Embed Size (px)
Citation preview

UK-SPAINOrganometallic Chemistry Symposium
17-19 September 2019University of AlcaláAlcalá de HenaresMadrid, Spain
Book of Abstracts

Coordinator and scientific editor: √ Marta E. G. Mosquera √ Juan Carlos Flores √ Gerardo Jiménez √ Cristina García-Yebra √ Jesús Cano √ Carlos Yélamos √ Vanessa Tabernero
Of the texts: their authors
Of this edition: Fundación General de la Universidad de Alcalá c/ Imagen, 1 y 3 28801 Alcalá de Henares (Madrid). SPAIN Tel. +34 91 879 74 30 – Email: [email protected] Website: HYPERLINK "http://www.fgua.es" www.fgua.es
All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, transmitted in any form, or by any means, electronic, mechanical, photocopying, recording or otherwise, without the prior written permission of the publishers.

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 3
General Index
Presentation .................................................................................................. 4
Programme .................................................................................................... 6
Contributions Index ....................................................................................... 11
Lord Lewis Prize Communication................................................................... 19
Invited talks ................................................................................................... 21
Contributions– Oral ............................................................................................................ 37– Flash .......................................................................................................... 53– Poster ........................................................................................................ 62
Authors Index ................................................................................................ 101

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 4
Presentation
The USOCS2019 Organizing Committee welcomes you to the first UK-Spain Organometallic Chemistry Symposium (USOC2019). In this meeting we will celebrate the full diversity of modern organometallic chemistry research from both countries. In addition, the symposium includes a plenary lecture from Professor Luis Oro, recipient of the RSC´s Lord Lewis Prize in 2018. This meeting will provide a forum in which established and early-career inorganic chemists working in Spain or the UK will have the opportunity of presenting their work and enjoying networking opportunities. Spanish and British leading researchers will deliver invited lectures and also, oral, flash and poster contributions have been programmed. We hope that this exciting schedule programmed will promote lively discussions and networking opportunities.
Eva Hevia Richard Layfield Marta E. G. Mosquera Strathclyde University Sussex University Alcalá University
Local Committee
Jesús CanoVanessa Tabernero
Juan Carlos FloresCristina García-Yebra
Marta E. G. Mosquera
Gerardo JiménezCarlos Yélamos

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 5
TUESDAY WEDNESDAY THURSDAY
9:00 IT4: Dominic S. Wright 9:00
9:30 IT5: Concepción Gimeno 9:30 IT11: Debbie Kays
10:00 IT6: Lucía Riera 10:00 IT12: Jesús Campos
10:30 Coffee Break 10:30 Flash presentations
11:00 IT7: Eva Hevia 11:15 Coffee break
11:30 IT8: Miquel Costas 11:45 Tatsumi Ochiai
12:00 Manfred Bochmann Antonio J. Martínez Martínez
Montserrat Diéguez Elena Cuéllar
Christopher J. Whiteoak Sanjiv Prashar
Sonia Infante 12:45 IT13: Charlotte Willians
Miguel Palenzuela
13:00 Lunch break 13:15 IT14: Pedro J. Pérez
13:45 Closing Remarks
15:00 Registration 15:00 IT9: Richard Layfield
15:30 15:30 IT10: María Giménez
16:00 Openning Ceremony 16:00 Joaquín García-Álvarez
16:15 IT1: Robert Mulvey Claire Brodie
Mª Ángeles Fuentes,
16:45 IT2: Ana Carmen Albéniz Alberto Hernán-Gómez
Martí Garçon
17:15 IT3: José Goicoechea 17:00 Coffee Break
17:30 17:00 Poster presentations
17:45 Lord Lewis Award Ceremony:
19:00 Luis Oro 19:00 University Visit
19:15 Welcome Mixer
21:00 21:00 Conference Dinner

Programme

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 7
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Programme
PROGRAMME
TUESDAY, 17 SEPTEMBER
16:00 OPENING CEREMONY
16:15 IT1: Robert Mulvey - University of Strathclyde, UK Alkali Metal Organometallics: Masters of Mediation
16:45 IT2: Ana Carmen Albéniz - Centro de Innovación en Química y Materiales Avanzados Universidad de Valladolid, ES
Faster Palladium-Catalyzed Coupling Reactions of Arenes: Cooperating Ligands and Co-Solvents
17:15 IT3: José Goicoechea - University of Oxford, UK From Chemical Curiosities to Versatile Reagents: Heavy Analogues of the
Cyanate Ion
17:45 LORD LEWIS AWARD CEREMONY PLENARY LECTURE OF THE AWARD-WINNING LOPL1: Luis Oro - Instituto de Síntesis Química y Catálisis Homogénea Universidad de Zaragoza - CSIC, ES From Organometallic Complexes to Homogeneous Catalysis: Forty years of
Platinum Group Metal Chemistry
19:15 WELCOME MIXER
WEDNESDAY, 18 SEPTEMBER
09:00 IT4: Dominic S. Wright - University of Cambridge, UK Main Group Macrocycles; Design Concepts and New Host-Guest Chemistry
09:30 IT5: Concepción Gimeno - Instituto de Síntesis Química y Catálisis Homogénea Universidad de Zaragoza, ES Synthesis and Applications of Novel Organogold Complexes

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 8
10:00 IT6: Lucía Riera – CSIC Universidad de Oviedo, ES Dearomatization and Ring-Opening of 2,2’-Bipyridine and
1,10-Phenanthroline Ligands
10:30 Coffee break
11:00 IT7: Eva Hevia - University of Strathclyde, UK Tailoring Organozinc Reagents for Chemical Cooperativity
11:30 IT8: Miquel Costas - Institut de Química Computacional i Catàlisi Universitat de Girona, ES
FeV complexes of relevance in enzymology and organic synthesis
12:00 OP1: Manfred Bochmann - University of East Anglia, UK Carbene complexes of Cu, Ag, Au as OLED photoemitters OP2: Montserrat Diéguez - Universitat Rovira i Virgili, ES Improved generations of catalysts for the synthesis of elusive chiral synthons
OP3: Christopher J. Whiteoak - Sheffield Hallam University, UK Access to Unusual Heterocyclic Compounds Utilizing a Key Cobalt-Catalyzed
C-H Functionalization Approach OP4: Sonia Infante - IMDEA Nanociencia, ES Organometallic tethered compounds with a coordinative bound capable of
hijacking/releasing a proton OP5: Miguel Palenzuela - Universidad de Alcalá, ES
Combined catalytic action of homometallic and heterometallic aluminum species to generate polymeric nanoparticles
13:00 Lunch break
15:00 IT9: Richard Layfield - University of Sussex, UK High-Temperature Lanthanide Single-Molecule Magnets
15:30 IT10: María Giménez - Centro Singular de Investigación en Química Biológica y Materiales Moleculares
Universidad de Santiago de Compostela, ES Developing sustainable carbon hybrid materials

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 9
16:00 OP6: Joaquín García-Álvarez - Universidad de Oviedo, ES Synergistic combination of metal-catalyzed organic reactions with enzymes or
organolithium reagents (RLi) in water and Deep Eutectic Solvents OP7: Claire Brodie - Durham University, UK Investigating the Reduction and Reactivity Chemistry of Cobalt Diphosphine
Complexes OP8: Mª Ángeles Fuentes - University of Oxford, UK Phosphine/borane frustrated Lewis pairs chemistry supported by xanthene
scaffolds OP9: Alberto Hernán-Gómez - Universitat de Girona, ES Catalytic Alkane Functionalization Via Iron-Carbene Insertion Reaction OP10: Martí Garçon - Imperial College London, UK
Unusual Bonds between Magnesium and Transition Metals: New Opportunities in Catalysis
17:00 Coffee break
17:30 Poster Session
19:00 Visit to the historic building of the University of Alcalá
21:00 Conference dinner Parador
THURSDAY, 19 SEPTEMBER
09:30 IT11: Debbie Kays - University of Nottingham, UK Small molecule activation and catalysis using coordinatively unsaturated
complexes
10:00 IT12: Jesús Campos - Instituto de Investigaciones Químicas Universidad de Sevilla - CSIC, ES Frustration versus Interaction in Bimetallic Systems
10:30 FLASH PRESENTATIONS FP1: Verónica Conejo-Rodríguez - Universidad de Valladolid, ES d8···d10 RhI···AuI Interactions in Rh 2,6-Xylylisocyanide Complexes with Au(CN)2
–: Bond Analysis and Crystal Effects
FP2: Anindita Chakraborty - University of Sussex, UK Lanthanide Cyclobutadienyl Sandwich Complexes FP3: Alice Johnson - University of Oxford, UK Bis(imino)pyridine Rhodium Complexes for Amine-Borane
Dehydropolymerisation

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 10
FP4: Marta Navarro - Universidad Rey Juan Carlos, ES Bimetallic Aluminium Scorpionates For Coupling of CO2 And Epoxides Into
Cyclic Carbonates Under Mild Conditions FP5: Matthias Dirk Boege – CSIC - Universidad de Zaragoza, ES
Structure Property Relationship of Materials with Gold (I)-Gold (III) Interactions FP6: Elena de la Torre Rubio - Universidad de Alcalá, ES Novel arene Ru (II) compounds with N-phenanthroline glycosylamine ligands as
potential anticancer agents FP7: Ana Carrasco - IMDEA Nanociencia, ES Potent Half-Sandwich Iridium (III) Complexes as Mitochondria-Targeted
Anticancer Drugs FP8: Francisco Villalba - Universidad de Valladolid, ES Coupling of Reactive Carbene Precursors: Isolation of Intermediate Palladium
Complexes FP9: Alberto Abéngozar - Universidad de Alcalá, ES
BN-Arenes: Synthesis, Reactivity and Properties
11:15 Coffee break
11:45 OP11: Tatsumi Ochiai - University of Edimburg, UK Homoleptic Trigonal Planar Uranium Complex Induced by High Pressure OP12: Antonio J. Martínez Martínez - Universidad de Huelva, ES Catalytic Upgrading of Light Hydrocarbons: Solid–State Molecular
Organometallic Nanoreactors OP13: Elena Cuéllar - Universidad de Valladolid, ES Deprotonation studies, luminescent properties and catalytic CO2 reduction
activity of Re(CO)3 complexes with pyrazole and (3-(2-pyridyl)pyrazole) OP14: Sanjiv Prashar – Universidad Rey Juan Carlos, ES Spanish Metallocenes with a British accent. Titanocene Functionalized
Mesoporous Silica Nanoparticles in the Fight against Cancer 12:45 IT13: Charlotte Willans - University of Leeds, UK Electrochemical Generation of Catalysts using Batch and Flow Technology
13:15 IT14: Pedro J. Pérez - Centro de Investigación en Química Sostenible Universidad de Huelva, ES
The reactivity of alkanes towards metal-carbene electrophiles
13:45 CLOSING REMARKS

ContributionsIndex

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 12
Lord Lewis Award
From Organometallic Complexes to Homogeneous Catalysis: Forty years of Platinum Group Metal Chemistry ................................................................................................................................ 20LOPL1: Luis ORO
Invited Talks
Alkali Metal Organometallics: Masters of Mediation ...................................................................... 22IT1: Robert MULVEY
Faster Palladium-Catalyzed Coupling Reactions of Arenes: Cooperating Ligands and Co-Solvents ....................................................................................................................................... 23IT2: Ana Carmen ALBÉNIZ
From Chemical Curiosities to Versatile Reagents: Heavy Analogues of the Cyanate Ion ............... 24IT3: José GOICOECHEA
Main Group Macrocycles; Design Concepts and New Host-Guest Chemistry ................................ 25IT4: Dominic S. WRIGHT
Synthesis and Applications of Novel Organogold Complexes .......................................................... 26IT5: Concepción GIMENO
Dearomatization and Ring-Opening of 2,2’-Bipyridine and 1,10-Phenanthroline Ligands ............ 27IT6: Lucía RIERA
Tailoring Organozinc Reagents for Chemical Cooperativity ........................................................... 28IT7: Eva HEVIA
FeV complexes of relevance in enzymology and organic synthesis ................................................... 29IT8: Miquel COSTAS
High-Temperature Lanthanide Single-Molecule Magnets ................................................................ 30IT9: Richard LAYFIELD
Developing sustainable carbon hybrid materials ............................................................................. 31IT0: María GIMÉNEZ
Small molecule activation and catalysis using coordinatively unsaturated complexes .................... 32IT11: Debbie KAYS
Frustration versus Interaction in Bimetallic Systems ....................................................................... 33IT12: Jesús CAMPOS
Electrochemical Generation of Catalysts using Batch and Flow Technology .................................. 34IT13: Charlotte WILLANS
The reactivity of alkanes towards metal-carbene electrophiles ........................................................ 35IT14: Pedro J. PÉREZ

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 13
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Oral Presentations
Carbene complexes of Cu, Ag, Au as OLED photoemitters .............................................................. 38OP1: Manfred BOCHMANN, A. S. ROMANOV, D. CREDINGTON, P. J. CONAGHAN, M. LINNOLAHTI
Improved generations of catalysts for the synthesis of elusive chiral synthons ................................ 39OP2: Montserrat DIÉGUEZ, Maria BIOSCA, Feliu MASERAS, Miquel A. PERICÀS, Oscar PÀMIES
Access to Unusual Heterocyclic Compounds Utilizing a Key Cobalt-Catalyzed C-H Functionalization Approach .............................................................................................................. 40OP3: Christopher J. WHITEOAK, Paula G. CHIRILA, Alex HAMILTON
Organometallic tethered compounds with a coordinative bound capable of hijacking/releasing a proton ............................................................................................................................................. 41OP4: Sonia INFANTE-TADEO, Ana M. PIZARRO
Combined catalytic action of homometallic and heterometallic aluminum species to generate polymeric nanoparticles .................................................................................................................... 42OP5: M. Teresa MUÑOZ, Tomás CUENCA, Marta E.G. MOSQUERA, Miguel PALENZUELA
Synergistic combination of metal-catalyzed organic reactions with enzymes or organolithium reagents (RLi) in water and Deep Eutectic Solvents ........................................................................ 43OP6: Joaquín GARCÍA-ÁLVAREZ, María Jesús RODRÍGUEZ-ÁLVAREZ, Luciana CICCO, Vito CAPRIATI, Nicolas RÍOS-LOMBARDÍA, Javier GONZÁLEZ-SABÍN
Investigating the reduction and reactivity chemistry of cobalt diphosphine complexes ................... 44OP7: Claire N. BRODIE, Martin J. HANTON, Andrei S. BATSANOV, Philip W. DYER
Phosphine/borane frustrated Lewis pairs chemistry supported by xanthene scaffolds .................... 45OP8: M. Ángeles FUENTES, Simon ALDRIDGE
Catalytic Alkane Functionalization Via Iron-Carbene Insertion Reaction ....................................... 46OP9: Alberto HERNÁN-GÓMEZ, Mónica RODRÍGUEZ, Teodor PARELLA, Miquel COSTAS
Unusual Bonds between Magnesium and Transition Metals: New Opportunities in Catalysis ....... 47OP10: Martí GARÇON, Andrew J. P. WHITE, Mark R. CRIMMIN
Homoleptic Trigonal Planar Uranium Complex Induced by High Pressure .................................... 48OP11: Tatsumi OCHIAI, Amy N. PRICE, Jacob J. SHEPHARD, Victoria BERRYMAN, Polly L. ARNOLD, Simon PARSONS, Nikolas KALTSOYANNIS
Catalytic Upgrading of Light Hydrocarbons: Solid–State Molecular Organometallic Nanoreactors ..................................................................................................................................... 49OP12: Antonio J. MARTÍNEZ-MARTÍNEZ, Stuart A. MACGREGOR, Andrew S. WELLER

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 14
Deprotonation studies, luminescent properties and catalytic CO2 reduction activity of Re (CO)3 complexes with pyrazole and (3-(2-pyridyl) pyrazole) ........................................................... 50OP13: Beatriz MERILLAS, Elena CUÉLLAR, Alberto DIEZ-VARGA, Miryam ASENSIO-BARTOLOMÉ, Gabriel GARCÍA-HERBOSA, Tomás TORROBA, José M. MARTÍN-ALVAREZ, Daniel MIGUEL, Fernando VILLAFAÑE
Spanish Metallocenes with a British accent. Titanocene Functionalized Mesoporous Silica Nanoparticles in the Fight against Cancer ....................................................................................... 51OP14: Álvaro SERRANO-PINDADO, Diana DÍAZ-GARCÍA, Miguel DÍAZ-SÁNCHEZ, Irene MENA-PALOMO, Santiago GÓMEZ-RUIZ, Sanjiv PRASHAR
Flash Presentations
d8···d10 RhI···AuI Interactions in Rh 2,6-Xylylisocyanide Complexes with Au(CN)2–:
Bond Analysis and Crystal Effects .................................................................................................... 53FP1: Verónica CONEJO-RODRÍGUEZ, Marconi N. PEÑAS-DEFRUTOS, Pablo ESPINET
Lanthanide Cyclobutadienyl Sandwich Complexes .......................................................................... 54FP2: Anindita Chakraborty, Richard A. Layfield
Bis(imino)pyridine Rhodium Complexes for Amine-Borane Dehydropolymerisation ...................... 55FP3: Alice JOHNSON, Antonio J. MARTÍNEZ-MARTÍNEZ, Andrew S. WELLER
Bimetallic aluminium scorpionates for coupling of CO2 and epoxides into cyclic carbonates under mild conditions ....................................................................................................................... 56FP4: Marta NAVARRO, Luis F. SÁNCHEZ-BARBA, Andrés GARCÉS, Juan FERNÁNDEZ-BAEZA, Agustín LARA-SÁNCHEZ
Structure Property Relationship of Materials with Gold(I)-Gold (III) Interactions ......................... 57FP5: Matthias DIRK BOEGE, Juergen HECK, M. Concepción GIMENO
Novel arene Ru (II) compounds with N-phenanthroline glycosylamine ligands as potencial anticancer agents .............................................................................................................................. 58FP6: Elena de la TORRE-RUBIO, Isabel de la CUEVA-ALIQUE, Lourdes GUDE, María-Selma ARIAS-PÉREZ, Eva ROYO
Potent Half-Sandwich Iridium (III) Complexes as Mitochondria-Targeted Anticancer Drugs ........ 59FP7: Ana C. CARRASCO, José Javier CONESA, Vanessa RODRÍGUEZ-FANJUL, Yang YANG, José L. CARRASCOSA, Peter CLOETENS, Eva PEREIRO, Ana M. PIZARRO
Coupling of Reactive Carbene Precursors: Isolation of Intermediate Palladium Complexes ......... 60FP8: Francisco VILLALBA, Ana C. ALBÉNIZ
BN-Arenes: Synthesis, Reactivity and Properties ............................................................................. 61FP9: Alberto ABENGÓZAR, Isabel VALENCIA, Patricia GARCÍA-GARCÍA, David SUCUNZA, Miguel Ángel FERNÁNDEZ-GONZÁLEZ, Luis Manuel FRUTOS, Antonio SALGADO, Adrián PÉREZ-REDONDO, Juan J. VAQUERO

