Embed Size (px)
Citation preview

Draft for public comment 26 October 2016 Page 1 of 140
WHO Prequalification: Sample Product Dossier for a
Quantitative Nucleic Acid Test to detect HIV-1 RNA
SIMU HIV-1 Quant System
PQDx 9876-543-22
THE Manufacturing Company
DRAFT SAMPLE DOSSIER FOR PUBLIC COMMENT
Disclaimer
This Product Dossier is entirely fictitious and has been produced for illustrative purposes only. Each manufacturer must determine what should be submitted
to fulfil WHO requirements.
Draft for public comment 26 October 2016

WHO Sample product dossier for SIMU HIV-1 QUANT
Page 2 of 140
© World Health Organization 2016
All rights reserved. Publications of the World Health Organization can be obtained from WHO Press, World Health Organization, 20 Avenue Appia, 1211 Geneva 27, Switzerland (tel.: +41 22 791 3264; fax: +41 22 791 4857; e-mail: [email protected]). Requests for permission to reproduce or translate WHO publications – whether for sale or for non-commercial distribution – should be addressed to WHO Press, at the above address (fax: +41 22 791 4806; e-mail: [email protected]). The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted lines on maps represent approximate border lines for which there may not yet be full agreement. The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors and omissions excepted, the names of proprietary products are distinguished by initial capital letters. All reasonable precautions have been taken by the World Health Organization to verify the information contained in this publication. However, the published material is being distributed without warranty of any kind, either expressed or implied. The responsibility for the interpretation and use of the material lies with the reader. In no event shall the World Health Organization be liable for damages arising from its use. Contact: Irena Prat, EMP Prequalification Team Diagnostics WHO - 20 Avenue Appia - 1211 Geneva 27 Switzerland

WHO Sample product dossier for SIMU HIV-1 QUANT
Draft for public comment 26 October 2016 Page 3 of 140
Table of contents
ABBREVIATIONS .............................................................................................................. 6
Part I – Introductory information 9
1 INTRODUCTION ............................................................................................. 9
1.1 Purpose of the sample product dossier .....................................................................9 1.2 Content of the sample product dossier .....................................................................9 1.3 Completeness of the sample product dossier ............................................................9 1.4 Format of the sample product dossier ......................................................................9 1.5 Feedback on the sample product dossier ................................................................ 10
2 INTENDED AUDIENCE ................................................................................... 10
3 THE PRODUCT DOSSIER ................................................................................ 11
3.1 WHO product dossier elements .............................................................................. 11 3.2 When to submit a product dossier .......................................................................... 11
Part II - The WHO sample product dossier 12
4 WHO PRODUCT DOSSIER CHECKLIST ............................................................. 12
5 THE PRODUCT .............................................................................................. 21
5.1 Regulatory versions of this product ........................................................................ 21 5.2 Product description including variants (configurations) and accessories .................. 21
5.2.1 Product description.................................................................................... 21 5.2.2 Intended use .............................................................................................. 24 5.2.3 Intended users ........................................................................................... 24 5.2.4 Photographs of kit ...................................................................................... 24 5.2.5 A general description of the principle of the test method or
instrument principles of operation ............................................................ 25 5.2.6 A description of the components of the test (e.g. reagents, assay
controls and calibrators) ............................................................................ 27 5.2.7 A description of the specimen collection and transport materials
provided with the product or descriptions of specifications recommended for use. .............................................................................. 27
5.2.8 For Instruments of automated assays: a description of the appropriate assay characteristics or dedicated assays .............................. 27
5.2.9 For automated assays: a description of the appropriate instrumentation characteristics or dedicated instrumentation. ............... 28
5.2.10 If applicable, a description of any software to be used with the product ....................................................................................................... 28
5.2.11 If applicable, a description or complete list of the various configurations or variants of product that will be made available ............ 28
5.2.12 If applicable, a description of the accessories, and other products that are intended to be used in combination with the diagnostic. ........... 28

WHO Sample product dossier for SIMU HIV-1 QUANT
Draft for public comment 26 October 2016 Page 4 of 140
5.3 Essential principles (EP) check list ........................................................................... 28 5.4 Risk analysis and control summary ......................................................................... 29
5.4.1 Risk analysis policy ..................................................................................... 29 5.4.2 Risk categories ........................................................................................... 29 5.4.3 Instrument ................................................................................................. 31 5.4.4 Risk/Benefit ................................................................................................ 32
6 DESIGN AND MANUFACTURING INFORMATION ........................................... 33
6.0 Design control ........................................................................................................ 33 6.1 Product design ....................................................................................................... 35
6.1.1 Design overview ......................................................................................... 35 6.1.2 Formulation and composition ................................................................... 37 6.1.3 Biological safety ......................................................................................... 38 6.1.4 Documentation of design changes ............................................................ 38
6.2 Manufacturing process ........................................................................................... 39 6.2.1 Overview of manufacture .......................................................................... 39 6.2.2 Sites of manufacture .................................................................................. 41 6.2.3 Key suppliers .............................................................................................. 41
7 PRODUCT PERFORMANCE SPECIFICATIONS AND ASSOCIATED VALIDATION AND VERIFICATION STUDIES ................................................................................ 44
7.0 Overview of testing procedures .............................................................................. 45 7.1 Analytical studies ................................................................................................... 47
7.1.1 Specimen types .......................................................................................... 47 7.1.2 Analytical performance characteristics ..................................................... 49
7.2 Stability (excluding specimen stability) ................................................................... 67 7.2.1 Claimed shelf-life (including transport challenge) ..................................... 67 7.2.2 In-use stability ............................................................................................ 70
7.3 Robustness studies ................................................................................................ 72 7.3.1 Cross-contamination .................................................................................. 72 7.3.2 Whole system failure ................................................................................. 73
7.4 Clinical evidence (clinical sensitivity and specificity) ................................................ 74
8 LABELLING ................................................................................................... 77
8.1 Labelling ................................................................................................................ 77 8.1.1 Instrument labelling ................................................................................... 77 8.1.2 Test Cartridge pouch packaging and labelling (Figure XXY) ....................... 77
8.2 Instructions for Use ................................................................................................ 77 8.3 Instrument manual ................................................................................................ 78
9 COMMERCIAL HISTORY ................................................................................ 79
9.1 Countries of supply ................................................................................................ 79 9.1.1 List of countries where product is currently supplied ............................... 79 9.1.2 Minimum and maximum price in 2015 ...................................................... 79 9.1.3 Training and support network ................................................................... 79
9.2 Adverse events and field safety corrective actions .................................................. 80 9.2.1 Table of adverse event reports .................................................................. 80

WHO Sample product dossier for SIMU HIV-1 QUANT
Draft for public comment 26 October 2016 Page 5 of 140
10 REGULATORY HISTORY ................................................................................. 81
11 QUALITY MANAGEMENT SYSTEM ................................................................. 82
11.1 Quality manual ...................................................................................................... 82 11.1.1 Quality manual system documents ........................................................... 82 11.1.2 Quality manual system procedures ........................................................... 82
11.2 Quality manual test certification ............................................................................ 82
ANNEX I: ESSENTIAL PRINCIPLES CHECKLIST ................................................................... 83
ANNEX II: RISK MANAGEMENT POLICY ......................................................................... 102
ANNEX III: RESIDUAL RISK STATEMENT ........................................................................ 104
ANNEX IV: DESIGN INPUTS FMEA ................................................................................. 105
ANNEX V: USER AND PATIENT FMEA ............................................................................ 111
ANNEX VI: INPUT REQUIREMENTS FOR THE HIV-1 QUANT TEST ................................... 113
ANNEX VII: THE MANUFACTURING COMPANY QUALITY MANUAL ........................ 120

WHO Sample product dossier for SIMU HIV-1 QUANT
Draft for public comment 26 October 2016 Page 6 of 140
Abbreviations
ANSI American National Standards Institute
ART antiretroviral therapy
BSA bovine serum albumin
CAPA corrective and preventive action
cDNA complementary DNA
CE Conformité Européenne (European Conformity)
CI confidence interval
CLSI Clinical and Laboratory Standards Institute
CO capture oligonucleotide
COSHH control of substances hazardous to health
Cp copies
CRFs circulating recombinant forms
CTS Common Technical Specifications
Ct cycle threshold
DHF design history file
DNA deoxyribonucleic acid
DNase deoxyribonuclease
DMR device master record
EDTA ethylenediaminetetraacetic acid
EIA enzyme immunoassay
EID early infant diagnosis
EP Essentials Principles
EQAS external quality assessment scheme
EXP expiry
FDA US Food and Drug Administration
FMEA failure mode and effects analysis
GHTF Global Harmonization Task Force
HBsAg hepatitis B surface antigen
HBV hepatitis B virus
HCV hepatitis C virus
HIV human immunodeficiency virus
HTLV human T-lymphotropic virus
IgA, IgG, IgM immunoglobulins A, G and M
ID identifier
IPC in-process control
IFU instructions for use

WHO Sample product dossier for SIMU HIV-1 QUANT
Draft for public comment 26 October 2016 Page 7 of 140
IMDRF International Medical Devices Regulators Forum
ISO International Organization for Standardization
IU international units
IVD in vitro diagnostic medical device
IVDD in vitro diagnostic devices Directive 98/79/EC
LoD limit of detection
MOH ministry of health
NA nucleic acid
NAT nucleic acid test
NIBSC National Institute for Biological Standards and Control
No. number
NPA negative per cent agreement
NR non-reactive
OD optical density
OEM original equipment manufacturer
OQ operational quality
PCR polymerase chain reaction
PMS post-market surveillance
POCT point-of-care test
POS positive
PPA positive percent agreement
QA quality assurance
QC quality control
QMS quality management system
qPCR quantitative PCR
R reactive
R&D research and development
RNA ribonucleic acid
RNAse ribonuclease
RT reverse transcriptase
ROW rest of world
SD standard deviation
SOP standard operating procedure
SP sample processing control
Taq Thermus aquaticus
TB tuberculosis
TNA total nucleic acid
TSE Transmissible Spongiform Encephalopathies

WHO Sample product dossier for SIMU HIV-1 QUANT
Draft for public comment 26 October 2016 Page 8 of 140
VL viral load
WHO World Health Organization
Acknowledgements
This document, WHO Prequalification: Sample product dossier for a Quantitative HIV-1 nucleic acid test, was developed as part of the Bill & Melinda Gates Foundation Umbrella Grant and the UNITAID grant for “Increased access to appropriate, quality-assured diagnostics, medical devices and medicines for prevention, initiation and treatment of HIV/AIDS, TB and malaria”. The first draft of this dossier was prepared in collaboration with Dr Craig Hill, consultant, Encinitas, USA and Dr John Saldanha, Immucor, Norcross, USA and with input and expertise from Dr Elliot Cowan consultant, Washington DC, USA; Dr Fatima Gruszka consultant, Paris, France; Dr Mark Lanigan, consultant, Geneva, Switzerland; and Mercedes Perez, WHO/HIS/EMP, Geneva, Switzerland. This document was produced under the coordination and supervision of Deirdre Healy, Robyn Meurant and Irena Prat, WHO/HIS/EMP, Geneva, Switzerland.
This document is now available for public consultation from 26 October 2016

WHO Sample product dossier for SIMU HIV-1 QUANT Introduction
Draft for public comment 26 October 2016 Page 9 of 140
Part I – Introductory information
1 Introduction
Purpose of the sample product dossier 1.1The purpose of this sample product dossier is to provide manufacturers with an example of a product dossier required for WHO Prequalification of an in vitro diagnostic (IVD). The product dossier should contain evidence submitted by the manufacturer to demonstrate to WHO that the diagnostic is of acceptable quality, is safe and performs optimally when used as intended by the manufacturer. Evidence will take the form, for example, of results of testing, certifications, SOPs, systems and any other documentation necessary to support quality, safety and performance. As such, this sample product dossier contains the results of testing, extracts of standard operating procedures (SOPs) and other information that may be of relevance in support of an application for prequalification of a simple, rapid molecular diagnostic test for HIV1 infection.
Content of the sample product dossier 1.2This dossier is based on a fictitious IVD, the SIMU Quantitative HIV-1 NAT assay and its fictitious manufacturer, THE Manufacturing Company. As the product and its manufacturer do not exist, any related aspects that have been described within the sample product dossier are purely for the purposes of demonstrating the type of information that may be included in a product dossier submitted to WHO for Prequalification.
Completeness of the sample product dossier 1.3Because of its invented nature, the information provided is an example only and does not necessarily contain the full level of detail that may be required to fulfil WHO Prequalification requirements. At times the information is presented in summary format. Additionally, the abbreviation “XXX” is used extensively to describe materials that do not exist, but again is incorporated to provide an example of the type of information that may be required. Further instructions are also provided in red coloured boxes to indicate where additional information may be expected.
This sample product dossier is still in development and not all prequalification requirements have been addressed. WHO will continue to incorporate these additional relevant sections. However, it is important to repeat that the purpose of this document is to provide examples to assist manufacturers. This sample product dossier can never be considered to represent all the evidence that may be needed to meet WHO Prequalification requirements. Alternative approaches to the studies presented in this sample dossier may also be acceptable. Each manufacturer is responsible for identifying the type and volume of evidence that will be sufficient to support its submission. WHO Prequalification staff are available to assist manufacturers at any point in the prequalification process. Staff may be contacted by email at [email protected].
Format of the sample product dossier 1.4The format of this sample product dossier, including the section numbering system, follows that contained in WHO document PQDx_018 “Instructions for Compilation of a Product Dossier”. This document can be found on the WHO website (www.who.int) at the following link:
http://www.who.int/diagnostics_laboratory/evaluations/100506_pqdx_018_dossier_instructns_v1.pdf

WHO Sample product dossier for SIMU HIV-1 QUANT Intended Audience
Draft for public comment 26 October 2016 Page 10 of 140
Feedback on the sample product dossier 1.5Comments on this sample product dossier and its utility are welcomed by WHO at [email protected].
2 Intended Audience This document has been created to assist manufacturers who wish to submit a product dossier for a quantitative nucleic acid test for HIV-1 RNA.

WHO Sample product dossier for SIMU HIV-1 QUANT The Product Dossier
Draft for public comment 26 October 2016 Page 11 of 140
3 The Product Dossier
WHO product dossier elements 3.1For the purposes of WHO Prequalification – Diagnostics, the product dossier is a selection of records and documents compiled by a manufacturer from their existing records and documents to provide evidence that the IVD submitted for WHO prequalification assessment conforms to the Essential Principles of Safety and Performance of Medical Devices, available at (http://www.imdrf.org/docs/ghtf/final/sg1/technical-docs/ghtf-sg1-n68-2012-safety-performance-medical-devices-121102.pdf) and meets other WHO , available at (http://www.who.int/diagnostics_laboratory/evaluations/en/).
During the WHO review of a product dossier, WHO will take into account the information that was previously submitted in the WHO document PQDx_015 “Prequalification of Diagnostics – Pre-submission form”, available at (http://www.who.int/diagnostics_laboratory/evaluations/140701_pqdx_049_dossier_checklist_v2.pdf?ua=1). Therefore, manufacturers should ensure that the content of the product dossier is consistent with the information submitted with the pre-submission form and that WHO be promptly notified of any changes in the information submitted with the respective pre-submission form at [email protected]. Furthermore, WHO communicates to the manufacturer any inadequacies identified in the pre-submission form that need to be addressed in the product dossier submission.
When to submit a product dossier 3.2Manufacturers should only submit a product dossier to WHO Prequalification when formally requested to do so by WHO. Dossiers that are submitted without a request from WHO will be returned to the manufacturer without review.
Manufacturers should ensure that the dossier contains all the information as is prescribed in PQDx_018 “Instructions for Compilation of a Product Dossier”. The prequalification procedure may be terminated if the dossier does not contain the prescribed information, the information supplied is inadequate to complete the prequalification assessment effectively or the requested information is not provided by the manufacturer within a specified time period.
WHO sample product dossier starts on next page

WHO Sample product dossier for SIMU HIV-1 QUANT
WHO Product Dossier Checklist
Draft for public comment 26 October 2016 Page 12 of 140
Part II - The WHO sample product dossier
4 WHO Product Dossier Checklist The attached product dossier contains information in support of the previously submitted Prequalification of Diagnostics – Pre-submission form (WHO document PQDx_015) for the following product:
PQDx Number: PQDx
Product Name: SIMU HIV-1 Quant System
Manufacturer Name: THE Manufacturing Company
PRODUCT DOSSIER CHECKLIST
WHO require manufacturers to complete the product dossier checklist1 as part
of the product dossier submission
Dossier Content Requirement Provided Location:
NOTE: The text below matches that of PQDx_018, Instructions for compilation of a product dossier.
Yes/No Volume/Section
Page number – Page number (illustrative)
1.1.1 Letter of Agreement
The Letter of Agreement is attached to the front page of the dossier and supports attestation of payment
Yes/No
The information concerning the product is the same on the Letter of Agreement and the Prequalification Dossier
Yes/No
4. Dossier Format Yes/No
4.1. Product Dossier Submission Format Yes/No
One printed copy and one electronic copy of product dossier submitted
Yes/No
A signed document attesting that the content of the electronic version is an exact duplicate of the printed copy was submitted
Yes/No
Dossier is clearly presented (bound or in a clearly marked set of ring-binders)
Yes Entire dossier
4.2. Layout and Order Yes Entire dossier
Proper formatting of 1 of 2, 2 of 2, etc., used Yes/No
The submission is clearly divided into sections as described and all pages are numbered
Yes Entire dossier
Table of contents included Yes Page 2
1 WHO document PQDx_049 Product dossier checklist is available at
http://www.who.int/diagnostics_laboratory/evaluations/140701_pqdx_049_dossier_checklist_v2.pdf?ua=1

WHO Sample product dossier for SIMU HIV-1 QUANT
WHO Product Dossier Checklist
Draft for public comment 26 October 2016 Page 13 of 140
Dossier Content Requirement Provided Location:
This checklist is attached to the front of the submission and used as a cross-reference
Yes/No
The physical pages of the dossier and the page numbers correspond Yes/No
There are appropriately named tab identifiers Yes/No
Standard A4 paper is used for all submissions Yes/No
Font sizes are easily legible Yes/No
4.2.1. Electronic Copy Requirements Yes/No
The electronic copy is in PDF form with no password required Yes/No
The electronic copy is organized in the same format as the printed copy
Yes/No
The name of the file is descriptive and doesn’t contain any of the noted special characters
Yes/No
4.3. Language and Units of Measurement Yes/No
English language and units of measure used Yes/No
Any translations must have been carried out by a certified translator Yes/No
5. Product Yes/No
5.1. Regulatory versions of this product Yes Page 21
All regulatory versions of the product are identified and the version being submitted for assessment is indicated
Yes/No
For all submissions, the regulatory version to which it relates is identified
Yes/No
5.2. Product description including variants (configurations) and accessories
Yes Page 21
The intended use of the diagnostic, testing population, user, and setting of use for the diagnostic is included
Yes Page 24
Photographs of all kit components, both packaged and individual, are included
Yes Page 24
A description of the principle of the assay method/instrument principles of operation are provided
Yes Page 25
A description of the components and reactive ingredients are included
Yes Page 27
A description of the specimen collection and transport materials are provided
Yes Page 27
A description of the appropriate assay and instrumentation characteristics are included
Yes Page 28
If applicable, there are descriptions of software to be used with the product, a list of variants/configurations of the product, and a description of accessories are included
Yes/No Page 28
5.3. Essential principles (EP) checklist Yes Page 28 and Annex I
A checklist in the form of a table that lists all relevant material is included
Yes/No
5.4. Risk analysis and control summary Yes Pages 29–32 and Annex II
There is a summary report of the risks identified during the risk analysis process
Yes/No

WHO Sample product dossier for SIMU HIV-1 QUANT
WHO Product Dossier Checklist
Draft for public comment 26 October 2016 Page 14 of 140
Dossier Content Requirement Provided Location:
A description of how risks have been controlled to an acceptable level
Yes/No
A signed conclusion with evidence that the remaining risks are acceptable is presented
Yes/No Annex III
There is evidence that the risk analysis is part of the manufacturer’s risk management plan
Yes/No
When applicable, specific standards/guidelines recommended by the WHO are identified
Yes/No
6. Design and manufacturing information Yes Pages 33–41
6.1. Product design Yes Page 35
6.1.1. Design Overview Yes Page 35
Information to provide a general understanding on design is provided
Yes/No
There is a flowchart of the design process Yes/No
A general description of the critical assay ingredients for use with the product is provided
Yes/No
If applicable, a controlling site is identified Yes/No
6.1.2. Formulation and composition Yes Page 37
For each of the ingredients, formulation/composition information is provided
Yes/No
Sources of IVD component materials are identified Yes/No
6.1.3. Biological safety Yes Page 38
There is a table, including all needed information, listing all biological components included in the product
Yes/No
If applicable, a determination of the residual risk of transmission/infection to the user is provided
Yes/No
There is information on how users of the device are informed of any residual risk
Yes/No
6.1.4. Documentation of design changes Yes Page 38
Records of each design change for the product submitted, with all pertinent information, is included
Yes/No
6.2. Manufacturing process Yes Page 39
6.2.1. Overview of manufacture Yes/No
A flow chart of the entire manufacturing process is included Yes/No
A site master file, with a diagram of the floor plan, is provided Yes Annex XXX
If applicable, certified copies of quality management system certificates are annexed to the dossier
Yes/No
There are details of each major step in the manufacturing process with all needed information included in the proper form
Yes/No
There is an overview of verification, validation, and quality control activities for all stages of design and manufacture
Yes/No
Batch release criteria for the product are included Yes Page 41, verification reports AXXXX and BXXXX
6.2.2. Sites of manufacture Yes/No

WHO Sample product dossier for SIMU HIV-1 QUANT
WHO Product Dossier Checklist
Draft for public comment 26 October 2016 Page 15 of 140
Dossier Content Requirement Provided Location:
All critical manufacturing sites for all necessary stages of manufacture are listed with all necessary information
Yes/No
6.2.3. Key suppliers Yes/No Page 41
All key suppliers are listed with all needed information Yes/No
If applicable, certified copies of the key suppliers’ certificates are annexed to the dossier
Yes/No
7. Product performance specifications and associated validation and verification studies
Yes/No Pages 44 and following
7.1. Analytical studies Yes/No
7.1.1. Specimen types Yes/No Pages 47–xx
The different specimen types that can be used with the product are identified
Yes/No
The studies and needed information for support of the specimen type are included
Yes/No
The studies and needed information for support of claims made for specimen types are included
Yes/No
7.1.2. Analytical Performance Characteristics Yes/No
7.1.2.1. Accuracy of Measurement Yes/No
7.1.2.1.1. Trueness of Measurement Yes/No
The studies and information needed to establish trueness of measurement are provided
Yes/No
7.1.2.1.2. Precision of Measurement Yes/No
7.1.2.1.2.1. Repeatability Yes/No
The studies and information needed to establish within-run variability are included
Yes/No
If applicable, studies to establish repeatability undertaken by non-laboratory personnel should be provided
Yes/No
7.1.2.1.2.2 Reproducibility Yes/No
Studies and information to establish the appropriate types variability are included
Yes/No
The use of specimens that represent the full range of expected analyte concentration are included
Yes/No
If applicable, provide studies to establish repeatability undertaken by non-laboratory personnel
Yes/No
7.1.2.2. Analytical sensitivity Yes/No Page 54 and Annex X (Results of analytical sensitivity Study)
The studies required to establish analytical sensitivity are included with all needed information
Yes/No
If applicable, the relevant parameters are provided and there are details on their derivation
Yes/No

WHO Sample product dossier for SIMU HIV-1 QUANT
WHO Product Dossier Checklist
Draft for public comment 26 October 2016 Page 16 of 140
Dossier Content Requirement Provided Location:
7.1.2.3. Analytical Specificity Yes/No Page 59 (Cross-reaction with unrelated medical conditions) and XXX (Exogenous interfering substances)
There are studies and information included that evaluate the effects of potentially interfering and cross-reacting substances/agents on the assay
Yes/No
7.1.2.4. Metrological Traceability of Calibrators and Control Material Values
Yes/No
There is detailed information about the traceability of values assigned to calibrators and trueness control materials
Yes/No
7.1.2.5. Measuring range of the assay Yes/No Pages Error! Bookmark not defined.–xx and Annexes X (Linearity study), XXX (Limit of detection study) and X (Method correlation study)
Studies and information that define the measuring range of the assay are included
Yes/No
7.1.2.6. Validation of Assay Cut-off Yes/No
Studies and information on how the assay cut-off time was determined are included
Yes/No
7.1.2.7. Validation of Assay Procedure-reading Time Yes/No
Studies and information on how the reading time was determined are included
Yes/No
7.2. Stability (Excluding Specimen Stability) Yes/No
The studies and information on stability are included Yes/No
When applicable, the manufacturer has looked to internationally accepted methods for determining stability of diagnostics and followed WHO recommendations for stability
Yes/No
7.2.1. Claimed shelf-life Yes/No Pages Error! Bookmark not defined.–xx and Annex XX (Real-time stability study SHST XXX); Annex XXZ (previous real-time stability testing)
Ensure that testing is done on at least three different lots manufactured under conditions equivalent to routine production conditions
Yes/No
The study protocol specifies acceptance criteria and testing intervals Yes/No
Accelerated studies/extrapolated data are acceptable for initial shelf-life claim, but have been/will be followed up with real-time stability studies
Yes/No
If applicable, the method used for accelerated studies is identified Yes/No

WHO Sample product dossier for SIMU HIV-1 QUANT
WHO Product Dossier Checklist
Draft for public comment 26 October 2016 Page 17 of 140
Dossier Content Requirement Provided Location:
The shelf-life is derived from the lot with the shortest real-time stability data
Yes/No
The conclusions clearly identify claimed shelf-life stability Yes/No
7.2.2. In-use stability Yes/No Page Error! Bookmark not defined. and Annex XXX (In-use stability study)
There are studies and information provided for the in-use stability of each assay component
Yes/No
For each component, there is testing on at least one lot Yes/No
The studies reflect routine use of the device Yes/No
The study protocol specifies acceptance criteria and testing intervals Yes/No
If applicable, supporting data for calibration stability claims is provided
Yes/No
Conclusions clearly identify the claimed in-use stability Yes/No
7.2.3.Shipping stability Yes/No Page 67
Information needed on shipping stability studies is included. These studies are of one lot to evaluate the tolerance of products to the anticipated shipping conditions
Yes/No
Studies are done under real/Simulated conditions that include variable conditions
Yes/No
The studies reflect the environmental conditions of the countries of supply, along with justification
Yes/No
The study protocol specifies acceptance criteria and testing intervals Yes/No
If applicable, the methods of Simulated conditions have been identified
Yes/No
The results and conclusions must clearly demonstrate that the product will be effective at the end of its claimed shelf-life after being subjected to the stressed conditions
Yes/No
7.3. Robustness studies Yes/No Pages 72–xx and Annexes X (Cross-contamination study) and X (whole system failure study)
There is a summary of all evidence for the robustness study(ies) Yes/No
The test environment and its relation to the intended environment are stated
Yes/No
There is a discussion of what tests were considered for the device and why they were/were not performed
Yes/No
There is a discussion to support why the evidence presented is sufficient to support the application
Yes/No
If applicable, there is reference to the studies and endpoints for performance studies that include human factors/usability end points
Yes/No
7.4. Clinical Evidence (Clinical or Diagnostic Sensitivity and Specificity)
Yes/No

WHO Sample product dossier for SIMU HIV-1 QUANT
WHO Product Dossier Checklist
Draft for public comment 26 October 2016 Page 18 of 140
Dossier Content Requirement Provided Location:
7.4.1. Clinical evaluation - manufacturer Yes/No Pages 74–xx and Annexes XX (Clinical specificity study) and X (Clinical sensitivity study)
All performance claims are supported by well-designed performance evaluations that have been carried out/coordinated by the manufacturer and these studies are included with all relevant information
Yes/No
7.4.2. Clinical evaluation - independent study Yes/No
There are details of at least one well-designed independent performance evaluation for the product under assessment
Yes/No
If applicable, publication details of the independent study(ies) is included
Yes/No
Testimonials are not included as evidence of performance Yes/No
8. Labelling Yes/No Page 77
The product dossier contains a complete set of labelling associated with the product with all four required inclusions
Yes/No
8.1. Labels Yes/No Page 77
Copies of all packaging labels for the assay are included and contain all required information
Yes/No
8.2. Instructions for Use Yes/No
A copy of the current instructions for use are included and these instructions include all relevant information
Yes/No Page 77 and Annex XX
8.3. Instrument manual Yes/No
If applicable, there is a copy of the instrument manual/associated operator manuals included
Yes/No
8.4. Any Other Instructional Materials Provided to the User Yes/No
If applicable, any other instructional material copies are provided Yes/No
9. Commercial History Yes/No
9.1. Countries of Supply Yes/No
There is a list of all countries in which the product under assessment is currently supplied and the year when supply started
Yes/No Page 79 and Annex XX
For each country, detailed information about the training and support network are provided with all needed information
Yes/No
The minimum and maximum global price of supply for the product for the last financial year are included (quote in USD)
Yes/No
9.2. Adverse events and field safety corrective actions Yes/No Page 80
A list of all adverse events within the last five years, with details of the corrective and preventive action taken, is provided
Yes/No
There is a list of all events within the last five years that required field safety corrective action
Yes/No
10. Regulatory history Yes/No Page 81 and Annex XX
WHO Sample product dossier for SIMU HIV-1 QUANT
WHO Product Dossier Checklist
Draft for public comment 26 October 2016 Page 19 of 140
Dossier Content Requirement Provided Location:
If applicable, include a list of National Regulatory Authorities that have provided current regulatory approval for the supply of the in vitro IVD under assessment, as well as the type of regulatory approval obtained, in the countries/regions of the product authority
Yes/No
Current evidence of the regulatory approval, such as certified copies, must be included
Yes/No
If applicable, there are details regarding any situations in which the product was rejected by a National Regulatory Authority, an application was withdrawn, or approval was withdrawn
Yes/No
Information relating to the export-only regulatory approvals are clearly identified
Yes/No
11. Quality Management System Yes/No Pages 82–xx and Annex VIII (Quality manual)
11.1. Quality manual system documents and procedures Yes/No
There is a copy of the current version of the manufacturer’s quality manual and all needed points are addressed
Yes/No
An organizational chart for the manufacturer is provided Yes/No
A complete list of all valid quality management systems documents, with needed information, is included
Yes/No
Documented procedure/s for the control of design and development changes are included
Yes/No
Documented procedure/s relevant to risk management planning and implementation are included
Yes/No
Documented procedure/s relevant to control of non-conforming goods are included
Yes/No
Documented procedure/s relevant to the control of the key suppliers are included
Yes/No
11.2. Quality Management System Documents Yes/No
If applicable, there is evidence of certification for the manufacturer of the product
Yes/No
The two previous inspection reports issued by the certification body are included
Yes/No
Essential Principles (EP) Checklist Yes/No Annex I, page 83
This checklist is filled in as per the description and examples provided in the instructions
Yes/No
1

WHO Sample product dossier for SIMU HIV-1 QUANT
WHO Product Dossier Checklist
Draft for public comment 26 October 2016 Page 20 of 140
Manufacturer Declaration:
The undersigned authorized contact person for the Manufacturer makes the following declarations on behalf of the Manufacturer and, in signing this product dossier checklist form, declares that he/she has the authority to bind the Manufacturer.
I declare that:
I am authorized to represent the manufacturer specified in this prequalification product dossier (the "Manufacturer") for the purposes of WHO Prequalification of Diagnostics Programme of the product specified in this product dossier (the "Product").
All the information provided in this product dossier is current and correct.
This product dossier contains all the information as is prescribed in WHO Publication PQDx 018 “Instructions for compilation of a Product Dossier”.
The Manufacturer will notify WHO of all changes and variations to the Product prior to implementation of the changes.
The Manufacturer will notify WHO of any changes to the regulatory approval status for the Product, such as suspension or withdrawal of regulatory approval, in all countries of manufacture and supply.
Name of the Authorized Contact Person for the Manufacturer: Alan Bloggs
Signature of the Authorized Contact Person for the Manufacturer: Alan Bloggs
Date: 01 September 2016
Please Note: The Checklist submitted to WHO must be signed and dated.
WHO Sample product dossier for SIMU HIV-1 QUANT
The Product
Draft for public comment 26 October 2016 Page 21 of 140
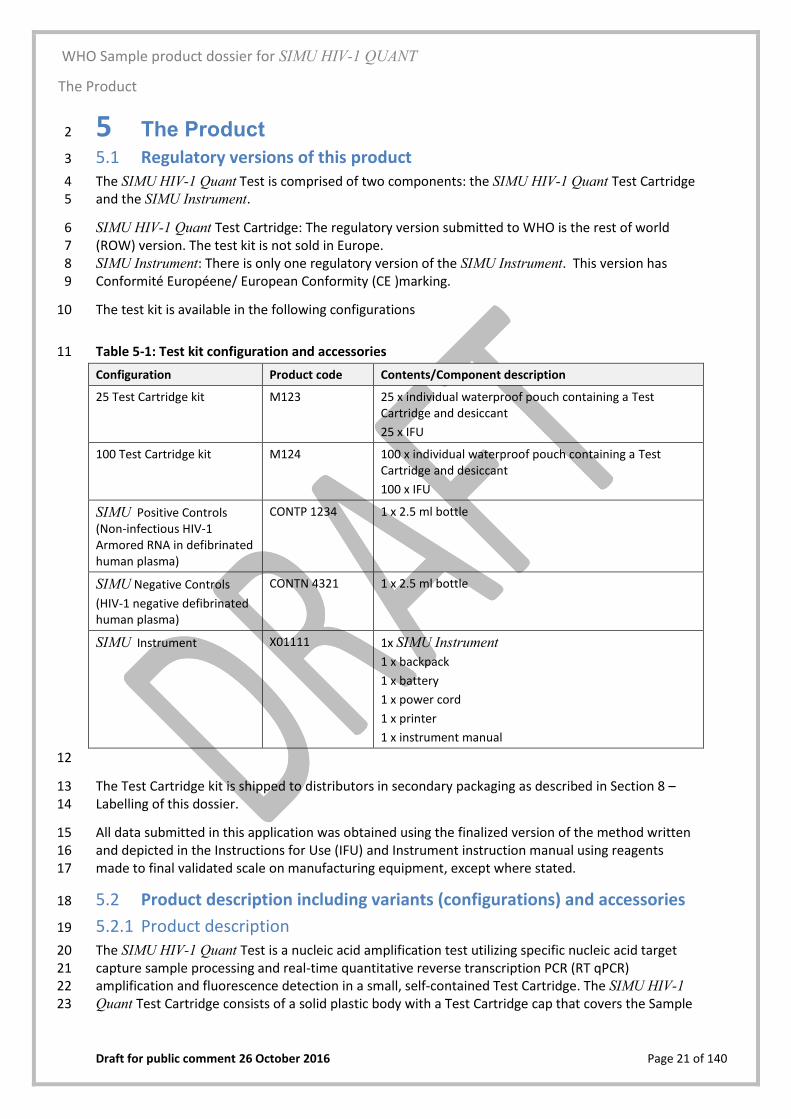
5 The Product 2
Regulatory versions of this product 5.13
The SIMU HIV-1 Quant Test is comprised of two components: the SIMU HIV-1 Quant Test Cartridge 4 and the SIMU Instrument. 5
SIMU HIV-1 Quant Test Cartridge: The regulatory version submitted to WHO is the rest of world 6 (ROW) version. The test kit is not sold in Europe. 7 SIMU Instrument: There is only one regulatory version of the SIMU Instrument. This version has 8 Conformité Européene/ European Conformity (CE )marking. 9
The test kit is available in the following configurations 10
Table 5-1: Test kit configuration and accessories 11
Configuration Product code Contents/Component description
25 Test Cartridge kit M123 25 x individual waterproof pouch containing a Test Cartridge and desiccant
25 x IFU
100 Test Cartridge kit M124 100 x individual waterproof pouch containing a Test Cartridge and desiccant
100 x IFU
SIMU Positive Controls (Non-infectious HIV-1 Armored RNA in defibrinated human plasma)
CONTP 1234 1 x 2.5 ml bottle
SIMU Negative Controls
(HIV-1 negative defibrinated human plasma)
CONTN 4321 1 x 2.5 ml bottle
SIMU Instrument X01111 1x SIMU Instrument
1 x backpack
1 x battery
1 x power cord
1 x printer
1 x instrument manual
12
The Test Cartridge kit is shipped to distributors in secondary packaging as described in Section 8 – 13 Labelling of this dossier. 14
All data submitted in this application was obtained using the finalized version of the method written 15 and depicted in the Instructions for Use (IFU) and Instrument instruction manual using reagents 16 made to final validated scale on manufacturing equipment, except where stated. 17
Product description including variants (configurations) and accessories 5.218
5.2.1 Product description 19
The SIMU HIV-1 Quant Test is a nucleic acid amplification test utilizing specific nucleic acid target 20 capture sample processing and real-time quantitative reverse transcription PCR (RT qPCR) 21 amplification and fluorescence detection in a small, self-contained Test Cartridge. The SIMU HIV-1 22 Quant Test Cartridge consists of a solid plastic body with a Test Cartridge cap that covers the Sample 23

WHO Sample product dossier for SIMU HIV-1 QUANT
The Product
Draft for public comment 26 October 2016 Page 22 of 140
Port (Figure 5-2). Each Test Cartridge is completely self-contained and prefilled with all reagents 24 needed for the test. The cartridge is used together with the SIMU Instrument (Figure 5-4). The 25 Instrument is a small and easily portable by handle or backpack. The Instrument is fully automated 26 after insertion of the Test Cartridge and operates with a rechargeable battery. All operations are 27 controlled on the Instrument and results can be read on the Instrument screen or output to a 28 computer or printer. The Instrument reads one Test Cartridge at a time, but multiple Instruments 29 can be combined together to increase throughput. The Instrument is classified as an in vitro 30 diagnostic device. 31
The SIMU HIV-1 Quant Test is capable of processing small volumes (50 µL) of plasma. The 32 appropriate volume of plasma is applied directly into the SIMU HIV-1 Quant Test Cartridge. Plasma 33 is added directly into the SIMU HIV-1 Quant Test Cartridge via the Sample Port using precision 34 pipetting. The amount of sample volume delivered into the Sample Port can be visualized through 35 the Sample Detection Port window on the Test Cartridge. Sample is added until it reaches the 36 gradation mark in the Sample Detection Port window. Once sample is introduced, the cap to the 37 port is closed and the Test Cartridge is loaded into the SIMU Instrument. The Test Cartridge consists 38 of several internal chambers including both liquid reagents and a buffer reservoir connected by 39 microfluidic channels. Air and liquid movement through the Test Cartridge is regulated by the 40 Instrument using valves within the cartridge. Once the Test Cartridge is loaded into the Instrument 41 and the test run is initiated, sample is delivered into the reaction chamber where the HIV RNA is 42 extracted. All excess sample and liquid waste produced during the test is sealed within the Test 43 Cartridge to reduce the possibility of contamination. Once the Test Cartridge is used, it cannot be 44 opened or reused. The real-time RT qPCR reaction takes place in the reactor chamber of the Test 45 Cartridge. Fluorescent signal generated from the sample and the sample processing control is 46 detected and interpreted by the system software. The results appear on the Instrument screen and 47 also as output to a computer or printer. The entire time to result after initiation of the test run is 48 45 minutes. 49

WHO Sample product dossier for SIMU HIV-1 QUANT
The Product
Draft for public comment 26 October 2016 Page 23 of 140
Figure 5-2. Diagrams of the SIMU HIV-1 Quant Test Cartridge 50
Top: external view; Bottom: cutaway showing internal components. 51
52
Specifications of the SIMU Instrument are given below in Table 5-3 53
Table 5-3: Specifications of the SIMU Instrument 54
Device dimensions/weight D 12 cm x W 23 cm x H 35 cm; 8.5 kg
Detection test fluorimeter
Languages English, French, Portuguese, Spanish
Memory 2 GB RAM, 100 GB Flash Storage
Software update Version 10.6.8
Operating conditions (°C, RH, Altitude) 12-45 °C, 5-95% RH, up to 3500 m
Storage conditions 2-55° C
Rated voltage 100-240 V, 50-60 Hz
Alternative charging options Can be run off 24 v DC power (e.g., solar panels or batteries)
Built-in voltage surge protection External uninterruptible power source and battery back-up 3 kg (product code 5555)

