Embed Size (px)
Citation preview

Comparação e alinhamento de sequências

Comparar sequências
• A comparação de sequências de proteínas ou DNA/RNA é uma ferramenta essencial na procura da existência de relações de semelhança entre o todo ou parte dessas sequências, e na avaliação da sua proximidade
• Alinhamento e comparação são problemas que podem ser expressos de forma matemática e para os quais existem algoritmos robustos, contudo:
• A parametrização do problema deverá reflectir o nosso conhecimento biológico (escolha das funções de score, gap penalties e outros parâmetros que afectam as soluções oferecidas pelos algoritmos)

Para quê comparar sequências?
• Identificação de regiões conservadas entre duas ou mais sequências evidencia zonas importantes para a estrutura e/ou função da proteína correspondente
• Estimativa da distância evolutiva entre os organismos dos quais provêm as sequências: maior disparidade das sequências deve resultar de uma maior divergência evolutiva
• Identificação de sequências numa base de dados que possuam semelhança significativa com uma determinada sequência de busca (identificação de homólogos)
• Identificação de uma sequência a partir de um fragmento

Comparar sequências não é trivial
(a) Sequências muito aparentadas: cadeias 𝑎 e β-hemoglobina humanas
(b) Sequências aparentadas: 𝑎-hemoglobina humana e leghemoglobina vegetal
(c) Sequências NÃO aparentadas: 𝑎-hemoglobina humana e GST-7 de C. Elegans
GST7_CEL
Idênticos: 18/41Similares: 17/41% identidade: 43%
Idênticos: 8/46Similares: 17/46% identidade: 17%
Idênticos: 13/45Similares: 12/45% identidade: 28%
gaps
gaps
Idêntico
Similar

Homologia vs. semelhançaOs termos homologia, semelhança e identidade têm significados distintos no contexto da análise de sequências biológicas:
•Homologia: descreve um parentesco evolutivo entre duas sequências que poderão corresponder a proteínas de funções homologas em diferentes organismos (exemplo: citocromo c humano e citocromo c bovino)
•Semelhança: descreve o grau de parecença entre duas sequências, independentemente do seu contexto ou significado biológico. É quantificada através de um método matemático de alinhamento e depende da escolha do “scoring scheme” (matriz de scoring)
•Identidade: fala-se normalmente em percentagem de identidade entre duas sequências, sendo definida como a razão entre o número total de resíduos idênticos e o número total de resíduos do alinhamento (incluindo gaps)

Comparar sequências: dot plotsUm “dot plot” é um modo de comparação de duas sequências baseado na construção de uma matriz de N linhas e M colunas, em que N e M são os comprimentos das duas sequências a comparar.
Seq
uênc
ia 1
Sequência 2

Comparar sequências: dot plotsUm “dot plot” é um modo de comparação de duas sequências baseado na construção de uma matriz de N linhas e M colunas, em que N e M são os comprimentos das duas sequências a comparar.
Seq
uênc
ia 1
Sequência 2
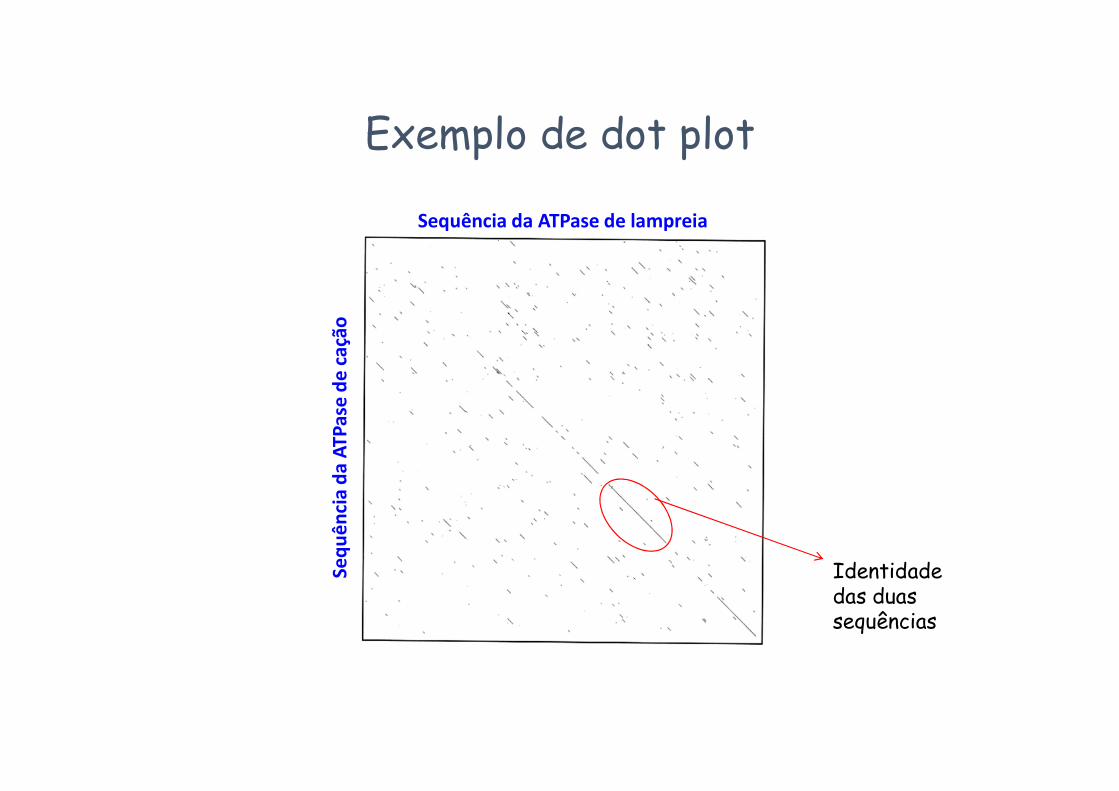
Exemplo de dot plot
Identidade das duas sequências
Sequência da ATPase de lampreia
Sequ
ênci
a da
ATP
ase
de c
ação

ACCTGCCCTGTCCAGCTTACATGCATGCTTATAGGGGCATTTTACAT
ACCTGCCGATTCCATATTACGCATGCTTCTGGGTTACCGTTCAGGGCATTTTACATGTGCTG
1+1+1+1+1+1+0+1+0+1+1=9A T G C
A 1 0 0 0T 0 1 0 0G 0 0 1 0C 0 0 0 1
“window size” (11)
Esquema de “scoring”(1 p/ posições iguais,0 p/ posições diferentes)
al
ATGCTTCTGGG
ATGCTTATAGG
Uma linha de pontos indica regiões similares

e no caso do score máximo (11) ser atingido:
Detecção de correspondências exactas entre regiões
Escolher um esquema de score
A T G CA 1 0 0 0T 0 1 0 0G 0 0 1 0C 0 0 0 1
ATGCTTATAGG
ATGCTTCTGGG
1+1+1+1+1+1+1+1+1+1+1=11
1)
Para cada par de janelas, calcular o score usando a matriz,
e um tamanho de janela
aATGCTTATAGG
ATGCTTCTGGG
1+1+1+1+1+1+0+1+0+1+1=9
Marcar um ponto Não marcar um ponto
Comparações usando “dot plots”
Neste caso o score de cut-off usado foi 11, mas podia ser um valor mais baixo
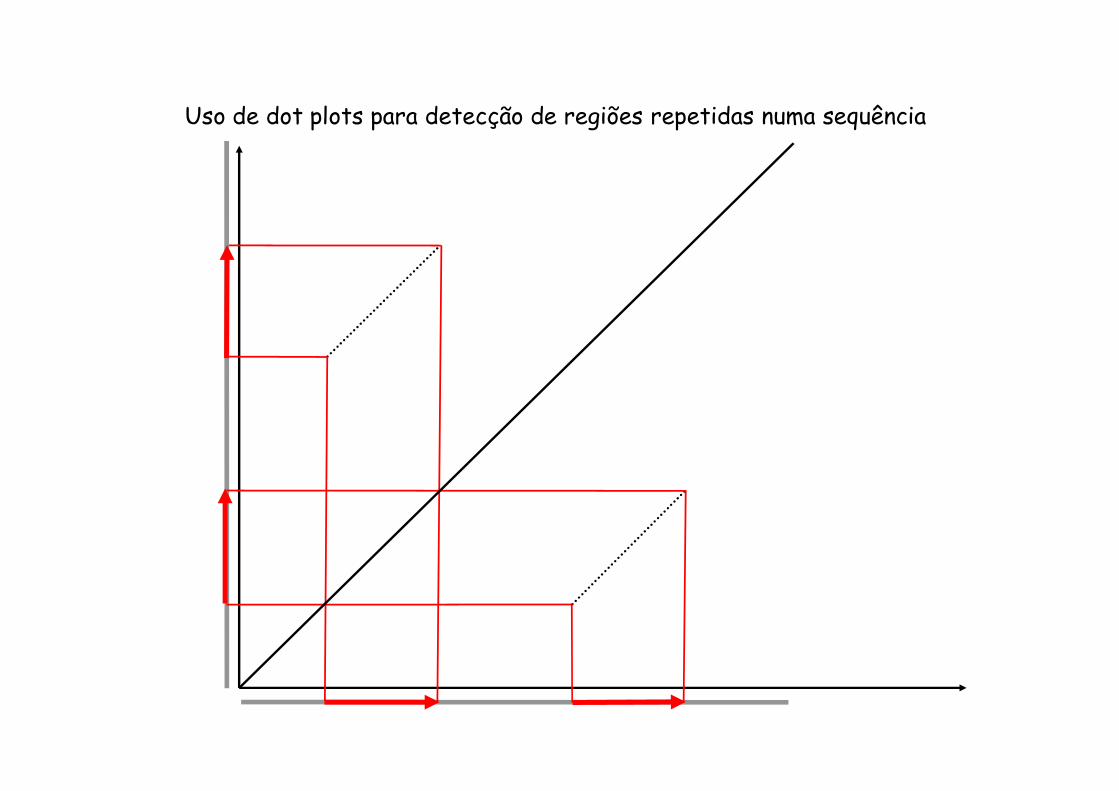
Uso de dot plots para detecção de regiões repetidas numa sequência

