Embed Size (px)
Citation preview

Vanderbilt University and Meharry Medical College
2011
Retreat Lake Barkley State Resort
Park, Cadiz, KY
Tuesday, October 25 and
Wednesday, October 26


Table of Contents • Park Directions, Retreat Schedule, and Miscellaneous Information
• Session 1 – Cardiovascular
• Session 2 – Neurological Disorders
• Session 3 – Neurological Diseases/Drug Discovery
• Session 4 – Cell Signaling
• Poster Session 1
• Keynote Address
• Session 5 – Channels and Transporters
• Poster Session 2
• Session 6 – Bioreactive Epoxides and B(a)P Toxicology
• Save the Date page for 2012 Retreat Information


Vanderbilt University and Meharry Medical College
2011 Retreat
Park Directions, Retreat Schedule,
and Miscellaneous Information
Lake Barkley State Resort Park, Cadiz, KY Tuesday, October 25 & Wednesday, October 26

2010 PHARMACOLOGY RETREAT
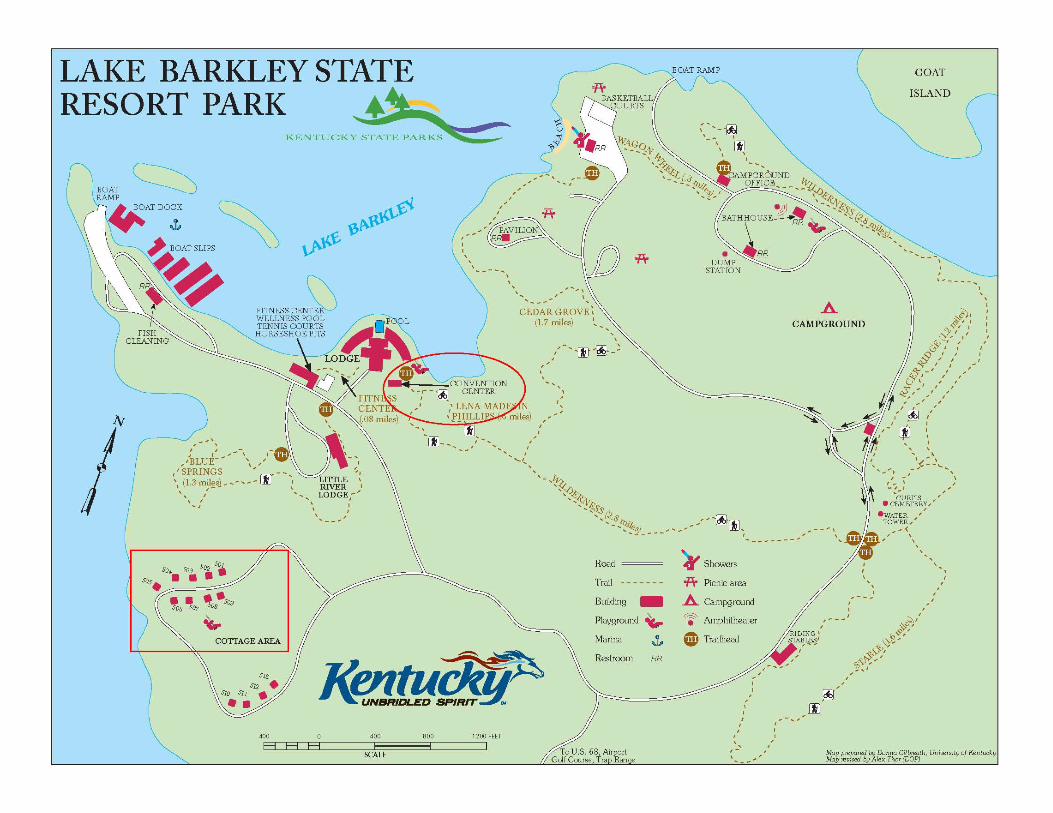
LAKE BARKLEY STATE RESORT PARK 3500 STATE PARK ROAD, CADIZ, KY 42211
TELEPHONE: 270-924-1131 TOLL FREE: 800-325-1708
MAP and DIRECTIONS Directions: From campus, take I-65 N Take a slight LEFT at I-24 W (signs for Clarksville/I-24 W) for approximately 72 miles Take EXIT 65 for KY-80/KY-68 (toward Hopkinsville/Cadiz). Turn LEFT at end of ramp (continue to follow KY-80/US-68 for approximately 9 miles). Turn RIGHT at Blue Springs Rd/KY-1489 (you will see a white sign w/red lettering for Mt. Pleasant Baptist Church). Proceed approximately 3 miles (past soccer fields and the golf course on the left). Turn RIGHT at the stop sign into Lake Barkley Resort State Park. Continue approximately 1 mile. Follow sign to Barkley Lodge (you will take a slight left, then bear right). The Convention Center is at the bottom of the hill.


2011 PHARMACOLOGY RETREAT LAKE BARKLEY STATE RESORT PARK
Tuesday Session – October 25, 2011
9:00 am – 9:45 am
Arrival/Registration (Continental Breakfast available in Convention Center)
9:45 am Welcome and Introductions Michele LeNoue-Newton, Co-Organizer, Vanderbilt University
10:00 am – 11:15 am Session 1 – Cardiovascular Session Facilitator: Jens Meiler
10:00 am – 10:15 am Characterization of Patient Specific Cardiomyocytes Derived from
Induced Pluripotent Stem Cells TK Feaster (Mentor: Charles Hong)
10:15 am – 10:30 am The Role of Inflammation and Adaptive Immunity in Aortic Stiffening Jing Wu (Mentor: David Harrison)
10:30 am – 10:45 am Understanding the role of PRDC in chamber specification during cardiomyocyte development Jeffery Bylund (Mentor: Antonis Hatzopoulos)
10:45 am – 11:00 am The Role of Canonical Wnt Signaling and Endothelial-to-Mesenchymal Transition During Cardiac Homeostasis and Repair Bryan Fioret (Mentor: Antonis Hatzopoulos)
11:00 am – 11:15 am Familial Atrial Fibrillation Mutation KCNQ1-S140G Exhibits Enhanced Sensitivity to Block by the IKs Selective Inhibitor HMR-1556 Courtney Campbell (Mentor: Al George)
11:15 am – 11:30 am Break!!
11:30 am – 12:30 pm Session 2 – Neurological Disorders Session Facilitator: Carrie Jones
11:30 am – 11:45 am Determining the role of M4 muscarinic acetylcholine receptor
activity in the cognitive deficits of schizophrenia Michael Grannan (Mentor: Carrie Jones)
11:45 am – 12:00 pm SLC1A1 and EAAT3: Investigating the Role of Glutamate Transport in Compulsive Behavior Isaac Zike (Mentor: Jeremy Veenstra-Vander Weele)
12:00 pm – 12:15 pm Elucidating the basis for pharmacoresistant epilepsy Lyndsey Anderson (Mentor: Al George)
12:15 pm – 12:30 pm Effects of 2-Chloroethyl Ethyl Sulfide (CEES) on Dopaminergic System Jessica Gadsden-Gray (Mentor: Salil Das)

2011 PHARMACOLOGY RETREAT LAKE BARKLEY STATE RESORT PARK
12:30 pm Group Photo (outside convention center, weather permitting)
12:30 pm – 3:00 pm Lunch & Outdoor Group Activities (weather permitting) 12:30 pm Lunch available in Convention Center
All Retreat Participants: Be sure to be back in the Conference Center between 2:45 and 3:00 pm. The next session of the Retreat will begin promptly at 3:00 pm. 3:00 pm – 4:00 pm Session 3 – Neurological Diseases/Drug Discovery Session Facilitator: Kevin Currie
3:00 pm – 3:15 pm High-Throughput Screening for Allosteric Modulators of the
Presynaptic Choline Transporter Elizabeth Ennis (Mentor: Randy Blakely)
3:15 pm – 3:30 pm Chemical Optimization & Pharmacological Characterization of mAChR5 PAMs Patrick Gentry (Mentor: Craig Lindsley)
3:30 pm – 3:45 pm Pharmacological and behavioral characterization of the novel GlyT1 inhibitor ACPPBII Michael Nedelcovych (Mentor: Ariel Deutch)
3:45 pm – 4:00 pm Heterodimerization of metabotropic glutamate receptors differentially regulates effects of allosteric modulators Shen Yin (Mentor: Jeff Conn)
4:00 pm – 4:15 pm Break!!
4:15 pm – 5:15 pm Session 4 – Cell Signaling Session Facilitator: Gregg Stanwood
4:15 pm – 4:30 pm Structural Basis of Arrestin‐1 Binding to Rhodopsin
Qiuyan Chen (Co-Mentors: Tina Iverson & Vsevolod Gurevich)
4:30 pm – 4:45 pm SNS regulation of IL-6 expression may influence breast cancer metastasis to bone and lung Matthew Karolak (Mentor: Florent Elefteriou)
4:45 pm – 5:00 pm GPR30 Regulates GLT-1 and GLAST expression in Primary Rat Astrocytes Brenya Griffin (Mentor: Eunsook Lee)
5:00 pm – 5:15 pm The Role of Type 3 Transforming Growth Factor β Receptor in Heart
Valve and Outflow Tract Development Jamille Robinson (Mentor: Joey Barnett)
5:15 pm – 5:30 pm Room Check-In
5:30 pm – 6:30 pm Poster Session 1 (group 1 presents) & Wine and Cheese Social

2011 PHARMACOLOGY RETREAT LAKE BARKLEY STATE RESORT PARK
6:30 pm Dinner
7:30 pm
Keynote Address:
“Smoking and Drinking: The role of nicotinic acetylcholine receptors in nicotine dependence and alcoholism” By Andrew Tapper, Ph.D. Assistant Professor of Psychiatry Brudnick Neuropsychiatric Research Institute UMass Medical School Worcester, MA
8:30 pm – whenever
Trivia Night

2011 PHARMACOLOGY RETREAT LAKE BARKLEY STATE RESORT PARK
Wednesday Session – October 26, 2011
7:00 am – 9:00 am
BREAKFAST AT THE INN Room Check out
9:30 am – 10:30 am Session 5 – Channels and Transporters Session Facilitator: Jeremy Veenstra-Vander Weele
9:30 am – 9:45 am Transient Receptor Potential Channels and Astrocyte Reactivity in
Neurodegeneration Karen Ho (Mentor: David Calkins)
9:45 am – 10:00 am Native immune system regulation of brain serotonin release and reuptake Nicole Baganz, Ph.D. (Mentor: Randy Blakely)
10:00 am – 10:15 am Structure-guided analysis of the VU591 binding site in ROMK
Thuy Nguyen (Mentor: Jerod Denton)
10:15 am – 10:30 am A forward genetic screen for novel regulators of dopamine transport in C. elegans Sarah Baas, Ph.D. (Mentor: Randy Blakely)
10:30 am – 11:30 am
Poster Session 2 (group 2 presents)
11:30 am – 12:15 pm Session 6 – Bioreactive Epoxides and B(a)P Toxicology Session Facilitator: Twum Ansah
11:30 am – 11:45 am Exposure to B(a)P in utero predispose LEH rat offspring to
cardiovascular dysfunction in later life George Jules (Mentor: Darryl Hood)
11:45 am – 12:00 pm Isolation and Structural Analysis of Leukotriene A Epoxides: Insights into the Mechanism of the Lipoxygenase-catalyzed Transformation Jing Jin (Mentor: Alan Brash) – alternative category: LIPID SIGNALING
12:00 pm – 12:15 pm Are oxidative metabolites of curcumin novel anti-cancer agents? Odaine Gordon (Mentor: Claus Schneider)
12:15 pm Closing Remarks By Vsevolod Gurevich and Clivel Charlton
12:30 pm – 3:30 pm CRUISE AROUND LAKE BARKLEY
FOR THOSE REGISTERED FOR THE CRUISE AROUND LAKE BARKLEY:
12:30 pm – 12:45 pm Board boat at dock at the Lodge 1:00 pm Departure Time; Cruise out onto Lake 1:00 pm – 3:15 pm Cruise Lake Barkley and Cumberland River 3:15 pm Arrive back at the Dock 3:30 pm Boats tendered at the dock, disembarkation, travel back to Nashville

Miscellaneous Information • The Retreat begins on Tuesday morning at 9:00 am with Registration and Continental
Breakfast. All scientific sessions will be held in the Cumberland/Ohio/Tennessee Rooms at the Convention Center at Lake Barkley. Scientific talks begin at 9:45 am.
• Check-In: For those spending the night, check in time at Lake Barkley is scheduled for 5:15 pm on Tuesday afternoon. There is NO PROVISION FOR EARLY CHECK IN.
• Check-Out: Check out time on Wednesday morning is 11:00 am. There is NO PROVISION FOR LATE CHECK OUT.
• WiFi: Lake Barkley offers free wireless internet access from the public areas in the lodge, lodge rooms, and meeting rooms. All Kentucky State Resort Parks wireless networks are 802.11b and 802.11g compliant. You should be able to access the Internet using any 802.11b or 802.11g wireless network card.
If your laptop computer does not have wireless network capability, you can check-out a wireless access device from the front desk of any resort park. A refundable deposit is required.
• Group Photo: Scheduled for 12:30 pm on Tuesday at a location to be determined.
• Poster Sessions: We have scheduled two poster sessions during this year’s Retreat, with half of the group presenting at each session. The Poster Sessions will be held in the Convention Center. ALL Posters can and should be put on the poster boards in the Convention Center any time throughout the day on Tuesday (but need to be ready before 5:00 pm).
Tuesday’s Poster Session is scheduled from 5:30 pm – 6:30 pm for Group 1. Wednesday’s Poster Session is scheduled from 10:30 am – 11:30 am for Group 2.
• Tuesday Free Time: We have planned for free time on Tuesday afternoon, following lunch. Our group will have use of the Fitness Center as well as the racquetball court, basketball, and tennis courts. They will also set up a volleyball net, washers and corn hole set. All Retreat participants are requested to be back in the Conference Center on time, so that we can begin the next session promptly at 3:00 pm.
• Tuesday Keynote Address: Andrew Tapper, Ph.D., from UMass Medical School, has agreed to be our Speaker at the Retreat following dinner on Tuesday evening. Dinner will be served promptly at 6:30 pm, with Dr. Tapper scheduled to begin his presentation at 7:30 pm.
• Wednesday Breakfast: There is a Buffet Breakfast on Wednesday morning in the restaurant beginning at 7:00 am. If you wish to eat breakfast, please sign the register at the cashier’s stand in the restaurant and help yourself. The charge for breakfast will be put on the Department’s tab.
Beginning at 9:00 am, we will have available inside the Convention Center a continental breakfast.
• Wednesday session begins at 9:30 am in the Convention Center.
• Wednesday Cruise Around Lake Barkley Event: We have planned a Cruise around Lake Barkley following the end of the Retreat on Wednesday. Lunch will be provided for you to take on the boat as well as soft drinks provided. Due to space limitations, you must be pre-registered for the cruise around the lake in order to participate.


Vanderbilt University and Meharry Medical College
2011 Retreat
Session 1: Cardiovascular
Session Moderated by: Jens Meiler
Presentations by:
TK Feaster Jing Wu
Jeffery Bylund Bryan Fioret
Courtney Campbell
Lake Barkley State Resort Park, Cadiz, KY Tuesday, October 25 & Wednesday, October 26

Characterization of Patient Specific Cardiomyocytes Derived from Induced Pluripotent Stem Cells
Tromondae K. Feaster, Jijun Hao, Charles C. Hong
Department of Pharmacology Division of Cardiovascular Medicine, Department of Medicine,
Vanderbilt Institute of Chemical Biology, Vanderbilt University School of Medicine, Nashville,
Tennessee 37232
Congenital heart disease (CHD) is the most common lethal birth defect in USA. In most cases, the causes of isolated, or non-syndromic, CHD remains unknown. While genetic models of CHD in mice have provided general understanding of how mutations in various genes involved in heart development can lead to heart defects, the molecular and cellular pathobiology of human CHD, and the critical gene-environment interactions involved remain poorly defined. Consequently, besides surgical interventions, which are often palliative, there is no rational medical therapy for treatment or prevention of CHD. A key hindrance to detailed mechanistic study of human CHD has been the lack of an appropriate cardiac tissue culture models. To meet to unmet need, we plan to utilize the revolutionary advances in the induced pluripotent stem cell (iPSC) technology to develop an in vitro cardiac tissue model for human CHD. We hypothesize that CHD has a genetic root cause, even in sporadic cases, involving interplay of a number of discrete genetic perturbations. We further hypothesize that that cardiac tissues engineered from pluripotent stem cells reprogrammed from somatic tissues of CHD patients will exhibit consistent and measurable biochemical, cell biological and functional differences from heart tissues engineered from normal controls. We specifically focus on hypoplastic left heart syndrome (HLHS), a spectrum of severe CHDs characterized by dramatic reductions in the sizes of heart chamber. Presently, it is unclear whether the constellation of HLHS structural changes is due to a defect inherent to cardiomyocytes or to defects in other cardiovascular tissues that disrupt heart chamber growth. Here, we generate iPSC from HLHS patients and healthy volunteers, generate cardiomyocytes from iPSC using the latest directed differentiation protocols, and characterize them using number of methodologies that were previously infeasible due to lack of tissue access.

The Role of Inflammation and Adaptive Immunity in Aortic Stiffening
Jing Wu1,2, Salim Thabet2, Wei Chen2, Anna Goldstein2, Meena Madhur4, Billy Hudson3, David Harrison2
1 Pharmacology Graduate Program, Department of Pharmacology; 2 Division of Clinical Pharmacology; 3 Center of Matrix Biology, Department of Nephrology, School of Medicine, Vanderbilt University; 4
Division of Cardiology, Department of Medicine, School of Medicine, Emory University
Clinical studies show an association between inflammation and arterial stiffness. We have previously demonstrated that T lymphocytes mediate experimental hypertension and vascular dysfunction in various models. In addition, T cells‐derived pro‐inflammatory cytokines such as IL‐17 and TGF‐β are known to induce fibrosis. We therefore propose that hypertensive stimuli induce T cell activation and the secretion of cytokines promote aortic stiffening in hypertension. Using masson trichrome staining, we initially observed profound collagen deposition in the aortic adventitia in C57Bl/6J mice receiving chronic angiotensin II infusion (490ng/kg/min, 14 days). We then biochemically determined the alteration of ECM proteins using hydroxyproline assay and ninhydrin assay. Angiotensin II treatment significantly increased collagen content in the aortas of C57Bl/6J mice by more than two fold (1.3±0.1 μg/mm v.s. 3.5 ±0.3 μg/mm, p<0.01) and this increase was further amplified in Recombination Activating Gene‐1 knockout (Rag1‐/‐) mice lacking mature lymphocytes (1.3±0.2 μg/mm v.s. 5.6±0.8 μg/mm, p<0.01). Although elastin content was not reduced, the elastin to collagen ratio was significantly depressed by angiotensin II infusion in both strains (23.4±1.2 v.s. 9.5±0.9, p<0.01; 25.5±3.5 v.s. 7.5±0.9, p<0.01). Consistently, angiotensin II increased aortic wall thickness (p<0.01), media to lumen ratio (p<0.01) while decreased extensibility index (p<0.01) in C57Bl/6J mice. These changes in pathology and mechanical properties were less impressive in Rag1‐/‐ mice (p<0.01 for all three parameters). Collectively, our data demonstrate that arterial stiffening might be caused by alteration in ECM proteins, vascular hypertrophy and loss of elasticity. There is clearly an involvement of inflammation and adaptive immunity in the pathogenesis of aortic stiffening.

