Embed Size (px)
Citation preview

White Paper
AuthorsDr. R. D. McDowall R. D. McDowall Limited, UK
P. A. Smith Agilent Technologies, Inc.
IntroductionUS Pharmacopeia (USP) general chapter <1058> on Analytical Instrument qualification (AIQ) was first implemented in 2008 and remained unchanged for nine years. During 2017, the USP implemented two updates to <1058>. These updates have a significant impact on AIQ, and as the only major pharmacopeia with a chapter dedicated to AIQ, changes to USP <1058> are of global significance.
To help regulated laboratories fully comply with 2017 <1058> requirements, Agilent has produced four White Papers with compliance consultant Bob McDowall, who has been closely involved with the development of <1058>. The series includes:
1. What Has Changed with the 2017 Version of USP <1058>?1
2. How to Comply with the 2017 Version of USP <1058>2
3. The Role of Analytical Instrument Qualification in Data Integrity with the 2017Version of USP <1058>3
4. What Does Performance Qualification Really Mean with the 2017 Version ofUSP <1058>?4
The changes implemented in the 2017 version of the general chapter5 were discussed in the first White Paper of this series: What has Changed with the New Version of USP <1058>?1. In this White Paper, we will look at the impact of these changes on the least understood phase of the 4Qs model: Performance Qualification (PQ).
What Does Performance Qualification Really Mean?

2
Evolution of the 4Qs model: impact of 2017 <1058>The 2008 and 2017 versions of USP <1058> both contain the 4Qs model for AIQ and software validation (for example, DQ, IQ, OQ, and PQ stages). When the need to define a User Requirements Specification (URS) and clarification of the different roles that the OQ and PQ stages have in AIQ are considered, the life cycle model shown in Figure 1 is produced.
This demonstrates the relationship between the instrument qualification stages, and shows that the instrument testing life cycle can be considered as a V model between the DQ, IQ, and OQ stages (see Figure 1).
A key differentiator from the 2017 version of <1058> is that the PQ stage satisfies two different requirements in the AIQ life cycle:
• Verify that the instrument is suitable for use under conditions of use.
• Demonstrate the continued suitability of the instrument (consistent performance).
The first requirement is satisfied through the inclusion of PQ testing during the initial instrument qualification/release and subsequent instrument qualification testing (for example, periodic and for cause OQ/PQ testing). While the second requirement is fundamental to successful implementation of 2017 <1058>, users must define PQ test plans that include periodic PQ testing in between periodic and for-cause qualification. PQ testing should no longer be considered as running an analytical method on the system. Therefore, even after release for operational use, the PQ phase extends throughout the use of the instrument. This is highlighted with the circle in Figure 1.
Historically, when AIQ was first implemented, following application of FDA guidance for manufacturing process validation6 to laboratory instruments, it was typically performed on an instrument before its initial “release for use” (for example, AIQ was perceived as a one-off activity). It is now understood that AIQ is a dynamic process that must be performed throughout the instrument lifetime of use. One of the significant changes in 2017 <1058> is highlighting the dynamic relationship between how an instrument is used and how it is tested (for instance, URS and OQ/PQ testing).
What does USP <1058> say about PQ?Let us start this discussion by looking at the specific wording of the 2017 version of USP <1058>, which defines PQ5 as:
"PQ is the documented collection of activities necessary to demonstrate that an instrument consistently performs according to the specifications defined by the user, and is appropriate for the intended use."
This definition is not the same as the 2008 USP <1058> PQ definition, as there is now alignment of the PQ with the laboratory requirements documented in the instrument URS. One of the problems associated with PQ is that few people know what it means. For example, most analytical scientists associate PQ with System Suitability Tests (SSTs) for chromatography instruments.
The reason it is incorrect to define PQ as an SST is that AIQ is instrument-specific and SSTs are method-specific:
"The PQ verifies the fitness for purpose of the instrument under actual conditions of use. After IQ and OQ have been performed, the instrument’s continued suitability for its intended use is demonstrated through continued PQ"5.
The fundamental question many laboratories have for chromatography instruments and 2017 <1058> is this: are SSTs alone sufficient for a PQ?
The answer is in the next paragraph of USP <1058>:
"The user must define the PQ plans, including test procedures, acceptance criteria, and frequency. Preventive maintenance plans and documentation of repairs and other changes are also a necessary part of the overall instrument qualification."
Figure 1. The 4Qs model as a V model.
Use outside existing qualification limits ormajor instrument upgrade
Regular or move or major upgrade
OQverifies
URS
Instrumentretirement
Initialqualification
Ongoingrequalification
Retirementand removal
Riskassessment
Designqualification
(DQ)
Installationqualification
(IQ)
Operationalqualification
(OQ)
Performancequalification
(PQ)
Laboratory userrequirementsspecification

3
Table 1 shows the most common approaches to satisfying PQ testing requirements for HPLC instruments.