Uso de dot plots para detecção de regiões repetidas numa sequência

Problema da comparação de strings
• Consideremos as duas sequências de caracteres:
GAATTCAGTTA
GGATCGA
• Pretendemos alinha-las de modo a obter um score máximo na comparação

O que se entende por “alinhar” ?
• Alinhar é estabelecer uma correspondência entre as duas sequências, o que pode ser feito inserindo espaços:
GAATTCAGTTA
GGATCGAG-GATCGAG--GATCGAG-G--ATCGAG-G--AT--CGA

GAATTCAGTTAG-G-ATCGA Score=2
O que se entende por “score” ?• Um score é um número que é associado a cada alinhamento
possível, e que pode ser definido de várias maneirasExemplo: associar um valor de 1 a cada posição idêntica nas duas sequências, e 0 a posições diferentes
A T G C
A 1 0 0 0
T 0 1 0 0
G 0 0 1 0
C 0 0 0 1
GAATTCAGTTAGGATCGA Score=4
GAATTCAGTTAG-GATCGA Score=3
GAATTCAGTTAG-G--AT--CGA Score=1
GAATTCAGTTAGGA-TC-G--A Score=6
Alinhamento Matriz de score

Como achar o score máximo ?• Podíamos tentar experimentar TODOS alinhamentos possíveis, e
escolher aquele que produzisse o score máximo ?...
NÃO !
• Para o caso apresentado, existem cerca de 220 alinhamentos , o que é mais de UM MILHÃO de possibilidades! Para duas sequências de 250 caracteres temos cerca de 10149
alinhamentos, um número computacionalmente inatingível!• Felizmente existem algoritmos que permitem achar o
alinhamento óptimo, ou seja aquela que maximiza o score, sem ter que pesquisar exaustivamente todos os alinhamentos possíveis

Alinhamento global vs. localAlinhamento global: as sequências A e B são comparadas na totalidade do seu comprimento, sendo as diferenças de comprimento da sequência compensadas com “gaps” (inserções)
Alinhamento local: consiste na identificação de regiões isoladas de elevada similaridade entre as duas sequências, independentemente do seu contexto.

Algoritmo de Needleman-WunschNeedleman, S.B & Wunsch, C.D (1970) J.Mol.Biol. 48:443
• É um algoritmo de programação dinâmica capaz de encontrar o alinhamento global óptimo de duas sequências
• Como ponto de partida necessitamos apenas de uma matriz com o score de alinhamento para cada par de aminoácidos (ou bases) e uma gap penalty (score de penalização para criação de um espaço na sequência)
• Este algoritmo fornece apenas o alinhamento óptimo, não permitindo identificar outros alinhamentos com scores próximos do óptimo e que poderão ser biologicamente relevantes (alinhamentos sub-optimais).

Algoritmo de Needleman-WunschExemplo:
Pretende-se alinhar as sequências GVTAH e AVTLI
•A matriz de score usada vai ser a BLOSUM50
Matriz BLOSUM50 Scores de match / mismatch que serão usados

d G V T A H
d
A
V
T
L
I
Algoritmo de Needleman-Wunsch
1) Construção da matriz de alinhamento
Outro score que será usado:
• Inserção de gap no alinhamento tem um score negativo = -1

d G V T A H
d
A
V
T
L
I
Matriz de alinhamentoTodos possíveis alinhamentos são caminhos nesta matriz
GVATHAVTLI
G V T A H
A 0 0 0 5 -2
V -4 5 0 0 -4
T -2 0 5 0 -2
L -4 1 -1 -2 -3
I -4 4 -1 -1 -4
Score= 0 + 5 + 5 + (-2) + (-4) = +4

d G V T A H
d
A
V
T
L
I
Matriz de alinhamentoTodos possíveis alinhamentos são caminhos nesta matriz
GVT-AHA-VTLI Score= 0 + (-1) + 0 + (-1) + (-2) + (-4) = -8
G V T A H
A 0 0 0 5 -2
V -4 5 0 0 -4
T -2 0 5 0 -2
L -4 1 -1 -2 -3
I -4 4 -1 -1 -4
Gap na sequência inferior
Gap na sequênciasuperior

d G V T A H
d
A
V
T
L
I
Matriz de alinhamentoTodos possíveis alinhamentos são caminhos nesta matriz
GVTAH----------AVTLI
Score= (-1) + (-1) + (-1) + (-1) + (-1) + (-1) + (-1) + (-1) + (-1) + (-1) = -10
G V T A H
A 0 0 0 5 -2
V -4 5 0 0 -4
T -2 0 5 0 -2
L -4 1 -1 -2 -3
I -4 4 -1 -1 -4

d G V T A H
d
A
V
T
L
I
Matriz de alinhamentoTodos possíveis alinhamentos são caminhos nesta matriz
GVT-AHAVTLI-
Score= 0 + 5 + 5 + (-1) + (-1) + (-1) = +7
G V T A H
A 0 0 0 5 -2
V -4 5 0 0 -4
T -2 0 5 0 -2
L -4 1 -1 -2 -3
I -4 4 -1 -1 -4

d G V T A H
d 0 -1 -2 -3 -4 -5
A -1
V -2
T -3
L -4
I -5
Algoritmo de Needleman-Wunsch2) Inserção dos valores da gap penalty
Neste caso a gap penalty tem valor -1

d G V T A H
d 0 -1 -2 -3 -4 -5
A -1 0 -1 -2 2 1
V -2 -1 5 4 3 2
T -3 -2 4 10 9 8
L -4 -3 3 9 8 7
I -5 -4 2 8 8 7
Algoritmo de Needleman-Wunsch3) Preenchimento da tabela, da esquerda para a direita e de cima para baixo, de acordo com seguinte regra:
( 1, 1) ( , )
( , ) max ( 1, ) ( , )
( , 1) ( , )
H i j S i j
H i j H i j S i
H i j S i
( 1, 1)H i j
( , 1)H i j
( 1, )H i j
( , )H i j
S(i,j) é o score da matriz de score (BLOSUM50 neste caso), e S(-,j) e S(i, -) scores para inserção de um gap horizonal ou vertical
G V T A H
A 0 0 0 5 -2
V -4 5 0 0 -4
T -2 0 5 0 -2
L -4 1 -1 -2 -3
I -4 4 -1 -1 -4
d G V T A H
d 0 -1 -2 -3 -4 -5
A -1 0 -1 -2 2 1
V -2 -1 5 4 3 2
T -3 -2 4 10 9 8
L -4 -3 3 9 8 7
I -5 -4 2 8 8 7
Scores da matriz BLOSUM50
Cada célula mantém a informação da proveniência do valor anterior (setas)

d G V T A H
d 0 -1 -2 -3 -4 -5
A -1 0 -1 -2 2 1
V -2 -1 5 4 3 2
T -3 -2 4 10 9 8
L -4 -3 3 9 8 7
I -5 -4 2 8 8 7
Algoritmo de Needleman-Wunsch4) Traçar o caminho desde o canto inferior direito, seguindo as setas. Cada movimento horizontal ou vertical corresponde a uma gap na sequência respectiva.
G V T — A H
A V T L I —
Alinhamento óptimo
Score: 7
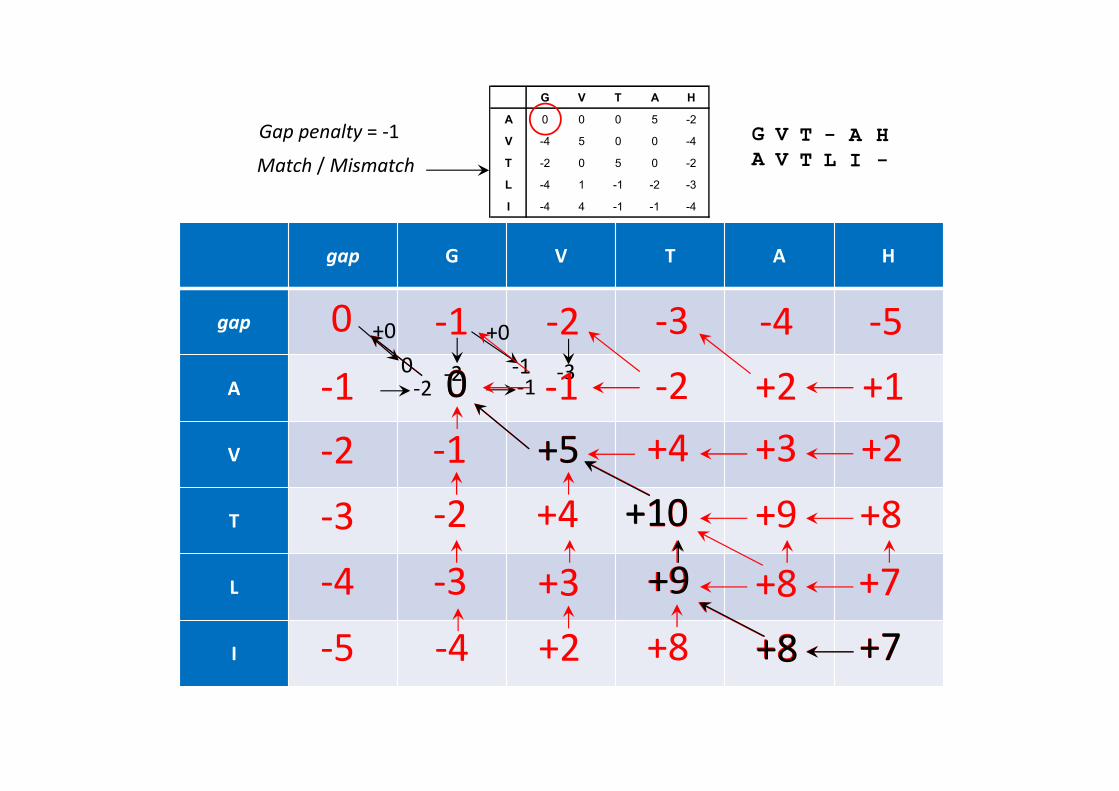
gap G V T A H
gap
A
V
T
L
I
0 -1 -2 -3
Gap penalty = -1
-4 -5
-1-2
-3
-4
-5
G V T A H
A 0 0 0 5 -2
V -4 5 0 0 -4
T -2 0 5 0 -2
L -4 1 -1 -2 -3
I -4 4 -1 -1 -4
Match / Mismatch
+0
-2-2
0 0+0
-1-3-1
-1 -2 +2 +1-1 +5 +4 +3 +2
-2 +4 +10 +9 +8
-3
-4
+3
+2
+9
+8+8 +7
+8 +7+7
H-
+8
AI
+9
-L
+10
TT
+5
VV
0
GA