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 15
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Poster Presentations
Heterobimetallic Lanthanide Isocarbonyl Complexes as Single-Molecule Magnets ....................... 63PP1: Richard COLLINS, Jinkui TANG, Richard A. LAYFIELD
Unusual ligand rearrangement of N-phosphinoguanidinato compounds ......................................... 64PP2: Fernando CARRILLO-HERMOSILLA, Estefanía HUERGO, Rafael FERNÁNDEZ-GALÁN, Alberto RAMOS, Antonio ANTIÑOLO, Antonio RODRÍGUEZ-DIÉGUEZ, Daniel GARCÍA-VIVÓ
Decarbonylation Processes in k3 -(N^C^C) Gold (III) Carboxylates ............................................... 65PP3: Estíbaliz MERINO, Hélène BEUCHER, Alexandre GENOUX, Thomas FOX Cristina NEVADO
Bipy and phen ring-opening at Re(I) carbonyl complexes ............................................................... 66PP4: Purificación CAÑADAS, Julio PÉREZ, Lucía RIERA
Reactivity of Phosphine-Stabilized Silylene Rhodium Complex ....................................................... 67PP5: N. ALMENARA, J. I. MIRANDA, A. RODRÍGUEZ-DIÉGUEZ, M. A. GARRALDA, M. A. HUERTOS
Novel Iridapyrazole based complexes, reactivity and catalytic activity ........................................... 68PP6: Itxaso BUSTOS, María Ángeles GARRALDA, Claudio MENDICUTE-FIERRO
Synthesis, structure, and properties of monopentamethylcyclopentadienyltitanium (III) dihalide complexes .......................................................................................................................................... 69PP7: Estefanía del HORNO, Reyes JIMÉNEZ-APARICIO, Miguel MENA, Adrián PÉREZ-REDONDO, José Luis PRIEGO, Carlos YÉLAMOS
Ligands with a pyridone or amido functionality: Assessment of their cooperating ability in the Pd-catalyzed direct arylation of arenes ............................................................................................ 70PP8: Cintya PINILLA, Vanesa SALAMANCA, Francisco VILLALBA, Ana C. ALBÉNIZ
Nano Base Metal Catalysts: Synthesis and Characterisation .......................................................... 71PP9: Alana SMITH, Phil DYER, Simon BEAUMONT, Xavier BAUCHEREL, Leon VAN DE WATER
Graphene-based Heterogeneous Cu-catalysts in Borylation Reactions ........................................... 72PP10: M. FRANCO FERNÁNDEZ, M. TORTOSA, M. B. CID
Synthesis and Characterization of Cyclopentadienyl Sulfur Niobium Complexes ........................... 73PP11: Elena ÁLVAREZ-RUIZ, Manuel GÓMEZ, Cristina HERNÁNDEZ-PRIETO, Avelino MARTÍN, Miguel MENA, Cristina SANTAMARÍA
Digold C-C Cross Coupling Reactions: Mechanistic Insights ......................................................... 74PP12: Juan MIRANDA-PIZARRO, Jesús CAMPOS, Ernesto CARMONA

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 16
The interaction of the light with ortho-palladated azlactones: different pathways .......................... 75PP13: Esteban P. URRIOLABEITIA, Carlos CATIVIELA, Pablo SÁNCHEZ, Alexandra POP, Cristian SILVESTRU
Synthesis of bis (amino acids) with cyclobutane skeleton through selective [2+2]-photocycloaddition of oxazolones and thiazolones ............................................................... 76PP14: Sonia SIERRA, David DALMAU, Alexandra POP, Cristian SILVESTRU, Carlos CATIVIELA, Esteban P. URRIOLABEITIA
Cyclobutadienyl Chemistry of the Rare-Earth Elements .................................................................. 77PP15: James DURRANT, Benjamin M. DAY, Richard A. LAYFIELD
Gold-catalyzed regiodivergent cyclization of alkynylcyclobutanes .................................................. 78PP16: M. Soledad GARRE, David SUCUNZA, Estibaliz MERINO, Enrique AGUILAR, Patricia GARCÍA-GARCÍA, Juan J. VAQUERO
New Advances in Rhodium-Catalyzed Olefin Hydrophosphination .................................................. 79PP17: Victor VARELA, Ana M. GEER, José A. LÓPEZ, Miguel A. CIRIANO, Cristina TEJEL
Stereoselective synthesis of versatile trifluoromethyl substituted borocyclopropanes ...................... 80PP18: Julia ALTAREJOS, David SUCUNZA, Ana CABALLERO, Pedro J. PÉREZ, Juan J. VAQUERO, Javier CARRERAS
Outher-Sphere Alkoxylation of Olefins promoted by Inner-Sphere Oxygen Activation .................... 81PP19: Cristina TEJEL, Paula ABRIL, M- Pilar del RÍO, Agustí LLEDÓS, José A. LÓPEZ, Miguel A. CIRIANO
Uranium complexes with bulky tetraaryloxide ligands .................................................................... 82PP20: Rory P. KELLY, Tatsumi OCHIAI, Francis Y. T. LAM, Megan L. SEYMOUR, Jordann A. L. WELLS, Laurent MARON, Polly L. ARNOLD
Magnetic Bistability in Coordination Nanohoops ............................................................................ 83PP21: María José HERAS OJEA, Lewis C. H. MADDOCK, Benjamin M. DAY, Fu-Sheng GUO, Jeff M. VAN RADEN, Daniel PIVIDORI, Jordi CIRERA, Ramesh JASTI, Karsten MEYER, Eliseo RUIZ, Richard A. LAYFIELD
Graphene-Supported Gold Nanoparticles Capped with NHC Ligands: A New Method to Synthesize an Active and Recyclable Catalysts for the Hydration of Alkynes ................................. 84PP22: David VENTURA-ESPINOSA, Santiago MARTÍN, José A. MATA
Coordination Versatility of Bidentate COC and Tridentate CNC Ligands Containing 1,2,3-Triazol-5-ylidenes .................................................................................................................... 85PP23: Gregorio GUISADO-BARRIOS, Lewis C. TOLLEY, Daniela I. BEZUIDENHOUT

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 17
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
β(Z)-vinylsilanes produced by hybrid graphene-based alkyne hydrosilylation NHC-rhodium catalyst .............................................................................................................................................. 86PP24: Beatriz SÁNCHEZ-PAGE, M. Victoria JIMÉNEZ, Jesús J. PÉREZ-TORRENTE, Patricia ÁLVAREZ, Javier BLASCO
An Organometallic Approach to Magnesium Aluminate Complexes for Rechargeable Battery Electrolytes ........................................................................................................................................ 87PP25: Stuart D. ROBERTSON, Scott A. BROWN
Green large-scale production of piezoelectric bioplastics ................................................................ 88PP26: Valentina SESSINI, Marta E. G. MOSQUERA
Imidazolium-2-Amidinates: role as ligands for main group metals and in C-Cl bond activation processes ........................................................................................................................................... 89PP27: David SÁNCHEZ-ROA, Marta E. G. MOSQUERA, Juan CÁMPORA, Tomás CUENCA, María FERNÁNDEZ-MILLÁN
Titanium (IV)-modified silica@magnetic nanocomposites. Synthesis and application as efficient nanocatalysts for sulfide oxidation with H2O2 .................................................................................. 90PP28: Joan MARTÍN VINUEZA, Gerardo JIMÉNEZ, Vanessa TABERNERO
New Heterometallic aluminium alkali metal compounds with oximate ligands ............................... 91PP29: Jesús Damián BURGOA, Tomás CUENCA, Marta E.G. MOSQUERA
Functional model of catecholase based on Schiff bases ................................................................... 92PP30: Aida Jaafar ARIAS, Aaron Terán MORE, Ana E. SÁNCHEZ PELÁEZ, Ángel GUTIÉRREZ ALONSO, María del Carmen TORRALBA MARTÍNEZ
Multifunctional molecular materials combining optical and liquid crystal properties .................... 93PP31: Brais GONZÁLEZ, Cristián CUERVA, José A. CAMPO, Mercedes CANO, Carlos LODEIRO
Monocatenar and dicatenar b-diketonylpyridinium salts: ionic liquid crystals .............................. 94PP32: M. Eugenia AZNÁREZ, Arturo PAREJA, Cristián CUERVA, José A. CAMPO, Mercedes CANO, Carlos LODEIRO
Phytohormones as bridging ligands in diruthenium complexes ....................................................... 95PP33: Isabel COLOMA, Miguel CORTIJO, Inés FERNÁNDEZ-SÁNCHEZ, Santiago HERRERO, Reyes JIMÉNEZ-APARICIO, José L. PRIEGO
New insights in the electronic structure of formamidinate-supported (Ru2 )5+ compounds by
Raman resonant spectroscopy ........................................................................................................... 96PP34: A. INCHAUSTI, A. LOBATO, S. HERRERO, R. GONZÁLEZ-PRIETO, R. JIMÉNEZ-APARICIO, M. TARAVILLO, V. G. BAONZA

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 18
[Ru2Cl(DArF)x(O2CMe)4-x] compounds as starting materials for macromolecules and RNA probes ....................................................................................................................................... 97PP35: A. de MARCOS GALÁN, A. INCHAUSTI, A. MANCHADO-PARRA, R. GONZÁLEZ-PRIETO, S. HERRERO, R. JIMÉNEZ-APARICIO
NMR studies of diruthenium-protein compounds ............................................................................. 98PP36: Aarón TERÁN MORE, Jose Manuel PÉREZ-CAÑADILLAS, Santiago HERRERO, Reyes JIMÉNEZ-APARICIO
Coordination capacity of thymine-1-acetate towards diruthenium complexes with open-paddlewheel structure .............................................................................................................. 99PP37: Miguel CORTIJO, Inés FERNÁNDEZ-SÁNCHEZ, Santiago HERRERO, Reyes JIMÉNEZ-APARICIO, Aarón TERÁN
Remote Functionalized Bidentate Phosphine-based Ruthenium(II) Catalysts for Ethanol/Methanol Upgrading to Advanced Biofuels ...................................................................................................... 100PP38: Folasade J. SAMA, Richard L. WINGAD, Duncan F. WASS

Lord Lewis Prize

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 20
LOPL1From Organometallic Complexes to Homogeneous Catalysis:
Forty years of Platinum Group Metal ChemistryLuis A. Oro
Department of Inorganic Chemistry-ISQCH, University of Zaragoza-CSIC, 50009-Zaragoza, Spain E-mail: [email protected]
An overview on a selection of research activities initiated long time ago at Zaragoza, after returning from a postdoctoral stay at Cambridge University under the supervision of Professor the Lord Lewis, will be presented. It was during that time that I became interested in organometallic chemistry. To receive the 2018 Lord Lewis Prize, at the first UK-SPAIN Organometallic Symposium, is an extraordinary privilege and honor.
During more than forty years of research, our group has synthesized hundreds of new organometallic and coordination compounds of rhodium, iridium, ruthenium and osmium, and studied their application as homogeneous catalysts on hydrogenation, hydrogen transfer, hydroformylation, hydrofunctionalization, C-C coupling and C-H activation, with particular emphasis on the understanding of the reaction mechanisms involving inner and outer-sphere catalysis. Carbon dioxide and ammonia activation by iridium complexes has also been studied. In addition, molecular architectures, cluster chemistry and polymetallic reactivity, particularly of rhodium and iridium complexes have been also subject of our interest.
In particular, our studies on the mechanism of a set of rhodium complexes with N-heterocyclic carbene (NHC) ligands in two specific atom-economy homogeneous reactions, vinyl selective H/D exchange and alkyne hydrothiolation, will be discussed in detail. The high steric hindrance and powerful electron-donor capacity of the bulky NHC´s used, along with ancillary donor ligands, seems to be determinant to get selective transformations and to facilitate valuable information about the mechanism of the mentioned reactions.
References[1] M Iglesias, F. J. Fernández-Alvarez, L. A. Oro, Coord. Chem. Rev., 2019, 386, 240-266.[2] M. Iglesias, L.A. Oro, Chem. Soc. Rev., 2018, 47, 2772-2808.[3] A. Di Giuseppe, R. Castarlenas, L.A. Oro, Top. Organomet. Chem., Springer-Verlag, 2018, 61, 31-67. [4] L. Rubio-Pérez, M. Iglesias, J. Munárriz, V. Polo, V. Passarelli, J.J. Pérez-Torrente, L.A. Oro, Chemical Science, 2017, 8, 4811-4822.[5] M. Iglesias, E. Sola, L.A. Oro, Top. Organomet. Chem., Springer-Verlag, 2016, 59, 31-58.[6] R. Castarlenas, A. Di Giuseppe, J.J. Pérez-Torrente, L.A. Oro, Angew. Chem. Int. Ed., 2013, 52, 211-222.[7] A. Di Giuseppe, R. Castarlenas, J.J. Pérez-Torrente, M. Crucianelli, V. Polo, R. Sancho, F.J. Lahoz, L.A. Oro, J. Am. Chem. Soc., 2012, 134, 8171-8183.[8] L.A. Oro, C. Claver, Ed., Iridium Complexes in Organic Synthesis, 424 pgs, Wiley-VCH, Wenheim. 2009.[9] C. Tejel, M.A. Ciriano, B.E. Villarroya, J.A. López, F.J. Lahoz and L.A. Oro, Angew. Chem. Int. Ed., 2003, 42, 529-532.[10] D.Carmona, M.P. Lamata, L.A. Oro, Coord. Chem. Rev., 2000, 200, 717-772.[10] Braunstein, L.A. Oro, P.R. Raithby, Ed. Metal Clusters in Chemistry, 1798 pgs, 3 volumes, Wiley-VCH, Wenheim. 1999.[11] M.A. Esteruelas, L.A. Oro, Chem. Rev., 1998, 98, 577-588

Invited talks

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 22
IT1Alkali Metal Organometallics: Masters of Mediation
Robert E. MulveyDepartment of Pure & Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow G1 1XL,
Scotland, UK E-mail: [email protected]
Alkali metal organometallics have played a seminal role in the development of chemistry for 100 years most often as lithium alkyl or lithium amide reagents. Brilliant bases (e.g., deprotonating C−H bonds) and nucleophiles (e.g., adding to unsaturated molecules) in organic synthesis, and transfer agents in transition metal and lanthanide/actinide chemistry (e.g., delivering ligands to other metals), these organolithium compounds and to a lesser extent the organic derivatives of the other common alkali metals sodium and potassium have proved reagents par excellence in both academia and technology.
Recently alkali metal mediation has taken on a new form in which an alkali metal plays a secondary but essential role in transformations carried out by bimetallic formulations. Synergistic communication with the primary metal partner (e.g., Mg, Zn, Al) can lead to reactivities and selectivities outside the scope of the conventional single metal systems. Showcasing some of our recent advances in this area, this presentation will discuss examples of both types of alkali metal mediation. In this International Year of the Periodic Table (IYPT), we ponder whether such bimetallic formulations could eventually realise a Pairiodic Table of Element Pairs (see Figure).
References[1] S. D. Robertson, M Uzelac, R. E. Mulvey, Chem. Rev., 2019, DOI:10.1021/acs.chemrev.9b00047.[2] V. A. Pollard, S. A. Orr, R. McLellan, A. R. Kennedy, E. Hevia, R.E. Mulvey, Chem. Commun., 2018, 54, 1233-1236.[3] V. A. Pollard, M. Ángeles Fuentes, A. R. Kennedy, R. McLellan, R. E. Mulvey, Angew. Chem. Int. Ed., 2018, 57, 10651-10655.[4] L. E. Lemmerz, R. McLellan, N. R. Judge, A. R. Kennedy, S. A. Orr, M. Uzelac, E. Hevia, S. D. Robertson, J. Okuda, R. E. Mulvey, Chem. Eur. J., 2018, 24, 9940-9948.

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 23
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
IT2Faster Palladium-Catalyzed Coupling Reactions of Arenes: Cooperating
Ligands and co-Solvents
Ana C. AlbénizIU CINQUIMA/Química Inorgánica. Universidad de Valladolid. 47071 Valladolid (Spain).
E-mail: [email protected]
Palladium-catalyzed C-C coupling reactions that directly functionalize C-H bonds have emerged as a powerful tool for C-C bond formation.[1] These reactions do not require a previous functionalization of every reaction partner and therefore conform to the principles of green chemistry, standing in a good position towards a more sustainable chemical synthesis. For this reason, the development of more efficient catalysts and conditions for these reactions is important, and their rational design can only be achieved by understanding the mechanistic aspects of these processes.
We have studied the direct arylation of arenes using [Pd(bipy-6-OH)(C6F5)Br] (1, bipy-6-OH = [2,2’-bipyridin]-6(1H)-one) as catalyst (Figure 1).[2] Our mechanistic studies unequivocally show that the chelating N-donor ligand bipy-6-OH, which has a basic keto group as substituent, facilitates the C-H activation step, which is turnover-limiting in the catalytic reaction. Experimental results and computational studies show this assistance occurs by a concerted mechanism in the metal coordination sphere, leading to a dramatic shortening of reaction times (Figure 1). The importance of choosing the reaction conditions, specially the solvent mixture for the less coordinating arenes will be discussed in detail.
Acknowledgements: Financial support of the Spanish MINECO (CTQ2016-80913-P) and the Junta de Castilla y León (VA062G18, VA051P17) is gratefully acknowledged.
References[1] C-H Activation, monographic issue, Chem. Rev. 2017, 117, Iss.13.[2] Salamanca, V.; Toledo, A.; Albéniz, A. C. J. Am. Chem. Soc. 2018, 140, 17851-17856

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 24
IT3From Chemical Curiosities to Versatile Reagents: Heavy Analogues
of the Cyanate Ion
Jose M. GoicoecheaDepartment of Chemistry, University of Oxford
E-mail: [email protected]
The 2-phosphaethynolate anion (PCO−; the phosphorus-containing analogue of cyanate) was first isolated by Becker and co-workers as a lithium salt in 1992.[1] Due to difficulties associated with its manipulation, the chemistry of this remarkable species laid dormant for decades. The report of a high yielding, multi-gram synthesis of [Na(dioxane)x][PCO] in 2014 rekindled the interest in this fundamental ion.[2] Since then, the reactivity of PCO− and its use in decarbonylative and deoxygenative processes has been extensively explored.[3] Following Becker’s original report on the isolation of PCO–, heavier analogues have also become synthetically accessible (PnCCh− where Pn = P, As; Ch = O, S, Se).[4, 5] These ions are rare insomuch as they contain highly reactive pnictogen–carbon multiple bonds, yet can be manipulated with ease due to their negative charges, which preclude common decomposition pathways (e.g. oligomerization), associated with neutral valence-isoelectronic species. This talk will survey recent studies on this family of anions paying particular attention to their structure and bonding and their use in the synthesis of novel molecular compounds, clusters and materials.
References:[1] G. Becker, W. Schwarz, N. Seidler, M. Westerhausen, Z. Anorg. Allg. Chem. 1992, 612, 72–82.[2] D. Heift, Z. Benkő, H. Grützmacher, Dalton Trans. 2014, 43, 831–840.[3] J. M. Goicoechea, H. Grützmacher, Angew. Chem. Int. Ed. 2018, 57, 16968–16994.[4] A. Hinz, J. M. Goicoechea, Angew. Chem. Int. Ed. 2016, 55, 8536–8541.[5] F. Tambornino, A. Hinz; R. Köppe, J. M. Goicoechea Angew. Chem. Int. Ed. 2018, 57, 8230−8234.