Understanding role of PRDC in chamber specification during cardiomyocyte
development
Jeffery Bylund
Stem cells have the potential to become any cell type in the body. Using this potential
to generate cardiomyocytes from stem cells holds promise for creating new clinical
treatments and pre-clinical models for heart disease. Derivation of cardiomyocytes from
stem cells is routinely done in labs across the country; however, these cardiomyocytes
are usually quite heterogeneous, containing several cardiomyocytic subtypes. This
heterogeneity poses a challenge to using them effectively for clinical or preclinical
applications. Gaining a better understanding of how specific classes of cardiomyocytes
develop will aid in the specific generation of more homogenous pools of cardiomyocytes.
Recent work in our lab has indicated that PRDC, a paracrine factor that inhibits BMP
signaling, plays a chamber specifying role in stem cell derived cardiomyocyte
generation. My thesis project aims to understand the mechanism behind PRDC’s
apparent chamber specificity and translate these findings into a human induced
pluripotent stem cell model.

The Role of Canonical Wnt Signaling and Endothelial-to-Mesenchymal Transition During Cardiac Homeostasis and Repair
Omonigho Aisagbonhi, Bryan Fioret, Meena Rai, Sergey Ryzhov, Nick Atria, Igor
Feoktistov and Antonis K. Hatzopoulos
Myocardial infarction (MI) is the most frequent cause of heart injury, leading to critical loss of cardiomyocytes. Cell death triggers a repair response that results in formation of fibrotic scar tissue that compromises cardiac function.
Our results show that canonical Wnt signaling is induced after MI in endothelial and peri-vascular cells during the granulation tissue phase. Our data further suggest that this pathway mediates an endothelial-to-mesenchymal transition (EndMT) that generates a significant portion of endothelial cells and myofibroblasts in and around the infarct area. My hypothesis is that EndMT is a key mechanism of cardiac repair after MI. Moreover, I hypothesize that timely modulation of Wnt/b-catenin signaling after MI may influence the fates of cells participating in cardiac fibrosis and neovascularization. This may provide an opportunity to minimize detrimental fibrosis or enhance beneficial angiogenesis.
My first Aim will investigate the contribution of canonical wnt signaling marked (wnt+) cells during cardiac repair after MI. We have generated TOP-MerCreMer-LacZ mice, a novel transgenic line designed for inducible lineage tracing of wnt+ cells and their progeny. Mice will receive acute MI and hearts will subsequently be isolated throughout the granulation tissue phase to better characterize the role and fate of wnt+ cells in the cardiac repair process.
To gain mechanistic insight into the role of wnt signaling after MI, I will stimulate or antagonize the canonical wnt pathway during different points of the granulation tissue phase. These experiments in Aim 2 will help to determine if the balance between fibrosis and angiogenesis can be altered to improve recovery as measured by histology and echocardiography. TOPGAL mice will be used as a direct readout of Wnt/b-catenin signaling.
Finally, the extent of EndMT contribution during cardiac homeostasis or repair will be explored in Aim 3. SCL-CreERT-LacZ mice will be used for their ability to exclusively label mature endothelial cells and their progeny in an inducible manner. Lineage tracing experiments will determine the dual role that EndMT may have in generating peri-vascular cells during homeostasis but contributing to fibrosis post-infarction.
These studies will provide new information on the biology of cardiac repair, opening the way to design novel approaches to optimize this process in order to attenuate ventricular remodeling and prevent heart failure.

Familial Atrial Fibrillation Mutation KCNQ1-S140G Exhibits Enhanced Sensitivity to Block by the IKs Selective Inhibitor HMR-1556 Courtney M. Campbell and Alfred L. George, Jr. Atrial fibrillation (AF) is the most common cardiac arrhythmia. The contribution of genetic factors to AF susceptibility has been emphasized by the discovery of mutations that enhance outward potassium current, predicted to shorten atrial action potential duration and predispose to re-entrant arrhythmia mechanisms. However, familial AF does not present during childhood suggesting that genetic predisposition alone is not sufficient to cause the disease. Therefore other factors, such as oxidative stress, acquired during life are presumed to interact with the genetically determined cellular substrate, a gene-environment interaction, for the full expression of the clinical phenotype. The effects of these AF-linked mutations on the electrophysiological properties of native cardiac myocytes and how these effects create an AF-prone substrate are unknown. Furthermore, whether and how this predisposition interacts with acquired AF susceptibility factors need to be determined. In Aim 1, we hypothesize that the expression of KCNQ1-S140G in rabbit atrial myocytes will cause shortening of APD when compared to the expression of wild-type (WT) KCNQ1 due to increased outward potassium current density. To test this hypothesis, we will transduce rabbit atrial myocytes with recombinant adenovirus to express WT or mutant channels. Using whole-cell electrophysiological recording, I will examine effects of the mutant channel on potassium current density and APD. These studies will test whether gain-of-function characteristics of KCNQ1-S140G observed in vitro cause increased current density and a shortened APD in atrial myocytes. To isolate IKs specific potassium current and potentially differentiate endogenous from exogenous potassium current in a myocyte, I will use an IKs specific channel inhibitor, HMR-1556. We hypothesized that potassium channel mutations predisposing to AF may have distinct pharmacological properties from wild-type channels. We tested this hypothesis by investigating the effects of HMR-1556, an IKs selective inhibitor, on a gain-of-function familial AF associated mutation, KCNQ1-S140G. We heterologously co-expressed human KCNQ1 or KCNQ1-S140G with human KCNE1 in Chinese hamster ovary (CHO) cells. Plasmids encoded fluorescent transfection markers (KCNQ1-DsRed; KCNE1-EGFP) enabling cells expressing both channel subunits to be selected for whole-cell patch clamp recording experiments. Cells were superfused with control (vehicle) or HMR-1556 (1 nM - 2 µM) solutions while being depolarized every 10 s to +40 mV from a holding potential of 80 mV followed by a voltage step to 30 mV. KCNQ1-S140G co-expressed with KCNE1 exhibited a previously documented constitutively active outward current in contrast to the slowly activating wild-type IKs. Mutant channels exhibited a 10-fold greater sensitivity to HMR-1556 as compared to wild-type IKs (IC50: 18.4 vs 214 nM; p<0.001). Mutant channels exhibited a 10-fold slower off-rate (time constant 514.0 ± 26.2 s) compared with wild-type IKs (time constant 55.8 ± 3.0 s; p<0.001), significantly slower on-rate at all drug concentrations and a steeper concentration dependence of on-rate than the wild-type channel. KCNQ1-S140G/KCNE1 channels exhibit greater sensitivity to block by the IKs-selective blocker, HMR-1556, than wild-type KCNQ1/KCNE1 channels. The differential pharmacological properties of KCNQ1-S140G/KCNE1 suggest a means potentially to differentiate mutant from wild-type current in a myocyte, a possible open state inhibition of IKs by HMR-1556, and a potential opportunity for genotype-specific treatment of familial atrial fibrillation.

Vanderbilt University and Meharry Medical College
2011 Retreat
Session 2: Neurological
Disorders
Session Moderated by: Carrie Jones
Presentations by: Michael Grannan
Isaac Zike Lyndsey Anderson
Jessica Gadsden-Gray
Lake Barkley State Resort Park, Cadiz, KY
Tuesday, October 25 & Wednesday, October 26

Determining the role of M4 muscarinic acetylcholine receptor activity in the cognitive deficits of schizophrenia
Michael Grannan
Recent studies indicate that selective activators of specific subtypes of muscarinic acetylcholine receptors (mAChRs) may provide a novel approach for treatment of cognitive impairments associated with many psychiatric and neurological disorders, including schizophrenia and Alzheimer’s disease (AD). For example, the M1/M4-preferring mAChR agonist xanomeline produces robust decreases in psychotic symptoms, and more importantly, aspects of the cognitive impairments in schizophrenic and AD patients. Over the last several years, Dr. Jones’ lab and others have developed a novel approach to selectively activating individual mAChR subtypes, especially M4, using highly selective positive allosteric modulators (PAMs). These compounds, represented by VU152100, provide an unprecedented opportunity to investigate whether the neurochemical and behavioral effects of mAChR agonists, such as xanomeline, thought to be important for antipsychotic activity and enhancement of cognition are mediated by M4. In the proposed studies, we will take advantage of these novel M4 PAMs along with mAChR KO mice to rigorously test the hypothesis that selective potentiation of M4 activity will produce effects in animal models of acute NMDA receptor blockade, or in a chronic NMDA receptor hypofunction model, specifically NR1 knockdown mice.

SLC1A1andEAAT3:InvestigatingtheRoleofGlutamateTransportinCompulsiveBehavior
IsaacZike3,JeremyVeenstra‐VanderWeele1,2,3,4
DepartmentofPsychiatry1,Pediatrics2,Pharmacology3andKennedyCenterfor
ResearchonHumanDevelopment4,VanderbiltUniversityMedicalSchool,NashvilleTennessee,37323
Obsessive‐compulsive disorder (OCD) affects 2‐3% of the worldwide population.Our current understandings of themolecular causes of OCD are inadequate to beable toattributeaspecificpathophysiology to thedisorder. Theonlyconsistentlyreplicated genetic finding is an association of OCD with the SCL1A1 gene, whichencodesEAAT3,aneuronalandepithelialglutamatetransporterprotein.Evidencefrom neuroimaging studies has identified abnormal activity patterns within thecortical‐striatal‐thalamic‐cortical (CSTC) loop of OCD patients with evidencepointingtowardthestriatumandincreasedEAAT3expression. Currently,thereisnotaconsensusmousemodelofOCDduetotheheterogeneityofthedisorder,butthe Sapap3 null mouse shows compulsive self‐grooming resulting in hair loss, aswell as increased anxiety‐like behavior, capturing two aspects ofOCD. SAPAP3 islocalizedtothepostsynapticdensityofexcitatoryneurons,andknockoutresultsindefective glutamatergic transmission at cortical‐striatal synapses. EAAT3 is alsoexpressed post0synaptically, with prominent expression in the CSTC pathwayimplicated inOCD. TheprimaryfunctionofallEAATs is toclearexcessglutamatefromthesynapticcleft,butEAAT3additionallyactstoprovidesubstratesforGABAandglutathionesynthesis.

Elucidating the basis for pharmacoresistant epilepsy Lyndsey Anderson Epilepsy is a common neurological disorder that is most commonly treated with anti-epileptic drugs (AEDs). Unfortunately, seizure control in approximately one-third of persons with epilepsy is inadequate with currently available AEDs. Various animal models of provoked seizures have been employed to study pharmacoresistant epilepsy, but no in vivo model mimicking spontaneous idiopathic epilepsy has been used for this purpose. We have a transgenic mouse line (Scn2a-Q54) expressing a gain-of-function mutation in the brain sodium channel Nav1.2 that exhibits spontaneous seizures refractory to conventional AEDs. Hippocampal CA1 pyramidal neurons from Q54 mice exhibit hyperexcitability caused by increased persistent sodium current evoked by the mutant Nav1.2 transgene. We recently demonstrated that ranolazine, an FDA approved drug which acts by inhibiting persistent sodium current, significantly reduced seizure frequency in 4 ½ week old (P32) male but not female Scn2a-Q54 mice. We hypothesize that the divergent response to acute ranolazine treatment between male and female Scn2a-Q54 mice is age-dependent and pharmacoresistance occurs when secondary epileptogenic mechanisms (e.g. hippocampal gliosis, neuron loss) become responsible for seizure susceptibility rather than the primary mutation. These secondary mechanisms arise from an accumulative effect of seizures provoked by the primary mutation and therefore should depend on the accumulated effect of seizure burden during the life of the animal. Female F1.Q54 mice exhibit a significantly greater frequency and severity of seizures than male mice at both P21 and P32, suggesting that females have a greater lifetime seizure burden. Consistent with this notion, we have observed an age-dependence of pharmacoresistance in Scn2a-Q54 mice. Acute ranolazine treatment significantly reduces seizure frequency in younger (P21) female Scn2a-Q54 mice and older (P42) male Scn2a-Q54 mice are resistant to acute ranolazine treatment. We predict that suppressing persistent sodium current which chronic ranolazine treatment throughout early adulthood can prevent the emergence of pharmacoresistant epilepsy later in life. To determine whether chronic ranolazine treatment prevents the development of pharmacoresistance, female Scn2a-Q54 mice were treated daily with ranolazine (0.5%) combined with chloramphenicol (0.15%) (to block ranolazine metabolism and prolong drug half-life) administered by supplementation in food. Following drug withdrawal, mice were re-challenged with acute ranolazine treatment. Preliminary results suggest that females exhibit sensitivity to acute ranolazine treatment following withdrawal of chronic ranolazine treatment. Results from experiments exploiting this novel in vivo model of spontaneous, drug resistant epilepsy will contribute new clues about the mechanisms of pharmacoresistant epilepsy and could inspire new treatment strategies for its prevention.

Effects of 2-Chloroethyl Ethyl Sulfide (CEES) on Dopaminergic System Jessica Gadsden-Gray1, Shyamali Mukherjee2, Salil K. Das1. 1Biochemistry and Cancer Biology, 2Professional and Medical Education, Meharry Medical College, Nashville, TN Mustard gas was used in both World Wars, the Gulf War, and on residents of Halabja. Sulfur mustard inhalation causes hemorrhagic inflammation of the tracheobronchial tree and severe pulmonary complications. Studies conducted with 2-chloroethyl ethyl sulfide (CEES) indicated it also crosses the blood- brain barrier. CEES decreases antioxidant defense systems and increases lipid peroxidation in brain by unclear mechanisms. This study’s purpose was to determine if CEES infusion caused abnormality in dopaminergic neurons. Western blotting and immunohistochemistry examined expression of α-synuclein, dopamine transporters, D2 receptors, and tyrosine hydroxylase. Results indicated that CEES caused neurotoxicity by increasing α-synuclein and tyrosine hydroxylase while decreasing D2 receptors and dopamine transporters. This research was supported by grants from the Department of the Army(W81XWH-06-2-0044) and NIH(5T32HL007735-12).


Vanderbilt University and Meharry Medical College
2011 Retreat
Session 3: Neurological
Diseases/Drug Discovery
Session Moderated by:
Kevin Currie
Presentations by: Elizabeth Ennis Patrick Gentry
Michael Nedelcovych Shen Yin
Lake Barkley State Resort Park, Cadiz, KY
Tuesday, October 25 & Wednesday, October 26

Elizabeth Ennis
Pharmacology Retreat 2011
High‐Throughput Screening for Allosteric Modulators of the Presynaptic Choline Transporter
Cholinergic signaling plays a critical role in autonomic function, motor control,
attention, learning and memory. Genetic variation in cholinergic signaling has been implicated in pediatric‐onset myasthenia, depression, attention‐deficit hyperactivity disorder (ADHD) and Alzheimer’s disease. The presynaptic, high affinity choline transporter (CHT) rapidly transports choline from the synapse following the molecule’s hydrolysis by acetylcholinesterase. Our lab cloned mouse and human CHTs and has generated specific CHT antibodies and transfected cell lines for an analysis of CHT mechanism, regulation, and the identification of novel ligands that could prove therapeutic in disorders with altered cholinergic signaling. To pursue the identification of novel CHT‐directed agents, we are conducting a high‐throughput screen (HTS) to identify orthosteric and allosteric inhibitors and potentiators. We propose that these compounds could initiate the development of novel therapeutic reagents to treat disorders with cholinergic dysfunction and bypass some of the dose‐limiting side effects of acetylchoinesterase inhibitors.

Chemical Optimization & Pharmacological Characterization of mAChR5 PAMs Patrick R. Gentry
The five subtypes of muscarinic acetylcholine receptors (mAChR1‐5 or M1‐5) are important to of a wide range of basic physiological functions, including cognitive, sensory, behavioral, motor, and autonomic processes. Recent advances in the discovery of highly subtype‐specific ligands for M1, M2, M3, and M4 has enabled researchers to begin pharmacological characterization of the discrete functions of these individual subtypes; however, discovery of M5‐selective ligands has remained challenging.
Consistent with M5 expression in midbrain dopamine pathways and throughout the cerebrovasculature, phenotypic studies with M5‐knockout mice have suggested that activation of M5 may be therapeutically useful for the treatment of chronic cerebrovascular diseases, acute ischemic stroke, Alzheimer’s disease, and drug addiction.
We recently reported the discovery of the first highly subtype‐selective mAChR5 positive allosteric modulators (PAMs) based on VU0238429, but their modest potency and poor physiochemical profiles limit their in vivo utility as pharmacological probes and thereby preclude their use as potential therapeutics. I aim to further optimize this novel series of M5 PAMs, characterize the lead compounds’ potency and subtype‐selectivity in vitro, and ultimately test the hypothesis that selective M5 activation modulates midbrain dopaminergic functions.

Pharmacological and behavioral characterization of the novel GlyT1 inhibitor ACPPBII
Michael Nedelcovych
Schizophrenia is a debilitating mental disorder with a worldwide prevalence of about 1 percent. Recent evidence suggests that enhancement of glutamatergic neurotransmission via N‐methyl‐D‐aspartate receptors (NMDARs) represents a novel target for pharmacological treatment of schizophrenia symptoms, in particular the negative symptoms and cognitive deficits which typically persist under currently available pharmacotherapies. NMDAR potentiation can be achieved by increasing synaptic concentrations of the receptor co‐agonist, glycine, via blockade of the type 1 glycine transporter (GlyT1). (R)‐N‐[3‐(49‐fluorophenyl)‐3‐(49‐phenylphenoxy)propyl]sarcosine (NFPS) is a potent GlyT1 inhibitor which has been shown to be effective in preclinical models of schizophrenia; it induces severe respiratory and motor side effects, however, which limit its therapeutic utility. Second generation non‐sarcosine‐derived GlyT1 inhibitors have since been developed that display reduced toxicity. Here, I will discuss the pharmacological and behavioral characterization of a novel non‐sarcosine‐derived GlyT1 inhibitor, ACPPBII. Specifically, I hypothesize that ACPPBII will differ significantly from NFPS in its in vitro pharmacological profile and its in vivo effect on glycine concentration in the rodent brain, and that these differences will correlate with a decreased side effect liability. Furthermore, I predict that ACPPBII will be a valuable tool for validating certain animal models of schizophrenia, especially with regard to their replication of social and cognitive deficits.

