Embed Size (px)
Citation preview

Use of Bioinformatics to Enhance Biophysical
Exploration of Ion Channels

Main points of talk:
Goals of project Introduction to ion channels & methods of study Introduction to K channels (functional & topological
classifications, KcsA structure) Bioinformatics & sequence-function prediction methods construction of a comprehensive alignment of the K
channel family permeation pathway Sequence-function predictions: Ca binding site in BK & K
channel selectivity analysis Strategy for exploration of Na, Ca channels Ion channel database proposal

Goal of project: Discover sequence-function relationships in ion channels and other membrane proteins by identifying K channel residues responsible for ion selectivity, conductance, and toxin affinities. This will be done by:
Gathering available experimental data (ex. from mutation experiments) regarding function of individual residues within K channels
Using bioinformatics (identifying structural features, sequence searches in databases, comparing sequence profiles, etc.) to check whether these functionalities can be projected within the channel class, throughout the K channel family, and across different ion channel families

Ion channels
Ion channels are excitable protein molecules in the lipid bilayer.
Passage of ions across cell membrane is essential for excitation and electrical signaling
Many ion channels are characterized by high conductance and ion selectivity
Na+ & Ca2+ channels have 4 homologous repeats of 6 TMS each. K+ channels have 4 identical subunits, of 2, 6, or 10 TMS each.

Methods of studying ion channels
Patch-clamp techniques help determine single channel activities to analyze channel electrophysiology
Studying protein sequences of ion channels and evaluation of data from mutation experiments
Using FRET (Fluorescence Resonance Energy Transfer) experiments to measure gating movements; determining contiguity of channel segments from NMR spectra; estimating channel topology from cysteine-scanning experiments
Using modeling programs to visualize channel activities (toxin binding, ion passage through pore, etc.)
Structural determination of ion channels (such as the gramicidin channel, MscL, certain porins, & KcsA K+ channel)

K+ channels
Characterized by wide variety in structure & function
Exhibit similar ion permeabilities: selectivity sequence is K+=Rb+>Cs+>Na+>Li+
Can be blocked by TEA subunits have 4 identical
subunits arranged around central pore. Each contains 2 (M1, M2), 6 (S1-S6), or 10 (S1-S10) TMS.
Figure 2. Postulated transmembrane topology for one voltage-gated K+ channel subunit. Cylinders represent helices.
S1
S2
S3
S5
S6
S4

K+ Channel classifications:
Table 1. Topological and functional characteristics of various K channel classes.
Channel Class Symbol Description #TM
Delayed rectifiers/transiently activating
DRK/A-type; Shaker, Shab, Shaw, Shal, Kv, Kh
Voltage-activated, highly K+-selective, low-conducting; stabilize and set rate of action potentials.
6
Small conductance Ca2+-
sensitive SK Ca2+-activated, voltage insensitive,
low conductance; contribute to afterhyperpolarization (AHP) activated by Ca2+-influx during APs.
6
Large-conductance Ca2+-sensitive
BK; slo; maxi-K Ca2+- and voltage-sensitive, high conductance; regulation of secretion in endocrine & exocrine glands.
10
Inward rectifiers IRK; Kir; BIR; ROMK Allow greater K+ influx than outflux; Maintain resting potential and control cell excitability
2
Plant inward rectifiers AKT; KAT; KST Inwardly rectifying and voltage-activated; K+-uptake from soil, mediate K+ influx leading to stomatal openings.
6
Cyclic nucleotide- sensitive
KCNG; SPIH; HAC; BCNG Hyperpolarization-activated with ow K+-selectivity; flagellar beating in sea urchin sperm and cardiac pacemaking.
6
Two domains/subunit TWIK; TOK; KCO1 Setting background membrane K+-conductance in many cell-types
TWIK:4; TOK:8; KCO1:4