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 25
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
IT4Main Group Macrocycles; Design Concepts and New Host-Guest Chemistry
Dominic S. WrightCambridge University
E-mail: [email protected]
Making large inorganic molecules in systematic ways that parallel the type of step-by-step synthetic approaches used in organic chemistry is a hard thing to do for many reasons. One particular problem when you step out of the comfort zone of carbon in the p-block are the lower bond energies encountered and the development of polarity, which lead to lower thermodynamic and kinetic stability. The P-N bond (famously, isoelectronic with C-C), however, has a high bond energy and a relatively low bond polarity and can therefore be employed as a basis for molecular design in the macrocyclic dimension, using simple synthetic strategies based on electrophilic and nucleophilic building blocks. This approach provides access to a range of new macrocycles which possess completely inorganic backbones, with a variety of (H-bond donor, or Lewis base donor) functionalities. This lecture details the development of this area, the types of macrocycle that can be formed and their unusual coordination chemistry.
EE'
EE'
R
RY
E''E'
E''E'
R'
R'
EE'
EE'
R
R
+ +
Y
E''E'
E''E'
R'
R'Y
__+
EE'
EE'
R
RHY
E''E'
E''E'
R'
R'YH
+X X
SynthonsMacrocycle
Electrophilic Nucleophilic
Al-Al N-N P-P B-B C-CSi-Si
N-O
As-O P-O
C-O
Si-OB-O
Al-O
P-N
Si-NB-N
Al-N
100 200 300 400 500 600
100
200
300
Total Bond Energy (k J mol-1)
Ioni
cC
ontri
butio
nto
the
Bond
Ener
gy(k
Jm
ol-1
)
C-N
References[1] J. Plajer, R. García-Rodríguez, C. G. M. Benson, P. D. Matthews, A. D. Bond, S. Singh, L. H. Gade, D. S. Wright, Angew. Chem., 2017, 56, 9087.[2] H.-C. Niu, A. J. Plajer, R. Garcia-Rodriguez, S. Singh, D. S. Wright, Chem. Eur. J., 2018, 24, 3073.[3] A. J. Plajer, Felix J. Rizzuto, H.-C. Niu, S. Lee, J. M. Goodman, D. S. Wright, Angew. Chem. Int. Ed., in press.

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 26
IT5Synthesis and Applications of Novel Organogold Complexes
M. Concepción GimenoDepartamento de Química Inorgánica. Instituto de Síntesis Química y Catálisis Homogénea (ISQCH),
CSIC-Universidad de Zaragoza, E-50009, Spain E-mail: [email protected]
Interest in the synthesis of new organogold complexes has revived in the last years because of the increasing applications of gold complexes, particularly within the areas of homogenous catalysis, material chemistry or medicine. Many novel organogold compounds have been synthesised in the search for new and more active catalysts, for a better understanding of the catalytic mechanism, including the isolation of key catalytic intermediates, or in the hunt of complexes with unique structure and properties. Several sort of ligands, which provide high stability to the final compounds, such as alkynyl or N-heterocyclic carbenes have helped to the development of this field. Moreover, other new types of ligands have been incorporated to the chemistry of this metal.
Alkynyl or propargyl ligands are excellent building blocks that offer the possibility for the synthesis of unusual gold complexes from a structural and bonding point of view, or in the construction of polymetallic complexes with unique luminescent properties [1]. The ease functionalisation of imidazolium salts with different fragments allows tuning the properties of NHC gold complexes. Thus, whereas the use of bulky substituents may confer great stability and rigidity for the use of the NHC gold complexes in catalysis [2], the presence of fluorophore groups could led to luminescence properties with potential applications in the fabrication of OLEDs [3], or the introduction of directing or water soluble groups could made the compounds of interest for biological applications [4].
References[1] N. J. Long, C. K. Williams, Angew Chem Int Ed. 2003, 42, 2586-2617.[2] N. Marion, S. P. Nolan, Chem. Soc. Rev. 2008, 37, 1776-1782.[3] R. Visbal, M. C. Gimeno, Chem. Soc. Rev. 2014, 43, 3551-3574.[4] M. Mora, M. C. Gimeno and R. Visbal, Chem. Soc. Rev. 2019, 48, 447-462.

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 27
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
IT6Dearomatization and Ring-Opening of 2,2’-Bipyridine and
1,10-Phenanthroline Ligands
Lucía Riera,1 Julio Pérez1,2 1Centro de Investigación en Nanomateriales y Nanotecnología (CINN), CSIC-Universidad de
Oviedo-Principado de Asturias, Avda de la Vega 4-6, 33940 El Entrego (Spain) E-mail: [email protected]
2Departamento de Química Orgánica e Inorgánica, Universidad de Oviedo, Julián Clavería 8, 33006 Oviedo
Coordination to a transition metal fragment dramatically modifies the reactivity of highly stable, usually inert molecules which have been employed as ligands for a long time, such as 2,2’-bipyridine (bipy) or 1,10-phenantrholine (phen). In this context, we have found new reactivity patterns of bipy and phen ligands by simple intramolecular addition of a nucleophile generated by deprotonation of a cis-coligand (in most cases an N-alkyl/arylimidazole).[1] C-C coupling products displaying dearomatized bipy or phen have been obtained under mild conditions, and the high reactivity towards electrophiles of some of these derivatives led to the opening of one pyridine ring of the bipy or phen ligands (see Scheme). The study of the dearomatization and ring-opening of pyridines mediated by transition metal complexes is expected to provide homogeneous models for the industrially relevant, yet poorly understood, hydrodenitrogenation process.[2] Metal-mediated C-N bond cleavage of pyridines had been previously accomplished only with a few, highly reactive early transition metal complexes.
References[1] R. Arévalo, M. Espinal-Viguri, M. A. Huertos, J. Pérez, L. Riera, Adv. Organomet. Chem., 2016, 65, 47-112.[2] M. Bachrach, T. J. Marks, J. M. Notestein, ACS Catal. 2016, 6, 1455-1476.

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 28
IT7Tailoring Organozinc Reagents for Chemical Cooperativity
Eva Hevia,1,2
1 Department of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral St, G11XL, Glasgow, UK. 2 Departement für Chemie und Biochemie, Universität Bern, Freiestrasse 3, 3012, Bern, Switzerland
E-mail: [email protected]; [email protected]
Organozinc reagents are amongst the most widely used organometallic reagents in synthesis. Their unique functional group tolerance makes them very valuable intermediates in transition-metal-mediated C-C bond forming reactions. However, the relative covalent character of their Zn-C bonds makes these reagents sluggish to react in other fundamental transformations such as deprotonative metallation or metal-halogen exchange.
This talk will present our most recent findings on the development of multimetallic strategies to enable direct zincation of a wide range of aromatic substrates via C-H or C-Halogen functionalization. Switching on cooperative effects, unprecedented reactivities can be realised, as illustrated in Scheme 1 for a new family of bimetallic reagents of type ZnEt2.2LiOR which efficiently promote I/Zn and Br/Zn exchanges at room temperature through the activation of both Et groups on Zn.[1] The first examples on exploiting cooperative effects to promote transition metal-free C-C bond forming reactions using arylzinc reagents will also be discussed.[2,3]
References[1] M. Balkenhohl, D. S. Ziegler, A. Desaintjean, L. J. Bole, A. R. Kennedy, E. Hevia, P. Knochel, Angew. Chem. Int. Ed. 2019, early view.[2] A. Hernán-Gómez, S. A. Orr, M. Uzelac, A. R. Kennedy, S. Barroso, X. Jusseau, S. Lemaire, V. Farina, E. Hevia, Angew. Chem. Int. Ed. 2018, 57, 10630.[3] M. Dell’Aera, F. Maria Perna, P. Vitale, A. Altomare, A. Palmieri, L. C. H. Maddock, L. J. Bole, A. R. Kennedy, E. Hevia, V. Capriati, manuscript submitted.

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 29
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
IT8FeV complexes of relevance in enzymology and organic synthesis
Miquel CostasInstitut de Química Computacional I Catàlisi, Universitat de Girona, Facultat de Ciències, Campus de Montilivi,
17003, Girona, Spain E-mail: [email protected]
High-valent iron compounds are very reactive species presumed to be key intermediates in a number of reactions of interest in biology, chemical synthesis and technology.[1-4] For instance high-valent iron-oxo species are key intermediates in challenging oxidation reactions such as C-H and C=C oxidation[2-3] and water oxidation,[5] and high-valent nitride and related species have been considered as possible intermediates in iron-mediated dinitrogen reduction to ammonia.[6-7]
The high reactivity of these species makes their preparation and characterization a challenging task. In the current contribution we will describe the generation and spectroscopic characterization of exceedingly reactive non porphyrinic Fe(V) species with terminal oxo ligands. Their relevance in biologically inspired alkane and alkene oxidation, and O-O bond formation reactions will be discussed.[8]
References[1] McDonald, A. R.; Que, L., Jr. Coord. Chem. Rev. 2013, 257, 414.[2] Groves, J. T. J. Inorg. Biochem. 2006, 100, 434.[3] Hohenberger, J.; Ray, K.; Meyer, K. Nat. Commun. 2012, 3, 720.[4] Nam, W.; Lee, Y.-M.; Fukuzumi, S. Acc. Chem. Res. 2014, 47, 1146.[5] Fillol, J. L.; Codolà, Z.; Garcia-Bosch, I.; Gómez, L.; Pla, J. J.; Costas, M. Nat. Chem. 2011, 3, 807-13.[6] Rittle, J.; Green, M. T. Science 2010, 330, 933.[7] Scepaniak, J. J.; Vogel, C. S.; Khusniyarov, M. M.; Heinemann, F. W.; Meyer, K.; Smith, J. M. Science 2011, 331, 1049.[8] a) Fan et al. J. Am. Chem. Soc. 2018, 140, 3916. b) Borrell M.; Andris, E.; Roithova, J.; Costas, M. Nat. Commun. 2019, DOI : 10.1038/s41467-019-08668-2. C) Dantignana et al. submitted.

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 30
IT9High-Temperature Lanthanide Single-Molecule Magnets
Richard A. LayfieldDepartment of Chemistry, School of Life Sciences, University of Sussex, Brighton, BN1 9QJ, U.K.
E-mail: [email protected]
Single-molecule magnets (SMMs) are coordination compounds that display magnetic memory effects.1 We have a particular interest in metallocene SMMs based on dysprosium, and, over the years, we have reported a few examples of such materials.2 Our work has shown that the [Cp]– ligands provide a strong axial crystal field that enhances the magnetic anisotropy of Dy3+, leading us to propose that a cation of the type [Cp2Dy]+ should be an interesting synthetic target.3 Recently, we described the properties of [(h5-Cp*)Dy(h5-C5
iPr5)][B(C6F5)4] ([1][B(C6F5)4]), the first SMM to show magnetic hysteresis above the boiling point of liquid nitrogen (Figure 1).4
Fig. 1 Molecular structure of 1, and frequency-dependence of the out-of-phase susceptibility, magnetic hysteresis at 2-75 K (sweep rate of 200 Oe s–1) and at 80 K (sweep rate of 25 Oe s–1) for [1][B(C6F5)4].
Having established the design principles required to optimize the SMM properties of dysprosium sandwich complexes, we now turn our attention to the cyclobutadienyl ligand, [Cb]2–. There were, hitherto, no lanthanide complexes of [Cb]2– ligands, hence investigations of such materials furnish an opportunity for fundamental advances in lanthanide organometallic chemistry. In addition, the greater formal charge and the four-fold symmetry provided by the [Cb]2– ligand provide a strategy for the development of SMMs. Our initial results on such systems will be described.5
AcknowledgementsRAL thanks the University of Sussex, the EPSRC, the ERC, the EU Marie Sklowdowska-Curie Actions and COST Actions, the Royal Society and the Newton Trust.
References[1] F.-S. Guo, A. K. Bar, R. A. Layfield. Chem. Rev. 2019, ASAP. (b) J. M. Frost, K. L. M. Harriman, M. Murugesu, Chem. Sci. 2016, 7, 2470. (c) J.-L. Liu, Y.-C. Chen, M.-L. Tong, Chem. Soc. Rev. 2018, 7, 2431.B. M. Day, F.-S. Guo, R. A. Layfield, Acc. Chem. Res. 2018, 51, 1880.[2] T. Pugh, N. F. Chilton, R. A. Layfield, Angew. Chem. Int. Ed. 2016, 55, 11082; (b) S. Gao et al. Chem. Eur.–J. 2016, 22, 12724.F.-S. Guo, B. M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki, R. A. Layfield, Science 2018, 362, 1400.B. M. Day, F.-S. Guo, S. R. Giblin, A. Sekiguchi, A. Mansikkamäki, R. A. Layfield, Chem.– Eur. J. 2018, 24, 16779.

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 31
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
IT10Developing sustainable carbon hybrid materials
Maria del Carmen Gimenez-Lopez 1,2
1Centro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CIQUS), Universidade de Santiago de Compostela, 15782 Santiago de Compostela, Spain.
2School of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: [email protected]
Recyclability and re-use of highly active nanocatalysts, often containing precious metals, is still an outstanding global challenge of increasing importance in both in the area of heterogeneous catalysis and in the area of energy conversion. We have recently reported on the design and preparation of magnetically separable catalytic carbon nanoreactors using carbon-coated magnetic nanoparticles, as well as, their testing in the reduction of nitrobenzene demonstrating a high activity and excellent durability, while their magnetic recovery enables significant improvement in the reuse of the nanocatalyst (Fig. 1). Thus, our strategy allows for the first time the simultaneous separation of heterogeneous catalyst from the products mixtures in a fast, easy and efficient way by simply applying a magnetic field contributing to the re-use of the catalytic material, while exploiting confinement effects.On the other hand, electrochemical devices based on electrocatalyst containing precious metals, such as Pt, are currently hindered by their short-term durability. As these precious elements are rapidly diminishing, the research community is forced to urgently address this major issue until more abundant efficient electrocatalysts are put forward. In this respect, hollow carbon nanostructures can provide an excellent mean for the fabrication of highly durable electrocatalyst materials through nanocatalyst confinement, allowing their sustainable use in electrochemical processes. These surprising and remarkable properties of the reported hybrid electrocatalyst materials has opened up a new strategy for the sustainable use of precious metals in electrocatalysis and other technological applications that require stabilization of metal nanoparticles under harsh conditions (Fig. 2).
Figure 1. A schematic Illustration of the recovery of catalytic carbon nanoreactors by magnetic separation from a liquid solution after a chemical reaction.
Figure 2. HRTEM image of the internal cavity of a electrocatalytic nanoelectrode with NPs positioned predominantly at the graphitic step-edges.
References [1] M. Aygun, T. W. Chamberlain, M. C. Gimenez-Lopez*, and A. N. Khlobystov*. Magnetically Recyclable Catalytic Carbon Nanoreactors. Advanced Functional Materials, 2018, 28, 34, 1802869.[2] M.C. Gimenez-Lopez*, A. Kurtoglu, D.A. Walsh and A.N. Khlobystov. Extremely Stable Platinum-Amorphous Carbon Electrocatalyst within Hollow Graphitized Carbon Nanofibers for the Oxygen Reduction Reaction. Advanced Materials, 2016, 28, 41, 9103.[3] A. La Torre, M.C. Gimenez-Lopez*, M.W. Fay, C.H. Lucas, P.D. Brown and A.N. Khlobystov. Dynamics of Gold Nanoparticles on Carbon Nanostructures Driven by van der Waals and Electrostatic Interactions. Small, 2015, 11, 23, 2756.

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 32
IT11Small molecule activation and catalysis using coordinatively
unsaturated complexesDeborah L. Kays
School of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, U.K. E-mail: [email protected]
The use of sterically demanding ligand frameworks allows the isolation of highly unsaturated transition metal complexes which show unusual bonding modes and reactivity towards small molecules.1,2 Stoichiometric quantities of low-coordinate iron(II) complexes react with carbon monoxide, selectively cleaving and homologating CO to afford unusual squaraines and iron carboxylates at ambient temperature and pressures.3 Recent investigations within our group have shown that these complexes are also efficient precatalysts for chemical reactions, for example in the cyclotrimerisation and hydrophosphination of isocyanates (Figure 1).4,5 Significantly, this reactivity can afford unusual reaction products such as phosphinodicarboxamides through coupling and P–H addition chemistry. Dehydrocoupling reactions between low-coordinate manganese(II) precatalysts and dimethylamine-borane highlight the importance of ligand choice, as small changes in the coordination environment give rise to significant differences in the reaction pathway.6 These and other recent investigations of small molecule activation and catalysis using coordinatively unsaturated complexes will be described.
N N
N
R
R
RO
OO +PHPh2
Ph2P NH
O
RPh2P N
R
O O
NH
Ror
M = FeM = Mn, Fe
M D
5 mol% catalyst0.5-5 mol% catalyst
Toluene Toluener.t. or 60 oC r.t. or 60 oC
RN=C=O(Ar2C6H3)2MLn
(Ar2C6H3)2MLn
Figure 1.
References[1] D. L. Kays, Dalton Trans., 2011, 40, 769.[2] P. P. Power, Chem. Rev., 2012, 112, 3482.[3] H. R. Sharpe, A. M. Geer, L. J. Taylor, B. M. Gridley, T. J. Blundell, A. J. Blake, E. S. Davies, W. Lewis, J. McMaster, D. Robinson and D. L. Kays, Nat. Commun., 2018, 9, 3757.[4] H. R. Sharpe, A. M. Geer, H. E. L. Williams, W. Lewis, A. J. Blake, D. L. Kays, Chem. Commun., 2017, 53, 937. [5] H. R. Sharpe, A. M. Geer, W. Lewis, A. J. Blake, D. L. Kays, Angew. Chem. Int. Ed., 2017, 56, 4845. [6] H. R. Sharpe, A. M. Geer, T. J. Blundell, F. R. Hastings, M. W. Fay, G. A. Rance, W. Lewis, A. J. Blake and D. L. Kays, Catal. Sci. Technol., 2018, 8, 229.

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 33
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
IT12Frustration versus Interaction in Bimetallic Systems
Jesús CamposInstituto de Investigaciones Químicas (IIQ), Centro de Innovación en Química Avanzada (ORFEO-CINQA).
Consejo Superior de Investigaciones Científicas (CSIC) and Universidad de Sevilla. Av. Américo Vespucio 49, 41092 Sevilla (Spain).
E-mail: [email protected]
In the early 80s Chisholm proposed that “all the types of reactions which have been studied for mononuclear transition metal complexes will also occur for dinuclear transition metal complexes”.[1] Almost 40 years later, continued research on the area of bimetallic systems has proven that claimed and gone beyond. Regarding catalytic applications, there are many important transformations that require the concerted action of pairs of active metal sites, paralleling what is often found in metalloenzymes. We recently started to investigate late-transition bimetallic systems characterized by the use of novel sterically hindered phosphine ligands bearing a terphenyl substituent.[2] We have focused on the competition between the formation of M-M bonds (in metal-only Lewis pairs (MOLPs))[3] versus M···M frustration (in metal-only frustrated Lewis pairs (MOFLPs))[4] in a variety of bimetallic pairs (see Figure). Our results pertaining the cooperative reactivity of these metallic pairs and their potential in catalysis will be discussed.
Rh
Me3P PMe3
R
R
RR
PAu
R'R'
NTf2
MP
R
RR
R+
M = Ir, Rh
Zn
F
F
FF
F
F
FF
F
F
Ir
iPrP
iPr
iPriPr
Cl
Pt
P(tBu)3
P(tBu)3vs
MOLP
MOFLP(Metal Only Frustrated Lewis Pair)
(Metal Only Lewis Pair)LEWIS BASES
LEWIS ACIDS
B
B
A
A
References[1] Chisholm, M. H. Chapter 2, Reactivity of Metal-Metal Bonds, ACS Symposium Series, 1981, 155.N.[2] (a) Moreno, J. J.; Espada, M. F.; Krüger, E.; López-Serrano, J.; Campos, J.; Carmona, E. Eur. J. Inorg. Chem. 2018, 2309; (b) Moreno, J. J.; Espada, M. F.; Campos, J.; López-Serrano, J.; Macgregor, S. A.; Carmona, E. J. Am. Chem. Soc. 2019, 141, 2205.[3] Hidalgo, N.; Maya, C.; Campos, J. Chem. Commun. 2019, Advance Article , DOI:10.1039/C9CC03008E[4] (a) Campos, J. J. Am. Chem. Soc. 2017, 139, 2944; (b) Hidalgo, N.; Bajo, S.; Moreno, J. J.; Navarro-Gilabert, C.; Mercado, B.; Campos, J. Dalton Trans. 2019, 48, 9138.