Alinhamento local: algoritmo de Smith-Waterman
O algoritmo de Smith-Waterman é uma versão modificada de N-W que permite encontrar o alinhamento local óptimo entre duas sequências.
0
( 1, 1) ( , )( , ) max
( 1, ) ( , )
( , 1) ( , )
H i j S i jH i j
H i j S i
H i j S i
Se o valor calculado partir das células anteriores for <0, é substituído pelo valor zero e o alinhamento termina nesse ponto. O alinhamento local inicia-se na célula de valor mais alto da matriz de alinhamento.
Smith, T.F. & Waterman, M.S (1981) J.Mol.Biol. 147:195-197

Para que este algoritmo funcione, é necessário que o score esperado para um alinhamento aleatório seja negativo, e que existam valores positivos na matriz de comparação
Alinhamento local: algoritmo de Smith-Waterman

Importância do alinhamento local
Muitas proteínas apresentam uma estrutura modular, tendo regiões com proveniências evolutivas distintas e relacionadas com diferentes famílias.
A comparação local de duas sequências permite mais facilmente reconhecer estas regiões, mesmo quando na sua globalidade as sequências são largamente discrepantes.
Exemplo:
Domínio Catalítico
Domínio Catalítico
PLAT – plasminogen activatorF12 – coagulation factor XII
Domínioshomólogos

Matrizes de score
•As matrizes de score, ou matrizes de substituição, permitem obter um score para cada par de aminoácidos comparados.
• Os valores destas matrizes reflectem as diferentes tendências que os aminoácidos têm de ser substituídos por outros.
• Existem diferentes tipos de matrizes de score, baseados em diferentes análises e diferentes pressupostos sobre os mecanismos de substituição
• O processo de inserção (criação de gaps) é geralmente tratado separadamente.
• Os dois tipos de matrizes mais usados são:
Matrizes PAM: baseadas na comparação, por alinhamento global, de famílias de sequências muito próximas
Matrizes BLOSUM: baseadas no alinhamento de regiões de elevada similaridade (blocos) entre diferentes grupos de proteínas.
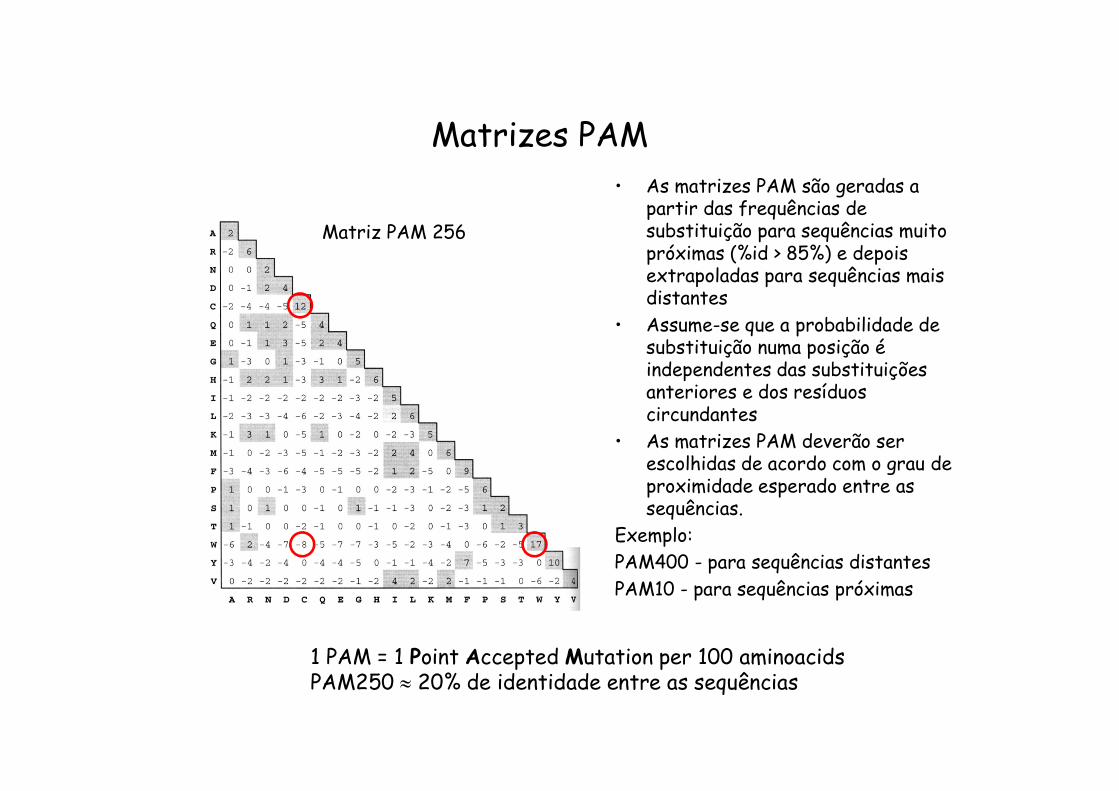
Matrizes PAM
1 PAM = 1 Point Accepted Mutation per 100 aminoacidsPAM250 20% de identidade entre as sequências
Matriz PAM 256
• As matrizes PAM são geradas a partir das frequências de substituição para sequências muito próximas (%id > 85%) e depois extrapoladas para sequências mais distantes
• Assume-se que a probabilidade de substituição numa posição é independentes das substituições anteriores e dos resíduos circundantes
• As matrizes PAM deverão ser escolhidas de acordo com o grau de proximidade esperado entre as sequências.
Exemplo:PAM400 - para sequências distantesPAM10 - para sequências próximas

Matrizes PAM
1 PAM = 1 Point Accepted Mutation per 100 aminoacidsPAM250 20% de identidade entre as sequências
Matriz PAM 256
• As matrizes PAM são geradas a partir das frequências de substituição para sequências muito próximas (%id > 85%) e depois extrapoladas para sequências mais distantes
• Assume-se que a probabilidade de substituição numa posição é independentes das substituições anteriores e dos resíduos circundantes
• As matrizes PAM deverão ser escolhidas de acordo com o grau de proximidade esperado entre as sequências.
Exemplo:PAM400 - sequências distantesPAM10 - sequências próximas
A 2R -1 6N 0 0 2D 0 -1 2 4C -2 -4 -4 -5 12Q 0 1 1 2 -5 4E 0 -1 1 3 -5 2 4G 1 -2 0 1 -3 -1 0 5H -1 2 2 1 -3 3 1 -2 6I 0 -2 -2 -2 -2 -2 -2 -2 -2 4L -2 -3 -3 -4 -6 -2 -3 -4 -2 2 6K -1 3 1 0 -5 1 0 -2 0 -2 -3 5M -1 0 -2 -3 -5 -1 -2 -3 -2 2 4 0 6F -3 -4 -3 -5 -4 -5 -5 -5 -2 1 2 -5 0 9P 1 0 0 -1 -3 0 -1 0 0 -2 -2 -1 -2 -4 6S 1 0 1 0 0 0 0 1 -1 -1 -3 0 -2 -3 1 2T 1 -1 0 0 -2 -1 0 0 -1 0 -2 0 -1 -3 0 1 2W -6 2 -4 -7 -8 -5 -7 -7 -3 -5 -2 -3 -4 0 -5 -2 -5 17Y -3 -4 -2 -4 0 -4 -4 -5 0 -1 -1 -4 -2 7 -5 -3 -3 0 10V 0 -2 -2 -2 -2 -2 -2 -1 -2 4 2 -2 2 -1 -1 -1 0 -6 -2 4
A R N D C Q E G H I L K M F P S T W Y V
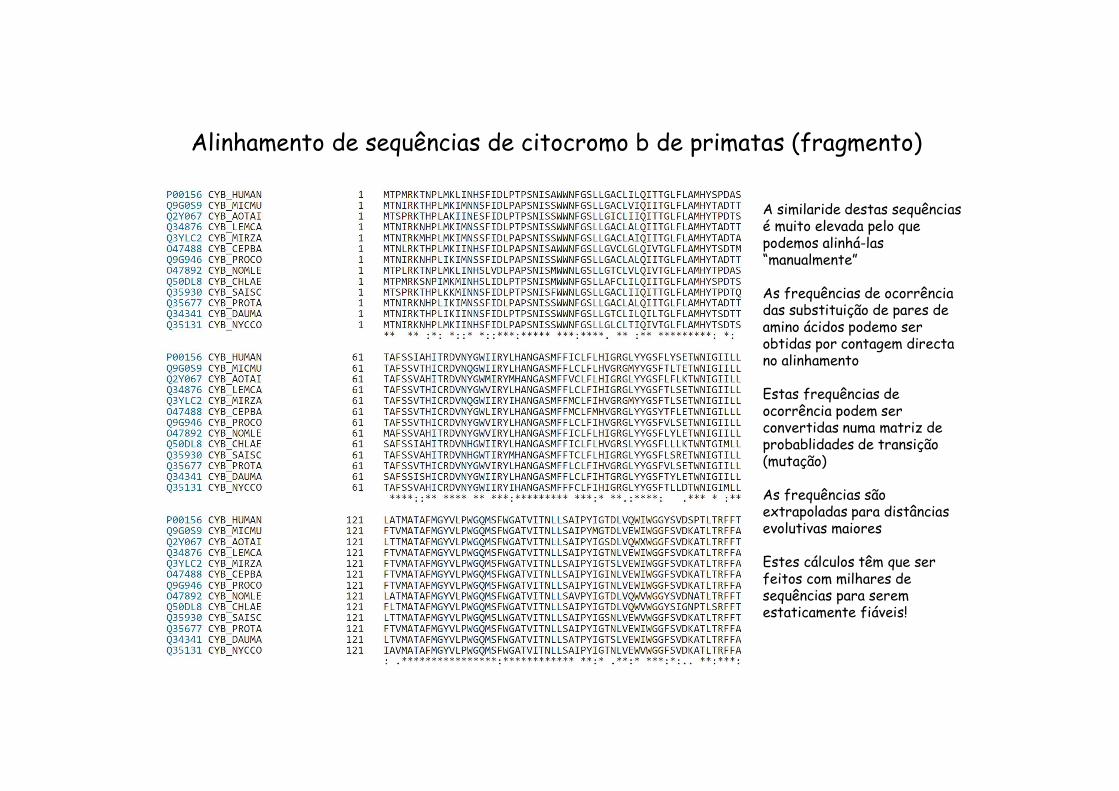
Alinhamento de sequências de citocromo b de primatas (fragmento)
A similaride destas sequênciasé muito elevada pelo que podemos alinhá-las “manualmente”
As frequências de ocorrênciadas substituição de pares de amino ácidos podemo ser obtidas por contagem directano alinhamento
Estas frequências de ocorrência podem ser convertidas numa matriz de probablidades de transição(mutação)
As frequências sãoextrapoladas para distânciasevolutivas maiores
Estes cálculos têm que ser feitos com milhares de sequências para seremestaticamente fiáveis!