WHO Sample product dossier for SIMU HIV-1 QUANT
The Product
Draft for public comment 26 October 2016 Page 24 of 140
5.2.2 Intended use 55
The SIMU HIV-1 Quant Test is a quantitative nucleic acid amplification test for the detection of 56 Human Immunodeficiency Virus (HIV) type 1 groups M, N and O RNA in plasma samples from 57 individuals suspected of HIV-1 infection. The results of this test are intended to be used in 58 conjunction with other markers of disease progress for the clinical management of HIV-1 patients. 59 The assay can quantitate 800-10,000,000 copies of HIV-1 RNA per ml. It assesses patient prognosis 60 by measuring the changes in RNA levels during the course of antiviral therapy. The SIMU HIV-1 61 Quant Test is for in vitro diagnostic use only. 62
This assay is not intended to be used for screening of blood donors nor as a part of a diagnostic 63 testing algorithm for HIV. 64
The assay can be used in laboratory environments. 65
5.2.3 Intended users 66
The SIMU HIV-1 Quant System is intended to be used by trained healthcare or laboratory 67 professionals or other health care workers who have received appropriate training. 68
5.2.4 Photographs of kit 69
Insert photographs of all kit configurations with all components and accessories 70 collectively and individual components and accessories, in and out of any 71
packaging. 72
Figure 5-4: Illustration of the SIMU Instrument and backpack 73
74

WHO Sample product dossier for SIMU HIV-1 QUANT
The Product
Draft for public comment 26 October 2016 Page 25 of 140
5.2.5 A general description of the principle of the test method or instrument 75
principles of operation 76
5.2.5.1 Sample handling and processing 77
The sample volume required is a minimum of 50 µL. When using EDTA anti-coagulated plasma, the 78 appropriate volume is transferred into the sample port by using a volumetric pipette or transfer 79 capillary tube. Once the specimen is added to the port, the chamber will fill to a line visible on the 80 chamber. Insufficient specimen volume can be clearly seen by the meniscus and additional volume 81 may be added. Once the specimen is added to the cartridge, and the cartridge cap is snapped closed, 82 the cartridge can be stored up to 24 hours from 20–45°C before inserting into the SIMU Instrument 83 and performing the test. 84
After delivering the sample to the Test Cartridge, the cartridge cap is snapped into place, eliminating 85 the chance of sample spillage or contamination of the Instrument. The Test Cartridge is inserted into 86 the SIMU Instrument, the patient name and ID number are entered using the keypad on the 87 Instrument touchscreen, and the start button is pressed to initiate the testing. The steps in the 88 following subsections are performed automatically by the SIMU Instrument within the Test 89 Cartridge. 90
5.2.5.2 RNA target processing 91
Once the start button is pushed, the instrument scans the bar code on the Test Cartridge and a 92 measured aliquot of the sample is injected into the reaction chamber. A protease and chaotropic 93 lysis buffer is injected into the chamber to solubilize the viral envelope, denature proteins, and 94 release viral RNA. The lysis buffer also contains a sample processing control (SP control). The SP 95 control consists of an Armored RNA construct that is co-processed and co-amplified with the HIV 96 targets and controls for the effectiveness of sample processing, degradation of enzymes, 97 amplification, and sample inhibition. The reaction chamber contains magnetic microparticles 98 conjugated with capture oligonucleotides complementary to highly conserved regions of the HIV 99 genome (Table 5-5). Selection of the target RNA sequence for HIV-1 was performed using regions 100 within the HIV-1 genome that have maximum sequence conservation among HIV-1 groups M, N, and 101 O. Failures with mono-target HIV-1 NAT assays have been documented for HIV-1 M strains (Chudy, 102 M. et. al., 2012, Transfusion 52(2): 431-439, Fearon M et al, 2016, Transfusion 56(4): 994-995). Dual 103 target primer and probe sequences were therefore selected (two different amplicons are generated 104 with different primer sets, with the amplicons being detected by different probes). The assay was 105 designed with two or more target regions for group M (the most frequent representative group) in 106 addition to amplification of group N and O. The HIV RNA and the SP control RNA are specifically 107 bound to the capture oligonucleotides on the magnetic microparticles. The particles are drawn to an 108 electromagnet and washed to remove extraneous components such as salts, proteins and cellular 109 debris. The waste wash solution is moved to the liquid waste chamber within the Test Cartridge. The 110 amplification reagents are described in Table 5.5. 111

WHO Sample product dossier for SIMU HIV-1 QUANT
The Product
Draft for public comment 26 October 2016 Page 26 of 140
Table 5-5: Capture, primer and probe sequences and the region they are derived from for HIV-1 M 112 (dual target)/N/O and SP Control 113
Oligonucleotide Sequence Seq ID
Region sequence derived from
HIV-1 forward primer 1 5’-xxxxxxxxxxxx-3’ xx xx
HIV-1 reverse primer 1
HIV-1 Taqman Probe 1
HIV-1 forward primer 2
HIV-1 reverse primer 2
HIV-1 Taqman Probe 2
HIV-1 capture
SP forward primer
SP reverse primer
SP Taqman probe
SP capture
114
5.2.5.3 Real-time HIV target amplification and detection 115
Target amplification and detection of the HIV RNA captured on the microparticles is performed using 116 real-time RT qPCR. Prior to PCR amplification, RNA is reverse transcribed into cDNA. Specific DNA 117 primers hybridize to the RNA target and form a DNA-RNA hybrid. An rTth DNA polymerase then 118 transcribes the RNA into its complementary cDNA by extending the oligonucleotide primer. Reverse 119 transcription is followed by a heat denaturation step to deactivate the reverse transcriptase and 120 separate the RNA-DNA hybrid to make the newly formed cDNA accessible for primer binding and 121 extension by PCR. 122
This real-time multiplex PCR reaction utilizes hydrolysis probes to detect the DNA produced during 123 the PCR reaction. The hydrolysis probes consist of oligonucleotides specific for regions of the HIV 124 cDNA molecules and have a reporter fluorophore covalently attached to one end of the probe and a 125 quencher at the other end. When the probes are intact, the reporter and quencher molecules stay 126 close to each other preventing the emission of fluorescence. Two specific probes with the same 127 fluorophore are used to target two different highly conserved regions of HIV-1 (Group M/N/O). A 128 third specific probe with a unique fluorophore is used to detect the SP control. The HIV target DNA 129 and cDNA created during the reverse transcription step bind specific primers and probes. rTth DNA 130 polymerase extends the primers and during the process cleaves the probe that separates the 131 reporter and quencher molecules allowing the reporter molecules to emit fluorescence. The 132 Instrument automatically repeats the PCR cycle for a designated number of cycles, with each cycle 133 doubling the amount of DNA amplicon and increasing the emission intensity of the individual 134 fluorophores. The amplification of the HIV-1 RNA and SP control is measured independently at 135 different wavelengths to quantitatively determine the presence or absence of the targets. Since the 136 SP control is added to each specimen at a known copy number, the Instrument can calculate the 137 RNA concentration in the test specimens by comparing the HIV-1 signal to the SP control signal for 138 each specimen. The test can quantitate HIV-1 RNA over the range of 800 to 10,000,000 copies/mL. 139 The Instrument system software interprets the results that are read on the Instrument screen or 140 downloaded to a computer or printer. An example of a test report is shown in Figure 5-6. 141
The table is provided as an example of how the data may be presented in the product dossier submitted to WHO. The sequence information is expected to be provided by the manufacturer. All proprietary information will be kept
confidential by WHO.

WHO Sample product dossier for SIMU HIV-1 QUANT
The Product
Draft for public comment 26 October 2016 Page 27 of 140
Figure 5-6: Example of SIMU HIV-1 Quant Test report 142
Sample ID 334-4567
HIV-1 Result 5530 copies/mL
Date/Time 2015-FEB 10
Test Cartridge ID 034289833
Test Cartridge Serial number 0023456
Software version 10.06.8
SP Control Pass
………
143
5.2.6 A description of the components of the test (e.g. reagents, assay controls 144
and calibrators) 145
The SIMU HIV-1 Quant consists of the dedicated components described in Table 5-7. 146
Table 5-7: Description of components of the assay 147
Configuration Product code Contents
25 Test Cartridge test kit M123 25 x individual waterproof pouches containing a Test Cartridge and desiccant
25 x IFU
100 Test Cartridge test kit M124 100 x individual waterproof pouches containing a Test Cartridge and desiccant
100 x IFU
148
The SIMU HIV-1 Quant Test Cartridge comes in two pack sizes (Table 5-7). The reagents for the 149 system are contained in the SIMU HIV-1 Quant System Test Cartridge. The Test Cartridge includes 150 all the materials needed for the test including probes, primers, control oligonucleotides and 151 microparticles. The nucleic acids are in an inorganic buffering solution containing bactericides and 152 fungicides. 153
Positive and negative external controls can also be ordered individually and should be used in 154 accordance with IFU instructions. 155
5.2.7 A description of the specimen collection and transport materials 156
provided with the product or descriptions of specifications 157
recommended for use. 158
The device has been validated for use with plasma specimens using EDTA as an anticoagulant. 159
Plasma specimens in EDTA tubes can be stored for 48 hours at 2-8°C or stored frozen indefinitely at 160 -20 °C or less. Avoid more than three freeze-thaw cycles. 161
5.2.8 For Instruments of automated assays: a description of the appropriate 162
assay characteristics or dedicated assays 163
See Section 5.2.1, Product Description 164

WHO Sample product dossier for SIMU HIV-1 QUANT
The Product
Draft for public comment 26 October 2016 Page 28 of 140
5.2.9 For automated assays: a description of the appropriate instrumentation 165
characteristics or dedicated instrumentation. 166
The SIMU Instrument is used for the SIMU HIV-1 Quant Test assay and is also able to run the entire 167 line of SIMU nucleic acid amplification assays. It is a small, free-standing bench-top instrument easily 168 portable by hand or backpack. The Instrument has one slot for Test Cartridge insertion and a simple 169 touch screen control panel interface for inputting patient information and initiation of the assay run. 170 Once the patient information is entered and the start button is pushed, the Instrument will scan the 171 Test Cartridge barcode and match the patient information to the sample. The Instrument is fully 172 automated after insertion of the Test Cartridge and is powered with an integrated rechargeable 173 battery. The battery operates up to 8 hours if there is an unexpected interruption to the power 174 supply. A supplemental rechargeable battery weighing 3 kg (product code 5555) is also available that 175 can power the Instrument for up to 24 hours. All operations are controlled on the Instrument and 176 results can be read on the Instrument screen or output to a computer or printer. The Instrument 177 runs one Test Cartridge at a time but multiple instruments can be combined together to increase 178 output and to run multiple different assays if required. 179
5.2.9.4 Data export and connectivity 180
The Instrument can transmit instrument and test information in real-time to computers, mobile 181 devices, and printers via wireless internet and USB connections The Instrument is water resistant 182 and able to withstand temperature extremes, dust, high altitude, and high humidity. The 183 instrumentation manual is attached in Annex XX. 184
5.2.10 If applicable, a description of any software to be used with the 185
product 186
Software is incorporated in the SIMU Instrument and no additional software is required. The test 187 software is controlled via the touchscreen interface and is used to: input patient information, initiate 188 the assay run, output data to other devices and other basic functions. The software automatically 189 controls all of the processes necessary for running the assay Test Cartridge once the run is initiated. 190 There is a countdown timer on the Instrument screen to indicate the time remaining prior to result 191 and an alarm function indicating when the test is complete. The software also analyses the results 192 and stores the data for future retrieval. The test data storage module can store 30,000+ test results. 193 The test data storage module also controls for software or hardware failure by displaying a test 194 failure error code. The assay software is designed and maintained in accordance with EN 62304 195 (Medical Device Software, 2006). The design history file (DHF) and instrument software 196 documentation is kept on-site. 197
5.2.11 If applicable, a description or complete list of the various 198
configurations or variants of product that will be made available 199
The SIMU HIV-1 Quant Test Cartridge comes in two configurations: 25 Test Cartridge Pack (M123) 200 and 100 Test Cartridge Pack (M124). 201
5.2.12 If applicable, a description of the accessories, and other products 202
that are intended to be used in combination with the diagnostic. 203
See Table 5-1 Test kit configuration and accessories. 204
Essential principles (EP) check list 5.3205
An EP checklist has been produced for the device, Annex I: Essential Principles Checklist 206

WHO Sample product dossier for SIMU HIV-1 QUANT
The Product
Draft for public comment 26 October 2016 Page 29 of 140
The evidence supporting performance requirements is submitted as Section 7 of this dossier and 207 other evidence of manufacturing and quality management is also provided throughout this dossier, 208 in agreement with GHTF document GHTF/SG1/n046 (Principles of Conformity Assessment for In 209 Vitro Diagnostic (IVD) Medical Devices ) and with Annex II of the draft Regulation on In Vitro 210 Diagnostic Medical Devices of the European Community (“the draft IVD regulation”). 211
Risk analysis and control summary 5.4212
5.4.1 Risk analysis policy 213
The risk management policy of THE Manufacturing Company is attached as Annex II: Risk 214 Management Policy. The SOPs lead to the Failure Mode and Effects Analysis (FMEA) output 215 documents attached (Annex IV: Design Inputs FMEA, Annex V: User and Patient FMEA, Test 216 Software FMEA), and to the risk statement (Annex III: Residual Risk Statement), which describes any 217 residual risks and their control by warning statements. The process and supplier management FMEA 218 for this product are of the same format as the submitted FMEAs for 'risk to users and patients' and 219 are kept on file but have not been submitted with this dossier. 220
The design input FMEA is prepared before any R&D and is updated as work progresses and the 221 requirements are satisfied, as is the risk FMEA for user and patient. 222
A series of control measures arises from the risk assessment – these are listed in the spreadsheets of 223 the risk analyses and also shown on the flow diagram of the manufacturing process. The 224 effectiveness of the controls and changes in eliminating the identified risk is shown in the FMEA. 225 Some of the risks – such as instability, common- (but not specific-) interfering agents are regarded as 226 routine and evaluated as a matter of course. This data is presented in Section 7 of this dossier. 227 Instability and allowable storage times and temperatures are evaluated for plasma, for the shelf-life 228 of the Test Cartridge itself using several independent manufactured lots, and for the usable life of 229 the Test Cartridge device once taken out of the pouch. 230
5.4.2 Risk categories 231
5.4.2.5 Erroneous test results 232
Types of erroneous results and their impact: 233
This quantitative assay has been designed to monitor viral load levels in response to antiretroviral 234 treatment. Consequently, the major categories of erroneous results that can result in harm to the 235 patient include: 236
Inaccurate quantitation. This may result in misclassification of the patient as responsive to 237 treatment, leading to a patient remaining on an ineffective antiretroviral regimen and further 238 leading to a serious decline in health and ultimately the death of the patient. An inaccurate result 239 may also lead to a patient being misclassified as non-responsive to treatment resulting in an 240 unnecessary change in treatment regimen. 241
Various types of instability that could lead to erroneous test results have been evaluated including 242 specimen type, shipping conditions, shelf-life of the device, life of the materials once opened, and 243 length of time after adding specimen to Test Cartridge for which the result is valid. Instrument 244 instability that could lead to erroneous results includes optics misalignment, software issues, high 245 dust levels, and electrical supply instability. Evaluations were performed with several independently 246 manufactured lots of Test Cartridges and Instruments, and erroneous results were rarely identified. 247 The test is intended for users with varying levels of laboratory skills, from inexperienced to highly 248

WHO Sample product dossier for SIMU HIV-1 QUANT
The Product
Draft for public comment 26 October 2016 Page 30 of 140
experienced, who are responsible for specimen collection and preparation, performing the test, and 249 interpreting the test result. 250
5.4.2.6 Labelling 251
Product labelling is an important tool in mitigating these risks. The product packaging and IFU utilizes 252 symbols per ISO 15223-1:2012 Medical Devices—Symbols to be used with medical device labels, 253 labelling and information to be supplied—Part 1: General requirements to instruct users on how to 254 effectively and safely use the device. Internationally recognized symbols) are used for certain IVD 255 characteristics which are useful for users who are not familiar with the language in which the IFU is 256 written, to simplify communication. Stability conditions are clearly described in the IFUs for Test 257 Cartridges and instrumentation. In addition, the device does not require any special storage 258 conditions, such as refrigeration. Specimen collection labelling is also important for instructing on 259 proper specimen collection and stability of the specimens. 260
Product labelling is also important for addressing the risks associated with use of the test and 261 includes language or illustrations describing the accuracy of the test and how to interpret the test 262 correctly. Product labelling and language is easily readable (Flesch-Kincaid grade <6). The test is 263 designed to be simple to use so that the intended use population will be able to perform the test 264 properly. 265
5.4.2.7 Interfering substances 266
Investigation of interfering substances should be relevant to the intended testing population. Risk 267 was initially mitigated through design with careful selection of primers and probes to minimize 268 potential inaccurate results. Residual risk was measured through analytical studies (see Section 269 7.1.2.3) and confirmed through clinical studies in the population of intended use. 270
Exogenous substances including protease inhibitors, hepatitis C virus (HCV), hepatitis B virus 271 medicines, opportunistic infection treatments, such as anti-fungal medicines and anti-mycobacterial 272 medicines, have been tested with this assay. Endogenous substances from patients with medical 273 conditions such as diabetes mellitus and those causing high levels of bilirubin and cholesterol have 274 also been tested. 275
Potential cross-reactions due to related organisms e.g. human T-lymphotropic virus (HTLV), were 276 investigated. Results of these studies can be found in section 7.1.2.3. 277
Evaluation of the test in the intended testing population supported findings of analytical testing, 278 indicating the test is robust and the risk of interfering substances is minimal. No impact on test 279 performance was observed and the assay met the pre-established acceptance criteria for the 280 studies. 281
The manufacturer should add/remove discussion of risk categories as 282 appropriate, such as those indicated below. 283
5.4.2.8 Design 284
A risk of infection to personnel from patient specimens due to leakage of specimen from the Test 285 Cartridge has been excluded by design to the greatest extent possible. This includes permanent 286 sealing of the Test Cartridge door after sample application, lysis and denaturation of the sample 287 during sample processing, and clear instructions on handling and disposal of used Test Cartridges. 288

WHO Sample product dossier for SIMU HIV-1 QUANT
The Product
Draft for public comment 26 October 2016 Page 31 of 140
5.4.2.9 Testing process 289
The manufacturer should summarize the risks and mitigations related to the 290 testing process, such as the specimen collection process, minimizing the 291
interventions required by the operator, amplicon contamination etc. 292
5.4.2.10 Stability 293
The manufacturer should summarize the risks and mitigations related to 294 stability, such as storage temperature of the device, on performance including 295
potential long-term storage by the user after purchase and before testing. 296
5.4.2.11 Specimen type 297
There are several risks related to specimen type used in the assay. The major risk for specimens is 298 that too low a volume may be added to the sample port of the Test Cartridge, which could result in a 299 false negative or falsely low quantitative result in the assay. This has been mitigated by design of 300 liquid gradation marks visible through the sample capillary port window. Insufficient specimen 301 volume can be clearly seen and additional volume may be added prior to closing the Test Cartridge 302 sample port cap. In addition, inadequate sample volume added to the port would result in a sample 303 processing control failure resulting in an invalid assay run report rather than a false negative result. 304
There is a small risk that users may use other types of anticoagulants other than EDTA for plasma. 305 This is mitigated by specific instructions in the IFU to only use EDTA. 306
There is a risk hazard of disease transmission during the handling of samples and the disposal of Test 307 Cartridges. The sample handling risk was minimized by the design of simple procedures to deliver 308 sample to the Test Cartridge and detailed instructions in the IFU. Contamination after sample 309 addition to the Test Cartridge has been minimized by designing the cartridge to completely contain 310 the specimen after the specimen intake door is closed. Used Test Cartridges cannot be opened or 311 reused and will not leak under normal usage. In addition, the sample processing reagents lyse and 312 denature the virus making it much less likely to be infectious. The used Test Cartridge should be 313 handled and disposed as biohazard waste. 314
The manufacturer should summarize the risks and mitigations related to 315 specimen type if applicable. 316
5.4.3 Instrument 317
The main risks from the Instrument are for the operator and population in which it is intended for 318 use. These have been outlined in the Instrument FMEA (Annex XX). The main risks identified arise 319 from: 320
Temperature, humidity, altitude and electromagnetic radiation which have been studied 321 extensively as part of the software development, transport and robustness studies and which 322 are mitigated with clear labelling on the Instrument. 323
Software issues such as confusing user prompts, incorrect mathematical algorithm, undetected 324 errors, timing failure, or incorrect storage of test results in memory. 325
Hardware issues such as electronic failure, physical trauma or vibration, electromagnetic 326 interference, battery reliability, component failure or incorrect manufacturing. 327

WHO Sample product dossier for SIMU HIV-1 QUANT
The Product
Draft for public comment 26 October 2016 Page 32 of 140
Transport stability and robustness studies have been undertaken to assess the impact of these 328 issues and compliance with the recognized standards are listed in the EP checklist. 329
System issues such as incorrect calibration, calibration failure, or inadequate training. These 330 have been mitigated by appropriate user manual instructions, and integrated QC steps in the 331 assembly of the Instrument. 332
5.4.4 Risk/Benefit 333
As described above, the main risks are related to erroneous test results, whether they occur through 334 producing or distributing the test, or use of the test. 335
The SIMU HIV-1 Quant Test was compared with the well-established laboratory based HIV viral load 336 assays and found to have a high degree of concordance (see Section 7.4) with the assays tested. 337
In conclusion, the remaining risks posed by potential erroneous results of the test are outweighed by 338 the individual and public health benefits projected from use of the test. 339
The Manufacturer should provide evidence that the risk analysis is part of the 340 manufacturer's risk management plan (inclusion of the relevant manufacturer’s 341
document). 342
The Manufacturer should provide identification of specific standards or 343 guidelines recommended by WHO, when applicable (for example, ISO 344
14791:2007 (E) “Medical devices-- Application of risk management to medical 345 devices”). 346

WHO Sample product dossier for SIMU HIV-1 QUANT
Design and Manufacturing Information
Draft for public comment 26 October 2016 Page 33 of 140
6 Design and Manufacturing Information 347
Design control 6.0348
i) The R&D Department of THE Manufacturing Company (987 Somewhere Street, Somewhere 349 in Europe EU-1234, Europe) is responsible for the control of design of the assay and 350 instrument. 351
ii) The R&D Department was responsible for all design aspects and design validation of the Test 352 Cartridge device at the manufacturing site at company headquarters. Headquarters 353 manufacturing undertakes all aspects of manufacture for Test Cartridges, apart from 354 developing the prototypes in conjunction with the R&D Department. 355
iii) Although manufacturing of the SIMU Instrument is outsourced to an ISO 9001:2015 certified 356 manufacturer of electronic equipment (Imaginative Diagnostic Designers, Top Street 357 Industrial Estate, Some Country, Europe), the design, specification and quality requirements 358 were provided by the R&D Department, and control of final design specifications rests with 359 this group (SOP XXX). 360
iv) Following the development plan there is a risk assessment followed by full staff training, 361 qualification of all the manufacturing processes, re-verification of Test Cartridge 362 performance against the specifications and revalidation of the Quality Assurance release-to-363 market requirements. 364
v) Packaging materials and desiccant are sourced locally. Every lot is quality assurance (QA) 365 inspected and validated for use prior to acceptance by the manufacturer. Any changes to 366 packaging materials are evaluated by risk assessment; any changes to packaging or their 367 labels in immediate contact with product such as the Test Cartridge plastic housing require a 368 revalidation of stability as described in SOP XXX. Any changes to secondary and shipping 369 packaging are re-validated for transport including drop and shock testing as per ASTM 370 D4169-14. 371
vi) Certified copies of certificates for the quality management system (ISO 13485), and the 372 environmental management system (ISO 14001:2015) for the design and manufacturing site 373 are attached as part of Section XXX. 374
vii) The design control system is shown in Figure 6-1. It will be understood that product design is 375 iterative so although the process is shown as a linear flow, each stage might be repeated 376 several times to optimise the whole design. 377
viii) Design change control begins as soon as the customer requirements document is 378 authorized; changes to requirements documents undergo full change control, controlled by 379 SOP XXXX, changes during phase 1 are under R&D change control SOP XXXX, which is 380 different from full design change control only in the extent of revalidation potentially 381 required. 382
ix) All performance factors are obtained with product manufactured following authorized, finalized documentation and QA parameters.
x) Data generated throughout the design input and R&D phases is collected as the DHF, and 383 the finalized specifications as the device master record. Risk analyses are initiated as shown 384 and reviewed regularly with direction from SOP XXXX. Design control review meetings are 385 also held regularly and the output of the meetings stored in the DHF. 386

WHO Sample product dossier for SIMU HIV-1 QUANT
Design and Manufacturing Information
Draft for public comment 26 October 2016 Page 34 of 140
xi) Training for manufacturing staff commences as soon as possible in the design phase, and 387 during R&D phase 2 at the latest. Process SOPs are qualified in the factory by manufacturing 388 staff under R&D supervision. All design work is performed and controlled in our R&D 389 laboratories, manufacturing up to and including devices used for design validation is done in 390 our main factory, where production for all our ROW products occurs. 391
xii) Following the development phase there is a risk assessment followed by full staff training, 392 qualification of all the manufacturing processes, re-verification of device performance 393 against the specifications and revalidation of the QA release-to-market requirements, all 394 being completed before validating the device in user’s hands in the intended environment of 395 use. 396
xiii) The IFU is written according to ISO 18113 series and printed professionally close to our 397 factory, with translation where necessary from the authorized English version by licensed 398 scientific/medical translators and subsequent approval by our local staff. 399
xiv) A Universal Device Identification (UDI) is applied according to IMDRF/UDIWG/N7FINAL: 2013 400 and Article 22 of the draft IVD regulation. Packaging materials are sourced locally, every lot 401 is QA inspected and validated for use prior to acceptance by the manufacturer. Any changes 402 to packaging materials are evaluated by risk assessment. 403
xv) Any changes to the design, including to labelling, are controlled within the change control 404 system and cascaded down to all our factories, with risk evaluations and re-qualifications as 405 appropriate at each factory. 406
xvi) Changes to the design are notified in compliance with mandatory WHO and regulatory 407 requirements. 408
409
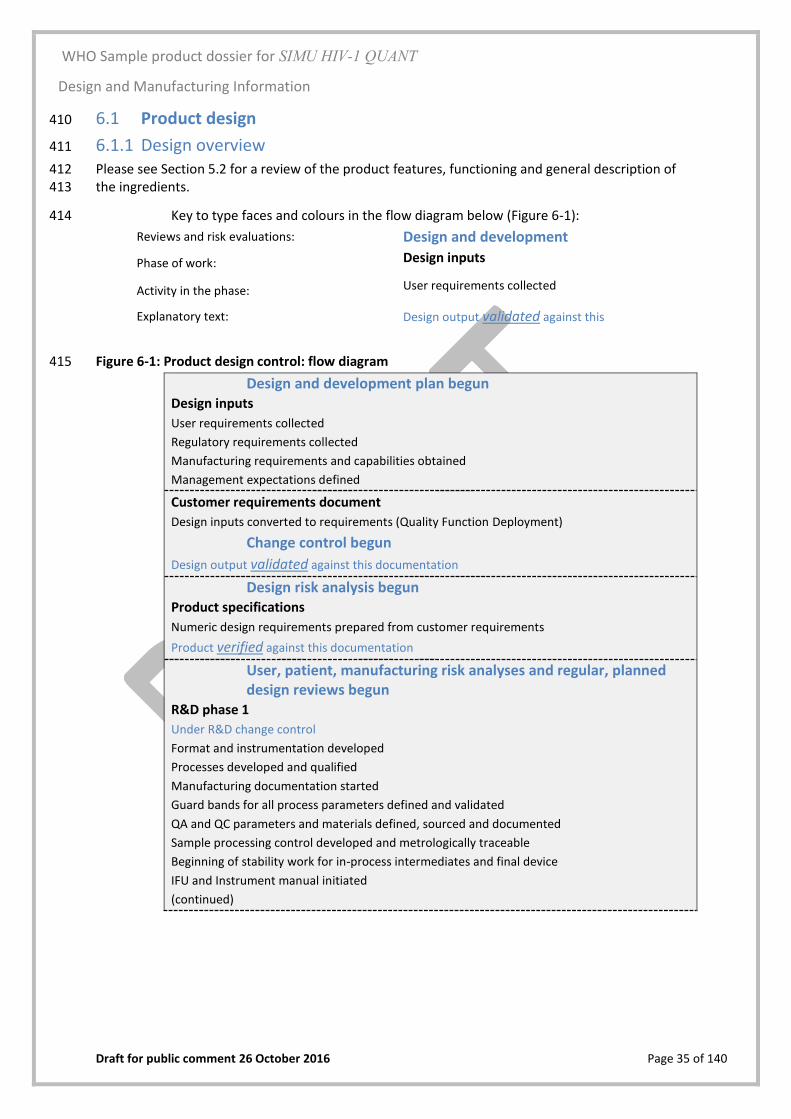
WHO Sample product dossier for SIMU HIV-1 QUANT
Design and Manufacturing Information
Draft for public comment 26 October 2016 Page 35 of 140
Product design 6.1410
6.1.1 Design overview 411
Please see Section 5.2 for a review of the product features, functioning and general description of 412 the ingredients. 413
Key to type faces and colours in the flow diagram below (Figure 6-1): 414
Reviews and risk evaluations: Design and development
Phase of work: Design inputs
Activity in the phase: User requirements collected
Explanatory text: Design output validated against this
Figure 6-1: Product design control: flow diagram 415
Design and development plan begun Design inputs
User requirements collected
Regulatory requirements collected
Manufacturing requirements and capabilities obtained
Management expectations defined
Customer requirements document
Design inputs converted to requirements (Quality Function Deployment)
Change control begun
Design output validated against this documentation
Design risk analysis begun Product specifications
Numeric design requirements prepared from customer requirements
Product verified against this documentation
User, patient, manufacturing risk analyses and regular, planned design reviews begun
R&D phase 1
Under R&D change control
Format and instrumentation developed
Processes developed and qualified
Manufacturing documentation started
Guard bands for all process parameters defined and validated
QA and QC parameters and materials defined, sourced and documented
Sample processing control developed and metrologically traceable
Beginning of stability work for in-process intermediates and final device
IFU and Instrument manual initiated
(continued)

WHO Sample product dossier for SIMU HIV-1 QUANT
Design and Manufacturing Information
Draft for public comment 26 October 2016 Page 36 of 140
R&D phase 2
Transitioning to factory Under design change control as documents are finalized
Instruments and material suppliers finalized and audited
Instrumental performance qualification, operational qualification in the factory
Pilot batches made, tested against presumed QC
Interfering substances evaluated
Efficacy of anti-microbials and fungicides proven
Process documentation finalized and approved
All aspects of device and specimen stability using devices made to approved specifications
IFU and Instrument manual finalized and approved
Any changes to the IFU other than addition of performance data require in-depth evaluation and revalidation
QA and QC specifications finalized and approved
R&D phase 3
Design verification using material made in the factory to approved documentation
Formal completion and documentation of the activities of phase 2 (R&D evaluation of all aspects of the product specification e.g. performance, repeatability, reproducibility, lot-to-lot variability, all aspects of stability)
R&D phase 4
Design validation using material made in the factory at scale to approved documentation
(User evaluation of all aspects of product specification and customer requirements: e.g. performance, repeatability, reproducibility, lot-to-lot variability, functionality of IFU, training manuals, software)
Normally done in three user-labs with three independent lots of reagent
IFU completed with data from validation work
e.g. reproducibility, specificity,
Final risk analyses and production of risk declaration Manufacture
Full scale manufacture under change control
On-market surveillance
Review of scientific literature for independent clinical evaluations

WHO Sample product dossier for SIMU HIV-1 QUANT
Design and Manufacturing Information
Draft for public comment 26 October 2016 Page 37 of 140
6.1.2 Formulation and composition 416
Formulation and composition information such as ingredients of buffers, amino 417 acid sequences for recombinant proteins etc. should be provided. Please note 418
that WHO will ensure that all proprietary information will be treated 419 confidentially. 420
Biochemical information on the critical reagents is shown in Table 6-2. 421
Table 6-2: Biochemical information on the critical reagents 422
Constituent Information
Armored RNA for SP control and external positive control
Armored HIV-1 RNA construct containing HIV-1 primer binding sequences and a unique probe binding region (non-infectious RNA in MS2 bacteriophage).
Primer oligonucleotides for HIV-1 Upstream and downstream primers to the gag and the l ong terminal repeat (LTR) region of HIV-1. See Table 5-5 for sequence information.
Probes Fluorescent and quencher labelled oligonucleotide probes specific for HIV-1 and SP control. See Table 5-5 for sequence and dye information.
Sample lysis buffer Sodium citrate dihydrate buffer containing 43% guanidine thiocyanate, 1.4 N-Lauroylsarcosine, 0.9% dithiothreitol, 7.8% proteinase K, 0.05% EDTA.
Wash solution contained within the device A pH buffered solution with sodium citrate dihydrate and <0.1% N-Methylisothiazolone-HCL.
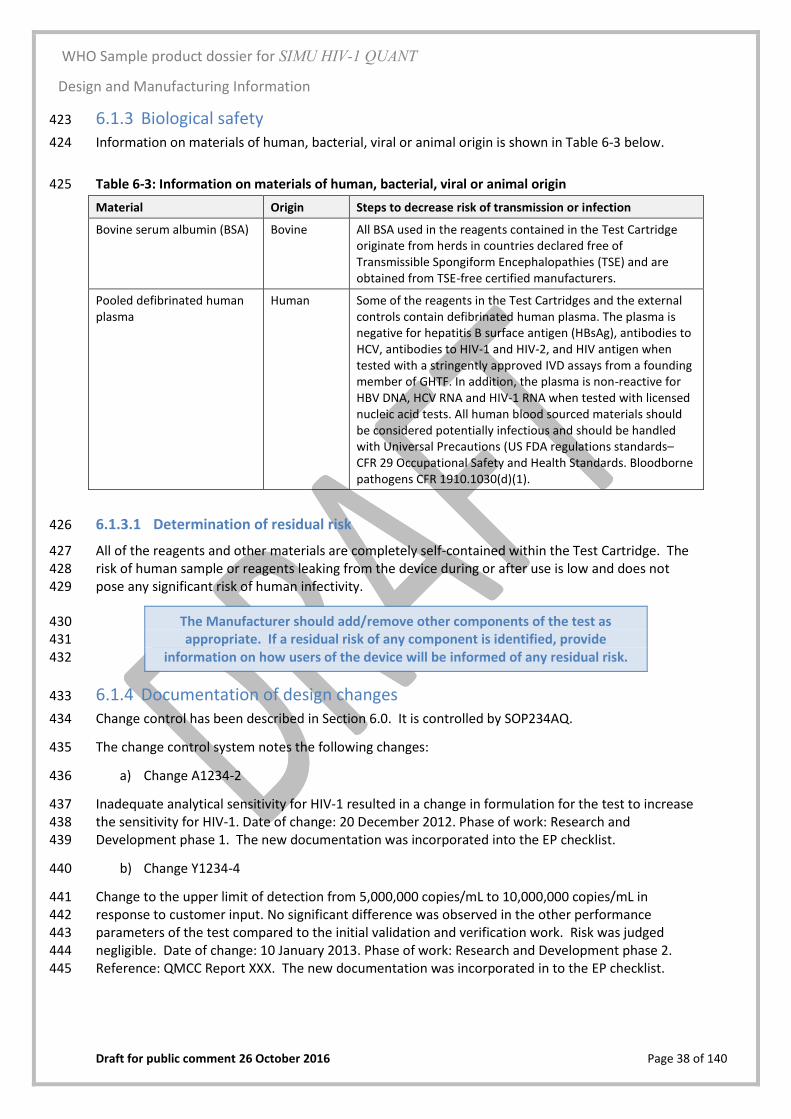
WHO Sample product dossier for SIMU HIV-1 QUANT
Design and Manufacturing Information
Draft for public comment 26 October 2016 Page 38 of 140
6.1.3 Biological safety 423
Information on materials of human, bacterial, viral or animal origin is shown in Table 6-3 below. 424
Table 6-3: Information on materials of human, bacterial, viral or animal origin 425
Material Origin Steps to decrease risk of transmission or infection
Bovine serum albumin (BSA) Bovine All BSA used in the reagents contained in the Test Cartridge originate from herds in countries declared free of Transmissible Spongiform Encephalopathies (TSE) and are obtained from TSE-free certified manufacturers.
Pooled defibrinated human plasma
Human Some of the reagents in the Test Cartridges and the external controls contain defibrinated human plasma. The plasma is negative for hepatitis B surface antigen (HBsAg), antibodies to HCV, antibodies to HIV-1 and HIV-2, and HIV antigen when tested with a stringently approved IVD assays from a founding member of GHTF. In addition, the plasma is non-reactive for HBV DNA, HCV RNA and HIV-1 RNA when tested with licensed nucleic acid tests. All human blood sourced materials should be considered potentially infectious and should be handled with Universal Precautions (US FDA regulations standards– CFR 29 Occupational Safety and Health Standards. Bloodborne pathogens CFR 1910.1030(d)(1).
6.1.3.1 Determination of residual risk 426
All of the reagents and other materials are completely self-contained within the Test Cartridge. The 427 risk of human sample or reagents leaking from the device during or after use is low and does not 428 pose any significant risk of human infectivity. 429
The Manufacturer should add/remove other components of the test as 430 appropriate. If a residual risk of any component is identified, provide 431
information on how users of the device will be informed of any residual risk. 432
6.1.4 Documentation of design changes 433
Change control has been described in Section 6.0. It is controlled by SOP234AQ. 434
The change control system notes the following changes: 435
a) Change A1234-2 436
Inadequate analytical sensitivity for HIV-1 resulted in a change in formulation for the test to increase 437 the sensitivity for HIV-1. Date of change: 20 December 2012. Phase of work: Research and 438 Development phase 1. The new documentation was incorporated into the EP checklist. 439
b) Change Y1234-4 440
Change to the upper limit of detection from 5,000,000 copies/mL to 10,000,000 copies/mL in 441 response to customer input. No significant difference was observed in the other performance 442 parameters of the test compared to the initial validation and verification work. Risk was judged 443 negligible. Date of change: 10 January 2013. Phase of work: Research and Development phase 2. 444 Reference: QMCC Report XXX. The new documentation was incorporated in to the EP checklist. 445

WHO Sample product dossier for SIMU HIV-1 QUANT
Design and Manufacturing Information
Draft for public comment 26 October 2016 Page 39 of 140
Manufacturing process 6.2446
6.2.1 Overview of manufacture 447
The flow of product manufacture of the Test Cartridge is shown below in Figure 6-4. 448
In-process quality checks are included – the reasons for each of these checks can be traced back 449 either to a risk management document (see Section 5.4 and annexes) or a routine technical 450 measurement for the process concerned. 451
Critical materials supplied to THE Manufacturing Company are marked in red borders and their 452 sources listed in Table 6-5. Incoming goods are checked against specifications and functional testing 453 is performed as decided from risk analyses and supplier auditing. 454
455

WHO Sample product dossier for SIMU HIV-1 QUANT
Design and Manufacturing Information
Draft for public comment 26 October 2016 Page 40 of 140
Figure 6-4: Manufacturing flow diagram for the SIMU HIV-1 Quant Test Cartridge and SIMU 456 Instrument system 457

WHO Sample product dossier for SIMU HIV-1 QUANT
Design and Manufacturing Information
Draft for public comment 26 October 2016 Page 41 of 140
6.2.1.2 Batch release criteria/Final lot release 458
Final lot release criteria for the Test Cartridge device is contained in the verification report AXXXX 459 and for the Instrument system in verification report BXXXX. The final product is inspected to meet 460 the final requirements outlined in the Quality Specifications for the SIMU HIV-1 Quant Test. 461
Each batch of critical reagents manufactured by THE Manufacturing Company such as 462 oligonucleotides and labelled probes are purified and undergo analytical testing followed by 463 functional testing to meet the requirements outlined in the Quality Specifications for the SIMU HIV-464 1 Quant Test. Critical reagents such as proteins supplied to THE Manufacturing Company undergo 465 analytical and functional testing to assure they meet specifications. The final device release testing 466 is performed using well-qualified panels of samples to assure the product meets lot release criteria. 467 Inter-lot homogeneity testing is also performed to assure consistent performance between product 468 lots. 469
6.2.2 Sites of manufacture 470
The entire Test Cartridge, including all oligonucleotides, is made in the European factory location of 471 THE Manufacturing Company located at 987 Somewhere Street, Somewhere in Europe EU-1234, in 472 accordance with the processes described above. A number of other devices are made at this site 473 under the same design control and quality systems, and in some cases using similar processes. 474 These devices include IVDs for the detection of: 475
Malaria antigens 476
HIV – antibody-antigen 477
Hepatitis C – antibody-antigen 478
Syphilis antibodies 479
Hepatitis B surface antigen 480
HIV-1/2 TNA (qualitative assay) 481
A site plan for the factory is included in Annex XXX 482
The company has 96 staff. A structure chart is included in the Quality Manual, appended as ANNEX 483 VIII. 484
The SIMU Instrument is manufactured at the ISO 9001:2015 certified facility listed in the key 485 suppliers list below in Table 6-5. 486
6.2.3 Key suppliers 487
Certified certificates for the manufacturers are attached in Annex XXX. 488
The constituents and their source for critical reagents and the SIMU Instrument are shown in Table 489 6-5. 490

