O
OH
+
Dwie możliwości przebiegu reakcji:
Reakcja: JEDNOCZĄSTECZKOWA, DWUETAPOWA, I rzędu
ETAP I - powolnyKARBOKATION
+ Nu1
JEDNOCZĄSTECZKOWY - decydujący o szybkości reakcji
- prod. pośr. reakcji
1
C NuC
H
CC
H
Nu2
KARBOKATION
+
ETAP II - szybkiDWUCZĄSTECZKOWY - nie wpływa na szybkość reakcji
CC
H+CC
2H Nu
E1
1E
1C Nuv = k
Reakcja: DWUCZĄSTECZKOWA, JEDNOETAPOWA, II rzędu
- nie jest to produkt pośredni reakcji STAN PRZEJŚCIOWY
+ Nu1
H Nu2
CC +
C NuC
H1 2
Nu+
CC
Nu
H
1
Nu2
E2
2E
2Nu1C Nuv = k
B IO
TE
HC N OL
O GAI
CHEMIAORGANICZNA
Mechanizmy reakcji
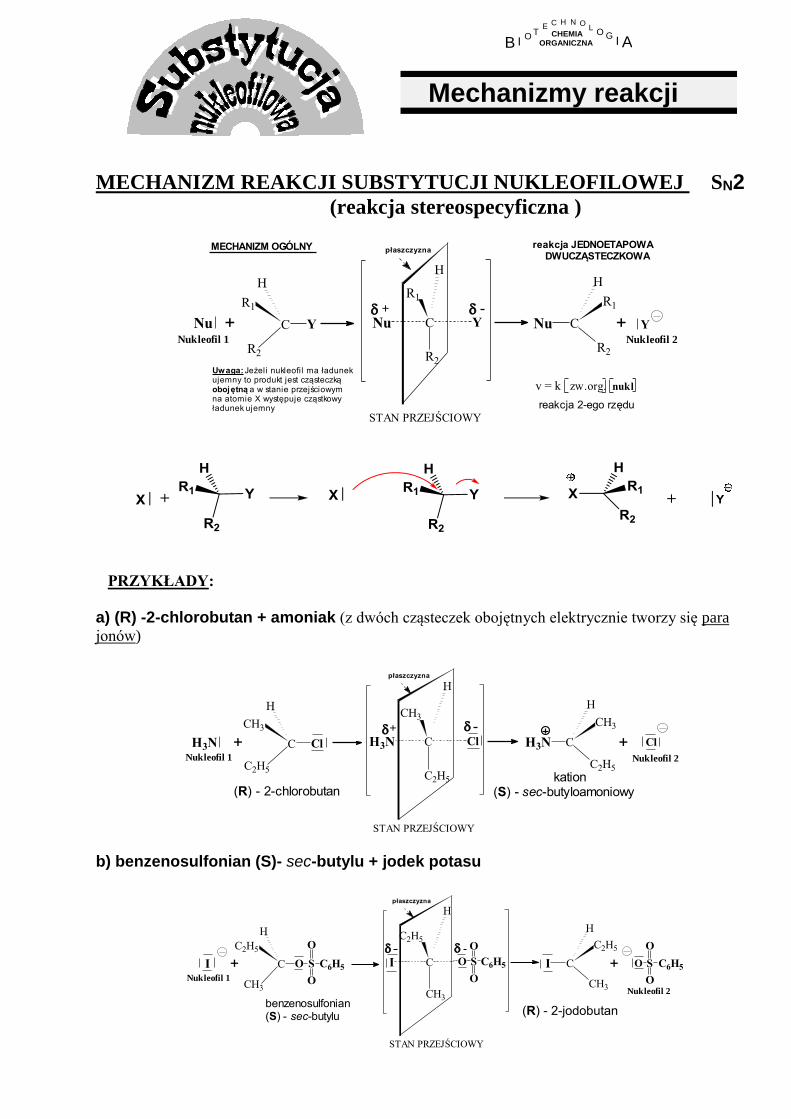
MECHANIZM REAKCJI SUBSTYTUCJI NUKLEOFILOWEJ SN2 (reakcja stereospecyficzna )
+
Nukleofil 1
Uwaga: Jeżeli nukleofil ma ładunekujemny to produkt jest cząsteczkąobojętną a w stanie przejściowymna atomie X występuje cząstkowyładunek ujemny
+Nu C
R2
R1
H
Y
płaszczyzna
+ YC
R1
H
Y
R2
Nu
STAN PRZEJŚCIOWY
C
R2
R1
H
Nu Nukleofil 2
v = k nuklzw.org.
MECHANIZM OGÓLNY reakcja JEDNOETAPOWA DWUCZĄSTECZKOWA
reakcja 2-ego rzędu
PRZYKŁADY:
a) (R) -2-chlorobutan + amoniak (z dwóch cząsteczek obojętnych elektrycznie tworzy się para
jonów)
+
Nukleofil 1
(R) - 2-chlorobutan
+H3N C
C2H5
CH3
H
Cl
płaszczyzna
+ ClC
CH3
H
Cl
C2H5
H3N
STAN PRZEJŚCIOWY
C
C2H5
CH3
H
H3N Nukleofil 2
kation(S) - sec-butyloamoniowy
b) benzenosulfonian (S)- sec-butylu + jodek potasu
(R) - 2-jodobutan
Nukleofil 2
C
CH3
C2H5
H
I
STAN PRZEJŚCIOWY
C
C2H5
H
O
CH3
I S C6H5
O
O
O S
O
O
C6H5+
płaszczyzna
C
CH3
C2H5
H
O S C6H5
O
O
I +
benzenosulfonian(S) - sec-butylu
Nukleofil 1

c) (S) – butan-2-ol + HBr aq (z pary jonów tworzą się dwie cząsteczki obojętne elektrycznie)
+
Nukleofil 1
kation(S) - sec-butylooksoniowy
+ H2OC
CH3
C2H5
H
OH
H
płaszczyzna
+Br C
C2H5
H
O
CH3
BrH
H
STAN PRZEJŚCIOWY
C
CH3
C2H5
H
Br
Nukleofil 2
(R) - 2-bromobutan
d) (R)-2-bromo-1metoksypropan + KCN (konfiguracja absolutna nie ulega zmianie)
Nukleofil 1
(R) - 2-bromo-1-meto- ksypropan
+CN C
CH3OCH2
CH3
H
Br
płaszczyzna
+ BrC
CH3
H
Br
CH2
CN
CH3O
STAN PRZEJŚCIOWY
C
CH2OCH3
CH3
H
CN Nukleofil 2
(R) - 3-metoksy-2-metylopro- pionitryl
Komentarz: Atakujący nukleofil i grupa odchodząca muszą znajdować się po przeciwnych
stronach płaszczyzny przechodzącej przez atom węgla i prostopadłej do osi wiązania węgiel – grupa
odchodząca. W stanie przejściowym tej JEDNOETAPOWEJ REAKCJI:
– wytwarza się słabe wiązanie pomiędzy atakującym nukleofilem i atomem węgla (kosztem wolnej
pary elektronowej tego nukleofila)
– wiązanie pomiędzy atomem węgla i grupą odchodzącą ulega osłabieniu i wzmożonej polaryzacji.