Similar approaches to the three options in the table have been used for all chromatography instruments. Each of these choices has apparent advantages and disadvantages. For example, the single largest disadvantage of option 1 is the risk that this approach may be rejected during an audit, while option 3 requires a large number of resources to achieve. Use of a holistic PQ method (option 2) has the advantage that the performance of each instrument can be evaluated in the same way, building up common performance data.
The clarification of PQ requirements in 2017 <1058> means that PQ is not a single activity, but an integration of planned testing (with frequency and acceptance criteria defined), maintenance activities, and checks in operational use, as documented in the instrument logs that will be discussed later. Reliance on only SST results is a weak implementation of PQ for chromatography systems. If your laboratory defines PQ as SST results, and an auditor or inspector challenges this interpretation, what would you do?
Linking the URS, OQ, and PQFrom the 2017 USP <1058> PQ definition shown earlier, PQ testing must relate to user requirements. The problem is that an OQ typically tests the user requirements directly through traceable standards, metrology measurements using calibrated equipment, and use of appropriate reference materials, designed to test the instrument performance and range of use.
In contrast, a PQ is usually application- or method-based (for example, OQ and PQ test different attributes of system performance, which is why both are required):
• OQ: Related to testing the instrument performance under standardized conditions, so that the correct operation of the instrument in the laboratory against the URS can be demonstrated. For example, for HPLC, flow rate accuracy and reproducibility can be measured directly as metrology measurements using a calibrated and traceable digital flow meter. The range of use (for example, maximum and minimum settings) is measured in the OQ phase.
• PQ: Addresses the suitability of the instrument under actual conditions of use between repetition of the OQ/PQ cycle. A PQ indirectly measures the laboratory user requirements. For example, flow rate accuracy, and reproducibility can be measured indirectly in a PQ using retention time windows and %RSD of retention time. Because the range of use is measured in the OQ phase, it does not need to be measured in the PQ.
The key issue is that there must be a laboratory URS upon which PQ (and OQ) tests should be based.
The earliest regulatory publication on AIQ is the 1994 paper by Furman; et al7 from the FDA, which includes a discussion of modular versus holistic qualification of chromatographic instruments. The argument was that if the performance of each module or component were within acceptance limits, in principle, the system could potentially fail due to the addition of errors or the system components not working correctly together. The authors proposed the inclusion of an overall system, or holistic test, in the qualification of chromatography instruments, as module testing alone would not detect this.
A holistic PQ test executed after an OQ, or even as part of the OQ, would provide a link between the functional and operational-based OQ and the method-based PQ, as shown in Figure 3.
Table 1. Example PQ approaches for an HPLC instrument.
Approach to PQ instrument testing Comments/Observations
1 PQ = System suitability tests (for example, PQ = SST)
• Validity of SSTs included in each method?• SSTs are method-specific• Auditor may not accept this interpretation
2 PQ = Holistic PQ method + SST (for example, the same PQ is used for all HPLCs)
• Simplifies PQ requirements• Builds on SST during use• Justify that PQ testing is representative of use
3 PQ = Specific PQ method + SST (for example, PQ is instrument/use specific)
• Complicates PQ requirements across a Laboratory (for instance, specific and different PQ requirements for each HPLC system)
• Builds on SST during use• Instruments dedicated to specific methods

4
Instrument complexity and PQ requirementsUSP <1058> includes three groups of instrument complexity, with the classification dependent on instrument complexity and use. Generally, the instrument compliance testing strategy for the three groups is:
Laboratories must now apply a risk assessment based on intended use to determine if the instrument is Group B or C.
It is also important to understand that compliance with 2017 <1058> is a dynamic process:
• More than one category: An instrument type can be in more than one <1058> category (A, B, or C), depending on use/intended use.
• Change of use: May change the group classification (A, B, or C in <1058>)
• Change of use: Will change the URS and may change the range of use and qualification requirements
As an example, an ultrasonic bath is sometimes used in sample preparation, to aid the dissolution of the sample. Subject to confirmation (by risk assessment), this would generally be expected to be Group A, with the operation confirmed by observation during use. However, if the ultrasonic bath includes a heater or timer, this may change the group classification. If these functions are used, the risk assessment would identify this, and the classification would change to Group B. The timer and temperature controller must be calibrated against their range of use following an SOP. If these functions are not used, they do not require calibration (for example, because their use is not specified in a procedure). It is unlikely use of these uncalibrated functions can be physically or electronically controlled, so how does a laboratory prevent their use? If they were to be used, this would signify use of uncalibrated instruments (for example, someone uses the temperature controller when it is not calibrated). Compliance would typically be achieved by labeling the status of the instrument (for example, temperature and timer not calibrated, do not use for compliance work), training, and documenting in the SOP the instrument
use. However, procedural control such as this will not be accepted indefinitely, impacting the user requirements of future instruments being bought (all instruments will need to be designed to satisfy data integrity requirements without procedural control).
