Embed Size (px)
Citation preview

UvA-DARE is a service provided by the library of the University of Amsterdam (http://dare.uva.nl)
UvA-DARE (Digital Academic Repository)
KLF2, a critical modulator in vascular disease
van Thienen, J.V.
Link to publication
Citation for published version (APA):van Thienen, J. V. (2009). KLF2, a critical modulator in vascular disease.
General rightsIt is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s),other than for strictly personal, individual use, unless the work is under an open content license (like Creative Commons).
Disclaimer/Complaints regulationsIf you believe that digital publication of certain material infringes any of your rights or (privacy) interests, please let the Library know, statingyour reasons. In case of a legitimate complaint, the Library will make the material inaccessible and/or remove it from the website. Please Askthe Library: https://uba.uva.nl/en/contact, or a letter to: Library of the University of Amsterdam, Secretariat, Singel 425, 1012 WP Amsterdam,The Netherlands. You will be contacted as soon as possible.
Download date: 07 Aug 2019

7 Simvastatin has an anti-inflammatory effect on macrophages via upregulation of an atheroprotective transcription factor KLF2
Tiina T. Tuomisto1*, Henri Lumivuori1*, Emilia Kansanen1, Sanna-Kaisa Häkkinen1, Mikko P. Turunen1, Johannes V. van Thienen2, Anton J. Horrevoets2, Anna-Liisa Levonen1, Seppo Ylä-Herttuala1,3,4
*Authors with equal contribution 1Department of Biotechnology and Molecular Medicine, A. I. Virtanen Institute, Kuopio University, Kuopio, Finland
2Department of Medical Biochemistry, Academic Medical Center, University of Amsterdam, Amsterdam, The Netherlands
3Department of Medicine, Kuopio University, Kuopio, Finland
4Gene Therapy Unit, Kuopio University Hospital, Kuopio, Finland
Cardiovasc Res. 2008 Apr 1;78(1):175-84.
Proefschrift JVvT page 125
Composite
Monday, November 24, 2008 16:54

126
Abstract
Aim. Statins have beneficial vascular effects beyond their cholesterol-lowering action. Since macrophages play a central role in atherogenesis, we characterized the effects of simvastatin on gene expression profile of human peripheral blood monocyte-macrophages (HPBM).
Methods. Gene expression profile was studied using Affymetrix gene chip analysis. Lentiviral gene transfer of Kruppel-like factor 2 (KLF-2) was used to further study its role in macrophages.
Results. Simvastatin treatment lead to downregulation of many proinflammatory genes including several chemokines (e.g. monocyte chemotactic protein-1 (MCP-1), macrophage inflammatory proteins-1a and β, interleukin-2 receptor-β), members of the tumor necrosis factor family (TNF) (e.g. lymphotoxin beta), vascular cell adhesion molecule-1 and tissue factor (TF). Simvastatin also modulated the expression of several transcription factors essential for inflammation: NF-kB relA/p65 subunit and ets-1 were downregulated, and an atheroprotective transcription factor KLF-2 was upregulated. The effects of simvastatin on MCP-1 and TF could be mimicked by KLF-2 overexpression using lentiviral gene transfer.
Conclusions. Simvastatin has a strong anti-inflammatory effect on HPBM including upregulation of the atheroprotective factor KLF-2. This may partly explain the beneficial effects of statins on cardiovascular diseases.
Proefschrift JVvT page 126
Composite
Monday, November 24, 2008 16:54

127
Chapter
Introduction
Inflammation and vessel wall macrophages play important roles in the pathogenesis of atherosclerosis. Lesion macrophages secrete a number of growth factors, cytokines and other molecules, such as matrix metalloproteinases (MMPs), which are involved in lesion progression: they activate T-cells, enhance SMC proliferation, and contribute to endothelial dysfunction, lesion rupture, and blood coagulation1. Therefore, to effectively prevent and treat cardiovascular events treatments targeting macrophages would be desireable.
The HMG-CoA inhibitors, statins, have other beneficial effects on atherogenesis in addition to their lipid-lowering action. These pleiotropic effects include e.g. the upregulation of the production of nitric oxide in endothelial cells (ECs), decreased proliferation of vascular smooth muscle cells (SMC), inhibition of platelet activation and increased fibrinolytic activity2;3. Importantly, statins have been shown to modulate the inflammatory process in the vessel wall. They reduce both the number and the activity of inflammatory cells within atherosclerotic plaques 4. Favorable effects include the modulation of cytokine secretion and signaling3;5, decreased monocyte-endothelial cell adhesion 6, decreased expression of tissue factor (TF) and MMPs 4 in macrophages, and inhibition of oxLDL-induced macrophage proliferation 7. Our recent finding that human lesion macrophages overexpress HMG-CoA reductase gene may explain why statins effectively reduce inflammation in the vessel wall 8.
Mechanisms underlying these pleiotropic effects remain incompletely understood. Studies implicate that the inhibition of isoprenoid synthesis [geranyl-geranyl-pyrophosphate (GGPP) and farnesyl-pyrophosphate (FPP)] mediates the effects of statins on GTP-binding proteins (e.g. Ras and Rho)2;3;9. For example, Rho regulates eNOS gene expression and controls SMC proliferation 10. Statins have been shown to influence the activity of some transcription factors: they inhibit the binding of NF-kB and activator protein AP-1 to nuclear proteins in SMCs and ECs 11. Additionally, simvastatin has been shown to block TNF-α-induced NF-kappaB transcriptional activity and IkappaB phosphorylation/degradation. Interestingly, statins have recently been shown to upregulate the expression of Kruppel-like factor-2 (KLF-2) in ECs 12. KLF-2 is a transcription factor identified from the endothelial “atheroprotective phenotype”: its overexpression inhibits pro-inflammatory and pro-thrombotic gene expression in endothelial cells, such as vascular cell adhesion molecule-1 (VCAM-1) and plasminogen activator inhibitor-1 (PAI-1) expression, and its overexpression enhances the expression of eNOS and thrombomodulin 12-15. KLF-2 expression in ECs is modulated by shear stress, and thus it might partially mediate the atheroprotective effects of steady laminar flow 16;17. Less is known about the role of KLF-2 in monocytes, but according to recent study, KLF-2 regulates the proinflammatory activation of monocytes, which suggests an anti-inflammatory role for KLF-2 in monocytes 18.
ETS-1 transcription factor takes part in vascular inflammation and remodeling. It regulates the expression of a number of vascular-specific genes, such as adhesion molecules, chemokines (e.g. MCP-1), and matrix metalloproteinases 19;20. Its expression can be induced
7
Sim
vastatin is anti-inflamm
atory in macrophages
Proefschrift JVvT page 127
Composite
Monday, November 24, 2008 16:54