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 34
IT13Electrochemical Generation of Catalysts using Batch and Flow Technology
Charlotte WillansUniversity of Leeds, UK
E-mail: [email protected]
We have developed electrochemical methods for facile preparation of metal-N-heterocyclic carbene complexes and other organometallic and coordination compounds.[1-3] Upon applying a potential, the ligand precursor is reduced at the cathode, the sacrificial metal anode oxidises and releases ions into solution, and the two components come together with the only theoretical by-product being hydrogen. This talk will discuss the translation of the batch methodology into flow technology for maximum efficiency and to enable broad use (Figure 1).[4] Our electrochemical technology proposes to provide clean, high-yielding metal complexes on-demand, without the formation of metal salt by-products or the requirement for isolation and purification prior to use.
Figure 1 Electrochemical flow-reactors which use a stacked-disk design. Metal plates, which alternate between cathode and anode, are separated by PTFE spacers featuring a serpentine flow channel.
The electrochemical flow reactors are being incorporated into an automated flow platform for rapid generation and screening of metal catalysts. I will discuss recent results in developing the flow platform.
References[1] M. R. Chapman, S. E. Henkelis, N. Kapur, B. N. Nguyen, C. E. Willans, ChemistryOpen 2016, 5, 351.[2] E. K. Bullough, M. A. Little, C. E. Willans, Organometallics 2013, 32, 570.[3] B. R. M. Lake, E. K. Bullough, T. J. Williams, A. C. Whitwood, M. A. Little, C. E. Willans, Chem. Commun. 2012, 48, 4887.[4] M. R. Chapman, Y. M. Shafi, N. Kapur, B. N. Nguyen, C. E. Willans, Chem. Commun. 2015, 51, 1282.

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 35
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
IT14The reactivity of alkanes towards metal-carbene electrophiles
Pedro J. PérezLaboratorio de Catálisis Homogénea, Unidad Asociada al CSIC, CIQSO-Centro de Investigación en Química
Sostenible and Departamento de Química, Universidad de Huelva, 21007 Huelva, Spain E-mail: [email protected]
Our group has been involved in the area of hydrocarbon C-H functionalization using the strategy of carbene transfer from diazo compounds,1 which along the years has been developed until methane could be reached.2 In this contribution an extensive study of relative reactivity of a number of alkanes,3 from C1 to C8, is presented as well as a simple model that allows the estimation of the reactivity based on a few simple rules.
References[1] Caballero, A.; Díaz-Requejo, M. M.; Fructos, M. R.; Olmos, A.; Urbano, J.; Pérez, P. J. Dalton Trans. 2015, 44, 20295-20307.[2] Caballero, A.; Despagnet-Ayoub, E.; Díaz-Requejo, M. M.; Díaz-Rodríguez, A.; González-Núñez, M. E.; Mello, R.; Muñoz, B. K.; Solo Ojo, W.; Asensio, G.; Etienne, M.; Pérez, P. J. Science, 2011, 332, 835–838.[3] Olmos, A.; Gava, R.; Noverges, B.; Bellezza, D.; Jacob, K.; Besora, M.; Sameera, W. M. C.; Etienne, M.; Maseras, F.; Asensio, G.; Caballero, A.; Pérez, P. J. Angew. Chem. Int. Ed. 2018, 57, 13848-13852.

Contributions

Oral Presentations

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 38
OP1Carbene complexes of Cu, Ag, Au as OLED photoemitters
Manfred Bochmann,1 A. S. Romanov1, D. Credington 2, P. J. Conaghan2, M. Linnolahti3
1 School of Chemistry, University of East Anglia, Earlham Road, Norwich, NR4 7TJ, UK 2 Department of Physics, Cavendish Laboratory, Cambridge University, Cambridge CB3 0HF, UK
3 Department of Chemistry, University of Eastern Finland, FI-80101 Joensuu, Finland E-mail: [email protected]
“Carbene-metal-amides” (CMAs) are a new class of photoemitters, with the potential for use in electronic displays such as smartphones. We found that Cu, Ag and Au complexes of cyclic (alkyl)(amino)carbenes (CAACs) ccoupled with selectron-rich anionic ligands are highly photo- and electro-luminescent [1,2]. Complexes of the structure (CAAC)M (carbazolate) 1 are thermally stable, soluble
in most organic solvents and sufficiently volatile to enable their incorporation into the emissive layer of organic light emitting diodes (OLEDs), fabricated by either solution processing or by thermal vapour deposition techniques. These are the first examples of photoemitters based on simple, linear geometry and are easily accessible by a Lego-brick type assembly. Their electronic structure makes them ideally suited to convert electrical energy into light with up to 100% efficiency. Compounds of type 1 operate very different from currently used cyclometallated iridium emitters, which rely on high spin-orbit coupling for energy conversion. For efficient devices short excited state lifetimes are also desirable.
We will discuss here the principles of molecular design, which enabled us not only to produce OLEDs with cutting edge efficiency, but also control the excited state lifetimes and the emission colours [3]. We also report the first example of efficient OLEDs based only on silver [4], as well as the synthesis and performance of carbene metal amides with dendrimer ligands for flexible displays.
This work was supported by the ERC. M. B. is an ERC Advanced Investigator Award holder (grant no. 338944-GOCAT).
References[1] D. Di, A. S. Romanov, et al., Science 2017, 356, 159–163. [2] A. S. Romanov, et al., Chem. Eur. J. 2017, 23, 4625-4637. [3] P. J. Conaghan, et al., Adv. Mater. 2018, 30, 1802285.[4] A. S. Romanov, et al., Adv. Optical Mater. 2018, 6, 1801347.

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 39
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
OP2Improved generations of catalysts for the synthesis of elusive chiral
synthonsMontserrat Diéguez,1 Maria Biosca1, Feliu Maseras2, Miquel A. Pericàs2, Oscar Pàmies1
1 Department de Química Física i Inorgànica, Universitat Rovira i Virgili, C/ Marcel.lí Domingo 1, 43007, Tarragona, Spain
E-mail: [email protected] 2Institute of Chemical Research of Catalonia (ICIQ). The Barcelona Institute of Science and Technology,
Av. Països Catalans, 16 43007, Tarragona, Spain
Asymmetric catalysis is a powerful and sustainable method for preparing chiral compounds. Most catalysts are only tested in benchmark substrates, eluding challenging ones that would yield to more appealing compounds. Here I present our recent finding in the design of improved catalyst libraries for the elusive and challenging asymmetric hydrogenation (AH) of cyclic b-enamides and tetrasubstituted olefins.[1] These reactions led to the formation of important chiral synthons, such as aminotetraline and aminochromanone derivatives, which can be found in numerous therapeutic agents and biologically active natural products (e.g. rotigotine, alnespirone,...). Despite this, they are underdeveloped compared to the AH of the most studied and solved a-enamides and trisubstituted olefins. Finally, I would also show the design of improved catalysts for the preparation chiral (poly)carbo- and heterocyclic compounds, with multiple stereocenters, by using straighthforward sequences of allylic substitution an either 1,6-enyne cyclization, or ring-closing metathesis or Pauson-Khand reactions.[2]
The families of catalysts developed presents the advantages of a simple and modular architecture, they have been synthesized in a few steps from unexpensive starting materials (e.g., sugars) and are solid and stable to air and therefore easy to handle. The combination of computational studies and NMR spectroscopy, together with the analysis of the catalytic results were crucial to identify the species responsible for the catalytic performance, to rationalize the catalysts’ structure, and to ensure the finding of optimal catalyst.[1,3]
References[1] a) J. J. Verendel, O. Pàmies, M. Diéguez, P. G. Andersson, 2014, 114, 2130–2169. b) M. Magre, O. Pàmies, M. Diéguez, M. ACS Catal. 2016, 6, 5186–5190. c) J. Margalef, O. Pàmies, M. Diéguez, Chem. Eur. J. 2017, 23, 813–822 (Selected to be in inside front cover and as a Hot paper). d) Biosca, M.; Magre, M.; Pàmies, O.; Diéguez, ACS Catal. 2018, 8, 10316-10320 (Highlighted in Synfacts. 2018, 14, 1277). d) M. Biosca, E. Salomó, P. Cruz-Sánchez, A. Riera, X. Verdaguer, O. Pàmies, M. Diéguez, Org. Lett. 2019, in press, DOI: 10.1021/acs.orrglett.8b04084.[2] For recent publications: a) R. Bellini, M. Magre, M. Biosca, P.-O. Norrby, O. Pàmies, M. Diéguez, C. Moberg, ACS Catal. 2016, 6, 1701-1712. b) b) Biosca, M.; Margalef, J.; Caldentey, X.; Besora, M.; Rodríguez-Escrich, C.; Saltó, J.; Cambeiro, X. C.; Maseras, F.; Pàmies, O.; Diéguez, M.; Pericàs, M. A. ACS Catal. 2018, 8, 3587-3601. c) M. Biosca, J. Saltó, M. Magre, P-O. Norrby, O. Pàmies, M. Diéguez, ACS Catal. 2019, in press.

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 40
OP3Access to Unusual Heterocyclic Compounds Utilizing a Key
Cobalt-Catalyzed C-H Functionalization ApproachChristopher J. Whiteoak1,2, Paula G. Chirila1, Alex Hamilton1,2
1Biomolecular Sciences Research Centre, Sheffield Hallam University, Sheffield, S1 1WB, United Kingdom, E-mail: [email protected]
2Department of Biosciences and Chemistry, Sheffield Hallam University, Sheffield, S1 1WB, United Kingdom
Since the publication of Matsunaga and Kanai in 2013 demonstrating the potential of Cp*CoIII-type catalysts in C-H functionalization protocols[1], the field has rapidly expanded and engaged the interest of a large number of researchers[2]. In general, these reports can be divided into two key areas; (a) linear couplings and (b) new/efficient routes to heterocycle formation. The latter of these areas is of significant interest as most of the small molecule drugs approved by the FDA in 2017 contained complex heterocyclic motifs[3] and as a result novel, improved methods for their synthesis is likely to be an impactful innovation. In this context, we have engaged in the use of Cp*CoIII catalysts, with readily available benzamide and related acetanilide substrates, for the preparation of some more unusual heterocyclic compounds through either a cascade reaction[4] or a two-step sequential one-pot protocol[5] using C-H functionalization as the key step in both cases. In addition to these synthetic results, studies of their mechanisms using DFT have revealed key aspects of the catalytic cycles, allowing for a fuller understanding of the observed selectivities and reactivities. In summary, this contribution showcases our recent research demonstrating the potential for rapidly building up molecular complexity exploiting efficient and sustainable first-row transition metal catalysis as the key tool.
References[1] T. Yoshino, H. Ikemoto, S. Matsunaga, M. Kanai, Angew. Chem. Int. Ed., 2013, 52, 2207.[2] For an overview see: P. G. Chirila, C. J. Whiteoak, Dalton Trans., 2017, 46, 9721.[3] L. M. Jarvis, Chem. Eng. News, 2018, 96, 26.[4] P. G. Chirila, J. Adams, A. Dirjal, A. Hamilton, C. J. Whiteoak, Chem. Eur. J., 2018, 24, 3584.[5] P. G. Chirila, L. Skibinski, K. Miller, A. Hamilton, C. J. Whiteoak, Adv. Synth. Catal., 2018, 360, 2324.

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 41
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
OP4Organometallic tethered compounds with a coordinative bound capable of
hijacking/releasing a protonSonia Infante-Tadeo, Ana M. Pizarro
IMDEA Nanociencia, Faraday 9, Ciudad Universitaria de Cantoblanco, Madrid, 28049, Spain E-mail: [email protected]
Osmium(II) and ruthenium (II) arene complexes with a “piano-stool” structure have received much attention in the last decade as possible antitumor agents. By tuning the four different building blocks: arene (R), chelating ligand (XY) and labile ligand (Z) as well as the metal (M) (Figure 1), [1] it is possible to produce complexes that ultimately cause cell death via novel strategies, such as triggering a metabolic shift and artificial catalysis.
Since the Warburg effect was postulated in the 1950s it is known that cancer cells favour anaerobic metabolic routes such as fermentation even in presence of oxygen. [2] As a result of a high production of lactate in the cytosol, and consequent extrusion of lactic acid excess, the pH of the extracellular microenvironment (pHe) is dropped to values as low as 6.5, facilitating metastasis. As a result, the internal pH (pHi) undergoes basification, which promotes proliferation.[3]
Our approach aims to interfere with the finely controlled pH balance of the tumour cell, inverting its pH gradient.
We have designed, synthesised and characterised novel families of half-sandwich tethered compounds of ruthenium(II) and osmium(II), that bear an alcohol-derivatised R=C6H5(CH2)3OH arene. The oxygen atom binds to the metal centre [Ru(II) or Os(II)] forming a coordination bond (Figure 2). This oxygen atom has the potential to capture a proton at a pH biologically relevant (pKa in the range 4.2-6.6). In addition, some compounds can undergo Metal-O cleavage upon protonation, activating the metal centre.
We hypothesize that these complexes could have a key role in biological systems with a reversed pH gradient, such as that found in tumour cells, since they have the capability to highjack and release a proton as a function of pH. We present Ru(II) and Os(II) complexes as candidates capable to act as proton shuttles working against the intracellular pH gradient and ultimately causing cancer cell death.
References[1] (a) Hearn, J. M.; Romero-Canelón, I.; Munro, A. F.; Fu, Y.; Pizarro, A. M.; Garnett, M. J.; McDermott, U.; Carragher, N. O.; Sadler, P. J., Potent organo-osmium compound shifts metabolism in epithelial ovarian cancer cells. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, E3800–E3805; (b) Coverdale, J. P. C.; Romero-Canelón, I.; Sanchez-Cano, C.; Clarkson, G. J.; Habtemariam, A.; Wills, M.; Sadler, P. J., Asymmetric transfer hydrogenation by synthetic catalysts in cancer cells. Nat. Chem. 2018, 10, 347-354.[2] Warburg, O., On the Origin of Cancer Cells. Science 1956, 123 (3191), 309-314.[3] Webb, B. A.; Chimenti, M.; Jacobson, M. P.; Barber, D. L., Dysregulated pH: a perfect storm for cancer progression. Nature Reviews Cancer 2011, 11 (9), 671-677.
Figure 1. General structure of “half-sandwich” complexes
Figure 2. Protonation/deprotonation reactivity as a function of pH

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 42
OP5Combined catalytic action of homometallic and heterometallic aluminum
species to generate polymeric nanoparticles
M. Teresa Muñoz, Tomás Cuenca, Marta E.G. Mosquera*, Miguel Palenzuela Departmento de Química Orgánica y Química Inorgánica. Instituto de Investigación en Química
Andrés M. del Rio. Universidad de Alcalá. Campus Universitario, 28805-Alcala de Henares, Madrid. E-mail: [email protected]
Aluminium derivatives play a prominent role in many catalytic polymerization reactions. In particular, aluminium alkoxide ligands are very active catalysts in ROP processes,1 also it is well known the use of aluminoxanes as co-catalysts in industrial olefin polymerization reactions.2 In our group we have studied the reactivity of aluminium aryloxide derivates that have shown to be very active for ROP polymerization process.3 As well, we are interested in the so-called aluminate derivatives, these heterometallic species contain aluminum and an alkaline metal and display a unique reactivity in different chemical processes such as C-H orthometalation and C-heteroatom bond formation.4 However their activity in polymerization reactions has been less explored. We have prepared a series of aluminates that when tested in polymerization reactions have shown to be active in vinyl polymerization processes.5 When the reaction is performed with a monomer such as Glycidyl Methacrylate (GMA), where the polymerization can occur via the methacrylate function (vinyl polymerization) or the oxirane group (ROP), we are able to perform the selective polymerization of one of the functional groups. Furthermore, using a strategy of a controlled crosslinking we have attained the formation of polymeric nanoparticles (Figure 1).
Figure 1
References [1] T. J. Dickerson, N. N. Reed, K. D. Janda, Chem. Rev. 2002, 102, 3325 [2] H. Sinn, W. Kaminsky, Adv. Organomet. Chem. 1980, 18, 99; E. Y.-X. Chen, T. J. Marks, Chem. Rev., 2000, 100, 1391 [3] G. Martínez, S. Pedrosa, V. Tabernero, M. E. G. Mosquera and T. Cuenca, Organometallics, 2008, 27, 2300; M. T. Muñoz, M. Palenzuela, T. Cuenca and M. E. G. Mosquera, ChemCatChem, 2018, 10, 936. [4] R. E. Mulvey, F. Mongin, M. Uchiyama and Y. Kondo, Angew. Chem., Int. Ed., 2007, 46, 3802; M. Uzelac and E. Hevia, Chem. Commun., 2018, 54, 2455; B. Haag, M. Mosrin, H. Ila, V. Malakhov and P. Knochel, Angew. Chem., Int. Ed., 2011, 50, 9794. [5] M. T. Muñoz, T. Cuenca and M. E. G. Mosquera, Dalton Trans., 2014, 43, 14377 M.T. Muñoz, C. Urbaneja, T. Cuenca, M.E G. Mosquera, Chem. Commun, 2011, 47, 11757.

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 43
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
OP6Synergistic combination of metal-catalyzed organic reactions with enzymes
or organolithium reagents (RLi) in water and Deep Eutectic SolventsMaría Jesús Rodríguez-Álvarez1, Luciana Cicco1,2, Vito Capriati2,*, Nicolas Ríos-Lombardía 3,
Javier Gonzalez-Sabín3,*, Joaquín García-Álvarez1,*
1Laboratorio de Compuestos Organometálicos y Catálisis (Unidad Asociada al CSIC), Departamento de Quími-ca Orgánica e Inorgánica (IUQOEM), Centro de Innovación en Química Avanzada (ORFEO-CINQA), Facultad
de Química, Universidad de Oviedo, Oviedo, Spain. E-mail: [email protected]
2Dipartamento di Farmacia-Scienze del Farmaco, Università di Bari “Aldo Moro”, Consorzio C.I.N.M.P.I.S, Via E. Orabona 4, I-70125 Bari, Italy.
3EntreChem SL, Vivero Ciencias de la Salud, Colegio Santo Domingo de Guzmán, Oviedo, Spain.
New cleaner and more efficient one-pot multistep cascades in green solvents [i.e. water or Deep eutectic Solvents (DESs)] are emerging as exciting alternatives to highly-costly and tedious step-by-step processes, which also minimise chemical waste, save time and simplify practical aspects. In this field, the sustainable combination of metal-catalyzed organic reactions with either enzymatic (bio-catalyzed) or main-group-mediated organic transformations are the most recalcitrant challenges to solve. In this communication, we present our recent findings in the coupling of metal-catalyzed organic reactions (isomerizations or cycloisomerizations) with: i) ω-transaminase (ω-TA) [1a] or ketoreductases (KREDs) in either water [1b-c] or DESs [1d]; and ii) the chemoselective addition of highly-polarized organometallic reagents (RLi) in DESs at room temperature and under air [2].
References[1] (a) N. Ríos-Lombardía, C. Vidal, M. Cocina, F. Morís, J. García-Álvarez, J. González-Sabín, Chem. Commun., 2015, 51, 10937; (b) N. Ríos-Lombardía, C. Vidal, E. Liardo, F. Morís, J. García-Álvarez J. González-Sabín, Angew. Chem. Int. Ed., 2016, 55, 8691; (c) M. J. Rodríguez-Álvarez, N. Ríos-Lombardía, S. Schumacher, D. Pérez-Iglesias, F. Morís, V. Cadierno, J. García-Álvarez, J. González-Sabín, ACS Catal., 2017, 7, 7753; (d) L. Cicco, N. Ríos-Lombardía, M.-J. Rodríguez-Álvarez, F. M. Perna, J. García-Álvarez, V. Capriati, Green Chem., 2018, 20, 3468.[2] (a) C. Vidal, J. García-Álvarez, A. Hernan-Gómez, A. R. Kennedy, E. Hevia, Angew. Chem. Int. Ed., 2016, 55, 16145; (b) L. Cicco, M.-J. Rodríguez-Álvarez, F. M. Perna, J. García-Álvarez, V. Capriati, Green Chem., 2017, 20, 3069.