Heterodimerization of metabotropic glutamate receptors differentially regulates effects of allosteric modulators
Shen Yin1,2, Rocio Zamorano1,2, P. Jeffrey Conn1,2, Colleen M. Niswender1,2
Departments of Pharmacology1 and the Vanderbilt Center for Neuroscience Drug Discovery2, Vanderbilt University Medical School, Nashville, Tennessee 37232
G-protein-coupled receptors (GPCR) represent the most common target of drugs on the market. Metabotropic glutamate receptors (mGluRs) are a group of Family C GPCRs that are activated by glutamate and play important roles in the central nervous system. There are eight mGluR subtypes, which can be further divided into three groups: group I (mGluR1 and 5), group II (mGluR2 and 3) and group III (mGluR4, 6, 7 and 8). Different mGluRs are expressed at distinct synaptic and extrasynaptic locations, but some can be co-expressed at certain regions in the brain. Our lab has been developing small molecule modulators of mGluRs that act via allosteric sites to specifically enhance or inhibit receptor function. This has provided fundamental advances in the discovery of highly selective small molecules that can regulate the activity of different mGluR subtypes. It has been recently discovered that, besides forming constitutive homodimers, mGluRs can also form intra- and inter-group heterodimers, which reveals further complexity of mGluR signaling and function. Therefore, it will be interesting and important to study how heterodimerization of mGluRs affects the ability of allosteric modulators to regulate receptor activity. We found that heterodimerization of mGluR2 and mGluR4 appears to differentially regulate the effect of mGluR4 positive allosteric modulators (PAMs). In HEK cells that express both mGluR2 and mGluR4, most mGluR4 PAMs lose their ability to potentiate mGluR4 response. In contrast, VU0155041, an mGluR4 PAM belonging to a distinct chemical scaffold, displays enhanced potentiation when mGluR2 is present. Our findings suggest that VU0155041 might bind to a different allosteric site compared to other mGluR4 PAMs. These observations further indicate that allosteric modulators of different chemical scaffolds may exhibit distinct abilities to modulate mGluRs in native tissue when different receptors are co-expressed.


Vanderbilt University and Meharry Medical College
2011 Retreat
Session 4: Cell Signaling
Session Moderated by:
Gregg Stanwood
Presentations by: Qiuyan Chen
Matthew Karolak Brenya Griffin
Jamille Robinson
Lake Barkley State Resort Park, Cadiz, KY
Tuesday, October 25 & Wednesday, October 26

Qiuyan Chen Iverson & Gurevich Lab
Structural Basis of Arrestin-‐1 Binding to Rhodopsin G protein coupled receptors (GPCR), as the largest and most diverse family of signaling proteins targeted by almost 50% of clinically used drugs. Arrestins are universal regulators of GPCR signaling, and recently the role of receptor-‐bound arrestins as scaffold proteins for multiple non-‐receptor partners has been established. While it is clear that the conformations of free and receptor-‐bound arrestin are different, there no direct information on the shape of the “active” arrestin in complex with the receptor. The detailed structure of the arrestin-‐receptor complex will improve our understanding of biological functions of arrestins and provide structural basis to generate custom-‐designed mutants that improve the balance of signaling in congenital and acquired disorders. Solution nuclear magnetic resonance (NMR) spectroscopy is a very powerful tool for high-‐resolution structure and studies of protein dynamics in near-‐physiological conditions. Recent technological developments in NMR for large biomolecules make NMR a very promising tool in elucidating the “active” arrestin conformation. Indeed, solution NMR had been successfully applied to mapping the binding sites in arrestin-‐1 for two polyanionic ligands (inositol hexaphosphate (IP6) and heparin) that mimic phosphorylated light-‐activated rhodopsin [2]. The next step is to study the arrestin–rhodopsin interaction using solution NMR. Rhodopsin is a membrane protein, which retains its structure and function only when embedded in membrane system. The traditional lipid vesicles are too large for solution NMR. Although detergent micelles are frequently used as membrane mimics in biophysical studies, they do not always reproduce the native environment of the proteins that may not retain their native structures and functions. Moreover, arrestin, which is a soluble protein, is unstable in the presence of detergent micelles. Thus, it is necessary to find a model membrane system where both rhodopsin and arrestin-‐1 are stable and for the complex tumbles fast enough for NMR studies. Bicelles, amphipols, and fluorinated surfactants are better membrane mimics than detergent micelles. Here we tested thirteen membrane mimics using near UV CD spectroscopy and NMR spectroscopy. Three bicelle systems were successfully identified to reproduce the near UV CD and NMR spectra of arrestin-‐1, similar to those observed in the absence of detergents or lipids. The same bicelle systems were also successfully used for the solubilization of phosphorylated rhodopsin from ROS membranes. References: 1. V. V. Gurevich and E. V. Gurevich (2010), Custom-‐designed proteins as novel therapeutic tools?The
case of arrestins. Expert Rev. Mol. Med. Vol. 12, e13. 2. T. Zhuang et al (2010), Elucidation of InositolHexaphosphate andHeparin Interaction Sites and
Conformational Changes in Arrestin-‐1 by Solution Nuclear Magnetic Resonance. Biochemistry, 49, 10473-‐10475.

SNS regulation of IL-6 expression may influence breast cancer metastasis to bone and lung
Matthew R. Karolak1,3, J. Preston Campbell1,3, Florent Elefteriou1,2,3
Departments of Pharmacology1, Cancer Biology2, and the Vanderbilt Center for Bone Biology3, Vanderbilt University Medical School, Nashville, Tennessee 37232
The sympathetic nervous system (SNS) plays an important role in maintaining body homeostasis. As an important endocrine organ, bone is subject to regulation by the SNS and bone forming cells express β2 adrenergic receptors (β2AR). We have shown that the cytokine milieu in the bone is modulated in mice treated with β1/2AR agonist isoproterenol (Iso). Specifically, receptor activator of NF-κB (RANK) ligand (RANKL) and interleukin-6 (IL-6) expression increases in bone following Iso treatment. Past in vivo and in vitro work has focused exclusively on the RANK/RANKL signaling axis. However, the presence of a positive feedback loop involving RANKL-induced tumor IL-6 expression and IL-6 induced tumor RANK expression has been proposed. This has led us to investigate stomal produced IL-6 in response to SNS activation and Iso treatment. IL-6 is a promiscuous soluble cytokine playing roles in innate and adaptive immune responses, cancer related cachexia, depression, osteoclastogenesis, and tumor cell growth. These functions position IL-6 central to important areas of cancer research. Preliminary data shows that IL-6 expression increases following Iso treatment ~30 fold in pre-osteoblast cells in vitro and ~90 fold in whole bone in vivo. Interestingly, IL-6 expression also increases ~15 fold in lung, another frequent site of breast cancer metastasis. In vivo observations using an orthotopic 4T1 mouse mammary carcinoma model of metastasis showed that chronic immobilization stress (CIS) as a method of increasing endogenous SNS outflow and Iso treatment increased metastasis to the bone and lung. In the CIS model, this could be blocked with propranolol, a β1/2AR antagonist. Intriguingly, CIS increased primary orthotopic 4T1 tumor and subcutaneous MDA-231 human tumor cell growth over controls. This may be IL-6 mediated. Future studies will be directed at elucidating the role of IL-6 in bone metastasis. We will characterize the effects of βAR stimulation on IL-6 expression in various bone cell types, CIS on IL-6 expression in whole bone, and the ability of IL-6 to affect RANK expression in breast cancer cells. We will also explore the effects of IL-6 in vivo on bone and lung metastasis. Finally, we will define the signaling mechanisms of RANK and potentially IL-6 on cancer cell properties such as migration, adhesion, cell growth and apoptotic resistance.

GPR30 Regulates GLT-1 and GLAST expression in Primary Rat Astrocytes Brenya Griffin
Glutamate is an abundant neurotransmitter within the CNS. Excessive glutamate in the synaptic cleft has been involved in many promoting many neurodegenerative diseases, such as manganism, alzheimer’s disease and epilepsy. Glutamate is actively transported from the synaptic cleft through glutamate transporters. The major of glutamate uptake facilitated by astroglial glutamate transporters, GLAST and GLT-1. GLT-1 and GLAST both play a central role in preventing excitatory neurotoxicity by taking up excess glutamate from the synapse into astrocytes. The impairment of astroglial glutamate transporters expression and function is associated with neurodegenerative disease and manganism. 17�-Estradiol (E2) and some selective estrogen receptor modulators (SERMs) such as tamoxifen (TX) exert neuroprotective effects in various experimental and clinical settings; however, the mechanisms involved in E2 -induced neuroprotection remains unclear. Our previous study has shown that GPR30, a G protein-coupled estrogen receptor regulates E2-mediated action on GLT-1/GLAST expression. Therefore, in the present study, we attempted to understand the cellular and molecular mechanisms by which GPR30 enhances the expression of GLT-1/GLAST in rat primary astrocytes. The administration of G1, a selective agonist for GPR30; significantly increased the expression of both GLT-1/GLAST as early as 2 h in primary astrocytes. This GPR30-enhancing expression of GLT-1 was also mimicked by TX, a SERM and partial agonist of GPR30. Treatment with G-15, a selective antagonist for GPR30, blocked G1 action on GLT-1/GLAST expression in astrocytes, demonstrating that G1 action on GLT-1/GLAST expression is mediated through GPR30. We also revealed that intracellular signaling proteins, MAPK and PI3K, mediated G1 action on GLT-1/GLAST expression. Treatment of astrocytes with G1 phosphorylated EGFR at tyrosine 1068 and EGFR inhibitor, AG1478, blocked G1 enhancing effect on GLT-1/GLAST expression, showing that EGFR pathway is involved in GPR30 regulation of GLT-1/GLAST expression. Moreover, G1-mediated increase of GLT-1/GLAST expression was completely blocked by protein kinase A (PKA) inhibitor H89, indicating cyclic-AMP pathway plays an important role in GPR30 regulation of GLT-1/GLAST expression. Co-treatment of G1, E2 and TX with Manganese, (Mn) attenuated the manganese (Mn)-induced NF-kB activation by blocking nuclear translocation of NF-kB. Taken together, the present data demonstrate that G1/GPR30 pathway increases expression of GLT-1 and GLAST via activation of multiple pathways including EGFR and cAMP pathways and that G1 may protect astrocytes by blocking Mn-induced pro-inflammatory NF-kappa B activation.

Jamille Robinson
The Role of Type 3 Transforming Growth Factor β Receptor in Heart Valve and Outflow Tract Development
Our laboratory has demonstrated a role for the Type III TGFβ receptor in valve development, and most recently, in adult cardiovascular function. My work has focused on addressing components of each of these roles. First, I have used a well characterized in vitro assay of valve development to address the roles of specific molecules in Type III TGFβ receptor signaling. In collaboration with other members of the laboratory I have shown that individual members of the Smad family of transcription factors are required, but not sufficient for the endothelial to mesenchymal cell transformation (EMT) that occurs in early valve development. Further, our laboratory has developed gene networks for early valve formation in the chick and mouse. I have selected specific genes from these networks and confirmed a role in EMT. Second, I have examined the effects of aging on the impaired cardiac function seen in heterozygous null mice. These mice demonstrate an 18% decrease in contractility and ejection fraction when compared to wildtype mice. These changes occur without alterations in wall thickness or the generation of fibrosis. Littermate male mice have been serially examined by echocardiography for one year to reveal any further decrement in cardiac function. Our preliminary data suggests that cardiac function, in the absence of additional stressors, remains stable. Currently analyses of gene expression and histologic changes, specifically in valve morphology, are underway.


Vanderbilt University and Meharry Medical College
2011 Retreat
Poster Session #1
Tuesday, October 25 5:30 pm – 6:30 pm
Lake Barkley State Resort Park, Cadiz, KY Tuesday, October 25 & Wednesday, October 26
2010 PHARMACOLOGY RETREAT LAKE BARKLEY STATE RESORT PARK
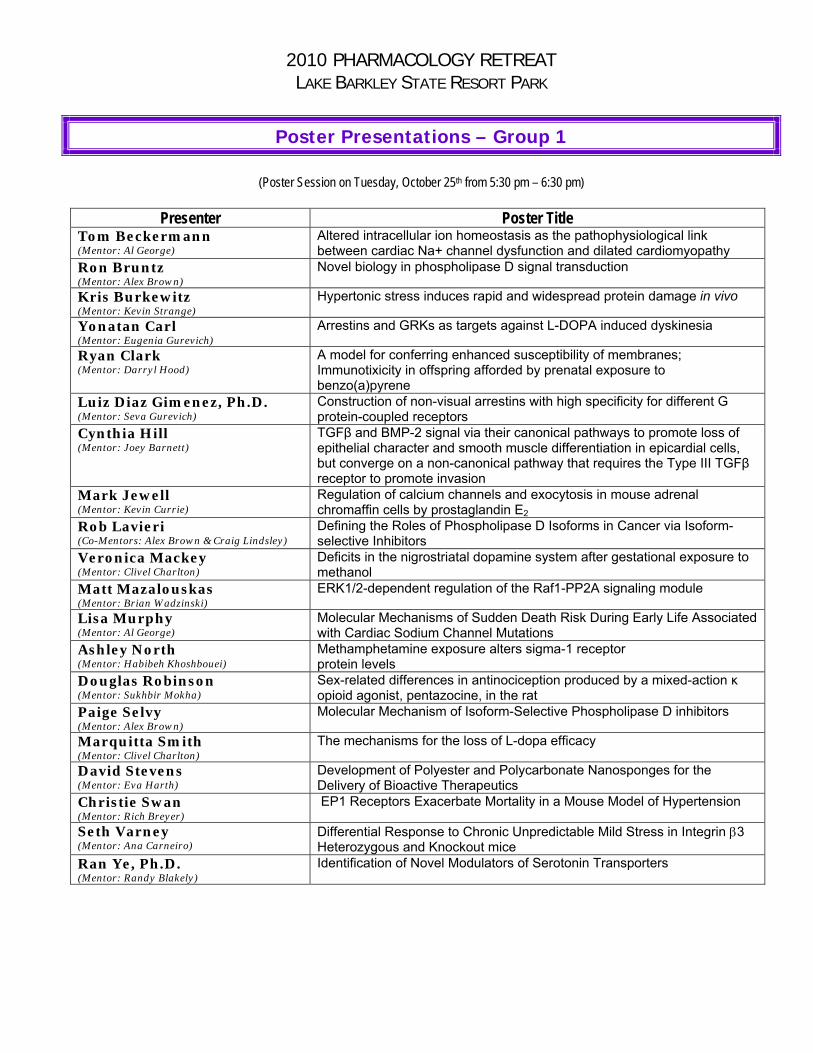
Poster Presentations – Group 1
(Poster Session on Tuesday, October 25th from 5:30 pm – 6:30 pm)
Presenter Poster Title Tom Beckermann (Mentor: Al George)
Altered intracellular ion homeostasis as the pathophysiological link between cardiac Na+ channel dysfunction and dilated cardiomyopathy
Ron Bruntz (Mentor: Alex Brown)
Novel biology in phospholipase D signal transduction
Kris Burkewitz (Mentor: Kevin Strange)
Hypertonic stress induces rapid and widespread protein damage in vivo
Yonatan Carl (Mentor: Eugenia Gurevich)
Arrestins and GRKs as targets against L-DOPA induced dyskinesia
Ryan Clark (Mentor: Darryl Hood)
A model for conferring enhanced susceptibility of membranes; Immunotixicity in offspring afforded by prenatal exposure to benzo(a)pyrene
Luiz Diaz Gimenez, Ph.D. (Mentor: Seva Gurevich)
Construction of non-visual arrestins with high specificity for different G protein-coupled receptors
Cynthia Hill (Mentor: Joey Barnett)
TGFβ and BMP-2 signal via their canonical pathways to promote loss of epithelial character and smooth muscle differentiation in epicardial cells, but converge on a non-canonical pathway that requires the Type III TGFβ receptor to promote invasion
Mark Jewell (Mentor: Kevin Currie)
Regulation of calcium channels and exocytosis in mouse adrenal chromaffin cells by prostaglandin E2
Rob Lavieri (Co-Mentors: Alex Brown & Craig Lindsley)
Defining the Roles of Phospholipase D Isoforms in Cancer via Isoform-selective Inhibitors
Veronica Mackey (Mentor: Clivel Charlton)
Deficits in the nigrostriatal dopamine system after gestational exposure to methanol
Matt Mazalouskas (Mentor: Brian Wadzinski)
ERK1/2-dependent regulation of the Raf1-PP2A signaling module
Lisa Murphy (Mentor: Al George)
Molecular Mechanisms of Sudden Death Risk During Early Life Associated with Cardiac Sodium Channel Mutations
Ashley North (Mentor: Habibeh Khoshbouei)
Methamphetamine exposure alters sigma-1 receptor protein levels
Douglas Robinson (Mentor: Sukhbir Mokha)
Sex-related differences in antinociception produced by a mixed-action κ opioid agonist, pentazocine, in the rat
Paige Selvy (Mentor: Alex Brown)
Molecular Mechanism of Isoform-Selective Phospholipase D inhibitors
Marquitta Smith (Mentor: Clivel Charlton)
The mechanisms for the loss of L-dopa efficacy
David Stevens (Mentor: Eva Harth)
Development of Polyester and Polycarbonate Nanosponges for the Delivery of Bioactive Therapeutics
Christie Swan (Mentor: Rich Breyer)
EP1 Receptors Exacerbate Mortality in a Mouse Model of Hypertension
Seth Varney (Mentor: Ana Carneiro)
Differential Response to Chronic Unpredictable Mild Stress in Integrin 3 Heterozygous and Knockout mice
Ran Ye, Ph.D. (Mentor: Randy Blakely)
Identification of Novel Modulators of Serotonin Transporters

Altered intracellular ion homeostasis as the pathophysiological link between cardiac Na+ channel dysfunction and dilated cardiomyopathy
Tom Beckermann
Mutations in the human heart Na channel NaV1.5 have been linked to a range of disorders affecting cardiac rhythm. More recently, NaV1.5 mutations have also been associated with familial dilated cardiomyopathy (DCM). This unexpected association creates an expanded clinical spectrum of conditions associated with cardiac sodium channelopathies and establishes a need to elucidate the relationship between cardiac Na channel dysfunction and DCM.
Intracellular ion homeostasis in myocytes is tightly regulated by multiple exchangers and transporters including the Na/Ca exchanger (NCX) and the Na/H exchanger (NHE). Through the NCX and the NHE, Na+ is directly coupled to Ca2+ and pH levels within the cell where an abnormally high intracellular [Na+] might affect Ca2+ and H+ handling. Therefore, any change of intracellular Na+ levels due to cardiac sodium channel dysfunction might influence cellular responses. Two NaV1.5 mutations associated with DCM, R814W and R222Q, are predicted to cause an inappropriate influx of Na+ during diastole. Diastolic Na+ influx has the potential of disturbing Na+ and Ca2+ homeostasis that in turn could lead to myocardial damage. We hypothesize that certain cardiac sodium channel mutations such as R814W and R222Q may disturb intracellular Ca2+ and H+ dynamics leading to myocardial failure.
The objective of this project is to establish a mechanistic relationship between Na channel dysfunction and impaired contraction as a consequence of altered ion and/or pH homeostasis in cardiac myocytes. We plan to test this by expressing WT, R814W, or R222Q cardiac sodium channels in isolated guinea pig cardiomyocytes using a lentiviral vector. We will then analyze and compare the effects of these channels on cardiac action potentials, intracellular Ca2+ measurements and pH measurements. We expect to see altered morphology of the cardiac action potential along with alterations in intracellular Ca2+ and H+ levels compared to myocytes expressing wild-type NaV1.5.