128
e.g. by proinflammatory cytokines and vasoactive peptides such as angiotensin II 19, but there are no studies implicating statin effects on its expression.
Although there is already evidence that statins have anti-inflammatory properties, the mechanism of action remains incompletely understood. Especially, very little is known about the global effects of statins on macrophage gene expression. In this study we show that simvastatin has a strong anti-inflammatory effect on human peripheral blood monocyte macrophages (HPBM), including downregulation of the expression of several cytokines, members of the TNF-family and TF. These effects are mediated by the effects on transcription factors: simvastatin reduces NF-kB signaling pathway and c-ets, and upregulates KLF-2, which implicate an atheroprotective role for KLF-2 also in macrophages. These findings may at least partly explain the beneficial pleiotropic effects of statins on cardiovascular diseases.
Methods
Cell culture studies Human peripheral blood monocytes (HPBM) were isolated from buffy coats from healthy blood-donor volunteers (Finnish Red Cross, Helsinki, Finland) using Ficoll-Paque gradient centrifugation 21. None of the blood donors were on statin therapy. During isolation, monocytes from 3-4 individuals were pooled. Adherent cells were cultured in standard medium (RPMI 1640-medium with 20% human serum [Cambrex], 1 % Penisillin and Streptomycin and 1 % L-Glutamine) for differentiation into macrophages. The macrophage-phenotype at day 7 after isolation was confirmed with the typical shape of macrophages and also by macrophage-immunostaining (mAB CD68, DAKO, Denmark), where macrophages presented >95% of the cell population (data not shown). Human monocytic THP-1 cells (ATCC TIB-202) were cultured in RPMI-1640 medium according to ATCCs instructions. Cells were stimulated by 0.1 mM phorbol 12-myristate 13-acetate (PMA) (Sigma, USA) to induce differentiation into macrophages (=”THP-1 macrophages”). The study protocol has been accepted by the Ethical Committee of the University of Kuopio. The investigation conforms with the principles outlined in the Declaration of Helsinki for use of human tissues. Simvastatin treatment Simvastatin was a gift from Merck & Co. The inactive lactone form of simvastatin was hydrolyzed to the corresponding β-hydroxy acid. The HPBM-macrophages were treated with statin at day 7 after the isolation and the THP-1 cells at day 4 after the PMA-stimulation. 12 hours prior to statin treatment the cell growth media were changed to serum-free media. The statin-treated (10 mM simvastatin in serum-free media) cells were collected at 12 h and 24 h for Affymetrix analyses. Statin-stimulated THP-1 macrophages were collected also at 48 h and 72 h time-points for TaqMan analyses. The simvastatin treatment was also carried at lower concentrations of simvastatin (0.025 - 1 mM) in THP-1 macrophages for TaqMan analysis.
Proefschrift JVvT page 128
Composite
Monday, November 24, 2008 16:54

129
Chapter
The toxicity of simvastatin was assessed in cell culture: 5x concentration of simvastatin (50 mM) had no effect on cell viability. Inhibition of protein prenylation The THP-1 macrophages were stimulated by 20 mM concentration of farnesyl transferase inhibitor (FTI-277, Sigma) and geranyl-geranyl-transferase inhibitor (GGTI-298, Sigma). FTI and GGTI were dissolved in DMSO. Control cells were incubated with an equivalent concentration of DMSO.
RNA isolation Total RNA was isolated from the cells using Trizol reagent (Gibco BRL, USA) according to manufacturers’ instructions. The quality of RNA was assessed by spectrophotometry (NanoDropÒ, USA) and by agarose gel electrophoresis. Microarray analyses For Affymetrix analyses three separate HPBM cell isolation and simvastatin experiments were performed (HPBMs from 3 individuals at each time, total n=9). Cells were collected at 12 h and 24 h after the statin treatment. Total RNA was isolated as above. cDNAs were prepared from RNAs (5 mg of RNA) with reverse transcriptase (Superscript II primed by a poly (T) oligomer/T7 promoter). cDNAs were subsequently used as a template to make biotin-labeled cRNA with an in vitro transcription reaction. cRNAs (15 mg) were hybridized to Affymetrix HGU133 Plus 2.0 oligonucleotide arrays, which was processed and scanned according to manufacturer’s instructions. Each array quantifies the expression of over 47 000 transcripts (including full-length mRNA sequences and ESTs) derived from build 133 of the UniGene database (www.affymetrix.com). Microarray data analysis Affymetrix GeneChip® Operating Software (GCOS) was used to generate .CEL files which were then converted into .DCP files using dChip (http://www.dchip.org) V1.3 software 22. The arrays were normalized to baseline array with median probe intensity, and gene expression data were generated calculating model-based expression values. The t-statistic is computed as (mean1 – mean2) / sqrt (SE(mean1)2 + SE(mean2) 2), and its p-value is computed based on the t-distribution and the degree of freedom is set according to Welch modified two-sample t-test. In this study, genes were considered differentially expressed if they changed more than 1.5-fold (90% confidence bound of the fold change), absolute difference of signals was >100, at least 40% of the samples were called present in both groups and False Discovery Rate (FDR) was ≤ 1%. Hierarchical clustering was performed by dChip using Pearson correlation with a centroid-linkage method 23. Gene function analysis was performed by using the gene ontology mining tool GoSurfer incorporated in dChip program. The .CEL files and pivot table .txt
7
Sim
vastatin is anti-inflamm
atory in macrophages
Proefschrift JVvT page 129
Composite
Monday, November 24, 2008 16:54