The risk assessment also identifies if the firmware of the Group B instrument includes:
• Calculations: Built-in calculations (and if they are used).
• User defined programs: The ability to create user-defined programs.
For Group B instruments, the risk assessment results in the following extra subclassification:
The extra requirements of B2 and B3
are associated with software testing (see White Paper 2: How to Comply with the 2017 Version of USP <1058>2). The primary way to document the successful operation of a Group B instrument (rather than test the software) is for a user to calibrate the instrument against an SOP. Depending on the instrument complexity, there may also be maintenance and verification/qualification tests performed by an individual external to the laboratory (for example, service provider or metrology department):
1. User calibration: Performed within the laboratory
2. External maintenance/calibration/ qualification: Performed by someone independent from the lab
AIQ
Group
Group A
Group B
Group C
Observe
Calibrate
Qualify
Strategy
The first question to ask is—what is the impact of A, B, or C classification on PQ requirements?
• Group A: The apparatus are monitored by observation, and do not require user calibration (for example, a nitrogen evaporator or volumetric glassware).
• Group A—PQ testing requirements: As long as the observation of successful operation is made under conditions of use, and there is an SOP associated with using the Group A apparatus, there are no additional PQ testing requirements. For volumetric glassware, for example, the SOP would state “examine before use”, and discard any unsuitable glassware (for example, damaged or chipped).
Therefore, for Group A (apparatus), no OQ or PQ testing is required, but this decision must be documented in a laboratory procedure.
Groups B and C refer to instruments of increasing complexity. With the 2008 <1058>, laboratories could define the instrument group by looking at the examples provided in 2008 <1058>, but these are not present in 2017 <1058>.
AIQ Group BB2: Qualification and verify calculations
B1: Qualification only (calibration)
B3: Qualification and control user programs

5
One of the challenges associated with AIQ is that different terms can be used by laboratories and regulators8. USP <1058> uses calibration (for Group B instruments) and qualification (for Group C instruments), while the US Code of Federal Regulation (CFR) uses the word calibration (for example, 21 CFR 211.68 (a), 211.160 (b)4 and 211.194(d)).
For Group B instruments, the question “What are the PQ Testing Requirements”, depends on the answer to the following question:
“Is the routine use of the instrument different from 1 and 2, above?"
If the answer to this question is No, then there are no additional PQ-specific testing requirements that need to be included in the PQ test plans.
The instrument life cycle process used in a laboratory needs to document/justify how the testing performed satisfies the OQ and PQ requirements of 2017 <1058>. For PQ, this must include defining PQ test plans, test plans, acceptance criteria, and test frequency. Historically, PQ may only have been considered as a PQ test protocol. Addressing this requirement is a laboratory responsibility.
Figure 2 shows the relationship between URS, OQ, and PQ for simple Group B instruments. For simple instruments (for example, a pH meter), daily or point-of-use calibration is the only testing performed. The color-graded box represents the fact that this calibration satisfies both OQ and PQ testing requirements. For more complex Group B instruments (for example, an analytical balance) two kinds of instrument calibration are performed:
• External calibration by a metrology department or service provider tests the range of use and OQ requirements.
• User-performed calibration satisfies PQ testing requirements.
For both options, there is a regulatory expectation that laboratories will perform periodic reviews of instrument performance (for example, calibration records).
For Group C instruments, the risk assessment results in the following subclassification:
With more complex Group C instruments, such as an HPLC, there is only an indirect relationship between PQ and OQ tests and the URS because testing involves a separation step that is method-based, as shown in Figure 3.
PQ Expicitly verifies URS
OQverifies
URS
External calibration(for example,
vendor or metrology)
Calibration performed
by laboratory
Operationalqualification
(OQ)
Performancequalification
(PQ)
Laboratory userrequirementsspecification
Very simple Group B instruments—user calibration only
(for example, a pH meter)
Figure 2. Relationship between PQ and the laboratory URS and OQ for Group B instruments.
AIQ Group C
C1: Qualification and nonconfigurable software
C2: Qualification and configurable software
C3: Qualification plus configurable and custom software
GAMP Category 3 software
GAMP Category 4 software
GAMP Category 3 software with 5 module
Figure 3. Relationship between PQ and the laboratory URS and OQ for complex instruments.