– pozostałe trzy wiązania sigma atomu węgla znajdują się w opisanej wyżej płaszczyźnie
(przechodzącej przez atom węgla i prostopadłej od osi słabych wiązań tego atomu z obydwoma
nukleofilami). W wyniku eliminacji grupy odchodzącej i wytworzenia wiązania z atakującym
nukleofilem następuje przejście wspomnianych wyżej trzech wiązań sigma na drugą stronę
rozpatrywanej płaszczyzny. Proces ten nazywa się INWERSJĄ KONFIGURACJI.
KONSEKWENCJE STEREOCHEMICZNE:
a) W każdej reakcji SN 2 zachodzi INWERSJA KONFIGURACJI. Z chiralnego substratu tworzy się
chiralny produkt
b) Inwersja konfiguracji NIE MUSI pociągać za sobą zmiany konfiguracji absolutnej (produktu w
stosunku do substratu). Konfiguracja absolutna atomu węgla jest bowiem zdeterminowana szeregiem
pierwszeństwa związanych z tym atomem podstawników. Szereg pierwszeństwa zaś, w trakcie reakcji
substytucji, może ulegać zmianie. Przypadek taki występuje w podanym wyżej przykładzie d).
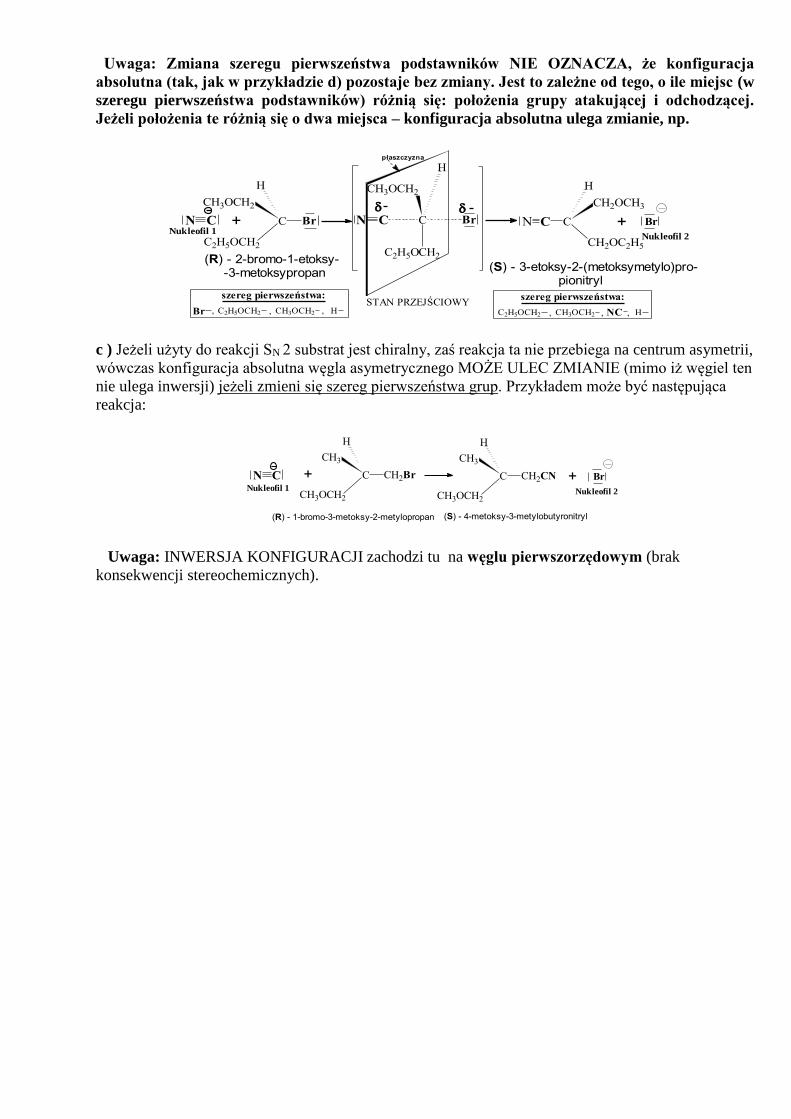
Uwaga: Zmiana szeregu pierwszeństwa podstawników NIE OZNACZA, że konfiguracja
absolutna (tak, jak w przykładzie d) pozostaje bez zmiany. Jest to zależne od tego, o ile miejsc (w
szeregu pierwszeństwa podstawników) różnią się: położenia grupy atakującej i odchodzącej.
Jeżeli położenia te różnią się o dwa miejsca – konfiguracja absolutna ulega zmianie, np.
szereg pierwszeństwa:
, H
szereg pierwszeństwa:
NCC2H5OCH2 ,, CH3OCH2HCH3OCH2 ,,, C2H5OCH2Br
Nukleofil 1
(R) - 2-bromo-1-etoksy- -3-metoksypropan
+CN C
C2H5OCH2
CH3OCH2
H
Br
płaszczyzna
+ BrC
CH3OCH2
H
Br
CH2
CN
C2H5O
STAN PRZEJŚCIOWY
C
CH2OC2H5
CH2OCH3
H
CN Nukleofil 2
(S) - 3-etoksy-2-(metoksymetylo)pro- pionitryl
c ) Jeżeli użyty do reakcji SN 2 substrat jest chiralny, zaś reakcja ta nie przebiega na centrum asymetrii,
wówczas konfiguracja absolutna węgla asymetrycznego MOŻE ULEC ZMIANIE (mimo iż węgiel ten
nie ulega inwersji) jeżeli zmieni się szereg pierwszeństwa grup. Przykładem może być następująca
reakcja:
(S) - 4-metoksy-3-metylobutyronitryl
Nukleofil 2
Br+C
CH3OCH2
CH3
H
CH2BrCN +
(R) - 1-bromo-3-metoksy-2-metylopropan
Nukleofil 1
C
CH3OCH2
CH3
H
CH2CN
Uwaga: INWERSJA KONFIGURACJI zachodzi tu na węglu pierwszorzędowym (brak
konsekwencji stereochemicznych).