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 44
OP7Investigating the Reduction and Reactivity Chemistry of Cobalt
Diphosphine ComplexesClaire N. Brodie1, Martin J. Hanton2, Andrei S. Batsanov1, Philip W. Dyer1,3,
1Department of Chemistry, South Road, Durham, DH1 3LE, UK 2Sasol Technology UK
3Centre for Sustainable Chemical Processes, Department of Chemistry, Durham University, DH1 3LE, UK E-mail: [email protected]
Recently, there has been significant interest in the synthesis and reactivity of chelate diphosphine cobalt(I) complexes. These systems have been explored as pro-catalysts to mediate coupling of ethylene with CO2, and have been shown to catalyse heterodimerisation of acrylates with 1,3-dienes. [1,2]
Building on these studies we initiated an investigation into the application of chelate diphosphine cobalt(I) complexes in regio- and stereo-selective linear α-olefin (LAO) to LAO oligomerisation reactions. The chelating diphosphine motif provides a readily tuneable metal scaffold, to engineer selective oligomerisation. Together, substituent variation at phosphorus and variation on the backbone allow tuning of both the electronic and steric properties of the resulting diphosphine cobalt systems.
Here, the synthesis and characterisation of the necessary precursor cobalt(II) [(P^P)CoX2] complexes and transformation to the target cobalt(I) derivatives is discussed. The reduction chemistry of [(P^P)CoX2] systems is thoroughly explored and found that the outcome of single electron reduction of such species using Zn metal is governed by steric and electronic effects imposed by the diphosphine (Scheme 1). The direct synthesis of chelating diphosphine complexes from cobalt(I) sources such as [(PPh3)3CoCl] is investigated. Structural, spectroscopic and spectrometric techniques are used to gain insight into the ligand–olefin selectivity relationship between a range of diphosphine ligands.
Scheme 1 Example reactivity of cobalt(II) diphosphine complexes
References[1] I. Knopf, M-A. Courtemanche and C. C. Cummins, Organometallics, 2017, 36, 4834.[2] S. M. Ying, V. Balasanthiran, V. Pagar, J. C. Gallucci and T. V. Rajanbabu, J. Am. Chem. Soc., 2017, 139, 18034.

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 45
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
OP8Phosphine/borane frustrated Lewis pairs chemistry supported by
xanthene scaffoldsM. Ángeles Fuentes, Simon Aldridge
Chemistry Research Laboratory, University of Oxford, Mansfield Rd, Oxford, OX1 3TA, UK E-mail: [email protected]
Frustrated Lewis pairs (FLP) chemistry has become a powerful tool with essential applications in main group chemistry and catalysis.[1] FLP species show remarkable reactivity towards small molecule activation and capture protocols. The Aldridge group has recently reported the use of FLP scaffolds based on phosphine/borane-dimethylxanthene systems for the reversible activation of dihydrogen. Similar systems are also capable to capture other small molecules, nitrous oxide as a key example, Figure 1.[2]
We have now explored further the potential of FLP dimethylxanthene chemistry with unsaturated substrates, such as alkynes. We have observed a reversible process operating in solution. In addition, catalytic hydroboration reactions of alkynes with boranes in the presence of FLP dimethylxanthene and mechanistic implications will be discussed.[3]
Figure 1. FLP dimethylxanthene chemistry with small molecules.
References[1] (a) Welch, G. C.; San Juan, R. R.; Masuda, J. D.; Stephan, D. W. Science 2006, 314, 1124. (b) Stephan, D. W.; Erker, G. Angew. Chem. Int. Ed. 2010, 49, 46; (c) Stephan, D.W. Acc. Chem. Res. 2015, 48, 306.[2] (a) Mo, Z.; Kolychev, E. L.; Rit, A.; Campos, J.; Niu, H.; Aldridge, S. J. Am. Chem. Soc. 2015, 137, 12227. (b) Mo, Z.; Rit, A.; Campos, J.; Kolychev, E. L.; Aldridge, S. J. Am. Chem. Soc. 2016, 138, 3306.[3] Vasko, P.; Zulkifly, I. A.; Fuentes, M. Á.; Mo, Z.; Hicks, J.; Kamer, P. C. J.; Aldridge, S. Chem. Eur. J. 2018, 24, 10531.

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 46
OP9Catalytic Alkane Functionalization Via Iron-Carbene Insertion Reaction
Alberto Hernán-Gómez1, Mònica Rodríguez1, Teodor Parella2 and Miquel Costas1
1Institut de Química Computacional i Catàlisi, IQCC and Departament de Química, Universitat de Girona Campus de Montilivi, 17003 Girona (Spain).
E-mail: [email protected] 2Facultat de Ciències, Universitat Autònoma de Barcelona E-08193 Bellaterra (Catalonia)
Saturated hydrocarbons are nature abundant reagents which upon functionalization constitute a potential vast and low cost feedstock for the synthesis of more valuable chemicals.[1] However, conversion of Csp3-H into new C-C bonds is a difficult challenge due to their kinetically inert nature. To overcome this lack of reactivity, highly reactive reagents (superacids or radicals) or forcing reaction conditions are required, albeit these approaches usually compromise the selectivity of the process. An alternative to this energetically demanding processes has emerged within the field of Metal-carbene compounds LM=CR2,
[2] although it is dominated by precious metals such as rhodium, silver and gold. In replacing these systems by earth-abundant metals, iron rises as an optimal choice since it is of low cost, high natural abundance and low toxicity. Despite these attractive properties, iron catalysts have shown modest activity being limited to less demanding processes, including insertion into relatively weak allylic and benzylic (BDE < 90 Kcal/mol) C(sp3)-H bonds.[3]
Herein we report an electrophilic iron compound [Fe(Fpda)(THF)]2 [Fpda = N,N’-bis(pentafluorophenyl)-o-phenylenediamide] which in combination with Lewis acids enables intramolecular functionalization of strong (BDE > 90 Kcal/mol) Csp3-H bonds under mild conditions. Mechanistic investigations point to a crucial role played by the Lewis acids in the formation of the metallocarbene species (rate determining step), which then proceeds via concerted insertion into the C-H bond.
References[1] Bergman, R.G. Nature 2007, 446, 391.[2] Díaz-Requejo, M.M.; Caballero, A.; Fructos, M. R.; Pérez, P. J. Alkane C- H Activation by Single-Site Metal Catalysis, Springer, Amsterdam, 2012, Chap.6.[3] a) Zhang, R.K.; Chen, K.; Huang, X.; Wohlschlager, L.; Renata, H.; Arnold, F.H. Nature, 2019, 565, 67. (b) Griffin, J. R.; Wendell, C. I.; Garwin, J. A.; White, M. C. J. Am. Chem. Soc. 2017, 139, 13624

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 47
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
OP10Unusual Bonds between Magnesium and Transition Metals:
New Opportunities in Catalysis
Martí Garçon1, Andrew J. P. White1, Mark R. Crimmin1,* 1Department of Chemistry, Imperial College London, MSRH White City Campus, London, W12 0BZ.
E-mail: [email protected]
The study of metal–metal bonds has received an increasing amount of attention in recent years.[1,2,3] Accessing new reactivity modes and cooperative effects different from the individual monometallic fragments is especially appealing. We have recently found that combining transition metals with main group fragments allows us to access novel complexes with remarkable bonding configurations and capable of unexpected reactivity.
While investigating the reactions between [Pd(PCy3)2] and β-diketiminate supported Mg- and Zn- complexes, we discovered well-defined intermetallic complexes with extremely unusual coordination geometries and unprecedented Pd–Mg bonds. (Fig. 1). A series of analogues with Pd, Ni and Pt have also been prepared and thoroughly studied by NMR, X-ray diffraction, DFT and neutron diffraction techniques, which allowed us to interrogate the addition of Mg–H bonds to transition metals.
Figure 1. Coordination modes of Mg-H bonds to Transition Metals and selected complexes that represent snapshots along the bonding continuum.
These complexes were found to react readily with inert C–F and C–H bonds of arenes. We have also been able to use catalytic amounts of the transition metal moiety without compromising the reactivity. For example, Mg–Mg reagents effect the C–H magnesiation of benzene when in the presence of [Pd(PCy3)2] as a catalyst[4]. The new C–Mg bonds can then be used for further functionalisation analogously to Grignard reagents. Future efforts will be devoted to developing systems that are catalytic respect both metals, allowing us to open new avenues for heterobimetallic catalysis.
References[1] S. T. Liddle (ed.). Molecular Metal-Metal Bonds. 1st ed, Wiley-VCH, Weinheim, Germany. 2015.[2] C. Jones. Nat. Chem. Rev., 2017, 1, 0059.[3] I. Resa, E. Carmona, E. Gutierrez-Puebla, A. Monge. Science, 2004, 305, 1136-1138.[4] M. Garçon, A. J. P. White, M. R. Crimmin. Chem. Commun. 2018, 54, 12326-12328.

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 48
OP11Homoleptic Trigonal Planar Uranium Complex Induced by High PressureTatsumi Ochiai1, Amy N. Price1, Jacob J. Shephard1, Victoria Berryman2, Polly L. Arnold1*,
Simon Parsons1*, Nikolas Kaltsoyannis2*1 EaStCHEM School of Chemistry, University of Edinburgh, The King’s Buildings, Edinburgh, EH9 3FJ (UK).
2 School of Chemistry, University of Manchester, Oxford Road, Manchester M13 9PL (UK). E-mail: [email protected]
Low-coordinate U(III) complexes are rare but their high symmetry and potential flexibility offers opportunities for in-depth studies of potentially unusual actinide electronic structure and physicochemical properties [1]. Surprisingly, three-coordinate uranium(III) complexes almost exclusively adopt trigonal pyramidal coordination geometries rather than trigonal planar. This could be explained by either covalent and electrostatics arguments, but the real reason for pyramidalization has not been established. Theoretical studies have shown that the energy difference between pyramidal and planar geometry is small [2] and, a totally planar U(III) amide was synthesized by exploiting steric repulsion of very bulky ligands [3]. This result indicates the geometries are flexible so pressure studies might provide new insight.
We will present the first studies of the effect of pressure on the geometry and electronic structure of U(III) complexes: the uranium(III) tris(aryloxide) complex [U(ODtbp)3] (ODtbp = O-2,6-tBu2C6H3) 1 and the uranium(III) tris(amide) complex [UN″3] (N″ = N(SiMe3)2) 2, the most studied UX3 complexes; and the only known sulfur analogue [U(SMes*)3] (SMes* = S-2,4,6-tBu3C6H2) 3, [4]. The structures of 1, 2 and 3 were determined at pressures up to 4.99 GPa, 4.09 and 5.26 GPa, respectively. 1 and 2 do not show planarization at high pressure (ΣO-U-O = 319° at 4.99 GPa, ΣN-U-N = 350° at 4.09 GPa). In sharp contrast, 3 becomes planar with increasing pressure (ΣS-U-S = 360° at 5.26 GPa), and shows acute U-S-C bond angles (av. = 79.6°) and short U···Ar distances (the shortest U···CAr distance = 2.856 Å) (Scheme 1). Computational analyses of the pressure-induced planarization and the effect and potential ramification in actinide reactivity of these weak interactions will also be presented.
Scheme 1. Transformation of pyramidal structure of 3 to planar structure of 3P
References[1] Arnold, P. L. Chem. Commun. 2011, 47, 9005. [2] Ortiz, J. V., Hay, P. J., Martin, R. L., J. Am. Chem. Soc. 1992, 114, 2736. [3] Goodwin, C. A. P., Tuna, F., McInnes, E. J. L., Liddle, S. T., McMaster, J., Vitorica-Yrezabal, I. J., Mills, D. P. Chem. Eur. J. 2014, 20, 14579. [4] Roger, M., Barros, N., Arliguie, T., Thuéry, P., Maron, L., Ephritikhine, M. J. Am. Chem. Soc. 2006, 128, 8790.

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 49
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Catalytic Upgrading of Light Hydrocarbons: Solid–State Molecular Organometallic Nanoreactors
Antonio J. Martínez-Martínez1,3, Stuart A. Macgregor2, Andrew S. Weller3
1 Research Centre for Sustainable Chemistry CIQSO, University of Huelva, Spain E-mail: [email protected]
2 Institute of Chemical Sciences, Heriot-Watt University, Edinburgh, UK 3 Department of Chemistry, University of Oxford, Oxford, UK
Low-temperature catalytic processes for the valorisation of light hydrocarbons (alkanes and alkenes) are desirable methods for the selective production of valuable chemical building blocks (aromatics, upgraded olefins).[1] Although a variety of systems for selective catalytic functionalisation of light hydrocarbons have been developed, methods that operate at low energy profiles with high selectivity represent a real challenge in heterogeneous catalysis. In this context, C–H bond and small molecule activation processes using Solid-State Molecular Organometallics (SMOM) offer new catalytic opportunities that harness the benefits of discrete molecular species placed in a well-defined solid-state microenvironment.[2] We will account our advances on the use of well-defined rhodium sigma-alkane and alkene complexes that behave as “molecular nanoreactors” for the catalytic isomerisation of light hydrocarbons at room temperature, ambient pressure and with high recyclability (Figure 1). The use of diverse chelating phosphines and various non-coordinating anions on these solid-state molecular organometallic catalysts allows to evaluate the influence of bite angle, confinement, and structure/activity relationships.[3] The combination of X-ray crystallography, NMR spectroscopy and periodic DFT calculations provide useful insights in catalyst speciation and reaction pathways.
Figure 1. Activation of light hydrocarbons over confined solid-state molecular organometallic catalysts.
References[1] Hartwig, J. F. Organotransition Metal Chemistry, University Science Books, USA, 2010. [2] Pike, S.; Thompson, A.; Algarra, A.; Apperley, D.; Macgregor, S.; Weller, A. Science, 2012, 337, 1648. [3] a) Chadwick, F.; McKay, A.; Martínez-Martínez, A.; Krämer, T.; Macgregor, S.; Weller, A. Chem. Sci., 2017, 8, 6014; b) Martínez-Martínez, A.; Tegner, B.; McKay, A.; Bukvic, A.; Rees, N.; Tizard, G.; Coles, S.; Warren, M.; Macgregor, S.; Weller, A. J. Am. Chem. Soc., 2018, 140, 14958; c) Unpublished results.
OP12

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 50
Deprotonation studies, luminescent properties and catalytic CO2 reduction activity of Re(CO)3 complexes with pyrazole and (3-(2-pyridyl)pyrazole)
Beatriz Merillas1, Elena Cuéllar1, Alberto Diez-Varga2, Miryam Asensio-Bartolomé2, Gabriel García-Herbosa2, Tomás Torroba2, Jose M. Martín-Alvarez1, Daniel Miguel1,
Fernando Villafañe1
1GIR MIOMeT-IU Cinquima-Química Inorgánica, Facultad de Ciencias, Campus Miguel Delibes, Universidad de Valladolid, 47011 Valladolid, Spain.
E-mail: [email protected] 2 Departamento de Química, Facultad de Ciencias, Universidad de Burgos, 09001 Burgos, Spain
The electrochemical reduction of CO2 might help to solve the problem of renewable energy. Nowadays, one of the most attractive molecular catalyst systems described is based on the use of complexes with diimines coordinated to the fac-ReI(CO)3 fragment. A feasible CO2 reduction mechanism has been already proposed for this system,[1] and one of its most interesting aspects lies in determining the influence of the electronic and stereo characteristics of both the diamine and of the “sixth” ligand. So far, the activity of complexes containing bidentate ligands with different donor groups has been scarcely studied. On the other hand, the relevant role played in the catalytic process by the presence of protons is obvious, so studies on ligands with acidic protons are getting relevant.
In the present work we describe the synthesis and characterization of complexes containing 3-(2-pyridyl)pyrazole (pypzH) as diimine ligand, and different pyrazoles (pz*H = pzH, dmpzH, indzH, pypzH) as the “sixth” ligand. Both ligands present acidic hydrogens. Cationic complexes fac-[Re(CO)3(pypzH)(pz*H)]+ are synthesized and subsequently deprotonated in order to obtain neutral fac-[Re(CO)3(pypz)(pz*H)] and anionic fac-[Re(CO)3(pypz)(pzH)]- complexes. The study of their photophysical properties allow to conclude that the complexes are phosphorescent, whereas their electrochemical properties, studied both in Ar and CO2 atmosphere, demonstrate their catalytic activity in the reduction of CO2. The luminescence and electrochemical measurements are supported by computational calculations.
References[1] (a) Y. Kuramochi, O. Ishitani, H. Ishida, Coord. Chem. Rev. 2018, 373, 333–356. (b) R. Francke, B. Schille, M. Roemelt, Chem. Rev. 2018, 118, 4631−4701.
OP13

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 51
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Spanish Metallocenes with a British accent. Titanocene Functionalized Mesoporous Silica Nanoparticles in the Fight against Cancer
Álvaro Serrano-Pindado, Diana Díaz-García, Miguel Díaz-Sánchez, Irene Mena-Palomo, Santiago Gómez-Ruiz, Sanjiv Prashar*
Departamento de Biología y Geología, Física y Química Inorgánica, E.S.C.E.T., Universidad Rey Juan Carlos, 28933 Móstoles, Madrid, Spain E-mail: [email protected]
The novel titanocene chemistry presented by Sanjiv Prashar, the only British Full Professor of Inorganic Chemistry in Spain, is a UK legacy from Professor Michael Lappert’s research group [1].
Since the discovery of cisplatin as a tumour agent, many organometallic compounds have been studied [2]. In this context, our group has worked for several years in the design of new metal-based drugs such as titanium and tin with promising results [3]. However, there are several problems with the use of metallodrugs in chemotherapy, namely solubility, stability, dosage etc. Mesoporous silica nanoparticles are a good alternative to overcome these problems due to their high biocompatibility, low toxicity and high loading capacity.
In this presentation, we describe the synthesis of a series of titanocene derivatives immobilized onto mesoporous silica nanoparticles (MSN) using two different routes, namely, simple grafting via protonolysis of the Ti-Cl bond and tethering using 3-mercaptopropyltriethoxysilane with which we have previously observed gives an increase in the biological activity [4]. All materials were characterized by different techniques, XRD, XRF, FTIR TEM, etc., and their cytotoxic activity tested against two breast cancer lines (MCF-7 and MDA-MB-231) to assess the behaviour of the different functionalized nanoparticles and possible improvements when using these nanostructured materials as biological vehicles.
References[1] http://blogs.rsc.org/dt/2014/10/21/the-influence-of-michael-lappert-on-the-chemistry-landscape/[2] K. Mjos, C. Orvig, Chem. Rev. 2014, 114, 8, 4540-4563.[3] Y. Ellahioui, S. Prashar, S. Gómez-Ruiz, Inorganics 2017, 5, 4.[4] S. Gómez-Ruiz, A. García-Peñas, S. Prashar, A. Rodríguez-Diéguez, E. Fisher-Fodor, Materials 2018, 11, 224.
OC14