PQ Impicitly verifies URS
OQverifies
URS
Baseline linkagewith OQ results
PQversus
OQ
Specifiesoperationalparameters
Confirmsoperational use
and functionality
Holistic PQ test using model compounds
Holistic and SSTresults trended
over time
Operationalqualification
(OQ)
Holisticperformancequalification
Operationalrelease
Performancequalification
(PQ)
Laboratory userrequirementsspecification
Ongoing PQ linked to prerelease holistic PQ and, implicitly, to last
OQ results
6
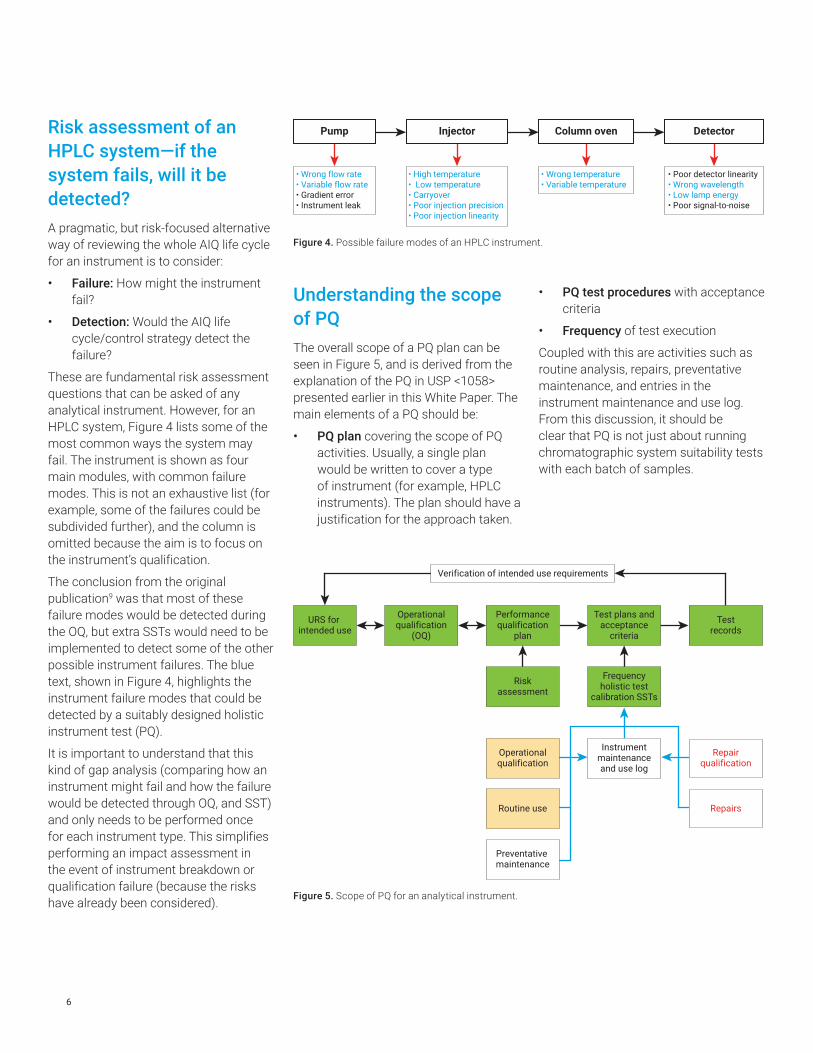
Risk assessment of an HPLC system—if the system fails, will it be detected?A pragmatic, but risk-focused alternative way of reviewing the whole AIQ life cycle for an instrument is to consider:
• Failure: How might the instrument fail?
• Detection: Would the AIQ life cycle/control strategy detect the failure?
These are fundamental risk assessment questions that can be asked of any analytical instrument. However, for an HPLC system, Figure 4 lists some of the most common ways the system may fail. The instrument is shown as four main modules, with common failure modes. This is not an exhaustive list (for example, some of the failures could be subdivided further), and the column is omitted because the aim is to focus on the instrument’s qualification.
The conclusion from the original publication9 was that most of these failure modes would be detected during the OQ, but extra SSTs would need to be implemented to detect some of the other possible instrument failures. The blue text, shown in Figure 4, highlights the instrument failure modes that could be detected by a suitably designed holistic instrument test (PQ).
It is important to understand that this kind of gap analysis (comparing how an instrument might fail and how the failure would be detected through OQ, and SST) and only needs to be performed once for each instrument type. This simplifies performing an impact assessment in the event of instrument breakdown or qualification failure (because the risks have already been considered).
Understanding the scope of PQ The overall scope of a PQ plan can be seen in Figure 5, and is derived from the explanation of the PQ in USP <1058> presented earlier in this White Paper. The main elements of a PQ should be:
• PQ plan covering the scope of PQ activities. Usually, a single plan would be written to cover a type of instrument (for example, HPLC instruments). The plan should have a justification for the approach taken.