WHO Sample product dossier for SIMU HIV-1 QUANT
Design and Manufacturing Information
Draft for public comment 26 October 2016 Page 42 of 140
Table 6-5: Constituents and their source for critical reagents 491
Constituent Certification if available
(certified copy provided in Annex XX)
Source
Primers In-house synthesis
Probes 13485:2003 Purchased from:
Probeandprod Inc. 2 Last Rd, San Francisco, CA USA
SP and External control Armored RNA
13485:2003 Purchased from:
Universal Oligos Inc. 456 Science Rd, San Diego, CA USA
M-MLV reverse transcriptase 13485:2003 Purchased from:
Enzymes R Us, 765 Rotor Rd, San Francisco, CA USA
rTth DNA polymerase 13485:2003 Purchased from:
Enzymes R Us, 765 Rotor Rd, San Francisco, CA USA
Magnetic Microparticles 13485:2003 Purchased from:
Colossal Health Limited Services
53 Some Street, Some Town, Europe
www.CHLS.co.xcvb
Plastic components for Test Cartridge assembly
ISO 9001:2015
ISO 14001:2015
Purchased from:
Impossible Mouldings, Malleable Mansions, ConriceVille Aus8765 www.mouldy.com.au
SIMU Instrument ISO 9001:2015 Purchased from:
Imaginative Diagnostic Designers, Top Street Industrial Estate, Some Country, Europe
Printed materials (IFU, labels) and translations
ISO 9001:2015 Purchased from:
IPrint Local street Small Town UK
492
The print shop is not quality certified but is regularly inspected according to our supplier 493 management SOP 1234. The translators they employ are certified by national authorities and all 494 translations are certified by an accredited organization. 495
Information on other suppliers providing essential but non-critical material are shown below in Table 496 6-6. 497

WHO Sample product dossier for SIMU HIV-1 QUANT
Design and Manufacturing Information
Draft for public comment 26 October 2016 Page 43 of 140
Table 6-6: Information on other suppliers providing essential but non-critical material 498
Material Supplier Certification
(certified copy provided in Annex XX)
Control of incoming goods
Packaging for shipment
Bags of Boxes
Long Way, Forest Ville Europe
www.moreWords.com
ISO 9001:2015 and 14001:2015
SOP XXXX : visual inspection
Instrumentation for automated device assembly
Best Systems Inc.
Make and Mend Road, Buzzville USA
www.besttest.com
ISO 9001:2015 SOP XXXX – full functional IQ OQ PQ and performance testing
Desiccant sachets used in Test Cartridge pouch
Drybyzones Ltd
Drought Rd Somewhere CH
www.DrybyzonesLtd.co.ch
ISO 9001:2015 SOP XXXX: visual, opened sachets, plus water content
Temperature logger
WetrackTemp Ltd
Desert Road,
USA
www.wetracktemp.co.ch
ISO 9001:2015 SOP XXXX : visual inspection
General laboratory chemicals
SuperPure chemicals Inc., Somewhere, 5600001
www.cleanascanbe.com
ISO 9001:2015 SOP XXXX: visual inspection and performance testing
499
500

WHO Sample product dossier for SIMU HIV-1 QUANT
Product Performance Specifications and Associated Validation and Verification Studies
Draft for public comment 26 October 2016 Page 44 of 140
7 Product Performance Specifications and Associated 501
Validation and Verification Studies 502
503
Please read the following important notes about the information provided in 504 this section. 505
NOTE 1. For the purposes of this sample product dossier, validation studies 506 have been presented in varying levels of detail in order to illustrate the 507 reporting requirements. When submitting a dossier for WHO Prequalification 508 assessment, the following should be provided either in the main dossier or as 509 annexes for EACH PERFORMANCE CLAIM: 510
• The complete study protocol 511
• The methods of data analysis 512
• The complete study report (signed and dated), including the study conclusion. 513 For each study presented, the site(s) and principal investigator(s) should be 514 identified. Similarly, the date each study was performed, the lot numbers used, 515 and a description of the study design (including justification of the number of 516 specimens tested) along with the statistical analysis, results and conclusions 517 should be provided 518
NOTE 2. The studies presented in this section should not be taken as 519 representing a complete list of studies that are expected to be performed and 520 submitted; they are provided as an illustrative sample of the type of 521 information that should be submitted. For each claim (including, but not limited 522 to: specimen type to be used, testing population, stability and other 523 performance characteristics), one or more validation studies must be 524 performed to support that claim. 525
NOTE 3. The data provided in this section must be produced using the final 526 version of the assay product submitted for prequalification. Where this has not 527 occurred, the version used should be stated and a justification for the inclusion 528 of the data provided. Depending on the nature of differences between product 529 versions, additional validation evidence may be required. 530
NOTE 4. Where possible, internationally accepted standards should be used to 531 guide not only the design of each study, but also the formulation of criteria for 532 judging the acceptance of the study and to inform the acceptance of the 533 outcomes. These are comprehensively noted in the Essential Principle Checklist 534 (Refer to Section 5.3 and Annex I of this dossier). 535
NOTE 5. The studies and suggested number of specimens for the evaluation of 536 the performance of SIMU HIV-1 Quant Test represent ideal study recruitment. 537
538

WHO Sample product dossier for SIMU HIV-1 QUANT
Product Performance Specifications and Associated Validation and Verification Studies
Draft for public comment 26 October 2016 Page 45 of 140
7.0 Overview of testing procedures 539
The results of all analytical and clinical performance testing, submitted in this dossier were 540 conducted on the final version of the SIMU HIV-1 Quant Test. Data for earlier Test Cartridge 541 versions and prototype systems and procedures are not included, but the data are retained in 542
design history files (held on-site at THE Manufacturing Company). 543
For each study presented, the site(s) and principal investigator(s) are identified, the date each 544 study was performed, the lot numbers used, and a description of the study design are given 545 along with the statistical analysis, results and conclusions. All efforts were undertaken to 546 eliminate the potential for bias, including but not limited to randomization of procedures that 547 may unfairly influence the result outcome, and careful selection of reference testing. 548 Additionally, validation studies were designed and executed in a manner to avoid the 549 potential for conflicts of interest. 550
Where possible, internationally accepted standards were used to guide the design of each 551 study and inform on the acceptance of the outcomes. These are comprehensively noted in the 552 Essential Principle Checklist (Refer Section 5.3 and Annex I of this dossier). 553
554
The results of both verification studies (performed by the manufacturer) and validation studies (performed either by or on behalf of the manufacturer in the setting of intended use) should be submitted in support of the performance of
the assay.
555
The following verification studies were performed by the manufacturer: 556
Analytical Performance 557
Analytical sensitivity (Limit of Detection, LoD) for HIV-1 group M, HIV-1 group O 558
Genotype/subtype detection 559
o At least 10 clinical isolates each of HIV-1 group M, subtypes A-F, CRFs, HIV-1 560 group O near the LoD were tested 561
Specificity was determined using a panel of repository samples representing 562 different infectious organisms unrelated to HIV infection tested with the SIMU HIV-563 1 Quant Test (including malaria, Human African trypanosomiasis, leishmaniasis, 564 Chagas disease, chikungunya virus, TB, HCV, HBV, HTLV-I/II) 565
Exogenous interfering substances - A panel of HIV-1 negative human EDTA plasma 566 samples containing drugs commonly used in countries with generalized HIV 567 epidemics were tested with the SIMU HIV-1 Quant Test 568
The precision of the SIMU HIV-1 Quant Test was determined for HIV-1 group M 569 using a material that was traceable to the third WHO International Standard for HIV-570 1 NAT 571
The linearity of the SIMU HIV-1 Quant Test was determined over the range 200–572 10,000,000 copies/mL of HIV-1 group M 573
Method correlations between the SIMU HIV-1 Quant System and two reference 574 method assays were performed 575

WHO Sample product dossier for SIMU HIV-1 QUANT
Product Performance Specifications and Associated Validation and Verification Studies
Draft for public comment 26 October 2016 Page 46 of 140
The stability of plasma specimens was tested up to temperatures of 40 oC, ≤ –20 oC 576 and after 3 freeze-thaw cycles 577
578
Clinical Performance 579
The clinical performance of the SIMU HIV-1 Quant Test (Reproducibility, Sensitivity, 580 Specificity and viral Load Correlation) was determined at three external sites located in 581 countries with generalized HIV epidemics. 582
The reproducibility of the SIMU HIV-1 Quant Test was evaluated using a panel 583 covering the dynamic range of the SIMU HIV-1 Quant Test (log10(3) – log10(7) 584 copies/mL). The study was designed to evaluate key variables contributing to total 585 precision variance, including lot, site/instrument, operator, day/run, and within-run. 586
Clinical sensitivity was determined by testing 200 frozen plasma samples with HIV-1 587 RNA concentrations >200 copies/mL and 200 fresh samples (EDTA plasma) collected 588 from HIV-1 positive individuals. The samples were tested for anti-HIV-1 and NAT 589 reactivity using assays that have been authorized for use by a Regulatory Authority 590 of the founding members of GHTF. The RNA concentration of the samples were 591 determined with a reference method assay. Clinical specificity was determined by 592 testing 300 frozen plasma samples and 100 fresh plasma samples from blood donors 593 shown to be negative for HIV-1/2 and non-reactive for HIV-1 RNA using assays that 594 have been authorized for use by a Regulatory Authority of the founding members of 595 GHTF. The viral loads of the HIV-1 positive samples were compared to the expected 596 viral loads using Deming Regression analysis. 597
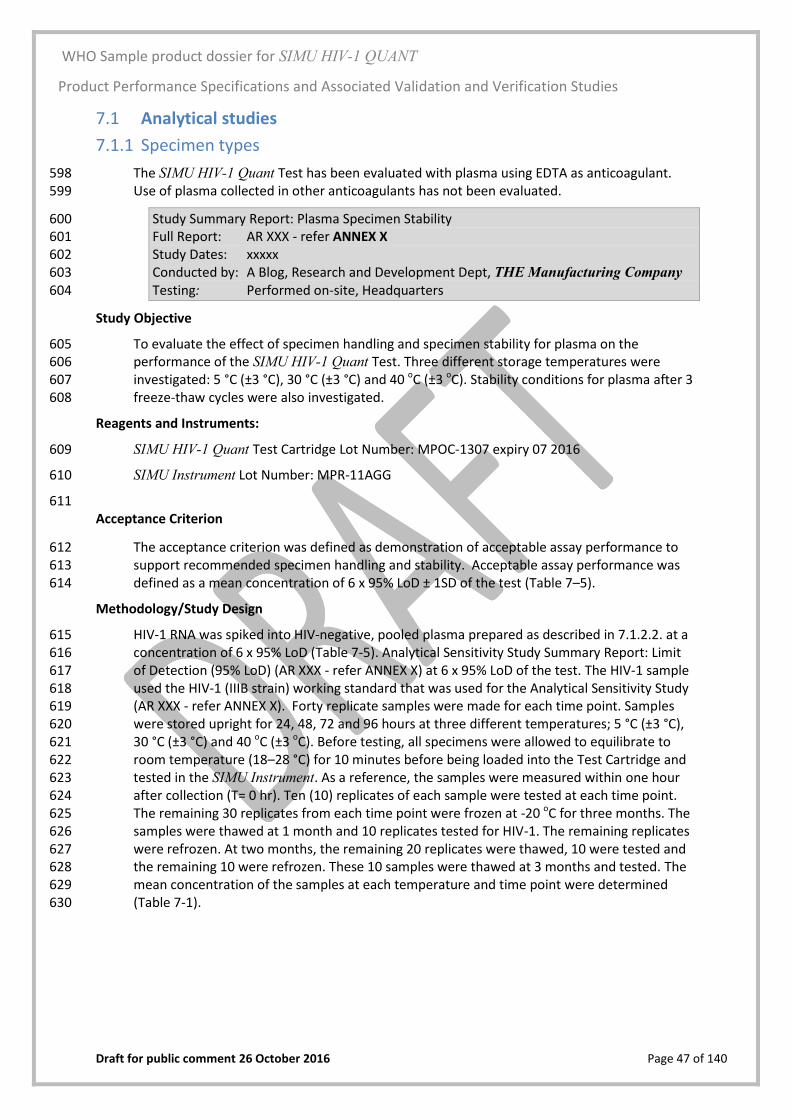
WHO Sample product dossier for SIMU HIV-1 QUANT
Product Performance Specifications and Associated Validation and Verification Studies
Draft for public comment 26 October 2016 Page 47 of 140
Analytical studies 7.1
7.1.1 Specimen types
The SIMU HIV-1 Quant Test has been evaluated with plasma using EDTA as anticoagulant. 598 Use of plasma collected in other anticoagulants has not been evaluated. 599
Study Summary Report: Plasma Specimen Stability 600 Full Report: AR XXX - refer ANNEX X 601 Study Dates: xxxxx 602 Conducted by: A Blog, Research and Development Dept, THE Manufacturing Company 603 Testing: Performed on-site, Headquarters 604
Study Objective
To evaluate the effect of specimen handling and specimen stability for plasma on the 605 performance of the SIMU HIV-1 Quant Test. Three different storage temperatures were 606 investigated: 5 °C (±3 °C), 30 °C (±3 °C) and 40 oC (±3 oC). Stability conditions for plasma after 3 607 freeze-thaw cycles were also investigated. 608
Reagents and Instruments:
SIMU HIV-1 Quant Test Cartridge Lot Number: MPOC-1307 expiry 07 2016 609
SIMU Instrument Lot Number: MPR-11AGG 610
611 Acceptance Criterion
The acceptance criterion was defined as demonstration of acceptable assay performance to 612 support recommended specimen handling and stability. Acceptable assay performance was 613 defined as a mean concentration of 6 x 95% LoD ± 1SD of the test (Table 7–5). 614
Methodology/Study Design
HIV-1 RNA was spiked into HIV-negative, pooled plasma prepared as described in 7.1.2.2. at a 615 concentration of 6 x 95% LoD (Table 7-5). Analytical Sensitivity Study Summary Report: Limit 616 of Detection (95% LoD) (AR XXX - refer ANNEX X) at 6 x 95% LoD of the test. The HIV-1 sample 617 used the HIV-1 (IIIB strain) working standard that was used for the Analytical Sensitivity Study 618 (AR XXX - refer ANNEX X). Forty replicate samples were made for each time point. Samples 619 were stored upright for 24, 48, 72 and 96 hours at three different temperatures; 5 °C (±3 °C), 620 30 °C (±3 °C) and 40 oC (±3 oC). Before testing, all specimens were allowed to equilibrate to 621 room temperature (18–28 °C) for 10 minutes before being loaded into the Test Cartridge and 622 tested in the SIMU Instrument. As a reference, the samples were measured within one hour 623 after collection (T= 0 hr). Ten (10) replicates of each sample were tested at each time point. 624 The remaining 30 replicates from each time point were frozen at -20 oC for three months. The 625 samples were thawed at 1 month and 10 replicates tested for HIV-1. The remaining replicates 626 were refrozen. At two months, the remaining 20 replicates were thawed, 10 were tested and 627 the remaining 10 were refrozen. These 10 samples were thawed at 3 months and tested. The 628 mean concentration of the samples at each temperature and time point were determined 629 (Table 7-1). 630

WHO Sample product dossier for SIMU HIV-1 QUANT
Product Performance Specifications and Associated Validation and Verification Studies
Draft for public comment 26 October 2016 Page 48 of 140
Table 7-1: Stability of plasma specimens with SIMU Quant HIV-1 Test 631
632
Results 633
The mean concentrations each time point and after 3 freeze-thaw cycles were all at 6 x 95% 634 LoD ± 1 SD of the test and met the acceptance criteria. 635
Conclusion: 636
This study provides evidence that plasma specimens can be exposed to 40 oC (± 3 oC) for up to 637 96 hours after collection and freeze-thawed up to three times without loss in concentration 638 when tested at a concentration of 6 x 95% LoD. 639
640
Temperature Virus Mean concentration (10 replicates) at time point (SD)
0 h 24 h 48 h 72 h 96 h
5 oC HIV-1
30 oC HIV-1
40 oC HIV-1
The table is for illustrative purposes. WHO expects manufacturers to provide all results for the studies
in an easy to read format.

WHO Sample product dossier for SIMU HIV-1 QUANT
Product Performance Specifications and Associated Validation and Verification Studies
Draft for public comment 26 October 2016 Page 49 of 140
7.1.2 Analytical performance characteristics 641
7.1.2.1 Accuracy of measurement 642
The manufacturer is expected to provide studies relating to trueness of 643 measurement, precision of measurement (including repeatability and 644
reproducibility in this section). 645
7.1.2.1.1 Trueness of measurement 646
Study Summary Report: Method Correlation 647 Full Report: AR XXX - refer ANNEX X 648 Study Dates: xxxxx 649 Conducted by: A Blog, Research and Development Dept, THE Manufacturing Company 650 Testing: Performed on-site, Headquarters 651
652
Study Objective 653
To demonstrate correlation between the SIMU HIV-1 Quant Test and the reference method 654 assay. 655
656
Reagents and Instruments
SIMU HIV-1 Quant Test Cartridge Lot Number: MPOC-1307 expiry 07 2016 657
SIMU Instrument Lot Number: MPR-11AGG 658
Acceptance Criteria
Bland Altman analysis of the SIMU HIV-1 Quant Test and each reference test. 659
Methodology/Study Design
The performance of the SIMU HIV-1 Quant Test was compared to two reference method 660 assays. Ninty (90) prospectively collected HIV-1 group M samples from individuals with HIV-661 1/AIDS and not on antiviral therapy were tested with the three tests. The sample viral loads 662 covered the dynamic range of the SIMU HIV-1 Quant Test (200 – 10,000,000 copies/mL). The 663 data were analysed by Bland Altman for valid, paired viral loads within the dynamic ranges of 664 both assays. 665
Results
Bland Altman analysis of 90 specimens tested with the SIMU HIV-1 Quant Test and reference 666 method 1 and the SIMU HIV-1 Quant Test and reference method 2 are shown in Figures 7-2 667 and 7-3. 668
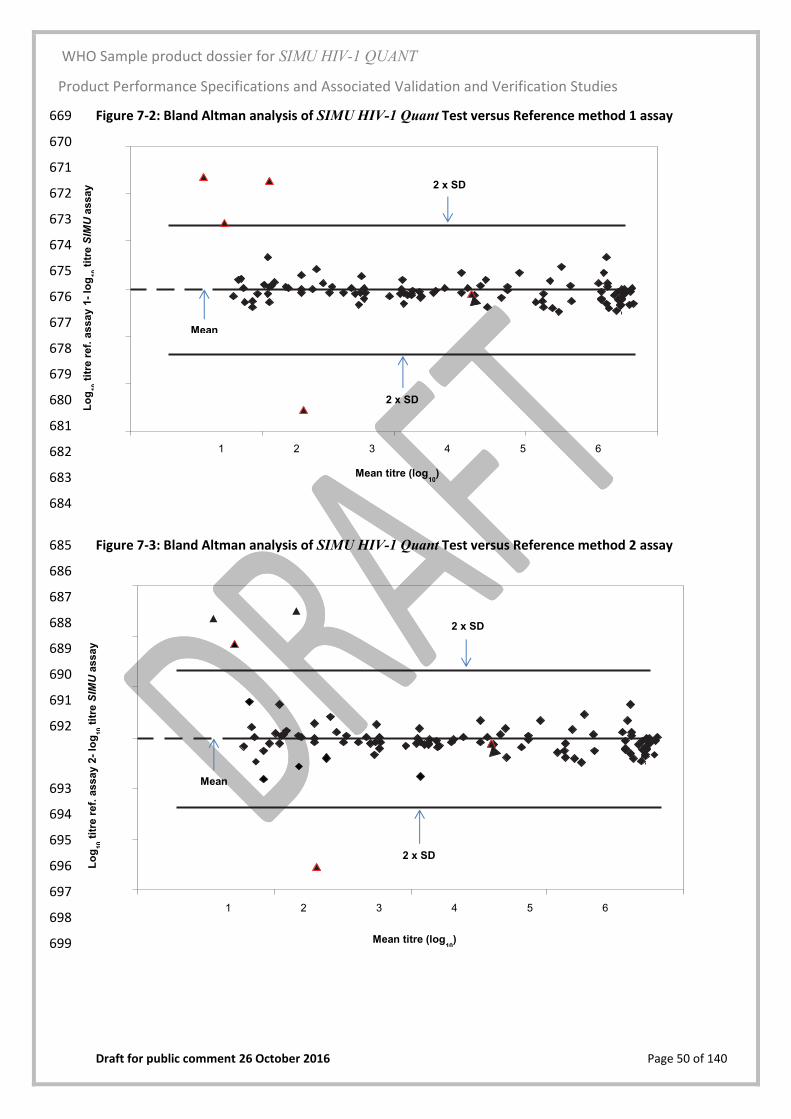
WHO Sample product dossier for SIMU HIV-1 QUANT
Product Performance Specifications and Associated Validation and Verification Studies
Draft for public comment 26 October 2016 Page 50 of 140
Figure 7-2: Bland Altman analysis of SIMU HIV-1 Quant Test versus Reference method 1 assay 669
670
671
672
673
674
675
676
677
678
679
680
681
682
683
684
Figure 7-3: Bland Altman analysis of SIMU HIV-1 Quant Test versus Reference method 2 assay 685
686
687
688
689
690
691
692
693
694
695
696
697
698
699
1 2 3 4 5 6
Mean titre (log10
)
Lo
g10 t
itre
ref.
assay 1
- lo
g10 t
itre
SIM
U a
ssay
Mean
2 x SD
2 x SD
1 2 3 4 5 6
Mean titre (log10
)
Lo
g10 t
itre
ref.
assay 2
- lo
g10 t
itre
SIM
U a
ssay
Mean
2 x SD
2 x SD

WHO Sample product dossier for SIMU HIV-1 QUANT
Product Performance Specifications and Associated Validation and Verification Studies
Draft for public comment 26 October 2016 Page 51 of 140
Conclusions 700
The SIMU HIV-1 Quant System showed good correlation with reference method 1 and 2 (the 701 differences in titres of the 90 samples between the SIMU Quant HIV-1 Test and the two 702 reference methods were all within ± 2 SD of the mean difference . 703
7.1.2.1.2 Precision of measurement 704
Study Summary Report: Precision 705 Full Report: AR XXX - refer ANNEX X 706 Study Dates: xxxxx 707 Conducted by: A Blog, Research and Development Dept, THE Manufacturing Company 708 Testing: Performed on-site, Headquarters 709
710
Study Objective 711
To demonstrate the precision of the SIMU HIV-1 Quant Test. 712
Reagents and Instruments 713
SIMU HIV-1 Quant Test Cartridge Lot Number: MPOC-1307 expiry 07 2016, MPOC-1409 714 expiry 09 2016, MOOC-1111 expiry 07 2016 715
SIMU Instrument Lot Number: MPR-11AGG 716
Acceptance Criteria 717
The log standard deviation (log SD) at each dilution level tested should be ≤log 0.25 for each 718 lot and for all lots combined. 719
Methodology 720
The SIMU working standard based on HIV-1 (lllB Strain) was used for this study. The working 721 standard has been quantified using Reference Method 1 to achieve traceability to the WHO 722 International Standard HIV-1 RNA, 1st International Standard. Therefore, one copy of HIV-1 723 RNA is equivalent to 1.67 International Units (IU) (Holmes HH, Davis C, Heath A, Hewlett I, Lelie 724 N. An international collaborative study to establish the 1st international standard for HIV-1 RNA for 725 use in nucleic acid-based techniques. J Virol Methods 2001;92: 141-150). This material has been 726 described in Annex XXX, Limit of Detection (95% LoD) studies. 727
Serial log dilutions of the working reagent were prepared in HIV-1 negative human EDTA 728 plasma. The preparation of negative human plasma has been described in Annex XXX, Limit of 729 Detection (95% LoD) study. Three reagent lots were analysed and 45 tests per reagent lot 730 were performed for each dilution level. The results for each reagent lot and for the three 731 reagent lots combined are shown in Table 7-4, together with the log SD for each lot and for 732 the combined lots. 733
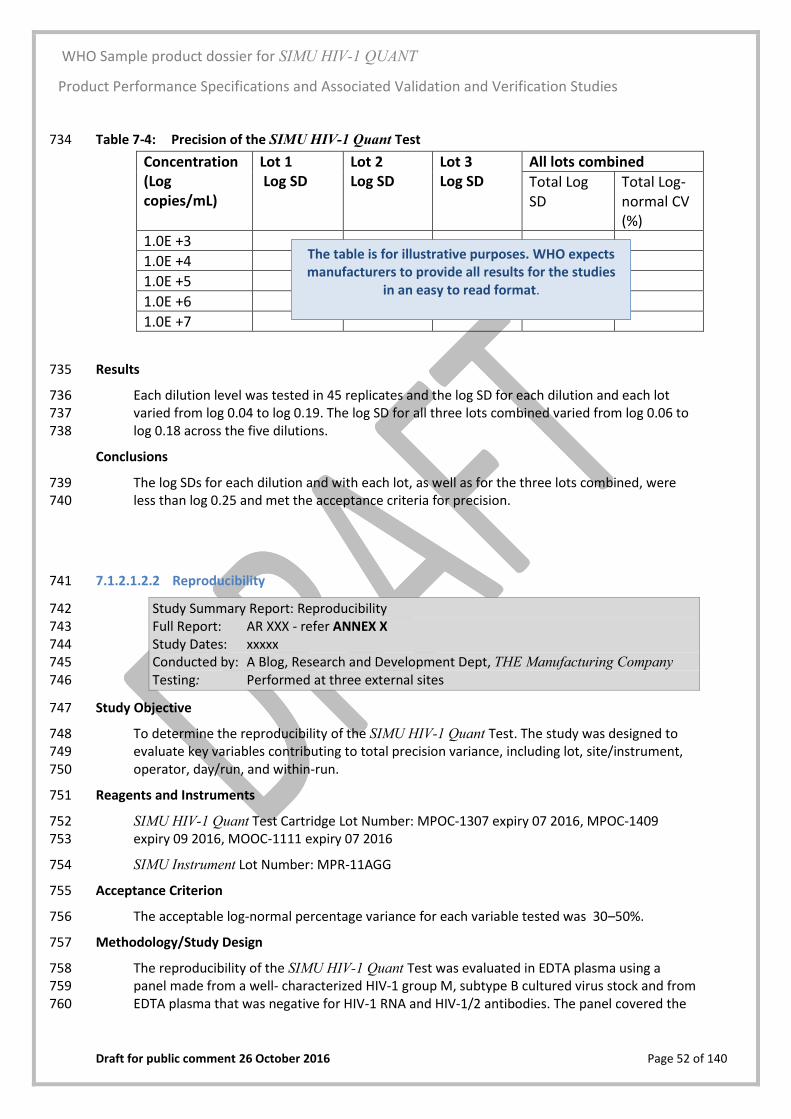
WHO Sample product dossier for SIMU HIV-1 QUANT
Product Performance Specifications and Associated Validation and Verification Studies
Draft for public comment 26 October 2016 Page 52 of 140
Table 7-4: Precision of the SIMU HIV-1 Quant Test 734
Concentration (Log copies/mL)
Lot 1 Log SD
Lot 2 Log SD
Lot 3 Log SD
All lots combined
Total Log SD
Total Log-normal CV (%)
1.0E +3
1.0E +4
1.0E +5
1.0E +6
1.0E +7
Results 735
Each dilution level was tested in 45 replicates and the log SD for each dilution and each lot 736 varied from log 0.04 to log 0.19. The log SD for all three lots combined varied from log 0.06 to 737 log 0.18 across the five dilutions. 738
Conclusions
The log SDs for each dilution and with each lot, as well as for the three lots combined, were 739 less than log 0.25 and met the acceptance criteria for precision. 740
7.1.2.1.2.2 Reproducibility 741
Study Summary Report: Reproducibility 742 Full Report: AR XXX - refer ANNEX X 743 Study Dates: xxxxx 744 Conducted by: A Blog, Research and Development Dept, THE Manufacturing Company 745 Testing: Performed at three external sites 746
Study Objective 747
To determine the reproducibility of the SIMU HIV-1 Quant Test. The study was designed to 748 evaluate key variables contributing to total precision variance, including lot, site/instrument, 749 operator, day/run, and within-run. 750
Reagents and Instruments 751
SIMU HIV-1 Quant Test Cartridge Lot Number: MPOC-1307 expiry 07 2016, MPOC-1409 752 expiry 09 2016, MOOC-1111 expiry 07 2016 753
SIMU Instrument Lot Number: MPR-11AGG 754
Acceptance Criterion 755
The acceptable log-normal percentage variance for each variable tested was 30–50%. 756
Methodology/Study Design 757
The reproducibility of the SIMU HIV-1 Quant Test was evaluated in EDTA plasma using a 758 panel made from a well- characterized HIV-1 group M, subtype B cultured virus stock and from 759 EDTA plasma that was negative for HIV-1 RNA and HIV-1/2 antibodies. The panel covered the 760
The table is for illustrative purposes. WHO expects manufacturers to provide all results for the studies
in an easy to read format.
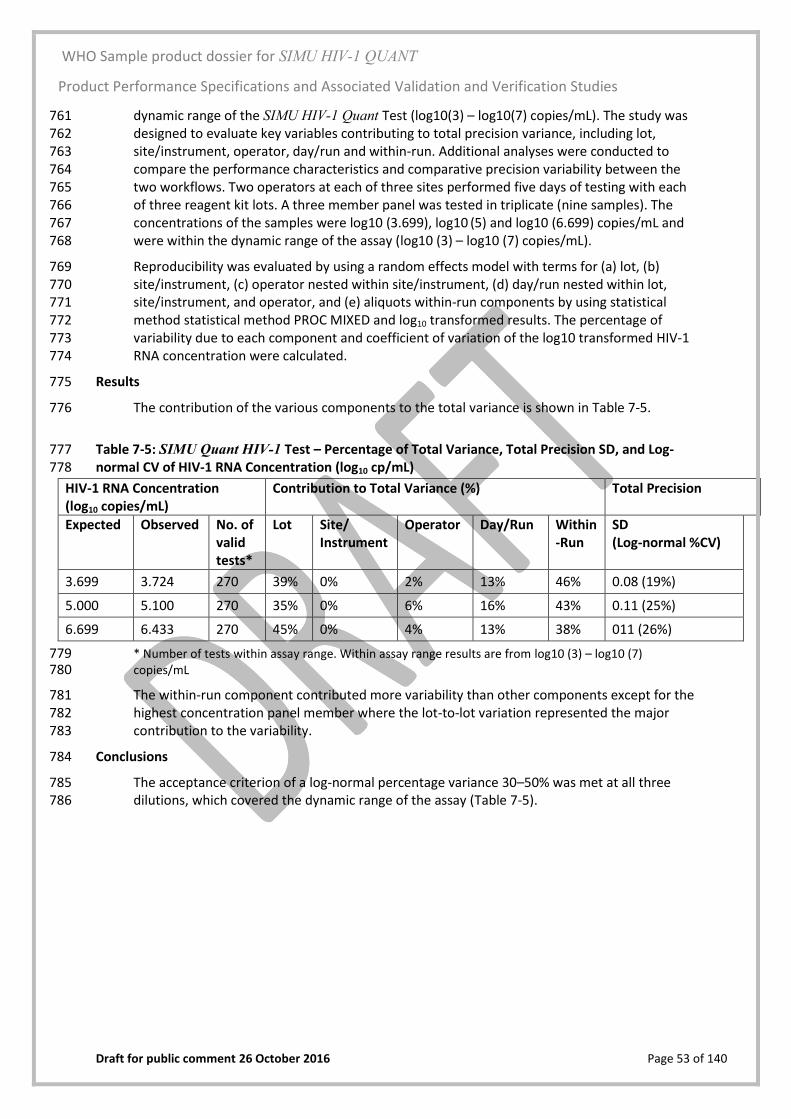
WHO Sample product dossier for SIMU HIV-1 QUANT
Product Performance Specifications and Associated Validation and Verification Studies
Draft for public comment 26 October 2016 Page 53 of 140
dynamic range of the SIMU HIV-1 Quant Test (log10(3) – log10(7) copies/mL). The study was 761 designed to evaluate key variables contributing to total precision variance, including lot, 762 site/instrument, operator, day/run and within-run. Additional analyses were conducted to 763 compare the performance characteristics and comparative precision variability between the 764 two workflows. Two operators at each of three sites performed five days of testing with each 765 of three reagent kit lots. A three member panel was tested in triplicate (nine samples). The 766 concentrations of the samples were log10 (3.699), log10 (5) and log10 (6.699) copies/mL and 767 were within the dynamic range of the assay (log10 (3) – log10 (7) copies/mL). 768
Reproducibility was evaluated by using a random effects model with terms for (a) lot, (b) 769 site/instrument, (c) operator nested within site/instrument, (d) day/run nested within lot, 770 site/instrument, and operator, and (e) aliquots within-run components by using statistical 771 method statistical method PROC MIXED and log10 transformed results. The percentage of 772 variability due to each component and coefficient of variation of the log10 transformed HIV-1 773 RNA concentration were calculated. 774
Results 775
The contribution of the various components to the total variance is shown in Table 7-5. 776
Table 7-5: SIMU Quant HIV-1 Test – Percentage of Total Variance, Total Precision SD, and Log-777 normal CV of HIV-1 RNA Concentration (log10 cp/mL) 778
HIV-1 RNA Concentration (log10 copies/mL)
Contribution to Total Variance (%) Total Precision
Expected Observed No. of valid tests*
Lot Site/ Instrument
Operator Day/Run Within-Run
SD (Log-normal %CV)
3.699 3.724 270 39% 0% 2% 13% 46% 0.08 (19%)
5.000 5.100 270 35% 0% 6% 16% 43% 0.11 (25%)
6.699 6.433 270 45% 0% 4% 13% 38% 011 (26%)
* Number of tests within assay range. Within assay range results are from log10 (3) – log10 (7) 779 copies/mL 780
The within-run component contributed more variability than other components except for the 781 highest concentration panel member where the lot-to-lot variation represented the major 782 contribution to the variability. 783
Conclusions 784
The acceptance criterion of a log-normal percentage variance 30–50% was met at all three 785 dilutions, which covered the dynamic range of the assay (Table 7-5). 786

WHO Sample product dossier for SIMU HIV-1 QUANT
Product Performance Specifications and Associated Validation and Verification Studies
Draft for public comment 26 October 2016 Page 54 of 140
7.1.2.2 Analytical sensitivity 787
7.1.2.2.1 Limit of detection (95% LoD) 788
Study Summary Report: Limit of Detection (95% LoD) 789 Full Report: AR XXX - refer ANNEX X 790 Study Dates: 02 August 2012 791 Conducted by: A Blog, Research and Development Dept, THE Manufacturing Company 792 Testing: Performed on-site, Headquarters 793
Study Objective 794
The LoD is the lowest viral load where the true detection rate is 95%. The LoD was 795 calculated with a PROBIT regression analysis. The objective of this study was to determine the 796 LoD of the SIMU HIV-1 Quant Test for HIV-1 group M subtype and HIV-1 group O. 797
Reagents and Instruments 798
SIMU HIV-1 Quant Test Cartridge Lot Number: MPOC-1307 expiry 07 2016 799
SIMU Instrument Lot Number: MPR-11AGG 800
Acceptance Criteria 801
The acceptance criterion for the HIV-1 group M and group O 95% LoD In plasma should be at 802 least 150 copies/mL. 803
Methodology/Study Design 804
The LoD of the SIMU HIV-1 Quant Test was determined by testing the 2nd International HIV-1 805 RNA WHO Standard, NIBSC Code 97/65041, HIV-1 subtype B, diluted in HIV-1-negative, 806 human, EDTA plasma. The LoD was determined for three reagent lots. Three dilution series 807 were analysed for each reagent lot. Seven dilutions covering the predetermined 95% hit rate 808 of the test were prepared (Table 7-6). A total of approximately 120 replicates per 809 concentration level were tested. The evaluation was performed according to CLSI Guideline 810 EP17-A Evaluation of Detection Capability for Clinical Laboratory Measurement Procedures; 811 Approved Guideline—Second Edition. 812
For the LoD studies, a pool of negative, human plasma was prepared and stored in 100 mL 813 aliquots at ≤ -20 oC. Since large volumes of plasma were required for the LoD studies, pooled 814 plasma was purchased from a commercial vendor. The plasma was negative for HBsAg, anti-815 HBc, anti-HCV, anti-HIV and non-reactive for HBV DNA, HCV RNA and HIV RNA using a NAT test 816 authorized for use by a Regulatory Authority of the founding members of GHTF. Before use, as 817 many aliquots of negative plasma as required were thawed rapidly in a 37 oC water bath with 818 agitation until the plasma was completely thawed. The thawed plasma was then cooled to 4–8 819 oC before being used for the dilution panels. 820
One copy of HIV-1 RNA is equivalent to 1.67 International Units (IU) ( Holmes HH, Davis C, 821 Heath A, Hewlett I, Lelie N. An international collaborative study to establish the 1st 822 International standard for HIV-1 RNA for use in nucleic acid-based techniques. J Virol Methods 823 2001;92: 141-150). 824
Analysis 825
For each virus, PROBIT analysis on the data combined across dilution series and reagent lots 826 was used to estimate the LoD, along with the lower and upper limit of the 95% confidence 827 interval. 828

WHO Sample product dossier for SIMU HIV-1 QUANT
Product Performance Specifications and Associated Validation and Verification Studies
Draft for public comment 26 October 2016 Page 55 of 140
Results 829
The concentration of HIV-1 RNA that can be detected with a positivity rate of greater than 830 95% as determined by PROBIT analysis, was 122 copies/mL or 203.7 IU/mL. The results for the 831 individual lots were 114 copies/mL (95% confidence interval: x–y copies/mL) for lot 1, 130 832 copies/mL (95% confidence interval: a–b copies/mL) for lot 2 and 122 copies/mL (95% 833 confidence interval: m–n copies/mL) for lot 3. The combined results for all three reagent lots 834 are shown in Table 7-6. A conversion factor one copy of HIV-1 RNA equivalent to 1.67 IU was 835 used for converting the results IU/mL (Holmes HH, Davis C, Heath A, Hewlett I, Lelie N. An 836 international collaborative study to establish the 1st international standard for HIV-1 RNA for 837 use in nucleic acid-based techniques. J Virol Methods 2001;92: 141-150). 838
Table 7-6: SIMU Quant HIV-1 Test – Reactivity rate of HIV-1 Group M RNA 839
HIV-1 group M RNA concentration (copies/mL)
No. reactive No. tested (valid) % Reactive
200 120
150 120
130 120
120 120
110 120
100 120
50 120
HIV-1 group M subtypes 840
In addition, dilutions of cell culture supernatants representing the sensitivity of the assay for HIV-1 841 group M subtypes A-H was also determined. Dilutions of cell culture supernatant representing HIV-1 842 group M, subtypes A-H (purchased from a commercial vendor) were prepared in HIV-1-negative 843 human EDTA plasma and were analysed with two reagent lots. For each HIV-1 subtype isolate, three 844 concentration levels, 100 copies/mL, 150 copies/mL and 200 copies/mL, were tested in 24 replicates 845 per reagent lot. The assignment of nominal concentrations to the cell culture stock materials was 846 performed by averaging the concentrations of two, commercial, HIV-1 quantitative tests, reference 847
method 1 and reference method 22. Hit rate analysis gave a positivity rate of greater than 95% for all 848
subtypes at 150 copies/mL or lower. The combined results for the two reagent lots are shown in 849 Table 7-7. 850
2 Reference method 1 and 2 are stringently regulated product types that have been approved by one of the
Regulatory Authorities of the Founding Members of GHTF.
The table is for illustrative purposes. WHO expects manufacturers to provide all results for the studies
in an easy to read format.