Novel biology in phospholipase D signal transduction Bruntz, RC and Brown, HA
Phospholipase D (PLD) enzymes catalyze the hydrolysis of phosphatidylcholine to liberate the bioactive lipid second messenger phosphatidic acid (PtdOH) from a choline headgroup. PLD and its product, PtdOH, are important regulators of cellular processes such as cell proliferation, cytoskeletal remodeling, and migration. In the absence of stimuli, PLD activity is highly regulated with PtdOH production remaining low. Several cancer types including breast, gastric, and renal show increased PLD activity compared to normal tissue. PLD activity is required for H-Ras induced transformation of rat fibroblasts and stable cells overexpressing PLD1 and PLD2 have increased anchorage-independent growth and upregulation of matrix metalloprotease secretion. Furthermore, preliminary data suggests that novel small-molecule PLD inhibitors are able to inhibit migration and invasion of cancer cells in vitro, further implicating the importance of PLD in oncogenic processes. Despite ubiquitous activation of PLD by extracellular stimuli and mediation of critical cellular processes, the molecular targets of PLD and PtdOH remain largely uncharacterized. This project has utilized a proteomic approach to identify novel protein-protein interactions in order to enhance understanding of the regulation and function of PLD. The current focus of this project is to investigate a role for PLD in the regulation of cellular bioenergetics and cell survival through interactions with enzymes responsible for adenosine triphosphate (ATP) synthesis and through an interaction with the anti-apoptotic kinase Akt.

Hypertonic stress induces rapid and widespread protein damage in vivo
Kris Burkewitz, Keith Choe, Kevin Strange
Abstract
Proteostasis is defined as the homeostatic mechanisms that maintain the function of all cellular proteins. We recently demonstrated that the capacity of the proteostasis network is a critical factor that defines the limits of cellular and organismal survival in hypertonic environments. The current studies were performed to determine the extent of protein damage induced by cellular water loss. Using worm strains expressing fluorescently tagged foreign and endogenous proteins and proteins with temperature-sensitive point mutations, we demonstrate that hypertonic stress causes aggregation and misfolding of diverse proteins in multiple cell types. Protein damage is rapid. Aggregation of a polyglutamine yellow fluorescent protein reporter is observable with <1 h of hypertonic stress, and aggregate volume doubles approximately every 10 min. Aggregate formation is irreversible and occurs after as little as 10 min of exposure to hypertonic conditions. To determine whether endogenous proteins are aggregated by hypertonic stress, we quantified the relative amount of total cellular protein present in detergent-insoluble extracts. Exposure for 4 h to 400 mM or 500 mM NaCl induced a 55-120% increase in endogenous protein aggregation. Inhibition of insulin signaling or acclimation to mild hypertonic stress increased survival under extreme hypertonic conditions and prevented aggregation of endogenous proteins. Our results demonstrate that hypertonic stress causes widespread and dramatic protein damage and that cells have a significant capacity to remodel the network of proteins that function to maintain proteostasis. These findings have important implications for understanding how cells cope with hypertonic stress and other protein-damaging stressors.

Arrestins and GRKs as targets against L-DOPA induced dyskinesia
Carl, Yonatan
L-DOPA induced dyskinesia (LID) represents the primary limiting side-effect of L-DOPA therapy against Parkinson's disease akinesia. My study aims to show the role that GRK3 and Arrestin 3 (Arr3) may play in the pathophysiology of LID, as well as the feasibility of GRK3 and arr3 as targets for gene therapy against LID. We have completed in vivo studies to quantify our ability to knockdown GRK3– we hope to show that, like GRK6 knockdown, GRK3 miRNA knockdown in the hemiparkinsonian striatum increases LID. To better understand the in vivo importance of Arr3 in the development of LID, we are now using a hemiparkinsonian mouse model in Arr3 KO mice. As in the rat behavioral model, LID manifests itself as increasing contralateral rotations with repeated L-DOPA challenge. We use the number of rotations as a measure of the degree of dysregulation to striatal dopaminergic signaling. In parallel with these behavioral studies of LID, there are parallel signaling studies to better understand what key players, downstream of arr3, are implicated in the development of dopaminergic signaling supersensitivity.

A MODEL FOR CONFERRING ENHANCED SUSCEPTIBILITY OF MEMBRANES; IMMUNOTIXICITY IN OFFSPRING AFFORDED BY PRENATAL EXPOSURE TO BENZO(a)PYRENE. Ryan Clark, Department of Neuroscience and Pharmacology, Environmental Health Disparities and Medicine, Center for Molecular and Behavioural Neuroscience, Meharry Medical College, Nashville, TN 37208, USA
Polycyclic aromatic hydrocarbons (PAHs) are a well‐studied group of environmental toxicants that have been demonstrated to be mutagenic, carcinogenic and neurotoxic. The mechanism as to how PAHs suppress the immune system is not well understood. Our investigation into the mechanism will begin with lipid rafts that are found in the plasma membrane that consist of a combination of cholesterol, glycosphingolipids, and protein receptors organized in glycolipoprotein microdomains. Our initial aim is to investigate whether exposure to B(a)P disrupts the homeostatic lipid raft composition, therefore, our hypothesis is in utero exposure to B(a)P alters lipid raft composition in offspring and enhances susceptibility to later‐life infection. Timed‐pregnant Long‐Evans Hooded dams were exposed by oral gavage to a single dose of B(a)P (600µg/kg BW) on embryonic days 14‐17. Offspring were born on E21 and mesenteric lymph nodes (MLN) were collected on P100. Lipid rafts from MLN were isolated using sucrose gradient centrifugation, and the lipid fractions were verified by dot blot analysis with CD59 and CD55. In parallel, fractions were analyzed for B(a)P metabolites. The preliminary results demonstrate that in utero B(a)P exposure alters homeostatic lipid raft composition in offspring mesenteric lymph nodes as evidence by the co‐elution of B(a)P metabolites with CD59 verified lipid‐raft fractions. The results lend credence to an overarching hypothesis that altering cholesterol domains within membranes may serve as a mechanism of membrane destabilization. Such destabilization may allow for the incorporation of host lipid raft proteins by viruses. Based on these fundamental concepts, our model predicts that in utero B(a)P‐exposure will disrupt the lipid raft organization of the membrane and may serve to render host T‐cells more susceptible to infection.
Supported by RISE
School of Graduate Studies and Research
2nd year student Dr. Darryl B. Hood 703 Watts Circle Nashville, TN 37209 (615)739‐6394

Construction of non-visual arrestins with high specificity for different G protein-coupled receptors.
Luis E. D. Gimenez and Vsevolod V. Gurevich.
Department of Pharmacology, Vanderbilt University Medical Center, Nashville, TN 37232.
Arrestins are a small family of proteins that participate in agonist-induced desensitiza-tion of G protein-coupled receptors (GPCRs). Arrestins bind to phosphorylated active receptors terminating G protein-mediated signaling, targeting of receptors to endocytic vesicles and initiating a second round of signaling. In vertebrates, arrestin-1 and arres-tin-4 are confined to photoreceptors regulating rhodopsin and opsin signaling. In con-trast, the ubiquitously expressed arrestin-2 and -3 interact with the widely varied binding interfaces offered by numerous GPCRs. We identified elements that determine the bind-ing of arrestin-3 (arr-3) to receptors. Based on previous work that established the amino acids implicated in defining the affinity of arrestin-1 for active rhodopsin or arrestin-2 for M2 acetylcholine receptors (M2R) and on the analysis of the evolutionary trends shown by the members of the arrestin family in different species, we introduced a set of muta-tions in arr-3. These mutations were targeted to specific sites on the N- and C-domains of arrestin that form part of the arrestin – receptor interface. Mutants were constructed on the A87V background that stabilizes the β strand “sandwich” in the N domain via hy-drophobic interactions. The dynamics of the association of different receptor types with the mutated arr-3 were assessed by live cell-based arrestin recruitment using biolumi-nescence energy transfer (BRET) between receptors fused to luciferase and the N-terminal fusion of the arrestin mutants with Venus (enhanced YFP). Mutants D260K and Q262P showed enhanced recruitment by M2R and D1 – D2 dopamine receptors (D1R and D2R respectively), as compared to β2-adrenergic receptors (β2AR). This preference profile was further enhanced when D260K and Q262P were combined. Conversely, Y239T induced enhanced binding of arr-3 to β2AR while diminishing binding to M2R, D1R and D2R. Mutation Q257Y reduced arrestin recruitment to D2R. Interestingly, when Y239T and Q257Y were combined, a mixed effect was obtained being null for β2AR, further enhancing D1R arr-3 recruitment over the other receptors and reducing D2R arrestin recruitment; hence the Y239T – Q257Y combination had opposite effects on D1R and D2R binding. Thus, the manipulation of the key residues on the receptor-binding surface significantly modifies receptor preference, paving the way to construc-tion of non-visual arrestins specific for particular GPCR subtypes. This opens the possi-bility of constructing non-visual “enhanced” arrestins specifically targeting the receptor of choice for research and therapeutic purposes.
Support: This study was supported by NIH grants GM081756, GM077561 and EY011500 (VVG)

TGFβ and BMP-2 signal via their canonical pathways to promote loss of epithelial character and smooth muscle differentiation in epicardial cells, but converge on a
non-canonical pathway that requires the Type III TGFβ receptor to promote invasion
Cynthia Hill
Over half of all cardiovascular disorders in the United States affect the coronary arteries. Understanding the role of cell populations and molecules that regulate coronary vessel development can reveal novel drug targets and therapeutic strategies to direct the repair or remodeling of coronary vessels. Given the importance of the epicardium during coronary vessel development we focused our efforts on identifying the actions of specific growth factors on epicardial cell behavior and differentiation. Since BMP-2 binds the Type III Transforming Growth Factor β Receptor (TGFβR3), a receptor critical for coronary vessel development, we examined the effects of BMP-2. 5nM BMP-2 induced loss of epithelial character, but failed to induce the smooth muscle markers, SM22α, SMαA, or calponin. In contrast, 250 pM TGFβ1 or TGFβ2 induce loss of epithelial character and smooth muscle differentiation The ALK5 inhibitor, SB431452, specifically blocked the actions of TGFβ1 or TGFβ2, but not BMP-2. Conversely, DMH1 (ALK2/3 inhibitor) blocked BMP-2, but had no effect on TGFβ. Thus, BMP-2 and TGFβ act through distinct pathways to drive the loss of epithelial character and smooth muscle differentiation. Using a modified Boyden chamber assay, BMP-2, TGFβ1, or TGFβ2 induced invasion of Tgfbr+/+, but not Tgfbr3-/- epicardial cells into a collagen gel. Restoring full length TGFβR3 in Tgfbr3-/- cells rescued deficits in invasion in response to BMP-2, TGFβ1, and TGFβ2. Expression of TGFβR3 missing the 3 C-terminal amino acids that are required to interact with the scaffolding protein GIPC1 did not rescue any of the deficits. Overexpression of GIPC1 alone in Tgfbr3-/- cells did not rescue invasion whereas knockdown of GIPC1 in Tgfbr3+/+ cells decreased invasion in response to ligand. Therefore TGFβ1, TGFβ2 and BMP-2 use distinct pathways to drive the loss of epithelial character in 2-dimensions, but use a converging pathway for invasion that requires TGFβR3.

Regulation of calcium channels and exocytosis in mouse adrenal chromaffin cells by prostaglandin E2. *M. L. JEWELL1, R. M. BREYER2, K. P. M. CURRIE3; 1Pharmacol., 2Medicine, Biochem. and Pharmacol., 3Anesthesiology, Pharmacol, Ctr. for Mol. Neurosci., Vanderbilt Univ., Nashville, TN Adrenal chromaffin cells express G protein coupled receptors (GPCRs) that can inhibit or potentiate catecholamine release. For example, P2Y purinergic and μ-opioid receptors produce G-mediated inhibition of voltage gated Ca2+ channel currents (ICa) and exocytosis. In this study we have investigated the effects of prostaglandin E2 (PGE2), the most widely expressed prostanoid, on stimulus-secretion coupling in chromaffin cells. PGE2 acts in an autocrine or paracrine fashion on specific GPCRs, designated EP receptors (EP1-EP4), to control blood pressure through effects on vascular smooth muscle, controling salt and water reabsorption in the kidney, and by modulating neurotransmitter/hormone release. It has been proposed that EP receptor subtype selective drugs (e.g. EP1 antagonists) could be developed as novel antihypertensives. Adrenal catecholamines help control blood pressure and heart function, and previous work suggests that PGE2 can alter catecholamine release. However, the cellular mechanisms, receptors, and even the net effect on catecholamine release remain unclear. Using PCR techniques we have identified mRNA for all four EP receptor subtypes in mouse adrenal glands. Using whole cell patch-clamp recording we show that PGE2 (100nM) produced robust inhibition of ICa (43 ±6%) that was reversed by a strong depolarizing prepulse, a signature of G-mediated inhibition of N- and P/Q- type Ca2+ channels. Sulprostone, a selective EP1/EP3 receptor agonist, also inhibited ICa. The inhibition produced by PGE2 was abolished in cells isolated from EP3 receptor knockout mice. P2Y receptor-mediated inhibition of ICa remained intact in cells from knockout mice indicating that the channels and G-mediated signaling pathways were not disrupted. To probe ICa and secretion simultaneously, we combined perforated patch-clamp recording and carbon fiber amperometry. Initial results indicate that even though PGE2 inhibited ICa (~60%), there was a net potentiation of catecholamine release (~50%). Activation of endogenous H1 receptors by histamine has been shown to potentiate exocytosis in bovine chromaffin cells by actions downstream of phosphatidylinositol (PI) hydrolysis. Our data suggest that in addition to inhibitory effects on EP3 receptors, PGE2 potentiates catecholamine release through actions on another EP receptor subtype, perhaps the EP1 receptor that can increase PI turnover. The overall impact of PGE2 on catecholamine release will reflect the net sum of these opposing actions and could be shifted by changes in relative receptor expression and/or receptor subtype selective drugs.

Defining the Roles of Phospholipase D Isoforms in Cancer via Isoform-selective Inhibitors Robert R. Lavieri, Sarah A. Scott, Paige E. Selvy, Kwangho Kim, J. Scott Daniels, H. Alex Brown and Craig W. Lindsley Phospholipase D (PLD) catalyzes the production of the lipid second messenger phosphatidic acid (PA). PLD expression and/or enzymatic activity are both elevated in a variety of human cancers. Inhibition of PLD enzymatic activity, via genetic or biochemical methods, leads to decreased cancer cell invasion and decreased cancer cell survival. The aforementioned evidence provided the impetus for our medicinal chemistry project focused on the development of isoform-selective PLD inhibitors. The development of such inhibitors is an essential step in advancing the study of PLD as a potential cancer drug target. A group from Novartis published a report in 2007 disclosing halopemide as a hit from a high throughput screen for PLD inhibitors. While we initiated our iterative analog synthesis with halopemide we have explored a broad chemical space. We utilized technology-enabled synthesis to develop a library of approximately 600 compounds. This effort has yielded the most potent, isoform-selective PLD inhibitors described to date. While halopemide inhibits both PLD isoforms relatively equally VU0359595 inhibits PLD1 1,700 times more potently than PLD2, and VU0364739 inhibits PLD2 75 times more potently than PLD1. In a triple negative breast cancer cell line, MDA-MB-231, VU0364739 significantly decreases cell viability whereas VU0359595 does not affect cell viability; indicating that a PLD2 selective inhibitor may be the optimal therapeutic agent in this particular malignancy. We are currently utilizing our novel, isoform-selective PLD inhibitors in order to more clearly characterize the role of PLD signaling in oncogenic processes such as the suppression of apoptosis, cell invasion and metastasis.

Deficits in the nigrostriatal dopamine system after gestational exposure to methanol Veronica R. Mackey, Muthian G, King J and Charlton C.G. Department of Neurobiology and Neurotoxicology, Meharry Medical College, Nashville, TN 37208 About 1 million Americans are diagnosed with sporadic Parkinson's disease (PD), the second most common neurodegenerative disorder after Alzheimer's disease. Individuals with PD experience symptoms such as bradykinesia, postural instability and resting tremor, as the result of the degenerated nigrostriatal (NS) dopamine (DA) neurons. The etiology of PD remains unknown, but it is possible that two stages are involved. The 1st stage involves the exposure to environmental toxicants during the fetal stage of life that alters the nigrostriatal neurons making them vulnerable, and the 2nd occurred when age-related stress further weaken the vulnerable neuronal set and causing the symptoms of PD. To test the 1st stage hypothesis, pregnant C57bl/6J dams were administered a sub-acute dose of methanol (MeOH), 4 mg/kg, twice daily during the period of neurogenesis of the NS DA neurons. This we hope will initiate the vulnerable stage in the offspring. Twelve wks after parturition the offspring were sacrificed and the striatum was dissected and tyrosine hydroxylase (TH), neurofilament (H), α-synuclein, and dopamine and its metabolites were measured. The brain was also fixed and Nissl stained; enzymatic activity of TH was measured. As compared to the PBS group the SN of the MeOH group showed intensely Nissl stained, elongated neurons with spindle-like shape. In addition, the extracellular regions showed wider and larger intracellular space and unstained intracellular zones that may be indicative of disruption of the extracellular matrix. Moreover, Western blot analysis revealed decreases in striatal TH, alpha-synuclein and neurofilament (H) protein levels, by about 40%, 36% and 41.3%, respectively, and prenatal MeOH decreased the levels of DA and increased DOPAC/DA ratio by 44% and 23%, respectively, in MeOH treated offspring. The study shows that MeOH is capable of interacting with the nigrostriatum and causing aberrations that may serve as the underpinning for PD-like changes that occur later in life. It is note worthy that the sparseness of the intracellular space in the MeOH SN group is indicative of interference with DA neuronal migration into the substantia nigra. This study may be a positive test for the “1st stage in PD concept”.