• PQ test procedures with acceptance criteria
• Frequency of test execution
Coupled with this are activities such as routine analysis, repairs, preventative maintenance, and entries in the instrument maintenance and use log. From this discussion, it should be clear that PQ is not just about running chromatographic system suitability tests with each batch of samples.
• High temperature• Low temperature• Carryover• Poor injection precision• Poor injection linearity
Injector
• Wrong temperature• Variable temperature
Column oven
• Poor detector linearity• Wrong wavelength• Low lamp energy• Poor signal-to-noise
DetectorPump
• Wrong flow rate• Variable flow rate• Gradient error• Instrument leak
Figure 4. Possible failure modes of an HPLC instrument.
Figure 5. Scope of PQ for an analytical instrument.
Verification of intended use requirements
Repairqualification
Repairs
Operationalqualification
(OQ)
Performancequalification
plan
Riskassessment
Operationalqualification
Routine use
Preventative maintenance
Frequencyholistic test
calibration SSTs
Instrumentmaintenanceand use log
Test plans andacceptance
criteria
Testrecords
URS for intended use

7
Frequency of PQ testsFor chromatographic analysis, a holistic PQ is recommended after the OQ, or should be included in the OQ to link the URS to the OQ and provide a baseline for the remaining PQ. The laboratory needs to determine the frequency of PQ tests and to integrate this into the regular OQ and preventative maintenance cycle. For example:
• Preventative maintenance visit by the service provider
• Annual OQ
• Holistic PQ after the OQ
• Periodic holistic PQ
• SST results gathered and trended each time an analysis is performed to meet the requirements of EU GMP Chapter 6, clauses 6.9 and 6.16, and FDA Guidance for Industry10
PQ: linking the layers of the data quality triangleA modified USP <1058> data quality triangle is shown in Figure 6. Note that only the lowest level of the triangle, AIQ, is instrument-specific, using traceable reference standards and calibrated test equipment. All remaining layers are method-specific. Therefore, if method tests, such as SST and holistic tests, are to be used for the PQ, they must show that the user requirements defined in the laboratory URS are being met each day a test is performed.
A new approach to the development and validation of analytical procedures (based on a life cycle approach) using quality by design (QbD) is the basis of the new draft USP general chapter <1220>, published in Pharmacopoeial Forum11. This provides a structured approach to development and validation of analytical procedures, including defining critical parameters that can be monitored by system suitability tests during routine use. This approach will help develop appropriate SST criteria for monitoring and trending instrument performance, and result in more robust analytical methods.
The great advantage of an integrated OQ and PQ approach, linked to documented laboratory user requirements, is that it is easy to defend. The rationale for the qualification approaches taken in both the OQ and PQ phases can easily be traced back to the URS, and the risk assessments undertaken and documented.
Figure 6. A modified USP <1058> data quality triangle.
Qualitycontrolcheck
samples
System suitability tests
Analytical methodvalidation
Analytical instrument qualificationDQ IQ OQ PQ
Methodbased
Instrumentbased

8
PQ Roles and responsibilitiesTable 2 shows the roles and responsibilities of people involved in PQ. Most roles are laboratory-based. In principle, an organization outside of the laboratory, such as a service provider or metrology department, could perform a holistic PQ test, but it might be argued by a regulator that this is not fully representative of conditions of use. In fact, PQ is typically a laboratory responsibility and should be carried out by them. When this is carried out on behalf of the laboratory, the people performing the work must receive appropriate training in the PQ testing that is performed.
The key responsibility in Table 2 is that of the subject matter experts to define scientifically sound PQ test acceptance criteria. The basis of a scientifically sound approach is to link the PQ acceptance criteria, derived from method validation, to operational limits derived from the appropriate USP general chapter, for example, for a UV HPLC detector, wavelength accuracy should be ±3 nm rather than ±2 nm from USP <621>, not USP <857>.
System suitability tests as part of a PQ Designed to satisfy Pharmacopeia requirements, such as USP <621> or EP 2.2.46, SSTs play a pivotal role in documenting the performance of the chromatography system at the analytical run level. A natural evolution of this is to consider how SSTs can contribute to a PQ test plan.
If SST results are to be used as part of PQ testing of a chromatograph, it is important that:
Method development parameters and acceptance criteria are defined when the method is developed.
• Method validation should confirm the suitability of SSTs for performance monitoring and provide traceability between the method validation and the use of SSTs. This should be done to monitor that the instrument is meeting its user requirements during operational use.
• Trending SST parameters, as required by EU GMP 6.9 and 6.16 and FDA Guidance for Industry10. The summaries of method testing will be part of the overall PQ acceptance criteria.
