Embed Size (px)
Citation preview

0
UNIVERSIDAD CENTRAL DE VENEZUELA FACULTAD DE AGRONOMIA
DEPARTAMENTO DE QUÍMICA Y TECNOLOGÍA CÁTEDRA QUÍMICA III
GUIA DE LABORATORIO
Maracay, enero de 2010

1
Personal de la Cátedra
Activo
Ing. Agro. Dra. Alejandra Ramírez Ing. Agro. MSc. Francisca Sosa Ing. Agro. MSc. Yasmin Román
Ing. Agro. Audrey Suárez Ing. Agro. Dra. Nora Techeira
Ing. Agro. MSc. Palmira Zambrano Ing. Agro. MSc. Fanny Molina
Jubilado
Ing. Agro. Dr. Ekcbert Schulz Ch. Lic. Qca. MSc. Ligia Ortiz
Lic. Qca. Gladys Quijada Fca. Carmen Sofía Bravo
Ing. Agro. Dra. Carmen Rivero Lic. Biol. Dra. Emperatriz Pacheco Lic. Qca. MSc. Feliciano R. Anzola
Ing. Qco. MSc. Lucia Graciani

2
PROLOGO
Los objetivos fundamentales de la asignatura QUÍMICA III, radican en capacitar al
estudiante en la planificación, ejecución de diferentes tipos de análisis químicos
así como la de interpretar sus resultados. Como una contribución al logro de este
objetivo, los Profesores de la Cátedra prepararon la presente Guía de Laboratorio,
producto de la experiencia acumulada en el transcurso de varios años de trabajo
docente. La Guía comprende por prácticas de laboratorio sus objetivos,
fundamento teórico y procedimiento mediante los cuales el estudiante aplica
métodos de análisis químico cuantitativo.
Sin duda este material, donde se presentan métodos gravimétricos, volumétricos,
espectrofotométricos y potenciométricos, todos de gran utilidad práctica, será una
valiosa ayuda para los estudiantes tanto desde el punto de vista formativo como
informativo.

3
ÍNDICE
Contenido Pag.
PROLOGO ……………………………………………………………………… 2
Normas para la realización del trabajo práctico ………………………... 5
Introducción al análisis cuantitativo …………..………………….……… 6
Métodos del análisis cuantitativo ……………………………………… 7
Métodos químicos ………………………………………………………… 7
Métodos físicos y fisicoquímicos ………………………………………… 8
Análisis gravimétrico …………………………………………………………… 8
Análisis volumétrico ………………..…………………………………………… 8
Métodos ópticos ………………………………………………………...……… 8
Métodos eléctricos …………………………………………………………..… 9
Operaciones básicas del análisis cuantitativo ………………………………. 9
Material de laboratorio …………………………………………………….… 12
Laboratorio 1: Balanza analítica, principios y usos ……………………….. 17
Principios de la balanza analítica ………………………..……………… 18
Métodos de pesada ……………………………………….……………… 20
Sensibilidad de la balanza analítica …………………………………..… 21
Construcción de la balanza analítica ………………………………...…. 22
Errores en la pesada ……………………………………………………… 23
Cuidados de la balanza analítica ………………………………………... 25
Parte experimental. Pesada de un objeto ……………………………… 26
Laboratorio 2: Determinación de humedad en diferentes muestras ..…… 27
Gravimetrías de precipitación ………………………………………….… 27
Gravimetrías de electrodeposición ………………………………...……. 27

4
Gravimetrías de volatilización …………………………………………… 27
Determinación del contenido de agua en muestras ………………...… 28
Parte experimental………………………………………………………… 29
Laboratorio 3: Preparación de soluciones estándares ………………….... 31
Soluciones patrones o estándares …………...…………………………. 32
Preparación de soluciones patrones o estándares ..………………….. 32
Manejo y conservación de soluciones estándares ………….………… 34
Indicadores …..………………………………………….…………………. 34
Valoraciones o titulaciones ácido-base ……………..………………….. 35
Observaciones ……………………………………………………………. 36
Parte experimental ……………………………………………………….. 38
Ejemplo del uso de los datos experimentales ………………………… 39
Laboratorio 4: Aplicación del análisis quelométrico ...……………………. 42
Parte experimental ……………………………………………………….. 44
Laboratorio 5: Aplicación de la potenciometría: ejecución de una
valoración potenciométrica ……………….……………………………………
45
Electrodo de referencia .………………………………………………… 46
Electrodo indicador ….…………………………………..………………. 46
Mediciones prácticas de pH .………………………………..…………… 47
Medidores de pH ………………………………..………………………… 49
Determinación del punto final ……….…………………………………… 49
Parte experimental ……………………………………………………….. 52
Laboratorio 6: Análisis espectrofotométrico de manganeso ..……………. 54
Espectrofotómetro ..……………………………………………………….. 56
Funcionamiento de un espectrofotómetro ……………………………… 58
Parte experimental ……………………………………………………….. 59

5
NORMAS PARA LA REALIZACIÓN DEL TRABAJO PRÁCTICO EN
LA ASIGNATURA QUÍMICA III.
A los fines de lograr una sistematización del manejo de la información, que el
estudiante obtendrá en el desarrollo del curso, se considera conveniente el
establecimiento de algunas normas que conlleven al logro cabal de los objetivos.
1. LLEVAR UN CUADERNO DE PRÁCTICAS.
Cada estudiante debe poseer un cuaderno de anotaciones de laboratorio en el
cual tomará nota durante la práctica, de las actividades realizadas. Este cuaderno
debe contener toda la información inherente a la ejecución de todas y cada una de
las prácticas y debe conservarse durante todo el semestre.
2. REDACTAR Y PRESENTAR INFORMES.
2.1. El informe debe cumplir los siguientes requisitos: estar escrito en tinta,
contener los datos exigidos y en forma indicada los cálculos necesarios para
obtenerlos.
2.2. Cuando uno de los alumnos, integrante de un equipo, no cumpla con su
deber en la realización del trabajo práctico y elaboración del informe, el otro
miembro del equipo queda en libertad de efectuar sólo la práctica y presentar el
informe que será calificado individualmente. Esta norma se aplica igualmente a los
casos de inasistencia.
2.3. Los informes, correspondientes a las prácticas realizadas, deben
entregarse en los plazos fijados. Pasado el lapso, no serán admitidos y por tanto la
práctica será considerada como no efectuada.
2.4. Entregado el Informe sólo será devuelto después de su corrección.

6
2.5. Los Informes corregidos deberán ser conservados en orden por los
alumnos para su presentación en casos necesarios.
2.6. La justificación de inasistencia para la recuperación de una práctica y
su correspondiente Informe, se regirá por el reglamento vigente en la Facultad de
Agronomía.
3. EVALUACIÓN DEL TRABAJO PRÁCTICO.
La evaluación de cada Informe será planificada por la Cátedra e incluirá:
Aplicación de los límites de precisión y exactitud aceptados para cada análisis,
Apreciación del profesor sobre el desempeño del alumno en la práctica y la nota
del quiz correspondiente.
INTRODUCCIÓN AL ANÁLISIS CUANTITATIVO
La Química Analítica es la ciencia que permite caracterizar las sustancias
químicas. En la práctica la Química Analítica recurre a diferentes métodos y
técnicas para obtener información acerca de la estructura y composición de la
materia. Estos métodos y técnicas constituyen el Análisis Químico.
La caracterización química completa de la composición de toda porción de materia
comprende información cualitativa y cuantitativa. En el Análisis Cualitativo, el
Analista está interesado en el descubrimiento e identificación de los componentes
que constituyen la muestra que se analiza. Los resultados de un análisis
cualitativo se expresan en palabras, nombres o símbolos de las clases o
agrupamientos especiales de átomos, iones o moléculas. En el Análisis
Cuantitativo, se determinan las cantidades relativas o absolutas de uno o varios
de los componentes. La información obtenida en un análisis cuantitativo se
expresa en números con indicación de las unidades respectivas.

7
El análisis cuantitativo, generalmente se basa sobre los mismos principios que el
análisis cualitativo, pero el método a seguir depende en gran parte, de la
naturaleza y de la cantidad relativa de los constituyentes presentes en el material
en análisis, de modo que siempre es necesario que un examen cualitativo preceda
al cuantitativo.
Una determinación cuantitativa completa consiste, en general, de cuatro pasos
fundamentales:
La obtención de una muestra representativa para el análisis.
La separación del componente o analito deseado en una forma mensurable.
La medición del componente buscado o analito.
El cálculo de los resultados y la generación de las conclusiones del análisis.
De estos cuatro pasos, la medición es el paso central en el análisis cuantitativo.
Los dos primeros tienen por objeto preparar la muestra para la medición deseada
y el cuarto, hacer significativos los resultados de la medición. Así toda
determinación cuantitativa se basa, fundamentalmente, en la medición de alguna
propiedad relacionada directa o indirectamente con la cantidad del analito
presente en la muestra. Aún cuando en el análisis cuantitativo se obtiene
información sobre las reacciones o propiedades químicas, el analista realmente
mide propiedades físicas antes, durante o después de la reacción, por lo tanto, en
definitiva lo único que el químico puede medir directamente en el laboratorio son
propiedades físicas o estructurales.
Métodos del análisis cuantitativo
Los métodos analíticos se pueden clasificar en:
1. Métodos químicos: se basan en una reacción estequiométrica:
CxRy yR xC
Si se agrega un exceso del reactivo R y se determina el peso del producto
CxRy se está en presencia del Análisis Gravimétrico.

8
Si determina el volumen de solución de concentración conocida del reactivo
(R) que reacciona estequiométricamente con el constituyente se está en
presencia del Análisis Volumétrico.
2. Métodos físicos y fisicoquímicos: En estos se mide una propiedad que esté
relacionada cuantitativamente con la cantidad del analito presente en la muestra,
tal como: color, refracción, ionización, etc. Cualquier método que use la energía
radiante para llegar a la concentración de un analito se define como Análisis
Óptico, en tanto que cuando se mide una propiedad eléctrica, el método es
definido como eléctrico.
Análisis gravimétrico
Una determinación gravimétrica implica la separación del analito deseado, en una
forma de composición química definida que pueda ser pesada con exactitud.
Según la forma como se separe el constituyente antes de su medición, el análisis
gravimétrico se ha clasificado en métodos de precipitación, electrodeposición y
volatización.
Análisis volumétrico
En una determinación volumétrica, se mide el volumen de una solución de
composición conocida (estándar) necesario para reaccionar cuantitativamente con
el analito de la muestra en análisis. Con base al tipo de reacción química que
ocurre entre el analito y la solución estándar, los métodos volumétricos pueden
clasificarse en: volumetrías de neutralización, precipitación, por formación de
complejos (complejometrías) y óxido–reducción.
Métodos ópticos
Estos métodos están basados en la interacción entre la energía radiante y la
materia. Según el tipo de interacción utilizada en el análisis, los métodos ópticos
pueden clasificarse en métodos de absorción, emisión, difracción y refracción.
También suelen clasificarse con base al intervalo de longitud de onda de la

9
energía radiante empleada, así se tienen los métodos de infrarrojo, ultra violeta,
visible y rayos x. Algunos de estos métodos se cuentan entre los más usados en
los laboratorios analíticos modernos.
Métodos eléctricos
Estos métodos de análisis incluyen todos aquellos en los que la medición primaria
es una cantidad eléctrica fundamental como voltaje, resistencia o intensidad de
corriente.
Operaciones básicas del análisis cuantitativo
Obtención de una muestra representativa
En un curso de análisis químico cuantitativo el alumno generalmente recibe las
muestras a punto para pesarlas o ya disueltas, es decir, que no necesita realizar la
operación de muestreo. Sin embargo, en situaciones reales se presenta la
necesidad de obtener y preparar, una muestra adecuada para el análisis. No
efectuar estos pasos en forma adecuada puede, eventualmente originar graves
dificultades que con frecuencia limitan la validez del resultado final.
Para que los resultados de un análisis sean confiables deben haber sido
realizados en una muestra representativa, es decir, una porción que corresponda
a la composición media de una gran cantidad del material bajo estudio. En la
práctica, el analista debe responder por la cualificación y cuantificación de grandes
cantidades de material sobre las cuales es imposible trabajar directamente, que
además puede ser heterogéneo. Es necesario entonces obtener pequeñas
cantidades de material representativas del conjunto para ser analizadas en el
laboratorio y que es lo que se conoce como muestra representativa. Para ello
deben cumplir ciertas operaciones que dependen de la naturaleza del material, se
señalarán aquí aquellas de aplicación general:
Toma de la muestra
La toma de la muestra depende básicamente de la homogeneidad del material
bajo estudio, lógicamente si un material es homogéneo (su composición es la