XXXXXXXXXXXXXAXXXXXXXXXXXXXX
XXXXXXXXXXXXXGXXXXXXXXXXXXXX
XXXXXXXXXXXXXEXXXXXXXXXXXXXX Tempo
XXXXXXXXXXXXXAXXXXXXXXXXXXXX
XXXXXXXXXXXXXWXXXXXXXXXXXXXX
Reve
rsão
Score=1
Score=0
Score=-6Pouco provável

% identidade Unidades PAM
99 1
95 5
90 11
85 17
80 23
75 30
70 38
66 47
60 56
55 67
50 80
45 94
40 112
35 133
30 159
25 195
20 246
15 328
% de identidade mínima para conseguir produzir um alinhamento
Distância PAM versus % de identidade

Probablidade da Fenilalanina não mutar:99% (1 PAM)32% (250 PAM)

Matriz PAM 1 – probabilidades de transição
As probabilidades estão multiplicadas por 10000

A 11 -26 -20 -19 -24 -20 -17 -16 -26 -22 -24 -26 -21 -27 -16 -14 -14 ### -26 -17R -26 14 -25 ### -26 -16 ### -30 -16 -22 -28 -13 -20 -29 -20 -18 -26 -17 -31 -26N -20 -25 14 -10 ### -20 -18 -19 -13 -21 -25 -15 ### -28 -24 -13 -16 -26 -20 -28D -19 ### -10 13 ### -19 -9 -19 -21 -26 ### -21 ### ### -29 -20 -22 ### ### -27C -24 -26 ### ### 15 ### ### -30 -25 -24 ### ### ### ### -27 -18 -28 ### -20 -23Q -20 -16 -20 -19 ### 14 -11 -25 -12 -27 -21 -18 -19 ### -18 -22 -23 ### ### -25E -17 ### -18 -9 ### -11 13 -21 -23 -22 -29 -21 -25 ### -23 -21 -25 ### -27 -24G -16 -30 -19 -19 -30 -25 -21 10 -30 ### -31 -26 -28 -28 -25 -16 -25 ### ### -23H -26 -16 -13 -21 -25 -12 -23 -30 15 -31 -23 -26 ### -23 -20 -24 -26 -26 -19 -23I -22 -22 -21 -26 -24 -27 -22 ### -31 14 -16 -23 -15 -17 -29 -26 -17 ### -24 -11L -24 -28 -25 ### ### -21 -29 -31 -23 -16 11 -27 -13 -18 -25 -28 -25 -23 -25 -18K -26 -13 -15 -21 ### -18 -21 -26 -26 -23 -27 11 -16 ### -25 -20 -19 ### -29 -30M -21 -20 ### ### ### -19 -25 -28 ### -15 -13 -16 18 -20 -28 -22 -20 ### ### -16F -27 -29 -28 ### ### ### ### -28 -23 -17 -18 ### -20 14 -29 -23 -29 -21 -12 -29P -16 -20 -24 -29 -27 -18 -23 -25 -20 -29 -25 -25 -28 -29 13 -16 -21 ### ### -23S -14 -18 -13 -20 -18 -22 -21 -16 -24 -26 -28 -20 -22 -23 -16 11 -13 -21 -25 -25T -14 -26 -16 -22 -28 -23 -25 -25 -26 -17 -25 -19 -20 -29 -21 -13 12 ### -24 -18W ### -17 -26 ### ### ### ### ### -26 ### -23 ### ### -21 ### -21 ### 20 -22 ###Y -26 -31 -20 ### -20 ### -27 ### -19 -24 -25 -29 ### -12 ### -25 -24 -22 15 -25V -17 -26 -28 -27 -23 -25 -24 -23 -23 -11 -18 -30 -16 -29 -23 -25 -18 ### -25 12
A R N D C Q E G H I L K M F P S T W Y V
Matriz PAM 1 – log odds scores

Família de matrizes de substituição — PAM 1, PAM 2, etc. — onde PAMn é adequada à comparação de sequências que distam entre si de n PAM PAM n = (PAM 1)n
PAM2 = PAM1 x PAM1PAM4 = PAM2 x PAM2PAM8 = PAM4 x PAM4PAM16 = PAM8 x PAM8PAM32 = PAM16 x PAM16PAM64 = PAM32 x PAM32PAM128 = PAM64 x PAM64PAM256 = PAM128 x PAM128
Família de Matrizes PAM
Não confundir com as matrizes PAM de substituição com as matrizes PAM de score.
Matrizes PAM de score são obtidas a partir das matrizes PAM de substituição calculando os logaritmos da razão das frequências observadas e esperadas (“log odds score”)..

Os valores de score são logaritmosSij = logb (pi Mij / pipj)
Sij = score (“log odds” ratio) Mij = score da matriz de transiçãopi , pj = probabilidades de ocorrência dos aminoácidosb = base do logaritmo (arbitrária)Odds ratio (pi Mij/pipj) – razão entre a probabilidade de ocorrência de uma transição, e a probabilidade de ocorrência dessa mesma transição para num modelo aleatório
GALHIVHGGVNLVH p1*p2*p3*p4*p5*p6*p7 = Pa (probabilidade de ocorrência do alinhamento)
dado que log(a*b) = log(a) + log(b) temos:
log(Pa) =log(p1*p2*p3*p4*p5*p6*p7) = log(p1) +log(p2) +log(p3) +log(p4) +log(p5) +log(p6) +log(p7)
Assim, se usarmos log(pi) como valor de score para cada para de resídudos, a soma destes valores produz o logaritmo do score total log(Pa) !