Many laboratories have implemented lean initiatives to reduce potentially unnecessary work. However, this must be balanced with scientific soundness, as required under the GMP regulations, such as 21 CFR 211.160(b), for example:
• Blank injection removal: Done to save time, but will limit troubleshooting of a problem, as there will be no chromatogram of the injection of mobile phase. A blank injection can determine if there is any carryover from the autosampler and the level of baseline flatness/noise in the detector response. This can be related back to the user requirements, as discussed later in this White Paper.
• Control samples: Similar considerations need to be made for the inclusion of an approved and well-characterized control sample, particularly for impurity characterization. For example, it is not uncommon in a post lean laboratory for chromatographic methods not to include a standard to serve as a comparison for the run. Since chromatography is a comparative analytical technique, this could be seen as problematic.
In principle, a risk-based rationale was applied when good chromatography practices were reviewed and cut back. The problem is that relying on leanly designed SST tests means there could be a higher risk of PQ failure and an inability to investigate out-of-specification (OOS) results adequately, or provide scientific evidence that an instrument failure did not affect analytical results (because there is no evidence, depending on what is performed in SST).
Figure 4 shows the common ways an HPLC system could fail. In operational use, some of these failure modes may not be detected, depending on how the instrument is used (for example, lamp emission lines provide a diagnostic wavelength check when a detector
Table 2. Roles and responsibilities for performance qualification.
Role Responsibilities
Process owner• Accountable/responsible for all qualification work for the instrument• Reviews and approves OQ test plan (or delegates this to specific role in company) • Reviews and approves PQ test plan and test procedures
Subject matter experts
• Write the instrument (and software) user requirements• Reviews and approves the OQ test plan• Writes the PQ plan and test procedures • Defines scientifically sound PQ test acceptance criteria linked to method performance• Executes and documents PQ tests
Quality assurance• Approves User Requirements Specification• Approves PQ test plan and test procedures• Reviews PQ test data and test results periodically
Qualification engineer• Performs and documents instrument preventative maintenance and repairs• Performs and documents OQ at defined intervals• May perform holistic PQ test if contracted to do so and appropriately trained
Note: These responsibilities and roles are provided for guidance.

9
is turned on) and the limits applied to system suitability tests (such as retention time windows).
Care must also be taken when using samples to evaluate the performance of a chromatograph, because recent FDA guidance12 suggests avoiding sample injections as a means of testing into compliance. All work needs to be included in documented procedures, and the data generated reviewed.
Holistic HPLC PQ testAs part of an overall approach to PQ, there should be a holistic test that can show that the user requirements are still being met. Analytical procedures should routinely be designed to be as robust as possible. However, the principle of a good holistic test is to design an analytical procedure that is sensitive to instrument performance (which is the opposite of normal analytical science). This is the approach used for Performance Verification Testing (PVT) of dissolution instruments, which is universally interpreted as a PQ for these instruments. Ideally, the procedure must use stable model compounds, with simple and stable chromatography to minimize analytical variance from the reference material or use. The performance of the procedure is dependent on instrument performance. The test is performed under actual conditions of use, and for an HPLC instrument, consists of the following:
• Two stable model compounds: Well behaved and well separated model compounds that have good peak shape when run in a simple chromatographic system
• Same absorbance maxima under test conditions, so that they have the same peak areas when run in the same injection
• Use a robust chromatography system with simple organic/aqueous mobile phase
• Analytical column: Use relatively short analytical columns to reduce run time for overall PQ test.
• Prepare standards in the mobile phase to minimize disturbance when injecting.
• Prepare standard solutions gravimetrically to avoid pipetting errors and minimize overall method variance.
• Use four solution concentrations (25, 50, 75, and 100 %) containing the two compounds to test that autosampler and detector reproducibility and linearity are prepared.
• Run sequence: Consists of a blank, injected once at the beginning of the sequence and at the end of each standard set.
• Inject each standard six times.
These overall holistic standards allow limits to be set for:
• Detector reproducibility and linearity
• Autosampler precision
• The combination of the pumping system and thermostatic control of the column
All parameters are measured with the instrument under actual conditions of system use.
SummaryTo date, there have been many different interpretations of what AIQ and PQ should contain. The 2017 version of USP <1058> provides some clarification of AIQ requirements and clarification of differences between the OQ and PQ qualification phases. However, as a guidance document, <1058> cannot be prescriptive, and it is a laboratory responsibility to document how their AIQ aligns with, or satisfies, <1058> requirements. Generally, PQ is the AIQ area where there is more diverse interpretation, and this White Paper provides clarification of PQ requirements. To support PQ and deeper understanding of AIQ requirements, Table 2, in the Appendix of this White Paper, lists some of the frequently asked questions related to AIQ and PQ requirements. The changes implemented in the 2017 USP <1058> and implications of those changes need to be understood and acted upon by laboratories, or they risk noncompliance.