10
misma en cualquier punto) bastarán pocos puntos de muestreo, si por el contrario
el material es heterogéneo (composición variable) deberá incrementarse el
número de puntos de muestreo a los fines de garantizar una muestra compuesta
que comprenda dicha variabilidad.
Cuando se muestran procesos de producción es conveniente tomar muestras a
intervalos regulares durante las fases del proceso que se desee controlar. Otro
factor importante es la homogeneidad en el tamaño de las partículas que forman el
material. Aquellos materiales con partículas de diferentes tamaños requieren
muestras de mayor tamaño.
En Agronomía, un aspecto importante es la toma de muestras representativas de
suelo a los fines del análisis químico, por ello es necesario tomar suelo en varios
puntos y mezclar luego en el laboratorio para obtener una muestra uniforme y
representativa. El número de puntos dependerá de la variabilidad espacial que se
observa en la zona bajo estudio.
Trituración de la muestra.
Reunidas las diferentes porciones tomadas como muestra, se procede a triturarlas
con el fin de reducir el tamaño de las partículas y llevarlas a las dimensiones
fijadas para cada análisis. Durante esta operación hay que evitar que la muestra
sea contaminada, por lo cual se deben utilizar implementos de buena calidad para
su ejecución.
Cuarteo.
En esta operación se trata de lograr una pequeña porción representativa mediante
homogeneización y cuarteo. El material triturado se apila formando un cono de
modo que el material caiga siempre en el vértice, al mismo tiempo se circunda el
cono hasta que toda la muestra haya sido mezclada. Se aplana el vértice del cono
y se divide en cuatro partes. Se toman los dos cuartos opuestos, se trituran y se
mezclan como anteriormente. Se apila y se cuartea nuevamente. Esta serie de
operaciones, triturar, mezclar, apilar y cuartear, se repiten hasta que la muestra
quede reducida a una cantidad alrededor de 200 a 300 gramos, que es la que

11
llega al laboratorio. A pesar de todas las manipulaciones anteriores, en el
laboratorio se repite la operación de cuarteo antes de usar la muestra para el
análisis.
Secado de la muestra.
Las muestras de materiales que llegan al laboratorio usualmente contienen agua,
sea como agua de hidratación o como agua adsorbida. La cantidad de agua por
unidad de peso del material no es constante y varía notablemente según los
cambios de humedad ambiental. Dos análisis del mismo material sin secado
previo, practicados en dos días diferentes, pueden no concordar entre sí, aún
cuando ambos se hayan efectuado en forma impecable. Por esta razón se deben
secar las muestras antes de analizarlas.
El secado se efectúa, generalmente, colocando la muestra en estufa a una
temperatura 105ºC – 110ºC. No obstante, el secado dependerá de la naturaleza y
características del material objeto de estudio, en algunos casos también influye las
características del analito que finalmente se determinará.
Pesada de la muestra.
Obtenida la muestra representativa, se pesa la cantidad necesaria para el análisis.
En esta fase deberá ser utilizada la balanza analítica.
Disolución de la muestra.
Excepto en algunos casos, antes de la separación y la medición del componente,
es necesario disolver la muestra pesada.
El agua y los ácidos minerales son los solventes más usados en el análisis
cuantitativo. Cuando se trabaja con muestras que se disuelven lentamente se
agiliza el proceso calentando a temperatura inferior al punto de ebullición del
solvente. Existen algunos casos donde es necesario utilizar compuestos que
actúan como fundentes.

12
Material de laboratorio
Papel de filtro: El papel de filtro que se utiliza en el análisis cuantitativo tiene
como característica principal no dejar residuos de cenizas. Existen diferentes
gradaciones desde el punto de vista de la retención. Es fundamental usar el grado
de papel apropiado para un tipo de precipitado dado. El papel de filtro usado y sus
características puede ser identificado de acuerdo a dos clasificaciones, la
americana y la inglesa:
Americana Inglesa Tipo de precipitado
589 – Cinta negra 41 Gelatinosos, cristales gruesos
589 – Cinta blanca 40 Cristales medianos
589 – 1 H 41-H Cristales finos
589 – Cinta azul Cristales finos
590 – Alta retención 44 Uso especial
El Desecador: Es un recipiente generalmente de vidrio grueso, que consta de dos
secciones separadas por un plato de porcelana perforado, sobre el cual se coloca
el recipiente que contiene la sustancia que se está desecando. La sección inferior
contiene una sustancia higroscópica (absorbe la humedad) tal como: CaCl2, P2O5,
H2SO4, CaSO4, gel de sílice, etc. La tapa, también de vidrio grueso, debe cerrar
herméticamente, pero debe poderse deslizar con suavidad; para lograr esto último
se untan los bordes del desecador y de la tapa, con una ligera película de
vaselina. La siguiente figura ilustra los desecadores más comunes:

13
Los materiales colocados en el desecador, sólo serán sacados justo en el
momento de ser pesados, de modo que no absorba humedad por exposición
prolongada al aire. Se debe recordar que toda sustancia seca tiene tendencia a
absorber H2O.
Al destapar el desecador, lo cual se hará únicamente deslizando la tapa hacia un
lado, debe tenerse presente que existe en el interior un vacío, que será llenado por
el aire exterior. Debe evitarse que esto ocurra con violencia, pues se inducirían
pérdidas del material objeto del análisis. El desecador se mantendrá siempre
tapado para preservar la sustancia desecante.
Pipetas: Las pipetas se usan para medir porciones o alícuotas de soluciones. Las
hay de dos tipos: volumétricas, calibradas para medir un volúmen específico; y
graduadas, para medir volúmenes variables de líquido. La pipeta se llena con la
solución a medir por succión y el vaciado se regula por admisión de aire en el
extremo superior.
Antes de llenar la pipeta, ésta debe lavarse con dos o tres pequeñas porciones de
la solución que se va a medir (esta operación se conoce como curado de la
pipeta). Para llenarla se introduce su parte inferior, aproximadamente 1 cm, en el

14
líquido a medir y se succiona lentamente de modo que el líquido sobrepase unos
dos centímetros por encima de la señal de aforo, se seca la parte externa con un
papel absorbente limpio y se enrasa. Para enrasar se sostiene la pipeta
verticalmente, con la punta apoyada en la pared del recipiente que contiene el
líquido, se elevan simultáneamente el recipiente (con la mano izquierda) y la
pipeta, de modo que ésta esté vertical y que la marca de aforo esté a la altura
visual del operador. A continuación se muestran los dos tipos más importantes:
La operación de llenado de una pipeta puede ser manual o con el auxilio de
instrumentos diseñados para ello como las propipetas. Existen sustancias que por
su nivel de riesgo no pueden ser pipeteadas manualmente.
Enseguida se deja caer el líquido medido en el recipiente adecuado, manteniendo
la pipeta vertical y su punta apoyada contra la pared inclinada del recipiente. El
destino del residual de la pipeta dependerá del tipo de pipeta (este es indicado en

15
el cuerpo de la misma) si es TD, el residual no está contemplado en el volumen
medido por lo que debe permanecer allí, si Ex, el residual si pertenece al volumen
medido por lo que debe ser expedido de la pipeta.
Buretas: Consisten en un tubo de vidrio, construido con elevada precisión, que
está calibrado y posee una válvula en su parte inferior que permite descargar
cualquier líquido contenido dentro del tubo. El volumen de líquido descargado es
dterminado por medición del volumen inicial contenido en la bureta, al cual se le
resta el volumen final al cual se ha llegado después de la descarga. La siguiente
figura ilustra la construcción de una bureta:
Las buretas más precisas se conocen como buretas tipo A, las cuales son
certificadas para una tolerancia dada en la medida, usualmente los valores son los
mostrados en el siguiente cuadro:

16
Capacidad
de la Bureta (mL)
Menor
graduación (mL) Tolerancia (mL)
5 0,01 0,01
10 0,02 0,02
25 0,1 0,03
50 0,1 0,05
100 0,2 0,1
Balón Aforado: Consiste en un recipiente de vidrio construido y calibrado en
forma precisa para contener una determinada cantidad de líquido (volumen
definido) a una temperatura definida:
Existen balones aforados de diferentes capacidades y usualmente los fabricantes
indican la tolerancia de la medida, en el caso de los balones tipo A la tolerancia
para cada capacidad es la que se indica en el cuadro mostrado a continuación:

17
Capacidad (mL) Tolerancia (mL)
1 0,02
5 0,02
25 0,03
50 0,05
100 0,08
250 0,12
1000 0,30
Laboratorio1. BALANZA ANALÍTICA, PRINCIPIOS Y USOS
OBJETIVOS.
Al finalizar las actividades programadas el estudiante debe ser capaz de:
Conocer los principios teóricos y prácticos que hacen de la balanza
analítica un instrumento de uso indispensable en el análisis químico.
Manejar adecuadamente la balanza analítica.
INTRODUCCIÓN.
La primera balanza analítica construída fue la llamada balanza analítica de brazos
iguales que consistía esencialmente en una cruz sustentada en su centro por un
soporte o fulcro, de forma que actuaba como una palanca sencilla:

18
En la figura las distancias L1 y L2 representan los brazos de la cruz. De cada
extremo de la cruz, en sitios equidistantes del punto de apoyo central (fulcro)
penden los platillos para colocar el objeto a pesar y las pesas. La posición de la
cruz respecto a la horizontal está indicada por el fiel op, unido rígidamente con la
cruz en su punto de apoyo.
Principios de la balanza analítica.
Un peso Pi aplicado en A se traduce en un momento Pi x L1 que tiende a hacer
girar la cruz en sentido contrario a las agujas del reloj. Análogamente un peso Pd
aplicado en B da por resultado una fuerza Pd x L2 que causa la rotación del fiel en
el sentido de las agujas del reloj. Es decir que, la balanza combina los principios
físicos de la palanca y el péndulo.
Cuando se igualan los momentos aplicados en A y B, se alcanza una posición de
equilibrio, es decir, el momento de la fuerza que tiende a darle movimiento en una
dirección está compensando exactamente por el que tiende a moverlo en sentido
opuesto. De este modo, podemos escribir que:
PiL1 = PdL2 (Ecuación 1)
en donde Pi y Pd representan las fuerzas de igual magnitud y sentido contrario,
cuando L1 y L2 son iguales. Si se considera la aceleración debida a la gravedad
(g), es posible escribir esta relación de la siguiente manera:

19
MigL1 = MdgL2
en donde Mi es la masa localizada a distancia L1 a la izquierda del fulcro, M2 y L2
representan a la derecha del fulcro y g la aceleración de la gravedad. Puesto que
la gravedad g afecta exactamente, en la misma extensión, a ambas masas
(conocida y desconocida) y las longitudes de los brazos son iguales tenemos que:
Mi = Md
Para esta igualdada se considera que el peso de la cruz está concentrado en el
punto 0 de modo que no contribuye a la rotación del fiel en ningún sentido.
Por tanto el uso de la balanza analítica implica la comparación de la masa
desconocida de un objeto con la de objetos de masa conocida. No siempre se
considera la distinción entre peso y masa; de ordinario la operación de comparar
las masas se llama pesada y los objetos de masa conocida con los cuales se
realiza la comparación se llaman pesas. Aunque en lo sucesivo los dos términos
se utilizan como sinónimos, estrictamente hablando es su masa a la que se hace
referencia.
Una evolución en la construcción de las balanzas analíticas llevó a la sustitución
de uno de los brazos por un mecanismo contrapesado que funciona a carga
constante, este tipo de balanza es la conocida como Balanza Monoplato, la figura
mostrada a continuación ilustra este tipo de balanza.
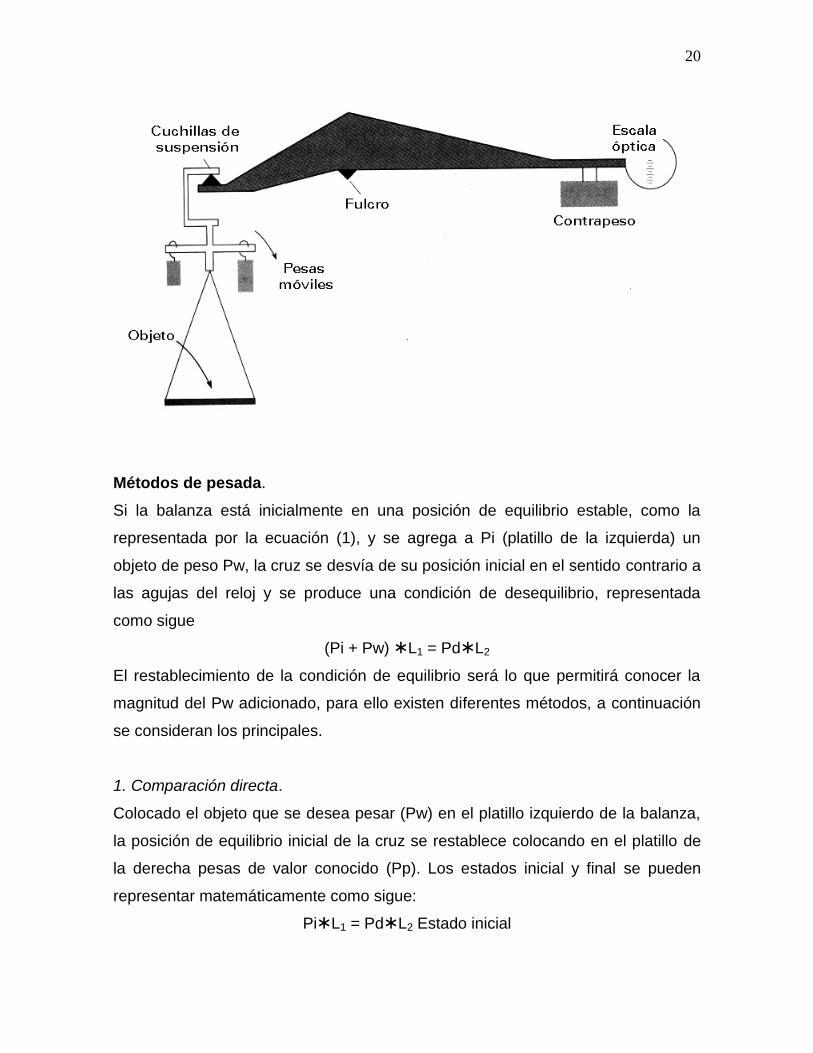
20
Métodos de pesada.
Si la balanza está inicialmente en una posición de equilibrio estable, como la
representada por la ecuación (1), y se agrega a Pi (platillo de la izquierda) un
objeto de peso Pw, la cruz se desvía de su posición inicial en el sentido contrario a
las agujas del reloj y se produce una condición de desequilibrio, representada
como sigue
(Pi + Pw) L1 = PdL2
El restablecimiento de la condición de equilibrio será lo que permitirá conocer la
magnitud del Pw adicionado, para ello existen diferentes métodos, a continuación
se consideran los principales.
1. Comparación directa.
Colocado el objeto que se desea pesar (Pw) en el platillo izquierdo de la balanza,
la posición de equilibrio inicial de la cruz se restablece colocando en el platillo de
la derecha pesas de valor conocido (Pp). Los estados inicial y final se pueden
representar matemáticamente como sigue:
PiL1 = PdL2 Estado inicial

21
(Pi + Pw) L1 = (Pd + Pp) L2 Estado final
Estas dos ecuaciones pueden combinarse:
PwL1 = PpL2
Dado que los brazos de la balanza son iguales:
Pw = Pp
Así, si se conoce el valor de las pesas, colocadas para restablecer el equilibrio, se
conoce el peso del objeto (Pw).
2. Sustitución.
Al colocar en una balanza de un solo plato (que funciona a carga constante) el
objeto (Pw) se produce una condición de desequilibrio que se compensa quitando
al peso inicial (Pi), una porción (Pp) equivalente al peso del objeto. De esta
manera se restablece el estado inicial de equilibrio. Pd no se modifica dado que tal
como se indicó esta balanza funciona a carga constante.
(Pi + Pw – Pp) L1 = PdL2 (equilibrio)
El peso del objeto (Pw) será igual a la porción Pp que tuvo que quitarse de Pi; esta
porción corresponde a objetos de peso conocido (pesas), lo cual hace posible
determinar por este método el peso del objeto. (Pp = valor de las pesas).
Sensibilidad de la balanza analítica.
Esta variable describe la magnitud del cambio en la posición de equilibrio de la
cruz como resultado de la adición de un peso dado a uno de los platillos. La
sensibilidad de la balanza se define como el número de divisiones de la escala
que se desplaza la posición de equilibrio para una sobrecarga de 1 mg. En la
práctica se prefiere expresar la sensibilidad a través del Factor de Sensibilidad, el
cual se define como el peso necesario para que la posición de equilibrio, se
desplace una unidad en la escala. Es decir, el Factor de Sensibilidad es el inverso
de la sensibilidad.
Es posible deducir una expresión matemática que permita describir como el diseño
de una balanza afecta su sensibilidad, dicha expresión es:

22
Tg = dP*
LW*
Donde
W = Sobrecarga
L= Longitud del brazo de la balanza
P= Peso del conjunto de la cruz
d= distancia del fulcro al centro de gravedad del conjunto de la cruz.
El ángulo es una medida de la sensibilidad de la balanza, pués es el ángulo que
se desplaza el fiel con relación a su posición inicial de equilibrio. Según la
expresión, la sensibilidad aumenta con la longitud del brazo y disminuye con el
peso de la cruz y con la distancia del fulcro al centro de gravedad del conjunto.
Sin embargo, conciliar estos factores de manera de lograr la máxima sensibilidad
pudiera resultar imposible, así por ejemplo, no se pueden combinar una ilimitada
longitud del brazo con un bajo peso de la cruz porque se haría difícil satisfacer los
requisitos de resistencia mecánica y de rigidez.
La cantidad P incluye peso de la cruz y todos sus aditamentos, es decir, toda la
carga soportada en el fulcro. El objeto que se pesa es parte de esa carga. En el
método de pesada por comparación directa, el peso del objeto (Pw) y las pesas
correspondientes (Pp) sirven para aumentar P. Por consiguiente, en éste método
la sensibilidad de la balanza disminuye conforme la carga aumenta. En una
pesada por sustitución el peso del objeto (Pw) es compensado al eliminar la
cantidad correspondiente de Pi, de modo que el peso del conjunto de la cruz (P)
es constante. Por lo tanto, en las balanzas donde se pesa por sustitución, es decir
la balanza monoplato, la carga y la sensibilidad son constantes.
Construcción de la balanza analítica.
Tal como se indicó en párrafos anteriores las balanzas pueden ser de dos platos o
de un plato, y sus componentes finales dependen de dicha arquitectura.
Las partes más críticas de una balanza analítica son los bordes de las cuchillas
que soportan las partes móviles, a saber: el fulcro (en el centro) y las cuchillas

23
donde van suspendidos los platillos. La construcción y operación de la balanza
están planeadas con miras a librar los bordes de las cuchillas de toda tensión
innecesaria; para ello se usan soportes que se manejan desde el exterior de la
balanza y se liberan únicamente durante los breves momentos en que se hacen
las observaciones de la posición de desviación de la cruz. En estado de apoyo los
bordes de las cuchillas no soportan carga y en consecuencia no pueden dañarse
por choques repentinos cuando, por ejemplo, se ponen o quitan objetos de un
platillo.
Toda balanza está provista de un medio de lectura para observar la posición y las
desviaciones del fiel. El método más simple es el de una escala fija, colocada
detrás de la punta del fiel. Hay otros medios de lectura que emplean amplificación
óptica de la escala; en algunos de ellos no hay fiel, en su lugar se sujeta
directamente a un extremo de la cruz una microescala de reflexión. La escala
puede estar graduada en unidades empíricas o directamente en unidades de
peso.
Errores en la pesada.
Algunas posibles fuentes de error en la pesada son:
1. Desigualdad de los brazos de la balanza
Es un error inherente a las balanzas de dos platos donde se pesa por el método
de comparación directa, donde todas las consideraciones sobre el equilibrio y
desequilibrio cambiarían si la longitud de los brazos no es igual. Con anterioridad
se representó matemáticamente la condición en que se restablece el punto de
reposo inicial de la balanza, cuando se pesa un objeto, de la siguiente manera:
Pw x L1 = Pp x L2
Donde las longitudes de los brazos de la balanza L1 y L2 son iguales; sólo en este
caso el peso del objeto es igual a la suma de las pesas analíticas:
2L
1LPpPw
En el método de pesada por sustitución, balanza de un sólo platillo, no se presenta
este tipo de error.

24
2. Empuje de aire
Según el principio de Arquímedes, el peso aparente de un objeto sumergido en un
fluido es menor que su peso real en el vacío, la diferencia es una cantidad de igual
magnitud al peso del fluido desplazado por el objeto. En las pesadas por
comparación o por sustitución tanto el objeto como las pesas experimentan un
empuje similar del aire; No obstante, estos dos empujes se cancelan entre sí
únicamente si el objeto y las pesas tienen la misma densidad de modo que
desplacen volúmenes iguales de aire. Es decir, que a medida que la diferencia de
densidad entre el objeto y las pesas es mayor, se incrementa la magnitud del error
debido al empuje del aire.
El peso del objeto observado en el aire se puede corregir al peso correspondiente
en el vacío mediante la siguiente expresión:
Pv = Pa + (Pa/do – Pa/dp) da
Donde:
Pv y Pa son los pesos del objeto en el vacío y en el aire respectivamente; do, dp y
da son las densidades del objeto, de las pesas y del aire respectivamente. Se
puede observar, que Pa/do es igual al peso del aire desplazado por el objeto y que
Pa/dp x da es el peso del aire desplazado por las pesas.
3. Efecto de la temperatura
Los cambios de temperatura pueden causar varios tipos de errores en la pesada.
Sin embargo, el más común y más grave se presenta cuando el objeto por pesar
no está a la misma temperatura de la balanza y el medio circundante; esta
diferencia de temperatura induce corrientes de aire por convección que conducen
a pesos falsos o a una conducta errática de la balanza que impide observar el
punto de reposo.
4. Otros efectos
Otra posible fuente de error es la adsorción de humedad presente en el ambiente
por parte del objeto que se pesa. También la electrificación de los envases de

25
vidrio usados para pesar causa errores debido a que induce oscilaciónes erráticas
de la balanza. Además, el platillo donde está el objeto electrificado es atraído por
el piso de la balanza, por lo que el peso aparente del objeto es demasiado grande.
Cuidados de la balanza analítica.
Debido a la importancia de la pesada en la exactitud del análisis químico, es
necesario observar en esta operación normas cuyo incumplimiento traería como
consecuencia resultados falsos. Esas normas son las siguientes:
1. La balanza debe estar colocada sobre una base firme, se mantendrá nivelada y
no debe moverse innecesariamente.
2. Debe evitarse la exposición directa al sol, pués esto es causa de perturbaciones
e irregularidades.
3. Cuando la balanza no está en funcionamiento, la cruz y los soportes estarán
suspendidos, lo que evita el desgaste de las cuchillas.
4. Los botones y perillas deberán manipularse cuidadosa y suavemente.
5. Nunca deben quitarse o añadirse pesas o sustancias, si la balanza está en
posición libre.
6. Las lecturas se harán siempre con la caja de la balanza cerrada, para evitar
errores debido a corrientes de aire.
7. El objeto a pesar debe colocarse siempre en el centro del platillo.
8. No deben colocarse directamente sobre los platillos sustancias u objetos que
puedan deteriorarlos. Las sustancias se deben pesar en recipientes apropiados:
vidrios de reloj, beakers pequeños, pesa filtros o crisoles. Los líquidos o sólidos
volátiles o higroscópicos se deben pesar en recipientes de cierre hermético como
son los pesafiltros de tapa esmerilada.
9. El objeto a pesar deberá estar a temperatura ambiente.
10. No debe sobrecargarse la balanza.
11. Cuando la balanza no está en uso se mantendrá bloqueada y cerrada.