T a b l e 2 . K c h a n n e l m u t a t i o n t a b l e c o n s t r u c t e d f r o m a v a i l a b l e e x p e r i m e n t a l d a t a f r o m K c s A ,D s h a k e r , K v 2 . 1 , a n d K v 3 . 1 c h a n n e l
T E A b l o c k a d e
Res
idue
#
Ksc
A (
C)
Dsh
aker
(S
)
Kv2
.1 (
D)
Kv3
.1 (
N)
Sid
e ch
ain
acce
ssib
ility
to p
ore
Aff
ects
cond
ucta
nce
Aff
ects
ion
sele
ctiv
ity/
perm
eatio
n
4AP
(1)/
CT
X(2
) bi
ndin
g
Ext
.
Int.
C-t
ype
inac
tiva
tion
S6-
H5
inte
ract
ion
1 L F F F2 I K K K3 T S S N4 Y I I I5 P P P P S i 1 8 , D 2 2
6 R D A I S 1 8 S i i( 2 )
3 S 3 3 , D 2 2
7 A A S G8 L F F F9 W W W W S 1 8 , 2 7 S i i i 1 3 S i v 2 7
1 0 W W W W S 1 8
1 1 S A A A1 2 V V T V1 3 E V I V D 7 , N v 4 7 , S 1 8 D v i 1 4 D 7 , 1 4 , 2 2 N - 1 81 4 T Y T T S v i i 1 6
1 5 A M M M D 2 2 , S 3 2
1 6 T T T T S 5 , 3 2 , D 2 2
1 7 T T T T1 8 V V V L D 7 , 1 9 , N 7 , S 1 8 N 1 1 D v i i i 5 , 1 2 ,
S i xD 7 , 2 2 S - 3 8 2 9 , C - 3 8 2 9
1 9 G G G G S x 1 6
2 0 Y Y Y Y S 1 8
2 1 G G G G S 1 6
2 2 D D D D S 1 8 , , D 1 9 , 2 4 D x i 2 3 D x i i 2 3 D 2 4 , S 2 6
2 3 L M I M S 1 8 , D 1 9 , 2 2 , 2 4 D - 3 8 x i i i 1 2
2 4 Y T Y Y S 1 8 , D 1 9 , 2 2 , 2 4 S 3 , 3 5 S ( 2 ) 3 S 3 , 8 , 3 8 , D 2 2 S x i v 3 5 , 2
62 5 P P P P S 1 8
2 6 V V K Q D 2 2 , 2 4
2 7 T G T T2 8 L F L W2 9 W W L S3 0 R G G G3 1 G K K M3 2 L I I L3 3 V V V V3 4 A G G G3 5 V S G A S 3 8
3 6 V L L L3 7 V C C C D x v 3 0 D 2 , 3 0
3 8 M V C A D x v i 2 9 S 1 D - 2 3 1 , S - 1 8 x v i i 2 9 , C 2 9
3 9 V I I L S x v i i i 3 8
4 0 A A A A4 1 G G G G4 2 I V V V4 3 T L L L4 4 S T V T D x i x 1 7 D ( 1 )
3 4 , S ( 1 )x x 3 1 S x x i 3 2
4 5 F I I I S x x i i 1 , 3 3
4 6 G A A A4 7 L L L M D 1 7 S 3 6 , 3 8
4 8 V P P P4 9 T V I V S 3 3 D ( 1 )
3 4
5 0 A P P P5 1 A V I V D ( 1 )
3 4
5 2 L I I I5 3 A V V V

Functional characteristics of K channels:
The S4 sequence in voltage-gated channels contains positively charged basic residues at every 3rd position and serves as a voltage-sensor
In certain channels, an extended N-terminal segment occludes the pore in a ball-and-chain mechanism causing inactivation
In IRK’s, an Asp residue in M2 affects channel blocking by Mg2+, which influences inward rectification and K selectivity
A C-terminal Ca2+-sensing domain is responsible for Ca2+- dependent activation in BK channels
In each subunit, S5 & S6 (in 6 or 10 TM channels), M1 & M2 (in 2 TM channels), and the linker joining them form part of the permeation pathway. Residues responsible for ion selectivity, toxin sensitivities, and channel conductance are suspected to be located here. Linkers contain K channel “signature” G[Y/F]G sequence