WHO Sample product dossier for SIMU HIV-1 QUANT
Product Performance Specifications and Associated Validation and Verification Studies
Draft for public comment 26 October 2016 Page 56 of 140
Table 7-7: LoD of the SIMU HIV-1 Quant Test for HIV-1 group M subtypes A–H: ≥95% Hit Rate 851 Analysis 852
Subtype Isolate no. Lowest concentration with ≥ 95% hit rate (copies/mL)
A 150
B 150
B 150
B 100
C 150
C 100
C 150
D 100
F 100
G 100
H 150
CRF01_AE 100
CRF01_AE 100
HIV-1 group O 853
Dilutions of a HIV-1 group O cell culture supernatant (isolate xxx) in human EDTA plasma were 854 analysed with two reagent lots. The nominal concentration of this material was assigned on the 855 results of reference method 1. Six concentration levels of 50, 100, 110, 120, 130 and 150 copies/mL 856 were tested in 24 replicates per reagent lot. The results of the two reagent lots were pooled. Hit rate 857 analysis showed that the SIMU HIV-1 Quant Test gave a greater than 95% positive rate at a 858 concentration of 120 copies/mL for HIV-1 group O. 859
Conclusions: 860
The LoD of the SIMU HIV-1 Quant Test was 122 copies/mL and met the acceptance criterion of a 861 95% LoD of 150 copies/mL. The LoD for the SIMU HIV-1 Quant Test for HIV-1 group M (150 862 copies/ml) was higher than those for other commercial quantitative tests where the LoD for HIV-1 863
group M is about 20 copies/mL (reference test assays3). This is not surprising since the SIMU HIV-1 864
Quant Test only uses 50 µL of specimen which is five-fold to ten-fold lower than that used for most 865 commercial tests. No significant lot-to-lot variation was detected with the three lots of the SIMU 866 HIV-1 Quant Test. 867
3 Reference methods should be stringently regulated product types that have been approved by one of the
Regulatory Authorities of the Founding Members of GHTF.
The table is for illustrative purposes. WHO expects manufacturers to provide all results for the studies
in an easy to read format.

WHO Sample product dossier for SIMU HIV-1 QUANT
Product Performance Specifications and Associated Validation and Verification Studies
Draft for public comment 26 October 2016 Page 57 of 140
Hit rate analysis showed that all the common subtypes of HIV-1 were detected at a greater than 95% 868 positive hit rate at 150 copies/mL or lower. The SIMU HIV-1 Quant Test detected HIV-1 group O at 869 120 copies/mL with a positive rate of greater than 95%. 870
7.1.2.2.2 Genotype detection 871
Study Summary Report: Genotype Detection 872 Full Report: AR XXX - refer ANNEX X 873 Study Dates: xxxxx 874 Conducted by: A Blog, Research and Development Dept, THE Manufacturing Company. 875 Testing: Performed on-site, Headquarters. 876
Study Objective 877
To determine the sensitivity of the SIMU HIV-1 Quant Test for HIV-1 group M subtypes and 878 HIV-1 group O. 879
Reagents and Instruments 880
SIMU HIV-1 Quant Test Cartridge Lot Number: MPOC-1307 expiry 07 2016 881
SIMU Instrument Lot Number: MPR-11AGG 882
Acceptable Criterion 883
Detection of all subtypes. 884
Methodology/Study Design 885
The performance of the SIMU HIV-1 Quant Test with the eight HIV-1 group M subtypes (A – 886 H) was evaluated by analysis of cell culture stock material of representatives for each HIV-1 887 group M subtype. The assignment of nominal concentrations to the cell culture stock 888 materials was performed by averaging the concentrations of the reference method 1 and 889 reference method 2. Each cell culture stock material was diluted to nominal concentrations of 890 approximately 5.00E+02, 5.00E+04 and 5.00E+06 copies/mL in EDTA plasma. The 891 concentrations were then tested in 10 replicates with the SIMU HIV-1 Quant Test using one 892 reagent lot. The mean log10 concentrations of all concentrations and subtypes were 893 compared to the respective log10 nominal concentrations. 894
The results are shown in Table 7-8. 895
Table 7-8: Results of HIV-1 group M subtype detection testing with SIMU Quant HIV-1 Test 896
Isolate Concentration (log10 copies/mL)
Test Nominal Test Nominal Test Nominal
A 2.450 2.699 4.450 4.699 6.450 6.699
B 2.600 2.699 4.600 4.699 6.600 6.699
C 2.577 2.699 4.577 4.699 6.577 6.699
D 2.712 2.699 4.712 4.699 6.712 6.699
CRF01_AE 2.668 2.699 4.668 4.699 6.668 6.699
F 2.788 2.699 4.788 4.699 6.788 6.699

WHO Sample product dossier for SIMU HIV-1 QUANT
Product Performance Specifications and Associated Validation and Verification Studies
Draft for public comment 26 October 2016 Page 58 of 140
G 2.489 2.699 4.489 4.699 6.489 6.699
H 2.559 2.699 4.559 4.699 6.559 6.699
897
The evaluation of the 8 HIV-1 subtype isolates by the SIMU HIV-1 Quant Test demonstrates 898 equivalent results for all tested representatives of the HIV-1 group M subtypes (Table 7-6). 899 Mean log10 concentration results for all subtypes were within ± 0.3 log10 of the assigned input 900 concentration. 901
HIV-1 group O 902
Ten HIV-1 group O cell culture samples and one patient specimen (high concentration, >1E 903 +06 copies/mL) were quantitated with a commercial, quantitative HIV-1 test (reference 904
method assay4). The samples were diluted concentrations of approximately 2.00E+02, 905
2.00E+03 and 2.00E+05 copies/mL in EDTA plasma. The concentrations were then tested in 10 906 replicates with the SIMU HIV-1 Quant Test using one reagent lot. 907
Results 908
The results are shown in Table 7-9. 909
Table 7-9: Results of HIV-1 group O isolate detection testing with SIMU Quant HIV-1 Test 910
Sample Concentration (log10 copies/mL)
Test Nominal Test Nominal Test Nominal
Cell culture 1 2.450 2.301 4.450 4.301 6.450 6.301
Cell culture 2 2.333 2.301 4.333 4.301 6.333 6.301
Cell culture 3 2.577 2.301 4.577 4.301 6.577 6.301
Cell culture 4 2.412 2.301 4.412 4.301 6.412 6.301
Cell culture 5 2.268 2.301 4.268 4.301 6.268 6.301
Cell culture 6 2.188 2.301 4.188 4.301 6.188 6.301
Cell culture 7 2.489 2.301 4.489 4.301 6.489 6.301
Cell culture 8 2.222 2.301 4,222 4.301 6,222 6.301
Cell culture 9 2.401 2.301 4.401 4.301 6.401 6.301
Cell culture 10 2.189 2.301 4.189 4.301 6.189 6.301
Patient yyy 2.56 2.301 4.56 4.301 6.56 6.301
911 The mean log10 concentrations of all concentrations were within ± 0.3 log10 of the respective log10 912 nominal concentration (Table 7-9). 913
4 Reference methods should be stringently regulated product types that have been approved by one of the
Regulatory Authorities of the Founding Members of GHTF.

WHO Sample product dossier for SIMU HIV-1 QUANT
Product Performance Specifications and Associated Validation and Verification Studies
Draft for public comment 26 October 2016 Page 59 of 140
HIV-1 group N 914
There are no quantitative assays for HIV-1 group N. Therefore, for HIV-1 group N, log-fold dilutions 915 of a cell culture isolate (from 1:102 to 1:106) were prepared in negative plasma and tested. Four 916 replicates at each dilution were tested. 917
Results 918
The results are shown in Table 7-10. 919
Table 7-10: Detection of dilutions of HIV-1 group N by SIMU Quant HIV-1 Test 920
HIV-1 Group N dilutions Mean concentration (copies/mL)
1:102 1E + 04
1: 103 1.9E + 02
1: 104 1.5E + 02
1: 105 Below level of quantitation
1:106 Below level of quantitation
Conclusion 921
The SIMU HIV-1 Quant Test was able to detect all the common subtypes of HIV-1 groups M 922 and O circulating in the region. A limited study was done with HIV-1 group N due to the lack of 923 these specific genotype isolates. Thus while this genotype was detected, these data should be 924 viewed with caution since some isolates of HIV-1 group N may not be detected with the SIMU 925 HIV-1 Quant Test. Since no HIV-1 group P isolates were available for testing, no claim for 926 detection of this group can be made for the SIMU HIV-1 Quant Test. 927

WHO Sample product dossier for SIMU HIV-1 QUANT
Product Performance Specifications and Associated Validation and Verification Studies
Draft for public comment 26 October 2016 Page 60 of 140
7.1.2.3 Analytical specificity 928
7.1.2.3.1 Cross-reaction from other medical conditions 929
Study Summary Report: Cross-reaction with specimens from individuals with medical 930 conditions unrelated to HIV infection 931 Full Report: AR XXX - refer ANNEX X. 932 Study Dates: xxxxx 933 Conducted by: A Blog, Research and Development Dept, THE Manufacturing Company. 934 Testing: Performed on-site, Headquarters. 935
936
Study Objective 937
To determine whether samples representing different categories of medical conditions 938 unrelated to HIV infection cross-react or interfere with the SIMU HIV-1 Quant Test. 939
Reagents and Instruments 940
SIMU HIV-1 Quant Test Cartridge Lot Number: MPOC-1307 expiry 07 2016 941
SIMU Instrument Lot Number: MPR-11AGG 942
Acceptance Criteria 943
There should be no cross-reactivity with the common infectious agents found in the intended 944 end-user region (Table 7-11). There should be no inhibition of the test by any of the agents. 945
Methodology/Study Design 946
Plasma specimens from individuals with infections that are common in regions where the HIV-947 1 Quant test would be used were tested for interference. These infections, unrelated to HIV, 948 are listed in Table 7-11. Such specimens can be obtained from commercial vendors and 949 national biological institutes. The specimens were tested with and without HIV-1 group M 950 added to a concentration of 250 copies/mL. 951
Results 952
The results are summarized in Table 7-11. 953
Table 7-11: Results of analytical specificity study, SIMU Quant HIV-1 Test 954
Medical condition Plasma without HIV-1 Plasma with added HIV-1
No. tested No. reactive (%) No. tested No. reactive (%)
Malaria 20 0 (0%) 20 20 (100%)
Leishmaniasis 10 0 (0%) 10 10 (100%)
Sleeping sickness 10 0 (0%) 10 10 (100%)
Chagas disease 10 0 (0%) 10 10 (100%)
HBV 10 0 (0%) 10 10 (100%)
HCV 10 0 (0%) 10 10 (100%)
HTLV-1 10 2 (20%) 10 10 (100%)
HTLV-2 10 1 (10%) 10 10 (100%)
Chikungunya virus 10 0 (0%) 10 10 (100%)
Tuberculosis 10 0 (0%) 10 10 (100%)
Parvovirus B19 10 0 (0%) 10 10 (100%)
Adenovirus type 5 10 0 (0%) 10 10 (100%)

WHO Sample product dossier for SIMU HIV-1 QUANT
Product Performance Specifications and Associated Validation and Verification Studies
Draft for public comment 26 October 2016 Page 61 of 140
Medical condition Plasma without HIV-1 Plasma with added HIV-1
No. tested No. reactive (%) No. tested No. reactive (%)
Hepatitis A virus 10 0 (0%) 10 10 (100%)
Yeast (Candida) reactive
10 0 (0%) 10 10 (100%)
Cytomegalovirus 10 0 (0%) 10 10 (100%)
Dengue virus 10 0 (0%) 10 7 (70%)
Epstein-Barr virus 10 0 (0%) 10 10 (100%)
Zika virus 10 0 (0%) 10 10 (100%) 955
There was no cross-reactivity with any of the organisms tested except for HTLV-I and HTLV-2 956 (highlighted), where a low level of cross-reactivity was observed, possibly due to overlap of 957 one of the HIV primer pairs or the probes with the HTLV-1 and HTLV-2 genome. The 958 acceptance criterion regarding cross-reactivity was met except for HTLV-1 and HTLV-2. 959
No inhibition was observed with any of the organisms except with Dengue virus (highlighted) 960 where inhibition caused a drop to a 70% reactive rate. A second study was done in which the 961 Dengue specimen was spiked with HIV-1 group M added to a concentration of 3x LoD, 7x LoD, 962 10x LoD and 15x LoD. Ten replicates of each spiked specimen were tested with the SIMU HIV-963 1 Quant Test System and the results are summarized in Table 7-10. The acceptance criteria 964 regarding inhibition were met for all agents with the exception of Dengue virus. 965
966 Table 7-12: Reactivity of Dengue specimen spiked with HIV-1 Group M at different concentrations 967
968
Concentration of spiked HIV-1 group M
Number tested Number reactive (%)
3x LoD 10 6 (60%)
7x LoD 10 7 (70%)
10x LoD 10 10 (100%)
15x LoD 10 10 (100%)
969
Conclusions: 970
The common infectious agents found in regions where the SIMU HIV-1 Quant Test could 971 potentially be used did not cross-react or interfere with the test with two exceptions. There 972 was some cross-reactivity with HTLV-I and HTLV-2 which may cause a misdiagnosis of HIV in 973 regions where HTLV-I and HTLV-2 are prevalent. Dengue virus reduced the sensitivity of the 974 SIMU HIV-1 Quant Test for HIV-1 group M. Therefore, individuals with a Dengue/HIV-1 co-975 infection may not have an accurate viral load determination below about 10x LoD (1,220 976 copies/mL), possibly resulting in incorrect information for a treatment decision. This 977 information will be highlighted as a limitation in the instructions for use. 978

WHO Sample product dossier for SIMU HIV-1 QUANT
Product Performance Specifications and Associated Validation and Verification Studies
Draft for public comment 26 October 2016 Page 62 of 140
7.1.2.3.2 Exogenous interfering substances 979
Study Summary Report: Exogenous interfering substances 980 Full Report: AR XXX - refer ANNEX X 981 Study Dates: xxxxx 982 Conducted by: A Blog, Research and Development Dept, THE Manufacturing Company 983 Testing: Performed on-site, Headquarters. 984
Study Objective 985
To determine whether potentially interfering substances, such as endogenous compounds 986 found in blood, as well as drugs and other exogenous compounds commonly used in the 987 region of potential end-users, interfere with the SIMU HIV-1 Quant Test. 988
Reagents and Instruments 989
SIMU HIV-1 Quant Test Cartridge Lot Number: MPOC-1307 expiry 07 2016 990
SIMU Instrument Lot Number: MPR-11AGG 991
Methodology/Study Design 992
HIV-1 negative human EDTA plasma samples were spiked with elevated levels of blood 993 analytes listed in Table 7-13. In addition, therapeutic drugs commonly used in the region of 994 potential end-users spiked at 3 x Peak Plasma Level (Cmax). The samples were tested with the 995 SIMU HIV-1 Quant Test. The testing was performed according to CLSI Guideline EP7-A2 996 Interference Testing in Clinical Chemistry; Approved Guideline—Second Edition using one lot 997 of reagent. 998
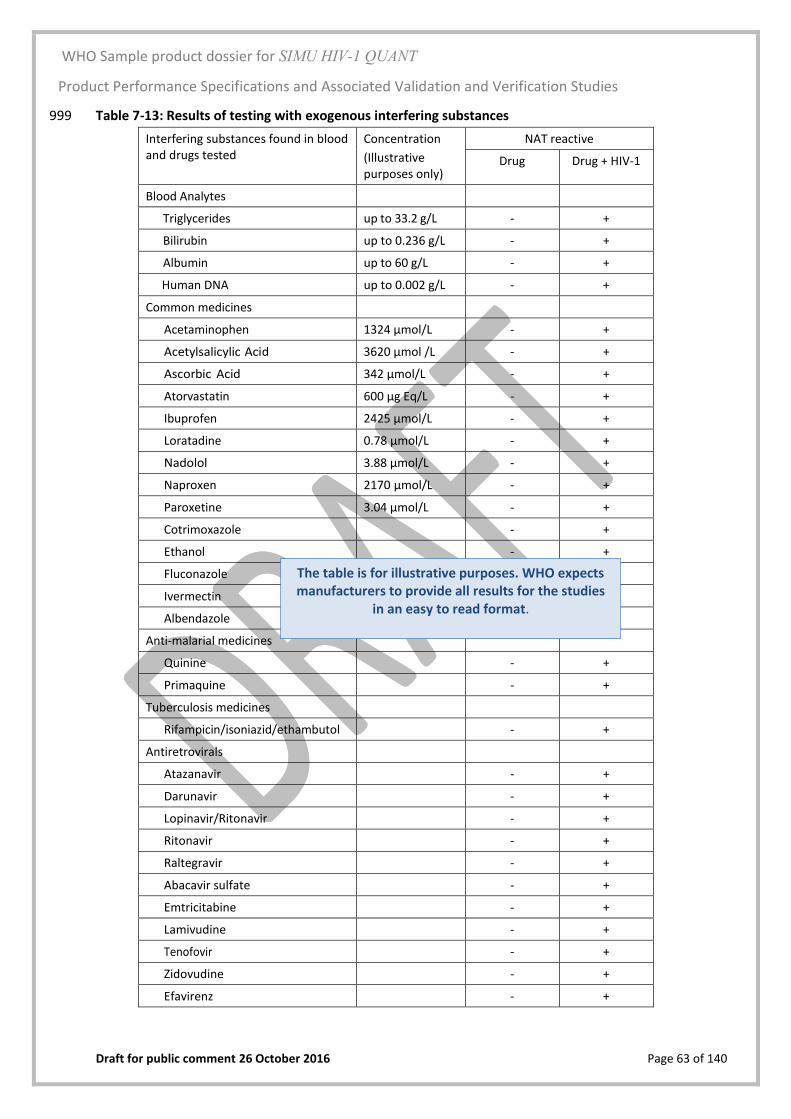
WHO Sample product dossier for SIMU HIV-1 QUANT
Product Performance Specifications and Associated Validation and Verification Studies
Draft for public comment 26 October 2016 Page 63 of 140
Table 7-13: Results of testing with exogenous interfering substances 999
Interfering substances found in blood and drugs tested
Concentration
(Illustrative purposes only)
NAT reactive
Drug Drug + HIV-1
Blood Analytes
Triglycerides up to 33.2 g/L - +
Bilirubin up to 0.236 g/L - +
Albumin up to 60 g/L - +
Human DNA up to 0.002 g/L - +
Common medicines
Acetaminophen 1324 µmol/L - +
Acetylsalicylic Acid 3620 µmol /L - +
Ascorbic Acid 342 µmol/L - +
Atorvastatin 600 µg Eq/L - +
Ibuprofen 2425 µmol/L - +
Loratadine 0.78 µmol/L - +
Nadolol 3.88 µmol/L - +
Naproxen 2170 µmol/L - +
Paroxetine 3.04 µmol/L - +
Cotrimoxazole - +
Ethanol - +
Fluconazole - +
Ivermectin - +
Albendazole - +
Anti-malarial medicines
Quinine - +
Primaquine - +
Tuberculosis medicines
Rifampicin/isoniazid/ethambutol - +
Antiretrovirals
Atazanavir - +
Darunavir - +
Lopinavir/Ritonavir - +
Ritonavir - +
Raltegravir - +
Abacavir sulfate - +
Emtricitabine - +
Lamivudine - +
Tenofovir - +
Zidovudine - +
Efavirenz - +
The table is for illustrative purposes. WHO expects manufacturers to provide all results for the studies
in an easy to read format.
WHO Sample product dossier for SIMU HIV-1 QUANT
Product Performance Specifications and Associated Validation and Verification Studies
Draft for public comment 26 October 2016 Page 64 of 140
Nevirapine
Anti-hepatitis medicines- HBV, HCV
Adefovir dipivoxil
Peginterferon alfa-2a and 2b
Entecavir
Telbivudine
Ribavirin
Sofosbuvir
Anti-herpes medicines
Aciclovir
Results 1000
All the blood analytes and therapeutic drugs tested were shown not to interfere with the 1001 quantitation of the SIMU HIV-1 Quant Test over the linear range of the assay (200-1002 10,000,000 copies/mL). 1003
Conclusions 1004
The exogenous substances tested do not affect the specificity or the accuracy of quantitation 1005 of the SIMU HIV-1 Quant Test. As new antimicrobial drugs come onto the market, they will 1006 be tested in a similar manner as part of an ongoing study. 1007

WHO Sample product dossier for SIMU HIV-1 QUANT
Product Performance Specifications and Associated Validation and Verification Studies
Draft for public comment 26 October 2016 Page 65 of 140
7.1.2.4 Traceability of calibrators and control material values 1008
See annex XXX 1009
7.1.2.5 Measuring range of the assay 1010
Study Summary Report: Linearity 1011 Full Report: AR XXX - refer ANNEX X 1012 Study Dates: xxxxx 1013 Conducted by: A Blog, Research and Development Dept, THE Manufacturing Company 1014 Testing: Performed on-site, Headquarters 1015
1016
Study Objective 1017
To demonstrate the linearity of the SIMU HIV-1 Quant Test. 1018
Reagents and Instruments 1019
SIMU HIV-1 Quant Test Cartridge Lot Number: MPOC-1307 expiry 07 2016, MOOC-1300 1020 expiry 07 2016 1021
SIMU Instrument Lot Number: MPR-11AGG 1022
Acceptance Criteria 1023
The SIMU HIV-1 Quant Test should demonstrate linearity over a range from 200 to 1024 10,000,000 copies/mL. Good correlation with the target HIV-1 viral loads should be 1025 demonstrated by Deming Regression analysis. 1026
Methodology/Study Design 1027
The SIMU working standard based on HIV-1 (lllB Strain) was used for this study. The working 1028 standard has been quantified using reference method 1 to achieve traceability to the WHO 1029 International Standard HIV-1 RNA, 1st International Standard. Therefore, one copy of HIV-1 1030 RNA is equivalent to 1.67 International Units (IU) (Holmes HH, Davis C, Heath A, Hewlett I, 1031 Lelie N. An international collaborative study to establish the 1st international standard for 1032 HIV-1 RNA for use in nucleic acid-based techniques. J Virol Methods 2001;92: 141-150). (1). 1033 This material has been described in Annex XXX, Limit of Detection (95% LoD) studies. Serial log 1034 dilutions of the working reagent were prepared in HIV-1-negative human EDTA plasma. The 1035 preparation of negative human plasma has been described in Annex XXX, Limit of Detection 1036 (95% LoD) study. Serial half-log dilutions were tested. Forty-five replicates of each dilution 1037 were tested. Two reagent lots were used to give a total of 90 tests per dilution. The results for 1038 one reagent lot are shown in the following Figure 7-14. 1039

WHO Sample product dossier for SIMU HIV-1 QUANT
Product Performance Specifications and Associated Validation and Verification Studies
Draft for public comment 26 October 2016 Page 66 of 140
Figure 7-14: Linear range of SIMU Quant HIV-1 Test 1040
Target concentration as log copies/mL
1041
Conclusion: 1042
This study provides evidence that the SIMU HIV-1 Quant Test has a linear range of 200 to 1043 10,000,000 copies/mL for HIV-1, group M, subtype B. Deming Regression analysis showed 1044 good correlation between the results of the SIMU HIV-1 Quant Test and the target viral loads 1045 (The slope and R2 value for the curve were 1.003 and 0.989 respectively). 1046
WHO Sample product dossier for SIMU HIV-1 QUANT
Product Performance Specifications and Associated Validation and Verification Studies
Draft for public comment 26 October 2016 Page 67 of 140
Stability (excluding specimen stability) 7.21047
Stability studies were performed with the SIMU HIV-1 Quant Test Cartridge in order to 1048 determine shelf-life (including storage and transport conditions) and in-use stability of kit 1049 components. Stability panels contained HIV-1-negative, human, EDTA plasma spiked with HIV-1050 1 group M (IIIB) virus to a concentration of 3 x LoD. The HIV-1 spiked specimens are traceable 1051 to the WHO International Standards for HIV-1 RNA for Nucleic Acid-Based Techniques. It is 1052 expected that the results will be comparable for HIV-1 group N and O because the 1053 oligonucleotides for these analytes have the same degradation profile (See “Validation of 1054 oligonucleotide stability” SLST XXX.xx) 1055
1056
7.2.1 Claimed shelf-life (including transport challenge) 1057
Study Summary Report: SIMU HIV-1 Quant Test Cartridge Transport and real-time 1058 stability study 1059 Protocol: SHST XXX - refer ANNEX XX 1060 Full Report: SHST XXX – refer ANNEX XXX Study report SIMU HIV-1 Quant Test Cartridge 1061 transport and real-time stability 1062 Study Dates: October 2014 – October 2015 1063 Lot number: MOOC-137 expiry 10 2015, MPOG -144 expiry 10 2015, MPOG-143 expiry 10 1064 2015 1065 IFU version: v1.0 1066 Conducted by: A Techie, Research and Development Dept., THE Manufacturing Company 1067 Testing: Performed on-site, Headquarters 1068
The objective was to follow the stability of the cartridge and external positive controls over at 1069 least 13 months at multiple temperatures to determine shelf-life of the device. The impact of 1070 temperature and humidity extremes on cartridge performance was also considered by 1071 preceding real-time testing with simulated transport challenges designed to mimic expected 1072 storage and transport conditions. Testing beyond 13 months would allow an understanding of 1073 when, in real-time, the device is likely to ‘fail’ and may allow an extension of the proposed 1074 shelf-life from the six months that was established in previous testing (Annex XXZ). 1075
Acceptance Criterion 1076
The acceptance criterion was defined as demonstration of acceptable assay performance to 1077 support recommended storage and transport conditions. Acceptable assay performance was 1078 defined as a detection rate of 100% at a concentration of approximately 3x LoD HIV-1 M and 1079 specificity of 100%. In addition, the Ct value was expected to deviate by no more than 20% 1080 over the testing period. The stability was taken as the time point prior to last time point to 1081 have met the acceptance criteria, e.g. if the device was stable to 13 months, the stability will 1082 be deemed to be 12 months. 1083
Methodology/Study Design 1084
Stability studies for the SIMU HIV-1 Quant Test evaluated shelf-life when stored at 6 °C, 1085 20 °C, and 45 °C. Stability panels contained HIV-1-negative, human, EDTA plasma spiked with 1086 HIV-1 group M (IIIB) virus to a concentration of 3 x LoD. The HIV-1 spiked specimens are 1087 traceable to the WHO International Standards for HIV-1 RNA for Nucleic Acid-Based. Sufficient 1088 volume of each stability panel member for the duration of the testing schedule was prepared. 1089
Testing was performed in triplicate for each panel member for each condition. Multiple 1090 instruments were used to allow simultaneous testing of cartridges at each time point. Three 1091 kit lots, from three independent production lots, using a predetermined sampling protocol 1092

WHO Sample product dossier for SIMU HIV-1 QUANT
Product Performance Specifications and Associated Validation and Verification Studies
Draft for public comment 26 October 2016 Page 68 of 140
SOP 199 were tested. At least 10% overage was allowed for unexpected requirements and re-1093 testing. 1094
SIMU HIV-1 Quant Test Cartridge and external positive controls evaluated the effect of 1095 shipping conditions on these devices including a freeze-thaw cycle (-20 °C to 2–8 °C), and 1096 temperature cycling from 20 °C to 55 °C under variable humidity conditions. Prior to 1097 temperature cycling conditions, cartridges and external positive controls were stored at 20 ± 5 1098 °C to mimic normal manufacturer storage conditions prior to shipping. Cartridges were 1099 removed from storage within one month of manufacture and subjected to a freeze-thaw cycle 1100 (-20 °C to 2–8 °C). After the freeze-thaw cycle, cartridges and external positive controls were 1101 subjected to the following temperature and humidity sequence: 1102
Ambient humidity (60% RH) 1103 Put at 20 ± 5 °C storage temperature for 24 ± 4 hours followed by 1104 55 ± 2 °C for 24 ± 4 hours, followed by 1105 20 ± 5 °C storage temperature for 24 ± 4 hours 1106
Followed by: 1107
Desert humidity (30% RH) 1108 Put at 20 ± 5 °C storage temperature for 24 ± 4 hours followed by 1109 55 ± 2 °C for 24 ± 4 hours, followed by 1110 20 ± 5 °C storage temperature for 24 ± 4 hours 1111
Followed by: 1112
Tropical humidity (85% RH) 1113 Put at 20 ± 5 °C storage temperature for 24 ± 4 hours followed by 1114 55 ± 2 °C for 72 ± 4 hours, followed by 1115 20 ± 5 °C storage temperature for 24 ± 4 hours 1116
Followed by: 1117
Ambient humidity (60% RH) 1118 20 ± 5 °C storage temperature for 24 ± 4 hours 1119 1120
The above complete sequence was used as opposed to separate cartridges or controls held at 1121 individual temperatures. Following simulation of transport conditions, cartridges were stored 1122 at 6 ± 2 °C, 20 ± 2 °C or 45 ± 5 °C. Testing was conducted at 0, 3, 6, 9, 12 and 13 months. At 1123 each time point, cartridges and external controls were brought to room temperature (20 ± 5 1124 °C) and cartridges were used to test both the stability panel and external positive controls. 1125
Results 1126
Results for Lot 1 are shown in Table 7-15. 1127
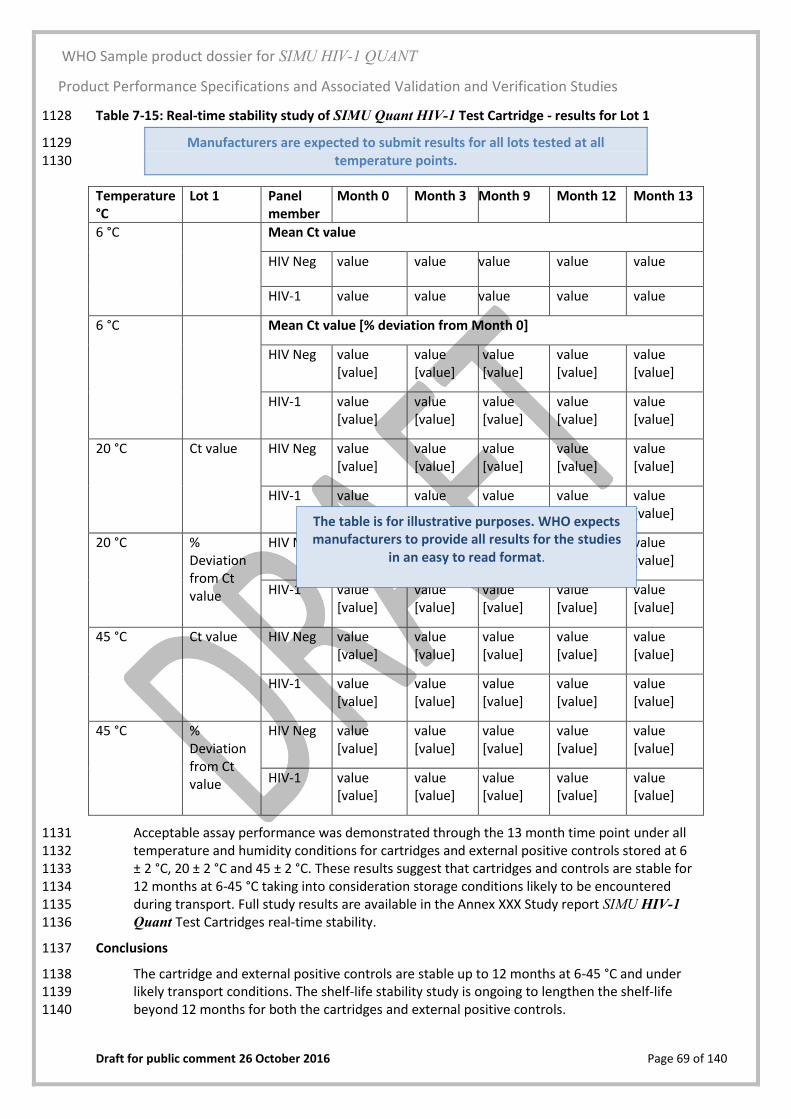
WHO Sample product dossier for SIMU HIV-1 QUANT
Product Performance Specifications and Associated Validation and Verification Studies
Draft for public comment 26 October 2016 Page 69 of 140
Table 7-15: Real-time stability study of SIMU Quant HIV-1 Test Cartridge - results for Lot 1 1128
Manufacturers are expected to submit results for all lots tested at all 1129 temperature points. 1130
Temperature °C
Lot 1 Panel member
Month 0 Month 3 Month 9 Month 12 Month 13
6 °C Mean Ct value
HIV Neg value value value value value
HIV-1 value value value value value
6 °C Mean Ct value [% deviation from Month 0]
HIV Neg value [value]
value [value]
value [value]
value [value]
value [value]
HIV-1 value [value]
value [value]
value [value]
value [value]
value [value]
20 °C Ct value HIV Neg value [value]
value [value]
value [value]
value [value]
value [value]
HIV-1 value [value]
value [value]
value [value]
value [value]
value [value]
20 °C % Deviation from Ct value
HIV Neg value [value]
value [value]
value [value]
value [value]
value [value]
HIV-1 value [value]
value [value]
value [value]
value [value]
value [value]
45 °C Ct value HIV Neg value [value]
value [value]
value [value]
value [value]
value [value]
HIV-1 value [value]
value [value]
value [value]
value [value]
value [value]
45 °C % Deviation from Ct value
HIV Neg value [value]
value [value]
value [value]
value [value]
value [value]
HIV-1 value [value]
value [value]
value [value]
value [value]
value [value]
Acceptable assay performance was demonstrated through the 13 month time point under all 1131 temperature and humidity conditions for cartridges and external positive controls stored at 6 1132 ± 2 °C, 20 ± 2 °C and 45 ± 2 °C. These results suggest that cartridges and controls are stable for 1133 12 months at 6-45 °C taking into consideration storage conditions likely to be encountered 1134 during transport. Full study results are available in the Annex XXX Study report SIMU HIV-1 1135 Quant Test Cartridges real-time stability. 1136
Conclusions 1137
The cartridge and external positive controls are stable up to 12 months at 6-45 °C and under 1138 likely transport conditions. The shelf-life stability study is ongoing to lengthen the shelf-life 1139 beyond 12 months for both the cartridges and external positive controls. 1140
The table is for illustrative purposes. WHO expects manufacturers to provide all results for the studies
in an easy to read format.

WHO Sample product dossier for SIMU HIV-1 QUANT
Product Performance Specifications and Associated Validation and Verification Studies
Draft for public comment 26 October 2016 Page 70 of 140
Real-time stability studies were conducted for the external positive and negative controls and 1141 can be found at Annex XXX. 1142
1143
The Manufacturer should refer to the WHO TGS-2 document “Establishing 1144 stability of an in vitro diagnostic for WHO Prequalification” for further 1145
information. 1146
http://www.who.int/diagnostics_laboratory/guidance/en/ 1147
7.2.2 In-use stability 1148
Study Summary Report: SIMU HIV-1 Quant System Test Cartridge In-Use Stability 1149 Protocol: AP XXX 1150 Full Report: AR XXX – refer ANNEX XXX Study report SIMU HIV-1 Quant Test Cartridge In-1151 Use Stability 1152 Study Dates: October 2014 1153 Lot number: MOOC-137 expiry 10 2015 1154 IFU version: v1.0 1155
Conducted by: J. Patrick, Research and Development Dept., THE Manufacturing 1156
Company 1157 Testing: Performed on-site, Headquarters 1158
Study Objective 1159
The objective was to determine the stability of the Test Cartridge and sample after removing 1160 the Test Cartridge from the pouch package and loading a plasma panel specimen into the 1161 device. The intended use population is trained users in a situation where the Test Cartridge 1162 could be subjected to uncontrolled conditions such as obtaining the sample in the Test 1163 Cartridge and delaying testing. 1164
Acceptance Criterion 1165
The acceptance criterion was defined as demonstration of acceptable assay performance to 1166 support recommended in-use conditions. Acceptable assay performance was defined as a 1167 detection rate of 100% at a concentration of approximately 3x LoD HIV-1 M and specificity of 1168 100%. The stability was taken as the time point prior to last time point to have met the 1169 acceptance criteria. 1170
Methodology/Study Design 1171
Stability panels contained HIV-1-negative, human, EDTA plasma spiked with HIV-1 group M 1172 (IIIB) virus to a concentration of 3 x LoD. The HIV-1 spiked specimens are traceable to the 1173 WHO International Standards for HIV-1 RNA for Nucleic Acid-Based. 1174
In-use stability studies for the SIMU HIV-1 Quant Test Cartridge were performed on one lot of 1175 the Test Cartridge by loading the Test Cartridges with the panel members described above and 1176 closing the cartridge port. Cartridges were tested immediately after opening the cartridge 1177 pouch and loading plasma specimen, and after storage at 20 ± 2 °C and 45 ± 5 °C for 1, 2, 6, 1178 and 25 hours at 60% humidity. Sufficient volume of each stability testing panel member for 1179 the duration of the testing schedule was prepared. Testing was performed in triplicate for 1180 each panel member for each condition. Multiple instruments were used to allow simultaneous 1181

WHO Sample product dossier for SIMU HIV-1 QUANT
Product Performance Specifications and Associated Validation and Verification Studies
Draft for public comment 26 October 2016 Page 71 of 140
testing of cartridges at each time point. At least 10% overage was allowed for unexpected 1182 requirements and re-testing. 1183
The Test Cartridges were divided into two groups and placed at either 20 ± 2 °C or 45 ± 5 °C. 1184 Cartridges from both groups were sampled at 0, 1, 2, 6, and 25 hours. 1185
1186
Results 1187
In-Use Stability acceptable assay performance was demonstrated through the 24 hour time 1188 point for Test Cartridges stored at 20 ± 5 °C and 45 ± 5 °C at 60% humidity. These results 1189 demonstrate in-use stability at 20–45 °C for up to 24 hours after injection of the sample into 1190 the Test Cartridge. Full study results are available in the Annex XXX Study report SIMU HIV-1 1191 Quant System Test Cartridge in-use stability. 1192
Conclusion 1193
The device can be used for testing up to 24 hours at temperatures ranging from 20–45 °C after 1194 opening the package and loading a EDTA plasma sample. 1195

WHO Sample product dossier for SIMU HIV-1 QUANT
Product Performance Specifications and Associated Validation and Verification Studies
Draft for public comment 26 October 2016 Page 72 of 140
Robustness studies 7.31196
7.3.1 Cross-contamination 1197
Study Summary Report: Cross-contamination 1198
Full Report: AR XXX - refer ANNEX X. 1199 Study Dates: xxxxx 1200 Conducted by: A Blog, Research and Development Dept, THE Manufacturing Company. 1201 Testing: Performed on-site, Headquarters. 1202
Study Objective 1203
To check for cross-contamination on the SIMU HIV-1 Quant Test 1204
Reagents and Instruments 1205
SIMU HIV-1 Quant Test Cartridge Lot Number: MPOC-1307 expiry 07 2016 1206 SIMU Instrument Lot Number: MPR-11AGG 1207
Acceptance Criteria 1208
All negative specimens should be non-reactive and all positive specimens should be reactive. 1209
Methodology/Study Design 1210
The cross-contamination study used HIV-negative, pooled plasma, prepared as described in 7.1.2.2. 1211 Analytical Sensitivity Study Summary Report: Limit of Detection (95% LoD)Full (AR XXX - refer ANNEX 1212 X). Twenty high viral load, HIV-1 plasma samples (1.00E+05 copies/mL) were purchased from a 1213 commercial vendor. The viral loads of these samples were determined by the vendor and confirmed 1214 with a reference method assay. The 20 positive samples and 20 aliquots of the pooled, negative 1215 plasma were tested with the SIMU HIV-1 Quant Test. The positive and negative samples were 1216 loaded alternatively on the Instrument. The testing was performed on a second day using a further 1217 20 HIV-1-positive and 20 negative plasma samples. 1218
Results 1219
All 40 negative samples showed no detectable target and all 40 HIV-1-positive samples showed 1220 detection of target at appropriate levels in the HIV-1 channel only, giving a 0% cross-contamination 1221 rate. 1222
Conclusion 1223
No cross-contamination was observed with the SIMU HIV-1 Quant Test. 1224