26
PARTE EXPERIMENTAL.
Materiales.
Balanza analítica
PROCEDIMIENTO:
1. Pesada de un objeto.
Previo a la introducción del objeto en la balanza chequee los siguientes aspectos:
que la balanza esté nivelada y que el punto de reposo corresponda al cero de la
escala. A continuación siga el siguiente orden:
Coloque el objeto en el platillo de la balanza.
Prepese el objeto mediante el uso sistemático de las pesas. La pesada en
balanza analítica requiere varias operaciones de tanteo hasta encontrar la
combinación correcta de pesas. Al hacer los tanteos debe seguirse un
procedimiento sistemático. Con el objeto colocado en el platillo de la
balanza se prueban las pesas en el siguiente orden: en primer lugar, los
cientos, luego las decenas, después las unidades y finalmente las
fracciones de gramo. Es importante revisar las especificaciones de manejo
de cada balanza en particular, pués las hay de prepesada directa.
Concluida la prepesada, libere la balanza y ajuste el micrométrico hasta
alcanzar la condición de equilibrio. Anote la lectura correspondiente al peso
del objeto.
Retire el objeto y devuelva los controles de peso a su condición inicial.

27
Laboratorio 2. DETERMINACIÓN DE HUMEDAD EN DIFERENTES
MUESTRAS
OBJETIVOS
Al finalizar las actividades programadas, el estudiante debe estar en capacidad de:
Determinar gravimétricamente el contenido de agua en una muestra.
Interpretar y utilizar los resultados de la determinación gravimétrica
realizada.
INTRODUCCIÓN
Uno de los tipos de análisis químico cuantitativo usado, con cierta frecuencia, en
los laboratorios analíticos son los llamados Métodos Gravimétricos. En este grupo
se reúnen todos aquellos donde la concentración del analito que se determina es
conocida gracias a una “pesada” como medida final. Ahora bien, en función de la
forma como se aisla un determinado analito, es posible establecer subtipos:
Gravimetrías de precipitación: el analito es precipitado gracias al uso de
una reacción química apropiada. El precipitado obtenido es pesado luego
de una serie de manipulaciones analíticas que garanticen su pureza,
composición e integridad.
Gravimetrías de electrodeposición: consisten en provocar mediante la
aplicación de una corriente eléctrica, la deposición de un metal sobre un
electrodo que ha sido pesado previamente. Concluido el proceso se pesa
nuevamente el electrodo, la diferencia de peso encontrada corresponderá a
la cantidad de metal que se depositó sobre dicho electrodo.
Gravimetrías de volatilización: El fundamento de este tipo de análisis
radica en la separación del componente bajo estudio, gracias a su
volatilización, bien por aplicación de calor, bien por aplicación de un
reactivo químico apropiado. El analito puede ser medido de manera directa
(mediante su recolección en un absorbente apropiado) o bien de manera
indirecta por determinación de la pérdida de peso que sufre la nuestra bajo

28
estudio. La medición indirecta es posiblemente la más ampliamente
aplicada.
Una de las aplicaciones más extendidas de este último tipo de gravimetría es la
determinación del contenido de humedad presente en una muestra, en el
entendido que debe ser considerada la presencia de algún otro compuesto volátil
en la muestra, esto llevaría lógicamente a la necesidad de las correcciones
pertinentes.
En este curso se aplicará la gravimetría de volatilización para determinar el
contenido de agua en muestras de origen vegetal.
Determinación del contenido de agua en muestras.
La mayoría de las muestras a procesar por un analista contienen agua, la cual
puede encontrarse en dos formas: Agua esencial, la cual puede ser agua de
constitución (forma parte de su estructura química) y Agua de hidratación (unida
por fuerzas covalentes y fácil de eliminar por aplicación de calor) y Agua no
esencial, que se encuentra simplemente adsorbida en la superficie de dichas
muestras, o ocluida en la masa de dichas muestras. La presencia de agua se hace
más importante cuando la muestra es de origen vegetal, ya que en estos
materiales el agua constituye más del 90% del peso fresco de los mismos. Ahora,
bien, el contenido de humedad en una muestra puede resultar muy variable por
cuanto depende de factores ambientales tales como la temperatura y humedad
relativa y de factores inherentes a la muestra misma, tales como su
higroscopicidad y el tamaño de las partículas, esto obliga a la necesidad de
conocer cual es el contenido de agua en una determinada muestra, previo a su
análisis, como única forma de obtener resultados analíticos reproductibles y
comparables.
Se trata entonces de secar la muestra, es decir, eliminar el agua que no esté
químicamente unida al material. Por esta razón conviene recalcar que existen
varios métodos cuya selección depende de la estabilidad del material bajo estudio
ante el tratamiento que cada método particular establece para la determinación.

29
Tal como se indicó previamente en este caso se usará el método gravimétrico por
volatilización y el agua contenida en la muestra se determinará por la pérdida de
peso que sufre el material cuando es sometido a la acción del calor, conviene
recalcar que éste método sólo es aplicable, cuando la pérdida de peso es debida
unicamente a la pérdida de agua. La temperatura a usar dependerá, como
también se mencionó anteriormente, de la estabilidad del material utilizado.
Generalmente la temperatura oscila entre 100 y 130º C por períodos también
oscilantes entre 1 y 3 horas.
En muchos casos, las condiciones utilizadas no garantizan la eliminación de
“Toda” el agua contenida en la muestra pero si el llegar a un contenido estándar
de humedad que permita la comparación sobre una base común (un contenido de
materia seca constante) de los resultados.
PARTE EXPERIMENTAL
Materiales:
Recipientes (usualmente cápsulas de porcelana).
Estufa (dotada de control de temperatura).
Desecador (debe contener un secante apropiado; CaCO3, CaCl2, etc.)
Muestras.
PROCEDIMIENTO.
1. Disponga de dos recipientes previamente normalizados (recipientes que fueron
llevados a peso constante).
2. Pese, en balanza analítica, en cada uno de los recipientes anteriores 1,0000 g
de la muestra que le suministrará el profesor (esto constituye su pesada original
en la muestra húmeda).
3. Coloque ambas muestras en una estufa a una temperatura entre 105 – 110º C
durante 1 hora.
4. Retire las muestras de la estufa y colóquelas en el desecador, deje enfriar
durante un mínimo de 20 minutos.

30
5. Pese cada muestra, en balanza analítica, para obtener la primera pesada de la
muestra seca (recipiente más muestra).
6. Repita el desecado en estufa a la misma temperatura y por un período de 20
minutos.
7. Retire las muestras de la estufa y lleve nuevamente el desecador para el
enfriamiento.
8.- Pese nuevamente y obtendrá su segunda pesada de la muestra seca
(recipiente más muestra). Este proceso deberá repetirse hasta peso constante,
Recuerde que usualmente se entiende que el material esté a peso constante
cuando la diferencia entre dos pesadas consecutivas es menor o igual a 5 mg.
9. Por diferencia obtenga el peso de su muestra y proceda a los cálculos con el
auxilio de las siguientes expresiones:
100 *Húmeda Muestra Peso
Seca Muestra Peso - Húmeda Muestra Peso Humedad de %
Humedad % - 100 Seca Materia de %
100 * Muestra Peso
Seca Muestra Peso Seca Materia de %
10. Informe sus resultados al profesor, utilice para ello el formato apropiado y de
respuesta a todo lo solicitado en dicho formato.

31
Laboratorio 3. PREPARACIÓN DE SOLUCIONES ESTÁNDARES
OBJETIVOS
Al finalizar las actividades programadas, el estudiante debe ser capaz de:
Analizar los principios teóricos usados en la preparación de las soluciones
estándares.
Utilizar las técnicas e instrumentos propios del análisis volumétrico.
INTRODUCCIÓN.
El análisis volumétrico consiste en determinar el volumen de una solución de
concentración conocida, que se requiere para la neutralización, precipitación total,
oxidación, reducción o formación de un complejo de analito objeto de análisis.
La operación experimental principal en cada determinación volumétrica es la
Titulacion, término que puede ser definido, como la adición controlada de una
solución a otra con la cual reacciona Cuantitativamente.
Como los resultados de una determinación volumétrica se obtienen a partir de la
cantidad de reactivo (concentración y volumen) que reacciona con la sustancia
que se determina, no puede usarse exceso del mismo, motivo por el cual debe
disponerse de algún medio que permita poner de manifiesto el punto en el cual la
reacción deseada se ha completado. Este punto, conocido en la titulación como
Punto de Equivalencia, puede ser definido como aquel punto en el cual la
cantidad de solución que se agrega para valorar es químicamente equivalente a
la cantidad de sustancia que se está valorando; en otras palabras, es el punto en
el cual la cantidad de sustancia añadida, es químicamente equivalente a la
cantidad de sustancia que se titula.
El punto de equivalencia puede reconocerse visualmente por un cambio
característico nítido dado por la misma solución estándar, por ejemplo, un cambio
de color (KMnO4); sin embargo, lo más frecuente es el uso de una sustancia
auxiliar llamada Indicador, la cual pone de manifiesto el mínimo exceso de
solución reactivo añadido. Se producen en ese momento, cambios apreciables

32
visualmente, tales como, la aparición de un enturbamiento, un cambio en la
coloración, etc.
El punto en el cual se produce el cambio del indicador se conoce como Punto
Final de la titulación y debería coincidir con el Punto de Equivalencia. La
diferencia entre el punto de equivalencia y el punto final se conoce como Error de
Titulación.
Soluciones patrones o estándares
Son soluciones a las que se les conoce exactamente la concentración de soluto
que contienen. Pueden ser clasificadas como primarias y secundarias.
Una solución estándar primaria es la que se prepara por medida directa del peso
de soluto y del volumen en el cual éste está disuelto; en cambio la solución
estándar secundaria, es aquella cuya concentración no puede calcularse
directamente del peso del soluto y del volumen de solución, sino que para ello
debe analizarse una porción de la misma.
Preparación de soluciones patrones o estándares
Las soluciones patrones se preparan por dos métodos: Directo e Indirecto.
Método Directo
Para preparar soluciones estándares por esta vía, es necesario que el soluto a
usar, pertenezca al grupo de las llamadas sustancias tipo primario o sustancias
volumétricas. Estas sustancias tipo primario, deben llenar algunos requisitos, a
saber:
Deben ser químicamente puras, es decir, que su composición corresponda
exactamente a su fórmula química.
Deben ser estables en la forma pura y en solución, es decir, no deben
alterarse durante la pesada ni durante la disolución, por lo cual no deben
ser fácilmente oxidables o higroscópicas y no deben absorber CO2 de la
atmósfera.
Deben reaccionar estequiométricamente con el analito que se analiza.

33
El peso de su Molc debe ser alto, de forma que se pueda pesar un número
razonable de miliMolesc sin errores significativos en la pesada.
Algunos ejemplos de sustancias tipo primario son: carbonato de sodio,
ácido benzoico, ftalato ácido de potasio. ácido oxálico dihidratado, oxalato
de sodio, bromato de potasio, etc.
Comprobada la existencia y disposición de la sustancia tipo primario requerida, las
soluciones estándares primarias se preparan de la siguiente manera: se pesa una
cantidad adecuada de la sustancia volumétrica en balanza analítica, se transfiere
cuantitativamente a un balón aforado, se disuelve con agua destilada y se
completa el volumen hasta la señal de enrase del balón. Cuando se trate de
soluciones muy exactas, al preparar la solución se debe tomar en cuenta la
temperatura a la cual el balón fue aforado.
Método indirecto
Si el soluto a utilizar no cumple los requisitos exigidos a las sustancias
volumétricas, como ocurre por ejemplo con la mayoría de los ácidos y álcalis, es
necesario preparar la solución estándar por vía indirecta. Para ello se procede de
la manera siguiente: se prepara una solución de concentración ligeramente
superior a la deseada, para lo cual se mide en un cilindro graduado o se pesa en
una balanza ordinaria un poco más de la cantidad de soluto calculado, se
transfiere a un balón aforado, se disuelve con el solvente (generalmente agua
destilada), y se completa el volumen hasta la señal de enrase del balón. A esta
solución se le determina la concentración exacta mediante un método de
valoración adecuado. Una vez valorada se rotula la solución con la concentración
determinada en la valoración: M, Mc, etc.
En caso de obtenerse una concentración muy superior a la que se requiere, es
posible ajustar la concentración mediante el añadido de solvente.