10
References1. What Has Changed with the
2017 Version of USP <1058>, Agilent Technologies White Paper, publication number 5991-9418EN, 2018.
2. How to Comply with the 2017 Version of USP <1058>. Agilent Technologies White Paper, publication number 5991-9419EN, 2018.
3. The Role of Analytical Instrument Qualification in Data Integrity with the 2017 Version of USP <1058>, Agilent Technologies White Paper, publication number 5991-9420EN, 2018.
4. What Does Performance Qualification Really Mean with the 2017 Version of USP <1058>? Agilent Technologies White Paper, publication number 5991-9421EN, 2018.
5. USP General Chapter on Analytical Instrument Qualification (AIQ), <1058> USP 40-NF 35, through 2nd Supplement, accessed online at: https://hmc.usp.org/about/general-chapters
6. FDA Guidance for Industry, Guidelines on General Principles of Process Validation, May 1987.
7. Furman; et al. Validation of Computerized Liquid Chromatographic Systems, J. AOAC Int. 1994, 77, 1314-1318.
8. Smith, P. Compliance in Regulated Laboratories, American Laboratory, March 2015.
9. Smith, P.; McDowall, R. D. Life Cycle Risk Assessment of HPLC Instruments, LC-GC Europe, February 2015.
10. FDA Guidance for Industry, Analytical Procedures and Method Validation for Drugs and Biologics, July 2015, reference ucm386366
11. Stimuli to the Revision Process, Proposed New General Chapter: The Analytical Procedure Life Cycle <1220>, Pharmacopeia Forum, PF 43(1), January-February 2017.
12. FDA Guidance for Industry, “Data Integrity and Compliance with cGMP”, April 2016.
Appendix 1 – Table of frequently asked questions about PQ and AIQQuestion Answer
Do I need to perform AIQ for all my analytical instruments?
Analytical instruments that are used to make quality decisions within a regulated environment, such as pharmaceutical-testing laboratories, must be suitable for their intended use. Without the ability to demonstrate this, the analytical results may be invalid or challenged during an audit. Performing an AIQ is the best way to address this need. Laboratories should consider and define an appropriate level of qualification for the decisions made on the analytical results, rather than justify why an AIQ is not required. The risk associated with providing any justification for not performing a task is that an auditor/regulator may not fully agree with the interpretation. Where AIQ requirements are not the same (for example, between quality control testing and research and development laboratories), it important to define and manage appropriate levels of AIQ for different kinds of laboratories, rather than applying a universal interpretation (for example, “AIQ is not required for analytical work in laboratory “x” because…”).
Can I can ignore USP <1058> because my company does not export to the USA?
Compliance with USP is a requirement for supply of pharmaceutical materials to the USA. Therefore, in principle, a company that does not supply to the USA does not need to comply. However, audits and inspections are frequently about managing regulatory expectations and, as USP is the only major pharmacopeia that includes a chapter dedicated to AIQ, it is influential beyond the USA. USP <1058> provides a valuable framework for AIQ that is simpler to understand than other frameworks, such as GAMP (for example, seven pages versus 352 pages for GAMP 5). Therefore, the contents of <1058> are influential, and should be considered as best practice and a regulatory expectation.
What do I need to include in an AIQ risk assessment?
Performing a risk assessment is now an intrinsic part of USP <1058> compliance requirements. For consistency of risk assessment application, a procedure needs to be defined and documented on this. The procedure should include three stages:• Identify if the instrument is group A, B, or C (based on intended use)• For all instruments, document how the instrument satisfies the URS• For group B instruments, identify:
• If any built in calculations are used need to be verified• If any user-defined programs are used need to be validated
• For group C instruments, identify GAMP categorization:• Group 3, Group 4, or Group 5
• Verify that the range of use matches the testing/URS or justify use.
Are there regulatory citations for laboratories not performing AIQ?
Yes. Although data integrity has dominated laboratory audits and regulatory inspections in recent years, there is increasing evidence that auditors are continuing to focus in greater detail on laboratory operations, including AIQ. White Paper 3 in this series includes a table of examples of laboratory noncompliance observations from FDA Warning Letters, FDA 483s and the EudraGMDP database (European equivalent to FDA warning letters). See White Paper 3 in References.
Why do I need to write a User Requirements Specification?
User Requirements Specification (URS) was not mentioned in the 2008 version of <1058>. However, the need to document a URS is a fundamental requirement of 2017 <1058>, which states that AIQ or software validation cannot be performed without a URS.
Do I need to perform both OQ and PQ for my analytical instruments?