130
tabdelimited files (GCOS) are available at GEO repository (www.ncbi.nlm.nih.gov/GEO) with series record GSE4883. Real-time quantitative RT-PCR (TaqMan) analyses TaqMan analyses were performed to validate gene expression changes of the selected genes. TaqMan analyses were performed from the same HPBM-samples as Affymetrix analyses and in THP-1 macrophages with protein prenylation inhibitors. Total RNA was converted to cDNA by in vitro transcription reaction (M-MuLV Reverse Transcriptase, Finnzymes, Finland). cDNAs (10-25 ng of cDNA depending on the gene) were used as templates for TaqMan qRT-PCR with ABI Assays-on-Demand on ABI Prism 7900 sequence detection system. The specific assays used were Hs00360439-g1 (KLF-2), Hs00234140-m1 (MCP-1), Hs00175225-m1 (TF), Hs00234142-m1 (MIP-1α), Hs00242737-m1 (LTB), and Hs00231279-m1 (p65/RelA). All samples were run in quarduplicate, and rRNA assay (Ribosomal RNA control reagent, ABI) was used as an internal control to normalize the RNA amount. Quarduplicates were averaged to calculate an expression value for each sample, and data was presented as mean expression value relative to the control ± standard deviation. To evaluate statistical significances, independent samples t-test was used for the appropriate parameters (Microsoft Excel). Western blot analyses HPBM and THP-1 cells treated with protein prenylation inhibitors were lysed in lysis buffer (50mM Tris, Ph 7,5, 150mM NaCl, 1mM EDTA, 1% Triton X-100, 0,5% sodium deoxylacholate, 0,1% SDS, 10% glycerol). Cells were incubated on ice for 10 min and centrifuged (10000 g) for 10 min at +4°C. Supernatants were transferred into new tubes and the protein concentration was determined using the BCA protein assay kit (Pierce, USA). For Western blot, sample (20 µg of protein) was separated in 12% SDS-PAGE gel. After electrophoresis proteins were transferred onto nitrocellulose membranes (Trans-Blot Transfer Medium, Bio-Rad Laboratories, CA). Membranes were blocked over night at +4°C (5% goat serum,Vector laboratories) in Tris-buffered saline with 0.1% Tween-20 (TBS/T), pH 7.6. Membranes were incubated with a diluted primary antibody (Anti-KLF-2, Abcam Cambridge, UK, 3 μg/ml) in 5% goat serum in TBS/T over night at +4°C. After washing with TBS/T, membranes were incubated with a diluted secondary antibody (Peroxidase-conjugated Affinipure Donkey Anti-Goat, Jackson Immunoresearch, USA, 8 ng/ml). Antigen-antibody complexes were detected by chemiluminecence (Supersignal West Dura Extended Duration Substrate, Pierce) and exposed to a high performance chemiluminescence film (Amersham Biosciences, UK).
Proefschrift JVvT page 130
Composite
Monday, November 24, 2008 16:54