Structure of the KcsA K+ channel: The KcsA channel structure was discovered and refined to 3.2 A accuracy by Doyle et al (1998).
The pore is constructed like an inverse teepee, with selectivity filter sequence at wide end
Selectivity filter is narrow (3 A) and 12 A long. Rest of pore is wider with inert hydrophobic lining (thus minimizing distance of strong ion-channel interactions and favoring high K+ throughput)
A large water-filled cavity near channel center combined with helix dipoles overcomes the electrostatic destabilization of a K+ ion in the pore
Figure 3. Structure of the KcsA K channel

The K+ selectivity filter is lined with main chain oxygen rings, which provide multiple closely-spaced binding sites. As a dehydrated ion enters the filter, the carbonyl oxygen atoms substitute for the water oxygen atoms. The filter is constrained at an optimal radius to coordinate a dehydrated K+ ion, but the Na+ ion is too small for the carbonyl oxygens to provide similar compensation.
Two K+ ions 7.3 A apart in the selectivity filter provide a force of repulsion which overcomes the strong ion-protein interaction to allow rapid conduction in an environment of high selectivity

Sequence-function prediction methods
Identification of and filtering for structural features: mask low-complexity regions (to prevent spurious hits); identify transmembrane regions, internal repeats; predict secondary structure
Identification of homologs: identify annotated domains from dedicated databases prior to a BLAST search (to reduce search space for remaining parts of protein); search complete sequence databases individually using subsequences separated by known domains; perform exhaustive, iterative database searches; combine search for sequence similarity with profile, motif, and pattern searches

Prediction of protein function: consider domain organization and distinct functions of individual domains (keeping in mind that many proteins are multifunctional), analyze database annotation if inconsistencies between different homologues are detected; perform cluster analysis of homologs to determine level of precision for functional prediction; identify similarities to proteins with known 3D structure to aid in model construction, which may provide further functional insights

Bioinformatics
Bioinformatics can be loosely defined as the use of computer technology to manage biological information. One of its goals is to construct and utilize tools in order to extract useful information pertaining to protein function from available sequence data
Bioinformatics will be used in this project to expedite the study of sequence-function relationships in K channels (primarily to identify residues responsible for ion selectivity, conductance, and toxin sensitivities) and to project these relationships across different types of ion channels.