WHO Sample product dossier for SIMU HIV-1 QUANT
Product Performance Specifications and Associated Validation and Verification Studies
Draft for public comment 26 October 2016 Page 73 of 140
7.3.2 Whole system failure 1225
Study Summary Report: Whole system failure 1226
Full Report: AR XXX - refer ANNEX X. 1227 Study Dates: xxxxx 1228 Conducted by: A Blog, Research and Development Dept, THE Manufacturing Company. 1229 Testing: Performed on-site, Headquarters. 1230
Study Objective 1231
To determine the whole system failure of the SIMU HIV-1 Quant Test. 1232
Reagents and Instruments 1233
SIMU HIV-1 Quant Test Cartridge Lot Number: MPOC-1307 expiry 07 2016 1234 SIMU Instrument Lot Number: MPR-11AGG 1235
Acceptance Criterion 1236
The whole system failure rate should be equal to or less than 1%. 1237
Methodology/Study Design 1238
The whole system failure rate was determined by testing 100 replicates of HIV-negative, pooled 1239 plasma, prepared as described in 7.1.2.2. Analytical Sensitivity Study Summary Report: Limit of 1240 Detection (95% LoD) (AR XXX - refer ANNEX X), spiked with HIV-1 group M at a concentration of 1241 approximately 3x LoD. The HIV-1 sample used the HIV-1 (IIIB strain) working standard that was used 1242 for the Analytical Sensitivity Study (AR XXX - refer ANNEX X). Twenty samples were tested on five 1243 consecutive days with the SIMU HIV-1 Quant Test. 1244
Results 1245
All 100 samples were quantitated to acceptable concentrations of HIV-1 copies/mL. 1246
Conclusion 1247
The SIMU HIV-1 Quant test is robust and met the acceptance criteria. 1248
WHO Sample product dossier for SIMU HIV-1 QUANT
Product Performance Specifications and Associated Validation and Verification Studies
Draft for public comment 26 October 2016 Page 74 of 140
Clinical evidence (clinical sensitivity and specificity) 7.41249
Study Summary Report: Clinical Specificity and Sensitivity 1250 Full Report: AR XXX - refer ANNEX X. 1251 Study Dates: xxxxx 1252 Conducted by: Three external sites 1253 Testing: Performed at three external sites in the region where the test will be used 1254
Study Objective 1255
To demonstrate the clinical specificity and sensitivity of the SIMU HIV-1 Quant Test using 1256 clinical specimens from HIV-negative and HIV-1-positive subjects. Both fresh and frozen EDTA 1257 plasma samples were tested at three test sites in the region where the test would be used. 1258
Reagents and Instruments 1259
SIMU HIV-1 Quant Test Cartridge Lot Number: MPOC-1307 expiry 07 2016 1260
SIMU Instrument Lot Number: MPR-11AGG 1261
Acceptance Criteria 1262
The acceptance criterion below is for illustrative purposes only. The 1263 manufacturer is expected to define acceptance criteria for each specimen 1264
during the planning stage of study and prior to commencement of testing[?]. If 1265 the results do not fall within the expected range, the manufacturer is expected 1266
to investigate why. 1267
The specificity was expected to be ≥ 99.5 % and 95% lower CI > 98.4%. The positive and 1268 negative percent agreement between the SIMU HIV-1 Quant Test and the expected results 1269 should be >99% (lower 95% CI >97%). The overall bias between the results for the SIMU HIV-1270 1 Quant Test and the test viral loads over the dynamic range of the two assays should be 1271
≤log10 0.3 copies/mL. 1272
Methodology/Study Design 1273
Clinical sensitivity was determined by testing 200 frozen plasma samples with HIV-1 RNA 1274 concentrations >200 copies/mL and 100 fresh sample in EDTA plasma collected from HIV-1 1275 positive individuals. The samples were tested for anti-HIV-1 and NAT reactivity using tests that 1276 have been authorized for use by a Regulatory Authority of the founding members of GHTF. 1277 The RNA concentrations of the samples were determined with a reference method assay. 1278
Clinical specificity was determined by testing 100 frozen plasma samples and 200 fresh plasma 1279 samples from blood donors shown to be negative for HIV-1/2 and non-reactive for HIV-1 RNA 1280 using tests that have been authorized for use by a Regulatory Authority of the founding 1281 members of GHTF. 1282
Both the HIV-1 negative and HIV-1 positive samples were collected in the region where the 1283 test would be used. 1284
Results 1285
A total of 300 evaluable HIV-negative and 300 HIV-1-positive patient specimens were included 1286 in clinical specificity and sensitivity analyses. Approximately 50% of the patient specimens 1287 were frozen. The demographic characteristics of the 300 evaluable HIV-1-positive specimens 1288 are summarized in Table 7-17. The CD4 cell counts of the subjects distributed approximately 1289 evenly across CD4 cell count categories (<200, 200-500, >500 cells/uL). Most of the subjects 1290

WHO Sample product dossier for SIMU HIV-1 QUANT
Product Performance Specifications and Associated Validation and Verification Studies
Draft for public comment 26 October 2016 Page 75 of 140
were female (60%) and between 18 to 29 years of age (66.7%). The ethnic distribution in the 1291 panel is comparable to that observed in the HIV-1 population of the region. 1292
1293
Table 7-15: Demographics of HIV-1 evaluable samples 1294
1295
The clinical sensitivity and specificity were calculated for the pooled results from all three sites and 1296 are summarized in Table 7-16. 1297
Table 7-16: SIMU HIV-1 Quant Test Clinical Sensitivity and Specificity 1298
1299
Sample Positive Negative Total Clinical sensitivity (95% CI)
HIV-1 positive 300 0 300 100% (98.4, 100)
Sample Positive Negative Total Clinical specificity (95% CI)
HIV-1 negative 3 297 300 99% (96.6, 99.7)
1300
The clinical sensitivity was 100% (98.4, 100) and the clinical specificity of the test was 99% 1301 (96.6, 99.7). Three of the 300 qualified HIV-1 negative samples gave false-positive results (all 1302 these false-positive results were obtained at site 2). 1303
Method comparison between SIMU HIV-1 Quant Test and the reference test was undertaken 1304 using Bland-Altman analysis. Of the 300 specimens in the HIV-1 positive testing panel a total 1305
Sample characteristics Category No. of HIV-1 positive individuals (%)
CD4 cell counts (cells/µL) <200 96 (32%)
200–500 120 (40%)
>500 84 (28%)
Sex Male 120 (40%)
Female 180 (60%)
Age 18–29 200 (66.7%)
30–49 50 (16.7%)
50–65 48 (16.0%)
>65 2 (0.6%)
Ethnicity African [value]
Asian [value]
Caucasian [value]
The table is for illustrative purposes. WHO expects manufacturers to provide all results for the studies
in an easy to read format.

WHO Sample product dossier for SIMU HIV-1 QUANT
Product Performance Specifications and Associated Validation and Verification Studies
Draft for public comment 26 October 2016 Page 76 of 140
of 250 paired specimen results (for which both test and reference results were within the 1306 linear range of both assays) were chosen for analysis. The SIMU HIV-1 Quant Test and 1307 reference test were considered to be equivalent if the differences in titres of the 250 paired 1308 samples between the SIMU Quant HIV-1 Test and the reference test were all within ± 2 SD of 1309 the mean. 1310
Bland-Altman analysis, described in XXX Study Report: Bland-Altman Analysis (Appendix XXX), 1311 suggests that the SIMU HIV-1 Quant Test and the reference test are equivalent (the 1312 differences in titres of the paired samples between the SIMU Quant HIV-1 Test and the 1313 reference testwere all within ± 2 SD of the mean. 1314
Conclusions 1315
The clinical sensitivity and specificity of the SIMU HIV-1 Quant Test as determined by three 1316 field evaluations were 99% and 100% respectively. The clinical sensitivity and specificity of the 1317 SIMU HIV-1 Quant Test as determined by three field evaluations were 100% and 99% 1318 respectively. The mean difference in the viral load of the SIMU HIV-1 Quant Test and the 1319 reference method over the dynamic ranges of the assays met the acceptance criterion. 1320

WHO Sample product dossier for SIMU HIV-1 QUANT
Labelling
Draft for public comment 26 October 2016 Page 77 of 140
8 Labelling 1321
8.1 Labelling 1322
8.1.1 Instrument labelling 1323
The Instrument label is attached to the front of the device. It includes the product name, serial 1324 number, manufacturing information, Li-Ion battery symbol, power type and consumption, and 1325 version number. It is resistant to water, alcohol based cleaning agents and friction. Only 1326 internationally recognized symbols are used (ISO 15223-1:2012). 1327
The Instrument box label is a well-fixed water resistant label (applied with high quality water 1328 resistant glue) and permanent printing is utilized. As per the International Air Transport Association 1329 lithium battery guidance the packaging is labelled with battery handling instructions. 1330
Temperature logger tags are included on the box. This allows THE Manufacturing Company to 1331 monitor shipments and storage as well as to trace and remediate errors. 1332
8.1.2 Test Cartridge pouch packaging and labelling (Figure XXY) 1333
8.1.2.1 Pouch packaging 1334
The retail pack for the device contains a single unit. 1335
Materials: Waterproof metallised wrapping 1336
heat sealable 1337
indentation to allow easy opening of the device packaging without the need for 1338 scissors. 1339
Design: Dimensions: Length = 5 cm, Width = 2.5 cm, Height = 1 cm (single device pack) 1340
Weight: 100 gm 1341
Pouch labelling: Only internationally recognized symbols are used (ISO 15223-1:2012). 1342 Well-fixed water resistant labels (applied with water resistant glue) and permanent printing 1343 are utilized. 1344
8.2 Instructions for Use 1345
A comprehensive IFU was developed and qualified by THE Manufacturing Company in compliance 1346 with the following regulatory requirements, standards and guidance documents: 1347
ISO 18113-1:2009. In vitro diagnostic medical (IVDs) - Information supplied by the 1348 manufacturer (labelling) - Part 1: Terms, definitions and general requirements. Geneva, 1349 Switzerland. International Organization for Standardization; 2009. 1350
ISO 18113-2:2009. In Vitro diagnostic medical devices – Information supplied by the 1351 manufacturer (labelling) – Part 2: In vitro diagnostic reagents for professional use. Geneva, 1352 Switzerland. International Organization for Standardization; 2009. 1353

WHO Sample product dossier for SIMU HIV-1 QUANT
Labelling
Draft for public comment 26 October 2016 Page 78 of 140
ISO 18113-3:2009. In vitro diagnostic medical devices – Information supplied by the 1354 manufacturer (labelling) - Part 3: In vitro diagnostic instruments for professional use Geneva, 1355 Switzerland. International Organization for Standardization; 2009. 1356
ISO 14971:2007 Medical devices - Application of risk management to medical devices. 1357 Geneva, Switzerland. International Organization for Standardization; 2007. 1358
ISO 15223-1:2012. Medical Devices - Symbols to be used with medical device labels, labelling 1359 and information to be supplied - Part 1: General requirements. Geneva, Switzerland. 1360 International Organization for Standardization; 2012. 1361
GHTF/SG1/N70:2011: Label and Instructions for Use for Medical Devices. Global 1362 Harmonization Task Force (GHTF) Steering Committee; 2011. 1363
IMDRF/UDI WG/N7 FINAL:2013: UDI Guidance Unique Device Identification of Medical 1364 Devices. International Medical Device Regulators Forum; 2013. 1365
Directive 98/79/EC of the European Parliament and of the Council of 27 October 1998 on in 1366 vitro diagnostic medical device. [2003] OJ L331/1. 1367
European Commission Enterprise and Industry Directorate-General. MEDDEV. 2.14/3 rev.1 1368 Guidelines on Medical Devices: IVD guidances: Supply of Instructions For Use (IFU) and other 1369 information for In-vitro Diagnostic (IVD) medical devices. A guide for manufacturers and 1370 notified bodies. Brussels, Belgium 2007. 1371
Backinger CL and Kingsley PA. Write it Right. U.S. Department of Health and Human Services, 1372 Public Health Service, Food and Drug Administration, Center for Devices and Radiological 1373 Health (CDRH), MD, USA; 1993. 1374
FDA. Final guidance for industry and FDA reviewer. Guidance on medical device patient 1375 labelling; MD, USA. Center for Devices and Radiological Health (CDRH); 2001. 1376
Health Canada. Draft Guidance for the labelling of in vitro diagnostic devices. Ontario, 1377 Canada. Health Canada; 1998. 1378
Readability Calculator. http://www.online-1379 utility.org/english/readability_test_and_improve.jsp. Mladen Adamovic, 2009. 1380
Flesch R. How to Write Plain English [web page]. 1381 http://www.mang.canterbury.ac.nz/writing_guide/writing/flesch.shtml . Christchurch, New 1382 Zealand. University of Canterbury; 1981. 1383
The IFU is attached in Annex XX. 1384
8.3 Instrument manual 1385
See Annex XXX 1386

WHO Sample product dossier for SIMU HIV-1 QUANT
Commercial History
Draft for public comment 26 October 2016 Page 79 of 140
9 Commercial History 1387
9.1 Countries of supply 1388
9.1.1 List of countries where product is currently supplied 1389
The SIMU HIV-1 Quant System has been supplied in South Africa, Kenya, India, China (PRC), 1390 Republic of Korea, Brazil, Mexico, and Peru since late 2015. A total of 1,110,000 tests have been 1391 supplied. 1392
9.1.2 Minimum and maximum price in 2015 1393
The price per test varies between US$ 5.00 and US$ 9.50. The majority of tests sold have been at 1394 US$ 7.50 per test. A number of tests (approximately 10,000) were donated to Nigeria for 1395 performance evaluation. 1396
9.1.3 Training and support network 1397
9.1.3.1 IFU 1398
The utility of the IFU has been subjected to intense testing by THE Manufacturing Company, as 1399 part of our risk analysis for instructions to users. The goal was that these instructions would prove 1400 sufficient to a range of users with varying skills levels, acknowledging the fact that the product will 1401 be used in a variety of settings. A report of this testing is held at Headquarters (refer DOC XXX). 1402
9.1.3.2 Online training 1403
A video training program for the SIMU HIV-1 Quant Test in English is available on the website of 1404
THE Manufacturing Company or on a CD by request, which gives training on the basic functions 1405 of the Instrument, and basic trouble shooting instructions. 1406
9.1.3.3 Extensive technical training for users familiar with running of the 1407
Instrument 1408
Extensive technical training is provided by THE Manufacturing Company for users who are already 1409 familiar with the running of the Instrument and focused on people with responsibility for a number 1410 of instruments. super 1411
9.1.3.4 Training the trainer 1412
Technical support is provided by the local distributor as required, and mobile agents are available in 1413 country for trouble shooting for the Instrument. Technical support training for local distributors is 1414 provided by staff of THE Manufacturing Company staff in all distribution countries. 1415
9.1.3.5 Software updates 1416
Software updates and fixes can be downloaded by internet connection. 1417
WHO Sample product dossier for SIMU HIV-1 QUANT
Commercial History
Draft for public comment 26 October 2016 Page 80 of 140
9.2 Adverse events and field safety corrective actions 1418
No field safety corrective action has been required since any version of the product has been 1419 marketed. 1420
9.2.1 Table of adverse event reports 1421
No adverse events have been reported as yet. 1422
Table 9-1: Table of adverse event reports 1423
QA reference Type of event Country of event Determined cause Action taken
(None reported)
1424

WHO Sample product dossier for SIMU HIV-1 QUANT
Regulatory History
Draft for public comment 26 October 2016 Page 81 of 140
10 Regulatory History 1425
Certified copies of the Import Medical Device Registration Certificate, issued by the China Food and 1426 Drug Administration, and the Korea Licence Holder Certificate, are also provided in ANNEX YYY. 1427

WHO Sample product dossier for SIMU HIV-1 QUANT
Quality Management System
Draft for public comment 26 October 2016 Page 82 of 140
11 Quality Management System 1428
11.1 Quality manual 1429
Refer to Annex VIII. 1430
11.1.1 Quality manual system documents 1431
Manufacturer to insert a complete list of all valid quality management system 1432 documents with document title and number relevant to the product under 1433
prequalification assessment 1434
11.1.2 Quality manual system procedures 1435
Refer to Annex XXX for documented procedures for the following areas: 1436
1. Control of design and development changes (SOP XXXX) 1437
2. Risk management planning and implementation (SOP XXXX) 1438
3. Control of non-conforming goods (SOP XXXX) 1439
4. Corrective and preventive actions (SOP XXXX) 1440
5. Recalls procedure (SOP XXXX) 1441
6. Control of key suppliers (SOP XXXX) 1442
11.2 Quality manual test certification 1443
THE Manufacturing Company holds ISO 13485:2003 Medical devices – Quality management systems 1444 – Requirements for regulatory purposes certification for the manufacture of the product under 1445 assessment, which is valid until 23 March 2018. A certified copy of the certificate is attached in 1446 Annex XX in addition to the two previous inspection reports issued by the certification. 1447

WHO Sample product dossier for SIMU HIV-1 QUANT Annex I: Essential Principles Checklist
Draft for public comment 26 October 2016 Page 83 of 140
Annex I: Essential Principles Checklist 1448
Notes 1449
This document applies to the following configuration of the SIMU HIV-1 Quant System. 1450
The Risk Management File is made up of the Risk Management Plan, Risk Management Report, and 1451 associated risk analyses. These documents are located in the DHF. 1452
Copies of testing results to applicable standards referenced below are located in the DHF. 1453
The test system is comprised of the SIMU Instrument with integrated software, SIMU HIV-1 Quant 1454 System Test Cartridge (consisting of 25 or 100 Test Cartridges) 1455
Check list of EP of GHTF/SG1/N068 for: SIMU HIV-1 Quant Test
SIMU HIV-1 Quant Test Product Codes: M123, M124
Document version # 02 Document date:
Written by:
Signatures Date
Approved by:
QA
Signature Date
Regulatory affairs
Signature Date
Agreed by:
R&D
Signature Date
Manufacturing
Signature Date
Changes from earlier versions:
Version Item modified Modification
01 Date 2011 01 01 N/A Document created according to SOP XXX
02 Date 2011 06 15 B6 B6 added for instrument readout
03 Date 2011 10 15 C9.3 C9.3 added for instrumentation
Please Note: The checklist submitted to WHO must include dates and signatures
1456
1457

WHO Sample product dossier for SIMU HIV-1 QUANT Annex I: Essential Principles Checklist
Draft for public comment 26 October 2016 Page 84 of 140
1458
THE Manufacturing Company SIMU HIV-1 Quant Test Product Codes M123 and M124
Essential principles Applicable/
Not Applicable (A/NA) Method(s) and references used to demonstrate conformity
Reference to supporting controlled documents of Manufacturer
A GENERAL PRINCIPLES
A.1 Medical devices should be designed and manufactured in such a way that, when used under the conditions and for the purposes intended and, where applicable, by virtue of the technical knowledge, experience, education or training, and the medical and physical conditions of intended users, they will perform as intended by the manufacturer and not compromise the clinical condition or the safety of patients, or the safety and health of users or, where applicable, other persons, provided that any risks which may be associated with their use constitute acceptable risks when weighed against the benefits to the patient and are compatible with a high level of protection of health and safety
A Application of recognized Standards:
ISO 13485:2003 Medical Devices – Quality management systems-Requirements for Regulatory Purposes.
ISO 14971:2007 Medical devices – Application of risk management to medical devices.
EN 13641:2002 Elimination or reduction of risk of infection related to in vitro diagnostic medical devices.
ISO 13485:2003 certificate held by QA department, certified copy submitted for Section 11.3 of this dossier.
“SIMU HIV-1 Quant Risk Management and Control Report” held by QA.
Risk analysis output documents “SIMU
HIV-1 Quant - design risk”, “SIMU HIV-1
Quant - processes”, “SIMU HIV-1 Quant risk-users”.
SIMU HIV-1 Quant Residual risk statement.
DHF, design input specification and design validation documentation (held by QA and in summary as Section 7 of this dossier).
Incoming goods analysis and control documentation held by QA “SOP XXX”.
Safety data sheets: Manufacturing department.
Regulation of Hazard control.
A.2 The solutions adopted by the manufacturer for the design and manufacture of the devices should conform to safety principles, taking account of the generally acknowledged state of the art. -When risk reduction is required, the manufacturer should control the risks so that the residual risk associated with each hazard is judged acceptable. -The manufacturer should apply the following principles in the priority order listed:
identify known or foreseeable hazards and
A Application of recognized Standards:
ISO 13485:2003 Medical Devices- Quality management systems-Requirements for Regulatory Purposes.
ISO 14971:2007 Medical devices – Application of risk management to medical devices.
ISO 18113:2009 In vitro diagnostic medical devices – Information supplied by the manufacturer (labelling) – Parts 1 and 4.
Risk management policy document “Policy XXX” and risk analysis SOPs “SOP XXX”, “SOP XXXX” held by Quality management (QM) Department
Risk analysis output documents “SIMU
HIV-1 Quant -design”, “SIMU HIV-1
Quant - processes”, “SIMU HIV-1 Quant -users”
“SIMU HIV-1 Quant Risk Management and Control Report” held by QA
WHO Sample product dossier for SIMU HIV-1 QUANT Annex I: Essential Principles Checklist
Draft for public comment 26 October 2016 Page 85 of 140
THE Manufacturing Company SIMU HIV-1 Quant Test Product Codes M123 and M124
Essential principles Applicable/
Not Applicable (A/NA) Method(s) and references used to demonstrate conformity
Reference to supporting controlled documents of Manufacturer
estimate the associated risks arising from the intended use and foreseeable misuse;
eliminate risks as far as reasonably practicable through inherently safe design and manufacture;
reduce as far as reasonably practicable the remaining risks by taking adequate protection measures, including alarms; and inform users of any residual risks.
SIMU HIV-1 Quant IFU.
A.3 Medical devices should achieve the performance intended by the manufacturer and be designed and manufactured in such a way that, during normal conditions of use, they are suitable for their intended purpose.
A Application of recognized standards:
ISO 13485:2003 Medical Devices - Quality management systems-Requirements for Regulatory Purposes.
ISO 14971:2007 Medical devices – Application of risk management to medical devices.
EN 13612:2002 Performance evaluation of in vitro diagnostic medical devices.
GHTF/SG5/N6:2012 Clinical Evidence for IVD medical devices – Key Definitions and Concepts.
GHTF/SG5/N8:2012 Clinical Evidence for IVD Medical Devices - Clinical Performance Studies for In Vitro Diagnostic Medical Devices.
Verification and Validation - Section 7 Studies
Risk management policy document “Policy XXX” and risk analysis SOPs “SOP XXX”, “SOP XXXX” held by QM department.
Risk analysis output documents “SIMU
HIV-1 Quant -risk-design”, “SIMU HIV-1
Quant - processes”, “SIMU HIV-1 Quant risk-users”.
“SIMU HIV-1 Quant Risk Management and Control Report” held by QA.
All verification and validation studies (refer Section 7 of this dossier). -
Traceability of SIMU HIV-1 Quant Control Cells.
A4 The characteristics and performances referred to in Clauses A1, A2 and A3 should not be adversely affected to such a degree that the health or safety of the patient or the user and, where applicable, of other persons are compromised during the lifetime of the device, as indicated by the manufacturer, when the device is subjected to the stresses which can
A Application of recognized standards:
ISO 18113:2009 In vitro diagnostic medical devices – Information supplied by the manufacturer (labelling) – Parts 1 and 4.
ISO 14971:2007 Medical devices – Application of risk management to medical devices.
All verification and validation studies (refer Section 7 of this dossier) including Stability studies (Section 7.2).
Risk management policy document “Policy XXX” and risk analysis SOPs “SOP XXX”, “SOP XXXX” held by QM department.
Risk analysis output documents “SIMU

WHO Sample product dossier for SIMU HIV-1 QUANT Annex I: Essential Principles Checklist
Draft for public comment 26 October 2016 Page 86 of 140
THE Manufacturing Company SIMU HIV-1 Quant Test Product Codes M123 and M124
Essential principles Applicable/
Not Applicable (A/NA) Method(s) and references used to demonstrate conformity
Reference to supporting controlled documents of Manufacturer
occur during normal conditions of use and has been properly maintained in accordance with the manufacturer’s instructions
ISO 23640:2011 In vitro diagnostic medical devices -- Evaluation of stability of in vitro diagnostic reagents.
CLSI EP25A:2009 Evaluation of Stability of In Vitro Diagnostic Reagents; Approved Guideline.
Stability and (user) Validation -Studies
HIV-1 Quant risk-design”, “SIMU HIV-1
Quant - processes”, “SIMU HIV-1 Quant -users”.
“SIMU HIV-1 Quant Risk Management and Control Report” held by QA.
SIMU HIV-1 Quant IFU.
A5 Medical devices should be designed, manufactured and packaged in such a way that their characteristics and performances during their intended use will not be adversely affected by transport and storage conditions (for example, fluctuations of temperature and humidity) taking account of the instructions and information provided by the manufacturer
A Application of recognized standards:
ISO 18113:2009 In vitro diagnostic medical devices – Information supplied by the manufacturer (labelling) – Parts 1 and 4.
ISO 14971:2007 Medical devices – Application of risk management to medical devices.
ISO 23640:2011 In vitro diagnostic medical devices -- Evaluation of stability of in vitro diagnostic reagents.
CLSI EP25A:2009 Evaluation of Stability of In Vitro Diagnostic Reagents; Approved Guideline.
Stability Studies
Stability studies (Section 7.2 of this dossier).
Risk management policy document “Policy XXX” and risk analysis SOPs “SOP XXX”, “SOP XXXX” held by QM department.
Risk analysis output documents “SIMU
HIV-1 Quant -risk-design”, “SIMU HIV-1
Quant risk - processes”, “SIMU HIV-1
Quant risk-users”.
“SIMU HIV-1 Quant Risk Management and Control Report” held by QA.
SIMU HIV-1 Quant IFU.
A6 All known and foreseeable risks, and any undesirable effects, should be minimized and be acceptable when weighed against the benefits of the intended performance of medical devices during normal conditions of use
A
Application of recognized standards:
GHTF/SG5/N6:2012 Clinical Evidence for IVD medical devices – Key Definitions and Concepts.
GHTF/SG5/N8:2012 Clinical Evidence for IVD Medical Devices - Clinical Performance Studies for In Vitro Diagnostic Medical Devices.
ISO 14971:2007 Medical devices – Application of risk management to medical devices.
Risk analysis output documents “SIMU
HIV-1 Quant -risk-design”, “SIMU HIV-1
Quant risk - processes”.
“SIMU HIV-1 Quant risk-users”.
“SIMU HIV-1 Quant Risk Management and Control Report” held by QA.

WHO Sample product dossier for SIMU HIV-1 QUANT Annex I: Essential Principles Checklist
Draft for public comment 26 October 2016 Page 87 of 140
THE Manufacturing Company SIMU HIV-1 Quant Test Product Codes M123 and M124
Essential principles Applicable/
Not Applicable (A/NA) Method(s) and references used to demonstrate conformity
Reference to supporting controlled documents of Manufacturer
B6 Devices with a diagnostic or measuring function
B6.1 Diagnostic devices and devices with a measuring function, should be designed and manufactured in such a way as to provide sufficient accuracy, precision and stability for their intended purpose of the device, based on appropriate scientific and technical methods. The limits of accuracy should be indicated by the manufacturer.
Application of recognized standards:
EN ISO 14971:2012
EN 13612:2002 (Clause 4.5)
ISO 8601:2004 (Section 4.1).
All verification studies (refer Section 7 of the dossier).
Process validation and software validation report (BXXXXX).
B6.2 The measuring, monitoring or display scale (including colour change and other visual indicators) must be designed and manufactured in line with ergonomic principles, taking account of the intended purpose of the device.
A Application of recognized standards:
EN ISO 14971:2012
EN 62366:2008.
SOPXXXX, SOPXXXX.
B6.3 Wherever possible values expressed numerically should be in commonly accepted, standardized units, and understood by the users of the device.
A Application of recognized standards:
EN ISO 18113-1:2011
EN ISO 18113-2:2011
EN ISO 18113-3:2011.
SIMU HIV-1 Quant IFU
C Essential Principles Applicable to IVD Medical Devices
C1 Chemical, physical and biological properties
C1.1 The IVD medical devices should be designed and manufactured in such a way as to ensure the characteristics and performance referred to in Section A. -Particular attention should be paid to the possibility of impairment of analytical performance due to incompatibility between the materials used and the specimens and/or analyte (measurand) to be detected (such as biological tissues, cells, body fluids and micro-organisms), taking account of its intended purpose
A Application of recognized standards:
ISO 13485:2003 Medical Devices - Quality management systems-Requirements for Regulatory Purposes.
ISO 14971:2007 Medical devices – Application of risk management to medical devices.
ISO 18113:2009 In vitro diagnostic medical devices – Information supplied by the manufacturer (labelling) – Parts 1 and 4.
Verification and Validation Studies- Section 7
All verification -studies (refer Section 7 of this dossier).
Risk analysis output documents “SIMU
HIV-1 Quant -risk-design”, “SIMU HIV-1
Quant risk - processes”.
“SIMU HIV-1 Quant risk-users”.
“SIMU HIV-1 Quant Risk Management and Control Report” held by QA.
SIMU HIV-1 Quant IFU (“Warnings for Users”, ”Specimen Collection, Preparation and Storage”).
C1.2 The IVD medical devices should be designed, manufactured and packaged in such a way as to
A Application of recognized standards:
ISO 14971:2007 Medical devices – Risk analysis output documents “SIMU
HIV-1 Quant -risk-design”, “SIMU HIV-1

WHO Sample product dossier for SIMU HIV-1 QUANT Annex I: Essential Principles Checklist
Draft for public comment 26 October 2016 Page 88 of 140
THE Manufacturing Company SIMU HIV-1 Quant Test Product Codes M123 and M124
Essential principles Applicable/
Not Applicable (A/NA) Method(s) and references used to demonstrate conformity
Reference to supporting controlled documents of Manufacturer
minimize the risk posed by contaminants and residues to the persons involved in the transport, storage and use of the devices and to patients, taking account of the intended purpose of the device. -
Application of risk management to medical devices.
ASTM D4169 – 14.
Quant - processes”, “SIMU HIV-1 Quant risk-users”.
“SIMU HIV-1 Quant Risk Management and Control Report” held by QA.
Packing and Shipping SOP XXX.
SIMU HIV-1 Quant -Robustness Studies (held on-site and Section 7.3).
Package Robustness Studies (held on-site).
C1.3 The IVD medical devices should be designed and manufactured in such a way as to reduce as far as reasonably practicable and appropriate the risks posed by substances that may leach or leak from the IVD medical device. -Special attention should be given to substances which are carcinogenic, mutagenic or toxic to reproduction.
A Application of recognized standards:
ISO 14971:2007 Medical devices – Application of risk management to medical devices.
Risk analysis output documents “SIMU
HIV-1 Quant -risk-design”, “SIMU HIV-1
Quant - processes”, “SIMU HIV-1 Quant risk-users”.
“SIMU HIV-1 Quant Risk Management and Control Report” held by QA.
SIMU HIV-1 Quant Robustness Studies (held on-site).
C1.4 IVD medical devices should be designed and manufactured in such a way as to reduce as far as reasonably practicable and appropriate risks posed by the unintentional ingress or egress of substances into or from the IVD medical device taking into account the device and the nature of the environment in which it is intended to be used
A Application of recognized standards:
ISO 14971:2007 Medical devices – Application of risk management to medical devices.
Verification and Validation studies – Section 7
Risk analysis output documents “SIMU
HIV-1 Quant -risk-design”, “SIMU HIV-1
Quant risk - processes”, “SIMU HIV-1
Quant risk-users”.
“SIMU HIV-1 Quant Risk Management and Control Report” held by QA.
SIMU HIV-1 Quant Cartridge Robustness Studies (held on-site).
C2 Infection and microbial contamination
C2.1 The IVD medical devices and manufacturing processes should be designed in such a way as to eliminate or to reduce as far as reasonably practicable and appropriate the risk of infection to user, professional or lay, or, where applicable, -other person . -The design should:
A Application of recognized standards:
ISO 13485:2003 Medical Devices - Quality management systems-Requirements for Regulatory Purposes
ISO 14971:2007 Medical devices – Application of risk management to medical
All verification studies (refer Section 7 of the dossier).
Risk analysis output documents “SIMU
HIV-1 Quant -risk-design”, “SIMU HIV-1
Quant risk - processes”, “SIMU HIV-1
Quant risk-users”.
WHO Sample product dossier for SIMU HIV-1 QUANT Annex I: Essential Principles Checklist
Draft for public comment 26 October 2016 Page 89 of 140
THE Manufacturing Company SIMU HIV-1 Quant Test Product Codes M123 and M124
Essential principles Applicable/
Not Applicable (A/NA) Method(s) and references used to demonstrate conformity
Reference to supporting controlled documents of Manufacturer
allow easy and safe handling ; and, where necessary: reduce as far as reasonably practicable and appropriate any microbial leakage from the IVD medical device and/or microbial exposure during use; and prevent microbial contamination of the IVD medical device or specimen where applicable, by the user, professional or lay, or other person
devices
EN 13641:2002 Elimination or reduction of risk of infection related to in vitro diagnostic medical devices
Verification and Validation Studies – Section 7
“SIMU HIV-1 Quant Risk Management and Control Report” held by QA.
Materials management requirements and policy kept in R&D (control materials contain potentially infectious components. Policy reflects requirements to source materials that pose the least possible risk of transmission of infection).
C2.2 IVD medical devices labelled either as sterile or as having a special microbiological state should be designed, manufactured and packaged to ensure they remain so when placed on the market and remain so under the transport and storage conditions specified by the manufacturer, until the protective packaging is damaged or opened
NA Not provided in a sterile state.
C2.3 IVD medical devices labelled either as sterile or as having a special microbiological state should have been processed, manufactured and, if applicable, sterilized by appropriate, validated methods.
NA Not provided in a sterile state.
C2.4 IVD medical devices intended to be sterilized should be manufactured in appropriately controlled (e.g. environmental) conditions.
NA Not provided in a sterile state.
C2.5 Packaging tests for non-sterile IVD medical devices should maintain the integrity and cleanliness of the device
A Application of recognized standards:
ISO 13485:2003 Medical Devices - Quality management systems -Requirements for Regulatory Purposes.
ISO 14001 Environmental management tests -- Requirements with guidance for use.
Dossier Sections 6&7.
Robustness studies (results available in R&D Department).
Control Specimen Antimicrobial Effectiveness study (results available in R&D Department).
Environmental Control SOP XXX.
C3 IVD medical devices incorporating materials of biological origin
C3.1 Where IVD medical devices include tissues, cells and A Application of recognized standards: No TSE transmissible agents in the device

WHO Sample product dossier for SIMU HIV-1 QUANT Annex I: Essential Principles Checklist
Draft for public comment 26 October 2016 Page 90 of 140
THE Manufacturing Company SIMU HIV-1 Quant Test Product Codes M123 and M124
Essential principles Applicable/
Not Applicable (A/NA) Method(s) and references used to demonstrate conformity
Reference to supporting controlled documents of Manufacturer
substances originating from animals, the processing, preservation, testing and handling of tissues, cells and substances of animal origin should be carried out so as to provide optimal safety for user, professional or lay, or other person In particular, safety with regard to viruses and other transmissible agents should be addressed by implementation of validated methods of elimination or inactivation in the course of the manufacturing process. -This may not apply to certain IVD medical devices if the activity of the virus and other transmissible agent are integral to the intended purpose of the IVD medical device or when such elimination or inactivation process would compromise the performance of the IVD medical device. National regulations may require that the manufacturer and/or the Regulatory Authority retain information on the geographical origin of the animals
ISO 14971:2007 Medical devices – Application of risk management to medical devices.
EN 13641:2002 Elimination or reduction of risk of infection related to in vitro diagnostic medical devices.
Section 4.2.4 ISO 13485:2003 Medical Devices - Quality management systems -Requirements for Regulatory Purposes.
Verification and Validation Studies.
but Management and Control Report” held by QA.
Materials management requirements and policy kept in R&D.
Policy reflects requirements to source materials that pose the least possible risk of transmission of infection.
Document control policy requires ongoing review of risk posed by scientific methods used to reduce or eliminate potential infectious agents in components for Tests already on the market.
Material specification sheets.
C3.2 Where IVD medical devices include human tissues, cells and substances, the selection of sources, donors and/or substances of human origin, the processing, preservation, testing and handling of tissues, cells and substances of such origin should be carried out so as to provide optimal safety for user, professional or lay, or other person In particular, safety with regard to viruses and other transmissible agents should be addressed by implementation of validated methods of elimination or inactivation in the course of the manufacturing process. –This may not apply to certain IVD medical devices if the activity of the virus and other
A Application of recognized standards:
ISO 14971:2007 Medical devices – Application of risk management to medical devices.
EN 13641:2002 Elimination or reduction of risk of infection related to in vitro diagnostic medical devices.
Section 4.2.4 ISO 13485:2003 Medical Devices - Quality management systems -Requirements for Regulatory Purposes.
Verification and Validation Studies.
The human plasma used for external controls is the only human material used in the kit. -see C3.1 for THE Manufacturing
Company’s policy on such materials.