A 10 -23 -17 -16 -21 -17 -14 -13 -23 -19 -21 -23 -18 -24 -13 -11 -11 -46 -23 -14R -23 14 -22 -41 -23 -13 -39 -27 -13 -19 -25 -10 -17 -26 -17 -15 -23 -14 -28 -23N -17 -22 14 -8 -41 -17 -15 -16 -10 -18 -22 -12 -38 -25 -21 -10 -13 -23 -17 -25D -16 -41 -8 13 -47 -16 -6 -16 -18 -23 -44 -18 -42 -48 -26 -17 -19 -48 -42 -24C -21 -23 -41 -47 15 -47 -47 -27 -22 -21 -48 -47 -46 -45 -24 -15 -25 -49 -17 -20Q -17 -13 -17 -16 -47 14 -9 -22 -9 -24 -18 -15 -16 -45 -15 -19 -20 -45 -43 -22E -14 -39 -15 -6 -47 -9 13 -18 -20 -19 -26 -18 -22 -47 -20 -18 -22 -52 -24 -21G -13 -27 -16 -16 -27 -22 -18 10 -27 -42 -28 -23 -25 -25 -21 -13 -22 -48 -46 -20H -23 -13 -10 -18 -22 -9 -20 -27 15 -28 -20 -22 -41 -20 -17 -21 -23 -23 -16 -21I -19 -19 -18 -23 -21 -24 -19 -42 -28 14 -13 -20 -12 -14 -26 -23 -14 -46 -21 -8L -21 -25 -22 -44 -48 -18 -26 -28 -20 -13 11 -24 -10 -15 -22 -25 -22 -20 -22 -15K -23 -10 -12 -18 -47 -15 -18 -23 -22 -20 -24 11 -13 -46 -22 -17 -16 -43 -26 -27M -18 -17 -38 -42 -46 -16 -22 -25 -41 -12 -10 -13 18 -17 -25 -19 -17 -45 -42 -13F -24 -26 -25 -48 -45 -45 -47 -25 -20 -14 -15 -46 -17 14 -26 -20 -25 -18 -9 -26P -13 -17 -21 -26 -24 -15 -20 -21 -17 -26 -22 -22 -25 -26 13 -13 -18 -46 -46 -20S -11 -15 -10 -17 -15 -19 -18 -13 -21 -23 -25 -17 -19 -20 -13 11 -10 -18 -22 -22T -11 -23 -13 -19 -25 -20 -22 -22 -23 -14 -22 -16 -17 -25 -18 -10 12 -44 -21 -15W -46 -14 -23 -48 -49 -45 -52 -48 -23 -46 -20 -43 -45 -18 -46 -18 -44 20 -19 -49Y -23 -28 -17 -42 -17 -43 -24 -46 -16 -21 -22 -26 -42 -9 -46 -22 -21 -19 15 -22V -14 -23 -25 -24 -20 -22 -21 -20 -21 -8 -15 -27 -13 -26 -20 -22 -15 -49 -22 12
A R N D C Q E G H I L K M F P S T W Y V
A 8 -8 -3 -3 -7 -4 -2 -1 -8 -5 -7 -8 -5 -10 -1 1 1 -16 -9 -2R -8 12 -6 -11 -10 -1 -10 -11 -1 -6 -10 2 -4 -11 -4 -3 -7 -1 -12 -9N -3 -6 10 3 -12 -3 -1 -3 2 -6 -9 0 -10 -10 -6 1 -1 -10 -5 -9D -3 -11 3 10 -17 -2 5 -3 -4 -9 -14 -4 -12 -17 -9 -3 -5 -18 -13 -9C -7 -10 -12 -17 14 -17 -17 -11 -9 -7 -18 -17 -16 -15 -10 -3 -9 -19 -4 -7Q -4 -1 -3 -2 -17 12 3 -8 3 -9 -6 -2 -4 -15 -3 -6 -6 -15 -14 -8E -2 -10 -1 5 -17 3 10 -4 -5 -6 -11 -4 -8 -17 -6 -4 -6 -20 -10 -7G -1 -11 -3 -3 -11 -8 -4 9 -10 -12 -13 -8 -10 -11 -7 -1 -6 -18 -16 -6H -8 -1 2 -4 -9 3 -5 -10 13 -10 -7 -6 -12 -7 -4 -6 -8 -9 -3 -7I -5 -6 -6 -9 -7 -9 -6 -12 -10 12 0 -7 1 -2 -10 -8 -2 -16 -7 4L -7 -10 -9 -14 -18 -6 -11 -13 -7 0 10 -10 2 -2 -8 -10 -8 -7 -8 -1K -8 2 0 -4 -17 -2 -4 -8 -6 -7 -10 9 -1 -16 -7 -4 -3 -13 -11 -10M -5 -4 -10 -12 -16 -4 -8 -10 -12 1 2 -1 16 -4 -9 -6 -4 -15 -13 0F -10 -11 -10 -17 -15 -15 -17 -11 -7 -2 -2 -16 -4 13 -12 -8 -10 -5 4 -9P -1 -4 -6 -9 -10 -3 -6 -7 -4 -10 -8 -7 -9 -12 11 -1 -4 -16 -16 -6S 1 -3 1 -3 -3 -6 -4 -1 -6 -8 -10 -4 -6 -8 -1 8 2 -6 -8 -7T 1 -7 -1 -5 -9 -6 -6 -6 -8 -2 -8 -3 -4 -10 -4 2 10 -15 -8 -2W -16 -1 -10 -18 -19 -15 -20 -18 -9 -16 -7 -13 -15 -5 -16 -6 -15 19 -6 -19Y -9 -12 -5 -13 -4 -14 -10 -16 -3 -7 -8 -11 -13 4 -16 -8 -8 -6 14 -9V -2 -9 -9 -9 -7 -8 -7 -6 -7 4 -1 -10 0 -9 -6 -7 -2 -19 -9 10
A R N D C Q E G H I L K M F P S T W Y V
A 4 -3 0 0 -3 -1 0 1 -3 -1 -3 -3 -2 -5 1 2 2 -9 -5 0R -3 9 -1 -4 -5 1 -3 -5 2 -3 -5 4 -1 -7 -1 -1 -2 2 -7 -4N 0 -1 5 3 -6 0 1 0 2 -3 -5 1 -3 -5 -2 1 0 -6 -3 -3D 0 -4 3 6 -8 1 5 0 0 -4 -7 -1 -5 -9 -3 0 -1 -10 -7 -4C -3 -5 -6 -8 13 -8 -8 -6 -5 -4 -9 -9 -8 -7 -5 0 -4 -11 -1 -3Q -1 1 0 1 -8 7 3 -3 4 -4 -3 0 -2 -8 0 -2 -2 -7 -6 -3E 0 -3 1 5 -8 3 6 -1 0 -3 -5 -1 -4 -9 -2 -1 -2 -11 -6 -3G 1 -5 0 0 -6 -3 -1 7 -4 -5 -7 -3 -5 -7 -2 1 -1 -10 -8 -3H -3 2 2 0 -5 4 0 -4 9 -5 -3 -1 -4 -3 -1 -2 -3 -4 -1 -4I -1 -3 -3 -4 -4 -4 -3 -5 -5 7 2 -3 2 1 -4 -3 0 -8 -3 5L -3 -5 -5 -7 -9 -3 -5 -7 -3 2 8 -5 4 1 -4 -5 -3 -3 -3 1K -3 4 1 -1 -9 0 -1 -3 -1 -3 -5 7 1 -8 -3 -1 -1 -6 -7 -5M -2 -1 -3 -5 -8 -2 -4 -5 -4 2 4 1 11 -1 -4 -3 -1 -7 -5 2F -5 -7 -5 -9 -7 -8 -9 -7 -3 1 1 -8 -1 11 -7 -5 -5 -1 7 -3P 1 -1 -2 -3 -5 0 -2 -2 -1 -4 -4 -3 -4 -7 8 1 0 -9 -8 -2S 2 -1 1 0 0 -2 -1 1 -2 -3 -5 -1 -3 -5 1 4 2 -3 -4 -2T 2 -2 0 -1 -4 -2 -2 -1 -3 0 -3 -1 -1 -5 0 2 5 -8 -4 0W -9 2 -6 -10 -11 -7 -11 -10 -4 -8 -3 -6 -7 -1 -9 -3 -8 18 -2 -10Y -5 -7 -3 -7 -1 -6 -6 -8 -1 -3 -3 -7 -5 7 -8 -4 -4 -2 12 -4V 0 -4 -3 -4 -3 -3 -3 -3 -4 5 1 -5 2 -3 -2 -2 0 -10 -4 7
A R N D C Q E G H I L K M F P S T W Y V
A 2 -1 0 0 -2 0 0 1 -1 0 -2 -1 -1 -3 1 1 1 -6 -3 0R -1 6 0 -1 -4 1 -1 -2 2 -2 -3 3 0 -4 0 0 -1 2 -4 -2N 0 0 2 2 -4 1 1 0 2 -2 -3 1 -2 -3 0 1 0 -4 -2 -2D 0 -1 2 4 -5 2 3 1 1 -2 -4 0 -3 -5 -1 0 0 -7 -4 -2C -2 -4 -4 -5 12 -5 -5 -3 -3 -2 -6 -5 -5 -4 -3 0 -2 -8 0 -2Q 0 1 1 2 -5 4 2 -1 3 -2 -2 1 -1 -5 0 0 -1 -5 -4 -2E 0 -1 1 3 -5 2 4 0 1 -2 -3 0 -2 -5 -1 0 0 -7 -4 -2G 1 -2 0 1 -3 -1 0 5 -2 -2 -4 -2 -3 -5 0 1 0 -7 -5 -1H -1 2 2 1 -3 3 1 -2 6 -2 -2 0 -2 -2 0 -1 -1 -3 0 -2I 0 -2 -2 -2 -2 -2 -2 -2 -2 4 2 -2 2 1 -2 -1 0 -5 -1 4L -2 -3 -3 -4 -6 -2 -3 -4 -2 2 6 -3 4 2 -2 -3 -2 -2 -1 2K -1 3 1 0 -5 1 0 -2 0 -2 -3 5 0 -5 -1 0 0 -3 -4 -2M -1 0 -2 -3 -5 -1 -2 -3 -2 2 4 0 6 0 -2 -2 -1 -4 -2 2F -3 -4 -3 -5 -4 -5 -5 -5 -2 1 2 -5 0 9 -4 -3 -3 0 7 -1P 1 0 0 -1 -3 0 -1 0 0 -2 -2 -1 -2 -4 6 1 0 -5 -5 -1S 1 0 1 0 0 0 0 1 -1 -1 -3 0 -2 -3 1 2 1 -2 -3 -1T 1 -1 0 0 -2 -1 0 0 -1 0 -2 0 -1 -3 0 1 2 -5 -3 0W -6 2 -4 -7 -8 -5 -7 -7 -3 -5 -2 -3 -4 0 -5 -2 -5 17 0 -6Y -3 -4 -2 -4 0 -4 -4 -5 0 -1 -1 -4 -2 7 -5 -3 -3 0 10 -2V 0 -2 -2 -2 -2 -2 -2 -1 -2 4 2 -2 2 -1 -1 -1 0 -6 -2 4
A R N D C Q E G H I L K M F P S T W Y V
PAM2 PAM50
PAM150 PAM256

PAM500
A 0 0 0 0 -1 0 0 1 0 0 -1 0 0 -1 0 0 0 -3 -1 0R 0 2 0 0 -2 1 0 -1 1 -1 -1 2 0 -2 0 0 0 2 -2 -1N 0 0 0 1 -2 1 1 0 1 -1 -1 0 -1 -2 0 0 0 -2 -1 0D 0 0 1 1 -2 1 1 1 1 -1 -1 0 -1 -2 0 0 0 -3 -2 -1C -1 -2 -2 -2 9 -2 -2 -1 -2 -1 -3 -2 -2 -1 -1 0 -1 -4 1 -1Q 0 1 1 1 -2 1 1 0 1 -1 -1 1 0 -2 0 0 0 -2 -2 -1E 0 0 1 1 -2 1 1 0 1 -1 -1 0 -1 -2 0 0 0 -3 -2 -1G 1 -1 0 1 -1 0 0 2 0 -1 -1 0 -1 -2 0 1 0 -3 -2 0H 0 1 1 1 -2 1 1 0 2 -1 -1 0 -1 -1 0 0 0 -1 0 -1I 0 -1 -1 -1 -1 -1 -1 -1 -1 1 2 -1 1 1 -1 0 0 -2 0 1L -1 -1 -1 -1 -3 -1 -1 -1 -1 2 3 -1 2 2 -1 -1 0 -1 1 1K 0 2 0 0 -2 1 0 0 0 -1 -1 2 0 -2 0 0 0 -1 -2 -1M 0 0 -1 -1 -2 0 -1 -1 -1 1 2 0 2 1 -1 -1 0 -2 0 1F -1 -2 -2 -2 -1 -2 -2 -2 -1 1 2 -2 1 6 -2 -1 -1 1 5 0P 0 0 0 0 -1 0 0 0 0 -1 -1 0 -1 -2 2 0 0 -3 -2 0S 0 0 0 0 0 0 0 1 0 0 -1 0 -1 -1 0 0 0 -1 -1 0T 0 0 0 0 -1 0 0 0 0 0 0 0 0 -1 0 0 0 -2 -1 0W -3 2 -2 -3 -4 -2 -3 -3 -1 -2 -1 -1 -2 1 -3 -1 -2 15 1 -3Y -1 -2 -1 -2 1 -2 -2 -2 0 0 1 -2 0 5 -2 -1 -1 1 6 -1V 0 -1 0 -1 -1 -1 -1 0 -1 1 1 -1 1 0 0 0 0 -3 -1 1
A R N D C Q E G H I L K M F P S T W Y V