ERK1/2-dependent regulation of the Raf1-PP2A signaling module Matthew D. Mazalouskas and Brian E. Wadzinski Raf1 (a.k.a. c-Raf) is a protein serine/threonine kinase that has an essential role in eliciting specific cellular responses by transferring upstream growth factor receptor signaling information to the downstream mitogen activated protein kinases (MAPKs) MEK1/2 and ERK1/2. Signal transduction via the Raf1-MEK1/2-ERK1/2 cascade is tightly controlled by several protein phosphatases. Protein serine/threonine phosphatase 2A (PP2A) functions as a key modulator of this signal transduction cascade by targeting multiple components in this pathway. The predominant form of PP2A in cells is a heterotrimeric holoenzyme composed of a structural A subunit, a regulatory B subunit, and a catalytic C subunit. Previous work from our lab has revealed that PP2A holoenzymes containing the Bα or Bδ regulatory subunit associate with Raf1 and function as a positive regulator of Raf1-MEK1/2-ERK1/2 signaling by directly dephosphorylating the inhibitory phospho-Ser259 residue in Raf1 [Adams D. et al. (2005) JBC 280:42644]. Not only are the cellular signals that regulate the Raf1-associated PP2A currently unknown, but also the mechanism by which ERK1/2 feeds back to regulate Raf1 is unclear. Therefore, to explore the possibility that ERK1/2 mediates its effects on Raf1 via the Raf1-associated PP2A, cells that stably express Bδ-FLAG, or empty vector control, were pretreated with the MEK1/2 inhibitor PD98059 before stimulation with epidermal growth factor (EGF). Immunoprecipitations from EGF-stimulated cells, pretreated with and without PD98059, indicate the association of the Bδ containing PP2A holoenzyme and Raf1 is affected by the presence of the inhibitor. Ongoing experiments are aimed at elucidating this feedback loop mechanism, shedding light on whether this loop positively or negatively regulates Raf1 signaling to downstream effectors and if ERK1/2 directly or indirectly targets the Raf1-associated PP2A.

Molecular Mechanisms of Sudden Death Risk During Early Life Associated with Cardiac Sodium Channel Mutations Lisa L. Murphy, Alfred L. George, Jr, M.D. Department of Pharmacology; 2Division of Genetic Medicine; Department of Medicine; Vanderbilt University Medical Center, Nashville, TN Mutations in SCN5A encoding the cardiac voltage-gated sodium channel NaV1.5 are associated with sodium channel dysfunction that can result in life-threatening cardiac arrhythmias such as in the long-QT and Brugada syndromes. SCN5A mutations have been associated with severe neonatal forms of long-QT syndrome (LQTS) and rare cases of intrauterine fetal demise. Our lab has demonstrated prominent expression of an alternatively spliced NaV1.5 mRNA transcript in fetal and neonatal human heart that differs from the adult isoform by several residues within a voltage-sensor domain (D1/S3-S4). We hypothesize that fetal NaV1.5 will provide a permissive background for fetal mutations and rare variants identified in SIDS that will result in exacerbation of cardiac sodium channel dysfunction. To test this hypothesis we investigated the functional consequences of a de novo SCN5A mutation, L409P that was identified in a 19-week fetus with LQTS and torsade de pointes that resulted in termination of the pregnancy. The fetus also demonstrated homozygosity for a common polymorphism R558. In vitro electrophysiological studies demonstrated that L409P in combination with R558 causes significant depolarized shifts in voltage-dependence of steady-state inactivation and activation, accelerated recovery from inactivation, and a 7-fold increase in persistent current (1.4% of peak current vs 0.2% for WT). Furthermore, when the mutation was expressed in the background of fetal NaV1.5, channel dysfunction was potentiated with a marked increase in persistent current (11% of peak current) and greater shifts in voltage- dependence. Future aims of this work will focus on the functional consequences of several other fetal mutations and rare variants identified in SIDS in the context of the fetal splice variant and we will also investigate the pharmacological effects of select sodium channel blockers on mutant fetal NaV1.5 channels. Most importantly, we will investigate how SCN5A mutations associated with early life arrhythmia susceptibility will affect action potential morphology and duration. These studies will elucidate the biophysical properties of fetal NaV1.5 and will give important new insight to arrhythmia susceptibility manifested during early life.

Methamphetamine exposure alters sigma-1 receptor protein levels
Ashley North, Jarod Swant, Sanika Chirwa, John Clark, Twum Ansah, Petra Prins, and Habibeh Khoshbouei
Methamphetamine, a substrate for the dopamine transporter, is a powerful psychostimulant that decreases dopamine uptake and releases dopamine via the reverse transport process. Chronic methamphetamine exposure is neurotoxic and can cause long-lasting alterations in synaptic transmission and behavioral sensitization- a phenomenon implicated in the development of psychosis in humans. Sigma receptors are found on dopaminergic neurons and have been hypothesized to modulate dopaminergic neurotransmission. It has been shown that methamphetamine preferentially interacts with sigma-1 receptor, and blockade of sigma-1 receptors diminishes methamphetamine-induced hyper-locomotion in rats. Our preliminary data suggest that chronic methamphetamine exposure is neurotoxic and can lead to long-lasting deficits in working memory in addition to alteration in hippocampal neurotransmission. Consistent with the literature, we found that chronic methamphetamine exposure is neurotoxic and decreases TH and DAT in the striatum and hippocampus of mice exposed to neurotoxic regimen of methamphetamine for two weeks. Interestingly, we discovered that methamphetamine cessation, but not active drug exposure leads to long-lasting deficits in short-term memory as measured by novel object and novel spatial recognition tasks. Similarly, we found that drug cessation, but not its active exposure affected hippocampal neurotransmission as determined by a decrease in the long-term potentiation (LTP) in conjunction with a decrease in paired pulse facilitation (PPF). We then, asked whether this chronic methamphetamine exposure can trigger compensatory neuroprotective mechanisms in the brain. Our data so far suggest that chronic methamphetamine exposure alters sigma-1receptor protein levels within the cortex, frontal cortex, striatum, cerebellum, and hippocampus of mouse brain. Importantly, methamphetamine-mediated changes in sigma-1receptor protein levels are structurally divergent. The underlying molecular mechanism is currently under investigation.

Sex-related differences in antinociception produced by a mixed-action κ opioid agonist, pentazocine, in the rat Douglas L. Robinson, Jr., Subodh Nag, and Sukhbir S. Mokha, Department of Neuroscience and Pharmacology, Meharry Medical College, Nashville, TN 37208 Research has shown that women have a higher prevalence of many pain syndromes such as irritable bowel syndrome, migraine, and temporomandibular joint pain. Sex-related differences in antinociception produced by mixed-action kappa opioid receptor (KOR) agonists, nalbuphine, pentazocine, and butorphanol have been reported in human studies during post-dental surgery treatment with women having a greater analgesic effect. The mixed-action kappa opioid agonists display affinity for the KOR and the mu opioid receptor (MOR). However, the extent to which the analgesic effect is mediated by the KOR or MOR still remains unknown. Hence, the current investigation examined whether there are sex-related differences in antinociception produced by pentazocine, and the relative contribution of KOR and MOR in mediating the antinociceptive effect of pentazocine. The heat-evoked tail withdrawal assay was conducted in rats and the tail flick latency (TFL) was recorded automatically. Stretched PE10 cannulae were implanted in the subarachnoid space of the lumbosacral spinal cord of Sprague- Dawley rats. Pentazocine (250 nmol/ 10 µL) administered intrathecally induced an increase in tail flick latency in estradiol treated ovariectomized animals (OVX+E), but not in OVX animals or males. In addition, intrathecally administered (5 minutes before pentazocine) nor-BNI or CTAP, KOR and MOR antagonists, respectively, abolished pentazocine’s antinociceptive effect. Thus, these data led us to conclude that pentazocine produces estrogen-dependent, sex-specific antinociception, which is mediated by KOR and MOR. These results suggest a potential use of pentazocine in women suffering from pain disorders during reproductive years.

Molecular Mechanism of Isoform‐Selective Phospholipase D inhibitors Paige E. Selvy, Craig W. Lindsley, Darryl J. Bornhop, H. Alex Brown Increasing evidence supports an integral role for the lipid second messenger phosphatidic acid (PtdOH) in cell signaling. In addition to altering curvature of biological membranes, PtdOH lies at the intersection between metabolism and signaling pathways and is involved in cell survival as well as proliferation pathways. Phospholipase D (PLD), an evolutionarily conserved phosphodiesterase that hydrolyzes phosphatidylcholine, is involved in generating signal‐mediated PtdOH molecular species. Aberrant PLD expression and activity have been implicated in metastatic cancers, neurological disorders, and microbial pathogenesis, making this enzyme a potential therapeutic target for which we recently developed potent and isoform‐selective pharmacological inhibitors. Here we define the mechanism of action for these novel small molecule inhibitors. Using backscattering interferometry (BSI), a novel method for measuring protein‐small molecule and protein‐lipid binding, we demonstrate that these compounds directly interact with PLD to allosterically block interfacial lipid binding and catalytic activity. This unanticipated finding represents a unique mechanism of phospholipase inhibition and a new paradigm for modulation of signaling pathways.

The mechanisms for the loss of L-dopa efficacy
Marquitta Smith
The mechanisms for the loss of 3,4-dihydroxyphenylalanine (L-dopa) efficacy and the presence of L-dopa-induced-dyskinesia (LID) during the treatment of Parkinson’s disease (PD) are unknown. Modifications related to the catecholamine (CA) metabolic pathway are seen as the primary considerations, and include changes in L-dopa and dopamine (DA) metabolism, the modulation of CA enzymes and the production of interfering metabolites. The goal of this project is to find out if exposure to L-dopa compromises the synthesis and metabolic capacity of the dopaminergic system in a model of PD. In this project we used a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of PD to study the effects of sub-chronic L-dopa on the striatal accumulation of L-dopa and the levels of DA, 3-O-methyldopa (3-OMD), 3,4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA). A group of MPTP mice were primed with 100 mg/kg of L-dopa twice a day for 14 days, and a matching group remained L-dopa naïve. Both groups treated with 100 mg/kg of L-dopa were sacrificed 30 min later and their striatal L-dopa, 3-OMD, DA, DOPAC and HVA were determined. In L-dopa naïve mice, the acute L-dopa increased brain L-dopa by 38.72% and DA by 176%, whereas in L-dopa primed mice, brain L-dopa was reduced by 17.03% and DA increased by only 30%. For the L-dopa naïve vs the L-dopa primed mice, 3-OMD was increased by 1,419 vs 3,186% and DOPAC by 236 vs 430%, and HVA by 325 vs 369%, respectively. The comparative study shows that sub-chronic exposure to L-dopa decreased the ability of the striatum to accumulate L-dopa and to produce an adequate level of DA, which is evidence to support a metabolic adaptation in the CA pathway in explaining the loss of L-dopa efficacy. The significant increase in 3-OMD supports previous data from our lab that indicates increased L-dopa methylation is due to the induction of COMT (catechol-o-methyltransferase) and MAT (methionine adenosyltransferase). The increase in DOPAC is indicative of increased oxidation of DA and likely the induction of monoamine oxidase (MAO).

Development of Polyester and Polycarbonate Nanosponges for the Delivery of Bioactive Therapeutics
David M. Stevens, Harry Watson, Ray Wang and Eva Harth
Vanderbilt University, Department of Chemistry and Pharmacology, Nashville, TN 37235‐1822
We present the preparation of polyester and polycarbonate “nanosponges” through an interchain cross‐linking process to generate delivery systems that are tailored for intravenous or oral delivery of biologically active cargo. Two cross‐linking chemistries were investigated such as the epoxide‐amine and the thiolene‐click chemistry to form particles that can be loaded after formation or during the cross‐linking process. Functionalized linear polycarbonate precursors are difficult to prepare and we present synthetic pathways that were employed and evaluated. We found that presence of the Sn(OTf)2 catalyst in ring‐opening polymerization reactions offers the most practical synthesis to gain access to polycarbonates with controlled molecular weights and narrow polydispersities. For the first time it was shown that these functionalized precursors can be transformed into supramolecular 3‐D network structures using either the thiol‐ene click or the epoxide amine cross‐linking reactions. Polyester particles are synthesized to function as targeted delivery systems for the “BH3 domain”, pro‐apoptotic peptides, of the BH3‐only protein to inhibit pro‐survival proteins of Bcl‐2 and trigger the apoptotic cascade.

EP1 Receptors Exacerbate Mortality in a Mouse Model of Hypertension
Christina E Swan, Kelli L Boyd, Roy Zent, and Richard M. Breyer.
C57BL/6 mice with a disrupted Prostaglandin E2 receptor EP1 allele (EP1-/-; n = 35) or control mice
(EP1+/+; n = 58) were uninephrectomized (Nphx), and 14 days later 50 mg deoxycorticosterone
acetate (DOCA) was administered s.c. concurrent with 1% NaCl in the drinking water. After an
additional seven days, angiotensin II (Ang II) was administered (1.5 ng/min/g s.c. Alzet minipump).
High mortality was observed in EP1+/+ mice (60%) compared to EP1-/- mice (24%, P = 0.004). Mean
intracarotid pressure (MAP) was elevated in all mice following the administration of Ang II, but was
significantly lower in EP1-/- mice vs EP1+/+ (EP1+/+ 128.8 ± 5.1 mmHg n=4, EP1-/- 102.4 ± 7.8
mmHg n=4, P = 0.03). 35% of EP1+/+ mice compared to 10% of EP1-/- mice died of aortic aneurysm
rupture . Overall, 65% of EP1+/+ mice and 40% of EP1-/- mice had formation of aortic aneurysms.
EP1+/+ mice developed severe edema at 5 days post-Ang II, while EP1-/- mice were protected
(EP1+/+ vs EP1-/-, P = 0.014). While moderate hypertensive damage was induced in the kidney of
both groups, no differences in albuminuria or renal histopathology were observed between groups.
Furthermore clinical biomarkers of renal damage, Kim-1 and Ngal mRNAs, were elevated to a
similar extent in the kidney of both groups following administration of Ang II. In an effort to
demonstrate the lower blood pressure observed in EP1-/- would translate into protection from
mortality, EP1+/+ and EP1-/- mice were treated with the antihypertensive agent hydralazine (200
mg/L) in the 1% NaCl drinking water beginning 3 days prior to the implantation of the Ang II osmotic
minipump. MAP measured in EP1+/+ and EP1-/- mice treated with hydralazine demonstrated
reduced blood pressure with antihypertensive treatment in EP1+/+ mice to a level comparable to
that seen in EP1-/- mice (EP1+/+ with and without hydralazine, P = 0.024. EP1-/- with and without
hydralazine, P = 0.762). The reduction in blood pressure significantly reduced mortality in EP1+/+
mice (P = 0.007) as well as aneurysm severity. These data suggest that the lower blood pressure
observed in EP1-/- mice could be the mechanism for protection against mortality. To investigate
which components of the model were necessary for induction of damage, EP1+/+ mice received
DOCA-NaCl + Ang II, Nphx + DOCA-NaCl, Nphx + Ang II, or Nphx + DOCA-NaCl + Ang II. Mortality of
60 % was observed in animals receiving the full model, while DOCA-NaCl + Ang II, Nphx + DOCA-
NaCl, and Nphx + Ang II groups had substantially reduced mortality of 25%, 10%, and 0%
respectively. Aneurysm formation was also reduced in all modified treatment groups, from 70% to
30% and no fluid retention was observed. These data suggest that all components of the model
(Nphx, DOCA-NaCl, and Ang II) are necessary for aortic aneurysm formation/rupture and fluid
retention. In summary, this model induces robust hypertension and mortality and may serve as a
novel experimental protocol for inducing aneurysms in mice. Though EP1 deletion did not affect
changes in renal function, EP1-/- mice have lower blood pressure post-Ang II than EP1+/+ mice and
are significantly protected against mortality.

Differential Response to Chronic Unpredictable Mild Stress in Integrin 3 Heterozygous and Knockout mice Seth Varney, Keith Polston, Tammy Jessen, Christa Gaskill, and Ana Carneiro Recent human genetic studies have associated variation in the Itgb3 gene encoding the integrin 3 subunit with blood serotonin content and autism. Additionally, integrin B3 expression levels are correlated with expression of the serotonin transporter (SERT), and functional interactions whereby integrin 3 as a component of the aIIa3 fibrinogen receptor modulates SERT activity have been observed in blood platelets. As integrin 3 is expressed in the brain as a component of the avb3 integrin heterodimer, the interaction between SERT and integrin av3 in the brain may represent a novel mechanism in the regulation of mood. As exposure to chronic stress, involved in the etiology of psychiatric disorders such as depression and anxiety, has been linked with serotonergic system function, we hypothesize that integrin 3 modulation of the brain’s serotonergic system will alter the depressive and anxious response to chronic stress. To examine this hypothesis, we exposed Itgb3 knockout and heterozygote mice to chronic unpredictable mild stress for six weeks before evaluating the behavioral phenotypes, monoaminergic system integrity, and expression levels of synapse components. Nest building behavior was evaluated weekly to assess anxiety-related behavior throughout the stress procedure. Here, we provide evidence that mice with genetically imposed reductions in integrin 3 expression exhibit basally altered behavioral, neurochemical, and biochemical phenotypes as well as a differential response to chronic stress.