WHO Sample product dossier for SIMU HIV-1 QUANT Annex I: Essential Principles Checklist
Draft for public comment 26 October 2016 Page 91 of 140
THE Manufacturing Company SIMU HIV-1 Quant Test Product Codes M123 and M124
Essential principles Applicable/
Not Applicable (A/NA) Method(s) and references used to demonstrate conformity
Reference to supporting controlled documents of Manufacturer
transmissible agent are integral to the intended purpose of the IVD medical device or when such elimination or inactivation process would compromise the performance of the IVD medical device
C3.3 Where IVD medical devices include cells and substances of microbial origin, the processing, preservation, testing and handling of cells and substances should be carried out so as to provide optimal safety for user, professional or lay, or other person. - In particular, safety with regard to viruses and other transmissible agents should be addressed by implementation of validated methods of elimination or inactivation in the course of the manufacturing process. -This may not apply to certain IVD medical devices if the activity of the virus and other transmissible agent are integral to the intended purpose of the IVD medical device or when such elimination or inactivation process would compromise the performance of the IVD medical device.
A Application of recognized standards:
ISO 14971:2007 Medical devices – Application of risk management to medical devices
EN 13641:2002 Elimination or reduction of risk of infection related to in vitro diagnostic medical devices
Section 4.2.4 ISO 13485:2003 Medical Devices - Quality management systems -Requirements for Regulatory Purposes.
Verification and Validation Studies.
No such materials in the device but -see C3.1 for THE Manufacturing Company’s policy on such materials.
C4 Environmental properties
C4.1 If the IVD medical device is intended for use in combination with other devices or equipment, the whole combination, including the connection test should not impair the specified performance of the devices. -Any restrictions on use applying to such combinations should be indicated on the label and/or in the IFU
A Application of recognized standards:
ISO 13485:2003 Medical Devices - Quality management systems-Requirements for Regulatory Purposes.
ISO 14971:2097 Medical devices – Application of risk management to medical devices.
Validation Studies Section 7
Risk analysis output documents “SIMU
HIV-1 Quant risk-design”,“SIMU HIV-1
Quant risk - processes”,“SIMU HIV-1
Quant risk-users”.
“SIMU HIV-1 Quant Test Risk Management and Control Report” held by QA.
Design specifications (held on-site).
SIMU HIV-1 Quant Test – IFU, SIMU

WHO Sample product dossier for SIMU HIV-1 QUANT Annex I: Essential Principles Checklist
Draft for public comment 26 October 2016 Page 92 of 140
THE Manufacturing Company SIMU HIV-1 Quant Test Product Codes M123 and M124
Essential principles Applicable/
Not Applicable (A/NA) Method(s) and references used to demonstrate conformity
Reference to supporting controlled documents of Manufacturer
HIV-1 Quant Instrument Users’ Manual.
C4.2 IVD medical devices should be designed and manufactured in such a way as to remove or reduce as far as reasonably practicable and appropriate:
C4.2.1 the risk of injury to user, professional or lay, or other person in connection with their physical and ergonomic features;
A Application of recognized standards:
ISO 13485:2003 Medical Devices - Quality management systems-Requirements for Regulatory Purposes.
ISO 14971:2097 Medical devices – Application of risk management to medical devices.
Validation - Section 7 Studies.
Risk analysis output documents, mostly
related to lancets -“ SIMU HIV-1 Quant -
risk-design”, “SIMU HIV-1 Quant risk -
processes”, “SIMU HIV-1 Quant risk-users”.
“SIMU HIV-1 Quant Risk Management and Control Report” held by QA.
Design specifications (held on-site).
All validation data (refer Section 7.4 of this dossier).
C4.2.2 the risk of use error due to the ergonomic features, human factors and the environment in which the IVD medical device is intended to be used;
A Application of recognized standards:
ISO 13485:2003 Medical Devices - Quality management systems-Requirements for Regulatory Purposes
ISO 14971:2097 Medical devices – Application of risk management to medical devices
ISO 18113:2009 In vitro diagnostic medical devices – Information supplied by the manufacturer (labelling) – Part 4
Validation - Section 7 Studies
Risk analysis output documents “SIMU
HIV-1 Quant -risk-design”, “SIMU HIV-1
Quant risk - processes”, “SIMU HIV-1
Quant risk-users”.
“SIMU HIV-1 Quant Risk Management and Control Report” held by QA.
Design specifications (held on-site).
SIMU HIV-1 Quant IFU.
All validation data (refer to Section 7.4 of this dossier).
C4.2.3 The risks connected with reasonably foreseeable external influences or environmental conditions, such as magnetic fields, external electrical and electromagnetic effects, electrostatic discharge, pressure, humidity, temperature or variations thereof;
A Application of recognized standards:
ISO 13485:2003 Medical Devices - Quality management systems-Requirements for Regulatory Purposes
ISO 14971:2007 Medical devices – Application of risk management to medical devices
Copy of subcontractor’s Declaration of Conformity (Annex IV) to DIRECTIVE 2004/108/EC (held on-site).
Routine audits of subcontractor’s compliance to required specifications, including safety testing.
Risk analysis output documents “SIMU
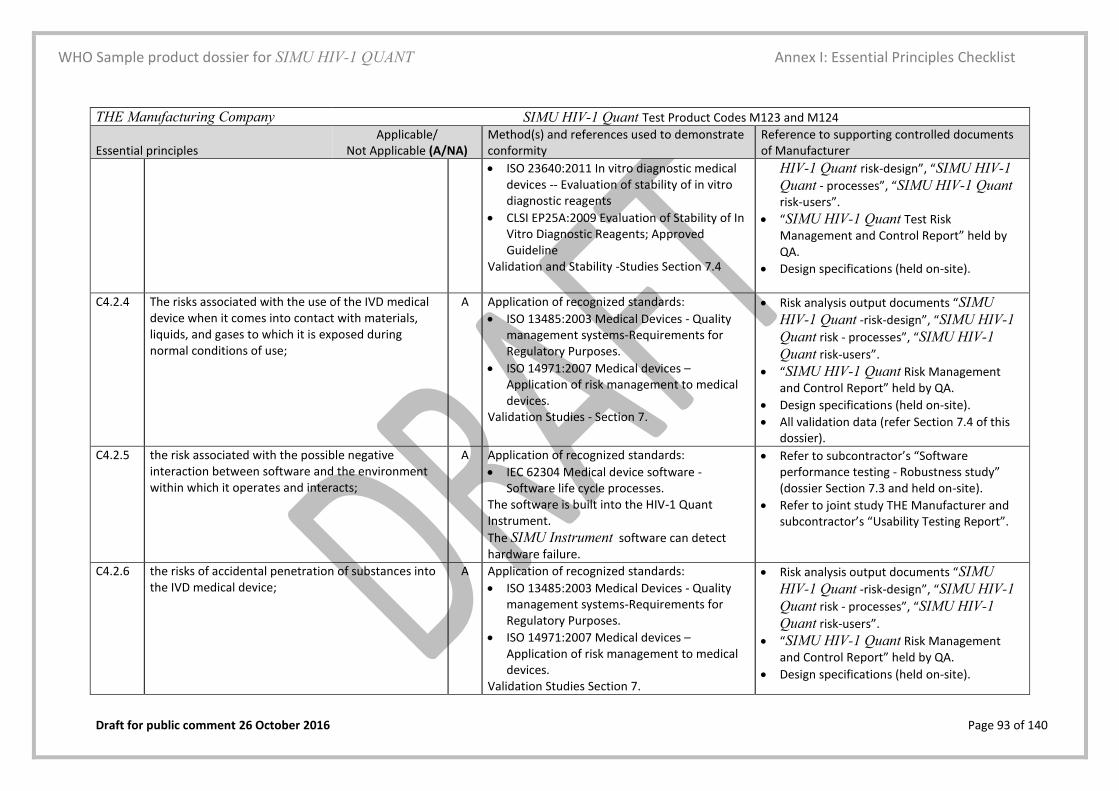
WHO Sample product dossier for SIMU HIV-1 QUANT Annex I: Essential Principles Checklist
Draft for public comment 26 October 2016 Page 93 of 140
THE Manufacturing Company SIMU HIV-1 Quant Test Product Codes M123 and M124
Essential principles Applicable/
Not Applicable (A/NA) Method(s) and references used to demonstrate conformity
Reference to supporting controlled documents of Manufacturer
ISO 23640:2011 In vitro diagnostic medical devices -- Evaluation of stability of in vitro diagnostic reagents
CLSI EP25A:2009 Evaluation of Stability of In Vitro Diagnostic Reagents; Approved Guideline
Validation and Stability -Studies Section 7.4
HIV-1 Quant risk-design”, “SIMU HIV-1
Quant - processes”, “SIMU HIV-1 Quant risk-users”.
“SIMU HIV-1 Quant Test Risk Management and Control Report” held by QA.
Design specifications (held on-site).
C4.2.4 The risks associated with the use of the IVD medical device when it comes into contact with materials, liquids, and gases to which it is exposed during normal conditions of use;
A Application of recognized standards:
ISO 13485:2003 Medical Devices - Quality management systems-Requirements for Regulatory Purposes.
ISO 14971:2007 Medical devices – Application of risk management to medical devices.
Validation Studies - Section 7.
Risk analysis output documents “SIMU
HIV-1 Quant -risk-design”, “SIMU HIV-1
Quant risk - processes”, “SIMU HIV-1
Quant risk-users”.
“SIMU HIV-1 Quant Risk Management and Control Report” held by QA.
Design specifications (held on-site).
All validation data (refer Section 7.4 of this dossier).
C4.2.5 the risk associated with the possible negative interaction between software and the environment within which it operates and interacts;
A Application of recognized standards:
IEC 62304 Medical device software - Software life cycle processes.
The software is built into the HIV-1 Quant Instrument.
The SIMU Instrument software can detect hardware failure.
Refer to subcontractor’s “Software performance testing - Robustness study” (dossier Section 7.3 and held on-site).
Refer to joint study THE Manufacturer and subcontractor’s “Usability Testing Report”.
C4.2.6 the risks of accidental penetration of substances into the IVD medical device;
A Application of recognized standards:
ISO 13485:2003 Medical Devices - Quality management systems-Requirements for Regulatory Purposes.
ISO 14971:2007 Medical devices – Application of risk management to medical devices.
Validation Studies Section 7.
Risk analysis output documents “SIMU
HIV-1 Quant -risk-design”, “SIMU HIV-1
Quant risk - processes”, “SIMU HIV-1
Quant risk-users”.
“SIMU HIV-1 Quant Risk Management and Control Report” held by QA.
Design specifications (held on-site).

WHO Sample product dossier for SIMU HIV-1 QUANT Annex I: Essential Principles Checklist
Draft for public comment 26 October 2016 Page 94 of 140
THE Manufacturing Company SIMU HIV-1 Quant Test Product Codes M123 and M124
Essential principles Applicable/
Not Applicable (A/NA) Method(s) and references used to demonstrate conformity
Reference to supporting controlled documents of Manufacturer
All validation data (refer to Section 7.4 of this dossier).
C4.2.7 the risk of incorrect identification of specimens/samples;
A Application of recognized standards:
ISO 13485:2003 Medical Devices - Quality management systems-Requirements for Regulatory Purposes.
ISO 14971:2007 Medical devices – Application of risk management to medical devices.
ISO 22870:2006 Point-of-care testing (POCT) — Requirements for quality and competence.
Validation Studies Section 7.
For clinical use, ISO 22870 applies.
Risk analysis output documents “SIMU
HIV-1 Quant -risk-design”, “SIMU HIV-1
Quant risk - processes”, “SIMU HIV-1
Quant risk-users”.
“SIMU HIV-1 Quant Risk Management and Control Report” held by QA.
Design specifications (held on-site).
All validation data (refer Section 7.4 of this dossier).
SIMU HIV-1 Quant IFU.
C4.2.8 the risks of reasonably foreseeable interference with other devices such as carry-over between IVD medical devices.
A Application of recognized standards:
ISO 13485:2003 Medical Devices - Quality management systems-Requirements for Regulatory Purposes.
ISO 14971:2007 Medical devices – Application of risk management to medical devices.
ISO 22870:2006 Point-of-care testing (POCT) — Requirements for quality and competence.
Validation Studies Section 7
For clinical use, ISO 22870 applies.
Risk analysis output documents “SIMU
HIV-1 Quant -risk-design”, “SIMU HIV-1
Quant risk - processes”, “SIMU HIV-1
Quant risk-users”.
“SIMU HIV-1 Quant Risk Management and Control Report” held by QA.
Design specifications (held on-site).
All validation data (refer Section 7.4 of this dossier).
SIMU HIV-1 Quant IFU,
C4.3 IVD medical devices should be designed and manufactured in such a way as to minimize the risks of fire or explosion during normal use and in single fault condition. -Particular attention should be paid to IVD medical devices whose intended use includes exposure to or use in association with flammable substances or substances which could cause
A Application of recognized standards:
ISO 13485:2003 Medical Devices - Quality management systems-Requirements for Regulatory Purposes.
Section 7.4.
Copy of subcontractor’s Declaration of Conformity (Annex IV) to DIRECTIVE 2006/95/EC (held on-site).
Routine audits of subcontractor’s compliance to required specifications, including safety testing.

WHO Sample product dossier for SIMU HIV-1 QUANT Annex I: Essential Principles Checklist
Draft for public comment 26 October 2016 Page 95 of 140
THE Manufacturing Company SIMU HIV-1 Quant Test Product Codes M123 and M124
Essential principles Applicable/
Not Applicable (A/NA) Method(s) and references used to demonstrate conformity
Reference to supporting controlled documents of Manufacturer
combustion.
C4.4 IVD medical devices should be designed and manufactured in such a way that adjustment, calibration, and maintenance, where such is necessary to achieve the performances intended, can be done safely
A Application of recognized standards:
ISO 13485:2003 Medical Devices - Quality management systems-Requirements for Regulatory Purposes.
Section 7.4
Refer to SIMU HIV-1 Quant Instrument Test IFU.
SIMU HIV-1 Quant Instrument Test User Manual “Section XYZ Calibration .
C4.5 IVD medical devices should be designed and manufactured in such a way as to facilitate the safe disposal of any waste substances
A Application of recognized standards:
ISO 14001:2004 Environmental management tests. Requirements with guidance for use.
ISO 14971:2007 Medical devices – Application of risk management to medical devices.
Risk analysis output documents “SIMU
HIV-1 Quant -risk-design”, “SIMU HIV-1
Quant risk - processes”, “SIMU HIV-1
Quant risk-users”.
“SIMU HIV-1 Quant Risk Management and Control Report” held by QA.
Design specifications (held on-site).
SIMU HIV-1 Quant IFU.
C5 Performance characteristics
C5.1 IVD medical devices should be designed and manufactured in such a way that the performance characteristics support the intended use, based on appropriate scientific and technical methods. -In particular, where appropriate, the design should address sensitivity, specificity, accuracy which is trueness and precision (repeatability and reproducibility), control of known relevant interference and limits of detection These performance characteristics need to be maintained during the lifetime of the IVD medical device as indicated by the manufacturer.
A Application of recognized standards:
ISO 13485:2003 Medical Devices - Quality management systems-Requirements for Regulatory Purposes.
ISO 23640:2011 In vitro diagnostic medical devices - Evaluation of stability of in vitro diagnostic reagents.
CLSI EP25A:2009 Evaluation of Stability of In Vitro Diagnostic Reagents; Approved Guideline.
EN 13612:2002 Performance evaluation of in vitro diagnostic medical devices.
CLSI EP07-A2 Interference Testing in Clinical Chemistry; Approved Guideline—Second Edition.
CLSI EP14-A2 Evaluation of Matrix Effects;
Design specifications (held on-site).
All validation and verification studies, and stability studies (refer to all of Section 7 of this dossier).
Lot release procedures “SOP QC xxx”.

WHO Sample product dossier for SIMU HIV-1 QUANT Annex I: Essential Principles Checklist
Draft for public comment 26 October 2016 Page 96 of 140
THE Manufacturing Company SIMU HIV-1 Quant Test Product Codes M123 and M124
Essential principles Applicable/
Not Applicable (A/NA) Method(s) and references used to demonstrate conformity
Reference to supporting controlled documents of Manufacturer
Approved Guideline—Second Edition.
FDA Guidance for Industry - Bioanalytical Method Validation.
EN 13975:2003 Sampling procedures used for acceptance testing of in vitro diagnostic medical devices - Statistical aspects.
Section 7 Performance and Stability Studies.
C5.2 Where the performance of devices depends on the use of calibrators and/or control materials, the traceability of values assigned to such calibrators and/or control materials should be assured through available reference measurement procedures and/or available reference materials of a higher order.
A Application of recognized standards:
ISO 15193:2009 In vitro diagnostic medical devices - Measurement of quantities in samples of biological origin - Requirements for content and presentation of reference measurement procedures.
ISO 17511:2003 In vitro diagnostic medical devices - Measurement of quantities in biological samples - Metrological traceability of values assigned to calibrators and control materials.
Refer Records SIMU HIV-1 Quant Sample Processing Control and Traceability Policy (held on-site).
C5.3 Wherever possible values expressed numerically should be in commonly accepted, standardized units, and understood by the users of the device.
A Application of recognized standards:
EN ISO 18113-1:2011
EN ISO 18113-2:2011
EN ISO 18113-3:2011.
C6 Protection against radiation A
C6.1 IVD medical devices should be designed, manufactured and packaged in such a way that exposure of user, professional or lay, or other person to the emitted radiation (intended, unintended, stray or scattered) is reduced as far as reasonably practicable and appropriate.
A Application of recognized standards:
EN 55011:2010
EN 61000-3-2:2009
EN 61000-3-3:2008
IEC CISPR 11:2010
EN 61326-1:2006
EN 61326-2-6:2007
EMC full compliance immunity report ABC123 EMCtech, Inc., EMC radiated and conducted emissions report EFC111222.
C6.2 When IVD medical devices are intended to emit potentially hazardous, visible and/or invisible
A Application of recognized standards:
EN 55011:2010 EMC full compliance immunity report
ABC123 EMCtech, Inc.

WHO Sample product dossier for SIMU HIV-1 QUANT Annex I: Essential Principles Checklist
Draft for public comment 26 October 2016 Page 97 of 140
THE Manufacturing Company SIMU HIV-1 Quant Test Product Codes M123 and M124
Essential principles Applicable/
Not Applicable (A/NA) Method(s) and references used to demonstrate conformity
Reference to supporting controlled documents of Manufacturer
radiation, they should as far as reasonably practicable and appropriate be designed and manufactured in such a way as to ensure that the characteristics and the quantity of radiation emitted can be controlled and/or adjusted; and fitted with visual displays and/or audible warnings of such emissions.
EN 61000-3-2:2009
EN 61000-3-3:2008
IEC CISPR 11:2010
EN 61326-1:2006
EN 61326-2-6:2007
EMC radiated and conducted emissions report EFC111222.
Test Verification Summary Report (10067786) EMCtech, Inc.
C7 IVD medical devices that incorporate software and standalone IVD medical device software
C7.1 For IVD medical devices which incorporate software or for standalone software that are IVD medical devices in themselves, the software must be validated according to the state of the art taking into account the principles of development life cycle, risk management, verification and validation.
A Application of recognized standards:
IEC 62304 Medical device software - Software life cycle processes.
IEC 62366 Medical devices - Application of usability engineering to medical devices standard.
ISO 14971, Medical devices – Application of risk management to medical devices.
The software is built into the SIMU HIV-1
Quant Instrument.
Refer to subcontractor’s “Software Validation and Verification Studies” (held on-site).
Refer to subcontractor’s “Software Risk Management Report” (held on-site).
C8 IVD medical devices connected to, or equipped with, an energy source
A
C8.1 IVD medical devices where the safety of the patient depends on an internal power supply in the IVD medical device should be equipped with a means of determining the state of the power supply.
A Design of the SIMU Instrument includes a screen that provides instructions, allowing confirmation it is turned on. A LED turns on
when the SIMU Instrument is “ON”.
C8.2 IVD medical devices should be designed and manufactured in such a way as to reduce as far as reasonably practicable and appropriate the risks of creating electromagnetic interference which could impair the operation of this or other devices or equipment in the usual environment.
A Application of recognized standards:
IEC 61010-2-101: Safety requirements for electrical equipment for measurement, control and laboratory use - Part 2-101: Particular requirements for in vitro diagnostic (IVD) medical equipment.
Copy of subcontractor’s Declaration of Conformity (Annex IV) to DIRECTIVE 2004/108/EC (held on-site).
C8.3 IVD medical devices should be designed and A Application of recognized standards: Copy of subcontractor’s Declaration of

WHO Sample product dossier for SIMU HIV-1 QUANT Annex I: Essential Principles Checklist
Draft for public comment 26 October 2016 Page 98 of 140
THE Manufacturing Company SIMU HIV-1 Quant Test Product Codes M123 and M124
Essential principles Applicable/
Not Applicable (A/NA) Method(s) and references used to demonstrate conformity
Reference to supporting controlled documents of Manufacturer
manufactured in such a way as to provide an adequate level of intrinsic immunity to electromagnetic disturbance to enable them to operate as intended.
EMC Testing for Medical Devices IEC 60601-1-2:2007 Electromagnetic Compatibility (EMC) Testing.
Conformity (Annex IV) to DIRECTIVE 2004/108/EC (held on-site).
C8.4 IVD medical devices should be designed and manufactured in such a way as to avoid, as far as reasonably practicable, the risk of accidental electric shocks to the user, professional or lay, or other person both during normal use of the device and in the event of a single fault condition in the device, provided the IVD medical device is installed and maintained as indicated by the manufacturer.
A The SIMU Instrument is battery operated (Li-ion rechargeable battery).
Application of recognized standards:
IEC 61010-2-101: Safety requirements for electrical equipment for measurement, control and laboratory use - Part 2-101: Particular requirements for in vitro diagnostic (IVD) medical equipment.
Copy of subcontractor’s Declaration of Conformity (Annex IV) to DIRECTIVE 2004/108/EC (held on-site).
C9 Protection against mechanical and thermal risks
C9.1 IVD medical devices should be designed and manufactured in such a way as to protect the user, professional or lay, or other person against mechanical risks connected with, for example, resistance to movement, instability and moving parts
A The SIMU HIV-1 Quant Instrument does not incorporate moving parts.
The SIMU HIV-1 Quant Instrument and rechargeable batteries are manufactured in accordance with the EC EMC standards.
Copy of subcontractor’s Declaration of Conformity (Annex IV) to DIRECTIVE 2004/108/EC (held on-site).
C9.2 Where there are risks due to the presence of moving parts, risks due to break-up or detachment, or leakage of substances, then appropriate protection means must be incorporated
A Application of recognized standards:
EN 61010-1:2010
IEC 61010-2-081:2009
EN 61010-2-101:2002
EN ISO 12100:2010.
Electrical safety report 14-445, rev 2 (ABS Laboratories, Inc.)
SOP XXXX (Held on-site).
C9.3 IVD medical devices should be designed and manufactured in such a way as to reduce to the lowest practicable level the risks arising from vibration generated by the devices, taking account of technical progress and of the means available for limiting vibrations, particularly at source, unless the vibrations are part of the specified performance.
A Application of recognized standards:
EN 61010-1:2010
IEC 61010-2-081:2009
EN 61010-2-101:2002
EN ISO 12100:2010.
Electrical safety report 14-445, rev 2 (ABS Laboratories, Inc.)
SOP XXXX (Held on-site).
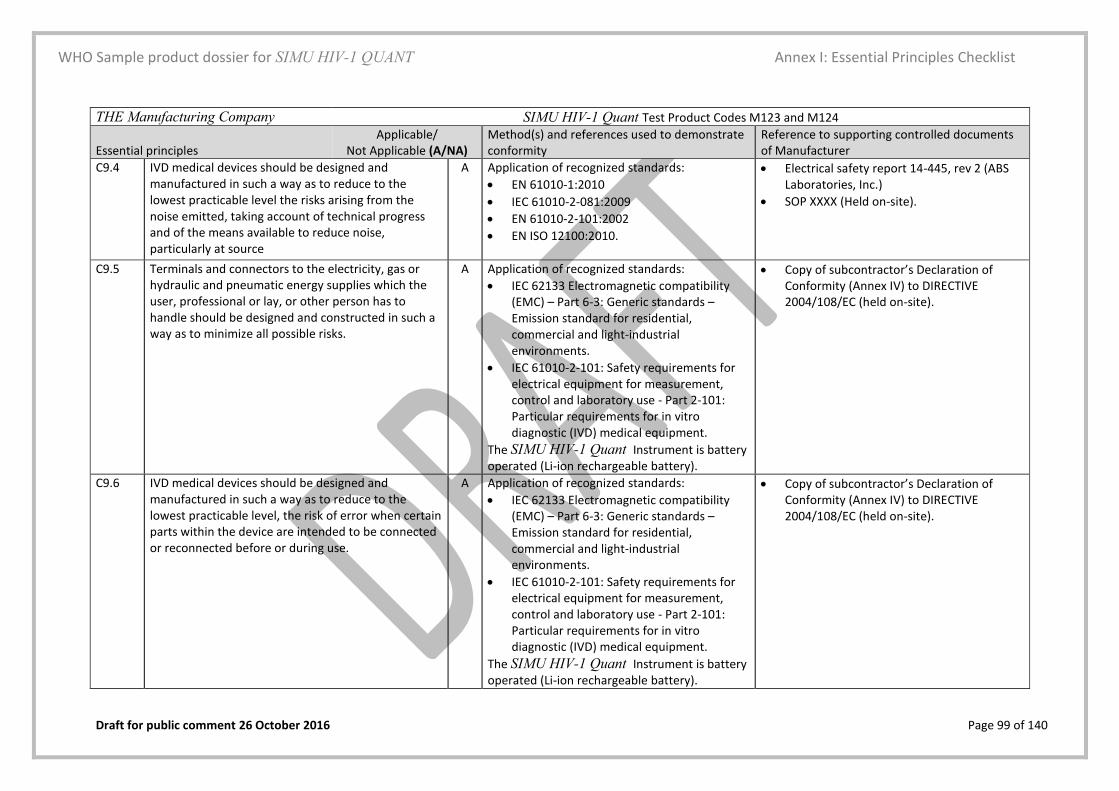
WHO Sample product dossier for SIMU HIV-1 QUANT Annex I: Essential Principles Checklist
Draft for public comment 26 October 2016 Page 99 of 140
THE Manufacturing Company SIMU HIV-1 Quant Test Product Codes M123 and M124
Essential principles Applicable/
Not Applicable (A/NA) Method(s) and references used to demonstrate conformity
Reference to supporting controlled documents of Manufacturer
C9.4 IVD medical devices should be designed and manufactured in such a way as to reduce to the lowest practicable level the risks arising from the noise emitted, taking account of technical progress and of the means available to reduce noise, particularly at source
A Application of recognized standards:
EN 61010-1:2010
IEC 61010-2-081:2009
EN 61010-2-101:2002
EN ISO 12100:2010.
Electrical safety report 14-445, rev 2 (ABS Laboratories, Inc.)
SOP XXXX (Held on-site).
C9.5 Terminals and connectors to the electricity, gas or hydraulic and pneumatic energy supplies which the user, professional or lay, or other person has to handle should be designed and constructed in such a way as to minimize all possible risks.
A Application of recognized standards:
IEC 62133 Electromagnetic compatibility (EMC) – Part 6-3: Generic standards – Emission standard for residential, commercial and light-industrial environments.
IEC 61010-2-101: Safety requirements for electrical equipment for measurement, control and laboratory use - Part 2-101: Particular requirements for in vitro diagnostic (IVD) medical equipment.
The SIMU HIV-1 Quant Instrument is battery operated (Li-ion rechargeable battery).
Copy of subcontractor’s Declaration of Conformity (Annex IV) to DIRECTIVE 2004/108/EC (held on-site).
C9.6 IVD medical devices should be designed and manufactured in such a way as to reduce to the lowest practicable level, the risk of error when certain parts within the device are intended to be connected or reconnected before or during use.
A Application of recognized standards:
IEC 62133 Electromagnetic compatibility (EMC) – Part 6-3: Generic standards – Emission standard for residential, commercial and light-industrial environments.
IEC 61010-2-101: Safety requirements for electrical equipment for measurement, control and laboratory use - Part 2-101: Particular requirements for in vitro diagnostic (IVD) medical equipment.
The SIMU HIV-1 Quant Instrument is battery operated (Li-ion rechargeable battery).
Copy of subcontractor’s Declaration of Conformity (Annex IV) to DIRECTIVE 2004/108/EC (held on-site).

WHO Sample product dossier for SIMU HIV-1 QUANT Annex I: Essential Principles Checklist
Draft for public comment 26 October 2016 Page 100 of 140
THE Manufacturing Company SIMU HIV-1 Quant Test Product Codes M123 and M124
Essential principles Applicable/
Not Applicable (A/NA) Method(s) and references used to demonstrate conformity
Reference to supporting controlled documents of Manufacturer
C9.7 Accessible parts of the IVD medical devices (excluding the parts or areas intended to supply heat or reach given temperatures) and their surroundings should not attain potentially dangerous temperatures under normal use
A See Section C9.3.
C10 Protection against the risks posed by IVD medical devices for self-testing -
NA This device is for professional use only. However a broad range of user skills have been considered in the design.
C11 Labels and Instructions for Use
C11.1 Users should be provided with the information needed to identify the manufacturer, to use the device safely and to ensure the intended performance, taking account of their training and knowledge. -This information should be easily understood
A Application of recognized standards:
ISO 14971:2007 Medical devices – Application of risk management to medical devices.
ISO 18113-1:2011 In vitro diagnostic medical devices - Information supplied by the manufacturer (labelling) - Part 1: Terms, definitions and general requirements.
EN ISO 18113-2:2011 In vitro diagnostic medical devices - Information supplied by the manufacturer (labelling) - Part 2: In vitro diagnostic reagents for professional use.
EN ISO 18113-3:2011 In vitro diagnostic medical devices - Information supplied by the manufacturer (labelling) - Part 3: In vitro diagnostic reagents for professional use.
Risk analysis output documents “SIMU
HIV-1 Quant -risk-design”, “SIMU HIV-1
Quant risk - processes”, “SIMU HIV-1
Quant risk-users”.
“SIMU HIV-1 Quant Test Risk Management and Control Report” held by QA.
SIMU HIV-1 Quant Test IFU, SIMU HIV-
1 Quant Instrument User Manual.
C12 Performance evaluation including analytical performance and, where appropriate, clinical performance
C12.1 For an IVD medical device a performance evaluation should be conducted in accordance with GHTF
A Application of GHTF Guidance:
GHTF/SG5/N6:2012 Clinical Evidence for Performance Evaluation Summary (refer
Section 7 of this dossier).
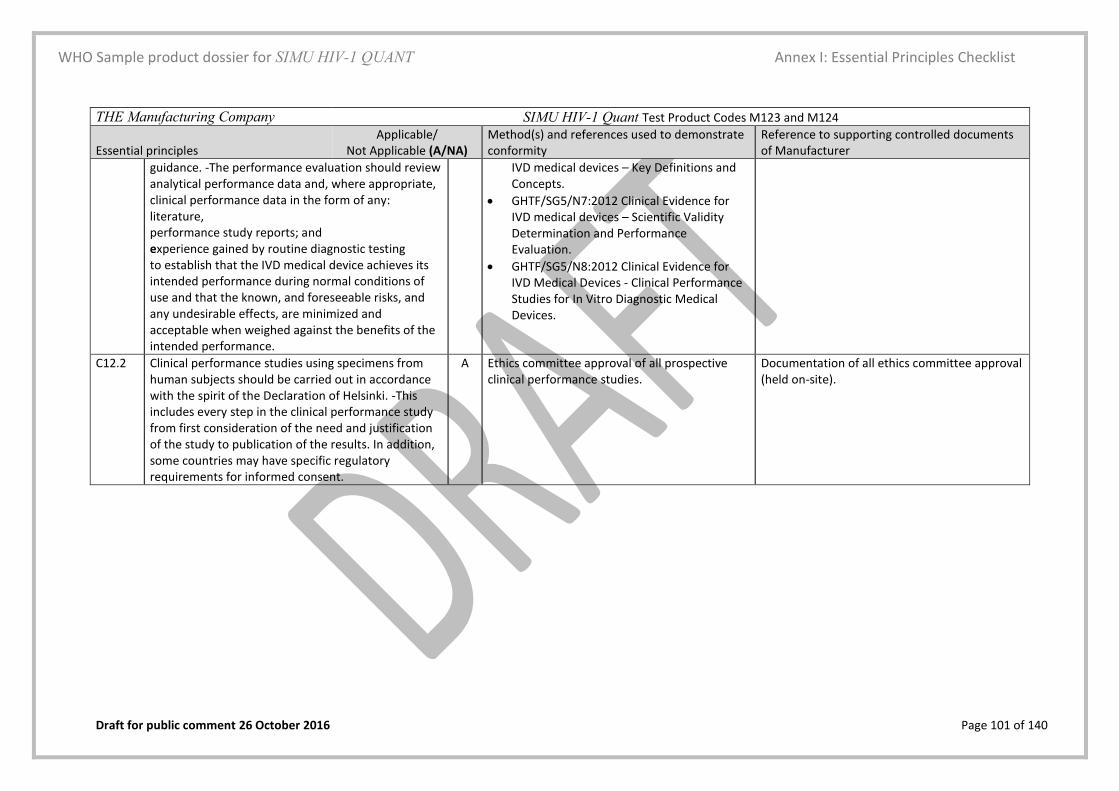
WHO Sample product dossier for SIMU HIV-1 QUANT Annex I: Essential Principles Checklist
Draft for public comment 26 October 2016 Page 101 of 140
THE Manufacturing Company SIMU HIV-1 Quant Test Product Codes M123 and M124
Essential principles Applicable/
Not Applicable (A/NA) Method(s) and references used to demonstrate conformity
Reference to supporting controlled documents of Manufacturer
guidance. -The performance evaluation should review analytical performance data and, where appropriate, clinical performance data in the form of any: literature, performance study reports; and experience gained by routine diagnostic testing to establish that the IVD medical device achieves its intended performance during normal conditions of use and that the known, and foreseeable risks, and any undesirable effects, are minimized and acceptable when weighed against the benefits of the intended performance.
IVD medical devices – Key Definitions and Concepts.
GHTF/SG5/N7:2012 Clinical Evidence for IVD medical devices – Scientific Validity Determination and Performance Evaluation.
GHTF/SG5/N8:2012 Clinical Evidence for IVD Medical Devices - Clinical Performance Studies for In Vitro Diagnostic Medical Devices.
C12.2 Clinical performance studies using specimens from human subjects should be carried out in accordance with the spirit of the Declaration of Helsinki. -This includes every step in the clinical performance study from first consideration of the need and justification of the study to publication of the results. In addition, some countries may have specific regulatory requirements for informed consent.
A Ethics committee approval of all prospective clinical performance studies.
Documentation of all ethics committee approval (held on-site).

WHO Sample product dossier for SIMU HIV-1 QUANT Annex II: Risk Management Policy
Draft for public comment 26 October 2016 Page 102 of 140
Annex II: Risk Management Policy 1459
THE Manufacturing Company DOC XXX 1460
Please Note: The following DOC XXX Risk Management Policy has been prepared for the 1461 purposes of this sample dossier, and is not a complete document. It is to provide an 1462 example of some aspects to be considered in such a document and must reflect the 1463 internal policies of the company. 1464 NOTE: all documents submitted to WHO must be signed and dated. 1465
Risk Management Policy 1466
Document version # 02 Document date: 2012 09 15
Written by:
Signatures Date
Approved by:
Head of QA
Signature Date
Head of manufacturing
Signature Date
Changes from earlier versions:
Version Item modified
Modification
01 2012 01 01 N/A Document created according to SOP x3w
02 2012 09 15 Para. 6 c) Reference to the proposed European in vitro diagnostic regulation added
03 … …
Introduction 1467
1. The company is committed to the effective management of risk at every level: 1468 1.1. Reducing as far as possible any risks associated with any product developed or 1469
manufactured by the company, at every stage of the product life cycle from design to in-use 1470 surveillance. 1471
1.2. Ensuring a safe environment for its employees and customers. 1472 1.3. Training to enable its employees to undertake their work effectively, efficiently and safely. 1473 1.4. Enhancing and protecting the local environment. 1474
2. The company will achieve these objectives 1475 2.1. By ensuring company management fully accepts the need for risk control, which cannot be 1476
balanced against financial profit. 1477 a) The Head of QA will be responsible for risk management and control throughout 1478
the company. 1479 b) The Head of Research & Development will be responsible for risk management 1480
and control in the design of product and process. 1481 c) The Head of Manufacturing will be responsible for risk management and control 1482
related to supply of product, safety in the facility, and respect for the 1483 environment. 1484
d) These responsibilities cannot be passed to subordinates. 1485

WHO Sample product dossier for SIMU HIV-1 QUANT Annex II: Risk Management Policy
Draft for public comment 26 October 2016 Page 103 of 140
2.2. By following the guidelines of the GHTF/SG3/N15R8 and 98/79/EC in a hierarchy of control 1486 methods: 1487
a) Inherent safety by design of any items we develop or manufacture, and their 1488 manufacturing processes, including risks related to security of supply. 1489
b) Protective measures for the user and patient in the device, and for our 1490 employees in the manufacturing processes. 1491
c) Information for safety, such as warnings, as a last resort for risks that cannot be 1492 removed by design of the products or the processes. 1493
2.3. By ensuring that risk management is integrated into the Quality management system of the 1494 company, to include risk management planning and plans for all aspects of our work and 1495 product development. 1496
2.4. By training of all employees in the importance of safe practices in all aspects of their work. 1497 2.5. By training specific employees in the regulations and methods of risk analysis and control. 1498
a) ISO 13485 Medical devices -- Quality management systems -- Requirements for 1499 regulatory purposes EN ISO 14971:2012, Medical Devices. Application of risk 1500 management to medical devices documentation, GHTF and IMDRF guidelines, 1501 FDA and ICH Q9 and Q10 guidance, 1502
b) Standard methods for the tools of risk analysis, including Failure Mode and 1503 Effects Analysis (FMEA), Fault Tree Analysis, Preliminary Risk Analysis, Risk 1504 Ranking and Filtering and Hazard and Operability Studies. 1505
3. The company will ensure consistent application of risk control methods through a set of SOP 1506 directed by this policy. The SOPs will include, but are not limited to: 1507
a) SOP and template for FMEA – who, how, when 1508 b) SOP for training in the use of risk management tools 1509 c) SOP for estimation of degrees of hazard and control measures 1510
This is not intended to be a complete policy document. 1511
1512

WHO Sample product dossier for SIMU HIV-1 QUANT Annex III: Residual Risk Statement
Draft for public comment 26 October 2016 Page 104 of 140
Annex III: Residual Risk Statement 1513
SIMU HIV-1 Quant Test, including SIMU Instrument 1514
THE Manufacturing Company 1515
Following optimisation of the design and performance characteristics by comprehensive testing in-1516 house and in the hands of intended users, as verified when testing populations representative of the 1517
intended patient population, THE Manufacturing Company considers the remaining risks to users 1518 and patients are minimal and acceptable for the SIMU HIV-1 Quant System. The testing is used for 1519 determining the infection status of a user in resource limited settings. Any unlikely, unexpected test 1520 result will be balanced by the major clinical value for patients who would otherwise have limited or 1521 no access to testing. 1522
The remaining risks to manufacture, continuity of supply, and to manufacturing staff are also 1523 minimal and acceptable in view of the training and safety measures in place for our staff and the 1524 supplier management and material testing specified for this product. 1525
Documented evidence to support these statements is filed on-site in the Risk Management and 1526 Control Report of the Risk Management File for this product. This report contains the assessment of 1527 all remaining risks after implementation of risk control measures. This report is subject to 1528 continuous review, based on experience with the device, to ensure the positive status of the benefit 1529 versus risk profile remains unchanged. 1530
Signed: Date: 1531
Head of QA, THE Manufacturing Company: responsible for risk management and control. 1532
Signed: Date: 1533
Head of R&D, THE Manufacturing Company: responsible for design and process risk control 1534
Signed Date: 1535
Head of Manufacturing, THE Manufacturing Company: responsible for risk management and 1536 control related to supply of product, safety in the facility, and respect for the environment. 1537
NOTE: Submitted documents must be signed and dated 1538

WHO Sample product dossier for SIMU HIV-1 QUANT Annex IV: Design Inputs FMEA
Draft for public comment 26 October 2016 Page 105 of 140
Annex IV: Design Inputs FMEA 1539
Design inputs FMEA: SIMU HIV-1 Quant Test
Product codes: M123, M124
FMEA Version: 4 Date: 2013 02 26
THE Manufacturing Company design input FMEA template version 2
Controlling SOP: Design input risk analysis SOP XXXX version 3
Version Control
Revision date Changes
1.0 2010 09 03 Initial, following finalization of customer requirements documentation
2.0 2012 12 20 Inadequate analytical sensitivity for HIV-1 resulted in a change in formulation for the test to increase the sensitivity for HIV-1 (Change A1234 – 2.)
3.0 2013 01 10 Change to the upper limit of detection from 5,000,000 copies/mL to 10,000,000 copies/mL in response to customer input. (Change Y1234 – 4.)
Approval of this version: Head of department or project leader (SOP XXXX)
Department Name Signature Date
Marketing Anne Smith 2013 02 29
Manufacturing Bill Jones 2013 02 26
QA Pete Brown 2013 02 27
R&D Joe White 2013 02 26
Present at FMEA meetings at least two representatives from each department (SOP 0 XXXX)
Department Name Signature Date
Marketing Anne Smith 2013 02 29
Manufacturing Tom Jackson 2013 02 26
QA Jean Roberts 2013 02 27
R&D Dave White 2013 02 26
Marketing Simon Richard 2013 02 29
Manufacturing John Jensens 2013 02 26
QA Sylvie Martin 2013 02 27
R&D Sam Dupont 2013 02 26
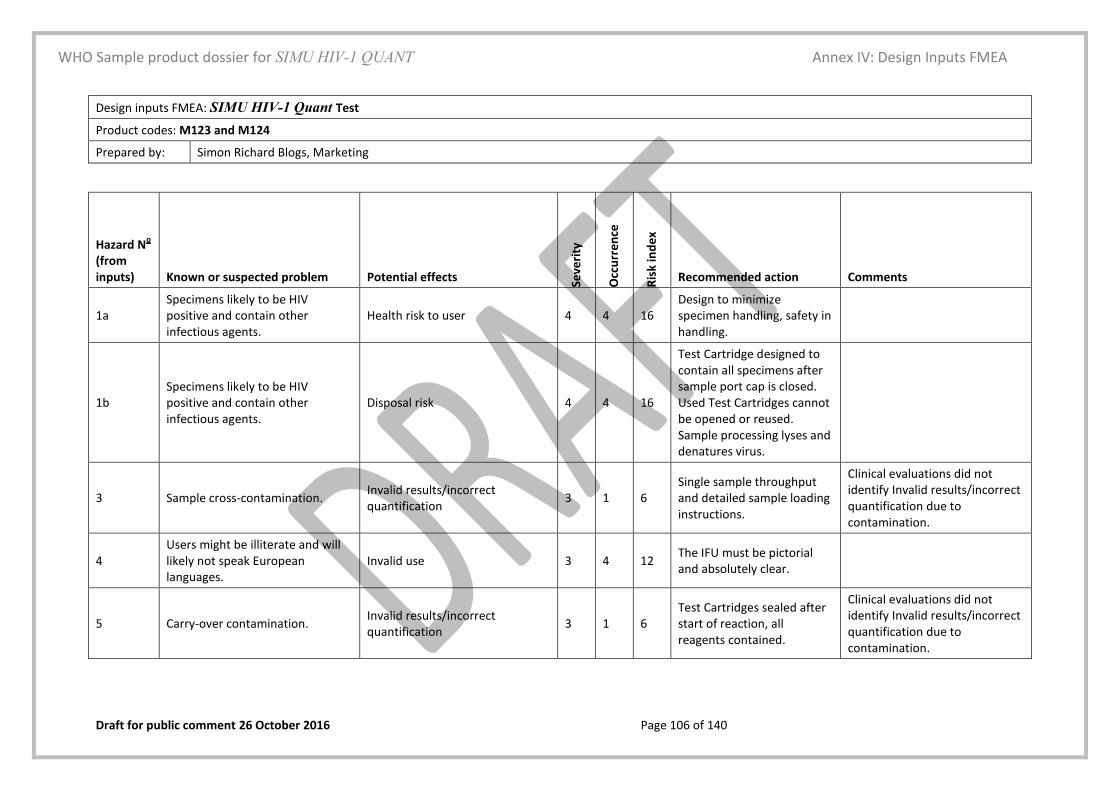
WHO Sample product dossier for SIMU HIV-1 QUANT Annex IV: Design Inputs FMEA
Draft for public comment 26 October 2016 Page 106 of 140
Design inputs FMEA: SIMU HIV-1 Quant Test
Product codes: M123 and M124
Prepared by: Simon Richard Blogs, Marketing
Hazard No
(from inputs) Known or suspected problem Potential effects Se
veri
ty
Occ
urr
en
ce
Ris
k in
de
x
Recommended action Comments
1a Specimens likely to be HIV positive and contain other infectious agents.
Health risk to user 4 4 16 Design to minimize specimen handling, safety in handling.
1b Specimens likely to be HIV positive and contain other infectious agents.
Disposal risk 4 4 16
Test Cartridge designed to contain all specimens after sample port cap is closed. Used Test Cartridges cannot be opened or reused. Sample processing lyses and denatures virus.
3 Sample cross-contamination. Invalid results/incorrect quantification
3 1 6 Single sample throughput and detailed sample loading instructions.
Clinical evaluations did not identify Invalid results/incorrect quantification due to contamination.
4 Users might be illiterate and will likely not speak European languages.
Invalid use 3 4 12 The IFU must be pictorial and absolutely clear.
5 Carry-over contamination. Invalid results/incorrect quantification
3 1 6 Test Cartridges sealed after start of reaction, all reagents contained.
Clinical evaluations did not identify Invalid results/incorrect quantification due to contamination.