A 10 -23 -17 -16 -21 -17 -14 -13 -23 -19 -21 -23 -18 -24 -13 -11 -11 -46 -23 -14R -23 14 -22 -41 -23 -13 -39 -27 -13 -19 -25 -10 -17 -26 -17 -15 -23 -14 -28 -23N -17 -22 14 -8 -41 -17 -15 -16 -10 -18 -22 -12 -38 -25 -21 -10 -13 -23 -17 -25D -16 -41 -8 13 -47 -16 -6 -16 -18 -23 -44 -18 -42 -48 -26 -17 -19 -48 -42 -24C -21 -23 -41 -47 15 -47 -47 -27 -22 -21 -48 -47 -46 -45 -24 -15 -25 -49 -17 -20Q -17 -13 -17 -16 -47 14 -9 -22 -9 -24 -18 -15 -16 -45 -15 -19 -20 -45 -43 -22E -14 -39 -15 -6 -47 -9 13 -18 -20 -19 -26 -18 -22 -47 -20 -18 -22 -52 -24 -21G -13 -27 -16 -16 -27 -22 -18 10 -27 -42 -28 -23 -25 -25 -21 -13 -22 -48 -46 -20H -23 -13 -10 -18 -22 -9 -20 -27 15 -28 -20 -22 -41 -20 -17 -21 -23 -23 -16 -21I -19 -19 -18 -23 -21 -24 -19 -42 -28 14 -13 -20 -12 -14 -26 -23 -14 -46 -21 -8L -21 -25 -22 -44 -48 -18 -26 -28 -20 -13 11 -24 -10 -15 -22 -25 -22 -20 -22 -15K -23 -10 -12 -18 -47 -15 -18 -23 -22 -20 -24 11 -13 -46 -22 -17 -16 -43 -26 -27M -18 -17 -38 -42 -46 -16 -22 -25 -41 -12 -10 -13 18 -17 -25 -19 -17 -45 -42 -13F -24 -26 -25 -48 -45 -45 -47 -25 -20 -14 -15 -46 -17 14 -26 -20 -25 -18 -9 -26P -13 -17 -21 -26 -24 -15 -20 -21 -17 -26 -22 -22 -25 -26 13 -13 -18 -46 -46 -20S -11 -15 -10 -17 -15 -19 -18 -13 -21 -23 -25 -17 -19 -20 -13 11 -10 -18 -22 -22T -11 -23 -13 -19 -25 -20 -22 -22 -23 -14 -22 -16 -17 -25 -18 -10 12 -44 -21 -15W -46 -14 -23 -48 -49 -45 -52 -48 -23 -46 -20 -43 -45 -18 -46 -18 -44 20 -19 -49Y -23 -28 -17 -42 -17 -43 -24 -46 -16 -21 -22 -26 -42 -9 -46 -22 -21 -19 15 -22V -14 -23 -25 -24 -20 -22 -21 -20 -21 -8 -15 -27 -13 -26 -20 -22 -15 -49 -22 12
A R N D C Q E G H I L K M F P S T W Y V
A 8 -8 -3 -3 -7 -4 -2 -1 -8 -5 -7 -8 -5 -10 -1 1 1 -16 -9 -2R -8 12 -6 -11 -10 -1 -10 -11 -1 -6 -10 2 -4 -11 -4 -3 -7 -1 -12 -9N -3 -6 10 3 -12 -3 -1 -3 2 -6 -9 0 -10 -10 -6 1 -1 -10 -5 -9D -3 -11 3 10 -17 -2 5 -3 -4 -9 -14 -4 -12 -17 -9 -3 -5 -18 -13 -9C -7 -10 -12 -17 14 -17 -17 -11 -9 -7 -18 -17 -16 -15 -10 -3 -9 -19 -4 -7Q -4 -1 -3 -2 -17 12 3 -8 3 -9 -6 -2 -4 -15 -3 -6 -6 -15 -14 -8E -2 -10 -1 5 -17 3 10 -4 -5 -6 -11 -4 -8 -17 -6 -4 -6 -20 -10 -7G -1 -11 -3 -3 -11 -8 -4 9 -10 -12 -13 -8 -10 -11 -7 -1 -6 -18 -16 -6H -8 -1 2 -4 -9 3 -5 -10 13 -10 -7 -6 -12 -7 -4 -6 -8 -9 -3 -7I -5 -6 -6 -9 -7 -9 -6 -12 -10 12 0 -7 1 -2 -10 -8 -2 -16 -7 4L -7 -10 -9 -14 -18 -6 -11 -13 -7 0 10 -10 2 -2 -8 -10 -8 -7 -8 -1K -8 2 0 -4 -17 -2 -4 -8 -6 -7 -10 9 -1 -16 -7 -4 -3 -13 -11 -10M -5 -4 -10 -12 -16 -4 -8 -10 -12 1 2 -1 16 -4 -9 -6 -4 -15 -13 0F -10 -11 -10 -17 -15 -15 -17 -11 -7 -2 -2 -16 -4 13 -12 -8 -10 -5 4 -9P -1 -4 -6 -9 -10 -3 -6 -7 -4 -10 -8 -7 -9 -12 11 -1 -4 -16 -16 -6S 1 -3 1 -3 -3 -6 -4 -1 -6 -8 -10 -4 -6 -8 -1 8 2 -6 -8 -7T 1 -7 -1 -5 -9 -6 -6 -6 -8 -2 -8 -3 -4 -10 -4 2 10 -15 -8 -2W -16 -1 -10 -18 -19 -15 -20 -18 -9 -16 -7 -13 -15 -5 -16 -6 -15 19 -6 -19Y -9 -12 -5 -13 -4 -14 -10 -16 -3 -7 -8 -11 -13 4 -16 -8 -8 -6 14 -9V -2 -9 -9 -9 -7 -8 -7 -6 -7 4 -1 -10 0 -9 -6 -7 -2 -19 -9 10
A R N D C Q E G H I L K M F P S T W Y V
A 4 -3 0 0 -3 -1 0 1 -3 -1 -3 -3 -2 -5 1 2 2 -9 -5 0R -3 9 -1 -4 -5 1 -3 -5 2 -3 -5 4 -1 -7 -1 -1 -2 2 -7 -4N 0 -1 5 3 -6 0 1 0 2 -3 -5 1 -3 -5 -2 1 0 -6 -3 -3D 0 -4 3 6 -8 1 5 0 0 -4 -7 -1 -5 -9 -3 0 -1 -10 -7 -4C -3 -5 -6 -8 13 -8 -8 -6 -5 -4 -9 -9 -8 -7 -5 0 -4 -11 -1 -3Q -1 1 0 1 -8 7 3 -3 4 -4 -3 0 -2 -8 0 -2 -2 -7 -6 -3E 0 -3 1 5 -8 3 6 -1 0 -3 -5 -1 -4 -9 -2 -1 -2 -11 -6 -3G 1 -5 0 0 -6 -3 -1 7 -4 -5 -7 -3 -5 -7 -2 1 -1 -10 -8 -3H -3 2 2 0 -5 4 0 -4 9 -5 -3 -1 -4 -3 -1 -2 -3 -4 -1 -4I -1 -3 -3 -4 -4 -4 -3 -5 -5 7 2 -3 2 1 -4 -3 0 -8 -3 5L -3 -5 -5 -7 -9 -3 -5 -7 -3 2 8 -5 4 1 -4 -5 -3 -3 -3 1K -3 4 1 -1 -9 0 -1 -3 -1 -3 -5 7 1 -8 -3 -1 -1 -6 -7 -5M -2 -1 -3 -5 -8 -2 -4 -5 -4 2 4 1 11 -1 -4 -3 -1 -7 -5 2F -5 -7 -5 -9 -7 -8 -9 -7 -3 1 1 -8 -1 11 -7 -5 -5 -1 7 -3P 1 -1 -2 -3 -5 0 -2 -2 -1 -4 -4 -3 -4 -7 8 1 0 -9 -8 -2S 2 -1 1 0 0 -2 -1 1 -2 -3 -5 -1 -3 -5 1 4 2 -3 -4 -2T 2 -2 0 -1 -4 -2 -2 -1 -3 0 -3 -1 -1 -5 0 2 5 -8 -4 0W -9 2 -6 -10 -11 -7 -11 -10 -4 -8 -3 -6 -7 -1 -9 -3 -8 18 -2 -10Y -5 -7 -3 -7 -1 -6 -6 -8 -1 -3 -3 -7 -5 7 -8 -4 -4 -2 12 -4V 0 -4 -3 -4 -3 -3 -3 -3 -4 5 1 -5 2 -3 -2 -2 0 -10 -4 7
A R N D C Q E G H I L K M F P S T W Y V
A 2 -1 0 0 -2 0 0 1 -1 0 -2 -1 -1 -3 1 1 1 -6 -3 0R -1 6 0 -1 -4 1 -1 -2 2 -2 -3 3 0 -4 0 0 -1 2 -4 -2N 0 0 2 2 -4 1 1 0 2 -2 -3 1 -2 -3 0 1 0 -4 -2 -2D 0 -1 2 4 -5 2 3 1 1 -2 -4 0 -3 -5 -1 0 0 -7 -4 -2C -2 -4 -4 -5 12 -5 -5 -3 -3 -2 -6 -5 -5 -4 -3 0 -2 -8 0 -2Q 0 1 1 2 -5 4 2 -1 3 -2 -2 1 -1 -5 0 0 -1 -5 -4 -2E 0 -1 1 3 -5 2 4 0 1 -2 -3 0 -2 -5 -1 0 0 -7 -4 -2G 1 -2 0 1 -3 -1 0 5 -2 -2 -4 -2 -3 -5 0 1 0 -7 -5 -1H -1 2 2 1 -3 3 1 -2 6 -2 -2 0 -2 -2 0 -1 -1 -3 0 -2I 0 -2 -2 -2 -2 -2 -2 -2 -2 4 2 -2 2 1 -2 -1 0 -5 -1 4L -2 -3 -3 -4 -6 -2 -3 -4 -2 2 6 -3 4 2 -2 -3 -2 -2 -1 2K -1 3 1 0 -5 1 0 -2 0 -2 -3 5 0 -5 -1 0 0 -3 -4 -2M -1 0 -2 -3 -5 -1 -2 -3 -2 2 4 0 6 0 -2 -2 -1 -4 -2 2F -3 -4 -3 -5 -4 -5 -5 -5 -2 1 2 -5 0 9 -4 -3 -3 0 7 -1P 1 0 0 -1 -3 0 -1 0 0 -2 -2 -1 -2 -4 6 1 0 -5 -5 -1S 1 0 1 0 0 0 0 1 -1 -1 -3 0 -2 -3 1 2 1 -2 -3 -1T 1 -1 0 0 -2 -1 0 0 -1 0 -2 0 -1 -3 0 1 2 -5 -3 0W -6 2 -4 -7 -8 -5 -7 -7 -3 -5 -2 -3 -4 0 -5 -2 -5 17 0 -6Y -3 -4 -2 -4 0 -4 -4 -5 0 -1 -1 -4 -2 7 -5 -3 -3 0 10 -2V 0 -2 -2 -2 -2 -2 -2 -1 -2 4 2 -2 2 -1 -1 -1 0 -6 -2 4
A R N D C Q E G H I L K M F P S T W Y V
A 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 -1 0 0R 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0N 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 -1 0 0D 0 0 0 0 0 0 0 0 0 0 0 0 0 -1 0 0 0 -1 -1 0C 0 0 0 0 5 0 0 0 0 0 -1 -1 -1 0 0 0 0 -1 0 0Q 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 -1 0 0E 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 -1 0 0G 0 0 0 0 0 0 0 0 0 0 0 0 0 -1 0 0 0 -1 -1 0H 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0I 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0L 0 0 0 0 -1 0 0 0 0 0 1 0 0 1 0 0 0 0 0 0K 0 0 0 0 -1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0M 0 0 0 0 -1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0F 0 0 0 -1 0 0 0 -1 0 0 1 0 0 2 0 0 0 1 2 0P 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 -1 0 0S 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0T 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 -1 0 0W -1 1 -1 -1 -1 -1 -1 -1 0 0 0 0 0 1 -1 0 -1 10 1 -1Y 0 0 0 -1 0 0 0 -1 0 0 0 0 0 2 0 0 0 1 2 0V 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 -1 0 0
A R N D C Q E G H I L K M F P S T W Y V