Flash Presentations

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 53
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
d8···d10 RhI···AuI Interactions in Rh 2,6-Xylylisocyanide Complexes with Au(CN)2
–: Bond Analysis and Crystal EffectsVerónica Conejo-Rodríguez, Marconi N. Peñas-Defrutos and Pablo Espinet
IU CINQUIMA/Química Inorgánica, Facultad de Ciencias, Universidad de Valladolid, 47071-Valladolid, Spain. E-mail: veró[email protected]
Rhodium(I) (d8) complexes [Rh(CNR)4](A) (A = anion) are known to have a tendency to produce colored oligomers [Rh(CNR)4]n(A)n (n = 2,3) with Rh···Rh interactions.[1a] Similarly, the dicyanoaurate(I) (d10) linear anion has the tendency to produce uncoloured [Au(CN)2
–]n oligomers via Au···Au interactions. Separately, these self-assemblies are responsible for fascinating photochemical properties.[1b] As a result of the intrinsic propensity of RhI (d8) and the AuI (d10) complexes to establish homometallic interactions, with interesting properties,[2] we have now envisaged the combination of these metal complexes resulting in new heterobimetallic complexes.
Figure 1. a) Synthesis of the new heterobimetallic complex 2 and its precursor 1. b) Crystallized Polymorphs of 2.
In this communication we report our studies on the combination of RhI and AuI complexes (Figure 1a), and the physical and photochemical properties of the new molecular assemblies observed.
Crystallization of 2 afforded three differently coloured crystals (Figure 1b), with the same composition but different X-ray structure. These structures display Rh···Au interactions (3.1–3.5 Å). The various contributions responsible for the crystal stabilizations have been studied with the help of the theoretical calculations (DFT-MO). The calculations also allow us to explain the colour tendencies observed in drop-cast film spectra.
References[1] a) Y. Chen, K. Li, H.O. Lloyd, W. Lu, S. S-Y. Chui,C-M. Che, Angew. Chem. Int. Ed., 2010, 49, 9968 –9971. b) G. Cui, X-Y. Cao, W-H. Fang, M. Dolg, W. Thiel, Angew. Chem. Int. Ed., 2013, 52, 10281.[2] a). V. W.-W.Yam,V. K.-M. Au,S.Y.-L. Leung, Chem. Rev., 2015, 115, 7589−7728.
FP1

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 54
Lanthanide Cyclobutadienyl Sandwich ComplexesAnindita Chakraborty1, Richard A. Layfield1*
1Department of Chemistry, School of Life Sciences, University of Sussex, Falmer, Brighton, BN1 9QJ (U.K.) E-mail: [email protected]
Carboyclic π-conjugated ligands such as cyclopentadienyl and cyclooctatetraene have long been used in rare-earth organometallic chemistry.1-3 However, rare-earth complexes of dianionic cyclobutadienyl ligands, [Cb]2-, have, until recently, remained unexplored. Our group recently reported the first rare-earth Cb complexes,4 finding that the potassium cyclobutadienyl [K2{h
4-C4(SiMe3)4}] reacted with [LnCl3(THF)3.5] [Ln = Y, Dy], to yield the bis-Cb sandwich complexes [Ln{h4-C4(SiMe3)4}{h4-C4(SiMe3)3-k-(CH2SiMe2}]2-, with a tuck-in silylmethyl ligand arising from deprotonation of an SiMe3 group (Figure 1). The dysprosium version of this compound is a single-molecule magnet (SMM).
Continuing our work on cyclobutadienyl coordination chemistry, we here present the reactivity of a wider range of rare-earth elements with the alkali metal salts [K2{h
4-C4(SiMe3)4}] (M = Na, K) (Figure 1). Single-crystal X-ray diffraction revealed the reactivity to be dependent on the alkali metal, with formation of a series of iso-structural compounds [Ln{h4-C4(SiMe3)4}{h4-C4(SiMe3)3-k-(CH2SiMe2}]2-
(Ln = Y, La, Pr, Dy and Lu) when M = potassium. In contrast, the new sodium cyclobutadienyl reacts with lanthanide salts to yield [Ln{h3-C4(SiMe3)4(H)}{h4-C4(SiMe3)3-k-(CH2SiMe2}]2- complex, which features a protonated h3-cyclobutenyl ligand and a tuck-in silylmethyl group.
Figure 1. Diverse rare-earth cyclobutadienyl complexes
References[1] W. J. Evans, Organometallics 2016, 35, 3088-3100.[2] B. M. Day, F.-S. Guo, R. A. Layfield, Acc. Chem. Res. 2018, 51. 1880-1889.[3] F.-S. Guo, B. M. Day, Y.-C. Chen, M.-L. Tong, A. Mannikkamaki, R. A. Layfield, Science 2018, 362. 1400-1403.[4] B. M. Day, F.-S. Guo, S. R. Giblin, A. Sekiguchi, A. Mannikkamaki, R. A. Layfield, Chem. Eur. J. 2018, 24, 16779-16782.
FP2

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 55
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Bis(imino)pyridine Rhodium Complexes for Amine-Borane Dehydropolymerisation
Alice Johnson, Antonio J. Martínez-Martínez, Andrew S. Weller
Chemistry Research Laboratory, Department of Chemistry, University of Oxford, Mansfield Road, Oxford, OX1 3TA, UK.
E-mail: [email protected]
The catalysed dehydropolymerisation of amine–boranes gives B–N oligomeric or polymeric materials of empirical formula (H2BNRH)n [1] which are (valence)isoelectronic with technologically pervasive polyolefins but virtually unexplored. Despite the significant potential for these new materials as precursors to B-N based ceramics and single layer hexagonal B-N thin films or as high-performance polymers, the mechanism for, and control of, their construction remains generally ill–defined when compared to analogous olefin polymerisation or other C–C bond forming processes.[2]
Bis(imino)pyridine complexes of rhodium have been prepared and studied in the catalytic dehydropolymerisation of amine-boranes. The bis(imino)pyridine framework allows the steric and electronic properties to be fine-tuned by varying the R groups and such ligands have also been reported to stabilise 14-electron rhodium species, essential for the formation of σ-complexes with amine boranes.[3] With an aim to gain further mechanistic insight, a study of the reactivity of the complexes with amine-boranes has been carried out, allowing the isolation and characterisation of reactive intermediates and providing key information into the initial binding step of the amine-borane to the metal centre. The kinetics of the rhodium catalysed dehydropolymerisation reactions have been studied and the resultant polymers have been characterised by GPC and TEM. The effect of changing the anion of the catalysts has also been explored. The nature of the active catalyst, whether homogeneous or heterogeneous, in the dehydropolymerisation reactions will be discussed.
References[1] H. C. Johnson, T. N. Hooper and A. S. Weller, Top. Organomet. Chem., 2015, 49, 153-220.[2] A. L. Colebatch and A. S. Weller, Chem. Eur. J., 2019, 25, 1379-1390.[3] E. L. Dias, M. Brookhart and P. S. White, Chem. Commun., 2001, 423-424.
FP3

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 56
Bimetallic Aluminium Scorpionates For Coupling of CO2 And Epoxides Into Cyclic Carbonates Under Mild Conditions
Marta Navarro1, Luis F. Sánchez-Barba1,*, Andrés Garcés1,*, Juan Fernández-Baeza2, Agustín Lara-Sánchez2
1Universidad Rey Juan Carlos, Departamento de Biología y Geología, Física y Química Inorgánica, Móstoles-28933-Madrid, Spain.
E-mail: [email protected] 2Universidad de Castilla-La Mancha, Departamento de Química Inorgánica, Orgánica y Bioquímica, Centro de
Innovación en Química Avanzada (ORFEO-CINQA) Campus Universitario, 13071-Ciudad Real, Spain
In recent years, carbon dioxide has attracted significant attention as a sustainable C1 feedstock for various types of chemical reactions, which help to mitigate global warming. Thus, one of the most successful and widely studied processes is the 100% atom economical cycloaddition of CO2 and epoxides to produce cyclic carbonates, given their important commercial applications such as electrolytes in lithium-ion batteries, sustainable polar aprotic solvents and chemical intermediates in organic synthesis.
Several catalytic systems have been developed during the last years, particularly, organoaluminium complexes have been reported as this metal represents a nontoxic, earth abundant and a effective Lewis Acid to produce highly very active catalytic systems. In this context, bimetallic aluminium complexes have been successfully described as catalyst for the synthesis of cyclic carbonates at ambient temperature and 1 bar CO2 pressure. Among these catalysts, we have recently reported very efficient systems based on scorpionate complexes in combination with quaternary ammonium salts.1
Here we report the simple synthesis of a new family of NNN-scorpionate bimetallic aluminium complexes as efficient catalyst to produce cyclic carbonates by coupling reaction of CO2 with different mono- and disubstituted as well as biobased epoxides using low catalyst loadings under mild conditions. (Scheme 1).
Scheme 1. Synthesis of cyclic carbonates by using catalyst [Me2Al(k2-pbpamd)-AlMe2]
References[1]. Martínez, J.; Castro-Osma, J. A.; Alonso-Moreno, C.; Rodríguez-Diéguez, A.; North, M.; Otero, A.; Lara-Sánchez, A. Chem. Sus. Chem. 2017, 10, 1175-1185.
FP4

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 57
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Structure Property Relationship of Materials with Gold(I)-Gold(III) Interactions.
Matthias Dirk Boege1, Juergen Heck2, M. Concepción Gimeno1
1 ISQCH, Universidad de Zaragoza/CSIC, Calle de Pedro Cerbuna 12, 50009 Zaragoza/Es, E-mail: [email protected]
2 Universität Hamburg, Martin-Luther-King-Platz 6, 20146 Hamburg/D
The luminescence of polymeric nanomaterials derived from alternatingly stacked cationic gold(I) and monoanionic coinage metal complexes can be modulated by choice of the metals and ligands [1]. There are only a few published examples of supramolecular compounds with AuI-AuIII-interactions of a rather short distance which either show [2,3] or do not show luminescence [4] in the solid state. A recent study demonstrates that the shift of the stretching vibration band of the triple bond of the ligand is a probe for changes of its electron density [5]. Thus, the research on the relations of the structure of the supramolecular compounds and their optical and vibracional properties is of high interest.
The mixed-valent nitrile gold compound [Au(NCCy)2][AuCl4] (Cy = cyclohexyl) [3] and its novel nearly isostructural isocyanide derivative [Au(CNCy)2][AuCl4] (crystal packing structure in Figure 1) display one-dimensional stacking with AuI-AuIII-bonds but do not have luminescent properties in the solid crystalline state.
The structural, optical and vibracional properties of the introduced compounds and others of the same class but with different isocyanide and anionic ligands will be presented and discussed.
Figure 1. Crystal packing structure of [Au(CNCy)2][AuCl4] (P21/n; R1 = 0.0258).
References[1] W. Liu, M. Xie, X. Chang, S. Cao, C. Zuo, W.-F. Fu, C.-M. Che, Y. Chen, W. Lu, Angew. Chem., Int. Ed., 2018, 57, 6279-6283.[2] M. Boege, J. Heck, Chem. Eur. J., 2016, 22, 6787-6792.[3] M. Bardaji, M. J. Calhorda, P. J. Costa, P. G. Jones, A. Laguna, M. Reyes Perez, M. D. Villacampa, Inorg. Chem. 2006, 45, 1059-1068. [4] Y.-D. Chen, L.-Y. Zhang, Y.-H. Qin, Z.-N. Chen, Inorg. Chem., 2005, 44, 6456-6462.[5] T. A. Engesser, C. Friedmann, A. Martens, D. Kratzert, P. J. Malinowski, I. Krossing, Chem. Eur. J., 2016, 22, 15085-15094.
FP5

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 58
Novel arene Ru(II) compounds with N-phenanthroline glycosylamine ligands as potential anticancer agents
Elena de la Torre-Rubio1, Isabel de la Cueva-Alique1, Lourdes Gude1, María-Selma Arias-Pérez1, Eva Royo1
1Departamento de Química Orgánica y Química Inorgánica, Instituto de Investigación Química Andrés M. del Río (IQAR), Universidad de Alcalá, 28805 Alcalá de Henares, Madrid, Spain.
E-mail: [email protected]
The search of new metal-based antitumor drugs with novel mechanisms of action different from those found in cisplatin, has become an emerging area of research. Many carbohydrates are known to be generally good binders for nucleic acids while Warburg effect is recognized as one of the hallmarks of cancer. Thus, functionalization of drugs with carbohydrates is an anticancer strategy that has gained great interest in recent years(1-3). On the other hand, polypyridyl compounds such as 1,10-phenanthroline are powerful bidentate metal chelating ligands, and a variety of metal compounds have been extensively studied for their anticancer properties, usually related with their ability to act as DNA intercalators and groove binders(4). Furthermore, they serve as scaffolds for several potent stabilizers of DNA G-quadruplexes, which are being investigated as potential targets for anticancer drug development(5). Herein, we report the synthesis and characterization of a new family of water soluble arene Ru(II) compounds of general formula [RuCl(p-cymene){(N-R)5-amino-1,10-phenanthroline}]Cl; R = Glc, Rham, Xyl, Man (glucose Glc, rhamnose Rham, xylose Xyl and mannose Man). A comparative study of their DNA interactions and cytotoxic activity with those of N-phenanthroline glycosylamine organic derivatives(6) is currently under investigation.
References[1] E.C. Calvaresi and P.J. Hergenrother, “Glucose conjugation for the specific targeting and treatment of cancer”, Chem. Sci. (2013) 4:2319.[2] M. Tanasova, V.V. Begoyan and L.J. Weselinski, “Targeting Sugar Uptake and Metabolism for Cancer Identification and Therapy: An Overview”, Curr. Topics in Med. Chem. (2018) 18:467.[3] A. Pettenuzzo, D. Montagner, P. McArdle and L. Ronconi, “An innovative and efficient route to the synthesis of metal-based glycoconjugates: proof-of-concept and potential applications”, Dalton Trans. (2018) 47:10721.[4] L. Salassa, “Polypyridyl Metal Complexes with Biological Activity”, Eur. J. Inorg. Chem. (2011) 2011:4931.[5] N. S. Ilyinsky, A. M. Varizhuka, A. D. Beniaminova, M. A. Puzanova, A. K. Shchyolkinaa, and D. N. Kaluzhnya, “G-Quadruplex Ligands: Mechanisms of Anticancer Action and Target Binding”, Mol. Biol. (2014) 48:778.[6] K. Duskova, S. Sierra, M.S. Arias-Pérez, L. Gude, “Human telomeric G-quadruplex DNA interactions of N-phenanthroline glycosylamine copper(II) complexes”, Bioorg. Med. Chem. (2016) 24:33.
FP6

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 59
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Potent Half-Sandwich Iridium(III) Complexes as Mitochondria-Targeted Anticancer Drugs
Ana C. Carrasco1, José Javier Conesa2, Vanessa Rodríguez-Fanjul1, Yang Yang3, José L. Carrascosa4, Peter Cloetens3, Eva Pereiro2, Ana M. Pizarro1
1 IMDEA Nanociencia, Faraday 9, 28049 Madrid, E-mail: [email protected]
2 MISTRAL beamline, ALBA Synchrotron Light Source, Cerdanyola del Vallès, 08290 Barcelona, Spain 3 ID16A beamline, European Synchrotron Radiation Facility (ESRF), 38043 Grenoble, France
4 Department of Structure of Macromolecules, Centro Nacional de Biotecnología/CSIC, 28049 Madrid, Spain
Organoiridium half-sandwich complexes are emerging as excellent candidates with potential medical applications. Ir(III)-Cp* complexes present exciting novel mechanisms of action, largely based on intracellular redox imbalance [1]. These organometallic complexes offer enormous scope due to their versatile structures; by modification of the different building blocks (cyclopendadienyl (Cp) ring, XY chelating ligand and Z monodentate ligand) in the general structure [η5-C5Me4R)Ir(XY)Z]0/+, we may affect dramatically the reactivity and thus the cytotoxic effect of Cp complexes of iridium(III) [2].We have designed a series of cyclopentadiendienyl Ir(III) complexes bearing a tethered pyridine that binds to the metal forming and additional chelating ring in the structure. The Ir-Npy(tether) bond is susceptible to cleavage in aqueous solution and in a biologically relevant time scale. The dissociation of the nitrogen would render the metal susceptible to macromolecule attack inside the cell, thereby activating the pro-drug, while Ir-Npy(tether) bond association protects the metal toward ligand substitution. In other words, in our structure the metal centre is sheltered against off-target reactivity of the metal, yet the reactivity of the pharmacophore stays intact.Preliminary cell viability results of our Ir(III) complexes, activatable by dissociation of the Ir-Npy(tether) bond, show that the cytotoxicity, as determined by the IC50 value, can be modified by suttle modifications in the XY ligand. The IC50 values presented by our Ir(III) family are strickingly low (nM range) in the human breast cancer cell line MCF7 [3]. Our data also show that the tether ring is a necessary feature of the Ir(III) organometallic structure to achieve sub-micromolar activity (Table 1) [3]. Additional cell-based studies using cryo-soft X-ray tomography (ALBA), X-ray fluorescence nanoimaging (ESRF) and cryo-TEM (CNB-CSIC) show that complex 1 targets and accumulate uniquevocally and solely in the mitochondria, revealing that this organelle is the target site of the iridium complexes [4]. Ir(III) tethered complexes could become a new family of mitochondria-targeting, potent activatable pro-drugs in cancer research.
References[1] Z. Liu, P.J. Sadler, Acc. Chem. Res., 2014, 47, 1174.[2] Z. Liu, L. Salassa, A. Habtermariam, A.M. Pizarro, G.J. Clarkson, P.J. Sadler, Inorg. Chem. 2011, 50, 5777.[3] A.C. Carrasco, V. Rodríguez-Fanjul, A.M. Pizarro, submitted.[4] J.J. Conesa, A.C. Carrasco, V. Rodríguez-Fanjul, Y. Yang, J.L. Carrascosa, P. Cloetens, E. Pereiro, A.M. Pizarro, submitted.
Table 1. Antiproliferative activity of complex [(h5:κ1-Cp*py)Ir(phpy)]PF6 (1). Cell viability as determined by IC50 data (concentration of drug required to inhibit cell growth by 50% compared to control) in several cell lines after 24 h of exposure to complex 1 and allowed to recover for 72 h. Cisplatin was used as positive control.
FP7

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 60
Coupling of Reactive Carbene Precursors: Isolation of Intermediate Palladium Complexes
Francisco Villalba, and Ana C. AlbénizIU CINQUIMA/Química Inorgánica. Universidad de Valladolid. 47011 Valladolid (Spain)
[email protected]; [email protected]
Palladium-catalyzed cross coupling reactions using N-tosylhydrazones or diazo compounds as a coupling partner have been extensively used for the last years. These reactions have special relevance because in many cases several C-C or C-X bond are formed by double functionalization of the carbene fragment.1 N-tosylhydrazones are able to generate by a non-stabilized diazo compound a very reactive palladium carbene complexes.2 The generally accepted mechanism for these catalytic reactions involve three fundamental steps A-C (Figure 1) but detailed information about the intermediates of each step is still scarce. We are interested in isolating and studying the intermediate complexes involved in steps A-C (Figure 1).
Starting from [Pd(L-L)(C6F5)(Br)] or [Pd(L-L)(C6F5)(NCMe)](BF4) complexes (L-L = bipy, dppe, dppf), we have obtained some palladium complexes in which the N-tosylhydrazonate is coordinated and may play a role in step B of the reaction.
Step C has been unambiguously proved by the isolation of intermediate 1 generated just after the migratory insertion according to the reaction of [Pd(L-L)(C6F5)(NCMe)](BF4) with the corresponding diazo compound (Figure 1). Complex 1 has been fully characterized by NMR spectroscopy and X-ray diffraction studies.
Figure 1. Fundamental steps (A-C) in Pd-catalyzed cross coupling reactions with precursors of carbene reagents. Complex 1 was isolated after migratory insertion step.
References[1] Xia, Y.; Qiu, D.; Wang, J. Chem. Rev. 2017, 117, 13810−13889.[2] Barluenga, J.; Valdés, C. Angew. Chem. Int. Ed. 2011, 50, 7486 – 7500.
FP8