WHO Sample product dossier for SIMU HIV-1 QUANT Annex IV: Design Inputs FMEA
Draft for public comment 26 October 2016 Page 107 of 140
Hazard No
(from inputs) Known or suspected problem Potential effects Se
veri
ty
Occ
urr
en
ce
Ris
k in
de
x
Recommended action Comments
6 Specimen amplification inhibitors. Inaccurate quantitation or invalid test results
4 1 4 Specific target capture specimen processing eliminates inhibitors.
Clinical evaluations did not identify invalid test results due to specimen amplification inhibition.
7 High dust levels may affect Instrument operation.
Instrument unreliability 2 3 6 Door for Test Cartridge bay seals before and after insertion of Test Cartridge.
8 Instrument optics misalignment. Unreliable or no results 4 1 4 Error code added to software that will appear on operator screen.
99999 Low air pressure at high altitude may affect system performance.
Unreliable results 4 1 4 Test Cartridge air intake channels modified.
Analytical testing in low pressure environment demonstrated reliable results.
… And so on, working through each section (user, regulatory, management, manufacturing) of the customer input documents (Annex XXX) thinking carefully about all possible hazards. This gives R&D a solid basis for work and usually results in innovation to meet customers’ needs; outputs from this FMEA lead to product requirement document Annex XXX. Update this FMEA if there are any design changes.
The results of FMEA are expressed in semi-quantitative terms (see risk grids and occurrence tables in Extracts from SOP XXX Preparation of design input 1540 FMEA below). The reason for this, rather than the attempted quantitation frequently seen, is provided by Shebl NA, Franklin BD, Barber N, “Is failure mode 1541 and effect analysis reliable?” J. Patient Saf. 5 (2009) 86-94, and Shebl NA et al., “Failure mode and effects analysis outputs: are they valid?” BMC Health 1542 Services Research 12 (2012) 150-160. However it is performed, the actions involved in generating FMEA early in the life of an IVD are invaluable and give 1543 evidence of consideration about the user, patient and design risks. 1544

WHO Sample product dossier for SIMU HIV-1 QUANT Annex IV: Design Inputs FMEA
Draft for public comment 26 October 2016 Page 108 of 140
Please Note: The following SOP XXX “Preparation of design input FMEA” has 1545 been prepared for the purposes of this sample dossier, and is not a complete 1546
document. It is to provide an example of some aspects to be considered in such 1547 a document and must reflect the internal policies of the company. 1548
NOTE: all documents submitted to WHO must be signed and dated. 1549
Extracts from SOP XXX Preparation of design input FMEA 1550
1. Health, safety and the environment 1551
There are no health and safety implications as this is a documentation SOP. 1552
2. Training Requirements 1553
Complete the sheet in Appendix 1 to indicate the trainee has read and understood this procedure 1554
Signatures by trainee and trainer required. 1555
3. Responsibilities 1556
The R&D project leader must: 1557
establish who will participate in the FMEA. This must include at least representation from 1558 the R&D project team, marketing, QA and manufacturing groups 1559
ensure that an FMEA is started at the time the first draft of the input documentation is being 1560 written 1561
ensure the FMEA is updated as required until product launch 1562
ensure that each version is approved as below 1563
after launch FMEA for design changes are addressed in SOP x/yy: the project is no longer an 1564 R&D responsibility. 1565
The heads of department or departmental project leaders must all 1566
review the content and approve each version of the FMEA. 1567
4. Procedure For A Design Input FMEA 1568
Ensure that the customer input responses are available 1569
Customers include: users, patients, distributors, funders, regulators, manufacturing, 1570 management, finance. 1571
Complete the general information relating to the FMEA requested at the top of the form. 1572
Complete each major section and subsection in the FMEA form. 1573
Work through the input requirements point by point and document any potential hazard, 1574 using previous experience, literature information, general knowledge. 1575
Record for each hazard the effect(s) that could be caused. 1576
o there may be more than one effect for each hazard 1577 o assess the severity of each effect 1578 o score the severity using the definitions in the Severity Table in Appendix 3 and 1579
record it the severity column. 1580
Complete the effect severity supporting information in Appendix 4. 1581
and so on 1582
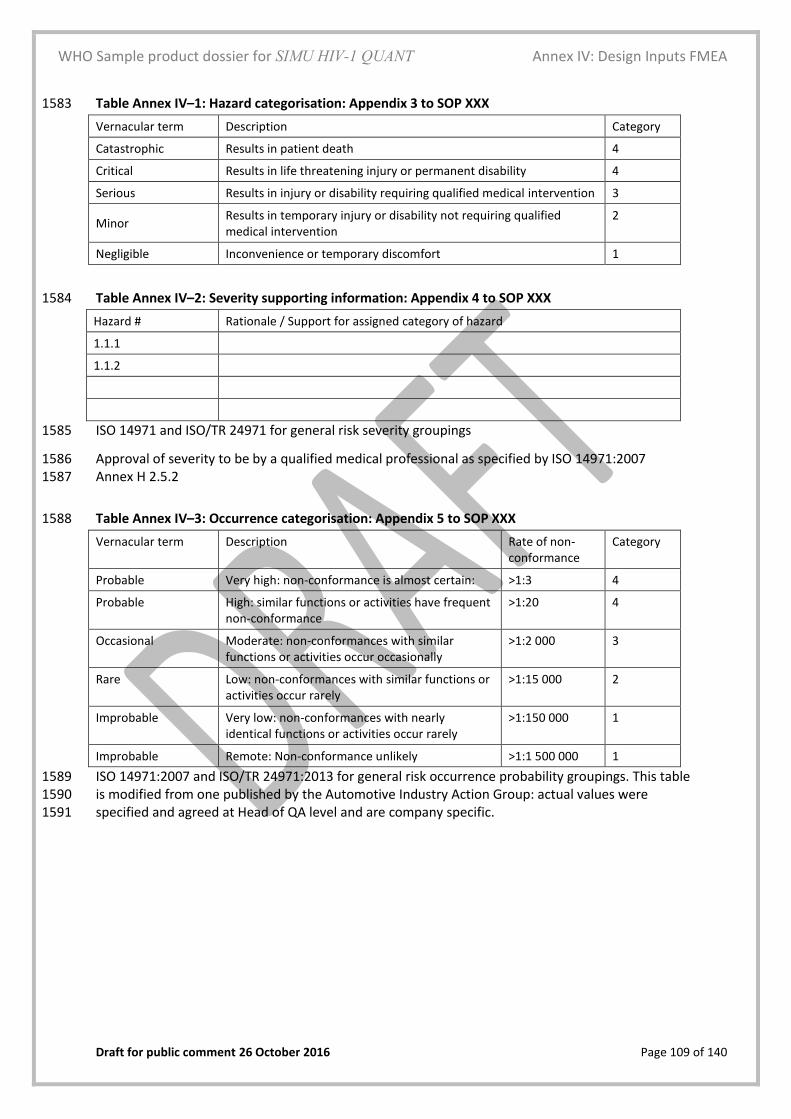
WHO Sample product dossier for SIMU HIV-1 QUANT Annex IV: Design Inputs FMEA
Draft for public comment 26 October 2016 Page 109 of 140
Table Annex IV–1: Hazard categorisation: Appendix 3 to SOP XXX 1583
Vernacular term Description Category
Catastrophic Results in patient death 4
Critical Results in life threatening injury or permanent disability 4
Serious Results in injury or disability requiring qualified medical intervention 3
Minor Results in temporary injury or disability not requiring qualified medical intervention
2
Negligible Inconvenience or temporary discomfort 1
Table Annex IV–2: Severity supporting information: Appendix 4 to SOP XXX 1584
Hazard # Rationale / Support for assigned category of hazard
1.1.1
1.1.2
ISO 14971 and ISO/TR 24971 for general risk severity groupings 1585
Approval of severity to be by a qualified medical professional as specified by ISO 14971:2007 1586 Annex H 2.5.2 1587
Table Annex IV–3: Occurrence categorisation: Appendix 5 to SOP XXX 1588
Vernacular term Description Rate of non-conformance
Category
Probable Very high: non-conformance is almost certain: >1:3 4
Probable High: similar functions or activities have frequent non-conformance
>1:20 4
Occasional Moderate: non-conformances with similar functions or activities occur occasionally
>1:2 000 3
Rare Low: non-conformances with similar functions or activities occur rarely
>1:15 000 2
Improbable Very low: non-conformances with nearly identical functions or activities occur rarely
>1:150 000 1
Improbable Remote: Non-conformance unlikely >1:1 500 000 1
ISO 14971:2007 and ISO/TR 24971:2013 for general risk occurrence probability groupings. This table 1589 is modified from one published by the Automotive Industry Action Group: actual values were 1590 specified and agreed at Head of QA level and are company specific. 1591

WHO Sample product dossier for SIMU HIV-1 QUANT Annex IV: Design Inputs FMEA
Draft for public comment 26 October 2016 Page 110 of 140
Table Annex IV–4: Occurrence supporting information: Appendix 6 to SOP XXX 1592
Hazard # Rationale / Support for assigned category of occurrence
1.1.1
1.1.2
Figure Annex IV–5: Risk Grid: Appendix 7 to SOP XXX 1593
Severity of Effect
4 3 2 1
Critical Serious Minor Negligible
Pro
bab
ility
of
Occ
urr
ence
4 Probable High High Med Low
3 Occasional High Med Low Low
2 Rare Med Low Low Low
1 Improbable Low Low Low Low
All items must be addressed: 1594
High Redesign or improve
Med Redesign and/or improve, or add a control method to reduce the categorisation to Low
Low Review with QA and marketing and/or improve if possible
1595
This risk grid applies to both design input FMEA and user and patient FMEA. It is modified to require 1596 that if a medium risk is found during manufacturing process FMEA that process must be redesigned. 1597
WHO Sample product dossier for SIMU HIV-1 QUANT Annex V: User and Patient FMEA
Draft for public comment 26 October 2016 Page 111 of 140
Annex V: User and Patient FMEA 1598
User and patient FMEA: SIMU HIV-1 Quant Test System
Product codes: M123 and M124
FMEA Version: 4 Date: 2013 02 26
THE Manufacturing Company design input FMEA template version 2
Controlling SOP: Design input risk analysis SOP 00967 version 3
Version Control
Revision date Changes
1.0 2010 09 05 Initial, following finalization of customer requirements documentation including FMEA
2.0 2012 12 20 Inadequate analytical sensitivity for HIV-1 resulted in a change in formulation for the test to increase the sensitivity for HIV-1 (Change A1234 – 2.)
3.0 2013 01 10 Change to the upper limit of detection from 5,000,000 copies/mL to 10,000,000 copies/mL in response to customer input. (Change Y1234 – 4.)
Approval of this version: Departmental Project leader (SOP 00967)
Department Name Signature Date
Marketing Anne Smith 2013 02 27
Manufacturing Bill Jones 2013 02 27
QA Pete Brown 2013 02 27
R&D Joe White 2013 02 27
Present at FMEA meetings at least two representatives from each department (SOP 00967)
Department Name Signature Date
Marketing Anne Smith 2013 02 24
Manufacturing Steve White 2013 02 24
QA Jean Roberts 2013 02 24
R&D Dave White 2013 02 24
Marketing Simon Richard 2013 02 24
Manufacturing Tom Weber 2013 02 24
QA Adrian Becker 2013 02 24
R&D Sharon Dupont 2013 02 24
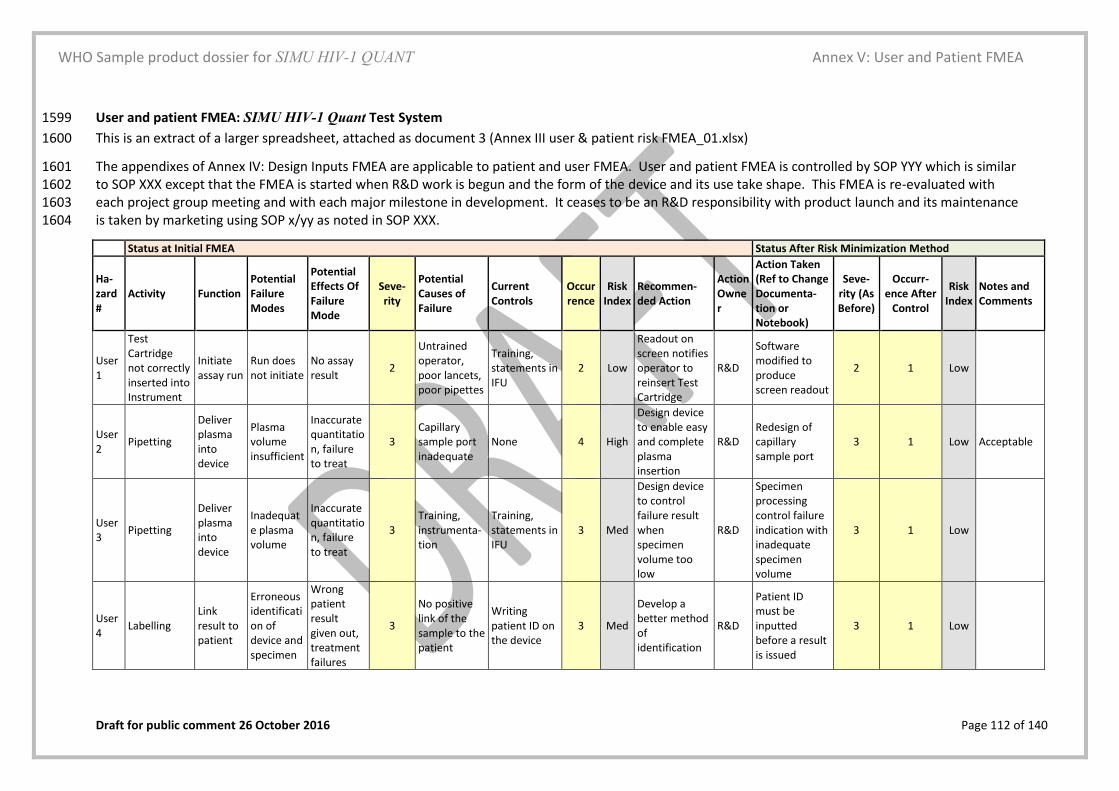
WHO Sample product dossier for SIMU HIV-1 QUANT Annex V: User and Patient FMEA
Draft for public comment 26 October 2016 Page 112 of 140
User and patient FMEA: SIMU HIV-1 Quant Test System 1599
This is an extract of a larger spreadsheet, attached as document 3 (Annex III user & patient risk FMEA_01.xlsx) 1600
The appendixes of Annex IV: Design Inputs FMEA are applicable to patient and user FMEA. User and patient FMEA is controlled by SOP YYY which is similar 1601 to SOP XXX except that the FMEA is started when R&D work is begun and the form of the device and its use take shape. This FMEA is re-evaluated with 1602 each project group meeting and with each major milestone in development. It ceases to be an R&D responsibility with product launch and its maintenance 1603 is taken by marketing using SOP x/yy as noted in SOP XXX. 1604
Status at Initial FMEA Status After Risk Minimization Method
Ha-zard #
Activity Function Potential Failure Modes
Potential Effects Of Failure Mode
Seve-rity
Potential Causes of Failure
Current Controls
Occurrence
Risk Index
Recommen-ded Action
Action Owner
Action Taken (Ref to Change Documenta-tion or Notebook)
Seve-rity (As Before)
Occurr-ence After
Control
Risk Index
Notes and Comments
User 1
Test Cartridge not correctly inserted into Instrument
Initiate assay run
Run does not initiate
No assay result
2
Untrained operator, poor lancets, poor pipettes
Training, statements in IFU
2 Low
Readout on screen notifies operator to reinsert Test Cartridge
R&D
Software modified to produce screen readout
2 1 Low
User 2
Pipetting
Deliver plasma into device
Plasma volume insufficient
Inaccurate quantitation, failure to treat
3 Capillary sample port inadequate
None 4 High
Design device to enable easy and complete plasma insertion
R&D Redesign of capillary sample port
3 1 Low Acceptable
User 3
Pipetting
Deliver plasma into device
Inadequate plasma volume
Inaccurate quantitation, failure to treat
3 Training, instrumenta-tion
Training, statements in IFU
3 Med
Design device to control failure result when specimen volume too low
R&D
Specimen processing control failure indication with inadequate specimen volume
3 1 Low
User 4
Labelling Link result to patient
Erroneous identification of device and specimen
Wrong patient result given out, treatment failures
3
No positive link of the sample to the patient
Writing patient ID on the device
3 Med
Develop a better method of identification
R&D
Patient ID must be inputted before a result is issued
3 1 Low

WHO Sample product dossier for SIMU HIV-1 QUANT
Annex VI: Input Requirements for the HIV-1 Quant Test
Draft for public comment 26 October 2016 Page 113 of 140
Annex VI: Input Requirements for the HIV-1 Quant Test 1605
Customer Requirements 1606
This document follows from the input requirements produced according to SOP-DDD of THE 1607
Manufacturing Company following market research with users, regulators, purchasers, funders. 1608
The notes in black are from the template in SOP-DDD and those added in green are compiled from 1609 customer responses and will form the basis of the numeric, measurable product requirements. 1610
1. Functional Requirements 1611
This section unchanged by user inputs – it is the highest managerial level description of the device 1612
1.1 What is the kit supposed to do? 1613
1.1.1 For the quantitative detection of viral RNA of HIV-1 in the general population from early 1614 to advanced infection. The test is to be used to identify virological failure in patients on 1615 ART, and enable early switching of failing patients to new ART regimens before 1616 accumulation of drug resistance mutations. The test should have a lower limit of 1617 detection of at least 800 copies/mL and an upper limit of detection of at least 1,000,000 1618 copies/mL. 1619
1.1.2 The test can be performed on EDTA plasma. 1620
1.1.3 Reference test: Comparable to established HIV-1 viral load (VL) technology available 1621
1.2 What inputs and what outputs? 1622
1.2.1 Inputs: EDTA plasma 1623
1.2.2 Outputs: printed, and results can be automatically transmitted to central databases 1624 using internet or they can be submitted by SMS. 1625
2. Management Requirements 1626
This section is unchanged – management has already made inputs according to SOP XXX prior to 1627 release of the design to other “customers”. 1628
2.1 Financial including staff allocation 1629
2.1.1 Staffing in R&D to be kept within budget constraints, no new staff to be taken on except 1630 as replacements. 1631
2.1.2 Total costs to be less than $100,000 per staff member per year. 1632
2.2 Administration and control structure 1633
2.2.1 The usual R&D, QA and manufacturing control tests, administered by the HIV team. 1634
2.3 Reporting methods and intervals 1635
2.3.1 By the normal project management group, with reporting to senior management 1636 through the design control committee at a minimum of two monthly intervals. 1637
3. Format 1638
3.1 Interfaces to other systems (measurement system – fluorescence, OD; result recording) 1639

WHO Sample product dossier for SIMU HIV-1 QUANT
Annex VI: Input Requirements for the HIV-1 Quant Test
Draft for public comment 26 October 2016 Page 114 of 140
3.1.1 The Instrument will have a readout screen for results and can interface to other 1640 computers via the internet or SMS. 1641
3.2 Methodology – RDT, EIA, or NAT 1642
3.2.1 NAT 1643
a) NAT to detect HIV-1 RNA. 1644 b) Sample processing and inhibition controls. 1645 c) External controls. 1646
3.3 Presentation of the product 1647
3.3.1 Instrument 1648
a) low throughput, one in one out 1649 b) battery powered with spare battery 1650 c) small footprint 1651 d) fully automated 1652 e) standard WIFI connection, USB port 1653 f) all operations controlled on the Instrument 1654 g) easily portable (backpack or handle) 1655 h) easy to clean/disinfect and fix (minimal moving parts) 1656
1657
3.3.2 IVD 1658
a) Package must be easily opened in the obvious and correct manner and 1659 easily, safely disposable. 1660
b) Package must completely protect the product against the environment 1661 expected in the operational sites. 1662
c) No parts of value for anything other than these contents. 1663
3.3.3 Labelling in accord with ISO 18113-5 and aligned with Jacobs et al., Malaria Journal 13 1664 (2014) 505-514 1665
a) Ensure place for Unique Device Identifier. 1666
4. Operating Requirements 1667
4.1 Expected physical environment – temperature, power availability, equipment, materials 1668 available (distilled water, pipette tips), transport capability, physical stability) 1669
4.1.1 From tertiary hospital settings at district level though to antenatal care clinics in 1670 community settings in resource limited Member States. No power, no equipment, no 1671 water: no external requirements allowable. Transport: limited capabilities. 1672
a) The expected environment can be particularly harsh dusty, not 1673 microbiologically clean. 1674
b) High altitudes and extremes of temperature, humidity and pressure can be 1675 expected. 1676
c) Transport to end-users and storage by users can be very haphazard and 1677 likely not under controlled conditions. 1678
d) Storage by distributors cannot be guaranteed. 1679 e) Timing might be impossible – no timers, watches, phones, therefore the 1680
instrument must be fully automated (minimal operator interventions). 1681

WHO Sample product dossier for SIMU HIV-1 QUANT
Annex VI: Input Requirements for the HIV-1 Quant Test
Draft for public comment 26 October 2016 Page 115 of 140
4.2 Expected technological environment (nature of the working area) 1682
4.2.1 To be used by health care workers in laboratories, antenatal care clinics and tertiary 1683 hospital settings. These workers may have had minimal training and are often nurses 1684 not lab technicians. 1685
a) This will have major consequences for documenting instructions, their 1686 interpretation and subsequent actions by users. 1687
b) Fail safe design – no confusion over role of each interactive design feature 1688 e.g. where to insert Test Cartridge, on/off switches etc. 1689
c) Obtaining plasma from patient blood may be the determining factor for 1690 where the test is used. A plasma separator or centrifuge must be 1691 accessible at the testing site or plasma samples must be obtained from 1692 other facilities. 1693
5. Performance Requirements 1694
5.1 Safety critical 1695
5.1.1 Specimen collection, use and disposal to be safe in completely untrained hands 1696
a) Plasma might well be contaminated with infectious agents other than HIV 1697 so a negative test result does not mean a used device is safe. 1698
c) Disposal facilities might be subject to scavenging, the used device must be 1699 tamperproof. 1700
d) Devices must be single-use; it must be impossible to attempt to reuse a 1701 device. 1702
5.2 Specimen types 1703
5.2.1 EDTA plasma 1704
a) Clear instructions regarding anticoagulants. 1705
5.3 Operating temperatures for the device 1706
5.3.1 From 12–40 °C 1707
a) Humidity and pressure are also an issue: both high and low must be 1708 accommodated. 1709
5.4 Sensitivity, specificity, precision at the cut-off, degree of confidence in these values, definition 1710 of populations 1711
5.4.1 To meet performance characteristics 1712
a) Must be suitable for all major subtypes present in HIV-1 endemic countries 1713 especially M, N, O and circulating recombinant form. 1714
5.5 Precision, trueness and accuracy: if quantitative 1715
a) LOD <800 copies/ml 1716 b) Precision <0.3 log 1717

WHO Sample product dossier for SIMU HIV-1 QUANT
Annex VI: Input Requirements for the HIV-1 Quant Test
Draft for public comment 26 October 2016 Page 116 of 140
5.6 Interfering factors 1718
5.6.1 To meet CLSI expectations and risk evaluated potential infections in HIV prevalent 1719 populations 1720
a) Cross-reactions due to malaria, Human African trypanosomiasis, 1721 Leishmaniasis and Chagas disease are all important. 1722
b) Interference from therapeutics for TB, malaria, HCV. 1723
5.7 Internal QA requirement (“run controls”) 1724
5.7.1 Internal controls: 1725
a) Amplification control (low and high) such as Armored RNA 1726 b) Volume adequacy control (sufficient volume) 1727 c) Sample processing control – housekeeping gene 1728 d) Control for software and hardware failure – display an ‘invalid result’ error 1729
code. 1730
5.8 Stability of device 1731
5.8.1 Transport stability: to meet ANSI packaging expectations and to survive shipment and 1732 storage in resource limited settings 1733
a) Temperatures in transport can reach 55 °C in some lorries and in airports 1734 b) Transport and storage can reach freezing temperature at high altitude and 1735
high latitude 1736 c) Vibrations and movement during transport in trucks 1737 d) Humidity during transport and storage. 1738
5.8.2 Shelf-life after manufacture under stated conditions for Test Cartridges and controls: at 1739 least 1 year following transport to the end-user with subsequent storage at up to 40 °C 1740
a) Note that transport temperatures can be extreme for long periods and 1741 variable 1742
b) Consider humidity and pressure too. 1743
5.8.3 Stability after first use: Not applicable, single-use, self-contained. 1744
5.8.4 Stability on-board: at least 6 hours between specimen addition to Test Cartridge and 1745 Test Cartridge being run in the Instrument. 1746
5.8.5 Life of the Instrument: 3-5 years. Ensure policy for replacement. 1747
5.8.6 Stability of result after test – time allowed prior to reading: resource restricted, timing 1748 might be an issue, long period of stability 1749
a) Printed results/USB export as txt file/wireless connection to mobile device, 1750 datacentre and visible on screen. If printing fails, memory on device will 1751 retain last results, but it is not a long-term results storage device. 1752
5.9 Stability of specimens 1753
5.9.1 Plasma samples 1754
a) Plasma: Usable for 48 hours at 2-8 °C and indefinitely when stored at 1755 <20 °C. 1756

WHO Sample product dossier for SIMU HIV-1 QUANT
Annex VI: Input Requirements for the HIV-1 Quant Test
Draft for public comment 26 October 2016 Page 117 of 140
5.10 Robustness or fault tolerance, “ruggedness”, “guard band studies” 1757
5.10.1 Insensitive to temperature, volume, bubbles (insufficient volume), humidity, variable 1758 pressure, light within wide limits, dust, resistant to mould, back-up battery power 1759 supply, tampering by the user, readable by healthcare workers without laboratory 1760 expertise 1761
5.11 Speed to result and batching of tests 1762
5.11.1 Rapid (less than 60 minutes), no batching. 1763
a) Need to have countdown on the Instrument screen to indicate the time 1764 remaining prior to result, and an alarm when the result is ready 1765
5.11.2 Capacity – tests per technician or Instrument per hour or per day 1766
a) Can run one test per hour. Machine recognizes the test from the barcode 1767 laser printed on the test 1768
b) Instrument available in a modular fashion to allow running of multiple 1769 specimens simultaneously. Instrument not dedicated to one analyte. 1770
6. Manufacturing Requirements 1771
This section will be company specific; guidance cannot be given beyond a suggestion of things to be 1772 considered. Examples for Sections 6.7 - 6.10 will be provided in other sections of the dossier. 1773
6.1 Where to be made 1774
6.1.1 Test to be made in-house, by THE Manufacturing Company, except for moulded 1775 parts 1776
6.1.2 Instrument: to be outsourced and control of key suppliers as per SOP XXX and audits 1777 every 12 months. 1778
6.2 Equipment, staff, space available or to be obtained 1779
6.2.1 Qualification of workspace, instruments, services. 1780
6.3 Cost of production 1781
6.3.1 No more than XXX. 1782
6.4 Cost of materials 1783
6.4.1 No more than YYY % of total cost of production. 1784
6.5 Constraints on materials to be used – safety, storage, environmental 1785
6.5.1 As usual in THE Manufacturing Company, but with attention to safe disposal and 1786 minimal waste in-use. 1787
6.6 Sourcing of raw materials 1788
6.6.1 Supplier qualification and monitoring processes as usual, SOP XXXX; biologicals to be 1789 developed and qualified in-house with the exception of the following: Outsource 1790 oligonucleotides, primers, probes, back-up supplier identified and audited. 1791
6.7 Storage, in-process stability of intermediates, product 1792

WHO Sample product dossier for SIMU HIV-1 QUANT
Annex VI: Input Requirements for the HIV-1 Quant Test
Draft for public comment 26 October 2016 Page 118 of 140
6.7.1 To meet SOP-BBB THE Manufacturing Company. 1793
6.8 Packaging 1794
6.8.1 Labelling to meet international expectations, IFU to be clearly written and validated as 1795 such. 1796
6.9 QA factors – “manufacturing capability” in the statistical sense 1797
6.9.1 Based on known factory capability analysis for other flow devices made by THE 1798
Manufacturing Company. 1799
6.10 In-process and release QA values 1800
6.10.1 To meet SOP-CCC. 1801
7. Maintainability and Support Requirements 1802
7.1 Maintenance requirements 1803
7.1.1 Internal controls to calibrate the machine, warning alert/error alerts on screen when 1804 controls out of specification 1805
7.1.2 Remote connectivity allows for software updates via cloud/GSM network or CD 1806 distribution through local distributor where internet not available 1807
7.1.3 Local mobile representatives and free phone number for product support 1808
7.1.4 No on-site service and maintenance required; broken instruments can be swapped with 1809 replacement instrument 1810
a) Availability of controls: External positive and negative controls included 1811 with the kits. Additional controls can be ordered from the manufacturer. 1812
7.2 Supportability requirements 1813
7.2.1 Will require considerable user feedback in the days after commercial launch to ensure 1814 that validation of the product was accurate and that the device can be used and 1815 interpreted in the expected environments 1816
a) Connectivity to allow automatic feedback of system errors, including user 1817 errors 1818
b) User feedback mechanisms provided as part of the user interface. 1819
7.3 Installation requirements: Regional technician to provide training and installation if required. 1820
8. Cultural and Political Requirements 1821
8.1 Cultural requirements – bovine, porcine materials. 1822
8.2 Political requirements – ensure ability of results from multiple users can feed into a central 1823 database for programme monitoring by e.g. MOH. 1824
9. Regulatory Requirements 1825
9.1 Compliance requirements – ISO, CE , WHO, FDA 1826
9.1.1 Environmental ISO 14001, safety, performance, registration 1827

WHO Sample product dossier for SIMU HIV-1 QUANT
Annex VI: Input Requirements for the HIV-1 Quant Test
Draft for public comment 26 October 2016 Page 119 of 140
a) National use and export regulations not covered by international 1828 regulation. 1829
9.1.2 Risk management ISO 14971:2007 1830
9.1.3 Material safety data sheets 1831
9.1.4 Electrical standards 1832
9.1.5 connectivity standards 1833
9.1.6 waste management standards. 1834
9.2 Clinical trials requirements ISO 14155: 2009 (not IVD but relevant) 1835
9.2.1 Helsinki Declaration. 1836
10. Usability and “Human Factors” Requirements 1837
10.1 Ease of use 1838
10.1.1 Critical for trained users but not laboratory technicians, no precision pipetting. 1839
10.2 Ease of learning – training – staff technical ability 1840
10.2.1 MUST be self-explanatory, must not be able to tamper with the regents or the 1841 Instrument. No result output without controls. 1842
10.3 Accessibility requirements – colour blindness, large font or pictures, 1843
10.3.1 Minimal manipulation – untrained users, requires simple operating dashboard. 1844

WHO Sample product dossier for SIMU HIV-1 QUANT
Annex VII: THE Manufacturing Company Quality Manual
Draft for public comment 26 October 2016 Page 120 of 140
Annex VII: THE Manufacturing Company Quality Manual 1845
THE Manufacturing Company 1846
Quality Manual 1847
1848
THE Manufacturing Company, 1849 987 Somewhere Street, 1850
Somewhere in Europe, EU-1234, 1851 Europe 1852
1853 (20 pages) 1854
Version 5: (08/2015) 1855
1856

WHO Sample product dossier for SIMU HIV-1 QUANT
Annex VII: THE Manufacturing Company Quality Manual
Draft for public comment 26 October 2016 Page 121 of 140
Document
Number
QM 001
Document Title THE Manufacturing Company Quality Manual
NAME TITLE SIGNATURE DATE
Author
Reviewer
Authorizer
1857
Effective Date:
1858
Document Version Updates 1859
Prepared by Version Summary of changes Date
Version 1 First release 01.01.2011
Version 2 …. …..
Version 3 Change of organization 01.01.2013
Version 4 Addition of manufacture of SIMU HIV 12 Qual 01.01.2014
Version 5 Update to reflect inclusion of WHO PMS
requirements
06.08.2015
1860
1861

WHO Sample product dossier for SIMU HIV-1 QUANT
Annex VII: THE Manufacturing Company Quality Manual
Draft for public comment 26 October 2016 Page 122 of 140
Letter of commitment 1862
Manufacturers are encouraged to include a letter of commitment signed by 1863 higher management as part of the Quality Manual 1864
Scope 1865
THE Manufacturing Company Quality System covers the Design, Manufacturing and Supply of in 1866 vitro diagnostic reagents and related systems in all areas and locations. 1867
Company Background 1868
THE Manufacturing Company is a venture capitalist supported ISO 9001-, ISO 13485- certified 1869 manufacturing company founded in 2001 and based in Europe. 1870
Manufacturers should add a description of the field of activities related to IVD, 1871 and other fields. 1872
Manufacturers should add a description of the range of products and 1873 customers. 1874
Quality System normative references 1875
THE Manufacturing Company Quality System meets the requirements of ISO 9001:2015, ISO 1876 13485:2003. The scope of the current valid ISO 13485:2003 Certificate obtained by THE 1877 Manufacturing Company is as follows: 1878
“The development, production, distribution of in-vitro diagnostics and supporting products for 1879 infection diseases”. 1880
In addition, THE Manufacturing Company Quality System meets the quality system requirements of 1881 the In-vitro Diagnostics Medical Devices Directive 98/79/EC (The IVD Medical Devices 1882 Regulations 2002 - UK Statutory Instrument No. 618),the Korea FDA and the China FDA, and the 1883 Indonesia National Agency of Food and Drug Control. 1884
Definitions and Abbreviations 1885
BPIC Inventory Control System 1886
DHF Design History File 1887
DMR Device Master Record 1888
IVDD In-vitro Diagnostics Medical Devices Directive 98/79/EC 1889
OEM Original Equipment Manufacturer 1890
PMS Post-market Surveillance 1891
QA Quality Assurance 1892
SOP Standard Operating Procedure 1893
WHO Sample product dossier for SIMU HIV-1 QUANT
Annex VII: THE Manufacturing Company Quality Manual
Draft for public comment 26 October 2016 Page 123 of 140
1. List of Major Procedures 1894
Procedure Document
number(s)
Document
owner Users
1. Document Management
2. Quality Record Management
3. Management Review Management
4. Management Responsibilities and Authorities
Regulation Management
5. Training Management
6. Infrastructure, Management
7. Equipment management
8. Working Environment Management
9. Design Management
10. Verification Management
11. Supplier Management
12. Purchasing Management
13. Logistics Management
14. Incoming Inspection Management
15. Production Process Management
16. Equipment Management
17. Product Identification and Traceability
Management
18. Monitoring and Measuring Device
Management
19. Product Inspection Management
20. Product Release Management
21. Customer Feedback and Complaint
Management
22. Process Monitoring and Measurement
Management
23. Internal Audit Management
24. Non-conforming Product Management
25. Corrective and Preventive Actions Management
26. Vigilance Management
1895

WHO Sample product dossier for SIMU HIV-1 QUANT
Annex VII: THE Manufacturing Company Quality Manual
Draft for public comment 26 October 2016 Page 124 of 140
2. Quality Management System 1896
General Requirements 1897
The Quality Manual and SOPs describe the processes for the quality 1898
Insert the company quality management system process structure chart here. 1899
Documentation Requirements 1900
General 1901
The Quality Management System Documentation includes the documents shown below: Policy 1902 Documents are generated by individual departments to give more detailed guidance of activities 1903 carried out by or managed by the particular department. 1904
Insert the company quality documents structure chart here. 1905
Quality Policies, Procedures and Processes define quality system requirements. 1906
A List of Documents held under the QMS should be appended here. 1907
Technical Documentation/Technical File/Design Dossiers form the total documentary evidence that 1908 a product conforms to the appropriate requirements of the various regulatory bodies as well as the 1909 Essential Principles. Technical Files and Design Dossiers provide a summary of the Technical 1910 Documentation. Preparation and maintenance of Technical Files and Design Dossiers is described in 1911 SOP XXX. 1912
Quality Manual 1913 The Quality Manual is maintained according to document control procedure. 1914
3. Control of Documents 1915
SOPs are written for systems and processes that may affect product quality or the quality of service. 1916 SOPs are controlled by the central Documentation Group and circulation is usually across 1917 Departments. SOPs are technical or administrative instructions intended to provide standard 1918 methods and/or procedures for conducting scientific tests, processes, manipulations or 1919 administrative procedures. The writing and departmental control of SOPs is described in SOP XXX 1920 & SOP XXX and the authorization, approval and change control of SOPs is described in SOP XXX. 1921 Production Documents are controlled by the central Documentation Group. 1922
Work Instructions/Local Procedures are written for tasks that are only used within a single working 1923 group. The control of Work Instructions and Local Procedures is detailed in SOP XXX. 1924
Technical Specifications are written for raw materials and packaging items which are manufactured 1925 specifically by a supplier for THE Manufacturing Company and/or where specific additional testing 1926 is required by either THE Manufacturing Company or the supplier, and also, if the item is 1927 manufactured from materials of human or animal origin, to ensure compliance with the EP. 1928
Technical Specifications are agreed with and authorized by the supplier together with 1929 representatives of THE Manufacturing Company. Writing, authorization and Control of Technical 1930 Specifications is defined in SOP XXX. 1931