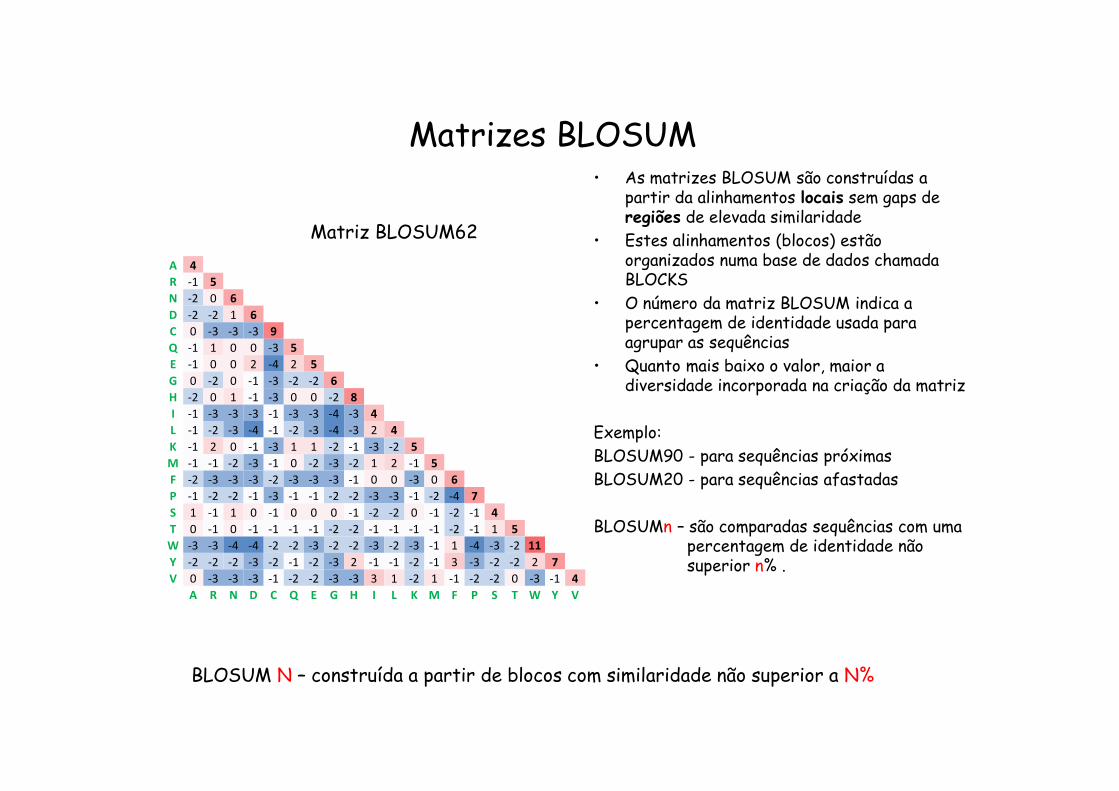
Matrizes BLOSUM
Matriz BLOSUM62 • As matrizes BLOSUM são construídas a partir da alinhamentos locais sem gaps de regiões de elevada similaridade
• Estes alinhamentos (blocos) estão organizados numa base de dados chamada BLOCKS
• O número da matriz BLOSUM indica a % de identidade usada para agrupar as sequências
• Quanto mais baixo o valor, maior a diversidade incorporada na criação da matriz
Exemplo:BLOSUM90 - para sequências próximasBLOSUM20 - para sequências afastadas
BLOSUM N – construída a partir de blocos com similaridade não superior a N%
Matrizes BLOSUM
Matriz BLOSUM62
• As matrizes BLOSUM são construídas a partir da alinhamentos locais sem gaps de regiões de elevada similaridade
• Estes alinhamentos (blocos) estão organizados numa base de dados chamada BLOCKS
• O número da matriz BLOSUM indica a percentagem de identidade usada para agrupar as sequências
• Quanto mais baixo o valor, maior a diversidade incorporada na criação da matriz
Exemplo:BLOSUM90 - para sequências próximasBLOSUM20 - para sequências afastadas
BLOSUMn – são comparadas sequências com uma percentagem de identidade não superior n% .
BLOSUM N – construída a partir de blocos com similaridade não superior a N%
A 4R -1 5N -2 0 6D -2 -2 1 6C 0 -3 -3 -3 9Q -1 1 0 0 -3 5E -1 0 0 2 -4 2 5G 0 -2 0 -1 -3 -2 -2 6H -2 0 1 -1 -3 0 0 -2 8I -1 -3 -3 -3 -1 -3 -3 -4 -3 4L -1 -2 -3 -4 -1 -2 -3 -4 -3 2 4K -1 2 0 -1 -3 1 1 -2 -1 -3 -2 5M -1 -1 -2 -3 -1 0 -2 -3 -2 1 2 -1 5F -2 -3 -3 -3 -2 -3 -3 -3 -1 0 0 -3 0 6P -1 -2 -2 -1 -3 -1 -1 -2 -2 -3 -3 -1 -2 -4 7S 1 -1 1 0 -1 0 0 0 -1 -2 -2 0 -1 -2 -1 4T 0 -1 0 -1 -1 -1 -1 -2 -2 -1 -1 -1 -1 -2 -1 1 5W -3 -3 -4 -4 -2 -2 -3 -2 -2 -3 -2 -3 -1 1 -4 -3 -2 11Y -2 -2 -2 -3 -2 -1 -2 -3 2 -1 -1 -2 -1 3 -3 -2 -2 2 7V 0 -3 -3 -3 -1 -2 -2 -3 -3 3 1 -2 1 -1 -2 -2 0 -3 -1 4
A R N D C Q E G H I L K M F P S T W Y V