34
Manejo y conservación de soluciones estándares
Para conservar soluciones estándares es necesario tomar ciertas precauciones a
saber:
Deben guardarse en recipientes herméticamente cerrados para prevenir la
evaporación del disolvente, lo cual traería como consecuencia un
incremento en la concentración de la solución.
Antes de utilizar la solución estándar, el recipiente que la contiene debe
agitarse para asegurar la uniformidad de la composición tanto de la porción
extraída, como del remanente en el recipiente.
No se deben regresar al recipiente porciones no usadas de la solución
extraída para evitar riesgos de contaminación de la solución estándar.
Algunas soluciones estándares deben ser protegidas de los gases
atmosféricos. Por ejemplo las soluciones de Hidróxido de Sodio, se diluyen
cuando el CO2 de la atmósfera se disuelve en la solución con la
consecuente formación de ácido carbónico, el cual a su vez reacciona con
el hidróxido de sodio. Otras soluciones estándares, como las de nitrato de
plata y permanganato de potasio, deben conservarse en frascos de vidrio
color ámbar, para prevenir la descomposición por efecto catalítico de la luz.
Indicadores
Las sustancias usadas como indicadores dependen del tipo de reacción que sirve
de base para la determinación. A continuación se hará referencia a aquellos
indicadores que permiten detectar el punto final de una reacción de neutralización
Los indicadores ácido-base son colorantes orgánicos con carácter de ácido
débil (ej. Fenolftaleina) o base débil (ej. Anaranjado de Metilo) que tienen la
particularidad de presentar colores diferentes en la forma ácida o básica, de esta
manera el punto final en una valoración ácido-base viene dado por un cambio
de color del indicador.
El cambio de color que experimenta el indicador es debido a cambios en la
configuración electrónica de sus moléculas. Si se considera un indicador ácido

35
orgánico débil, de fórmula general HI. Este ácido ioniza según la siguiente
reacción:
IHHI
En este ejemplo la molécula sin disociar (HI), es incolora, al producirse la
dlsociación genera el ion I- que es coloreado debido a la reagrupacion de los
átomos para formar una estructura quinóica.
En solucion acuosa la ionización del ácido es tan pequeña que no se puede
apreciar el color; sin embargo, el agregar una sustancia alcalina, la reacción con el
ión hidrógeno desplaza el equilibrio anterior hacia la derecha y se incrementa la
concentración del ión I- hasta un punto en que se hace visible el color. La
constante de ionización del indicador (denominada constante del indicador) es:
[HI]
]][I[HKa
Análogamente un indicador que sea una base débil, de fórmula general IOH, se
ioniza según:
OHIIOH
La constante de ionización es:
[IOH]
]][OH[IKb
En solución acuosa predomina el color de las moléculas de IOH, pero la adición de
un ácido, aumenta la concentración de I+ y por lo tanto cambia el color.
Valoraciones o Titulaciones Ácido-Base
Fundamento teórico
En esta práctica, se realizará la preparación y valoración de soluciones ácidas y
básicas. En teoría una valoración ácido–base o viceversa, implica un proceso de
neutralización que, expresado en términos de una reacción iónica, indica la unión
de iones hidronio que provienen del ácido, con los iones aceptores de protones de
la base (CO3=, OH-, etc.)

36
Ejemplo:
O22H-
OH O3H
O2H 3HCO3CO O3H
Cuando tal reacción ocurre como resultado de una titulación, se usa el término
Acidemetría si la solución estándar usada es un ácido y Alcalimetría si el
estándar usado es una solución alcalina.
Observaciones
1. En las valoraciones debe utilizarse una bureta limpia, previamente curada, es
decir, enjuagada con tres porciones de 3 mL de valorante cada una. Estas
porciones de valorante se desechan. Esto evita diluir la solución que se usa como
valorante. Para la sustancia a valorar (muestra) debe usarse una fiola limpia y
preferiblemente seco.
2. El pico de la bureta debe estar lleno de solución, por lo tanto es necesario
eliminar las burbujas de aire que hayan quedado.
3. El pico de la bureta debe colocarse de forma tal, que se eviten la siguientes
situaciones:
Producción de salpicaduras a las paredes de la fiola.
Escurrimiento de solución por las paredes de la fiola.
La existencia de contacto directo entre el pico y la solución que se valora.
De esta manera se asegura la máxima oportunidad de reacción entre el valorante
añadido y la sustancia valorada.
4. A medida que se añade el valorante, debe agitarse la fiola para que las
sustancias reaccionen en forma inmediata.

37
5. La velocidad recomendada para añadir el valorante es de 10 mL por minuto. Así
se evitan los errores debidos a la adherencia del valorante a la bureta. Además, si
la velocidad es mayor, se corre el peligro de sobrepasar el punto final; para reducir
esta posibilidad se aconseja que la adición de, al menos el último mililitro de
valorante, se realice gota a gota. En algunos casos puede ser conveniente añadir
sólo una gota; para ello se gira la llave de la bureta de forma que fluya sólo una
gota y permanezca en el pico de la bureta. Este pequeño volumen puede ser
transferido a la mezcla reaccionante de dos formas diferentes: una, tocando
cuidadosamente el pico de la bureta con la pared de la fiola y la otra, mediante
una varilla de vidrio se transfiere la gota al seno de la mezcla reaccionante.
6. Si la fiola que contiene la mezcla reaccionante se coloca sobre un trozo de
papel blanco (fondo blanco), el cambio de color se hace más evidente que cuando
se observa sobre un fondo oscuro. En algunos casos (especialmente para
analistas poco experimentados) es necesario observar el cambio de color
del indicador con relación al de otra fiola testigo en vez de hacer un juicio
independiente de cada valoración.
7. Es necesario lavar las paredes internas de la fiola de titulación con un frasco
lavador (que contiene agua destilada), justo antes del punto final, para estar
seguros de que toda la solución problema (muestra), reacciona con toda la
solución que se ha dejado caer desde la bureta.
8. Al terminar el proceso de valoración debe descartarse la solución que queda en
la bureta. El líquido sobrante NUNCA debe volverse al recipiente. La bureta debe
enjuagarse varias veces con agua antes de guardarla.
9. Las principales fuentes de error en los métodos de valoración son:
a. Errores en la preparación de las soluciones.
Errores en la pesada

38
Pérdida de soluto pesado cuando se transfiere al recipiente donde se va
a disolver.
b. Errores en la valoración.
Goteo de la bureta.
Errores al pipetear.
Errores de lectura en la bureta.
Selección incorrecta del indicador.
PARTE EXPERIMENTAL
Reactivos Materiales
Ácido sulfúrico conc. Bureta de 50 mL
Anaranjado de Metilo Balones aforados de 100 mL
Carbonato de Sodio (p.a.) Pipetas de 10 mL
PROCEDIMIENTO
Las titulaciones pueden efectuarse por el método Directo o por el método Indirecto
(retroceso). En el primer caso la solución estándar o patrón se añade a la solución
problema desde la Bureta hasta que se alcanza el Punto Final, punto en el cual
debe suspenderse la adición del titulante. Lógicamente, las últimas porciones de
reactivos deben añadirse lentamente (gota a gota) para no sobrepasar el citado
punto. En el segundo caso se añade un exceso de la solución patrón y a posterior
se determina el exceso añadido por una valoración con otra solución patrón.
En esta práctica se procederá a realizar una titulación por el método directo;
siguiendo los pasos siguientes:
1. Calcule la cantidad de soluto necesaria para preparar las solución asignada de
carbonato de sodio, se debe conocer el peso molecular carga de dicha sustancia.
2. Prepare la solución estándar primaria: haciendo uso de una balanza analítica
pese la cantidad de “sustancia tipo” calculada en (1) en un beaker, disuelva con
una pequeña porción de agua destilada y trnasfiera cuantitativamente a un balón
aforado de 100 mL limpio. Posteriormente enrase cuidadosamente. Rotule la

39
solución con la concentración exacta (M, Mc, etc.), indique además la fecha de
preparación de la solución
3. Valoración de la solución problema ácida:
3.1. Prepare una solución Testigo, colocando en una fiola 50 mL de agua
destilada tres gotas del indicador anaranjado de metilo y una gota de solución
patrón de carbonato de sodio, preparada en el punto anterior.
3.2. Mida con una pipeta 10,00 mL de solución problema ácida y colóquelos en
una fiola de 250 mL. Agregue 25 mL de agua destilada y tres gotas del indicador
anaranjado de metilo.
3.3. Titule con la solución patrón de carbonato de sodio, hasta alcanzar el punto
final (compare el color con el Testigo). Anote el volumen gastado.
3.4 Efectúe por lo menos tres titulaciones.
4. Calcule la Mc exacta de la solución ácida, hasta la cuarta cifra decimal.
Ejemplo del uso de los datos experimentales:
1. Cálculo de la Molaridad de carga de la solución de carbonato de sodio
asignada:
Con los gramos de soluto pesados y el volumen de solución a preparar (100 mL),
se procede así:
mMolc de Na2CO3=
32
pesado soluto
CONa c
MolP
g
Ejp:
mMolc de Na2CO3= cmMol10,16
cmMol.g0,53
g0,5383
Donde:
100mL3
CO2
Na de cmMol 10,16Mc
Mc = 0,1016 mMolc.mL -1

40
La solución estándar de Na2CO3 se rotulará así: Na2CO3 0,1016 Mc + Fecha de
preparación
2. Cálculo de la Mc exacta de la solución problema ácida
Con los datos de las cuatro titulaciones realizadas construir una tabla, donde se
indique el volumen de solución patrón gastado en cada titulación, y el volumen de
solución problema ácida, que en este caso es 10 mL.
Ejp.:
Titulación Na2CO3 (mL) Sol. Problema ácida (mL)
1 10,40 10,00
2 10,30 10,00
3 10,40 10,00
4 10,50 10,00
Promedio 10,40 10,00
Para el cálculo de la Mc promedio exacta, se calcula con cada volumen gastado en la
titulación la Mc y se promedia. Para ello, se utiliza la siguiente expresión:
V1 Mc1 = V2 Mc2
Donde:
V1 = Volumen de solución Na2CO3 gastado
V2 = Volumen solución ácida
Mc1= Molaridad carga Na2CO3
Mc2= Molaridad de la solución ácida (desconocida)
Titulación 1.
10,40 mL 0,1016 mMolc / mL = 10,00 mL Mc2
Despejando:
Mc2 = mL/cmMol0,1057mL10,00
mL/cmMol0,1016*mL10,40

41
Titulación 2.
10,30 mL 0,1016 mMolc / mL = 10,00 mL Mc2
Despejando:
Mc2 = mL/cmMol0,1046mL10,00
mL/cmMol0,1016*mL10,40
Titulación 3.
10,40 mL 0,1016 mMolc / mL = 10,00 mL Mc2
Despejando:
Mc2 = mL/cmMol0,1057mL10,00
mL/cmMol0,1016*mL10,40
Titulación 4.
10,50 mL 0,1016 mMolc / mL = 10,00 mL Mc2
Despejando:
Mc2 = mL/cmMol0,1067mL10,00
mL/cmMol0,1016*mL10,40
La solución así valorada se rotula de la forma siguiente:
Solución ácida 0,1057 Mc + Fecha de preparación