You must document how your AIQ satisfies OQ and PQ requirements of the 2017 <1058>. Because OQ and PQ test different attributes of the instrument performance, both are required. Details of specific OQ and PQ requirements are dependent on the analytical technology/complexity of the instrument, and the relationship between how the instrument is tested and the conditions of use.

11
What’s the difference between an OQ and a PQ?
OQ and PQ requirements are defined within USP <1058>, but to simplify:OQ: Verifies the instrument, satisfies user requirements and range of usePQ: Demonstrates the instrument continues to work under conditions of use
My chromatography methods include System Suitability Tests (SSTs), do I also need to perform a separate PQ?
Yes. Because SSTs are method-specific, although they contribute to documenting the ongoing instrument performance. On their own, SSTs are not considered fully compliant with PQ requirements of 2017 <1058>. You must document how your AIQ satisfies <1058> PQ requirements and be able to successfully explain this during an audit. This means two key PQ requirements:• That when tested under conditions of use, the instrument is suitable• The continued performance of the instrument is tested and documented
What are the risks associated with using SSTs as a PQ?
Perception of risk is difficult to quantify. The fundamental risk associated with the argument: SST = PQ is that an auditor may not agree with this interpretation or that this interpretation is compliant with USP <1058>. Integrated, well designed AIQ and life cycle processes add to the quality of the analytical results generated and support robust defence of the results (reducing audit risk).
Is PQ a regulatory requirement now?Yes. The 4Q life cycle for AIQ includes PQ as a requirement. This has always been the case, but the 2017 update of USP <1058> has brought this into greater regulatory focus and helped clarify the different roles of OQ and PQ. However, organizations must define in their own policy documents as to how their AIQ processes satisfy USP <1058> requirements, including OQ and PQ requirements.
How often should a PQ be performed? Users are responsible for PQ test plans. Therefore, it is difficult to give absolute guidance, and users must define the frequency of PQ test plans.
Who is responsible for performing a PQ?The laboratory is responsible for the quality of all qualification work performed, irrespective of who performs it. Users must define PQ test plans, but other groups external to the laboratory can perform PQ testing as long as testing is approved by the users, and the people performing the work are appropriately trained.
If a user (or service provider) makes repairs such as replacement of HPLC pump seals or the detector UV lamp, what requalification is required?
When an instrument is repaired, the performance of the instrument must be demonstrated before it can be used to perform analysis. This could be a full qualification or only qualification of the system components related to the repair (repair qualification, RQ). To support RQ, an approved procedure must be in place that documents the required qualification after an instrument repair, before it can be returned to use. For example, replacement of the pump seals in the laboratory will not affect the performance of the HPLC detector, and replacement of the lamp will not affect the performance of the pump. For any repairs not documented in the procedure, either a full qualification is required, or a risk assessment must be performed to document and justify the RQ required.
Where do I test the range of use of the instrument?
Testing the operating range of the instrument that is used is a basic compliance requirement of documenting the suitability for use, and one that has resulted in laboratory citations for not being performed. The OQ must test the URS. If the range of use is not tested, there is risk of a regulatory citation. Because of this risk, where the OQ does not test the range of use, extra work is usually performed by the laboratory to supplement the OQ work. It is better to configure the OQ to bracket the range of use, where possible.
Are there any compliance risks associated with “Hot Swapping” components of a system to keep it operational?
Any changes made to an instrument must be made under conditions where the change is documented and approved (for example, change control). The framework a laboratory uses to document and justify the continued suitability and consistent performance of an instrument is important. To perform a detailed impact assessment (where the potential impact of an instrument failure on analytical results is investigated), it is necessary to have appropriate information about the instrument failure. If “Hot Swaps” are used by a laboratory, then the procedure followed should include details of how an impact assessment is performed.
What do I need to ensure that AIQ is compliant with <1058>?
AIQ is a requirement for laboratories. Complying with and appropriating AIQ represents best practice for all analytical laboratories, irrespective of industry. You should:• Understand <1058> requirements and their interpretation• Perform a gap analysis between 2017 <1058> and your AIQ• Check that range of use is documented and tested in AIQ• Prioritize gaps including defining PQ test plans
Have confidence in your data integrity program with Agilent CrossLab, the industry leader in instrument and software qualification and computer system validation services.

For more information, visit:
www.agilent.com/chem/qualification
Contact:
www.agilent.com/chem/contactus
This information is subject to change without notice.
© Agilent Technologies, Inc. 2019 Printed in the USA, November 15, 2019 5991-9421ENDE4151157407




















![What Does Technology Assisted Review Really Mean? [Infobite]](https://img.dokumen.tips/doc/110x75/5481f6175906b50f058b4605/what-does-technology-assisted-review-really-mean-infobite.jpg)