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 61
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
BN-Arenes: Synthesis, Reactivity and PropertiesAlberto Abengózar1, Isabel Valencia1, Patricia García-García1, David Sucunza1, Miguel Ángel
Fernández-González2, Luis Manuel Frutos2, Antonio Salgado3, Adrián Pérez-Redondo1, Juan J. Vaquero1
1Departamento de Química Orgánica y Química Inorgánica, Instituto de Investigación Química “Andrés M. del Río” (IQAR), Universidad de Alcalá, Madrid (Spain).
E-mail to [email protected] 2Departamento de Química Analítica, Química Física e Ingeniería Química, Instituto de Investigación Química
“Andrés M. del Río” (IQAR), Universidad de Alcalá, Madrid (Spain) 3Centro de Espectroscopía de Resonancia Magnética Nuclear, Universidad de Alcalá, Madrid (Spain)
The chemistry of organoboron compounds has been primarily dominated by their use as powerful reagents in synthetic organic chemistry. Recently, a common applied strategy for the incorporation of boron as part of a functional target structure, is the replacement of a C=C bond in an aromatic ring by an isoelectronic B-N bond. These novel compounds retain aromaticity and are structurally analogous to their all-carbon counterparts, but have altered properties induced by the presence of a dipole [1].
In this context, we have developed an efficient synthesis of previously unknown helical BN-benzo[c]phenanthrene [2], BN-phenanthrene [3] and BN-chrysene in only three-four steps from commercial materials. Moreover, different methods for selective C-H functionalization of the BN-phenanthrene core have been found. Thus, treatment with an organolithium and a carbonyl compound leads to C3-substituted BN-arenes through an unprecedented reaction pathway which is initiated by the organolithium coordination to the boron atom of the BN-heterocycle.
References[1] C. R. McConnell, S.-Y. Liu, Chem. Soc. Rev., 2019 (DOI: 10.1039/c9cs00218a). [2] A. Abengózar, P. García-García, D. Sucunza, A. Pérez-Redondo, J. J. Vaquero, Chem. Commun., 2018, 54, 2467. [3] A. Abengózar, P. García-García, D. Sucunza, L. M. Frutos, O. Castaño, D. Sampedro, A. Pérez-Redondo, J. J. Vaquero, Org. Lett., 2017, 19, 3458.
FP9

Poster Presentations

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 63
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Heterobimetallic Lanthanide Isocarbonyl Complexes as Single-Molecule Magnets
Richard Collins1,2, Jinkui Tang3,4, Richard A. Layfield*1
1 Department of Chemistry, School of Life Sciences, University of Sussex, Brighton, BN1 9QJ, UK, 2 School of Chemistry, The University of Manchester, Manchester, M13 9PL, U.K.,
3 State Key Laboratory of Rare Earth Resource Utilization, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, P. R. China,
4 University of Science and Technology of China, Hefei 230026, P. R. China E-mail: [email protected]
4f/3d isocarbonyl complexes have previously been shown to display single-molecule magnet (SMM) behaviour with large energy barriers to magnetic reversal (Figure 1).[1] Isocarbonyl bridging ligands present an opportunity to explore the structural and electronic effects of bridging systems in relation to SMMs, as polymetallic complexes may provide higher blocking temperatures and energy barriers.
Here, research is focused on utilising the heavier d-block metal tungsten, in combination with 4f elements to investigate the effect on both the structural and magnetic properties in order to expand on the understanding of isocarbonyl SMMs. (Figure 2).[2,3]. The exhibited physical, electronic and magnetic properties are probed through the characterisation of a number of new CO-bridged 4f/5d complexes.
Figure 1: A 4f/3d carbonyl bridged dimer, DyFp, demonstrating SMM behaviour
Figure 2: 4f/5d isocarbonyl bridged complexes, crystal structure, out-of-phase susceptibility and magnetic hysteresis of DyW
References[1] T. Pugh, N. F. Chilton, R. A. Layfield, Angew. Chem. Int. Ed., 2016, 55, 1 – 5 [2] A. E. Crease, P. Legzdins, J. Chem. Soc., Dalton Trans., 1973, 1501-1507[3] R. Collins, R. A. Layfield, Manuscript in preparation, 2019
PP1

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 64
Unusual ligand rearrangement of N-phosphinoguanidinato compoundsFernando Carrillo-Hermosilla1, Estefanía Huergo,1 Rafael Fernández-Galán,1 Alberto Ramos,1
Antonio Antiñolo, 1 Antonio Rodríguez-Diéguez,2 Daniel García-Vivó3
1 Dpto. de Química Inorgánica, Orgánica y Bioquímica, Centro de Innovación en Química Avanzada (ORFEO-CINQA) Fac. de CC. y TT. Químicas. Universidad de Castilla-La Mancha. 13071-Ciudad Real (Spain)
E-mail: [email protected] 2 Dpto. de Química Inorgánica, Facultad de Ciencias, Universidad de Granada. 18071-Granada (Spain)
3 Departamento de Química Orgánica e Inorgánica/IUQOEM, Universidad de Oviedo. E-33071 Oviedo (Spain)
The ongoing demand for novel ligands in organometallic and coordination chemistry has prompted the development of a series of bidentate N,P-donor ligands in the last decades. In the last years our group has focused on the development of catalytic methods to prepare guanidines and the coordination chemistry of guanidinato ligands with transition metals and main group elements.[1] As an extension of this work and encouraged by the lack of examples of anionic N-phosphinoguanidinato compounds, here we report the synthesis of new examples of this kind of interesting ligands (1) from trisubstituted guanidines and their reactivity towards metal alkyls. These reactions afford phosphinimine-amidinato derivatives (3), via an unprecedented rearrangement of an initial N-phosphinoguanidinato intermediate (2).[2]
References[1] C. Alonso-Moreno, A. Antiñolo, F. Carrillo-Hermosilla, A. Otero, Chem. Soc. Rev. 2014, 43, 3406-3425. [2] R. Fernández-Galán, A. Ramos, E. Huergo, A. Antiñolo, F. Carrillo-Hermosilla, A. Rodríguez-Diéguez, D. García-Vivó, Chem. Commun. 2019, 55, 2809-2812.Financial support acknowledgment: MINECO Project nº CTQ2016-77614-P
PP2

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 65
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Decarbonylation Processes in k3-(N^C^C)Gold(III) CarboxylatesEstíbaliz Merino1,2, Hélène Beucher1, Alexandre Genoux1, Thomas Fox1, Cristina Nevado1
1Department of Chemistry, University of Zurich, Winterthurerstrasse 190, Zurich CH 8057, Switzerland 2 Current address: Department of Organic and Inorganic Chemistry, Chemical Research Institute Andrés M.
del Río (IQAR), University of Alcala, 28805, Alcalá de Henares, Spain E-mail: [email protected]
Metallocarboxylic acids are key intermediates in various transition metal mediated processes that involve CO and H2O, including the water-gas shift reaction (WGSR). Most metal carboxylates undergo thermal decomposition releasing CO2 with concomitant formation of the corresponding metal hydride. Recently, the synthesis of gold(III)-CO and dinuclear (m-CO2-k
2C,O)-gold(III) complexes supported by a k3-(C^N^C) ligand and their preference for decarboxylation pathways has been described [1].
Here, we present a new class of k3-(N^C^C)AuIII carboxylates stabilized by a k3-(N^C^C) pincer ligand template. For the first time, a h1-AuIII-C(O)-OH complex has been characterized under cryogenic conditions as a result of a nucleophilic attack of an ammonium hydroxide onto a dinuclear m-CO2-k
3-(N^C^C)AuIII precursor. An unusual decarboxylation process is observed by thermal decomposition in contrast to typical decarboxylation pathways observed in related metallocarboxylic acids, highlighting the importance of the ligand template in modulating the reactivity of metallocarboxylate species and the mechanistic diversity in their transformations [2].
References[1] D.-A. Roşca, J. Fernandez-Cestau, J. Morris, J. A. Wright, M. Bochmann, Sci. Adv. 2015, 1, e1500761.[2] H. Beucher, E. Merino, A. Genoux, T. Fox, C. Nevado, Angew. Chem. Int. Ed. 2019 DOI: 10.1002/anie.201903098.
PP3

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 66
Bipy and phen ring-opening at Re(I) carbonyl complexes.Purificación Cañadas1, Julio Pérez1,2, Lucía Riera2
1Departamento de Química Orgánica e Inorgánica, Universidad de Oviedo, Julián Clavería 8, 33006 Oviedo E-mail: [email protected]
2Centro de Investigación en Nanomateriales y Nanotecnología (CINN), CSIC-Universidad de Oviedo-Principa-do de Asturias, Avda de la Vega 4-6, 33940 El Entrego (Spain)
2,2’-Bipyridine (bipy) and 1,10-phenanthroline (phen) have been widely employed in all areas of coordination chemistry; however, prior to our work, examples of dearomatization of transition metal-coordinated bipy and phen under mild conditions were hardly known.[1] We had previously found that for [Re(CO)3(bipy)(N-MeIm)]OTf (N-MeIm= N-methylimidazole), the deprotonation of the central CH group of the imidazole ligand triggered the nucleophilic attack onto the C6 atom of bipy, which became dearomatized. Afterwards, two successive methylations of the dearomatized pyridyl moiety promoted the cleavage of a C-N bond, leading to a pyridyl ring-opening product (see Scheme 1a).[2]
Herein we report the analogous reactivity of cis-[Re(CO)2(N-N)(N-RIm)(PMe3)]OTf (N-N= bipy, phen; N-RIm= N-alkylimidazole) compounds, in which ring opening of a pyridyl moiety of a phen ligand is achieved for the first time (see Scheme 1b).
Scheme 1. Reactivity of [Re(CO)2(L)(N-N)(N-MeIm)]OTf (L= CO, PMe3; N-N= bipy, phen) compounds
References[1] a) S. Leelasubcharoen, K,-C. Lam, T. E. Concolino, A. L. Rheingold, K. H. Theopold, Organometallics 2001, 20, 182-187; b) L. M. Kobriger, A. K. McMullen, P. E. Fanwicck, I. P. Rothwell, Polyhedron 1989, 8, 77-81; c) C. Weetman, M. S. Hill, M. F. Mahon, Polyhedron 2016, 103, 115-120[2] M. A. Huertos, J. Pérez, L. Riera, J. Am. Chem. Soc. 2008, 130, 5662-5663.
PP4

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 67
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Reactivity of Phosphine-Stabilized Silylene Rhodium ComplexN. Almenara1, J. I. Miranda2, A. Rodríguez-Diéguez3, M. A. Garralda1, M. A. Huertos*,1,4
1Facultad de Química, Universidad del País Vasco (UPV/EHU), 20018, San Sebastián, Spain 2SGIker NMR Facility, Universidad del País Vasco (UPV/EHU), 20018 San Sebastián, Spain
3Departamento de Química Inorgánica, Universidad de Granada, 18071, Granada, Spain 4IKERBASQUE, Basque Fundation for Science, 48013, Bilbao, Spain
Metal silylene complexes are good candidates to study small molecule activations due to the high reactivity of the silicon atom [1]. This kind of complexes have been proposed as intermediates in hydrosilylation of multiple bonds [2].
Treatment of Wilkinson’s catalyst with bis(thioether)-dihydrosilyl ligand in the presence of a halogen-extractor (NaBArF
4), led to the formation of the cationic complex {Rh(H)2[Si(PPh3)(o-C6H4SMe)2](PPh3)}[BArF
4] 1. DFT calculations of 1 shows that the LUMO is mostly centered on the silicon atom.
Phosphine-stabilized silylene complex 1 reacts with water, methanol and isopropanol to give the alkoxysilyl-Rh(III) complexes (2, 3 and 4). Moreover, the dihydrido-silylene-Rh(III) compound 1 reacts with benzophenone leading to the formation of diphenylmethoxysilyl-Rh(III) complex, {Rh(H)[Si(OCHPh2)(o-C6H4SMe)2](PPh3)}[BArF
4] 5, which has been identified as intermediate in the mechanism of hydrosilylation of ketones proposed by Gade [2].
References[1] Meltzer, A.; Inoue, S.; Präsang, C.; Driess, M. J. Am. Chem. Soc. 2010, 132, 3038-3046.[2] Schneider, N.; Finger, M.; Haferkemper, C.; Bellemin-Laponnaz, S.; Hofman, P.; Gade, L. H. Angew. Chem. Int. Ed. 2009, 48, 1609–1613.
2
f
1
3 & 4
(
5
PP5

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 68
Novel Iridapyrazole based complexes, reactivity and catalytic activityItxaso Bustos1, María Ángeles Garralda*1, Claudio Mendicute-Fierro1
1 UPV/EHU, Faculty of Chemistry, San Sebastián-Donostia, 20018 E-mail: [email protected]
Metallacycles have been studied thoroughly in the last decades. Aromatic metallacycles containing a transition metal are very interesting due to their unique properties. Aromatic iridacycles such as iridabenzenes[1] or different heteroatom-containing ones[2] have been an important research object lately.
Metallapyrazoles were first reported by our group, that synthesised an iridapyrazole (complex 1)[3]. In this work, the iridapyrazole complex has been synthesised via a new method.
As for the reactivity of (1), this was tested towards acids and bases affording protonated and deprotonated species respectively. Besides, we were able to carry out the methylation of one of the iridapyrazole’s nitrogen. We also achieved to study how of other ligands affects (1) by replacing the original chloride ligand with nitrogen containing ligands, phosphines, olefins, thiocyanate and iodide. Finally, some of these complexes proved to have a good performance in the methanolysis of ammonia- and amino-borane substrates.
References[1] Bleeke, J.R. ; Acc. Chem. Res. 2017, 40, 1035.[2] Grieb, A.L. et al. ; J. Organomet. Chem., 2012, 713, 163.[3] Zumeta, I. et al.; Inorg. Chem. 2016, 55, 10284
PP6

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 69
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Synthesis, structure, and properties of monopentamethylcyclopentadienyltitanium(III) dihalide complexes
Estefanía del Horno1, Reyes Jiménez-Aparicio2, Miguel Mena1, Adrián Pérez-Redondo1, José Luis Priego2, Carlos Yélamos1
1Departamento de Química Orgánica y Química Inorgánica, Instituto de Investigación Química “Andrés M. del Río” (IQAR), Universidad de Alcalá 28805 Alcalá de Henares, Madrid (Spain)
E-mail: [email protected] 2Departamento de Química Inorgánica, Universidad Complutense de Madrid 28040 Madrid (Spain)
Low-valent early transition-metal complexes are receiving increasing attention due to their implication as catalysts in bond-forming reactions useful in organic synthesis [1]. Traditionally, the preparation of these compounds requires strong reducing agents, such as alkali/alkaline-earth derivatives or metal amalgams and alloys, but salt-free reduction methods exhibit clear advantages. We have recently reported that thermolysis or hydrogenolysis of [Ti(h5-C5Me5)Cl2Me] leads to the clean formation of the titanium(III) compound [{Ti(h5-C5Me5)Cl(m-Cl)}2] along with volatile by-products [2].
In this communication, we will describe the synthesis, structure and magnetic properties of monopentamethylcyclopentadienyltitanium(III) dihalide complexes [{Ti(h5-C5Me5)X(m-X)}2] (X = Cl, Br, I), as well as their reactivity with Lewis bases or lithium reagents. For instance, the treatment of the chloride compound [{Ti(h5-C5Me5)Cl(m-Cl)}2] with isocyanides leads to new dinuclear titanium(III) adducts (see Figure). Subsequently, the tert-butylisocyanide ligands undergo a reductive coupling reaction to give a titanium(IV) iminoacyl derivative.
References[1] (a) E.P. Beaumier, A.J. Pearce, X.Y. See, I.A. Tonks, Nat. Rev. Chem. 2019, 3, 15-34. (b) H. Tsurugi, K. Mashima, Acc. Chem. Res. 2019, 52, 769-779.[2] M. García-Castro, C. García-Iriepa, E. del Horno, A. Martín, M. Mena, A. Pérez-Redondo, M. Temprado, C. Yélamos, Inorg. Chem. 2019, 58, 5314-5324.Financial support acknowledgments: MCIU (PGC2018-094007-B-I00) and Universidad de Alcalá (CCG2018/EXP-008).
PP7

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 70
Ligands with a pyridone or amido functionality: Assessment of their cooperating ability in the Pd-catalyzed direct arylation of arenes.
Cintya Pinilla, Vanesa Salamanca, Francisco Villalba, Ana C. AlbénizIU CINQUIMA / Química Inorgánica, University of Valladolid, 47011-Valladolid, Spain.
E-mail: [email protected]
An alternative method to the conventional Pd-catalyzed cross coupling reactions is the C-H activation of the arene used as reagent. Most of the previous literature reports use an external base (acetate or carbonate) to assist the C-H cleavage step, usually rate-limiting, through the TS in Figure 1.[1] We have achieved the acceleration of the catalytic direct arylation of arenes by incorporation of the base on the ligand structure (a keto group) so it assists the C-H cleavage through the TS* in Figure 1. Thus, the use of [2,2-bipyridin]-6(1H)-one reduces the reaction time in the case of pyridine (Ar2 = Py, Eq. 1) or allows the direct arylation of other arenes (Ar2 = toluene, etc, Eq. 1).[2]
Figure 1.
In this work we have tested other realated ligands which have a pyridone or an amide group and could act as a base because of the keto-enol tautomerism (Eq. 1). These ligands have been studied in different catalytic direct arylations of arenes (pyridine, toluene and ethyl benzoate), and their efficiency evaluated. The synthesis and study of well-defined palladium complexes with the corresponding ligands allows to explain their behaviour as catalyst.
References[1] (a)Davies,D. L.; Donald, S. M. A.; Macgregor, S. A., J. Am. Chem. Soc., 2005, 127, 13754. (b) García-Cuadrado, D.; Braga, A. A.C.; Maseras, F.; Echavarren, A. M., J. Am. Chem. Soc., 2006, 128, 1066. (c) Ackermann, L., Chem. Rev. 2011, 111, 1315.[2] Salamanca, V; Toledo, A; Albéniz, A.C., J. Am. Chem. Soc., 2018, 140, 17851.
PP8

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 71
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Nano Base Metal Catalysts: Synthesis and CharacterisationAlana Smith1, Dr Phil Dyer1, Dr Simon Beaumont1, Dr Xavier Baucherel2,
Dr Leon van de Water2
1Department of Chemistry, Durham University, South Road, Durham, DH1 3LE, E-mail: [email protected]
2Johnson Matthey PLC, PO Box 1, Billingham, Cleveland, TS23 1LB
Grafting discrete molecular organometallic catalysts directly to a metal oxide support offers a new strategy for combining the selectivity and activity of homogeneous catalysis with the robustness and ease of separation of their heterogeneous counterparts.1 Here, the metal oxide acts as a discrete rigid ligand to the metal centre of the bound organometallic species – an approach known as Surface Organometallic Chemistry (SOMC).2 Since the oxide surface forms an element of the metal’s primary coordination sphere, the oxide directly affects the catalyst’s formation, stability, selectivity and activity.
Here, an extension to this approach is taken in which appropriately prepared/tuned oxide supports (e.g. silica) are treated with hydrocarbyl organometallic complexes, a process which results in surface-bound organometallic species via ligand protonation. The reactivity of the silica surface can be controlled either thermally (calcination) or chemically, both of which will alter the number and density of reactive silanol (Si-OH) sites and hence, the loading of the organometallic species. Subsequent thermal or reduction processes are being explored as methods for the generation of surface-stabilised metallic particles, which are potential catalysts in their own right. It is envisaged that initial modification of the oxide support will also influence the lability and rates of agglomeration of the surface metallic species and hence the growth of metal nanoparticles.
This poster focusses principally on the use of hydrocarbyl organocobalt precursors, which were chosen to provide silica-supported cobalt nanoparticles free from heteroatom poisons.
References[1] C. Copéret, M. Chabanas, R. P. Saint-Arroman, J-M. Basset, Angew. Chem. Int. Ed., 2003, 42, 2, 156-181[2] C. Copéret, F. Allouche, K. W. Chang, M. Conley, M. F. Delley, A Fedorov, I. Moroz, V. Mougel, M. Pucino, K. Searles, K. Yamamoto, P. Zhizhko, Angew. Chem. Int. Ed., 2018, 57, 22, 6398-6440
PP9