Construction of a comprehensive alignment of K channel
families: The following procedure was used for construction:
All K channel classes and isoforms to be included in the alignment were selected (K channel classes: DRK & A-type, Ca2+-activated, inward rectifiers, KCNG, EAG & ERG, plant K channels, 2 domains/subunit, and the KcsA K channel)
DRK/A was identified as the channel class with highest sequence similarity to KcsA and S5 & S6 TMS in these channels were located
Profiles of the DRK S5 & S6 TMS were constructed Corresponding TMS in all other K channel classes (called TM1 and
TM2) were located Linker segments in each channel type were identified as the
difference between TM1 and TM2 and aligned
Procedure for K channel alignment construction Selection of isoforms: A BLAST
search was performed on the NCBI non-redundant database using representatives from each of the channel classes. Channels were selected on the basis of completeness in database records and published literature
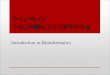
Identification of class closest to KcsA: The DRK/A class was chosen because it produced ungapped alignments with KcsA. S5 & S6 TMS in DRK were identified based on alignments with KcsA. All available DRK/A channel sequences were found and profiles for TM1 and TM2 were constructed (Figures 4, 5, & 6).
Figure 4.KcsA-DRK/AS5 alignmentand profile.
KcsA WRAAGAATVLLVIVLLAGSYLAVLAKv1.1 MRELGLLIFFLFIGVILFSSAVYFAKv1.2 MRELGLLIFFLFIGVILFSSAVYFAKv1.3 MRELGLLIFFLFIGVILFSSAVYFAKv1.4 MRELGLLIFFLFIGVILFSSAVYFAKv2.1 YHEVGLLLLFLSVGISIFSVLIYSVKv2.2 YNELGLLILFLAMGIMIFSSLVFFAKv2.3 YEEVGLLLLFLSVGISIFSTIEYFAkv3.1 TNEFLLLIIFLALGVLIFATMIYYAKv3.2 TNEFLLLIIFLALGVLIFATMIIYAKv3.3 TNEFLLLIIFLALGVLIFATMIIYAKv3.4 TNEFLLLIIFLALGVLIFATMIIYAKv4.1 ASELGFLLFSLTMAIIIFATVMFYAKv4.2 ASELGFLLFSLTMAIIIFATVMFYAKv4.3 ASELGFLLFSLTMAIIIFATVMFYAKv6.2 TREFGLLLLFLCVAMALFSPLVYLAKv9.1 YKEVGLLLLYLSVGISIFSVVAYTIkH1 FKELGLLLMYLAVGIFVFSALGYTMkH2 TREFGLLLLFLCVAIALFAPLLYVI
Profile: WRAAGAATVLLVIVLLAGSYLAVLAMHELLLLIFF VVGVILFASAVYFIYK V F MLS FMAIVI VIIFSMTS F LIY SL MSV TME YA VM A M PVM TF T A G V C F A
Procedure for K channel alignment construction (contd.)
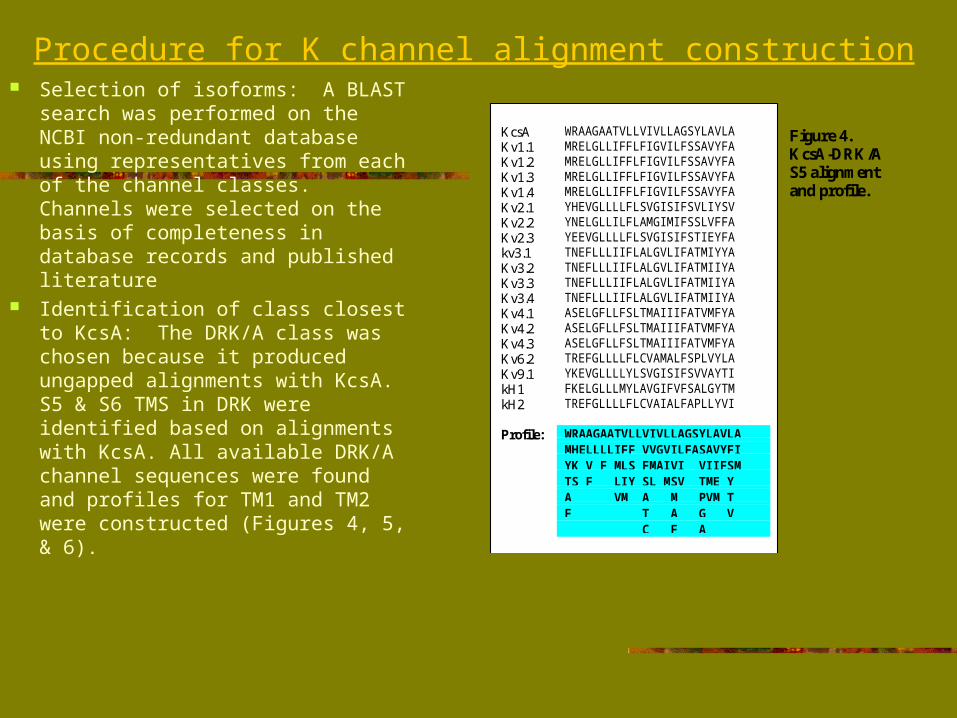
Location of TM1 & TM2 in other K channels: The S5 & S6 profiles were used as probes to locate TM1 & TM2 in other K channels. A binary scoring scheme was implemented.
Identification of linker segments: Linker sequences joining TM1 & TM2 were extracted from each channel, arranged in order of length, and aligned.
Profile Score
Score
0001010010010000100000000 = 5/25IAIVSVMVILISIVIFCLETLPELKDDKDFTGTVHRIDN
WRAAGAATVLLVIVLLAGSYLAVLAMHELLLLIFF VVGVILFASAVYFIYK V F MLS FMAIVI VIIFSMTS F LIY SL MSV TME Y testA VM A M PVM T sequence
F T A G V C F A Profile
Residue position ontransmembrane
segment
KcsA + DRK/A S5profile
KcsA + DRK/A S6profile
12345678910111213141516171819202122232425
QYATMFWHRKSENAQDEALVFIQTLMGALMFIALMITLMVILMVFYSLMVFYLIACSLTVFITLMNVAVFGILMVAISLMFVAILMVFFGASPYASTVIACLMVAILEMVGYVFYSLTVFIALMV
PQASTVFWGILMSGQRMAFVILTFVINAGASTFVGLASTVILMASCVACSFVIMVILMACSMGVITILASTVLFVILMAVGFLMPVTVIPATHPASVILILQAFVILN
Figure 5. Profiles of S5 and S6 constructed from KcsA and DRK/Asequences.
Figure 6. Profilescoring.