B IO
TE
HC N OL
O GAI
CHEMIAORGANICZNA
Mechanizmy reakcji
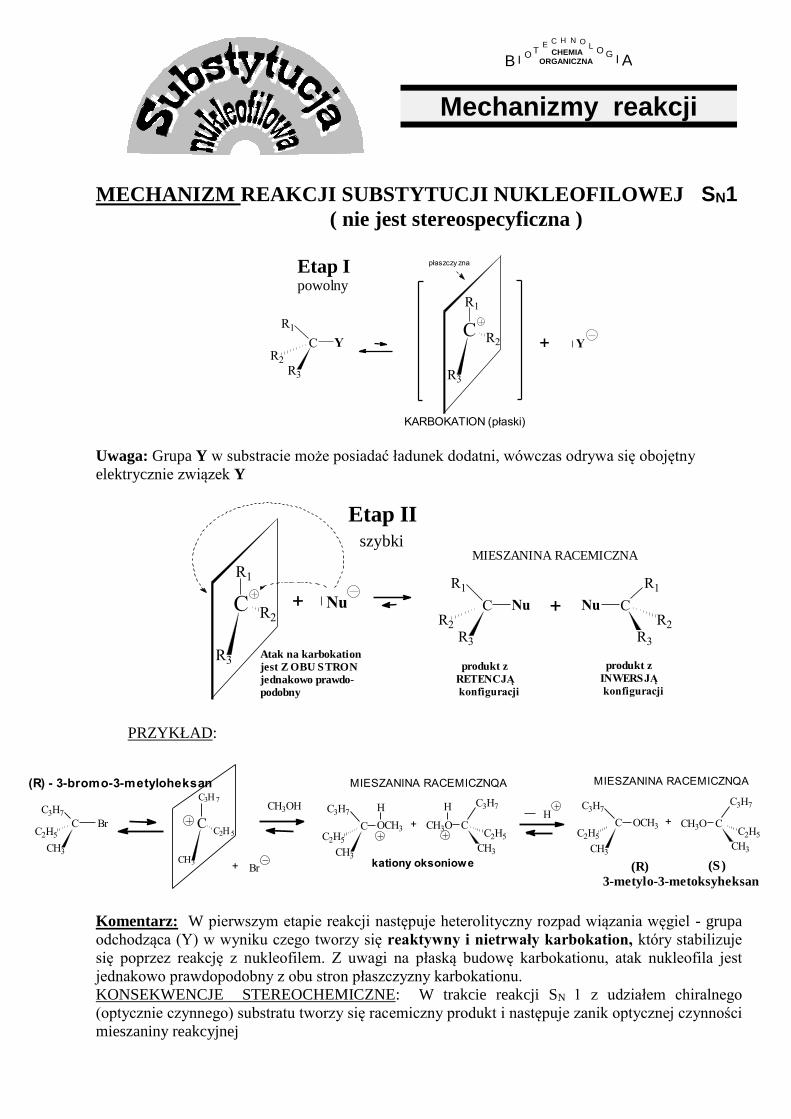
MECHANIZM REAKCJI SUBSTYTUCJI NUKLEOFILOWEJ SN1 ( nie jest stereospecyficzna )
Etap Ipowolny
Y+
płaszczy zna
KARBOKATION (płaski)
C
R3
R2
R1
C
R1
R3
R2
Y
Uwaga: Grupa Y w substracie może posiadać ładunek dodatni, wówczas odrywa się obojętny
elektrycznie związek Y
Etap II szybki
Nu+C
R3
R2
R1
C
R1
R3
R2
Nu C
R1
R3
R2
Nu+
Atak na karbokation
jest Z OBU STRON
jednakowo prawdo-
podobny
produkt z
RETENCJĄ
konfiguracji
produkt z
INWERSJĄ
konfiguracji
MIESZANINA RACEMICZNA
PRZYKŁAD:
C
C3H7
CH3
C2H5
Br
(R) - 3-bromo-3-metyloheksan
C
CH3
C2H 5
C3H 7
+ Br
MIESZANINA RACEMICZNQA
kationy oksoniowe
CH3OH
C
C3H7
CH3
C2H5
OCH3
H
C
C3H7
CH3
C2H5
CH3O
H
+ + C
C3H7
CH3
C2H5
CH3OC
C3H7
CH3
C2H5
OCH3
MIESZANINA RACEMICZNQA
H
(R) (S)
3-metylo-3-metoksyheksan
Komentarz: W pierwszym etapie reakcji następuje heterolityczny rozpad wiązania węgiel - grupa
odchodząca (Y) w wyniku czego tworzy się reaktywny i nietrwały karbokation, który stabilizuje
się poprzez reakcję z nukleofilem. Z uwagi na płaską budowę karbokationu, atak nukleofila jest
jednakowo prawdopodobny z obu stron płaszczyzny karbokationu.
KONSEKWENCJE STEREOCHEMICZNE: W trakcie reakcji SN 1 z udziałem chiralnego
(optycznie czynnego) substratu tworzy się racemiczny produkt i następuje zanik optycznej czynności
mieszaniny reakcyjnej

B IO
TE
HC N OL
O GAI
CHEMIAORGANICZNA
Mechanizmy reakcji
MECHANIZM REAKCJI ELIMINACJI E-1 ( nie jest stereospecyficzna )
C
R1
R3C
R2
Y
R4
H
C
R3C
R2
R1
HR4
KARBOKATION (płaski)
płaszczy zna
+ Y
Etap Ipowolny
Uwaga: Grupa Y w substracie może posiadać ładunek dodatni, wówczas odrywa się obojętny
elektrycznie związek Y
C
R3C
R2
R1
HR4
MIESZANINA IZOMERÓW
(E) i (Z)
Ponieważ istnieje możliwość
rotacji wokół wiązania C - C
więc atom wodoru może zostać
oderwany zarówno po jednej jak
i po drugiej stronie płaszczyzny
+ Nu
Etap II szybki
C C
R1
R2
R3
R4
+ C C
R1
R2
R4
R3
Reakcja REGIOSELEKTYWNA (reguła ZAJCEWA): Jeżeli w cząsteczce występują dwa lub
więcej atomów węgla związanych z protonem, wówczas PROTON ODRYWANY JEST OD
ATOMU WĘGLA O NAJWYŻSZEJ RZĘDOWOŚCI.
Komentarz: W pierwszym etapie reakcji następuje heterolityczny rozpad wiązania węgiel - grupa
odchodząca (Y) w wyniku czego tworzy się reaktywny i nietrwały karbokation, który stabilizuje się
poprzez odszczepienie protonu od atomu węgla . Tworzy się alken lub mieszanina
stereoizomerycznych alkenów ( E i Z)
PRZYKŁAD:
C
C2H6C
H
CH3
HCH3 +
C C
CH3
H
CH3
C2H5
+
C C
CH3
H
C2H5
CH3
HSO4C2H5CHCHCH3
O
CH3
H H
H2O

B IO
TE
HC N OL
O GAI
CHEMIAORGANICZNA
Mechanizmy reakcji
MECHANIZM REAKCJI ELIMINACJI E-2 ( reakcja stereospecyficzna )
C
R1
R2
C
R4
R3
C C
R2
Y
H
R3
R4
R1
Nu
C CR1
Y
H
R3
Nu
R4
R2
STAN PRZEJŚCIOWY
+ HNu + Y
+
Uwaga! Jeżeli nukleofil ma ładunek ujemny, wówczas w stanie przejściowym na atomie
odrywającym proton występuje cząstkowy ładunek ujemny
Reakcja jest REGIOSELEKTYWNA (reguła ZAJCEWA): Jeżeli w cząsteczce występują dwa lub
więcej atomów węgla związanych z protonem, wówczas PROTON ODRYWANY JEST OD
ATOMU WĘGLA O NAJWYŻSZEJ RZĘDOWOŚCI.