42
Laboratorio 4. APLICACIÓN DEL ANÁLISIS QUELOMÉTRICO
DETERMINACIÓN DE LA DUREZA TOTAL EN AGUAS
OBJETIVOS: Al finalizar las actividades programadas, el estudiante será capaz de:
Reconocer la importancia de la presencia de la dureza en aguas.
Emplear el método de titulación quelométrica para la determinación de la
dureza del agua
INTRODUCCIÓN: El término dureza del agua se refiere a la cantidad de calcio y magnesio disueltos en el agua. Estos minerales tienen su origen en las formaciones rocosas calcáreas, y pueden ser encontrados, en mayor o menor grado, en la mayoría de las aguas naturales. La dureza es producida por los carbonatos y bicarbonatos de calcio y de magnesio. Los carbonatos de estos elementos son ligeramente solubles en agua pura (Kps MgCO3 = 3,5x10-8 y Kps CaCO3 = 4,5x10-9 ), pero cuando ésta contiene anhídrido carbónico, los disuelve fácilmente formando bicarbonatos. Esta dureza se llama, temporal, ya que se suprime al hervir el agua. La dureza que se origina por la presencia de sulfatos y cloruros de cationes divalentes y que no les afecta la ebullición se llama dureza permanente. El calcio y magnesio causan dos grandes problemas: 1. Cuando el agua se calienta, éstos precipitan fuera de la solución, y forman una costra dura, de apariencia rocosa (sarro). Esta costra acelera la corrosión (arruinando equipos de calefacción de agua y sistemas de riego), restringe el flujo de agua, y reduce la transferencia de calor. 2. Cuando se combinan con el jabón, reaccionan para formar un cuajo, que interfiere con el efecto de la limpieza, seca la piel, y forma depósitos en cañerías y ropas. La dureza es indeseable en algunos procesos, tales como el lavado doméstico e industrial, provocando que se consuma más jabón, al producirse sales insolubles. En calderas y sistemas enfriados por agua, se producen incrustaciones en las tuberías y una pérdida en la eficiencia de la transferencia de calor. Además le da un sabor indeseable al agua potable. Por las razones antes expuestas, la dureza del agua debe ser removida antes de que el agua tenga uso apropiado para las industrias de bebidas, lavanderías, acabados metálicos, teñido y elaboración de textiles, entre otras. Las normas internacionales, establecen como límite máximo permisible 300 mg L-1 de dureza. En México, la norma (NOM 127 SSA1 1994) establece como límite máximo permisible 500 mg L-1. La mayoría de los suministros de agua potable

43
tienen un promedio de 250 mg L-1 de dureza. Para el agua utilizada en calderas, el límite es de 0 mg L-1 de dureza. Esta norma, establece además, el procedimiento de potabilización para la remoción de dureza por medio de ablandamiento químico o intercambio iónico. En función a lo expuesto anteriormente el agua se clasifica como: 0-75 mg L-1 CaCO3 agua suave 75-150 mg L-1 CaCO3 agua poco dura 150-300 mg L-1 CaCO3 agua dura > 300 mg L-1 CaCO3 agua muy dura De lo anterior se establece que la dureza está conformada como sigue: Dureza Total = Dureza de Calcio + Dureza de Magnesio (expresada como mg L-1 de CaCO3) Los iones Ca+2 y Mg+2 se pueden determinar directamente con una solución estándar de la sal disódica del ácido etilendiaminotetracético (Na2H2Y), conocida comercialmente con los nombres de Tritriplex, Complexona o Verseno. El ácido etilendiaminotetracético (EDTA) se representa como H4Y. Es un ligando que forma con el calcio y el magnesio complejos 1:1, sin tomar en cuenta la valencia de los cationes, razón por la cual al efectuar los cálculos no se trabaja con concentraciones molares de carga sino molares. Mg+2 + HY3- MgY2- + H+
Ca+2 + HY3- CaY2- + H+
Como indicador del punto final se usa el Negro de Eriocromo T, el cual en soluciones de pH 9-10 tiene color azul y forma con el calcio y el magnesio quelatos de color rojo vino, de menor estabilidad que los complejos CaY2- y MgY2-, conforme a la siguiente reacción: Mg+2 + HIn2- MgIn- + H+ Ca+2 + HIn2- CaIn- + H+
Azul Rojo vino Durante la valoración, la solución de Ca y Mg debe estar tamponada a pH 9-10. A pH superiores precipita Ca(OH)2 y Mg(OH)2, que no pueden ser redisueltos con EDTA. Si el pH es menor, disminuye la estabilidad de los quelatos CaY2- y MgY2-, por lo tanto, la reacción no será cuantitativa. Además una elevada acidez dificulta la observación del punto final, por cuanto la ionización del indicador depende del pH. Cuando se titula directamente el calcio y el magnesio con solución patrón de verseno, a pH 8-10 (“Buffer” de NH4OH / NH4Cl) y se emplea Negro de Eriocromo

44
T como indicador, antes de llegar al punto de equivalencia la solución es de color Rojo vino, debido a la formación de los complejos Calcio-Negro de Eriocromo T (CaIn-) y Magnesio-Negro de Eriocromo T (MgIn-). En el punto final, al agregar el primer exceso de valorante, el color de la solución se torna azul, debido a que los complejos CaIn- y MgIn- se han convertido en CaY2- y MgY2- (incoloro), por lo que predomina el color del indicador libre (HIn2-), que al pH de la solución es azul. PARTE EXPERIMENTAL Materiales y Reactivos:
- Solución estándar de verseno 0,0100 M (EDTA 0,0100 M) - Solución tampón o reguladora (NH4OH / NH4Cl) - Indicador Negro de Eriocromo T - Pipeta volumétrica de 25 mL. - Cilindro de 10 mL. - Fiola de 250 mL. - Bureta.
PROCEDIMIENTO
1) Tomar con una pipeta volumétrica una muestra de agua de 50 mL.
2) Transferir a una fiola de 250 mL.
3) Añadir 2 mL. de la solución reguladora de pH 10 y mezclar.
4) Mientras la solución se está agitando, añadir 3 gotas del indicador Negro de Eriocromo T. La muestra debe tomar un color vino rojizo.
5) Desde una bureta deje caer la solución de EDTA 0.0100 M, agitando
continuamente hasta que desaparezcan los últimos matices rojizos. En el punto final la muestra cambia de color rojo a azul.
6) Anotar el volumen de EDTA gastado.
7) Efectuar otras dos valoraciones y realizar los cálculos correspondientes.
8) Calcular la dureza total según la siguiente ecuación:
PM CaCO3 = 100 mg.mmol-1

45
Laboratorio 5. APLICACIÓN DE LA POTENCIOMETRÍA:
EJECUCIÓN DE UNA VALORACIÓN POTENCIOMÉTRICA
OBJETIVOS
Al finalizar las actividades programadas, el estudiante debe ser capaz de:
Identificar las partes elementales de un potenciómetro.
Manejar un potenciómetro.
Construir una curva de titulación potenciométrica.
Determinar el punto final en una valoración potenciométrica.
Comparar el uso de indicadores orgánicos y el método
potenciométrico para establecer el punto final.
INTRODUCCIÓN
Las valoraciones potenciométricas son aquellas en las que se registra el
cambio de la fuerza electromotriz (f.e.m.) en función del valorante agregado.
La técnica de valoración potenciométrica ofrece respecto a otros métodos de
valoración las siguientes ventajas.
Es aplicable a sistemas químicos coloreados, con los que serían
inutiles los métodos visuales ordinarios para la determinación del punto
final.
Es especialmente útil cuando no se dispone de un indicador interno.
Elimina decisiones subjetivas concernientes a los cambios de color de los
indicadores del punto final, al igual que la necesidad de correcciones por
valoración en blanco del indicador.
Es especialmente útil para valoraciones en medio no acuoso.
Es muy versatil, puede usarse en reacciones de neutralización, de
precipitación, complejométricas y de óxido-reducción, si se dispone de
los electrodos apropiados.
La curva de valoración en una reacción de neutralización consiste en un
gráfico de pH en función del volumen de valorante añadido. Una determinación
de este tipo requiere, además del equipo usual para un análisis volumétrico,

46
de una celda galvánica formada por un electrodo de referencia estable y por
un electrodo indicador sensible al ión hidrógeno.
Electrodo de referencia: Es un electrodo cuyo potencial se mantiene
constante durante una medición o una valoración potenciométrica. El
electrodo de referencia de uso mas común es el electrodo de calomel, que esta
compuesto de Hg metálico y cloruro mercurioso sólido (calomel) en contacto con
una solución acuosa saturada de KCI con la cual esta en equilibrio.
-2Cl (s)2Hg
-2e (s)2Cl2Hg
La ecuación de Nernst correspondiente es:
2-][CI log
2
0,059 - Hg ,2CI2Hg E Hg ,2CI2Hg E
Como se observa la f.e.m. del electrodo de calomel depende solo de la
concentración de ión cloruro (o de su actividad). Debido a que el Hg2Cl2 es muy
insoluble la concentración de ión cloruro a su vez depende casi en su totalidad de
la cantidad de KCI usado en la preparación del electrodo. El potencial del
electrodo de calomel saturado es de 0,2415 voltios frente al electrodo normal
de hidrógeno a 25°C (ENH). Otro electrodo de referencia muy importante es el de
Ag/AgCI, éste es análogo al de calomel, salvo que en lugar de Hg y Hg2Cl2
contiene Ag y AgCI y la semi-reacción pertinente es:
-Cl (s) Ag
-e (s)AgCl
También en este caso el potencial del electrodo de referencia depende
exclusivamente de la concentración (actividad) del ión Cl-1 en equilibrio con la
Ag(s), AgCl(s). Cuando se prepara con solución saturada de KCI su potencial es
+0,197 V frente al ENH a 25°C.
Electrodo indicador: Es un electrodo cuyo potencial depende de la
concentración de la solución que se valora. En su superficie se efectúa un
intercambio electrónico proporcional a la concentración de la(s) especie(s) en
solución, que intervienen en la reacción.

47
Un electrodo indicador para una valoración potenciométrica debe reunir tres
características generales, a saber:
Su potencial debe estar relacionado por la ecuación de
Nernst con la concentración (actividad) de la especie que se determina.
Deberá responder de un modo rápido y reproducible a variaciones
de la concentración (actividad) de la sustancia que interesa.
Deberá poseer una forma física que permita efectuar mediciones con
comodidad.
Ahora bien, de todos los electrodos sensibles a los iones hidrógeno, el
electrodo de vidrio es único, porque el mecanismo de su respuesta a los H+
se basa en una reacción de intercambio iónico y no en un proceso de
transferencia electrónica; por consiguiente, dicho electrodo no está sujeto a
interferencias derivadas de la presencia de agentes oxidantes y reductores en
la solución problema. Un modelo de electrodo de vidrio puede estar constituido
por un bulbo de un vidrio especial muy sensible a la actividad del H+ en solución,
que esta soldado al fondo de un tubo de vidrio ordinario. Dentro del tubo hay una
solución acuosa diluida de HCI, generalmente 0,1 M.
En la solución clorhídrica esta sumergido un alambre de Ag revestido de una
capa de AgCI. El alambre de plata se prolonga hacia arriba para proveer
contacto eléctrico con el circuito externo.
Los electrodos comerciales de vidrio, hechos de vidrio de cal y sosa (2,2% Na2O;
6% CaO y 72% SiO2) responden bien a valores de pH entre 0-9. A pH mas altos
este electrodo exhibe un tipo de error llamado error alcalino, en el cual el valor
de pH observado es menor que el verdadero valor. Este error se debe a que
otros cationes, como el Na+, K+ Li+, compiten con el H+ por los sitios de
intercambio en la membrana de vidrio.
Mediciones prácticas de pH
El pH se determina por una medición de potencial. La celda galvánica que se
emplea ordinariamente para mediciones prácticas del pH está constituida por un

48
electrodo de vidrio en combinación con el electrodo de calomel. La representación
de esta celda esla siguiente:
Ag AgCl(s),HCl(0,1M) Membrana Solución
Hg2Cl2(s),KCl(s) Hg de vidrio Problema
Cada línea vertical denota un límite de fase a través del cual se desarrolla una
f.e.m. o potencial. Por ello la f.e.m. total de esta celda galvánica esta compuesta
de cinco partes, a saber:
1 El potencial del electrodo Ag/AgCI.
2 El potencial entre la solución de HCI en el interior del electrodo de
vidrio y la pared interna de la membrana de vidrio.
3 El potencial entre la pared externa de la membrana de vidrio y la
solución de pH desconocido (Solución Problema)
4 El potencial entre la solución problema y el electrodo de calomel.
5 El potencial del electrodo de calomel.
Mediante cuidadosos estudios experimentales se ha concluido que la f.e.m. de
esta celda (Ecelda) está relacionada con el pH de la solución problema por la
expresión:
Ecelda = K + 0,059pH
Donde K comprende los potenciales 1°, 2°, 4° y 5° enumerados
anteriormente; sin embargo, K es difícil de conocer con exactitud debido a que
el 4° potencial es incierto; por consiguiente, todas las determinaciones
prácticas de pH involucran necesariamente un procedimiento de calibrado en
el cual se compara el pH de una solución problema con el pH de una solución
amortiguadora estándar.
Si se introduce una porción de la solución amortiguadora estándar en la celda
galvánica y se mide la fuerza electromotriz de la celda, se tiene que:
Ecelda(s) = K + 0,059 (pH)s
Donde el subíndice s corresponde al amortiguador; analogamente, si se introduce
en la celda una solución de pH desconocido, se obtiene:

49
Ecelda(x) = K+ 0,059(pH)x
Donde x se refiere a la solución problema. De ambas expresiones es posible
obtener el valor de K, esto permite combinar dichas expresiones y despejar el
valor (pH)x:
0,059
s)celda(E -x )celda(E s(pH) x (pH)
Este valor ha sido adoptado por la Oficina Nacional de Standards
de Estados Unidos como la definición funcional del pH de la muestra problema.
Para que esta definición sea válida, el valor de K debe permanecer constante;
para lograrlo, en la práctica se elige un amortiguador cuyo pH este lo mas cerca
posible del pH de la muestra problema.
Medidores de pH
El medidor de pH de lectura directa es esencialmente un voltímetro de tubo de
vacío en el cual se imprime la f.e.m. de la celda a través de una resistencia muy
alta; la corriente resultante se amplifica y pasa por un amperímetro cuya esfera
esta calibrada directamente en unidades de pH. Su exactitud es generalmente de
±0,1 unidades de pH y es particularmente útil para valoraciones potenciométricas
ácido-base, donde lo mas importante es la variación del pH en función del
valorante agregado que un valor particular de pH.
Determinación del punto final
El éxito de la potenciometría radica en que en la región del punto de
equivalencia se registren cambios drásticos en la concentración o actividad de
las especies químicas involucradas, lo que se refleja en cambios bruscos de la
f.e.m. en esa región, suficientes para establecer el punto de equivalencia con
precisión. Los datos experimentales obtenidos son:
Volumen (V) del valorante, en mililitros.
Potencial del electrodo indicador medido frente al electrodo de referencia,
o directamente el pH de la solución si se utiliza un medidor de pH.

50
Los procedimientos que se describen a continuación, para la determinación del
punto final, son de uso general:
1. El punto final puede determinarse a partir de la representación gráfica de la
fuerza electromotriz obtenida durante la titulación en función del volumen de
agente valorante agregado; el punto final en este caso coincide con el punto de
inflexión de la curva, es decir, con el punto en que la pendiente es máxima.
En realidad el punto de equivalencia de la reacción química solo será igual al
punto de inflexión si la curva de titulación es simétrica alrededor de este punto;
esto ocurre cuando reaccionan un número igual de iones o moléculas de la
solución y del agente valorante (figura 1a).
2. Ahora bien, no siempre es fácil localizar el punto final por una simple
inspección de la curva, por esto se recurre, en la mayoría de los casos, a la
representación diferencial. Para ello se lleva a un gráfico la primera derivada de
E o de pH, es decir E/ V (cambio de potencial por unidad de
cambio de reactivo) o en una titulación ácido base pH/ V, como función del
promedio de reactivo añadido. El máximo de la curva corresponde al punto de
inflexión y por ende al punto final (figura 1b).
Para construir esta gráfica se procede de la siguiente manera:
a. Si los incrementos de volumen cerca del punto de equivalencia son
iguales, se puede tomar como E/ V la diferencia en milivoltios entre cada par
consecutivo de lectura; pero si los V son desiguales, es necesario normalizar los
datos.
b. El volumen V, al que corresponde cada valor de E/ V es el promedio de
los volúmenes correspondientes a dos lecturas consecutivas de potencial.
c. La gráfica consta de dos curvas que deben extrapolarse hasta el punto de
intersección de ambas; el volumen en el punto de equivalencia se obtiene si
se traza una perpendicular desde el punto de intersección hasta la abscisa.
El siguiente Cuadro ilustra los valores que es necesario obtener para cada una de
las gráficas usadas para la obtención del punto final:
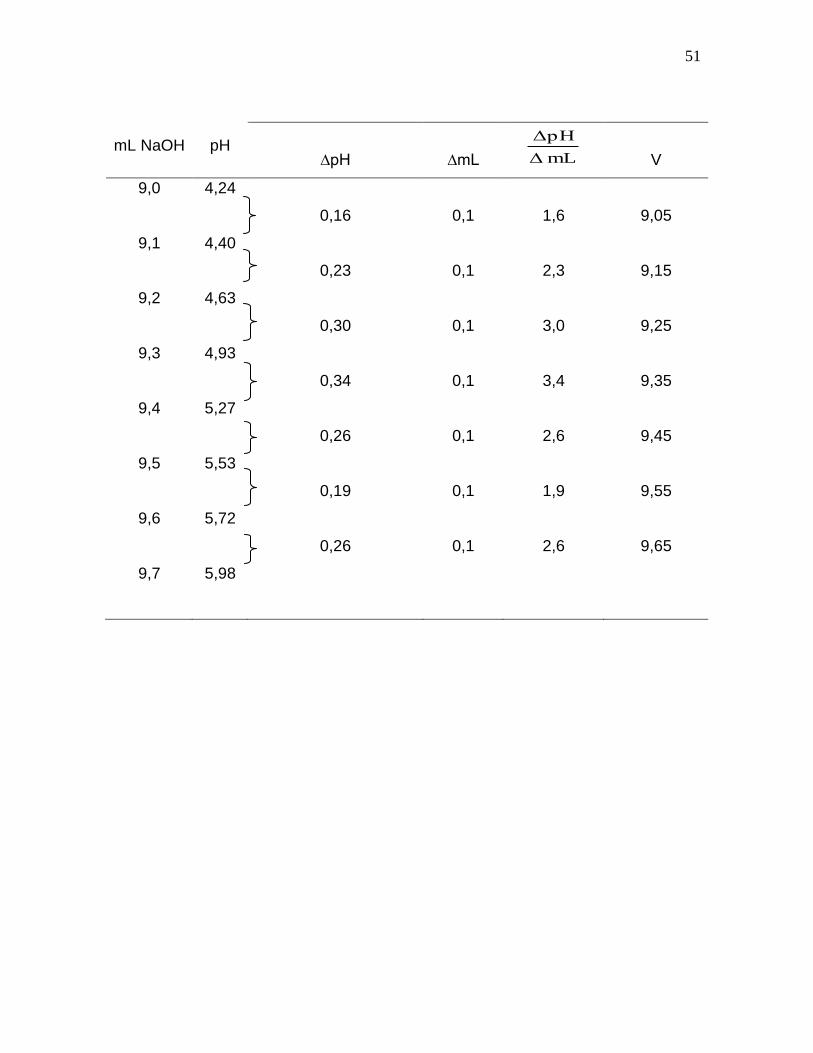
51
mL NaOH
pH
∆pH
∆mL mLΔ
ΔpH
V
9,0 4,24
0,16 0,1 1,6
9,05
9,1 4,40
0,23 0,1 2,3 9,15
9,2 4,63
0,30 0,1 3,0 9,25
9,3 4,93
0,34 0,1 3,4 9,35
9,4 5,27
0,26 0,1 2,6 9,45
9,5 5,53
0,19 0,1 1,9 9,55
9,6 5,72
0,26 0,1 2,6 9,65
9,7 5,98

52
Figura 1. Gráficos para la determinación de Punto Final
PARTE EXPERIMENTAL
Materiales Reactivos
Pipeta de 10 mL Solución 0,10 Mc de NaOH
Bureta de 25 mL Solución problema de HCI
Agitador magnético Solución Fenolftaleina
Barras magnéticas
Medidor de pH (potenciómetro)

53
PROCEDIMIENTO
1. Calibre el medidor de pH, use un amortiguador estándar de pH 4 y 7. Siga
las instrucciones de manejo del instrumento.
2. Pipetee 10,0 mL de solución de HCI a un beaker de 100 mL. Ponga el
beaker sobre la plataforma del agitador magnético. Introduzca en la solución la
barra magnética y los electrodos. Al girar, la barra no debe rozar los
electrodos. Añada agua destilada hasta asegurar la completa inmersión de
dichos electrodos.
3. Llene la bureta con la solución de NaOH. Anote el volumen inicial.
4. Agregue a la solución ácida 2 o 3 gotas de fenolftaleina. Empiece a agitar.
Anote el pH inicial de la solución
5. Deje caer sobre la solución ácida 2 mililitros del valorante básico. Espere a que
la lectura del pH no cambie más de 0,02 de unidad de pH por minuto
(aproximadamente 30"). Anote tanto la lectura de la bureta como la del pH. Repetir
la operación con 4; 6; 8; 8,5; 9 mL; a partir de 9 mL efectuar incrementos
consecutivos de 0,1 mL hasta 11 mL, luego adicionar 11,5, 12, 13 y 14 mL, anotar
an cada caso el pH. En este proceso anotar el volumen de NaOH donde ocurre
punto final (cambio de color de la solución).
6. Haga un gráfico de pH (ordenada) contra mililitros de NaOH (abscisa).
Indique la región correspondiente al cambio de color de la fenolftaleina.
Determine el punto final de la valoración, mediante los métodos descritos para tal
fin.
7. Compare y discuta la diferencia entre los puntos finales determinados por el
método visual (uso de fenolftaleina) y por el método potenciométrico.

54
Laboratorio 6. ANÁLISIS ESPECTROFOTOMÉTRICO DE
MANGANESO
OBJETIVOS
Al finalizar las actividades programadas, el estudiante debe ser capaz de:
Describir las partes básicas de un espectrofotómetro.
Manejar un espectrofotómetro.
Ejecutar un análisis espectrofotométrico de manganeso.
Interpretar los resultados del análisis efectuado.
INTRODUCCIÓN
Existen varios métodos analíticos basados en la absorción de la energía
radiante. Dichos métodos son de considerable utilidad práctica en el análisis
químico, pues se pueden aplicar a la determinación de cantidades muy pequeñas
de sustancias (10-6 M), con una exactitud del 1 al 2 por ciento. Los métodos
analíticos basados en la absorción de la luz por la materia, implican la medición
de intensidades de radiación electromagnética. Esquemáticamente este
proceso se representa por el siguiente diagrama:
La absorción selectiva de radiación electromagnética, cuando esta pasa a
través de una solución, hace que el haz emergente difiera del incidente. En la
zona de la radiación visible se puede apreciar fácilmente este cambio. El Cuadro
1 ilustra la modificación que sufre la radiación por la absorción en una solución.
El valor de la intensidad transmitida I, está influido por la intensidad de la luz
incidente Io, por el grosor (b) de la solución de la muestra y por la
concentración (c) del soluto absorbente. En este sentido, la Ley de Lambert-Beer
relaciona estos factores y se expresa matemáticamente mediante la ecuación:
Radiación Incidente Intensidad =Io
Radiación Emergente Intensidad =I
Solución Problema Concentración = C
Camino óptico = b

55
c.b.εAI
Iolog
Donde:
A, el término logarítmico de la ecuación, se conoce como Absorbancia.
, representa la Absortividad Molar,
c, es la concentración expresada en moles/litro y
b, el espesor de la cubeta o celda, expresado en cm.
De esta manera las unidades de son L.moL-l cm-l
Cuadro 1. Modificación de la radiación por efecto de la absorción en una muestra
Radiación Absorbida Radiación transmitida
(nm) Color color
380 - 450 Violeta Amarillo verdoso
450 - 480 Azul Amarillo
480 - 490 Verde azulado Anaranjado
490 - 500 Azul verdoso Rojo
500 - 560 Verde Púrpura
560 - 575 Amarillo verdoso Violeta
575 - 590 Amarillo Azul
590 - 625 Anaranjado Verde azulado
625 - 780 Rojo Azul verdoso
La absortividad puede ser expresada en otras unidades, las cuales dependen de
las unidades usadas para la concentración, en estos casos se representa por la
letra “a” minúscula.
Ahora bien, cuando se cumple la Ley de Lambert-Beer, es una constante para
diferentes concentraciones de un mismo soluto y "c" (moles/litro) es
directamente proporcional a la cantidad de luz absorbida por la solución. El