131
Chapter
KLF-2 overexpression studies The lentiviral vector constructs and the preparation of the viruses have been described elsewhere 17;24. As controls for lentivirus overexpressing KLF-2 we used respective virus without KLF-2 and no-virus-containing control. THP-1 derived macrophages were transduced overnight using MOI 5 and MOI 10. After 7 days RNA was isolated for TaqMan analyses of KLF-2, MCP-1 and TF expression as above. Costimulations of simvastatin and lentiviral overexpression of KLF-2 were performed with THP-1 macrophages. 7 days after lentiviral overexpression of KLF-2 (MOI 10) and/or statin stimulation (day 6, 10 mM simvastatin) RNA was isolated and used for the analysis of KLF-2, MCP-1 and TF expression as above. Results
Simvastatin attenuates the expression of inflammatory genes in HPBMs To examine the transcriptional response to simvastatin by HPBMs we performed genome wide gene expression analysis using Affymetrix gene chips at time-points 12 h and 24 h after the statin treatment. From the ~47000 transcripts on the Affymetrix gene chip, a total of 589 genes showed statistically significant changes in expression either at 12h or 24h as compared to the control group using a value of 1.5 for the lower bound of the 90% confidence interval for the fold change as a cutoff. When analyzed separately, 280 genes were either up- or downregulated at 12h and 502 genes at 24h. As a general phenomenon, we found more downregulated genes that upregulated genes after the statin treatment.
To assess the relationships and coordinate expression profiles between the regulated genes we performed a cluster analysis of the pooled data set. The analysis showed ten well-defined clusters, which contained several genes that were related (e.g. the same gene family or the functional group), for example "inflammatory gene cluster" (cluster 3), "cell structure" (cluster 2), "cholesterol-metabolism-cluster" (cluster 5), "signal transduction with downregulation" (cluster 4) and "signal transduction with upregulation" (cluster 6), "KLF-cluster" (cluster 8), and "metallothionein cluster" (cluster 10) (Figure 1). We found that the inhibition of HMG-CoA reductase in macrophages and the subsequent reduction in the cholesterol synthesis lead to compensatory changes at 24 h in several genes functioning in the cholesterol metabolism: HMG-CoA reductase was induced 1.85-fold, as well as enzymes participating in the HMG-CoA synthesis (Acetyl-CoA synthase 1.73-fold; HMG-CoA synthase 2.82-fold). We also found induction of downstream enzymes, such as farnesyl transferase (1.53) and squalene epoxidase (1.96). Additionally, LDL-receptor gene was induced 1.79-fold. A similar pattern of changes in gene expression (upregulation at 24 h) was also seen in cholesterol-metabolism-related genes in cluster analysis: the above-mentioned genes formed a separate cluster (cluster 5) (Figure 1). Therefore, we concluded that the simvastatin treatment was efficient enough to affect cholesterol and prenyl metabolism, and the Affymetrix system was sensitive enough to detect these changes in gene expression.
7
Sim
vastatin is anti-inflamm
atory in macrophages
Proefschrift JVvT page 131
Composite
Monday, November 24, 2008 16:54
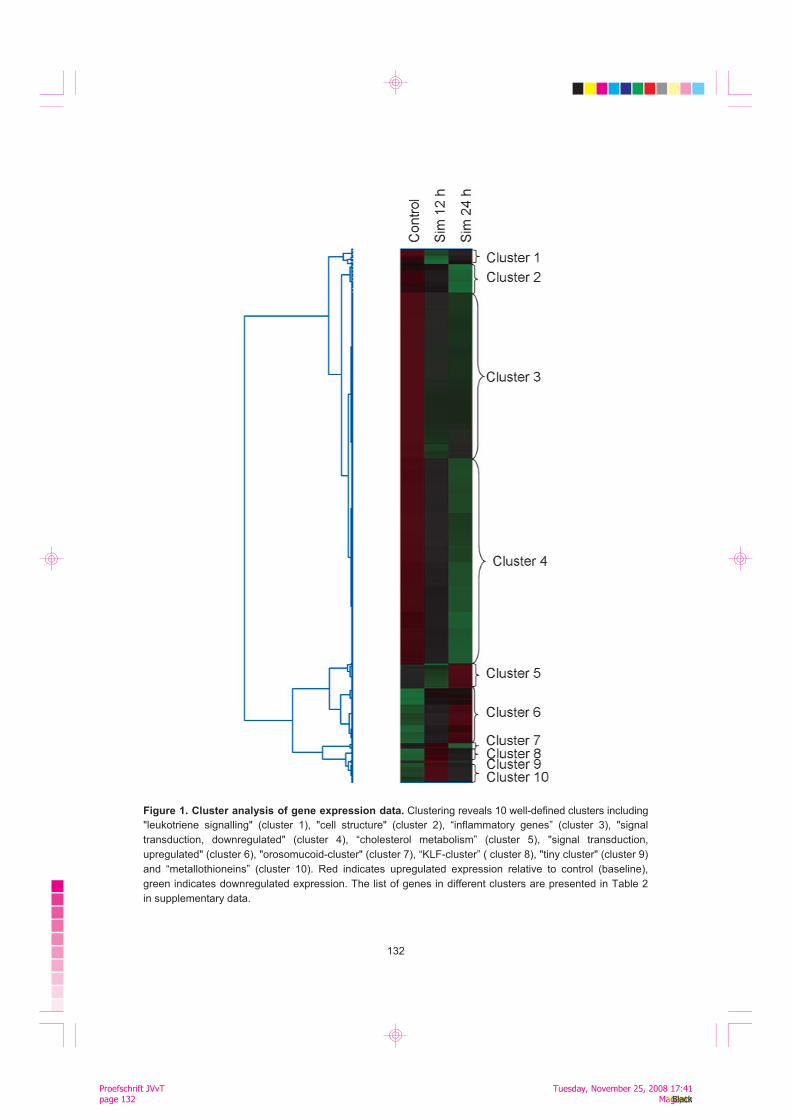
132
Figure 1. Cluster analysis of gene expression data. Clustering reveals 10 well-defined clusters including
"leukotriene signalling" (cluster 1), "cell structure" (cluster 2), “inflammatory genes” (cluster 3), "signal
transduction, downregulated" (cluster 4), “cholesterol metabolism” (cluster 5), "signal transduction,
upregulated" (cluster 6), "orosomucoid-cluster" (cluster 7), “KLF-cluster” ( cluster 8), "tiny cluster" (cluster 9)
and “metallothioneins” (cluster 10). Red indicates upregulated expression relative to control (baseline),
green indicates downregulated expression. The list of genes in different clusters are presented in Table 2
in supplementary data.
Proefschrift JVvT page 132
Cyan
Magenta
Yellow
Black
Cyan
Magenta
Yellow
Black
Tuesday, November 25, 2008 17:41