Identification of Novel Modulators of Serotonin Transporters Ran Ye, Kathryn Lindler, David Airey, Qiao Han, Ana Carneiro, Randy D. Blakely Serotonin transporter (SERT, Slc6a4) mediates the clearance and reuptake of serotonin (5‐HT) from serotonergic synapses in the brain. Serotonin‐selective reuptake inhibitors (SSRIs), acting on blocking the binding and translocating of 5‐HT through SERT, are some of the most widely prescribed antidepressants. Earlier studies from our and other labs have shown that SERT is tightly regulated by several associated proteins including Hic‐5, PP2A, NOS, integrin beta3, etc. In this study we use several approaches to identify novel modulators and interacting partners of SERT. First, co‐immunoprecipitation assays from mouse midbrain extracts controlled by SERT knockout midbrain tissue showed that components of multiple biological pathways such as actin cytoskeleton, focal adhesion and calcium binding are associated with SERTs. We were able to validate several SERT binding proteins via Western blotting from co‐immunoprecipitations and immunohistochemical staining of mouse midbrain Raphe Nuclei. Secondly, we explored proteins that specifically interact with SERT N‐ and C‐termini by GST fusion protein pull‐down assays followed by proteomic identification. We discovered a small GTPase Rab5c as novel SERT interacting protein. Lastly, using a genomic approach based on BXD recombinant inbred mice, we identified protocadherin 15 (Pcdh15) as a novel regulator of both 5‐HT and SERT levels in the brain. Our results demonstrated that serotonin transporters are actively engaged with multi‐protein complexes encompassing both pre‐ and post‐synaptic terminals. The interactions between SERT and its binding partners may have significant impact on the formation and signal transduction of serotonergic synapses. The novel modulators of brain SERT and serotonin levels we identified may also provide potential new targets for treating neurological and psychiatric disorders.

Vanderbilt University and Meharry Medical College
2011 Retreat
Keynote Address "Smoking and
Drinking: The role of nicotinic acetylcholine
receptors in nicotine dependence and
alcoholism"
presented by Andrew R. Tapper, Ph.D.
Assistant Professor of Psychiatry Brudnick Neuropsychiatric Research Institute
UMass Medical School, Worcester, MA
Lake Barkley State Resort Park, Cadiz, KY Tuesday, October 25 & Wednesday, October 26

Vanderbilt University and Meharry Medical College
2011 Retreat
Andrew R. Tapper, Ph.D.
Nicotine addiction elicited by smoking tobacco is responsible for over 3 million deaths annually making it the largest cause of preventable mortality in the world. Nicotine is a naturally occurring alkaloid found in tobacco and is the primary addictive component of cigarette smoke. Vaporized nicotine is rapidly absorbed through the lungs where it enters the blood stream. Within seconds of inhaling, nicotine base readily crosses the blood-brain barrier where it gains access to neuronal nicotinic acetylcholine receptors (nAChRs) expressed throughout the central nervous system (CNS). In its protonated form, nicotine mimics the endogenous neurotransmitter, acetylcholine, and can activate nAChRs, utilizing the cholinergic system which, under normal conditions, plays an important role in reward, anxiety, cognition, attention, and many other physiological processes. This ability of nicotine to “hijack” nAChRs is thought to underlie the molecular basis of nicotine addiction. It is becoming increasingly clear that nicotine dependence begins with activation of nAChRs. However, which nAChR subtypes are involved in the addictive properties of nicotine? Recent work has highlighted the idea that different subtypes may mediate different dependence-related behaviors. For example, activation of one particular nAChR subtype may be responsible for the rewarding properties of nicotine whereas chronic activation of a separate subtype may be responsible for withdrawal symptoms upon nicotine cessation. A primary goal of Dr. Tapper’s lab is to identify specific nAChR subtypes critical for behaviors associated with addiction including reward, tolerance, sensitization, and withdrawal. Acute nicotine exposure elicits many physiological effects including reward, hypothermia, and, at high enough concentrations, seizures. However, smokers expose themselves to nicotine chronically. It is this chronic exposure that produces long term physiological and behavioral changes associated with dependence. A second goal of the lab is to identify circuits and gene products that undergo adaptations because of chronic nAChR activation (or desensitization) and trigger a nicotine dependent state.
Dr. Tapper received his BS and MS degrees from the University of California, Riverside in 1995 and 1996, respectively, and his PhD degree from the Department of Pharmacology at Vanderbilt University in 2001 in Al George’s lab. He completed his postdoctoral work in the Department of Biology at California Institute of Technology in 2006 and thereafter joined UMass Medical School, where his current position is Assistant Professor of Psychiatry.

Vanderbilt University and Meharry Medical College
2011 Retreat
Session 5: Channels and Transporters
Session Moderated by: Jeremy Veenstra-Vander Weele
Presentations by:
Karen Ho Nicole Baganz, Ph.D.
Thuy Nguyen Sarah Baas, Ph.D.
Lake Barkley State Resort Park, Cadiz, KY
Tuesday, October 25 & Wednesday, October 26

Transient Receptor Potential Channels and Astrocyte Reactivity in Neurodegeneration
Karen Ho and David Calkins
Astrocytes are important mediators of neuronal homeostasis in health and react
early in pathogenesis for most neurodegenerative disorders. One such disorder is
glaucoma, which blinds through the degeneration of retinal ganglion cells (RGCs) and
their axons in the optic nerve. In the retina and optic nerve head, astrocytes form a
dense plexus around bundles of unmyelinated RGC axons, and become hypertrophic in
response to ocular stressors such as elevated pressure.
Calcium-dependent cascades are also important in the pathogenesis of this
disease, whereby elevated intraocular pressure and injury at the optic nerve head can
induce an increase in calcium in RGCs. Calcium conductance is amplified by activation
of the transient receptor potential vanilloid (TRPV) family of cation-gating channels.
Previous research has shown that TRPV1 activation increases intracellular calcium in
RGCs, contributing to their death following exposure to elevated pressure in vitro.
Astrocytes also express TRPV1, suggesting a role for this channel in regulating calcium
dynamics. Since calcium mediates a variety of intracellular events that underlie many
aspects of astrocytic function, including cytoskeletal reorganization, it is critical to
understand how TRPV1 contributes to changes in astrocyte function due to ocular
stress in diseases such glaucoma.

Abstract - Baganz, Nicole L. Native immune system regulation of brain serotonin release and reuptake Mood disorders are a major cause of lost productivity worldwide and are linked to alterations in serotonergic (5-HT) neurotransmission. The antidepressant-sensitive serotonin transporter (SERT, SLC6A4) tightly regulates synaptic 5-HT levels through high-affinity clearance of 5-HT following release and is also important for 5-HT recycling to refill vesicular stores. Previous work from our lab (Zhu et al., 2005 and 2007; Steiner et al., 2009) has implicated signaling pathways linked to interleukin-1 receptor (IL-1R) that act through p38 mitogen activated protein kinase (MAPK) to modulate SERT in vitro and in vivo. Because these signaling pathways are ubiquitously expressed in the nervous system and periphery, elucidating their cell-autonomous role in controlling SERT requires the use of interventions targeted specifically to serotonergic neurons. To initiate this effort, we have engineered mice that provide for constitutive and temporal excision of p38 MAPK and IL-1R genes in serotonergic neurons and, in the current proposal, I initiate a series of studies using the constitutive models to interrogate these pathways in SERT regulation. My initial studies indicate that animals positive for both ePet-1::Cre and floxed p38 MAPK transgenes are viable with no evidence of developmental pertubations in gross brain structure, the presence of serotonergic neurons or the abundance of SERT protein. Additional preliminary studies indicate that the double transgenics prevent synaptosomal SERT regulation by peripheral LPS administration. Through a use of in vitro and in vivo methods, including molecular, physiological, and behavioral analyses properties in singly (control) and doubly transgenic mice, I seek to establish the mechanisms by which systemic immune system activation can trigger changes in serotonergic function.

Structure-guided analysis of the VU591 binding site in ROMK
Thuy Nguyen
The Renal Outer Medullary potassium (K+) channel, ROMK or Kir1.1, is expressed almost exclusively in the nephron where it critically regulates systemic sodium, potassium, and water balance. Its unique physiological functions and emerging genetic evidence suggest that ROMK may be a viable drug target for a novel class of diuretic. We recently began a drug discovery campaign employing high-throughput screening and medicinal chemistry to develop the first small-molecule inhibitors with which to explore ROMK’s therapeutic potential. One inhibitor, termed VU591, inhibits ROMK with submicromolar affinity and is highly selective for ROMK over Kir2.1, Kir2.3, Kir4.1, Kir6.2/SUR1B, Kir7.1, Slo1/β1, and Kv1.3 K+ channels, and more than 65 other potential off targets. Patch clamp analysis, medicinal chemistry, and molecular modeling suggest that VU591 blocks the intracellular pore of ROMK by interacting with at least two channel subunits. Using a comparative homology model of ROMK based on the X-ray structure of Kir2.2, all putative solvent-accessible, pore-lining residues that could mediate VU591 interactions were identified. We are currently using scanning mutagenesis and electrophysiology to assess their role in VU591 block of ROMK. These studies should provide novel insights into the molecular structure of the channel and provide the first glimpse of a small-molecule binding site in ROMK.

In Vivo Analysis of Sequences Required for Synaptic Localization of the Presynaptic Dopamine Transporter
Sarah Baas, Andrew Hardaway, Shannon Hardie, Sarah Whitaker, Randy Blakely
The monoamine neurotransmitter dopamine (DA) modulates brain circuitry relevant to cognition, reward, motor control, and arousal. Defects in DA signaling have been implicated in risk for addiction, attention-deficit hyperactivity disorder (ADHD), schizophrenia, and Parkinson’s disease. The presynaptic dopamine transporter (DAT) is a major control point for DA signaling and a key target for psychostimulants, including cocaine and amphetamine. DAT function is believed to be partly regulated through mechanisms supporting active shuttling between endosomal and cell surface membranes, and an extensive regulatory network providing post-translational control of DAT is beginning to emerge, although to date mostly based on in vitro data. We have chosen to pursue the goal of elucidating DAT regulatory mechanisms in the powerful model system Caenorhabditis elegans. The molecular machinery required for DA signaling is highly conserved in C. elegans and has been demonstrated to play an important role in egg-laying, locomotion, touch response, and defecation. The C. elegans DAT, DAT-1, modulates these behaviors by reuptake of DA from the synapse, limiting receptor availability and synaptic spillover of DA and enabling DA re-release following DA recycling to the presynaptic terminal. Our work has shown that DAT-1 activity is particularly important to ensure normal swimming behavior. Whereas wild-type worms thrash in water for up to 20 minutes at a relatively constant rate (~1 Hz), dat-1 mutants paralyze in a few minutes, a phenotype known as swimming-induced paralysis (swip). In addition to forward genetic approaches, we are using quantitative methods to visualize the cellular localization DAT-1 by ratiometric comparison of DAT-1:GFP fluorescence in synapses, cell bodies, and dendrites using confocal microscopy. Using these methods, DAT-1 trafficking was found to depend on the DAT-1 C-terminus, as a
25 truncation resulted in somatic retention of the transporter and SWIP. Mutagenesis efforts are underway to determine the minimal motif necessary for DAT-1 export. Recent data suggests that the ER export protein SEC24 participates in biogenic amine transporter folding and trafficking through a distal C-terminal motif and we are also conducting mutagenesis studies to determine the role of this motif in proper DAT-1 trafficking. Through these efforts, we hope to identify mechanisms required for proper DAT-1 export and synaptic localization localization and then translate our findings to mammalian DA neurons. Supported by NIH awards T32 MH065215 to S.B., F31 MH093102 to A.H., and DA027739 to R.D.B.


Vanderbilt University and Meharry Medical College
2011 Retreat
Poster Session #2
Wednesday, October 26 10:30 am – 11:30 am
Lake Barkley State Resort Park, Cadiz, KY Tuesday, October 25 & Wednesday, October 26

2010 PHARMACOLOGY RETREAT LAKE BARKLEY STATE RESORT PARK
Poster Presentations – Group 2
(Poster Session on Wednesday, October 26th from 10:30 am – 11:30 am)
Presenter Poster Title Katherine Betke (Mentor: Heidi Hamm)
GPCR Mediated Modulation Of Synaptic Transmission
Preston Campbell (Mentor: Florent Elefteriou)
Chronic Stress Increases Breast Cancer Metastasis to Bone via β2AR on Osteoblasts
Ryan Ceddia (Mentor: Rich Breyer)
Characterization of the Role of the Prostaglandin E Receptor 3’s Role in Diabetes
Jason Downey (Mentor: Rich Breyer)
Antagonists of Prostaglandin E2 Receptors for the Treatment of Hypertension
Craig Goodwin (Mentor: Steven Fesik)
Discovery and Characterization of a p21-Activated Kinase 1 (PAK1) inhibitor by fragment-based screening and rational design
Ericka Holmstrand, Ph.D. (Mentor: Randy Blakely)
Biochemical and behavioral consequences of overexpression of the murine high-affinity choline transporter
Kari Johnson (Mentor: Jeff Conn)
Group II metabotropic glutamate receptor activation induces long-term depression of excitatory transmission in the substantia nigra pars reticulata
Michele LeNoue-Newton (Mentor: Ben Spiller)
Alpha4 inhibition of PP2Ac degradation requires both MID1 and PP2Ac binding domains
Tu Mai (Mentor: David Robertson)
The Role of the Kidneys in the Osmopressor Response
Keri McLean (Mentor: Sukhbir Mokha)
Activation of GPR30 and ERα rapidly attenuates antinociception produced by the activation of the opioid receptor-like 1 receptor in the rat spinal cord
Alex Nackenoff (Mentor: Randy Blakely)
Reverse Engineering SSRIs
Rene Raphemot (Mentor: Jerod Denton)
Discovery of an inward rectifying potassium channel inhibitor with preference for Kir2.3 Kir3.X and Kir7.1
Sara Savage (Mentor: John Penn) The PGF2 FP receptor mediates retinal angiogenic cell behaviors in vitro
Greg Sliwoski (Mentor: Jens Meiler)
High throughput screening for modulators of the human Y4 receptor
Cierra Spencer (Mentor: Alex Brown)
Lipid modulation of mammalian PLD activity in vitro
Sydney Stoops (Mentor: Craig Lindsley)
Understanding the Mechanism of Action of Small Molecules that Restore E-Cadherin Expression
Mengnan Tian (Mentor: Robert Macdonald)
The Intronic GABRG2 Mutation, IVS6+2TG, Associated with CAE Altered 2 Subunit mRNA Intron Splicing, Generated a Mutant Proten that Affected GABAA Receptor Biogenesis
Guy Watkins (Mentor: Brian Wadzinski)
Monoubiquitination promotes calpain cleavage of α4 altering PP2A stability and MAP phosphorylation
Summer Young (Mentor: Heidi Hamm)
Ethyl 4-(1-benzylindolin-3-yl)benzoate, an indole based protease activated receptor-4 antagonist
Zack Zurawski (Mentor: Heidi Hamm)
Identification of key residues for interaction with upon the t-SNARE proteins SNAP-25 and syntaxin1a

Chronic Stress Increases Breast Cancer Metastasis to Bone via 2AR on Osteoblasts
Campbell JP, Karolak M, Ma Y, Yang X, Sterling J, Elefteriou F
Increased sympathetic nervous (SNS) outflow, a hallmark of mental stress, controls multiple processes in the bone, such as hematopoietic stem cell trafficking, osteoblast proliferation, osteoclastogenesis, and inflammation via 2-adrenergic receptors (2AR) and cytokines also involved in cancer metastasis, such as SDF-1, RANKL, and IL-6. These observations suggest the involvement of SNS activation in cancer osteotropism, tumor burden, and the formation of osteolytic lesions, which may underlie the increased mortality and recurrence observed clinically with prolonged psychosocial stress. To address this hypothesis, we used an established model of osteolytic metastasis in which MDA-MB-231 human breast cancer cells are injected via left cardiac ventricle into athymic nude. The effects of chronic stress, simulated with restraint stress, were compared to Isoproterenol (ISO), a AR agonist acting independently of the hypothalamic-pituitary-adrenal axis (HPA). Osteolytic lesions, tumor burden and BV/TV were assessed with faxitron, 3D-microtomography, and histology. Both chronic stress and ISO treatment before MDA-MB-231 inoculation increased the number of bone lytic lesions (p<0.05), while treatment after bone metastasis increased the size of the lytic lesions (p<0.05). Furthermore, the effects of restraint stress were rescued by treatment with a commonly prescribed b-blocker, propranolol, suggesting that chronic stress alters the bone microenvironment (BMM) through the 2AR to make it favorable for cancer cell bone colonization and growth. In vitro, direct stimulation of the 2AR in MDA-MB-231 cells produced no effect on migration, cell growth or PTHrP, Rank and CXCR4 expression. In contrast, stimulation of the 2AR in bone marrow stromal cells, calvarial and MC3T3 osteoblasts increased Rankl but not Sdf1 expression (p>.005). The conditioned media of ISO-treated osteoblasts increased MDA-MB-231 migration (p<0.005), which could be blocked by OPG but not by the CXCR4 antagonist AMD3100. These results suggest that the increase in osteolysis and metastasis observed following SNS activation in vivo are due predominantly to a bone stromal effect. These findings support the hypothesis that depression, via sympathetic activation, make the BMM permissive to cancer metastasis, growth, or recurrence, and imply that b-blockers may reduce both bone tumor burden and relapse in breast cancer patients.

Characterization of the Role of the Prostaglandin E Receptor 3’s Role in Diabetes
Ceddia RP, Swan CE, Gannon M, Breyer RM
Diabetes is a diseased caused by defects in insulin production or action resulting in an impaired ability to clear glucose from the blood. Previous studies suggest that Prostaglandin E2 (PGE2) has an inhibitory effect on insulin release. The PGE2 E-Prostanoid receptor 3 (EP3), a Gi coupled receptor, is highly expressed in the pancreas. We hypothesize that PGE2 inhibits glucose stimulated insulin release through the EP3 receptor and that EP3 loss/blockade will improve glucose sensitivity. Wild-type and EP3 null animals showed no difference in glucose sensitivity at 16 weeks of age; however, at 38 weeks of age, EP3 null animals cleared glucose more efficiently than wild-type. We will investigate the glucose-mediated changes in insulin levels in these animals to determine whether EP3 loss improves insulin secretion, insulin sensitivity or both. Furthermore, we will use islet perifusion to determine the relevant EP3-evoked signal transduction pathways modulating insulin secretion. Successful completion of these studies will determine the role of EP3 receptors i) on inhibition of insulin secretion by pancreatic -cells ii) the role of EP3 on glucose sensitivity and iii) allow the identification of the relevant signal transduction pathways mediating each of these physiologic effects.