Komentarz: Cząsteczka musi mieć możliwość przyjęcia takiej konformacji, w której grupa odchodząca
(Y) oraz proton odrywany przez atakujący nukleofil, przyjmują względem siebie położenie
antyperiplanarne ( ELIMINACJA „TRANS” ). W stanie przejściowym tej JEDNOETAPOWEJ
REAKCJI:
wytwarzają się słabe wiązania między : a) atakującym nukleofilem a protonem
b) atomami węgla i słabe wiązanie
ulegają osłabieniu wiązania między: a) atomem węgla i grupą odchodzącą
b) atomem węgla i odrywanym protonem
W wyniku jednoczesnej eliminacji grupy odchodzącej od atomu węgla oraz protonu od atomu
węgla między obydwoma atomami węgla tworzy się wiązanie .
KONSEKWENCJE STEREOCHEMICZNE: W trakcie reakcji E-2 z udziałem chiralnego
(optycznie czynnego) substratu zawierającego asymetryczne atomy węgla i (oraz proton przy
asymetrycznym atomie węgla ), tworzy się alken o konfiguracji ściśle zdeterminowanej konfiguracją
obu atomów węgla oraz antyperiplanarną konfiguracją stanu przejściowego.
PRZYKŁADY:
A) (2S,3R)-2-fenylo-3-jodo-3-metylopentan + amoniak
CH3
C2H5
C+
(Z) - 2-fenylo-3-metylo-2-penten
I+ NH4+
STAN PRZEJŚCIOWY
C C
I
H
C6H5
NH3
CH3
C2H5
CH3
NH3
(2S,3R)-2-fenylo-3-jodo--3-metylopentan
C C
C2H5
I
H
C6H5
CH3
CH3
C
CH3
C6H5
Uwagi :1) W substracie występują 3 atomy węgla (I-rzędowy - w grupie metylowej, II-rzędowy - w
grupie etylowej i III-rzędowy - asymetryczny). Zgodnie z regułą Zajcewa w przewadze powstaje

produkt eliminacji protonu od III-rzędowego atomu węgla .
2) Z enancjomeru powyższego substratu - czyli stereoizomeru (2R,3S) powstanie także alken o
konfiguracji (Z), natomiast ze stereoizomerów: (2R,3R) i (2S,3S) powstanie alken o konfiguracji (E)
B) (2R,3R)-2-bromo-3-metylopentan + metanolan sodu
C
C2H5
CH3
C C
CH3
Br
H
CH3
C2H5
H
(2R,3R)-2-bromo--3-metylopentan
OCH3
C C
Br
H
CH3
OCH3
C2H5
CH3
H
STAN PRZEJŚCIOWY
+ CH3OH + Br
(E) - 3-metylo-2-penten
H
C
CH3
Uwagi :1) W substracie występują 2 atomy węgla (I-rzędowy - w grupie metylowej, i III-rzędowy -
asymetryczny). Zgodnie z regułą Zajcewa w przewadze powstaje produkt eliminacji protonu od III-
rzędowego atomu węgla .
2) Z enancjomeru powyższego substratu - czyli stereoizomeru (2S,3S) powstanie także alken o
konfiguracji (E), natomiast ze stereoizomerów: (2R,3S) i (2S,3R) powstanie alken o konfiguracji (Z)
C) cis -1-bromo-2-metylocykloheksan + alkoholowy roztwór KOH
Kierunek eliminacji ZGODNY z regułą Zajcewa
1-metylocykloheksen
CH3
Br
CH3
H
H
H
OH
D) trans -1-bromo-2-metylocykloheksan + alkoholowy roztwór KOH
OH
Br
H
CH3
H
H
CH3
3-metylocykloheksen
Kierunek eliminacji NIEZGODNY z regułą Zajcewa
E) (Z) -1-bromopropen + amidek sodu F) (E) -1-bromopropen + amidek sodu
CC
CH3
HH
Br
NH2
C C CH3H
propyn
CC
H
CH3H
Br NH2
REAKCJA NIE ZACHODZI
G) (Z) -2-bromo-2-buten + amidek sodu H) (E) -2-bromo-2-buten + amidek sodu
CC
CH3
HCH3
Br NH2
CH3 C C CH3
2-butyn
CH2 C CCH3
HNH2
CC
H
CH3CH3
Br
1,2-butadien

B IO
TE
HC N OL
O GAI
CHEMIAORGANICZNA
Szybkość reakcji i konkurencja
Czynniki wpływające na szybkość reakcji SN i E:
1. Zasadowość grupy odchodzącej
Reguła 1 Reakcje SN lub E przebiegają tym łatwiej im niższa jest zasadowość grupy odchodzącej
PRZYKŁADY:
1-propoanol
A) reakcja NIE PRZEBIEGA gdyż grupą odchodzącą musiałby być anion OH-,który jest MOCNĄ zasadą
KICH3CH2CH2OH
alkohole NIE REAGUJĄ z halogenkami nieorganicznymi
+HI
CH3CH2CH2OH CH3CH2CH2I H2O reakcja PRZEBIEGA ponieważ w pierwszym etapienastępuje protonowanie cząsteczki alkoholu z wy-tworzeniem kationu oksoniowego, w którym grupąodchodzącą jest już SŁABA ZASADA - cząsteczkawody (nukleofilem jest anion jodkowy)
CH3CH2CH2OH
H
CH3CH2CH2OH
B)
H ICH3CH2CH2I
kation oksoniowy
1-propoanol
alkohole REAGUJĄ z halogenowodorami (reakcja SN)
KIC) CH3CH2CH2O S C6H5
O
Obenzenosulfonian propylu
O S C6H5
O
O
CH3CH2CH2I + reakcja PRZEBIEGA ponieważ gru-pą odchodzącą jest anion benzeno-sulfonianowy - SŁABA ZASADA(nukleofilem jest anion jodkowy)
sulfoniany alkilowe REAGUJĄ z halogenkami nieorganicznymi (reakcja SN)
+CH3O
CH3CH2CH2I CH3CH2CH2OCH3 I reakcja PRZEBIEGA ponieważ grupą odchodzącąjest już SŁABA ZASADA - anion jodkowy(nukleofilem jest anion metoksylanowy)
D)1-jodopropan
halogenopochodne alkilowe I-rzędowe REAGUJĄ z metanolanami litowców
(reakcja SN)
CH3OH++CH3O
CH3CH2CH
CH3
Cl CH3CH CHCH3 Cl
reakcja PRZEBIEGA ponieważ grupą odchodzącą jest już SŁABA ZASADA - anion chlorkowy. Za-chodzi tu reakcja ELIMINACJI.(nukleofilem jest anion metoksylanowy)
E)2-chlorobutan
halogenopochodne alkilowe II-rzędowe REAGUJĄ z metanolanami litowców
(reakcja E)
Uwaga! W przypadku, gdy trudno jest ocenić zasadowość grupy odchodzącej można rozpatrywać
kwasowość sprzężonego z tą grupą kwasu a następnie wykorzystać regułę: im słabsza zasada tym
mocniejszy kwas z nią sprzężony. Przykładowo: anion wodorosiarczkowy (SH-) jest mocną zasadą,

gdyż sprzężony z nim kwas - siarkowodór jest kwasem słabym. Z tego wynika, że tiole (o wzorze R-
SH ) nie ulegają reakcjom substytucji ani reakcjom eliminacji.