133
Chapter
Simvastatin treatment led to a reduced mRNA expression of many inflammatory genes (Table 1). These genes include several chemokines, such as MCP-1 and macrophage inflammatory protein-1a (MIP-1a) and -b, RANTES, CXC9, and CXC10; several members of TNF superfamily, such as TRAIL and lymphotoxin beta (LTB); interleukin receptors, adhesion molecules, such as VCAM-1 and ICAM-3; and TF. Several inflammatory genes and mediators of inflammatory gene signaling had similar expression patterns in macrophages, as demonstrated by cluster analysis. The cluster number 3, which includes genes with downregulated gene expression, contains almost entirely inflammatory genes, and includes the above mentioned members of the cytokine and TNF-families.
The expression changes in cytokines MCP-1 and MIP-1α, TF, and a member of TNF-family (LTB) were further confirmed by TaqMan analyses in HPBM as well as in THP-1 derived macrophages (Figure 2A and B). Downregulation of MCP-1 and TF sustained for 48 and 72 h in THP-1 cells (results not shown).
Simvastatin attenuates inflammatory signalling in macrophages: downregulation of NF-kB relA/p65 subunit mRNA expression and upregulation of KLF 2 expression In Affymetrix analysis, simvastatin treatment lead to reduced expression of several genes that mediate inflammatory signaling in cells, including several guanylate binding proteins (GBP 1-5), Ras and Rho-proteins; and several transcription factors, such as NF-kB relA/p65 protein, PPARd, and v-ets. The expression of KLF-2 was strongly induced (3.9-fold upregulation at 12 h, 9.3-fold at 24 h). Additionally, the KLF family members KLF-3 and -4 had increased expression levels. Most of the inflammatory signaling-related genes clustered together with other inflammatory genes to cluster 3, which shows decreasing expression pattern, whereas KLFs 2-4 formed a separate cluster with increasing expression patterns (cluster 8) (Figure 1).
In Affymetrix analysis, several members of the NF-kB signaling pathway showed a trend to reduced RNA expression levels, for example NF-kB /rel protein RelA/p65 (-2.0-fold decrease at 24 h), RelB and NF-kB p50, but all the changes were not statistically significant. Additionally, several members of the signaling cascades leading to NF-kB activation had downregulated expression, e.g. protein kinase C (-1.4) and Akt and Cot (-1.4). Also JAK-STAT (STAT -1.6 fold) signaling pathway was donwnregulated.
The expression changes of relA/p65 and KLF-2 were further studied with TaqMan in HPBM and THP-1 macrophages (Figure 2), and KLF-2 protein expression by Western blot analysis (Figure 2). TaqMan analysis confirmed the downregulated levels of relA/p65 mRNA in HPBM cells (Figure 2A), as well as the induction of KLF-2 expression in HPBM and THP-1 macrophages (Figure 2A and B). There was also an increase in the KLF-2 protein (Figure 2C). The effect of simvastatin on KLF-2, TF and MCP-1 expression was detected also at concentration of 1 mM simvastatin as studied by TaqMan analysis (data not shown). However, at lower concentrations of simvastatin (0.025- 0.1 mM) the effect was not as remarkable.
7
Sim
vastatin is anti-inflamm
atory in macrophages
Proefschrift JVvT page 133
Composite
Tuesday, November 25, 2008 17:25
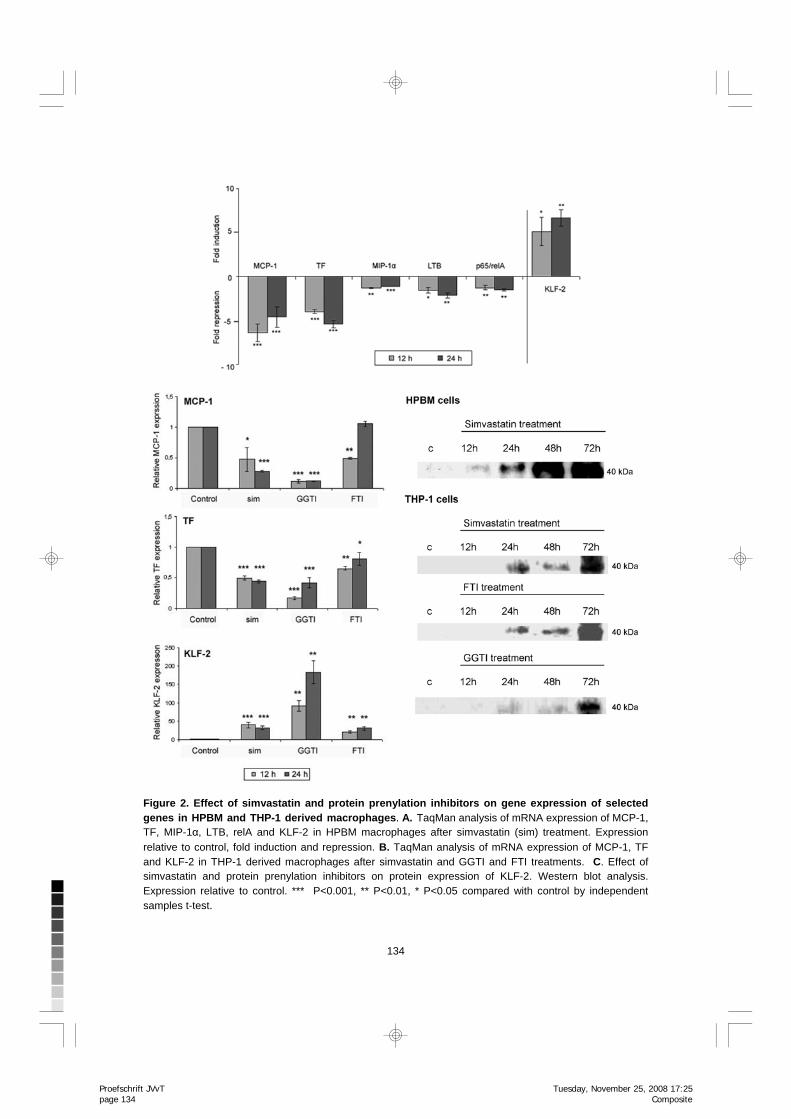
134
Figure 2. Effect of simvastatin and protein prenylation inhibitors on gene expression of selected genes in HPBM and THP-1 derived macrophages. A. TaqMan analysis of mRNA expression of MCP-1, TF, MIP-1α, LTB, relA and KLF-2 in HPBM macrophages after simvastatin (sim) treatment. Expression relative to control, fold induction and repression. B. TaqMan analysis of mRNA expression of MCP-1, TF and KLF-2 in THP-1 derived macrophages after simvastatin and GGTI and FTI treatments. C. Effect of simvastatin and protein prenylation inhibitors on protein expression of KLF-2. Western blot analysis. Expression relative to control. *** P<0.001, ** P<0.01, * P<0.05 compared with control by independent samples t-test.
Proefschrift JVvT page 134
Composite
Tuesday, November 25, 2008 17:25