Antagonists of Prostaglandin E2 Receptors for the Treatment of Hypertension Downey, J.D., Breyer, R.M. In contrast to blockade of all prostanoid production with NSAIDs, pharmacological blockade of PGE2 pressor receptors EP1 and EP3 may decrease systemic blood pressure and protect from hypertensive renal damage. To test this hypothesis, EP1 and EP3 small molecule antagonists were synthesized and their pharmacology, pharmacokinetics, and physiologic effects on mean arterial pressure (MAP) were assessed. Two lead compounds, DG-041 and JD-200, were synthesized as subtype-selective antagonists of the pressor EP receptors. DG-041 binds the mEP3 receptor with a Ki of 449 ± 130.0 pM and potently blocks mEP3 signaling with an IC50 of 420.0 pM in an LVIP2.0zc cell-based CRE reporter assay. JD-200 blocked signaling through mEP1 with an IC50 of 18.8 ± 11.5 nM in a fluorescent calcium flux cell-based assay. In vivo mouse pharmacokinetic experiments demonstrated that DG-041 is orally bioavailable with an elimination half-life of 1.2 h. JD-200 had an elimination half-life of 28 min. Preliminary studies demonstrate that while acute infusion of the EP1/EP3 agonist sulprostone caused a transient increase in MAP (+33.3 mmHg) in the EP1-/- mouse, pretreatment with DG-041 blocked the pressor effect of acute sulprostone infusion (+4.60 mmHg); the vasopressor effect of acute infusion of an unrelated pressor, α-adrenergic agonist phenylephrine, was not different (vehicle +42.0 mmHg, DG-041 +39.0 mmHg). Taken together these results suggest that these EP1 and EP3 receptor antagonists will be suitable for use in mouse models of hypertension.

Discovery and Characterization of a p21Activated Kinase 1 (PAK1) inhibitor by fragmentbased screening and rational design Craig M. Goodwin, Jason P. Burke, Ed Olejniczak, Alex Waterson, Olivia Rossanese, Stephen W. Fesik The PAK family kinases regulate cell survival, proliferation, and motility and are frequently upregulated in many cancers including breast, colon, and pancreatic. Induction of PAK1 expression can promote tumorigenesis in non‐cancerous cell lines, whereas silencing can inhibit tumor cell line proliferation and motility. Here we describe our efforts to discover small‐molecule inhibitors of PAK1 kinase that exhibits both high potency and selectivity. Utilizing previous literature on kinase inhibitor design, we have rationally designed a prototypical “hinge‐binding” compound that demonstrates micromolar binding affinity. Using Saturation Transfer Difference‐NMR (STD) to assay for binding, we have conducted a fragment‐based screen to also identify compounds that bind in the presence of our hinge‐binding compound. Binding affinity has been measured using SPR, fluorescence anisotropy, and DiscoveRx, a commercial binding assay. We will use medicinal chemistry to covalently link the original hinge‐binder and an optimized second‐site fragment from the screen. The resulting compound will be tested for activity in a biochemical kinase assay and also tested in a panel of breast cancer cells for inhibition of growth and motility.

Biochemical and behavioral consequences of overexpression of the murine high-affinity choline transporter
Ericka C. Holmstrand, David Lund, Jane Wright, Rolicia F. Martin, Hideki Iwamoto, and Randy D. Blakely
The presynaptic, high-affinity choline transporter (CHT) supplies choline, a necessary precursor to acetylcholine (ACh), to cholinergic neurons. Uptake through CHT is necessary to maintain sustained cholinergic firing in the central and peripheral nervous systems, and the function of CHT is regulated via subcellular trafficking of the transporter. Rates of vesicular release of ACh are tightly coupled to the total capacity of high-affinity choline uptake (HACU) in cholinergic terminals. Previous work from our lab has identified activity dependent deficits in HACU and the ability to support elevated ACh signaling in both homozygous and heterozygous CHT knockout mice (Ferguson et al, PNAS 2004; Bazalakova et al Genes, Brain and Behavior, 2007; Lund et al, Neuroscience 2010). In order to evaluate the effects of increased expression of CHT, a transgenic mouse line was created using a bacterial artificial chromosome (BAC) construct to insert multiple copies of the CHT gene into the C57BL/6 genome. Increased genomic CHT content was confirmed by quantitative PCR, and comparison of BAC-CHT transgenics with wild-type littermates by Western blotting or immunohistochemistry reveals significantly increased protein expression in all brain regions examined, with expression patterns consistent with the localization of cholinergic neurons. Furthermore, synaptosomal choline uptake is significantly higher in tissue from BAC-CHT vs WT animals under both basal (3 mM KCl) and depolarizing (20 mM KCl) conditions, and brain slice release studies reveal enhancement of endogenous but not relabeled Ach release in the BAC-CHT mice, consistent with a selective impact of extra transporters on the reserve capacity of cholinergic neurons for neurotransmitter release. Behaviorally, CHT overexpressing animals displayed decreased open arm activity on the elevated plus maze and decreased alternation on the Y-maze, a test of spatial memory. BAC-CHT mice also display increased sensitivity to scopolamine in the open field test of motor activity, whereas CHT heterozygous CHT mice display reduced sensitivity. These findings indicate that the BAC-CHT mice provide an opportunity to explore the molecular plasticities that follow from constitutive elevations in uptake and how these changes can influence behavior.

Group II metabotropic glutamate receptor activation induces long-term depression of excitatory transmission in the substantia nigra pars reticulata
Kari A. Johnson, Colleen M. Niswender, P. Jeffrey Conn, Zixiu Xiang
Activation of group II metabotropic glutamate receptors (mGluR2 and mGluR3) has been implicated as a therapeutic strategy for treating both motor symptoms and progressive neurodegeneration in Parkinson’s disease (PD). Modulation of excitatory transmission in the basal ganglia represents a possible mechanism by which group II mGluR agonists could confer antiparkinsonian effects. Previous electrophysiological studies have identified effects of mGluR2/3 activation on excitatory transmission at various synapses in the basal ganglia, including the excitatory synapse between the subthalamic nucleus (STN) and the substantia nigra pars reticulata (SNr). Specifically, brief activation of group II mGluRs by a selective agonist caused a reversible depression of excitatory transmission in the SNr. Using whole-cell patch clamp studies of putative GABAergic SNr neurons in rat midbrain slices, we have found that a more prolonged activation of group II mGluRs by the selective agonist LY379268 induces a long-term depression (LTD) of evoked excitatory postsynaptic current (EPSC) amplitude. Bath application of 30-100 nM LY379268 for ten minutes induced a 40-55% reduction in EPSC amplitude, and excitatory transmission remained depressed at this level for at least 45 minutes after agonist washout. The effect of LY379268 was concentration-dependent and was completely blocked by the group II mGluR-preferring antagonist LY341495 (500 nM). To determine the relative contributions of mGluR2 and mGluR3 to the LTD induced by LY379268, we tested the ability of LY379268 (100 nM) to induce LTD in wild type mice and mice lacking mGluR2 or mGluR3. LY379268 induced similar LTD in wild type mice and mGluR3 knockout mice, whereas LTD was absent in mGluR2 knockout mice. These studies suggest a novel role for mGluR2 in the long-term regulation of excitatory transmission in the SNr and invite further exploration of mGluR2 as a therapeutic target for treating the motor symptoms of PD.

Alpha4 inhibition of PP2Ac degradation requires both MID1 and PP2Ac binding domains Michele LeNoue-Newton1, Guy Watkins1, Ping Zou2, Katherine Germane3, Lisa McCorvey1, Brian Wadzinski1, and Benjamin Spiller1,3
Department of 1Pharmacology, 2 Molecular Physiology and Biophysics, 3Microbiology and Immunology, Vanderbilt University, Nashville, TN Protein phosphatase 2A (PP2A) is serine/threonine phosphatase that is regulated through a variety of mechanisms, including posttranslational modifications and association with regulatory proteins. Alpha4 binds to and regulates PP2A catalytic subunit (PP2Ac) stabilizing it through protecting PP2Ac from polyubiquitination and degradation. Alpha4 is a multi-domain protein with a C-terminal domain that binds MID1, an E3 ubiquitin ligase, and an N-terminal domain containing the PP2Ac binding site and a UIM consensus motif, shown to be important in protecting PP2Ac from polyubiquitination. We will present the structure of the N-terminal domain of Alpha4 determined by x-ray crystallography and show that it is a flexible tetratricopeptide repeat-like protein. We also demonstrate that both the MID1 binding domain and the PP2Ac binding domain of Alpha4 are essential to protect PP2Ac from degradation and polyubiquitination. The actual mechanism by which Alpha4 protects PP2Ac from degradation appears to involve contributions from multiple domains of Alpha4 indicating a multi-part mechanism of action that has yet to be fully elucidated.

The Role of the Kidneys in the Osmopressor Response
Tu Mai
PI: David Robertson
Abstract
Water ingestion induces a robust increase in blood pressure in patients with
baroreflex impairment. To better understand this phenomenon, we use a
modified sino-aortic denervated mouse as our model. Water infused into the
duodenum of these mice stimulates an increase in blood pressure that is similar
in magnitude and time course to that in patients. Clinical and mouse studies have
provided evidence that increased sympathetic outflow underlies this pressor
effect. However, the physiological and molecular mechanisms mediating this
effect remain unknown. Previous studies in our lab have shown that the osmo-
sensitive cation channel TRPV4 might play an important role in the osmopressor
response. TRPV4 is highly expressed in the kidneys, the principal regulator for
systemic blood pressure and osmolality. To investigate the potential role of the
kidneys in mediating the water effect, water is given into the duodenum of
denervated, nephrectomized mice. We hypothesize that the kidneys are one of
the main players of the osmopressor response and therefore, no pressor
response will be observed in nephrectomized animals, in contrast to a vigorous
response in sham control animals.

Activation of GPR30 and ERα rapidly attenuates antinociception produced by the activation of the opioid receptor-like 1 receptor in the rat spinal cord. Keri M.B. McLean, Subodh Nag, Sukhbir Mokha. Department of Neuroscience and Pharmacology, Meharry Medical College, Nashville, TN 37208-3599.
Activation of the opioid receptor-like 1 (ORL1) receptor has been shown to produce antinociception at the level of the spinal cord and the trigeminal system. We have previously shown that the antinociceptive effect of intrathecally (i.t.) administered orphanin FQ (OFQ), the endogenous ligand for the ORL1 receptor, is attenuated by estrogen in females [Claiborne et al. J Neurosci. 26(50):13048-53, 2006]. In addition, we have shown that estrogen induces downregulation of the ORL1 receptor mRNA, thus highlighting the involvement of a genomic mechanism mediating the action of estrogen [Flores et al. Neurosci. 118(3):769-78, 2003]. However, the expression of membrane estrogen receptors (mERs) such as ERα, ERβ, and GPR30 in the spinal cord suggests that estrogen can also produce its effects non-genomically. We have previously shown that selective activation of mERs, using a membrane impermeable analog of estradiol (E2BSA), attenuates ORL1-mediated antinociception in males and ovariectomized (OVX) female rats. The purpose of this study is to determine which estrogen membrane receptor (GPR30 or ERα) plays a role in attenuating antinociception produced by the activation of the ORL1 receptor in the spinal cord. G-1 (GPR30 agonist), or PPT (ERα agonist) were injected intrathecally into the lumbosacral spinal cord of rats through an implanted PE-10 cannula and their effects were tested on OFQ-induced antinociception using a nociceptive heat-induced tail-flick assay. With comparable baseline latencies in all experimental groups, OFQ (10 nmol/10 µl) significantly increased tail flick latencies (TFL). G-1 or PPT injected immediately prior to OFQ immediately attenuated this effect. We also show that GPR30 antagonist, G-15, reversed the rapid effect observed in the G-1 treated group. These results suggest that activation of GPR30 or ERα is sufficient to rapidly attenuate the antinociceptive effect produced by the activation of ORL1 receptor in the spinal cord. Thus, this study provides a rationale basis for a potential non-genomic mechanism that may underlie the observed estrogen-induced attenuation of ORL1-mediated antinociception. This study was supported, in part, by the NIH R25 GM 59994-09, T32NS061201, U54N541071, RR03032, and SC1NS063951 grants.

Alex Nackenoff Retreat Talk
Reverse Engineering SSRIs
The primary objective of my thesis is to elucidate the neurological effects of SSRIs, specifically those that accompany antidepressant therapeutic efficacy. SSRIs bind to a sizable number of non‐trivial targets other than SERT, and do so at physiologically relevant concentrations. My first aim is to identify if any of these off‐target binding events are contributing to antidepressant therapeutic efficacy. To accomplish this, I will utilize mice bred with a point mutation at SERT I172M that confers insensitivity to many SERT blocking drugs. Therefore, given normal SSRI dosing, SSRIs will no longer be able to effectively antagonize SERT, but will remain in the CNS and able to bind these other targets. By selectively removing the serotonergic component of SSRIs, any further behavioral/biochemical modifications can be attributed to the off‐target effects of SSRIs. I will chronically dose both WT and M172 mice with Citalopram, Paroxetine (an SSRI insensitive to the I172M point mutation; positive control), and saline (negative control). I will then assay for behavioral efficacy using a modified version of the Novelty Induced Feeding Suppression paradigm. Afterwards, using microarray/RNASeq, I will compare the gene expression profile of WT and M172 mice, which will provide the first identification of serotonin‐specific effects and off‐target effects of chronic SSRI administration. I will also analyze the induction of hippocampal neurogenesis following chronic SSRI administration, which previous research has shown that hippocampal neurogenesis is both potentiated by chronic SSRI administration and required for SSRI antidepressant behavioral efficacy. In sum, I plan to investigate the role of serotonin on SSRI antidepressant behavioral efficacy and the requisite neurostructural and biochemical alterations. Abbreviations: CNS – Central nervous system; SERT‐ serotonin transporter; SSRI‐ serotonin selective reuptake inhibitor;

Discovery of an inward rectifying potassium channel inhibitor with preference for Kir2.3
Kir3.X and Kir7.1
Rene Raphemot1, Daniel Lonergan2, Thuy T. Nguyen1, Thomas J. Utley1, Rocco Gogliotti1,
Corey R. Hopkins1, L. Michelle Lewis1, Craig W. Lindsley1, C. David Weaver1, Jerod S.
Denton1,2.
1Pharmacology, 2Anesthesiology, Vanderbilt University School of Medicine, Nashville, TN
Inward rectifying potassium (Kir) channels play key physiological roles in diverse cell types and
may represent novel drug targets for disease. In cardiac myocytes, Kir channels contribute to the
resting membrane potential and the late repolarization phase of the action potential. Homomeric
Kir2.3 and heteromeric Kir3.1/3.4 channels are expressed almost exclusively in the atria and are
postulated drug targets for atrial fibrillation, a common arrhythmia that predisposes patients to
stroke. Efforts to assess the utility of Kir2.3 and Kir3.1/3.4 channels as therapeutic targets have
been hampered by the lack of drug-like compounds targeting these channels. We therefore
performed a fluorescence-based high-throughput screen for small-molecule inhibitors of Kir3.X
channel function. Several novel inhibitors were discovered. One compound, termed VU573,
inhibits Kir3.X channels with a half-maximal inhibition concentration of 1.9 μM. Subsequent
electrophysiology-based counterscreens revealed that VU573 inhibits Kir2.3 and Kir7.1 with
similar potency, but is less active toward Kir1.1, Kir2.1, and Kir4.1. VU573 is the first selective
inhibitor for Kir2.3 and most potent Kir7.1 inhibitor known to date. We are currently using
medicinal chemistry to improve the potency and selectivity of VU573 for Kir2.3 and Kir3.X and
understand the structural requirements for inhibition of these channels.

The PGF2 FP receptor mediates retinal angiogenic cell behaviors in vitro Sara R. Savage1, Rong Yang1, Susan E. Yanni2, John S. Penn1. 1Pharmacology, Vanderbilt University Medical Center, Nashville, TN; 2Retina Foundation of the Southwest, Dallas, TX. Purpose: Prostanoids contribute to the pathological angiogenesis that characterizes many diseases, including neovascular eye conditions. We have previously shown that the prostanoid PGF2 is increased in Müller cells and retinal microvascular endothelial cells exposed to disease-relevant stimuli. PGF2 is thought to affect the angiogenic process by binding to its high-affinity receptor, FP. The purpose of the present study was to investigate the role of FP receptor activation in VEGF production by Müller cells and in VEGF-induced proliferation and tube formation by retinal microvascular endothelial cells. Methods: Müller cells derived from COX-2 null mice were treated with increasing concentrations (0.1-10 µM) of the FP agonist Latanoprost. VEGF message and protein were assessed by RT-PCR and ELISA, respectively. Human retinal microvascular endothelial cells (HRMEC) were treated with increasing concentrations (0.1-10 µM) of Latanoprost, and cell proliferation was assessed by BrdU incorporation. HRMEC were treated with increasing concentrations of the FP antagonist AL-8810 (0.1-10 µM), and tubulogenesis was assessed in Matrigel assays. Results: COX-2 null mouse Müller cells treated with 10 µM Latanoprost demonstrated a 1.3-fold increase in VEGF mRNA expression. Similarly, treatment with 10 µM Latanoprost led to a two-fold increase in the level of VEGF protein secreted by Müller cells (p < 0.03). Increasing concentrations of Latanoprost led to a dose dependent increase in HRMEC proliferation (by approximately 180% at a 10 µM dose). HRMEC treated with 10 µM AL-8810 demonstrated a 50% reduction in tube formation. Conclusions: Preliminary investigation has demonstrated that FP activation or inhibition influences the behaviors of two retinal cell types that are known to play critical roles in the pathological ocular angiogenesis characteristic of retinopathy of prematurity, diabetic retinopathy, and age-related macular degeneration. These studies suggest that the FP receptor may be a rational therapeutic target in neovascular eye disease.

High throughput screening for modulators of the human Y4 receptor
Gregory Sliwoski, Jens Meiler & Dave Weaver labs
Obesity is a growing health concern that can lead to cardiovascular disease and diabetes. Current treatments for obesity include invasive surgery and various medications that are limited in their treatment potential due to side effects and lack of generalizability to younger populations. My research is aimed at the discovery of novel therapeutics for obesity through satiety signals not currently targeted by available treatments. Human pancreatic polypeptide binds to the Y4 receptor to relay satiety signals that originate in the gut and intestines following the ingestion of food. Injection of pancreatic polypeptide has been shown to reduce the food intake in obese mice and humans. Modulators of this receptor may prove to be effective and practical treatments for human obesity. Using a calcium flux assay technique with cells expressing the human Y4 receptor, 32,000 compounds from the Vanderbilt compound library were screened in high‐throughput fashion. Compounds that directly activated the receptor or potentiated the effect of pancreatic polypeptide were collected and a total of 28 hits were identified after validation. Of these 28 compounds, 23 showed selectivity for the modulation of pancreatic polypeptide versus bradykinin or histamine.

Cierra Spencer 2011 Retreat Abstract Title: Lipid modulation of mammalian PLD activity in vitro Phospholipase D (PLD) is an enzyme that converts phosphatidylcholine to the lipid signaling molecule phosphatidic acid. Phosphatidyl‐4,5‐bisphosphate (PIP2) and oleate have previously been identified as lipid stimulators of mammalian PLD in vitro. Variation in liposome composition was observed to cause changes in PLD1 and PLD2 activity in vitro, leading to the investigation of the effects of various phospholipids and fatty acids on PLD activity. All of the identified lipid stimulators are most effective when PIP2 is also present resulting in a synergistic response. Isoform selective differences have been observed, but phosphatidic acid, diacylglycerol, and phosphatidylbutanol can stimulate both isoforms.