Poniżej przedstawiony jest szereg zasadowości typowych grup występujących w związkach
organicznych :
Szereg zasadowości czynników nukleofilowych:
Grupa I - Br - Cl - ArSO3
- ArOH R-OH H2O F - ArNH2 RCOO - HS - CN - R-NH2 HO - RO - RCC -
pKb 24 23 21 20,5 20,4 16 15,7 11,8 10 9 7 5 4 -1,7 -2,5 -11
z a s a d o w o ś ć
2. Nukleofilowość czynnika atakującego
Reakcje SN2 przebiegają z różną szybkością w zależności od rodzaju czynnika nukleofilowego. Różne
czynniki nukleofilowe wykazują różną aktywność w reakcji substytucji co jest warunkowane m.in.
rozmiarami nukleofila, koncentracją ładunku ujemnego oraz stopniem jego solwatacji przez cząsteczki
rozpuszczalnika. Porównując stałe szybkości reakcji bromku metylu z różnymi nukleofilami w
wodzie, w stosunku do stałej szybkości reakcji tego związku z wodą otrzymano następujące wielkości
liczbowe charakteryzujące reaktywność poszczególnych nukleofili:
n = log(k/ko)
gdzie: k - stała szybkości reakcji bromku metylu z wybranym nukleofilem
ko- stała szybkości reakcji bromku metylu z wodą
Nukleofil woda F - CH3COO - Cl - Br - N3 - OH - ArNH2 I - CN - SH -
n 0 2 2,72 3,5 3,9 4 4,2 4,5 5 5,1 5,1
Wielkość n nazywa się NUKLEOFILOWOŚCIĄ czynnika nukleofilowego. Stanowi miarę jego
skłonności do ulegania reakcji SN2. Czynniki o dużej nukleofilowości nazywamy mocnymi
nukleofilami. Do mocnych nukleofili - oprócz podanych w prawej części powyższej tabeli, zalicza się
aniony RO - , amoniak i aminy, karboaniony (np. anion acetylenkowy), anion NO2 - . Do słabych
nukleofili należą: m.in. woda i alkohole. Szybkość reakcji ze słabymi nukleofilami (woda, alkohole)
jest tak mała, że praktycznie można przyjąć, że reakcja SN2 z tymi nukleofilami nie przebiega.
Podsumowaniem powyższych rozważań może być następujące stwierdzenie:
Reguła 2 Szybkość reakcji SN2 wzrasta ze wzrostem nukleofilowości czynnika atakującego. Ze
słabymi nukleofilami reakcja ta praktycznie nie przebiega.
W reakcjach SN1 tworzy się bardzo reaktywny wysokoenergetyczny karbokation, który łatwo reaguje
z nukleofilami niezależnie od ich nukleofilowości. Dlatego drugi etap reakcji SN1 jest etapem
szybkim. W etapie decydującym o szybkości reakcji SN1 (etap dysocjacji) nukleofilowość czynnika
atakującego nie odgrywa roli ponieważ czynnik nukleofilowy nie bierze w nim udziału.
W reakcjach eliminacji istotne jest powinowactwo czynnika atakującego do protonu (czyli jego
zasadowość) zaś nukleofilowość nie odgrywa tu również żadnej roli (wynika to z definicji
nukleofilowości)
Reguła 3 Szybkość reakcji SN1 i E nie zależy od nukleofilowości czynnika atakującego.

3. Budowa substratu organicznego
Wpływ rzędowości związku organicznego na przebieg reakcji SN2
Wraz ze wzrostem rzędowości atomu węgla, na którym przebiega reakcja SN2 szybkość reakcji maleje
ponieważ utrudniony jest dostęp nukleofila do atomu węgla. W przypadku atomów drugorzędowych
reakcja SN2 przebiega jeszcze stosunkowo łatwo, jednakże dla związków trzeciorzędowych szybkość
reakcji jest tak mała, że można przyjąć, iż reakcja praktycznie nie przebiega. W tym przypadku
znacznie łatwiejszy jest dostęp do atomu wodoru przy węglu , co powoduje, że - o ile tylko
zasadowość czynnika atakującego jest wystarczająco duża - przebiega głównie reakcja eliminacji. Gdy
zasadowość i nukleofilowość czynnika atakującego jest mała, reakcją dominującą może być reakcja
substytucji SN1.
Reguła 4 Szybkość reakcji SN2 maleje wraz ze wzrostem rzędowości atomu węgla, na którym
przebiega substytucja. Na trzeciorzędowym atomie węgla reakcja SN2 praktycznie NIE PRZEBIEGA.
Wpływ rzędowości związku organicznego na przebieg reakcji SN1
Im wyższa jest rzędowość atomu węgla związanego z grupą odchodzącą tym większe jest wzajemne
oddziaływanie grup związanych z tym atomem węgla, co prowadzi do występowania w cząsteczce
pewnych naprężeń. W procesie tworzenia się karbokationu wzrastają kąty między wiązaniami (od
wartości ok. 1090 do wartości ok. 1200) a tym samym wzrasta odległość i maleją oddziaływania
między grupami co powoduje, że naprężenia wewnętrzne maleją. Zatem skłonność cząsteczki do
dysocjacji wiązania: węgiel - grupa odchodząca, powinna wzrastać wraz ze wzrostem rzędowości
atomu węgla. Drugim elementem, który należy wziąć tu pod uwagę jest trwałość karbokationów, która
- jak wiadomo - wzrasta ze wzrostem ich rzędowości z uwagi na stabilizujący wpływ grup alkilowych.
Oba wymienione czynniki powodują że:
Reguła 5 Szybkość reakcji SN1 wzrasta wraz ze wzrostem rzędowości atomu węgla, na którym
przebiega substytucja.
Wpływ rzędowości związku organicznego na przebieg reakcji E-2
Wraz ze wzrostem rzędowości atomu węgla, na którym przebiega reakcja SN2 szybkość reakcji maleje
ponieważ utrudniony jest dostęp nukleofila do atomu węgla. W przypadku atomów drugorzędowych
reakcja SN2 przebiega - trzeciorzędowych szybkość ta jest tak mała, że można przyjąć, że reakcja
praktycznie nie przebiega. W tym przypadku znacznie łatwiejszy jest dostęp do atomu wodoru przy
węglu , co powoduje, że - o ile tylko zasadowość czynnika atakującego jest wystarczająco duża -
przebiega głównie reakcja eliminacji. Gdy zasadowość czynnika atakującego jest mała, reakcją
dominującą może być reakcja substytucji SN1.