56
gráfico de la absorbancia (A) en función de la concentración (c), es una línea
recta que pasa por el origen.
Existe otra variable que se define en términos de un cociente establecido entre la
intensidad de la radiación emergente y la intensidad de la radiación incidente, esta
razón de intensidades Io
I se denomina transmitancia y se representa por la
letra T. La transmitancia se relaciona con la absorbancia según:
I
Iolog A , por lo tanto se tiene que:
%T
100log
T
1log A
Esto a su vez puede ser escrito de la siguiente manera:
log%T - 2 A
Los métodos de absorción de energía radiante se clasifican comúnmente según
los instrumentos y las técnicas empleadas en las medidas. De este modo en
los Métodos Fotométricos, generalmente aplicados en la región visible del
espectro, se usa el Fotómetro o Colorímetro; éste es un instrumento sencillo
constituido por un detector fotoeléctrico, una fuente de luz, cubetas y filtros para
restringir la radiación. En los Métodos Espectrofotométricos se emplean
Espectrofotómetros, que son utilizados comúnmente en el trabajo analítico en
la región visible, ultravioleta e infrarroja, con el uso de monocromadores para la
selección de la radiación a utilizar.
Espectrofotómetro
Un Espectrofotómetro es un instrumento que mide la absorción de energía
radiante, está constituido esencialmente por una fuente de luz, un monocromador
de prisma o red, un detector fotoeléctrico de radiación y otros accesorios
adecuados. La representación esquemática del instrumento es la siguiente:
Diafragma (Rendija) Monocromador
Cubeta (Muestra)
Detector (Fotocélula)
Fuente de Radiación
Registrador
1 2 3 4
5
6

57
Descripción de Componentes
1.- Fuente de radiación: A fin de que suministre la radiación adecuada para
las medidas de absorción, la fuente debe cumplir ciertos requisitos:
Debe proporcionar la radiación en el intervalo de longitud de onda
necesaria en el análisis.
La radiación proporcionada debe tener intensidad suficiente para que las
mediciones den resultados con la precisión y exactitud requerida.
Debe proporcionar energía de intensidad constante, de manera que las
medidas sean reproducibles.
Las fuentes de energía mas comunes para la región visible de la luz son las
lámparas con filamento de Wolframio, que producen radiación continua entre
250 y 2.500 nm. Para la región ultravioleta se usa la lámpara de descarga de
hidrógeno, que emite radiación entre 180 y 350 nm.
2.- Regulador de intensidad: La intensidad de la radiación incidente se regula
mediante el uso de diafragmas o rendijas.
3.- Selector de longitud de onda ( ): Al restringir la radiación del espectro al
sector que es mas fuertemente absorbido por la sustancia en análisis, se
aumenta la sensibilidad de la determinación, puesto que así se observa un
mayor cambio de absorbancia en función de la concentración. Los dispositivos
empleados con esta finalidad son los monocromadores; éstos aíslan un haz de
alta pureza espectral. Un monocromador sirve para resolver la radiación en sus
longitudes de onda componentes y aísla una de estas en bandas muy estrechas.
Existen tres tipos principales de monocromadores: de filtro, de prisma y de rejilla.
4.- Recipientes para la muestra: Las cubetas o celdas deben estar hechas de un
material transparente a la radiación de la región espectral de interés, es decir que
no absorba en dicha región. Para las determinaciones en la región visible se usan
cubetas de vidrio, cuarzo o sílice fundido. En la región ultravioleta no se puede
utilizar el vidrio, porque este material no deja pasar la luz ultravioleta. Las

58
cubetas pueden ser cilíndricas o rectangulares y, por lo general su diámetro "b" es
de 1 cm.
5.- Detector de radiación: La mayoría de los dispositivos utilizados para este
proposito convierten la energía radlante en energía eléctrica que luego puede
ser medida. Entre los detectores mas usados para la región visible y ultravioleta
se tiene la célula fotovoltáica o fotoelemento y la célula fotoeléctrica o fototubo.
6.- Registrador: Permite obtener la información definitiva acerca de la cantidad de
radiación absorbida por una muestra particular.
Funcionamiento de un espectrofotómetro
Si se sigue el esquema mostrado anteriormente y la descrpción de componentes,
es posible indicar que el funcionamiento de un espectrofotómetro se produce en la
forma siguiente: Un haz de energía radiante sale de 1, su intensidad se regula en
2 y el ancho de la banda se selecciona en 3, para obtener luz monocromática o
luz de una cierta longitud de onda ( ). Esta luz monocromática pasa a través de
la solución en 4, donde es parcialmente absorbida. La intensidad de la luz que
emerge de la solución es recibida por el detector, célula fotoeléctrica, en 5 y
finalmente la fotocorriente que se genera en el detector es amplificada y
registrada en 6 por medio de un sistema que incluye un galvanómetro o
potenciómetro.
En esta práctica se utilizará el Spectronic 20 (Figura 2), éste es un
espectrofotómetro de lectura directa, de un solo haz y su sistema monocromador
consiste en una red de reflexión, lentes y un par de rendijas fijas. Debido a que
la red produce una dispersión que es independiente de la longitud de onda, se
obtiene una anchura de banda constante de 20 nm a lo largo de la región en
que se opera. El instrumento emplea un solo fototubo. Su intervalo normal de
funcionamiento es de 350 a 650 nm, aunque éste puede ser extendido hasta los
900 nm por el uso de un fototubo sensible al rojo y un filtro rojo. Tiene una
escala que permite realizar lecturas directas de transmitancia y absorbancia.

59
Figura 2. Diagrama del espectrofotómetro Spectronic 20
PARTE EXPERIMENTAL
Fundamento del análisis
Gran cantidad de iones inorgánicos, en disolución acuosa, absorben suficiente
energía radiante en la región visible (380-780 nm); ésto permite su
determinación espectrofotométrica directa, incluso en muy bajas
concentraciones. Ejemplos de estos iones son: permanganato, cromato y
dicromato.
En este sentido, es posible determinar espectrofotométricamente el manganeso
presente en diversos materiales, tales como acero, plantas, suelos y otros,
después de su transformación en permanganato.
Para ello, es necesario someter la muestra a un proceso de disolución
adecuado. Una vez disuelto el manganeso (Mn2+) debe oxidarse
cuantitativamente a permanganato (Mn04-). Por ser el permanganato un ion
Fuente de Radiación
Lentes
Rendija de Entrada
Monocromador De Red
Rendija de Salida
Cubeta o Celda
Filtro
>650 nm
Fototubo

60
intensamente coloreado es posible determinarlo por absorción en la región visible.
En resumen:
-4MnO
oxidante
2Mn
disolvente Mn
(color violeta) (Fuerte absorción entre
500-530 nm).
Materiales y Reactivos
- Balones aforados de 100 mL
- Bureta 50 mL
- Spectronic 20,
- Solución estándar KMnO4
PROCEDIMIENTO
Cada equipo de trabajo efectuará el siguiente protocolo práctico
1. Preparación de una serie de soluciones estándares de KMn04
2. Construcción del espectro de absorción para el KMn04.
3. Construcción de la curva estándar para el KMn04
4. Análisis de dos muestras problema por uso de la curva estándar.
1. Preparación de la serie de soluciones estándares de KMn04
a. Mida exactamente un volumen de 50 mL de solución estándar de
KMn04 0,1000 Mc, transfiéralo a un balón aforado de 250 mL, diluya con
agua destilada y enrase. Rotule: Solución Madre KMnO4.
b. En un balón aforado de 100 mL deje caer 2,0 mL de solución
madre de KMn04. Diluya con agua destilada y enrase. Rotule: Solución 1.
c. Repita el proceso “b” pero mida 3,0 mL; 3,5 mL; 4,0 mL; 5,0 mL y 5,5 mL.
Rotule: Solución 2, 3, 4, 5 y 6 respectivamente.
d. Calcule la molaridad de carga, molaridad y mg L-1 de Mn de cada una de
las soluciones así preparadas.

61
2. Construcción del espectro de absorción para el KMn04
a. Tome la solución número 3 (preparada con 3,5 mL de solución madre de
KMn04) y determine la transmitancia en función de la longitud de onda
desde 440 nm hasta 620 nm. Haga lecturas para las siguientes longitudes
de onda 440, 460, 480, 500, 520, 525, 530, 535, 540, 560, 580, 600 y 620
nm. Convierta los valores de transmitancia en absorbancia.
b. Trace la curva correspondiente, según el modelo mostrado en la Figura 3
señale la longitud de onda óptima, la cual corresponde, generalmente, aI
máximo de absorbancia.
Figura 3. Modelo de un Espectro de Absorción
3. Construcción de la Curva Estándar
Determine la absorbancia a la serie de soluciones patrones preparada en la
sección 1; la determinación deberá realizarse a la óptima. Trace la curva
correspondiente, ésta deberá concordar con una línea recta (Figura 4).
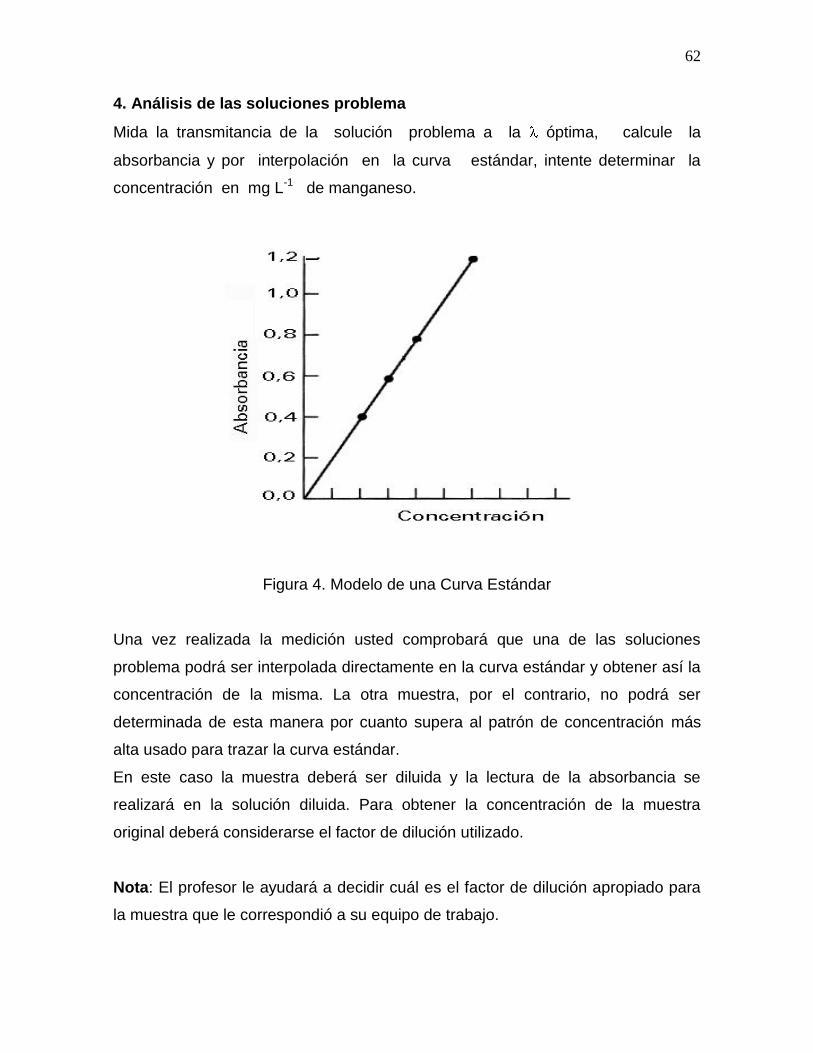
62
4. Análisis de las soluciones problema
Mida la transmitancia de la solución problema a la óptima, calcule la
absorbancia y por interpolación en la curva estándar, intente determinar la
concentración en mg L-1 de manganeso.
Figura 4. Modelo de una Curva Estándar
Una vez realizada la medición usted comprobará que una de las soluciones
problema podrá ser interpolada directamente en la curva estándar y obtener así la
concentración de la misma. La otra muestra, por el contrario, no podrá ser
determinada de esta manera por cuanto supera al patrón de concentración más
alta usado para trazar la curva estándar.
En este caso la muestra deberá ser diluida y la lectura de la absorbancia se
realizará en la solución diluida. Para obtener la concentración de la muestra
original deberá considerarse el factor de dilución utilizado.
Nota: El profesor le ayudará a decidir cuál es el factor de dilución apropiado para
la muestra que le correspondió a su equipo de trabajo.





