Understanding the Mechanism of Action of Small Molecules that Restore E-Cadherin Expression Sydney L. Stoops
E-cadherin is a transmembrane protein that maintains intercellular contacts and
cellular polarity in epithelial tissues. The down-regulation of E-cadherin is thought to aid in the induction of an epithelial-to-mesenchymal transition (EMT) resulting in an increased potential for invasion into surrounding tissues and entry into the bloodstream. Loss of E-cadherin has been observed in a variety of human tumors resulting from somatic mutations, chromosomal deletions, proteolytic cleavage of E-cadherin, and most commonly silencing of the CDH1 gene promoter.
A novel High Throughput Screen was developed to identify small molecules that restored E-cadherin expression in the SW620 cell line followed by medicinal chemistry employing iterative analog library synthesis to better identify the structure-activity relationship (SAR). Preliminary optimization of the screening hit has shown it is possible to synthesize small molecules that have an improved ability to restore E-cadherin expression compared to the initial screening hits. This restoration of protein has been confirmed by visualization of E-cadherin at the membrane via immunofluorescent microscopy. Further biological analysis of profiled analogs has shown a minimal effect on cell proliferation, but a decrease in cellular invasion.
Recent endeavors have been taken to elucidate the mechanism of action of these small molecules to restore E-cadherin expression. Quantitative PCR analysis has shown that treatment with selected small molecules increases mRNA expression ~50 fold after 16 hours suggesting the small molecules are altering transcription of the CDH1 gene. This was supported by experiments conducted using a plasmid construct containing a 1400bp fragment of the E-cadherin promoter and luciferase reporter. After transfection, the cells were treated with selected compounds, lysed, and luciferase activity was measured. It was shown that selected active small molecules had a significant increase in luciferase activity as compared to DMSO or a dead small molecule, which were used as controls. More specifically, this suggests that the small molecules are specifically effecting transcription within the 1400bp promoter region of the CDH1 gene. Future work will include truncating the promoter region within the plasmid to narrow down the portion of the promoter region targeted by these small molecules, in hopes that specific transcription binding sites will be present within the region.
Elucidation of the mechanism of action will aid in identifying the novel molecular target. Such information would allow for further development of more efficacious and potent small molecules as well as further research to understand the importance of this interaction in the role of EMT and as a potential therapeutic target. In addition, combinatorial treatments with sub-therapeutic doses of standard of care chemotherapeutics in the clinic and our small molecules will be screened for synergistic effects in proliferation and apoptosis assays.

THE INTRONIC GABRG2 MUTATION, IVS6+2TG, ASSOCIATED WITH CAE ALTERED
2 SUBUNIT MRNA INTRON SPLICING, GENERATED A MUTANT PROTEN THAT
AFFECTED GABAA RECEPTOR BIOGENESIS
Mengnan Tian (田梦楠)1, 2 and Robert L. Macdonald1, 2, 3
Departments of Neurology1, Pharmacology2 and Molecular Physiology and Biophysics3 Vanderbilt University Medical Center
Nashville, TN 37212
The GABRG2 intronic mutation, IVS6+2TG, was identified in an Australian family with childhood absence epilepsy (CAE) and febrile seizures (Kananura et al., 2002). In affected family members, the GABRG2 intron 6 splice donor site was found to
be mutated from GT to GG. We generated wildtype and mutant 2S subunit bacterial artificial chromosomes (BACs) and expressed them in HEK 293T cells. The BACs containing the endogenous hGABRG2 promoter were expressed also in transgenic mice.
The wildtype and mutant mRNA splicing patterns were determined in both transfected cells and transgenic mouse brain. The mutation abolished intron splicing at the donor site, activated a cryptic splice site during intron splicing, generated partial intron 6 retention and produced a frame shift in exon 7 that generated a premature translation-termination codon (PTC). The same splice donor site was recognized also in BAC transgenic mouse brain. The resultant mutant GABRG2(IVS6+2TG) mRNA was degraded by nonsense mediated mRNA decay (NMD), but the mRNA escaping NMD was translated as a
truncated protein (2-PTC subunit) containing the first 6 GABRG2 exons and a novel frame shifted 29 amino acid C terminal tail. It was detected in both BAC transfected
HEK293T cells and mutant BAC transgenic mouse brain. The 2-PTC subunit is homologous to the mollusk protein AChBP, but is not secreted from cells. It was
retained in the ER and not expressed on the membrane. It oligomerized with and
subunits with a low efficiency and decreased surface and total levels of and subunits. It also increased the ER chaperone BIP level in the cells, indicating possible ER stress. These results suggested that the GABRG2 mutation, IVS6+2TG, reduced GABAA receptor-mediated inhibition by reducing GABRG2 transcript level and
generating a dominant negative mutant subunit that decreased GABAA receptor biogenesis.

Monoubiquitination promotes calpain cleavage of α4 altering PP2A stability and MAP phosphorylation
Guy R. Watkins1, Ning Wang1, Matthew D. Mazalouskas1, Rey J. Gomez1, Chris R. Guthrie2, Brian C. Kraemer2,3, Susann Schweiger4, Benjamin W. Spiller1, and Brian E. Wadzinski1*
Multiple neurodegenerative disorders are linked to aberrant phosphorylation of microtubuleassociated proteins (MAPs). Protein phosphatase 2A (PP2A) is the major MAP phosphatase; however, little is known about its regulation at microtubules. α4 binds the PP2A catalytic subunit (PP2Ac) and the microtubule‐associated E3 ubiquitin ligase MID1, and through unknown mechanisms can both reduce and enhance PP2Ac stability. We show MID1‐dependent monoubiquitination of α4 triggers calpain‐mediated cleavage and switches α4’s activity from protective to destructive, resulting in increased tau phosphorylation. This regulatory mechanism is important in MAP‐dependent pathologies as levels of cleaved α4 are decreased in Opitz Syndrome (OS) and increased in Alzheimer’s disease (AD), disorders characterized by MAP hypophosphorylation and hyperphosphorylation, respectively. These findings indicate that regulated inter‐domain cleavage controls the dual functions of α4, and dysregulation of α4 cleavage likely contributes to OS and AD.

Ethyl 4-(1-benzylindolin-3-yl)benzoate, an indole based protease activated receptor-4 antagonist Summer Young1, Matthew Duvernay1, JT Brogan2, Craig Lindsley1,2, and Heidi Hamm1 Department of Pharmacology1, Department of Chemistry2
Following blood vessel injury platelets are essential for cessation of blood loss (hemostasis) by activating
and forming a platelet plug at the site of injury. However, in pathological states the platelet plug continues to grow culminating in thrombosis (vessel occlusion). The subsequent ischemic damage in the affected tissue can manifest as critical limb ischemia, myocardial infarction, stroke, or pulmonary embolism. Given the central role platelets play in thrombosis, several therapeutics are targeted towards receptors and proteins that are critical for platelet activation. Aspirin, Clopidigrel (Plavix), and Eptifbatide (Integrilin) are a few examples of therapeutics designed to inhibit platelet activation. Unfortunately current anti-platelet therapeutics carry an additional risk of bleeding.
Thrombin is a serine protease formed during coagulation and is necessary for formation of a clot, thus making it an attractive target for anti-coagulant therapy. A major mechanism through which thrombin aides in clot formation is through activation of protease activated receptors (PARs) on platelets. Activation PAR1 and PAR4 by thrombin results in platelet agglutination, crosslinking with fibrin, and activation of integrins, which causes platelet adhesion to other cells, thus building the foundations of a clot. Protease activated receptors (PARs) are a unique class of GPCRs which are activated by enzymatic cleavage of the N-terminus which results in an unmasking of a new N-terminus which then serves as the endogenous agonist for the receptor. A key difference between PAR1 and PAR4 is that PAR1 contains a hirudin-like domain that is able to bind thrombin at subnanomolar concentrations whereas PAR4 lacks this domain thus establishing PAR1 as the high affinity thrombin receptor and PAR4 as the low affinity thrombin receptor. Vorapaxar, a PAR1 antagonist, was a promising candidate for next generation anti-platelet therapeutics but recently this target also displayed an increased risk for bleeding in man. Unsurprisingly, the search continues to discover an anti-platelet/anti-thrombotic therapeutic without the risk of bleeding.
Our lab believes that PAR4, not PAR1, is the better target for anti-platelet therapeutics. In order to test this hypothesis we sought to use a PAR4 antagonist in isolated human platelets. YD-3 is a selective non-peptide PAR4 antagonist that is not commercially available and the current synthetic scheme is lengthy (7 steps total). So that we may test the role of PAR4 in human platelets, we synthesized YD-3 in two steps using commercially available reagents. A significant drawback to our synthetic route (as well as the original synthesis of YD3) is the formation of an inactive isomer. Not only does the presence of an inactive isomer require additional purification steps, it’s also formed preferentially over YD3 (3:1) effectively lowering the total yield of active compound. In order to eliminate the inactive compound we removed of N(2) of the YD3 indazole scaffold thereby producing a new compound. We demonstrate that the new compound, ‘SEY3’, retains activity toward PAR4 and shares a similar potency profile to that of YD3 when measuring PAR-4 mediated platelet activity. Specifically, SEY3 is effective at inhibiting PAR4 but not PAR1 mediated platelet aggregation, indicating a degree of selectivity. Additionally, we measured PAR1-AP and ADP mediated GPIIbIIIa activation and p-selectin expression using flow cytometry and by analyzing inhibitory concentration response curves using YD-3 and SEY3 we determined both molecules show similar off target effects at PAR1 and purinergic receptors (at �M concentrations of antagonists). Our future goals are, to use our synthetic route to create a library of small molecule PAR4 antagonists with improved selectivity for PAR4, and improved pharmacodynamic properties for use in additional physiologic assays and animal models.

“Identification of key residues for interaction with upon the t-SNARE proteins SNAP-25 and syntaxin1a” Zurawski, Zach
Negative regulation of exocytosis is a key cellular process occurring in a wide
range of secretory cells, acting to limit the release of neurotransmitters and peptide hormones into the extracellular space. It has been shown that activated G-protein coupled receptors (GPCRs), such as the alpha-2a adrenoreceptor, are capable of mediating the inhibition of neurotransmitter release at not only the synapses between neurons, but also the release of insulin granules from beta cells in the islets of Langerhans. Activation of the alpha-2a adrenoreceptor by noradrenaline generates free G protein βγ subunits, which can then bind to the SNARE proteins syntaxin1a (Stx1a), SNAP-25, and VAMP-2 to inhibit vesicle fusion in a competitive manner with the calcium sensor synaptotagmin. To determine residues on SNARE proteins required for the interaction with Gβγ subunits, our group has utilized a peptide-mapping approach in which purified Gβγ subunits interact with membrane-immobilized peptides derived from the primary sequences of SNAP-25 and Stx1a. This screen has highlighted a number of residues upon Stx1a and SNAP-25 potentially required for interaction with Gβγ, amongst them a series of charged residues on SNAP-25 in close proximity to the transmembrane regions. To identify the individual contribution of these residues on the full-length SNAP-25, we have adopted an alanine scanning mutagenesis approach. Residues of interest on SNAP-25b were mutagenized to Ala, with the resultant constructs expressed in E. coli as GST fusion proteins. To determine if these mutant SNAP-25 proteins were still capable of binding Gβγ, we performed GST-pull downs with these constructs and purified Gβ1γ2 and analyzed Gβγ bound via immunoblot. From this, it was determined that mutagenesis of the charged SNAP-25 residues identified in the peptide screen has reduced but not eliminated the ability of SNAP-25 to bind Gβγ, demonstrating a role for multiple residues on SNAP-25 in the interaction. For identification of key Gβγ-interacting residues upon Stx1a, co-flotation assays are being performed where recombinant human t-SNAREs consisting of full-length SNAP-25 and Stx1a will be incorporated into lipid vesicles and incubated with recombinant human Gβγ subunits. Residues on Stx1a identified as key to the interaction in the peptide mapping approach will be mutagenized to Ala, expressed as proteins, incorporated into vesicles, and subjected to co-flotation in the same manner. These studies will improve our understanding of G-protein mediated regulation of vesicle exocytosis through identification of the Gβγ binding regions on each component of the vesicle fusion machinery.

Vanderbilt University and Meharry Medical College
2011 Retreat
Session 6: Bioreactive
Epoxides and B(a)P Toxicology
Session Moderated by:
Twum Ansah
Presentations by: George Jules
Jing Jin Odaine Gordon
Lake Barkley State Resort Park, Cadiz, KY
Tuesday, October 25 & Wednesday, October 26

Title: Exposure to B(a)P in utero predispose LEH rat offspring to cardiovascular dysfunction in later life.
Authors: George E. Julesa, Siddharth Pratapb, Aramandla Rameshc, and Darryl B. Hooda,1
ABSTRACT
In utero exposure of fetuses to benzo(a)pyrene [B(a)P], a polycyclic aromatic hydrocarbon, can
result in the impairment of brain development and function. Although other developmental
indices are thought to be affected, the true nature of the effect of B(a)P exposure in utero is
relatively unclear. To investigate the effects of in utero B(a)P exposure on fetal cardiovascular
development, timed-pregnant Long Evans Hooded (LEH) rats were exposed to B(a)P (0, 600 and
1200 µg/kg/BW) by oral gavage on embryonic (E) days 14-17. We used high performance
liquid chromatography (HPLC) to establish the pre-weaning profile of B(a)P metabolites from
fetal heart tissue of B(a)P-exposed offspring. Although there were no significant effects of in
utero B(a)P exposure on the number of pups born per litter, the pre-weaning growth curves, or
the initial and final heart to body weight ratios, the blood pressure of the B(a)P exposed offspring
were significantly elevated compared to controls. Quantitative real time PCR also showed a in
the level of mRNA expression for angiotensin (AGT) and endothelial nitric oxide synthase
(NOS3/eNOS) both of which supported the significant upregulation of the AGT gene indicated
by microarray analysis. Ingenuity Pathway Analysis (IPA) and Expression Analysis Systematic
Explorer (EASE) software identified potential signaling and molecular pathways that could
explain the elevated blood pressure observed in the B(a)P-exposed offspring. Our findings
suggest that in utero exposure to B(a)P predispose offspring to functional deficits in
cardiovascular development which leads to cardiovascular dysfunction in later life.

Isolation and Structural Analysis of Leukotriene A Epoxides: Insights into the Mechanism of the Lipoxygenase-catalyzed Transformation Jing Jin, Yuxiang Zheng, William E. Boeglin and Alan R. Brash Department of Pharmacology, Vanderbilt University, Nashville, TN 37232 A pivotal role of Leukotriene A (LTA) epoxides is established in formation of bioactive mediators, including the leukotrienes, eoxins, lipoxins, resolvins, maresins and (neuro)protectins. LTA biosynthesis is attributed to a lipoxygenase (LOX)-catalyzed reaction with fatty acid hydroperoxides (HPETEs). Due to their extreme instability, LTA-type intermediates of LOX catalysis have not been isolated and their structure has not been analyzed directly. We hypothesize that transformation of the fatty acid hydroperoxide to LTA epoxide depends on participation of the lipoxygenase non-heme iron in catalyzing both the initial hydrogen abstraction and in facilitating cleavage of the hydroperoxide moiety. This postulate implies that the hydrogen abstracted and the hydroperoxide lie in suprafacial relationship, which in turn, dictates that the cis or trans epoxide configuration of the LTA product depends on the pro-R or pro-S chirality of the H-abstraction (an inherent property of the specific lipoxygenase) and the R or S chirality of the HPETE substrate. To test this hypothesis, we have developed methods for isolation and direct structural analysis of LTA epoxides. We expressed human 15-LOX-1 in E. coli and purified the protein by nickel affinity chromatography by using an N-terminal His-tag. Reaction of 15S-HPETE with purified human 15-LOX-1 was performed in a biphasic hexane/pH 7.5 aqueous system. After 1.5 min of vortex mixing at 0 °C, UV spectroscopy of the hexane phase showed a decrease of substrate and appearance of a new chromophore with max at 280 nm characteristic of the LTA epoxide. Rapid esterification (diazomethane) and RP-HPLC (pH 8) or SP-HPLC (with 0.5% TEA) produced 14,15-LTA4 methyl ester in 10-20 microgram quantities. Subsequent NMR analysis showed that the epoxy configuration of the generated 14,15-LTA4 is trans, which is consistent with our hypothesis. The results demonstrate the feasibility of enzymatic synthesis, isolation and characterization of LTA epoxides and provide the experimental basis for analysis of LTA epoxide intermediates in formation of eoxins, lipoxins, resolvins, and other novel products. This work was supported by NIH grant GM-15431.

ARE OXIDATIVE METABOLITES OF CURCUMIN NOVEL ANTI-CANCER AGENTS?
Odaine Gordon, Claus Schneider
Division of Clinical Pharmacology, Department of Pharmacology, Vanderbilt University Medical School, Nashville, TN, 37232, U.S.A
Curcumin, the polyphenolic yellow pigment from the turmeric plant, is the subject of several dozen ongoing clinical trials for its efficacy in inflammatory diseases including cancers and diabetes. Curcumin has been shown to be effective in animal cancer models, in part by targeting the pro-inflammatory NF-κB and cyclooxygenase-2 pathways, although the molecular-chemical mechanism of action of curcumin is largely unclear. Here we present evidence that curcumin undergoes a previously unrecognized (aut-)oxidative transformation. The novel products include reactive electrophilic intermediates and a dioxygenated bicyclopentadione as the final stable metabolite. We isolated and identified one of the intermediates as a reactive epoxide and showed that it formed covalent adducts with cellular nucleophiles (GSH, N-acetylcysteine). We hypothesize that the products and intermediates of oxidative transformation are mediators of some of the biological effects of curcumin, and specifically, that their reactivity with activated thiols is the mechanistic basis for the inhibition of NF-κB and induction of antioxidant signaling by curcumin. We detected abundant levels of the bicyclopentadione in plasma and intestinal mucosa of mice after oral administration of curcumin suggesting that oxidative transformation is a prominent reaction in vivo. Further exploration of the cellular targets of the reactive intermediates and final bicyclopentadione will increase our understanding of the mechanism of action of the cancer chemopreventive agent curcumin.

Vanderbilt University and Meharry Medical College
Retreat
SAVE THE DATE!!!
2012 Retreat Dates:
Wednesday, October 17 &
Thursday, October 18





