WHO Sample product dossier for SIMU HIV-1 QUANT
Annex VII: THE Manufacturing Company Quality Manual
Draft for public comment 26 October 2016 Page 125 of 140
Product Structure & Routing Specifications are a document set for a reagent or finished pack that 1932 includes the bill of materials for a manufacturing operation and the associated department 1933 instructions. Product Structure & Routing Specifications are used in nitrocellulose striping, coating, 1934 drying, labelling and final packing of product. The relevant SOP describing writing, authorization 1935 and control of these documents is SOP XXX. 1936
Quality Assurance Product Release Specifications detail the testing that is carried out by Product 1937 Quality Assurance team to ensure that the claims for the product detailed in the Instruction for Use 1938 (IFU) are met. Writing and authorization of these documents is detailed in SOP XXX. 1939
Device Master Records (DMRs) reference the location of all documents containing the design 1940 specifications, manufacturing procedures, quality assurance requirements and labelling of THE 1941 Manufacturing Company manufactured and marketed products. The preparation, authorization and 1942 maintenance of DMRs is described in SOP XXX. Manufacturing activities are conducted in 1943 accordance with these documents. 1944
Controlled documents are authorized and issued following the appropriate and correct SOPs. The 1945 distribution and revision of most of the controlled documents is by the central Documentation 1946 Group. The Planning department is responsible for the control and maintenance of Product Structure 1947 and Routing Specifications. 1948
Changes are proposed, processed, authorized and implemented using the procedure known as THE 1949 Manufacturing Company Change Management System. This includes changes to a production 1950 process, production documentation, technical documentation, QA specification, IFU, other printed 1951 material, labels, or other documentation which may affect either the performance claim of the 1952 product, the manufacturing process or product presentation. The change procedure is defined in SOP 1953 XXX. Changes to quality documents (e.g. correction of content-related errors) may be made 1954 according to SOP XXXX-Change. 1955
Certain changes to THE Manufacturing Company products and THE Manufacturing Company 1956 Quality System must be notified to the various Competent Authorities or their recognized 1957 assessment body (for example, the Notified Body under the provisions of the IVD Directive 1958 (98/79/EC) for CE marked products). The process for determining and notifying of ‘significant 1959 changes’ is outlined in SOP XXXX-Change. 1960
In addition, copies of applicable national or international standards/regulations are controlled by 1961 Regulatory Affairs. 1962
Control of Records 1963
Records are maintained to provide evidence of conformity to requirements and of the effective 1964 operation of the quality management system and to ensure the traceability of the operations. Records 1965 are completed according to SOP XXX, and must be legible and readily identified. 1966
The major record types and retention times are defined in SOP XXX. 1967
Quality records are maintained in such a way that they are readily retrievable and unlikely to 1968 deteriorate. 1969
4. Management Responsibility 1970
Management Commitment 1971
The senior management demonstrates its commitment to the development and implementation of the 1972 quality management system requirements by: 1973
Establishing and maintaining a quality system. 1974
WHO Sample product dossier for SIMU HIV-1 QUANT
Annex VII: THE Manufacturing Company Quality Manual
Draft for public comment 26 October 2016 Page 126 of 140
Defining the quality policy and quality objectives (quality plan) and communicating these 1975 throughout the organization. 1976
Ensuring the availability of resources. 1977
Conducting management reviews (SOP XXX). 1978
Customer Focus 1979
The senior managers ensure that customer requirements are defined throughout all stages of product 1980 realization. Performance against these is reviewed to improve customer satisfaction via the 1981 Complaint Handling process (SOP XXX), Customer Requirements (SOP XXX), Design Reviews, 1982 Post-Market Surveillance (SOP XXX) and CAPA Oversight / Management Review (SOP XXX). In 1983 addition THE Manufacturing Company Marketing Executive communicates within the company and 1984 with customers to provide further definition of customer needs. 1985
5. Quality Policy and Objectives 1986
The Quality Policy of THE Manufacturing Company (THE Manufacturing Company) is to improve 1987 health care by providing high quality, safe and effective diagnostic products. 1988
This is achieved 1989
a) Through the processes of design, development, manufacture and distribution carried 1990 out by competent and empowered staff. 1991
b) By striving for continuing quality improvement. 1992
c) By recognizing that the involvement and commitment of all THE Manufacturing 1993 Company personnel is essential. 1994
d) By providing appropriate training and an organizational structure that promotes 1995 empowerment. 1996
e) By assessment of and effective communication with original equipment 1997 manufacturers (OEM) suppliers. 1998
f) By ensuring appropriate investment in technology and systems. 1999
Effectiveness is measured by 2000
g) Customer feedback through the enquiry and complaint system. 2001
h) Product availability. 2002
i) Quality metrics. 2003
j) Employee feedback. 2004
The Quality Policy is communicated to all employees via the annual Quality System training; the 2005 policy is reviewed at management Review Meetings (SOP XXX). 2006
Planning 2007
Quality Objectives 2008 The requirements for quality are defined within the relevant Quality System Procedures, including 2009 Technical Specifications, and QA Product Release Specifications and the Design & Development 2010

WHO Sample product dossier for SIMU HIV-1 QUANT
Annex VII: THE Manufacturing Company Quality Manual
Draft for public comment 26 October 2016 Page 127 of 140
Plan for new products. The activities include identification and acquisition of resources and skills to 2011 achieve the required quality. 2012
Senior managers establish Quality objectives as part of the Quality Plan for each year. Each 2013 employee has a job description that includes the quality accountabilities of the position. In addition, 2014 an employee may have specific quality goals. 2015
Quality Management System Planning 2016 Senior managers review data relating to the quality system requirements and objectives at the six-2017 monthly Management Review and at the monthly meetings of the CAPA Oversight Review Board 2018 and Europe Site Management Team. This includes identification and acquisition of resources and 2019 skills to achieve the Quality Policy. Key metrics of the quality system are reviewed at these 2020 meetings. SOP XXX details the aspects of the quality system that are reviewed. 2021
Responsibility, Authority and Communication 2022
Insert the company organizational flowcharts. 2023
Senior managers are responsible for ensuring that responsibilities and authorities are defined and 2024 communicated. 2025 Senior managers include especially the following responsibilities: 2026
Site Director 2027
This position has overall responsibility for the site. 2028
Functional Managers in R&D, QA and Finance 2029
These positions report into their respective directorates within the company but also report to the 2030 Site Director on a day-to-day operational basis. 2031
The position holder has overall responsibility for THE Manufacturing Company Quality Policy and 2032 for the quality of all products produced by THE Manufacturing Company, and is chairman of the 2033 CAPA Oversight Review Board and Management Review Board. 2034
Operations Manager 2035
The prime accountability of the Operations Manager is to ensure that THE Manufacturing Company 2036 laboratories meet their cost, quality and service level goals and that the facilities and functions are 2037 effectively managed. This is achieved through delegated responsibilities to line reports. 2038
Financial Controller 2039
The principal responsibilities of this position are to provide the executive and line management of 2040 THE Manufacturing Company and other Companies managed from the site with accurate and timely 2041 financial information on which informed business decisions can be made. The position holder is also 2042 responsible for safeguarding the Companies' assets through the maintenance of a system of internal 2043 accounting controls and ensuring that the Companies are at all times compliant with all statutory 2044 financial requirements. 2045
The Financial Controller is responsible to the Site Director for the operation and development of 2046 electronic information systems. This includes the purchase and maintenance of hardware; systems 2047 security; data security; software licensing; and software development. Basic user training is 2048 provided in the use of systems. The position holder is also responsible for the development of 2049 improved business processes, in collaboration with other managers. 2050

WHO Sample product dossier for SIMU HIV-1 QUANT
Annex VII: THE Manufacturing Company Quality Manual
Draft for public comment 26 October 2016 Page 128 of 140
Quality Assurance (QA) Manager 2051
The position holder is responsible for ensuring that THE Manufacturing Company Quality System is 2052 in compliance with regulations and is operating to assure quality in all products and services. 2053
It is the responsibility of this position holder to ensure that THE Manufacturing Company Quality 2054 Systems requirements are communicated to all THE Manufacturing Company managers and 2055 employees and that the actions required to maintain compliance with ISO 9001: 2000, ISO 2056 13485:2003 and the requirements of the In-vitro Diagnostics Medical Devices Directive 98/79/EC 2057 (IVDD) and other relevant regulations are highlighted to appropriate personnel. 2058
The QA Manager is responsible for ensuring that the performance of THE Manufacturing Company 2059 Quality System is reviewed in order to improve its effectiveness. 2060
Product release for sale is the responsibility of the respective Product QA Manager, but may be 2061 delegated to appropriate authorized personnel from within Quality Assurance. 2062
Other responsibilities include the Internal Audit System, training personnel on the Quality Policy 2063 and Quality System, defining criteria for release of products, assessment and release of incoming 2064 raw materials and packaging items, issuing Quality Holds and Recalls due to internal failure and/or 2065 customer complaints and authorization of the IVDD Declaration of Conformity. 2066
The QA Manager reports to the Director, Immunoassay and Clinical Chemistry Quality Assurance. 2067
Regulatory Affairs Manager 2068
The Regulatory Affairs Manager is responsible to the QA Manager for ensuring that Site 2069 Management is aware of legal and regulatory requirements, which affect the company and its 2070 products. 2071
The Regulatory Affairs Manager has defined regulatory responsibilities for labelling and product 2072 safety, including defining the products already on the market to which the IVDD applies, and the 2073 classification of these products according to the various regulations in country of export. The 2074 position holder has sign off authority for non-conformances and all changes to THE Manufacturing 2075 Company products. 2076
The position holder facilitates any changes in processes and procedures that are needed in order to 2077 meet new regulatory requirements. 2078
It is the responsibility of the Regulatory Affairs Manager to ensure the knowledge of national and 2079 international regulatory requirements affecting THE Manufacturing Company’s products is 2080 maintained up-to-date within the Regulatory Affairs department. This includes ensuring the 2081 requirements are met for both existing and new products. Where appropriate, new requirements are 2082 incorporated into THE Manufacturing Company Quality System documentation. 2083
The Regulatory Affairs Manager is responsible for Product Support and has vigilance 2084 responsibilities under the various regulations. 2085
Services QA Manager 2086
The Services QA Manager reports to the QA Manager. The position holder is responsible for the 2087 management of documentation systems and administration of systems such as change management. 2088 The position holder is also responsible for the environmental quality through the use of effective 2089 sampling and monitoring, and the assessment and release of incoming goods using appropriate 2090 sampling and test methods. These responsibilities are met through the management of accountable 2091 teams. 2092

WHO Sample product dossier for SIMU HIV-1 QUANT
Annex VII: THE Manufacturing Company Quality Manual
Draft for public comment 26 October 2016 Page 129 of 140
Product QA Manager 2093
The Product QA Manager reports to the Quality Assurance Manager and is responsible for the 2094 management of the trend analysis process to ensure that production departments carry out effective 2095 trend reviews and that appropriate action is taken. Other responsibilities include ensuring that Test 2096 Method Validation is completed according to plans and approving all product related validation 2097 plans and reports. 2098
Human Resources Manager 2099
Reporting to the Site Director, the Human Resources Manager has responsibility for manpower 2100 resourcing, training, development and education, employee relations and reward. 2101
Manufacturing Services Manager 2102
Reporting to the Site Director, the Manufacturing Services Manager is responsible for all 2103 manufacturing operations. 2104
Supply Chain Manager 2105
Reporting to the Site Director, the Supply Chain Manager is responsible for ensuring that all the 2106 Manufacturing areas know what to manufacture, for planning orders on OEM suppliers and for the 2107 availability of purchased materials and other items to support the forecasted demands. 2108
The Supply Chain Manager is supported by a team of Planners and Purchasing staff. Responsibility 2109 for the different aspects of planning and procurement are allocated to these staff. These 2110 responsibilities extend to include the control and design of Packaging Materials. Plans include both 2111 quantities and due dates, and consist of both short- and mid-term requirements. Short-term 2112 requirements include specific lots with priorities and expediting requirements. Mid-term 2113 requirements focus on ensuring overall capacity requirements for both labour and plant are 2114 communicated in a timely manner to the manufacturing area, Managers and OEM suppliers. 2115 Procurement activities aim to optimise quality and costs while maintaining high service level 2116 performance. 2117
Health, Safety and Environmental Manager 2118
Reporting to the Operations Manager, the Health, Safety & Environmental Manager is responsible 2119 for providing a comprehensive health and safety service to all functions of the company. The 2120 position holder is responsible for ensuring that the demands of relevant national and international 2121 legislation are met, taking into consideration what is reasonably practicable in relation to the level of 2122 risk. 2123
In addition to the above responsibilities the position holder advises on environmental issues that 2124 affect the company's operation. 2125
The position holder is also the Dangerous Goods Safety Advisor for the company and advises on the 2126 shipping of items that are classified as dangerous under the various mandatory codes and 2127 regulations. 2128
Research & Development department Manager 2129
The Research and Development Manager is responsible for all aspects of assay projects within 2130 Research and Development including: 2131
a) The development of new products. 2132
b) The introduction and approval of new and improved materials and processes to be 2133 used for the manufacture of existing products. 2134

WHO Sample product dossier for SIMU HIV-1 QUANT
Annex VII: THE Manufacturing Company Quality Manual
Draft for public comment 26 October 2016 Page 130 of 140
c) Approval of changes to manufacturing processes. 2135
d) The authorization of production documentation relating to these projects. 2136
e) The content of the IFU. 2137
f) Validation of new instruments and generation of validation protocols for running THE 2138 Manufacturing Company products. 2139
g) Management of THE Manufacturing Company Marketing Executive who is the 2140 company’s principal customer contact for developing Customer Requirements and 2141 Marketing Plans. 2142
h) In addition, the position holder has responsibility to the Site Director for the 2143 resolution of technical problems related to the performance of products already on the 2144 market. 2145
Facilities Manager 2146
Reporting to the Operations Manager, the Facilities Manager is responsible for installation, service, 2147 maintenance and repair of all manufacturing machinery, building services, utilities, environmental 2148 control equipment and qualification of equipment, utilities and facilities. 2149
The position holder is also responsible for the calibration policy of quality critical devices and for 2150 providing traceable primary reference devices. The Facilities Manager is responsible for ensuring 2151 that the Company complies with all legal and statutory requirements in respect of electrical, 2152 mechanical and building services. 2153
Internal Communication 2154 The senior managers are responsible for communicating customer, statutory and regulatory 2155 requirements to all members of the company. This is achieved by means of All-Employee 2156 Meetings, Team Briefs, Communication Meetings and annual Quality System training. 2157
Management Review 2158
General 2159 Monitoring and review of THE Manufacturing Company Quality System is performed by the 2160 Management Review Board (SOP XXX), which includes the Site Director and senior managers 2161 from Operations, R&D and Quality. 2162
At least twice per calendar year the Management Review Board reviews the adequacy and 2163 effectiveness of THE Manufacturing Company Quality System, opportunities for improvement and 2164 the need for changes to the quality management system, including the Quality Policy and Quality 2165 objectives. 2166
Review Input 2167 Inputs to Management Review include review of internal audits, Complaint Issues, review of 2168 process and product conformance, Post-Market Surveillance Reviews, issues from Corrective and 2169 Preventive Action, Supplier issues, review of training and Regulatory issues and other changes that 2170 could affect the Quality System. 2171
Review Output 2172 Outputs of the Management Review Board are documented, including summaries of data evaluation, 2173 analyses, and recommendations and actions relating to the effectiveness of the Quality System and 2174 its processes. 2175
WHO Sample product dossier for SIMU HIV-1 QUANT
Annex VII: THE Manufacturing Company Quality Manual
Draft for public comment 26 October 2016 Page 131 of 140
6. Resource Management 2176
Provision of Resource 2177
THE Manufacturing Company identifies resources required through the budgeting process and 2178 inputs from the senior managers to maintain the Quality System and to ensure Customer and 2179 Regulatory requirements are met. 2180
Human Resources 2181
General 2182 All staff have a job description, describing the primary objective of the position, major 2183 accountabilities, supervisory responsibilities, education and background experience required for the 2184 job and an outline training plan (SOP XXX). 2185
Competence, Awareness and Training 2186 It is a policy of THE Manufacturing Company that all personnel take responsibility for the quality of 2187 their work. 2188
All staff will receive the necessary training to enable them to perform their job effectively and safely 2189 (SOP XXX). This training will be both formal and informal and will be provided by the Human 2190 Resources Department, external trainers or local area supervisors as appropriate. 2191
It is the responsibility of individual managers to assess whether their staff require re-training 2192 following changes to procedures. 2193
Direct job-related training will be supplemented by educational and developmental opportunities in 2194 line with the business needs. 2195
Job-related, functional training of all critical operating tasks will be provided by a formal system in 2196 accordance with SOP XXX. This system also covers other non-critical operating tasks, which are of 2197 a functional nature. 2198
Training on the processes and procedures is completed by reading SOPs and signing off on the 2199 Electronic Training Management System. If competency assessment is required this is documented 2200 by means of a Training Assessment Form as described in SOP 5/265. 2201
All departments identify individual training needs on an annual basis via the performance appraisal 2202 process and these are consolidated by the Human Resources Department into a company training 2203 plan. 2204
Annual Quality System training is given to all employees and includes consideration of how they 2205 can contribute to the achievement of the quality objectives. 2206
Infrastructure and Work Environment 2207
Systems have been established to provide an infrastructure to ensure conformity to product 2208 requirements. Where special arrangements are required e.g. Category III working, these are defined 2209 in specific SOPs and training is given. 2210
Equipment is qualified and is maintained to ensure continuing process capability. Monitoring the 2211 requirement for maintenance is carried out either locally according to the SOP for the equipment 2212 concerned or by the Engineering Department. 2213
Environmental monitoring is carried out, according to SOP XXX, to ensure that product 2214 performance is not impacted by the environment. 2215
WHO Sample product dossier for SIMU HIV-1 QUANT
Annex VII: THE Manufacturing Company Quality Manual
Draft for public comment 26 October 2016 Page 132 of 140
Facility cleaning is defined by the Cleaning Contract specification for Contract Cleaners, and 2216 cleaning of work surfaces by SOP XXX. The monitoring of facility cleaning is described in SOP 2217 XXX. 2218
Clothing procedures for staff working in manufacturing areas are detailed in SOP XXX. 2219
7. Product Realization 2220
Planning of Product Realization 2221
Stock and work in progress is considered before determining the net requirements. Firm orders for 2222 finished goods are raised on the system and become the driver for the manufacture of lower local 2223 components, bulks and the purchase of raw materials. Purchase orders are also placed for OEM 2224 products. 2225
The Design Control Procedures and responsibilities of personnel within the system are described in 2226 SOP XXX. Projects are approved and design goals set within the budgeting process. Products are 2227 then developed according to a procedure that ensures the product has been designed to meet the 2228 design inputs. A Design History File (DHF) is compiled during the development of a product, and is 2229 updated throughout the life cycle of the product. The methods of how a product is manufactured and 2230 released for sale are detailed in SOPs for the processes used. 2231
Customer-related processes 2232
Determination of requirements related to the product 2233 A Customer Requirements Document (CRD) and Product Requirements Document (PRD) are 2234 written for new or redeveloped products (SOP XXX). The CRD defines the physical and 2235 performance requirements of a device based on input from internal and external customers. The 2236 PRD translates customer requirements into a technical product description that is measurable and 2237 verifiable. These include statutory and regulatory requirements. 2238
Determination of Requirements Related to the Product 2239 The ongoing review of requirements to ensure customer needs are met is through the Product 2240 Coordination Group, chaired by THE Manufacturing Company Marketing Executive and through 2241 annual Post-Market Surveillance Reviews (SOP XXX). 2242
At each Design Review, and for changes made during the product life cycle, the appropriate 2243 requirements of the IVDD will be considered. Risk Analysis (User, Patient) is carried out according 2244 to SOP XXX. 2245
Customer Communication 2246 Customers are provided with information relating to the products through THE Manufacturing 2247 Company Marketing Executive and the organization. Information on products is also communicated 2248 to customers through Product Information Letters, Technical Bulletins, letters to customers in 2249 response to complaints informing the customer of the outcomes of complaint investigations, and 2250 communication to the customer where product requirements have not been met. 2251
Design and Development 2252
Design and Development Planning 2253 The design and development activities fall into the key project stages listed below. These are 2254 defined in the Design and Development Plan (SOP X/XXX). At each stage there may be several 2255 organizational functions involved. 2256
During the course of the project there will be regular reviews of the development activities against 2257 Design Input requirements. 2258
WHO Sample product dossier for SIMU HIV-1 QUANT
Annex VII: THE Manufacturing Company Quality Manual
Draft for public comment 26 October 2016 Page 133 of 140
The Design and Development Template is developed into the project plan by the Project Manager 2259 and is circulated to Project Group Members. As the project progresses, the plan is updated/revised 2260 as necessary and re-issued. Progress with the relevant parts of the Design and Development Plan are 2261 noted as the project progresses. 2262
During the initial planning stage, appropriate and adequate resource is assigned to each activity and 2263 is reviewed during the course of the project. 2264
Having received the approval to develop a product, the Project Group is the controlling body for a 2265 project. Regular meetings (normally monthly) are held, chaired and minuted by a Project Manager; 2266 copies of these minutes are distributed to Project Group members and appropriate members of the 2267 Site Management Team. Review of progress against the project plan is made by this group. 2268
Detailed issues are addressed outside the Project Group at R&D Technical meetings and Operations 2269 meetings. Technical Meetings run by R&D are normally held monthly and include members from 2270 operations. Minutes of these meetings form part of the DHF and are circulated appropriately. The 2271 functional leader for a specific area will report back to the next project on resulting issues. 2272
Design and Development Inputs 2273 The Design Inputs are defined according to SOP XXX. The product requirements (SOP XXX) are 2274 documented and agreed by Marketing and R&D. The needs of the user and patient are considered 2275 during this process. 2276
A summary of the regulatory requirements relevant to the product in the countries into which it will 2277 be sold is prepared according to SOP XXX. 2278
The conformity assessment route to be followed will be determined according to the risk class that 2279 the product falls into (Refer GHTF/SG1/N045:2008 Principles of IVD Medical Devices 2280 Classification). Where a product may fall into several classes according to the risk classification 2281 rules, the conformity assessment route must be determined by the highest risk class that may apply. 2282
Products are then designed and developed to meet their documented specifications with regular 2283 reviews of the product against the Design Inputs. 2284
Current statutory safety requirements are addressed by performing Control of substances hazardous 2285 to health (COSHH) assessments, providing Material Safety Data Sheets and in the "Warnings & 2286 Precautions" section in the package inserts. Each of these activities is covered by the Design and 2287 Development Plan or project minutes. 2288
Design and Development Outputs 2289 The design output is routinely monitored through Design Review, and Technical and Project Group 2290 meetings to ensure that it meets the requirement of the Design Inputs. 2291
Design outputs in the form of documentation and data such as Process Operating Instructions 2292 (POIs), QA Specifications, SOPs and stability reports, is reviewed before release. 2293
Design and Development Review 2294 An independent review of the design output against the Design Input requirements takes place at 2295 various stages during product development, including design input, design verification, and design 2296 validation. 2297
For products that will be CE marked under the IVD Directive, Product QA cannot release the first 2298 batch for sale until the Declaration of Conformity is signed. 2299
Design and Development Verification 2300 During the development process R&D undertake evaluations using materials of clinical origin and if 2301 available appropriate external standards to ensure that the product meets the Product Specification 2302 (SOP XXX). A Design Verification review is performed according to SOP XXX. 2303

WHO Sample product dossier for SIMU HIV-1 QUANT
Annex VII: THE Manufacturing Company Quality Manual
Draft for public comment 26 October 2016 Page 134 of 140
Design and Development Validation 2304 Design validation is carried out by undertaking Performance Evaluation Trials under the direction of 2305 the Project Manager. The new product is evaluated against the CRD using clinical samples (SOP 2306 XXX). Results are analysed and assessed by R&D team. Details of how the evaluation is planned 2307 and carried out can be found in SOP XXX. 2308
For products classified by the IVDD as Annex II List A, the requirements of the Common Technical 2309 Specifications are met as part of the design validation. 2310
Control of Design and Development Changes 2311 During the development phase of a new product, any changes that are proposed are reviewed, 2312 agreed by all signatories, and incorporated into the Product Specification. 2313
The Design Change Control Procedure is defined in SOP XXX. 2314
Once design validation has been undertaken, changes are managed through a controlled procedure 2315 for the review, approval and implementation of change, detailed in SOP XXX. 2316
The correct transfer of product design into POIs is checked by R&D and Manufacturing personnel 2317 during the Pre-Production Phase (SOP XXX). 2318
Process validation is a key part of design and development in order to ensure quality critical 2319 processes are sufficiently well defined and controlled. 2320
The DHF (SOP XXX) is completed throughout the design phase. It is a compilation of records that 2321 describe the design history of the finished device and includes the Design and Development Plan, 2322 the Product Specification and the dossier from each review meeting. 2323
Purchasing 2324
Purchasing Process 2325 Materials that form part of the manufacturing process for THE Manufacturing Company products 2326 are identified by means of an item coding system. Ad hoc purchases for R&D purposes, facilities 2327 maintenance and administration are dealt with separately. Where possible THE Manufacturing 2328 Company item codes are linked to supplier catalogue codes and both are stated on purchase orders. 2329 Where this combination is not possible, Technical Specifications are agreed with suppliers and 2330 referenced to THE Manufacturing Company item code. These specifications define the dimensional 2331 and quality requirements together with any other parameters critical to the performance and 2332 acceptability of the item being purchased. 2333
Where a finished product is purchased from an OEM supplier, the process for evaluation and 2334 approval of the product is defined in SOP XXX. 2335
Suppliers are selected on the basis of ability to supply the required items or services at the right 2336 quality in the most timely and economic manner. 2337
Suppliers are the subject of a rating/approval scheme (SOP XXX) that assesses either the suitability 2338 to supply (for a new supplier) or the effectiveness of supply (for existing suppliers). Ratings are 2339 regularly reviewed and updated and the results of the review process determine the priority of the 2340 requirement to audit. 2341
Suppliers are approved following assessment of their Quality System (SOP XXX). A list of 2342 Approved Suppliers is maintained. 2343
Purchasing Information 2344 Requirements are communicated to suppliers by means of authorized Purchase Orders. Purchasing 2345 documents are controlled by Standard Operating Procedures. 2346

WHO Sample product dossier for SIMU HIV-1 QUANT
Annex VII: THE Manufacturing Company Quality Manual
Draft for public comment 26 October 2016 Page 135 of 140
Purchase Orders will contain references, as appropriate, to THE Manufacturing Company item 2347 codes, Supplier Catalogue Codes, agreed Technical or Purchasing Specifications or legal 2348 agreements between THE Manufacturing Company and the Supplier. If necessary, copies of 2349 specifications, drawings etc. will be referenced in the Purchase Order document and that document 2350 will be annotated accordingly. Review and authorization of Purchase Orders is carried out as 2351 described in SOP XXX. Purchase orders are quality records and are retained for traceability 2352 purposes. 2353
Verification of Purchased Product 2354 Inspection and verification of incoming goods, including raw materials, packaging components and 2355 instrumentation is carried out according to documented procedures (SOP XXX). 2356
Testing is only performed at THE Manufacturing Company when quality critical material is 2357 involved and it is not possible to be sure that adequate controls are exercised at source. If 2358 components are required urgently for production, there are systems to allow production to progress 2359 before testing has been completed, but the final product cannot be released before the testing has 2360 been satisfactorily completed. 2361
Production and Service Provision 2362
Control of Production and Service Provision 2363 Manufacturing processes are controlled in accordance with POIs or Production Specifications. Some 2364 processes, such as filling, are monitored and controlled using statistical process control techniques 2365 throughout the operation. 2366
Product structure and routing information to support the production of products is defined by 2367 Product Structure & Routing Specifications that are controlled by the Planning Section. 2368
Products are manufactured using software, facilities, utilities, equipment, materials and processes 2369 that have undergone appropriate validation (SOP XXX). 2370
Transfer of manufacture of critical reagents between sites is detailed in SOP XXX. 2371
All materials from incoming goods through intermediate manufacturing stages to final product are 2372 handled in accordance with written, approved specifications and Standard Operating Procedures 2373 (SOPs XXX). 2374
The intention of THE Manufacturing Company is that premises are, as far as possible, suited to their 2375 intended use, being secure, of adequate size and design to allow product security by minimizing the 2376 risk of mix-up occurring. Safety, ease of cleaning and adequate pest control receives high priority. 2377
Procedures are followed to maintain labelling integrity and to prevent labelling errors. 2378
The packing process, including the information printed by THE Manufacturing Company on the 2379 labels and packaging, is controlled by Product Structure & Routing specifications. 2380
Final product can only be released for distribution if it has been approved for issue by Product QA. 2381
Product packaging and shipping containers are designed to protect the products from damage during 2382 normal conditions of storage, handling and distribution. 2383
Records relating to the identification and quantity of product shipped and the identity of the 2384 consignee are retained. 2385
Product installation and Servicing Activities are not undertaken by THE Manufacturing Company. 2386
Validation of Processes for Production and Service Provision 2387 The approach to validation is defined in SOP XXX. 2388
WHO Sample product dossier for SIMU HIV-1 QUANT
Annex VII: THE Manufacturing Company Quality Manual
Draft for public comment 26 October 2016 Page 136 of 140
Equipment qualification is performed to ensure that equipment used in the manufacture of quality 2389 critical processes has been designed to meet the needs of that function and will operate consistently 2390 to meet the needs of the intended process (SOP XXX). 2391
Process validation is performed to establish a high level of confidence that quality critical processes 2392 are sufficiently well controlled so as to ensure a consistently high level of quality in terms of 2393 efficacy and product stability (SOP XXX). 2394
Test Method Validation is performed to establish that the performance characteristics of the method 2395 meet the requirements of the intended use of the test (SOP XXX). 2396
Software validation is completed according to SOP XXX. 2397
Identification and Traceability 2398 Materials used as part of the manufacturing process for products are identified by a part code and 2399 the manufacturer's lot number or THE Manufacturing Company allocated batch number. All 2400 intermediate products and bulks are identified by unique THE Manufacturing Company lot numbers 2401 assigned by Manufacturing. 2402
Final components and kits are labelled with the component or kit product code together with 2403 individual batch numbers. The Batch Histories that are maintained by the Quality Assurance 2404 department can be used to trace the history of the product or component batch to the Production 2405 Records kept by Manufacturing. 2406
Material/component/product status is computer driven and under the control of Purchasing Quality 2407 Services/Product QA. 2408
Inspection and testing activities that verify the specified requirements for that product are met are 2409 defined in the relevant Quality and System Procedure, DMR and quality control specifications. 2410
In-Process inspection and testing of THE Manufacturing Company products is carried out by 2411 manufacturing personnel and quality control personnel following procedures described in 2412 Production and QC Specifications. Quality control personnel also perform in-process inspection and 2413 testing following QC Specifications. 2414
Depending on the results of the inspection and testing, products may be passed, referred or rejected. 2415
Final inspection, and testing where appropriate, of THE Manufacturing Company products is carried 2416 out by Quality control personnel following QC Specifications. 2417
Product release for sale is the responsibility of the respective Product QA Manager, but may be 2418 delegated to appropriate authorized personnel from within Quality Assurance. 2419
For CE marked products classified as Annex II List A products, the Product QA Manager is 2420 responsible for ensuring that the verification of manufactured product by the Notified Body is 2421 received before the batch is released for sale (SOP XXX). 2422
Production records containing the results of inspection and testing by Manufacturing and quality 2423 control personnel are held within the Manufacturing area before archiving. 2424
Batch Histories (Secondary Manufacturing records and Quality control testing, batch release by QA 2425 of each product batch) are held accessible as original hard copy for up to 3 months beyond the 2426 expiry of the product. Beyond that point QA records are archived in the form of microfiche. The 2427 personnel responsible for the release of product are defined in appropriate SOPs and indicated on 2428 the Batch Histories. 2429
Preservation of Product 2430 Storage areas preclude adverse environmental effects on stored materials and allow orderly stock 2431 control by providing effective separation of different materials and materials of different status. 2432

WHO Sample product dossier for SIMU HIV-1 QUANT
Annex VII: THE Manufacturing Company Quality Manual
Draft for public comment 26 October 2016 Page 137 of 140
Appropriate facilities are available for the storage and handling of materials that are potentially 2433 infectious. 2434
Control of Monitoring and Measuring Devices 2435
All quality influencing equipment is subject to regular calibration and is traceable to national 2436 standards. 2437
The management of routine calibration work will, for the most part, be the responsibility of the 2438 Facilities Manager. This responsibility will be shared by responsible personnel in each area who 2439 ensure that only properly calibrated equipment is in routine use. 2440
The frequency with which equipment is calibrated and the tolerances that are allowed before 2441 adjustments are made will be as recommended in manufacturer's user guides and/or service 2442 manuals. In deciding on the optimum retest cycle and adjustment limits the Engineering Department 2443 will take account of the reliability and criticality of the relevant equipment. 2444
Calibration records will be kept adjacent to the relevant equipment in the various operating areas 2445 and/or centrally by the Engineering Department as appropriate. 2446
Implementation of this policy is controlled in SOP XXX. 2447
Where products contain calibrators or quantified controls, traceability to an independent standard 2448 where available is ensured through design input and the QA Release Specifications. 2449
8. Measurement, Analysis and Improvement 2450
General 2451
Procedures have been designed / developed and implemented to monitor 2452
If customers’ requirements for products and service delivery have been met. 2453 The effectiveness of the Quality Management System. 2454 The ongoing improvement of the Quality Management System. 2455
Monitoring and Measurement 2456
Customer Satisfaction 2457 Procedures have been developed and implemented that define the process for the determination of 2458 product and service quality levels. 2459
End-user satisfaction is monitored by the distributors in country. The distributors provide feedback 2460 through the complaint management system. A review is held annually for each product (SOP XXX) 2461 as part of Post-Market Surveillance. 2462
Internal Audit 2463 Internal audits are coordinated by the Audit Co-ordinator within QA and are performed by trained 2464 internal auditors from a range of departments. Audits are performed by personnel independent of 2465 the process or area being audited. 2466
Audits are performed of specified activities (e.g. Hold/Corrective Action, Control of Change, 2467 Calibration) and of specific areas, to cover all relevant aspects of IS0 9001, ISO 13485 and the 2468 IVDD. The procedure followed is described in SOP XXX. 2469
The progress with corrective actions arising from internal audits involves visits to the appropriate 2470 area. Failure to meet agreed dates is highlighted to the Corrective and Preventive Action (CAPA) 2471 Oversight Review Board. 2472
WHO Sample product dossier for SIMU HIV-1 QUANT
Annex VII: THE Manufacturing Company Quality Manual
Draft for public comment 26 October 2016 Page 138 of 140
Monitoring and Measurement of Processes and Product 2473 Processes have been designed and implemented to ensure that the data on records relating to the 2474 Quality Management System and product quality are gathered, evaluated and analysed for trends. 2475 This trend data is then used to implement corrective and/or preventive actions. 2476
In-process materials are sampled, inspected and tested by trained personnel in accordance to 2477 documented, standardized and validated test procedures. Requirements are stated in the applicable 2478 testing procedures and sampling plans. 2479
Material conformance is based on test and/or inspection results and work order review. Disposition 2480 is assigned by trained personnel. Materials are held until all tests and inspections have been 2481 completed and approved. Any non-conforming in-process materials are controlled, segregated where 2482 practicable, and the non-conformity documented. 2483
Final sampling, testing, inspection and disposition of product are done by trained quality control 2484 personnel in accordance to documented, standardized and validated test procedures. Requirements 2485 are stated in the applicable testing procedures and referenced sampling plans. 2486
Final product test records that certifying that all required tests have been satisfactorily completed are 2487 required prior to disposition by authorized personnel. 2488
Appropriate statistical techniques are used for packaging materials inspection, and in-process 2489 inspection and testing during manufacture. These are defined in SOPs XXX. 2490
Where appropriate, sampling methods are reviewed in the light of non-conformances in product or 2491 processes or other appropriate information. 2492
Assessment of product performance for Design Verification is performed using techniques that are 2493 the accepted norm in the IVD industry. 2494
Product trends are reviewed according to SOP XXX. 2495
Product is released to distribution upon final approval or, in cases where regulatory requirements 2496 have to be met, upon regulatory approval or consent (e.g. in case of notifications). The stability of 2497 products is monitored after release for sale (SOP XXX). 2498
Post-market surveillance and measurement 2499
A post-market surveillance and reporting system (SOP XXX and Recall SOP XX) is followed that is 2500 aligned with the responsibilities outlined in the WHO document Post-Market Surveillance (PMS) of in 2501 vitro diagnostics. 2502
The aim of the system is to ensure that non-conformities, changes and complaints are identified and 2503 examined, including a risk analysis (as per figure 2 pg 14 of PMS guidance) and that necessary 2504 action is taken. 2505
Any customer complaints are to be documented by the team member in charge of complaints 2506 reporting, as per SOP XXX. 2507
It is the responsibility of the Safety Officer to perform a risk analysis. The responsibilities of all staff 2508 involved in PMS are outlined in SOP XXX. 2509
It is the responsibility of the QM manager to ensure that an analysis of risks and causes and 2510 initiation of a CAPA is undertaken as per SOP XXX. The timeframe for the investigation depends 2511 on the severity of the adverse event as defined by Table 4 pg 25 of PMS guidance. 2512
Recalls 2513 Recalls are measures to avert the risks to customers due to products that have deviated from the 2514 intended product specifications. Product recalls involve the following actions 2515
WHO Sample product dossier for SIMU HIV-1 QUANT
Annex VII: THE Manufacturing Company Quality Manual
Draft for public comment 26 October 2016 Page 139 of 140
Notification to the relevant competent authority prior to a recall (if appropriate). 2516
Notification of customers and users. 2517
Returns. 2518
Destruction. 2519
Revision of IFUs. 2520
The recalls procedure is described in SOP XXX. THE Manufacturing Company follows: 2521
MEDDEV 2 12-1 rev. 8 Vigilance, European Commission guidelines on a 2522
medical devices vigilance system. 2523
GHTF/SG2/N54R8:2006 Medical Devices Post-Market Surveillance: Global 2524
Guidance for Adverse Event Reporting for Medical Devices. 2525
GHTF/SG2/N008R4:1999 Guidance on How to Handle Information Concerning 2526
Vigilance Reporting Related to Medical Devices. 2527
GHTF/SG2/N57R8:2006 Medical Devices Post-Market Surveillance: Content of 2528
Field Safety Notices. 2529
Control of Non-conforming Product 2530
The control of non-conforming raw material or components and non-conforming final product is the 2531 responsibility of appropriate Quality Assurance personnel. The procedures followed are described in 2532 SOP XXX. The procedure for quarantining non-conforming material is detailed in SOP XXX. 2533
Appropriate corrective action following a product failure after release is initiated and coordinated by 2534 the Quality Assurance Manager. The procedures followed are described in SOP XXX. 2535
The review and disposition of non-conforming product is carried out according to SOP XXX. 2536
If product is reworked, the specific requirements of SOP XXX are followed. It is the responsibility 2537 of the Regulatory Affairs Manager or agreed deputy to ensure regulatory requirements are met 2538 before authorizing a non-conformance report. The procedure for the complete rejection of product 2539 and subsequent disposal is described in SOP XXX. 2540
Analysis of Data 2541
Procedures have been implemented to collect and analyse appropriate data. Data is evaluated for the 2542 reviews by Site Management. 2543
Improvement 2544
PMS and related activities to meet the requirements of the IVDD (Annex III (5), Annex IV (3.1) or 2545 Annex VII (3.1)) is carried out according to SOP XXX for ALL products, regardless of the regulatory 2546 version. 2547
Continual Improvement 2548 Data on trends in non-conformances related to the Quality Management System, suppliers, product 2549 and processes are input to the Management Review Meeting to determine that appropriate corrective 2550 / preventive action is taken. 2551
Corrective Action 2552 Customer complaints are received, investigated, reported, and replied to by the Product Support 2553 Group. SOP XXX describes the procedures followed and includes the requirements for vigilance 2554 reporting under the various regulations. 2555
The investigation of the cause of non-conforming product following customer complaints is detailed 2556 in SOP XXX 2557

WHO Sample product dossier for SIMU HIV-1 QUANT
Annex VII: THE Manufacturing Company Quality Manual
Draft for public comment 26 October 2016 Page 140 of 140
The procedure for investigations and defining of Corrective Action following a non-conformance or 2558 a Customer Complaint is detailed in SOP XXX. Subsequent to implementation of that Corrective 2559 Action Plan, the effectiveness of the action is reviewed. 2560
The following SOPs outline the activities associated with corrective action and preventive actions: 2561
Control of non-conforming products (SOP XXX) 2562
Control of non-conforming incoming goods (SOP XXX) 2563
Control of key suppliers (SOP XXX) 2564
Customer complaints (SOP XXX) 2565
Internal audits (SOP XXX) 2566
Risk management policy (SOP XXX) 2567
Preventive Action 2568 A variety of sources of information are reviewed in order to detect, analyse and eliminate potential 2569 causes of non-conformities. The information reviewed includes Non-Conformance reports, 2570 Investigations and Corrective Actions reports from internal and external audits, change notes, trend 2571 analysis, and complaints. 2572
Preventive action is progressed or monitored by the Management Review Board (SOP XXX). 2573 2574
Control of Key Suppliers 2575 Suppliers are subject to an approval evaluation before being included in the approved list of 2576 suppliers. The suppliers of quality related products and key components (as identified by the risk 2577 analysis) are subject to yearly evaluation (using the rating scale defined in SOP XXX). The 2578 evaluation is organized by the purchasing team and overseen by the quality management 2579 department. Yearly evaluation is conducted on a risk-based analysis and frequency may be 2580 decreased as set out in SOP XXX. Suppliers are evaluated on a number of different indicators: 2581
Quality management 2582
Commercial aspects and pricing 2583
Service 2584
Ability to meet deadlines and quantity requirements. 2585
Internal feedback is also used for continual yearly evaluations. 2586
--------------------------------------END OF DOCUMENT---------------------------------------------------





