Wpływ rzędowości związku organicznego na przebieg reakcji E-1
Wpływ rzędowości atomu węgla związanego z grupą odchodzącą jest tu taki sam jak w przypadku
reakcji E-2 i SN1 (skłonność do ulegania reakcji E-1 wzrasta ze wzrostem rzędowości atomu węgla)
Reguła 6 Wraz ze wzrostem rzędowości atomu węgla wzrasta skłonność do ulegania reakcjom
ELIMINACJI.

4. Zasadowość czynnika atakującego
Reakcja E-2 przebiega tym szybciej im większa jest zasadowość atakującego czynnika
nukleofilowego, ponieważ większe powinowactwo czynnika nukleofilowego do protonu ułatwia jego
oderwanie od węgla Bardzo mocne zasady - takie jak jon amidkowy (NH2 -) powodują np. przebieg
reakcji eliminacji we wszystkich fluorowcopochodnych niezależnie od rzędowości. W przypadku
słabych zasad reakcja eliminacji przebiega na ogół w niewielkim stopniu. Można tu sformułować
kolejną ważną regułę rządzącą reakcją dwucząsteczkowej eliminacji:
Reguła 7 Szybkość reakcji E-2 wzrasta wraz ze wzrostem zasadowości czynnika nukleofilowego
Rozważanie wpływu zasadowości czynnika atakującego na szybkość reakcji E-1 nie ma sensu
ponieważ w przypadku mocnych zasad szybciej od reakcji E-1 przebiega reakcja E-2.
Szybkość reakcji eliminacji nie zależały od nukleofilowości czynnika atakującego i podobnie:
Reguła 8 Szybkość reakcji SN NIE ZALEŻY od zasadowości czynnika atakującego.
Konkurencja między reakcjami SN i E :
W reakcji związku organicznego z czynnikiem nukleofilowym reakcje SN i E konkurują ze sobą.
Względna zawartość obu produktów zależy od trzech czynników: rzędowości atomu węgla
związanego z grupą odchodzącą oraz zasadowości i nukleofilowości czynnika atakującego. Wpływ
tych czynników na poszczególne reakcje SN i E został dokładnie opisany wyżej. W odniesieniu do
konkurencji reakcji SN i E wpływ ten można opisać następująco:
WZGLĘDNA ZAWARTOŚĆ PRODUKTU ELIMINACJI w mieszaninie
poreakcyjnej WZRASTA ze wzrostem rzędowości atomu węgla związanego z
grupą odchodzącą oraz ze wzrostem zasadowości czynnika nukleofilowego,
MALEJE zaś ze wzrostem nukleofilowości reagującego nukleofila.
Ilustracją wpływu rzędowości atomu węgla może być reakcja bromków butylowych z
etanolanem sodu:
a) w przypadku bromku n-butylu w reakcji powstaje 86% produktu substytucji (SN2) i 14%
produktu eliminacji (E-2) b) w przypadku bromku sec-butylu w reakcji powstaje 17% produktu substytucji (SN2) i 83%
produktu eliminacji (E-2) c) w przypadku bromku t-butylu w reakcji powstaje 100% produktu eliminacji (E-2)
Ilustracją wpływu zasadowości czynnika nukleofilowego może być:
1) reakcja bromku t-pentylu z etanolanem sodu, w której tworzy się 100% produktu eliminacji
2) reakcja tego samego bromku z etanolem w której tworzy się 40% produktu eliminacji i 60%
produktu substytucji
B IO
TE
HC N OL
O GAI
CHEMIAORGANICZNA
Konkurencja
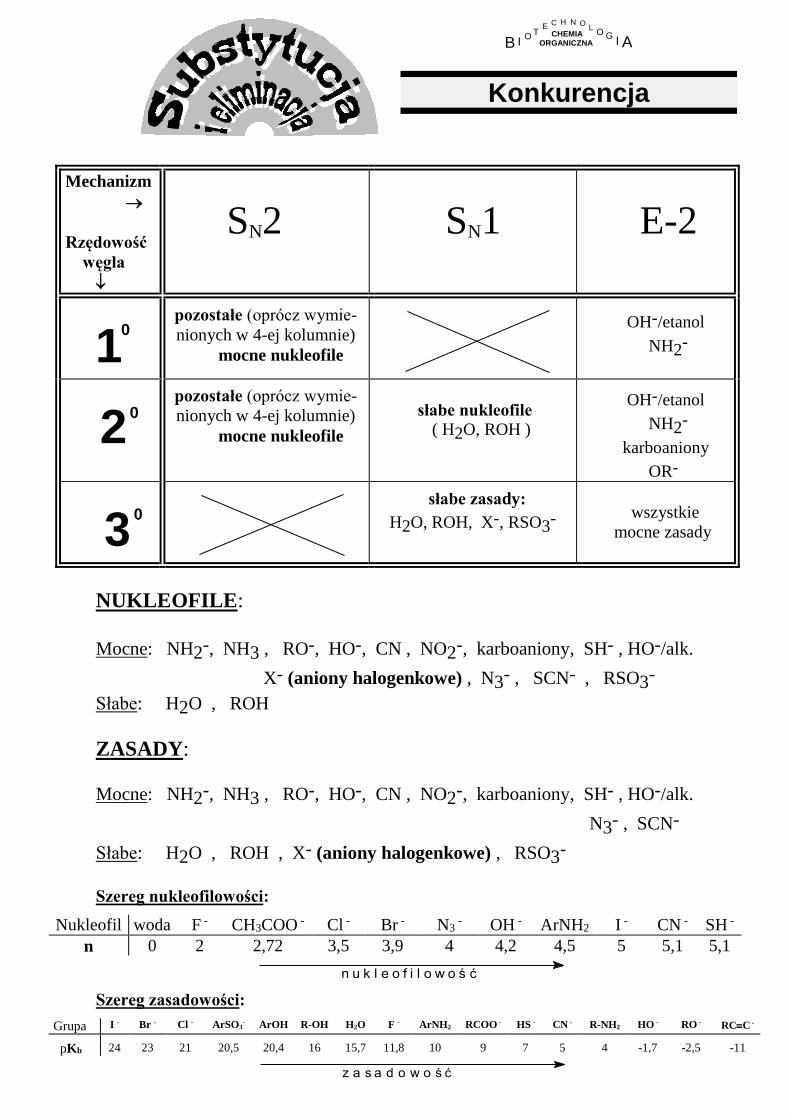
Mechanizm
Rzędowość
węgla
SN2
SN1
E-2
10
pozostałe (oprócz wymie-
nionych w 4-ej kolumnie)
mocne nukleofile
OH-/etanol
NH2-
2
0
pozostałe (oprócz wymie-
nionych w 4-ej kolumnie)
mocne nukleofile
słabe nukleofile ( H2O, ROH )
OH-/etanol
NH2-
karboaniony
OR-
3
0
słabe zasady:
H2O, ROH, X-, RSO3-
wszystkie
mocne zasady
NUKLEOFILE:
Mocne: NH2-, NH3 , RO-, HO-, CN , NO2-, karboaniony, SH- , HO-/alk.