135
Chapter
The inhibition of protein prenylation has the same effect on KLF-2, MCP-1 and TF expression as simvastatin THP-1 macrophages were treated with protein prenylation inhibitors GGTI (inhibitor of geranyl-geranyl transferase) and FTI (inhibitor of farnesyl transferase). Both GGTI and FTI treatments lead to the downregulation of MCP-1 and TF mRNA expression, and to the upregulation of KLF-2 expression (Figure 2B). Moreover, simvastatin, GGTI and FTI treatments had similar effects on KLF-2 protein expression (Figure 2C). MCP-1 and TF are KLF-2 target genes in macrophages To test whether MCP-1 and TF are KLF-2 target genes in macrophages, we performed lentiviral-mediated KLF-2 overexpression in THP-1 macrophages. Lentiviral transduction led to very strong (~6000 and ~12000-fold at 7 days for MOI 5 and MOI 10, respectively) expression of KLF-2 in macrophages (Figure 3A). In macrophages overexpressing KLF-2 the expression of MCP-1 was repressed -5.9-fold (MOI 10) and TF -1.9-fold (MOI 10) relative to control virus (Figure 3A). We also studied the effects of lentiviral KLF-2 overexpression in combination with statin treatment in THP-1 macrophages (Figure 3B). However, simvastatin treatment had no additional effects over KLF-2 overexpression.
Discussion
This gene expression array study of simvastatin effects on macrophages shows that simvastatin has a global anti-inflammatory effect on macrophages, which includes attenuated expression of several pro-inflammatory cytokines, TNF family members, TF, adhesion molecules and molecules mediating inflammatory signaling. All these molecules play important roles in atherogenesis. Interestingly, also some less known TNF-family members, such as TRAIL and LTB, were attenuated by simvastatin.
Clustering of gene expression data to several well-defined clusters suggests that the effects on simvastatin on gene expression are mediated by several different pathways. The potency of the cluster analysis in finding groups of genes with similar regulatory mechanisms is demonstrated by the clustering of genes coding for proteins participating in cholesterol and lipid metabolism to a separate cluster (cluster 5): this can be explained by the control of their expression by sterol regulatory elements: cholesterol deprivation induces their expression via SREBP1 25. Additionally, many cytokines and other inflammatory proteins had similar decreasing expression profiles (cluster 3), which might suggest that the effects of simvastatin on proinflammatory gene expression are mediated by the same mediators. Additionally, the KLFs 2-4 formed a separate cluster (cluster 8), as well as metallothioneins (cluster 10). Several metallothioneins (MT 1E, X, G and 2A), which participate in the metal homeostasis as well as the control of REDOX-balance, inflammation and cell proliferation 26, are induced at 12h by simvastatin in a similar manner. However, their role in macrophages has not been established, and there is no previous information about their regulation by statins.
7
Sim
vastatin is anti-inflamm
atory in macrophages
Proefschrift JVvT page 135
Composite
Tuesday, November 25, 2008 17:25

136
The anti-inflammatory effect of simvastatin can be at least partly explained by its impact on proinflammatory signal transduction. Several members of the NF-kB signaling pathway had reduced mRNA expression levels, for example NF-kB complex proteins p65/RelA, RelB and p50. Additionally, several members of the signaling cascades leading to NF-kappaB activation had downregulated expression, e.g. Janus kinases, protein kinase C, Akt and Cot. It has been previously shown that statins inhibit the binding of NF-kB to nuclear proteins 11, and inhibit the phosphorylation and degradation of the NF-kB inhibitory protein IkB 27. We show here that simvastatin downregulates mRNA expression of the NF-kB subunits in HPBM cells. Besides NF-kB signaling, a potent pro-inflammatory transcription factor c-ets, which mediates e.g. MCP-1 and phospholipase expression 19;20, as well as the family of guanylate binding proteins (GBPs 1-5), and JAK-STAT signaling pathway were downregulated by simvastatin.
We show here that KLF-2, as well as its family members KLF-3 and KLF-4, are induced in macrophages by simvastatin. KLFs are recently characterized zinc finger transcription factors that have several important functions; they modulate cell differentiation, organ development and proliferation 16. KLF-2, -4,-5 and -6 have particular importance in the vasculature, and they have functional as well as pathological roles 16. KLF-2 has been shown to be essential for the vascular development 16, and it has been identified as part of the
Figure 3. Effect of lentiviral KLF-2 overexpression in THP-1 macrophages. A. TaqMan analyses of KLF-2, MCP-1 and TF expression levels at 7 days after transduction with MOI 5 and MOI 10. Bars show expression levels relative to respective virus controls. *** P<0.001, ** P<0.01, * P<0.05 compared to control by independent samples t-test. B. Costimulation of THP-1 macrophages with simvastatin and lentiviral overexpression of KLF-2. TaqMan analyses of gene expression of KLF-2, MCP-1 and TF at 7 days after lentiviral overexpression of KLF-2 (MOI 10) and/or statin stimulation (day 6, 10 mM simvastatin). Expression relative to control, fold induction or repression.
Proefschrift JVvT page 136
Composite
Tuesday, November 25, 2008 17:25