Observations from K channel alignment
Region of highest conservation is pore helix-selectivity filter. KcsA structure here contains a hydrophobic cluster (interactions serve to stabilize twisting backbone) and a hydrophilic cluster (formation of intersubunit H-bonds creates a cuff-like sheet around selectivity filter, allowing passage to dehydrated K+ ions)
Region of maximum sequence and length variation is in the turret segment. This part constitutes the extracellular entryway of channel and interacts with different toxins depending on channel class

Sequence-function predictions:a) Identification of putative Ca2+-binding region of BK
Sequence analysis showed this channel class contained a long C-terminal domain absent in other K channels
BLAST searches using the C-terminal segment yielded other Ca2+-binding proteins such as troponin & calmodulin
Alignment of this segment with troponin showed sequence similarity in troponin’s known Ca2+-binding region
Hydropathy plots showed this segment was likely to be present in the cytoplasm
Dslo (1175 aa); TNC-1 (150 aa) Smith-Waterman Score: 49; 22.6% identity in 62 aa overlap 10 20 30TNC-1 EMIAEFKAAFDMFDADGGGDISVKELGTVMR .... ..:: ..:..:. : .. :.:DROKCH PSNDDPTLADKEAILASLNIKAMTFDDTIGVLSQRGPEFDNLSATAGSPIVLQRRGSVYG 900 910 920 930 940 950
40 50 60 70 80 90TNC-1 MLGQTPTKEELDAIIEEVDEDGSGTIDFEEFLVMMVRQMKEDAKGKSEEELAECFRIFDR . :. :. .. .:.:.... : : .:.DROKCH ANVPMITELVNDGNVQFLDQDDDDDPDTELYLTQPFACGTAFAVSVLDSLMSTTYFNQNA 960 970 980 990 1000 1010
100 110 120 130 140TNC-1 NADGYIDAEELAEIFRASGEHVTDEEIESLMKDGDKNNDGRIDFDEFLKMMEG