X- (aniony halogenkowe) , N3- , SCN- , RSO3-
Słabe: H2O , ROH
ZASADY:
Mocne: NH2-, NH3 , RO-, HO-, CN , NO2-, karboaniony, SH- , HO-/alk.
N3- , SCN-
Słabe: H2O , ROH , X- (aniony halogenkowe) , RSO3-
Szereg nukleofilowości:
Nukleofil woda F - CH3COO - Cl - Br - N3 - OH - ArNH2 I - CN - SH -
n 0 2 2,72 3,5 3,9 4 4,2 4,5 5 5,1 5,1
Szereg zasadowości:
Grupa I - Br - Cl - ArSO3- ArOH R-OH H2O F - ArNH2 RCOO - HS - CN - R-NH2 HO - RO - RCC -
pKb 24 23 21 20,5 20,4 16 15,7 11,8 10 9 7 5 4 -1,7 -2,5 -11
z a s a d o w o ś ć
n u k l e o f i l o w o ś ć

B IO
TE
HC N OL
O GAI
CHEMIAORGANICZNA
Konkurencja
Fluorowcopochodne typu ALLILOWEGO i BENZYLOWEGO
Mechanizm
Rzędowość
węgla
SN2
SN1
E-2
10
pozostałe (oprócz wymie-
nionych w 4-ej kolumnie)
mocne nukleofile
słabe nukleofile ( H2O, ROH )
OH-/etanol
NH2-
2
0
pozostałe (oprócz wymie-
nionych w 4-ej kolumnie)
mocne nukleofile
słabe nukleofile ( H2O, ROH )
OH-/etanol
NH2-
karboaniony
OR-
3
0
słabe zasady:
H2O, ROH, X-, RSO3-
wszystkie
mocne zasady
NUKLEOFILE:
Mocne: NH2-, NH3 , RO-, HO-, CN , NO2-, karboaniony, SH- , HO-/alk.
X- (aniony halogenkowe) , N3- , SCN- , RSO3-
Słabe: H2O , ROH
ZASADY:
Mocne: NH2-, NH3 , RO-, HO-, CN , NO2-, karboaniony, SH- , HO-/alk.
N3- , SCN-
Słabe: H2O , ROH , X- (aniony halogenkowe) , RSO3-
B IO
TE
HC N OL
O GAI
CHEMIAORGANICZNA
Reakcje fluorowcopochodnych
aromatycznych
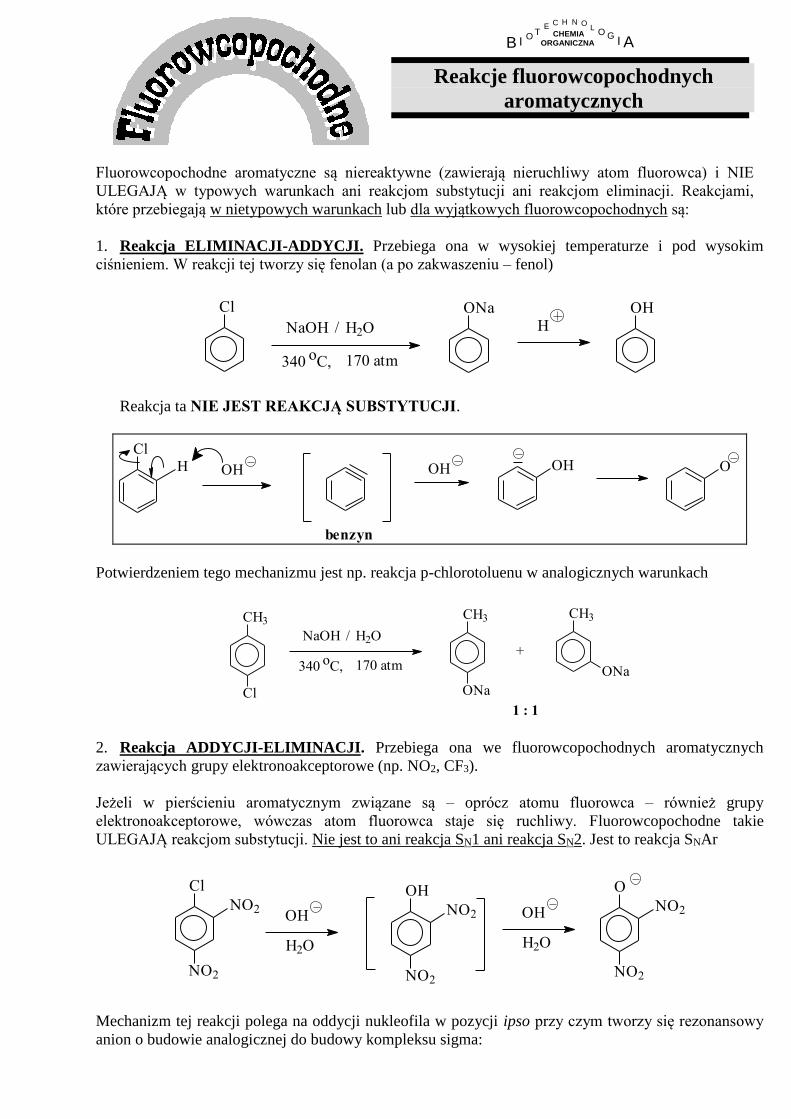
Fluorowcopochodne aromatyczne są niereaktywne (zawierają nieruchliwy atom fluorowca) i NIE
ULEGAJĄ w typowych warunkach ani reakcjom substytucji ani reakcjom eliminacji. Reakcjami,
które przebiegają w nietypowych warunkach lub dla wyjątkowych fluorowcopochodnych są:
1. Reakcja ELIMINACJI-ADDYCJI. Przebiega ona w wysokiej temperaturze i pod wysokim
ciśnieniem. W reakcji tej tworzy się fenolan (a po zakwaszeniu – fenol)
Cl
340 oC, 170 atm
NaOH H2O/
ONaH
OH
Reakcja ta NIE JEST REAKCJĄ SUBSTYTUCJI.
OH
Cl
H
benzyn
OHOH O
Potwierdzeniem tego mechanizmu jest np. reakcja p-chlorotoluenu w analogicznych warunkach
CH3
Cl
340 oC, 170 atm
NaOH H2O/
CH3
ONa
+
CH3
ONa
1 : 1 2. Reakcja ADDYCJI-ELIMINACJI. Przebiega ona we fluorowcopochodnych aromatycznych
zawierających grupy elektronoakceptorowe (np. NO2, CF3).