137
Chapter
“atheroprotective phenotype” of ECs. It is induced by steady laminar flow28;29, and inhibited by TNF-α28 and IL-1β 14 in ECs. It has several anti-inflammatory properties, such as reduction of adhesion molecule expression 14. Additionally, KLF-2 has been shown to regulate the thrombotic function of ECs, e.g. the expression of thrombomodulin, eNOS and PAI-1 as wells as cytokine-mediated production of TF 13. Global analysis of gene expression changes after adenoviral overexpression of KLF-2 in ECs showed that induction of KLF-2 resulted in the regulation of endothelial transcription programs controlling inflammation, thrombosis/hemostasis, vascular tone, and blood vessel development 12. Moreover, another microarray study showed that KLF-2 acts as a general transcriptional switch point between the quiescent and activated status of ECs where lentiviral overexpression of KLF-2 resulted in changes in several key functional pathways such as cell migration via VEGFR2, inflammation and hemostasis 30.
Recently, it has been shown that statins upregulate KLF-2 expression in ECs, which might partly explain the atheroprotective effects of statins 12;15. The statin effect on KLF-2 was dependent on the ability of statins to inhibit Rho-pathway, as adenoviral overexpression of Rho-protein decreased KLF-2 expression 15.
Very little is known about the role of KLF-2 in macrophages. A recent study showed that KLF-2 regulates proinflammatory activation of monocytes: overexpression of KLF-2 inhibited LPS-activated production of cytokines and reduced phagocytic activity 18. Interestingly, the expression of KLF-2 was reduced in circulating monocytes in patients with CAD 18. In this study we showed that KLF-2 is induced by simvastatin in mature human macrophages. We
Figure 4. Simvastatin has a strong anti-inflammatory e f f e c t o n H P B M macrophages. The inhibition of NF-kB and ets signaling and the upregulation of KLF-2 signalling by simvastatin leads to attenuated expression of many pro-inflammatory genes (see also Table 1).
7
Sim
vastatin is anti-inflamm
atory in macrophages
Proefschrift JVvT page 137
Composite
Tuesday, November 25, 2008 17:25

138
also showed that the mechanism of induction of KLF-2 by simvastatin in macrophages is dependent on the inhibition of protein prenylation. Additionally, by lentiviral overexpression of KLF-2 we showed that MCP-1 and TF are KLF-2 target genes in macrophages, and KLF-2 represses their expression. Moreover, several KLF-2 endothelial cell target genes 12, e.g. CXCL10, CCL5 and TNF had changed expression levels in simvastatin-stimulated macrophages, which suggests them to be potential KLF-2 target genes in macrophages as well. Taken together, this suggests an anti-inflammatory and vasculoprotective role for KLF-2 in macrophages.
In addition to the induction of KLF-2, we showed that also its family members KLF-3 and KLF-4 are induced by simvastatin. However, the functions of KLF-3 and KLF-4 are poorly known and further studies are needed to clarify their role in macrophages and other cell types.
The inhibition of protein prenylation by inhibitors of geranyl-geranylation and farnesylation had similar effects on KLF-2, MCP-1 and TF expression as simvastatin. Isoprenoid binding to Ras and Rho proteins is essential for the initiation of G-protein signaling, which controls for example cell proliferation and stress responses. Farnesylated proteins include e.g. Ras, whereas Rho and Rac activation are dependent on geranyl-geranylation9. The similar effects achieved by protein prenylation inhibitors and simvastatin suggest that the simvastatin effects might be dependent on the inhibition of protein prenylation. It would be interesting to study whether alterations in cellular cholesterol content correlate with these effects and whether similar effects on cellular transcriptional machinery could be achieved via other means that can alter cellular cholesterol levels.
In conclusion, we show here that simvastatin has a strong anti-inflammatory effect on macrophages including attenuated expression of several cytokines, TNF family members and some proinflammatory signaling molecules. Most interestingly, transcription factor KLF-2 is upregulated by simvastatin in macrophages. These findings support the notion that statins have other antiatherogenic actions beyond their effects on plasma cholesterol. Reference List
1. Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685-1695.
2. Palinski W, Napoli C. Unraveling pleiotropic effects of statins on plaque rupture. Arterioscler Thromb Vasc Biol. 2002;22:1745-1750.
3. Ray KK, Cannon CP. Intensive statin therapy in acute coronary syndromes: clinical benefits and vascular biology. Curr Opin Lipidol. 2004;15:637-643.
4. Aikawa M, Rabkin E, Sugiyama S, Voglic SJ, Fukumoto Y, Furukawa Y et al. An HMG-CoA reductase inhibitor, cerivastatin, suppresses growth of macrophages expressing matrix metalloproteinases and tissue factor in vivo and in vitro. Circulation. 2001;103:276-283.
5. Waehre T, Yndestad A, Smith C, Haug T, Tunheim SH, Gullestad L et al. Increased expression of interleukin-1 in coronary artery disease with downregulatory effects of HMG-CoA reductase inhibitors. Circulation. 2004;109:1966-1972.
Proefschrift JVvT page 138
Composite
Tuesday, November 25, 2008 17:25