DROKCH LTLIRSLITGGATPELELILAEGEGLRGGYSTVESLSNRDRCRVGQISLYDGPLAQFGEC 1020 1030 1040 1050 1060 1070
Figure 7. Dslo (DROKCH) aligned with troponin (TNC-1). Maximumsequence similarity was observed in the above region. Ca2+-binding loopson TNC-1 are highlighted in yellow, proposed Ca2+-binding site inDROKCHAN-1 in magenta.
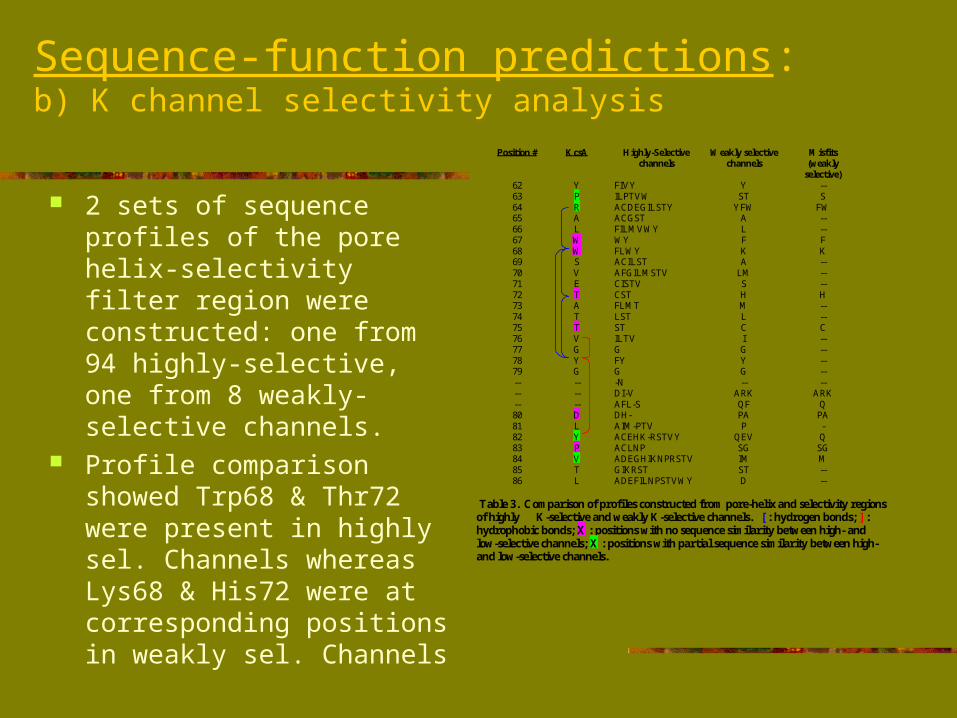
Sequence-function predictions:b) K channel selectivity analysis
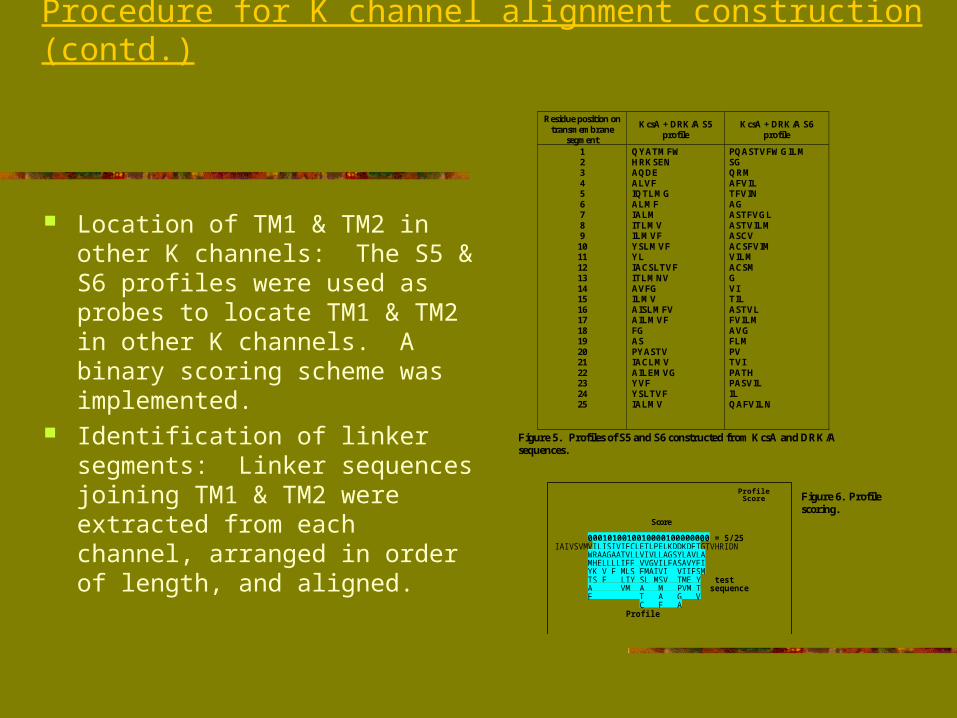
2 sets of sequence profiles of the pore helix-selectivity filter region were constructed: one from 94 highly-selective, one from 8 weakly-selective channels.
Profile comparison showed Trp68 & Thr72 were present in highly sel. Channels whereas Lys68 & His72 were at corresponding positions in weakly sel. Channels
Position # KcsA Highly-Selectivechannels
Weakly selectivechannels
Misfits(weakly
selective)626364656667686970717273747576777879------80818283848586
YPRALWWSVETATTVGYG------DLYPVTL
FIVYILPTVWACDEGILSTYACGSTFILMVWYWYFLWYACILSTAFGILMSTVCISTVCSTFLMTLSTSTILTVGFYG-NDI-VAFL-SDH-AIM-PTVACEHK-RSTVYACLNPADEGHIKNPRSTVGIKRSTADEFILNPSTVWY
YST
YFWALFKA
LMSHMLCIGYG--
ARKQFPAP
QEVSGIMSTD
--S
FW----FK------H----C----------
ARKQ
PA-Q
SGM----
Table 3. Comparison of profiles constructed from pore-helix and selectivity regionsof highly K-selective and weakly K-selective channels. [ : hydrogen bonds; ] :hydrophobic bonds; X : positions with no sequence similarity between high- andlow-selective channels; X : positions with partial sequence similarity between high-and low-selective channels.