Exemplo de entrada na base de dados BLOCKS
http://blocks.fhcrc.org/
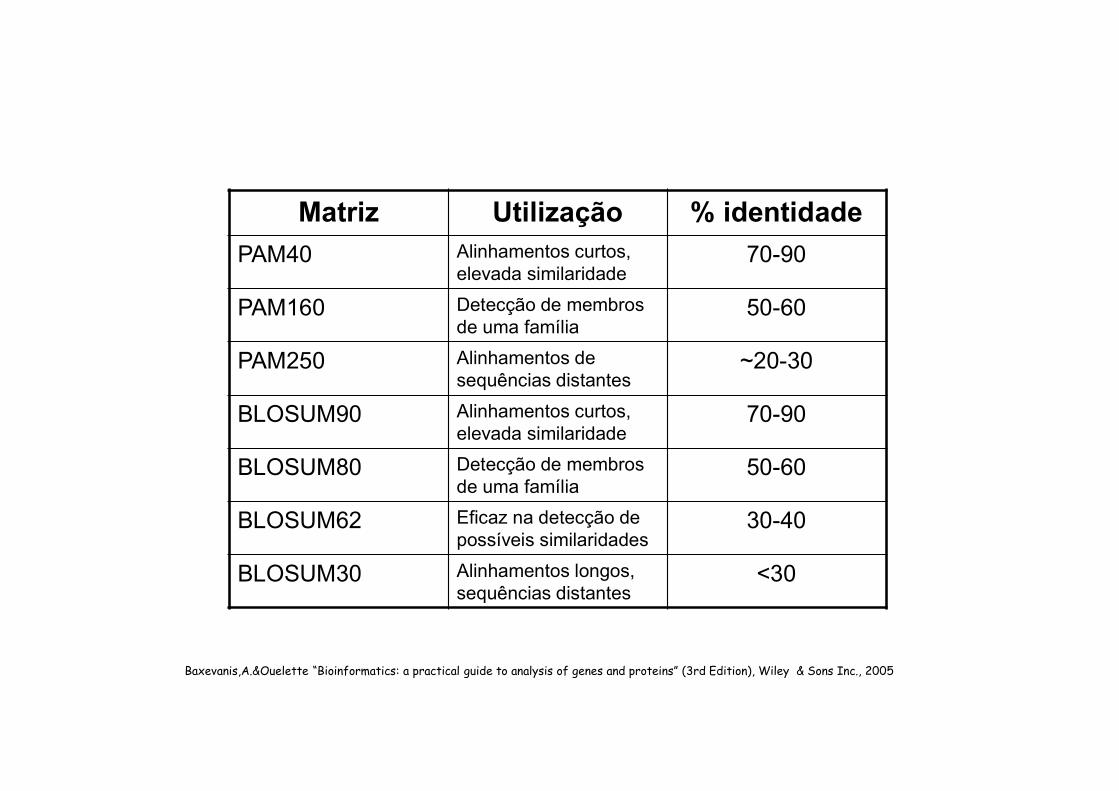
Matriz Utilização % identidade
PAM40 Alinhamentos curtos, elevada similaridade
70-90
PAM160 Detecção de membros de uma família
50-60
PAM250 Alinhamentos de sequências distantes
~20-30
BLOSUM90 Alinhamentos curtos, elevada similaridade
70-90
BLOSUM80 Detecção de membros de uma família
50-60
BLOSUM62 Eficaz na detecção de possíveis similaridades
30-40
BLOSUM30 Alinhamentos longos, sequências distantes
<30
Baxevanis,A.&Ouelette “Bioinformatics: a practical guide to analysis of genes and proteins” (3rd Edition), Wiley & Sons Inc., 2005

Matrizes de probabilidades de transição para nucleótidos
Transições: A G , C T
Transversões: A T , G T
A C , G C
A T G C
A 0.99
T 0.0033 0.99
G 0.0033 0.0033 0.99
C 0.0033 0.0033 0.0033 0.99
Frequências de mutação uniformes (1 PAM)
A T G C
A 0.99
T 0.0020 0.99
G 0.0060 0.0020 0.99
C 0.0020 0.0060 0.0020 0.99
Transições mais frequentes (3x) que transversões
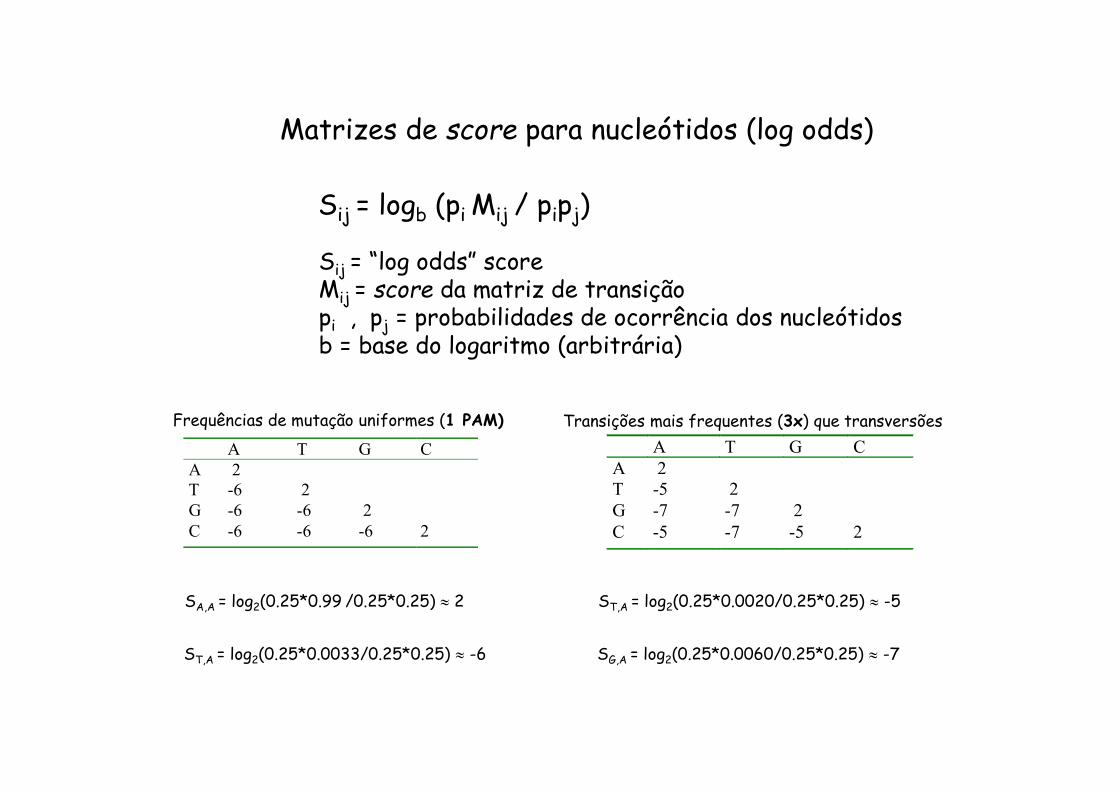
A T G CA 2T -6 2G -6 -6 2C -6 -6 -6 2
Frequências de mutação uniformes (1 PAM) A T G C A 2 T -5 2 G -7 -7 2 C -5 -7 -5 2
Transições mais frequentes (3x) que transversões
Matrizes de score para nucleótidos (log odds)
Sij = logb (pi Mij / pipj)
Sij = “log odds” scoreMij = score da matriz de transiçãopi , pj = probabilidades de ocorrência dos nucleótidosb = base do logaritmo (arbitrária)
SA,A = log2(0.25*0.99 /0.25*0.25) 2
ST,A = log2(0.25*0.0033/0.25*0.25) -6
ST,A = log2(0.25*0.0020/0.25*0.25) -5
SG,A = log2(0.25*0.0060/0.25*0.25) -7

Gap penaltiesA inserção ou deleção de porções de uma sequência são eventos raros que conduzem a divergências de comprimento entre sequências homólogas.
Estas diferenças de comprimento implicam a necessidade inserir espaços (“gaps”) num alinhamento, mas esta inserção tem que ser pesadamente penalizada para estar de acordo com a raridade destes eventos.
Existem diferentes esquemas de penalização dos gaps (“gap penalties”),mas todos passam pela atribuição de um score negativo que está geralmente relacionado com o comprimento do gap. Esquemas mais comuns:
• Constante: o tipo mais simples, consistem a atribuir uma penalização constante cada vez que é criado um gap num alinhamento
• Linear: a penalização é proporcional ao comprimento total dos gaps criados no alinhamento, não dependendo do seu número
• Afim (affine gap penalties): as penalizações possuem um termo constante para cada gap criado, e um termo proporcional ao comprimento do gap criado.

Affine gap penaltiesUma representação mais realista do processo de evolução das proteínas deveria penalizar de modo diferente a criação e a extensão de um gap. Para entender este facto, devemos considerar que os alinhamentos entre sequências tem tendência a conter poucos gaps, mas quase sempre com vários resíduos de comprimento.
Se atribuirmos uma penalidade c para a criação de um gap e uma penalidade e para a sua extensão, temos:gp = c + n x e ,em que n é o comprimento do gap.
Não existe uma teoria rigorosa para a escolha de valores para este parâmetros!
Valores usuais:c = -10, e = -2 (FASTA)c = -5… -10, e = -1, -2 (BLAST)c = -12, -2 (Smith-Waterman)
O valor óptimo das gap penalties depende da matriz de score usada!
Reese, JT & Pearson, WR (2002) Bioinformatics, 18:1500-1507

Alinhamentos sub-óptimos: o programa LALIGN
• Neste caso o alinhamento 1 é o alinhamento local óptimo, e os alinhamentos 2 e 3 são alinhamentos sub-óptimos identificados pelo programa LALIGN
• Muitas vezes a análise de alinhamentos sub-óptimos permite a identificação de regiões de similaridade entre duas sequências, não imediatamente reconhecíveis num alinhamento óptimo
3 2 1
f9
f12
http://www.ch.embnet.org/software/LALIGN_form.html
![Adm Serv Info - AULA 7 Out17 [Modo de Compatibilidade]...&rqkhflphqwr 7iflwr (vwi uhodflrqdgr j h[shulrqfld dfxpxodgd h j lqwxlomr grv lqglytgxrv e dtxhoh wlsr gh frqkhflphqwr vreuh](https://img.dokumen.tips/doc/110x75/5ff9c15e6173fa29053d1417/adm-serv-info-aula-7-out17-modo-de-compatibilidade-rqkhflphqwr-7iflwr.jpg)





![ATOS INTERNOS 768 - Bertioga · 7zf 1zn_ 5jwjnwf ij (frutx 'jwyntlf (*5 hvwh vreuh dtxhoh gd dotqhd ³f´ vxfhvvlydphqwh h shuvlvwlqgr r hpsdwh uhdol]du vh i vruwhlr](https://img.dokumen.tips/doc/110x75/5c01991f09d3f26f1e8cfcad/atos-internos-768-7zf-1zn-5jwjnwf-ij-frutx-jwyntlf-5-hvwh-vreuh-dtxhoh.jpg)