139
Chapter
6. Yoshida M, Sawada T, Ishii H, Gerszten RE, Rosenzweig A, Gimbrone MA, Jr et al. Hmg-CoA reductase inhibitor modulates monocyte-endothelial cell interaction under physiological flow conditions in vitro: involvement of Rho GTPase-dependent mechanism. Arterioscler Thromb Vasc Biol. 2001;21:1165-1171.
7. Senokuchi T, Matsumura T, Sakai M, Yano M, Taguchi T, Matsuo T et al. Statins suppress oxidized low density lipoprotein-induced macrophage proliferation by inactivation of the small G protein-p38 MAPK pathway. J Biol Chem. 2005;280:6627-6633.
8. Tuomisto TT, Korkeela A, Rutanen J, Viita H, Brasen JH, Riekkinen MS et al. Gene expression in macrophage-rich inflammatory cell infiltrates in human atherosclerotic lesions as studied by laser microdissection and DNA array: overexpression of HMG-CoA reductase, colony stimulating factor receptors, CD11A/CD18 integrins, and interleukin receptors. Arterioscler Thromb Vasc Biol. 2003;23:2235-2240.
9. Denhardt DT. Signal-transducing protein phosphorylation cascades mediated by Ras/Rho proteins in the mammalian cell: the potential for multiplex signalling. Biochem J. 1996;318:729-747.
10. Laufs U, Liao JK. Targeting Rho in cardiovascular disease. Circ Res. 2000;87:526-528. 11. Dichtl W, Dulak J, Frick M, Alber HF, Schwarzacher SP, Ares MP et al. HMG-CoA reductase
inhibitors regulate inflammatory transcription factors in human endothelial and vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2003;23:58-63.
12. Parmar KM, Nambudiri V, Dai G, Larman HB, Gimbrone MA, Jr., Garcia-Cardena G. Statins exert endothelial atheroprotective effects via the KLF2 transcription factor. J Biol Chem. 2005;280:26714-26719.
13. Lin Z, Kumar A, Senbanerjee S, Staniszewski K, Parmar K, Vaughan DE et al. Kruppel-like factor 2 (KLF2) regulates endothelial thrombotic function. Circ Res. 2005;96:e48-e57.
14. Senbanerjee S, Lin Z, Atkins GB, Greif DM, Rao RM, Kumar A et al. KLF2 Is a novel transcriptional regulator of endothelial proinflammatory activation. J Exp Med. 2004;199:1305-1315.
15. Sen-Banerjee S, Mir S, Lin Z, Hamik A, Atkins GB, Das H et al. Kruppel-like factor 2 as a novel mediator of statin effects in endothelial cells. Circulation. 2005;112:720-726.
16. Suzuki T, Aizawa K, Matsumura T, Nagai R. Vascular implications of the Kruppel-like family of transcription factors. Arterioscler Thromb Vasc Biol. 2005;25:1135-1141.
17. Dekker RJ, van Thienen JV, Rohlena J, de Jager SC, Elderkamp YW, Seppen J et al. Endothelial KLF2 links local arterial shear stress levels to the expression of vascular tone-regulating genes. Am J Pathol. 2005;167:609-618.
18. Das H, Kumar A, Lin Z, Patino WD, Hwang PM, Feinberg MW et al. Kruppel-like factor 2 (KLF2) regulates proinflammatory activation of monocytes. Proc Natl Acad Sci U S A. 2006;103:6653-6658.
19. Oettgen P. Regulation of vascular inflammation and remodeling by ETS factors. Circ Res. 2006;99:1159-1166.
20. Zhan Y, Brown C, Maynard E, Anshelevich A, Ni W, Ho IC et al. Ets-1 is a critical regulator of Ang II-mediated vascular inflammation and remodeling. J Clin Invest. 2005;115:2508-2516.
21. Pietarinen-Runtti P, Lakari E, Raivio KO, Kinnula VL. Expression of antioxidant enzymes in human inflammatory cells. Am J Physiol Cell Physiol. 2000;278:C118-C125.
22. Li C, Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci U S A. 2001;98:31-36.
23. Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95:14863-14868.
24. Makinen PI, Koponen JK, Karkkainen AM, Malm TM, Pulkkinen KH, Koistinaho J et al. Stable RNA interference: comparison of U6 and H1 promoters in endothelial cells and in mouse brain. J Gene Med. 2006;8:433-441.
7
Sim
vastatin is anti-inflamm
atory in macrophages
Proefschrift JVvT page 139
Composite
Tuesday, November 25, 2008 17:25

140
25. Wang X, Sato R, Brown MS, Hua X, Goldstein JL. SREBP-1, a membrane-bound transcription factor released by sterol-regulated proteolysis. Cell. 1994;77:53-62.
26. Haq F, Mahoney M, Koropatnick J. Signaling events for metallothionein induction. Mutat Res. 2003;533:211-226.
27. Hilgendorff A, Muth H, Parviz B, Staubitz A, Haberbosch W, Tillmanns H et al. Statins differ in their ability to block NF-kappaB activation in human blood monocytes. Int J Clin Pharmacol Ther. 2003;41:397-401.
28. Dekker RJ, van Soest S, Fontijn RD, Salamanca S, de Groot PG, VanBavel E et al. Prolonged fluid shear stress induces a distinct set of endothelial cell genes, most specifically lung Kruppel-like factor (KLF2). Blood. 2002;100:1689-1698.
29. van Thienen JV, Fledderus JO, Dekker RJ, Rohlena J, van Ijzendoorn GA, Kootstra NA et al. Shear stress sustains atheroprotective endothelial KLF2 expression more potently than statins through mRNA stabilization. Cardiovasc Res. 2006;72:231-240.
30. Dekker RJ, Boon RA, Rondaij MG, Kragt A, Volger OL, Elderkamp YW et al. KLF2 provokes a gene expression pattern that establishes functional quiescent differentiation of the endothelium. Blood. 2006;107:4354-4363.
Proefschrift JVvT page 140
Composite
Tuesday, November 25, 2008 17:25





