Sequence-function predictions:b) K channel selectivity analysis (contd.)
In KcsA, Trp68 & Thr72 form H-bonds with Tyr78 (of GYG). This bond, which might be a determinant of K channel selectivity, is preserved only in highly K-sel. Channels
Molecular dynamics calculations show that ease of movement through selectivity filter is modulated by tightness of restraint in the pore helix region.
Figure 9. The intersubunit hydrogen bonds thatplay a role in ion selectivity are shown
Figure 8. The protein backbone structure of all four subunits of KcsAare in yellow. H-bonds formed between three residues in every subunitare in blue. A:W68, A:T72, and D:Y78 illustrate one set of bonds.

Homology modeling & Fragment cluster analysis A homology model of the Shaker channel was built using
MODELLER and evaluated using PROCHECK by R. Shealy. Models of other K channels can be similarly constructed and might provide insights into the functioning of K channels
A fragment clustering analysis technique was applied on the A chain of KcsA by G. Hunter. The KcsA permeation pathway sequence showed a reasonable fit within the bounds of the available fragment cluster set. An investigation into which proteins from the PDB are similar in sequence and structure to the KcsA permeation pathway still remains to be made.
Na+ & Ca2+ channel analysisThe strategy for exploring properties of these channels is similar to what is being done with K channels:
Select all Na & Ca channel isoforms to be included in study
Identify channel class from each family with highest sequence homology to KcsA and identify S5 & S6 in these (each of the 4 repeats in Na and Ca channels have to be analyzed separately)
Construct profiles of S5 and S6 segments Use these profiles to locate corresponding TMS in all
other channel classes in each family Identify the linkers from each domain, arrange in order of
length, align, and study observable characteristics
Proposal for construction of an ion channel database
This will be a queryable HTML database, initially containing voltage-gated K channels (other types of channels will be added once a working format has been constructed)
Existing ion channel-related databases on the web contain limited information on selected channel types and are not as comprehensive as the one we are constructing

Proposal for construction of an ion channel database (contd.)
We have created a web query form and an indexing program called Ndjinn. Ndjinn can be used to search, index, and retrieve records from the database - it works by indexing the entire text of files contained in multiple database entries
Upon its completion, the database will be incorporated into the Biology Workbench. This will enable users to access DNA and protein sequences, and obtain mutational, electrophysiological, and functional data of ion channels from one source.