Jeżeli w pierścieniu aromatycznym związane są – oprócz atomu fluorowca – również grupy
elektronoakceptorowe, wówczas atom fluorowca staje się ruchliwy. Fluorowcopochodne takie
ULEGAJĄ reakcjom substytucji. Nie jest to ani reakcja SN1 ani reakcja SN2. Jest to reakcja SNAr
OH
Cl
NO2
NO2
H2O
OH
NO2
NO2
O
NO2
NO2
H2O
OH
Mechanizm tej reakcji polega na oddycji nukleofila w pozycji ipso przy czym tworzy się rezonansowy
anion o budowie analogicznej do budowy kompleksu sigma:

Br OH
NO2
NO2
Br OH
NO2
NO2
OHBr
NO2
NO2
. . .
(anion taki powstaje, ponieważ ładunek jest tu zdelokalizowany na grupy nitrowe, co zwiększa jego
trwałość oraz dlatego, że na atomie węgla ipso występuje duży deficyt elektronowy implikowany
obecnością grup nitrowych w pozycji orto i para ).
Z anionu tego eliminuje się następnie grupa łatwiej odchodząca (w tym wypadku anion bromkowy) w
wyniku czego odtwarza się sekstet elektronowy.
B IO
TE
HC N OL
O GAI
CHEMIAORGANICZNA
Zastosowanie w syntezie
Łatwość ulegania fluorowcopochodnych reakcjom substytucji nukleofilowej ma duże
znaczenie w syntezie organicznej, gdyż pozwala na uzyskanie wielu rodzajów innych
związków organicznych, które trudno byłoby otrzymać na innej drodze. Fluorowcopochodne
- łatwe do uzyskania z węglowodorów - stanowią więc bardzo wygodne i tanie substraty.
Poniższy schemat ilustruje różnorodność zastosowań reakcji substytucji
fluorowcopochodnych
R X
OR1
OH
HC C
CN
NO2
NH3
R1 NH2
SH
R1SO3
R1COO
N3
SCN
H2O
R1OH
R OR1
R OR1
H
R OH
R OH2
R SCN
R N3
R OOCR1
R OSO2R1
R SH
R C CH
R CN
R NO2
R NH3
R NH2R1
kationy amoniowe
nitrozwiązki
nitryle
alkiny terminalne
alkohole
eterytiole
sulfoniany alkilowe
estry alkilowe
azydki alkilowe
tiocyjaniany alkilowe
kationy oksoniowe

B IO
TE
HC N OL
O GAI
CHEMIAORGANICZNA
Fluorowcopochodne typu allilowego i
benzylowego
Fluorowcopochodne typu allilowego wykazują większą skłonność do ulegania reakcjom
substytucji w stosunku do odpowiednich fluorowcopochodnych nasyconych. Przyczyny tego
zjawiska są następujące:
a) większa skłonność do ulegania reakcji dysocjacji wiązania C - X spowodowana
względną trwałością tworzącego się rezonansowego karbokationu:
CH3 CH CH CH2 Br CH3 CH CH CH2 CH3 CH CH CH2 + Br
Utworzony karbokation może być atakowany przez nukleofil w jednym z dwu centrów
elektrofilowych zlokalizowanych zgodnie z powyższymi strukturami granicznymi. W reakcji
tworzy się zatem mieszanina produktów: a) produktu substytucji na atomie węgla związanym z
grupa odchodzącą b) produktu przegrupowanego. Zjawisko powstawania produktu
przegrupowanego w reakcjach fluorowcopochodnych typu allilowego nazywa się
PRZEGRUPOWANIEM ALLILOWYM.
b) polaryzacja wiązania C - X indukuje polaryzację wiązania i pojawienie się centrum
elektrofilowego na węglu
CH CHR CH2 X
Nukleofil może atakować jedno z dwu zaznaczonych centrów elektrofilowych. W przypadku
ataku na węgiel związany z grupą odchodzącą mamy do czynienia z reakcją SN2. W przypadku
ataku na węgiel tworzy się produkt PRZEGRUPOWANY:
CH CHR CH2 X
Nu
CH CHR CH2
Nu
+ X
Dla tego rodzaju substytucji stosuje się oznaczenie: SN2’
W przypadku fluorowcopochodnych typu benzylowego tworzy się rezonansowy karbokation,
w którym jednak struktury niearomatyczne mają - z uwagi na względnie wysoką energię -
nieduży udział. W wyniku reakcji tworzy się zatem jedynie produkt substytucji w pozycji
benzylowej, zaś produkty przegrupowania nie są obserwowane.
CH2CH2 X+CH2CH2CH2CH2 X
NuCH2 Nu
Zwiększona reaktywność fluorowcopochodnych typu allilowego i benzylowego ujawnia się
m.in. w tym, że ze słabymi nukleofilami reagują nawet halogenki pierwszorzędowe (reakcja
biegnie wówczas wg mechanizmu SN1.

B IO
TE
HC N OL
O GAI
CHEMIAORGANICZNA
Ruchliwość fluorowca
Skłonność atomu fluorowca (w cząsteczce fluorowcopochodnej) do ulegania rekcjom
substytucji lub eliminacji określa się terminem: ruchliwość fluorowca. Im łatwiej (szybciej)
przebiegają wymienione reakcje tym ruchliwość ta jest większa. Przykładowo powiemy, że we
fluorowcopochodnych allilowych, trzeciorzędowych i benzylowych atom fluorowca jest bardzo
ruchliwy, podczas gdy fluorowcopochodne typu winylowego lub fluorowcopochodne aromatyczne
zawierają nieruchliwy atom fluorowca.
Za doświadczalne kryterium ruchliwości fluorowca można przyjąć szybkość wytrącania osadu
halogenku srebra w reakcji fluorowcopochodnej z alkoholowo-wodnym roztworem azotanu srebra.
+R X AgNO3 AgX ( )
Fluorowcopochodne AROMATYCZNE i WINYLOWE nie strącają osadu halogenku srebra
nawet na gorąco (po podgrzaniu).
Fluorowcopochodne PIERWSZORZĘDOWE strącają osadu halogenku srebra po podgrzaniu.
Fluorowcopochodne DRUGORZĘDOWE strącają osadu halogenku srebra na zimno po upływie
pewnego czasu.
Fluorowcopochodne TRZECIORZĘDOWE, BENZYLOWE i ALLILOWE strącają osadu
halogenku srebra na zimno natychmiast.
Reakcja ta jest wykorzystywana w chemii analitycznej (jako tzw. reakcja probówkowa) do
szybkiego określania ruchliwości fluorowca. Efektem wizualnym w tej reakcji jest wytrącanie się
serowatego roztworu halogenku srebra (białego chlorku, kremowego bromku lub żółtego jodku).
Przykładowo: dwa izomery:
CH2 BrCH3 Br
różnią się tym, że pierwszy z nich nie wytrąca osadu w reakcji z alkoholowo-wodnym roztworem
AgNO3 nawet na gorąco, zaś drugi z nich strąca kremowy osad AgBr na zimno natychmiast.