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 72
Graphene-based Heterogeneous Cu-catalysts in Borylation ReactionsM. Franco Fernández, M. Tortosa, M. B. Cid
Departamento de Química Orgánica, Universidad Autónoma de Madrid, Cantoblanco, Madrid. E-mail: [email protected]
Keywords: Heterogeneous Catalysis ● Boron ● Copper ● Graphene ● Green Chemistry
The increasing interest for the green chemistry over the last years has driven researchers to the development of more sustainable techniques. One of these, is the employment of heterogeneous catalysts, which have the advantages of being less toxic, easy to handle and reusable.1 Graphene derivatives, due to its unique properties, are considered to be ideal two-dimensional supports to improve the catalytic properties of metal nanoparticles.2 In this context, we have developed a cheap, robust, effective, reusable and stable Cu2O catalyst supported on graphene nanoplatelets (Graphenit-Cu(I)),3 which successfully catalyses different reactions.
Figure 1. Graphical representation of the Graphenit-Cu(I)
We present the study of the properties and catalytic activity of this material in C-B bond formation reactions compared to other supported copper catalysts. The material have shown a good catalytic activity in the borylation reaction of halides. Unlike inactive Cu2O, Graphenit-Cu(I) catalyses the reaction of a wide range of substrates including alkyl and aryl halides without using any ligand and can be recycled up to 7 times without losing effectiveness.
Figure 2. Catalytic activity of the material in the borylation reaction.
The different techniques employed during the study of the catalyst such as TXRF, XRD and SEM, have demonstrated that the Cu (I) present on the material is bench-stable and it remains attached to the surface after the reaction. The synergistic effect between the metal and the surface is being studied using theoretical calculations.
Acknowledgements: Financial support from Spanish Government (CTQ2016-78779-R) and the European Research Council (ERC Starting Grant DAUBOR) is gratefully acknowledged.
References[1] Hübner, S.; de Vries, J. G.; Farina, V. Adv. Synth. Catal. 2016, 358, 3–25.[2] Navalon, S.; Dhakshinamoorthy, A.; Alvaro, M.; Garcia, H. Coord. Chem. Rev. 2016, 32, 99.[3] De Angelis, S.; Franco, M.; Triminì, A.; González, A.; Sainz, R.; Degennaro, L.; Romanazzi, G.; Carlucci, C.; Petrelli, V.; De la Esperanza, A.; Goñi, A.; Ferritto, R.; Aceña, J. L.; Luisi, R.; Cid, M. B. Chem. Asian J. Submmited
PP10

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 73
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Synthesis and Characterization of Cyclopentadienyl Sulfur Niobium Complexes
Elena Álvarez-Ruiz, Manuel Gómez, Cristina Hernández-Prieto, Avelino Martín, Miguel Mena, Cristina Santamaría
Departamento de Química Orgánica y Química Inorgánica. Instituto de Investigación Química “Andrés M. del Río” (IQAR), Universidad de Alcalá, Campus Universitario, E-28805 Alcalá de Henares, Madrid, Spain.
E-mail. [email protected]
The chemistry of soluble transition-metal sulfides species [1] revealing well-defined and characterized structures are attractive candidates for a better understanding of most of the metalloproteins of biological significance [2] such as nitrogenase, hydrogenase...In the last years there has been a growing interest not only in the crucial role of the metal centres but also in the careful selection/design of suitable auxiliary ligands. The presence of the cyclopentadienyl ligand affords an accessible entry to di- and polynuclear soluble transition-metal sulfide complexes, [3] where the distinguishing chemical and electronic properties of the cyclopentadienyl ring make possible further studies.
A trimetallic cluster [Nb3(h5-C5Me5)3Cl3(m3-Cl)(m-S)3(m3-S)] (2) has been synthesized from the reaction
of [Nb(h5-C5Me5)Cl4] (1) with (Me3Si)2S, as sulfur source. The trinuclear nature of complex 2 has been established by single crystal X-ray diffraction analysis. Thermal treatment of 2 with SiH3Ph generated the dinuclear niobium(IV) complex [Nb2(h
5-C5Me5)2Cl2(m-S)2] (3) in a quantitative way. A series of dinuclear niobium(IV) derivatives [Nb2(h
5-C5Me5)2R2(m-S)2] can easily be obtained from the reaction of 3 with 2 equiv of the corresponding alkylating reagents.
Financial support for this work was provided by the Ministerio de Ciencia, Innovación y Universidades (PGC2018-094007-B-I00) and the Universidad de Alcalá (CCG2018/ EXP-026). C. H.-P. thanks the Universidad de Alcalá for a predoctoral fellowship.
References[1] T. B. Rauchfuss, Inorg. Chem. 2004, 43, 14-26.[2] This field has been extensively reviewed. See, for example: Y. Ohki, K. Uchida, M Tada, R. E. Cramer, T.Ogura, T. Ohta, Nature Communications, 2018, 9, 3200.[3] M. Gómez, J. I González-Pérez, C. Hernández-Prieto, A. Martín, M. Mena, C. Santamaría, Inorg. Chem. 2019, 58, 5593-5602.
PP11

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 74
Digold C-C Cross Coupling Reactions: Mechanistic Insights
Juan Miranda-Pizarro, Jesús Campos, Ernesto CarmonaInstituto de Investigaciones Químicas (IIQ), Departamento de Química Inorgánica and Centro de Innovación en
Química Aplicada (ORFEO-CINQA). Consejo Superior de Investigaciones Científicas (CSIC), Avda. Américo Vespucio 49, 41092 Sevilla, Spain, E-mail: [email protected]
The development of dialkyl biarylphosphines1 by Buchwald and coworkers in the late 90s have had a profound impact in homogeneous catalysis, which recently motivated us to explore the related dialkyl terphenylphosphines (PR2Ar’; Ar’ = C6H3-2,6-Ar2) analogues as bulkier and highly tuneable ligands2. In this context, electrophilic gold compounds stabilized by terphenylphosphines constitute a scarcely investigated territory considering the prevalence of related species based on biaryl phosphines. For instance, recent results from our group on [(PR2Ar’)Au(I)]+ systems, include the first transition metal-only frustrate Lewis pair3 or the first alkyl-bridged digold compounds4.
In this work we took advantage of the kinetic stabilization provided by terphenyl phosphines to examine the C-C coupling reaction that takes place at bridging digold compounds alike [Au2(µ-R)(PMe2Ar’)2][B(C6F5)4]. The use of gold compounds as catalysts for C-C coupling reactions, which presumably operate by Au(I)/Au(III) redox cycles, has emerged as a promising approach in recent years5, although the precise mechanisms are yet to be understood. Here we provide kinetic and low temperature NMR spectroscopy studies in order to understand the mechanism of these transformations that seem to operate without accessing the +3 oxidation state at gold. Our results indicate that C-C coupling events arise from the combination of a cationic bridging digold fragment and a terminal alkyl gold species.
References[1] D. W. Old, J. P. Wolfe, and S. L. Buchwald, J. Am. Chem. Soc. 1998, 120, 9722-9723.[2] L. Ortega-Moreno, M. Fernandez-Espada, J. J. Moreno, C. Navarro-Gilabert, J. Campos, S. Conejero, J. López-Serrano, C. Maya, R. Peloso, E. Carmona, Polyhedron 2016, 116, 170 – 181.[3] J. Campos, J. Am. Chem. Soc. 2017, 139, 8, 2944-2947.[4] M. F. Espada, J. Campos, J. López-Serrano, M. L. Poveda, E. Carmona, Angew. Chem. Int. Ed. 2015, 54, 15379 – 15384.[5] A. Zeineddine, L. Estévez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune, D. Bourissou, Nat. Commun. 2017, 8, 565.
PP12

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 75
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
The interaction of the light with ortho-palladated azlactones: different pathways
Esteban P. Urriolabeitia1, Carlos Cativiela1, Pablo Sánchez1, Alexandra Pop2 Cristian Silvestru2
1 ISQCH, CSIC-University of Zaragoza, Pedro Cerbuna 12, 50009 Zaragoza, Spain, E-mail: [email protected]
2 SOOMCC, Faculty of Chemistry and Chemical Engineering, Babes-Bolyai University, 400028 Cluj-Napoca, Romania
4-Arylidene-5(4H)-oxazolones, -imidazolones and -thiazolones have been regioselectively palladated at the ortho position of the 4-arylidene ring through C‒H bond activation promoted by Pd(OAc)2. The reaction is general for the three substrates and tolerates electron-withdrawing and -releasing substituents at different positions of the 4-arylidene moiety (ortho-, meta-, para). Azlactones structurally analogous to the chromophore of the Kaede protein have also been studied. The interaction of the light with these ortho-palladated complexes will be presented here. Two types of behaviors have been found, depending of the ancillary ligands accompanying the oxazolones or imidazolones around the Pd(II) center.
On the one hand, mononuclear derivatives with labile ligands such as acetonitrile, or with chelating ligands such as acetylacetonate, are fluorescent when irradiated with visible light. The fluorescence of these complexes is compared with that of the free ligands, and the influence of the Pd(II), the nature of the heterocycle, the substituents in the ortho-palladated heterocyclic ligand, and the auxiliary ligands on the fluorescence is discussed. On the other hand, ortho-palladated dinuclear clam-shell derivatives with carboxylate bridges react with visible light affording cyclobutanes through [2+2]-photocycloaddition of the C=C bonds of the arylidene unit. This Pd-templated photocycloaddition allows fully stereoselective access to new derivatives of diaminotruxillic acids, not achievable by conventional photochemical procedures. The scope of the photocycloaddition and the release of the organic ligand through different methods will also be presented.
PP13

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 76
Synthesis of bis(amino acids) with cyclobutane skeleton through selective [2+2]-photocycloaddition of oxazolones and thiazolones
Sonia Sierra1, David Dalmau1, Alexandra Pop2, Cristian Silvestru2, Carlos Cativiela1 y Esteban P. Urriolabeitia1
1 ISQCH, CSIC-University of Zaragoza, Pedro Cerbuna 12, 50009 Zaragoza, Spain, E-mail: [email protected]
2 SOOMCC, Faculty of Chemistry and Chemical Engineering, Babes-Bolyai University, 400028 Cluj-Napoca, Romania
The [2+2]-photocycloaddition of alkenes to give cyclobutanes is experiencing nowadays a clear rebirth, due to its synthetic possibilities and sustainable character. Cyclobutanes show a well-established pharmacological activity and a high synthetic potential. But, when the alkene is highly substituted, it is often difficult to control the selectivity of this reaction in solution and many isomers are obtained.[1]
In this communication we present the regio- and stereoselective synthesis in solution of bis-amino acids with cyclobutane core: the 1,3-diaminotruxillic and 1,2-diaminotruxinic acid derivatives have been obtained from [2+2]-photocycloaddition of (Z)-4-aryliden-5(4H)-oxazolones or thiazolones. When the reaction is performed under direct irradiation the 1,3-diaminotruxillic compounds are selectively obtained by head-to-tail coupling of two (Z)-oxazolones[2] or thiazolones. The fate of the reaction changes totally when irradiation is performed in the presence of the triplet sensitizer [Ru(bpy)3](BF4)2, and 1,2-diaminotruxinic acid derivatives are obtained as single regio- and stereoisomers by head-to-head coupling of two (E)-oxazolones. We have fully determined the mechanism of the reaction (DFT), the role of the Ru-complex and the ultimate reasons of the selectivity, which reside in the different spin state (singlet vs triplet) of the oxazolone in the excited state.
Figure 1. Synthesis of 1,3-diaminotruxillic and 1,2-diaminotruxinic derivatives.
References[1] N. Hoffmann, Chem. Rev. 2008, 108, 1052.[2] E. P. Urriolabeitia et al., ACS Sustainable Chem. Eng. 2017, 5, 8370-8381.
PP14

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 77
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Cyclobutadienyl Chemistry of the Rare-Earth ElementsJames Durrant1,2, Benjamin M. Day1, Richard A. Layfield1.
1The University of Sussex, Department of Chemistry, School of Life Sciences, Brighton, BN1 9RH, UK. E-mail: [email protected]
2The University of Manchester, School of Chemistry, Manchester, M13 9PL, UK.
Cyclic π-conjugated organo-ligands are very well established in rare-earth organometallic chemistry, particularly cyclopentadienyl (Cp) and cyclooctatetraenyl (COT) ligands. [1] Our group has shown how Cp ligands can be extremely effective in the development of high-temperature single-molecule magnets (SMMs), with the slow magnetic relaxation properties being due to the strong axial crystal field. For example, the SMM [Dy(CpiPr5)(Cp*)][B(C6F5)4] shows magnetic hysteresis up to 80 K, the first to do so above the boiling point of liquid nitrogen. [2] We are now focused on replacing the monoanionic [Cp]-ligands with dianionic cyclobutadienyl ligands, [Cb]2-, with a view to improving the SMM properties even further. The first rare-earth Cb complexes were reported recently by our group, however the chemistry and magnetism of these compounds is essentially unexplored. [3] We now describe our further investigations into the fundamental chemistry of rare-earth cyclobutadienyl compounds and their potential applications in molecular magnetic materials.
Figure 1: Cyclobutadienyl complexes of the rare-earth elements.
References[1] W. J. Evans, Organometallics, 2016, 35, 3088-3100.[2] F.-S. Guo et al., Science, 2018, 362, 1400-1403.[3] B. M. Day et al., Chem. Eur. J., 2018, 24, 16779-16782.
PP15

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 78
Gold-catalyzed regiodivergent cyclization of alkynylcyclobutanesM. Soledad Garre1, David Sucunza1, Estibaliz Merino1, Enrique Aguilar2,
Patricia García-García1, Juan J. Vaquero1
1 Departamento de Química Organica y Química Inorganica, Campus Científico-Tecnologico, Facultad de Farmacia, Universidad de Alcalá (IRYCIS), Autovía A-II, Km 33.1, 28805 Alcalá de Henares, Madrid, Spain
E-mail: [email protected] 2 Departamento de Química Orgánica e Inorgánica, Instituto Universitario de Química Organometálica “En-
rique Moles”, Universidad de Oviedo, C/Julián Clavería, 8, 33006 Oviedo, Spain
Natural products based in cores of 5 or 6-membered heterocyclic rings fused with cyclobutanes are frequently found. Besides, these products also exhibit versatile biological activities.[1] Consequently, the development of methodologies for their synthesis is of significant interest.
In this work, these cores have been selectively prepared by cyclization of alkynylcyclobutane precursors with a pendant nucleophilic group, initiated by activation of the triple bond with gold catalysts.[2]
Thus, cyclobutane-fused 5-membered heterocyclic derivatives can be obtained from an alkynylcyclobutane having an appended hydroxyl group, by a selective 5-exo cycloisomerization,[3] whereas cyclobutane-fused 6-membered heterocyclic compounds can be prepared from an alkynylcyclobutane with a pendant amide group, by a selective 6-endo cycloisomerization. In addition, computational studies have been performed, which account for the observed selectivity.
References[1] F. Yao-Yue, G. Xin-Hua, Y. Jian-Min, Sci. China Chem. 2016, 59, 1126-1141. [2] Y. Yoshinori, D. G. Iliya, T. P. Nitin, J. Tienan, Chem. Commun. 2009, 5075-5087.[3] M. S. Garre, D. Sucunza, E. Aguilar, P. García-García, J. J. Vaquero, J. Org. Chem., 2019, 84, 5712-5725.
PP16

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 79
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
New Advances in Rhodium-Catalyzed Olefin HydrophosphinationVictor Varela1, Ana M. Geer2, José A. López1, Miguel A. Ciriano1, Cristina Tejel1,
1Departamento de Química Inorganica, Instituto de Síntesis Química y Catalisis Homogénea,(CSIC-UZ) Pedro Cerbuna 12, 50009-Zaragoza (Spain)
E-mail: [email protected] 2 Department of Chemistry, University of Virginia, Charlottesville, Virginia 22904 (United States)
Carbon-phosphorus (C-P) bond formation is a key step in the synthesis of organophosphanes, which play essential roles as ligands for transition metal complexes, in organocatalysis, as building blocks for medicinal and supramolecular chemistry, and have extensive commercial applications.[1] However, classical methodologies for the preparation of organophosphanes suffer from safety concerns (handling corrosive and flammable compounds), use of protective groups that introduce additional synthetic steps, lack of selectivity, and limited scope.[2] Therefore, there has been a growing interest in the development of sustainable and more efficient synthetic strategies, among which the net addition of a P-H bond to unsaturated substrates is one of the most attractive protocols due to the safety, selectivity, and 100% atom-economy provided by this approach. Consequently, key intermediates in these reactions are hydrido-phosphanido complexes arising from P-H bond activation reactions, whose ability to act as catalysts in olefins’ hydrophosphination is poorly developed in the case of rhodium.[3]
In this communication we showcase some clues that account for the feasibility (or not) of a series of mononuclear rhodium complexes to undergo the oxidative-addition of the P-H bond, as well as the reactivity of the new hydrido-phospanido complexes, having a terminal phosphanido ligand, towards electrophiles.
In addition, the high catalytic activity of some of them in the hydrophosphination of activated olefins will be illustrated. Moreover, DFT-studies on these reactions have revealed a new one-step outer-sphere mechanism (see figure for the transition state in the case of dimethyl fumarate). The relevant role of the hydride ligand in these reactions will also be discussed.
References[1] D. W. Allen, Organophosphorus Chem. 2016, 45, 1-50.[2] J.-L. Montchamp, Pure Appl. Chem. 2019, 91, 113–120.[3] L.-B. Han, T. D. Tilley, J. Am. Chem. Soc. 2006, 128, 13698-13699, A. M. Geer, A. L. Serrano, B. de Bruin, M. A. Ciriano, C. Tejel, Angew. Chem. Int. Ed. 2015, 54, 472-475.
PP17

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 80
Stereoselective synthesis of versatile trifluoromethyl substituted borocyclopropanes
Julia Altarejos1, David Sucunza1, Ana Caballero2, Pedro J. Pérez2, Juan J. Vaquero1, Javier Carreras1
1 Departamento de Química Orgánica y Química Inorgánica, Instituto de Investigación Química “Andrés M. del Rio” (IQAR), Universidad de Alcalá, 28805 Alcalá de Henares, Spain
E-mail: [email protected]. 2 Laboratorio de Catálisis Homogénea, Centro de Investigación en Química Sostenible (CIQSO),
Departamento de Química, Universidad de Huelva, 21007, Huelva, Spain.
Cyclopropane rings are present in numerous natural and synthetic products with important biological activities.[1] Functionalized cyclopropanes also find applications in drug discovery since the 1960s and are routinely included in SAR studies in order to modulate their activity, metabolism or conformational rigidity.[2] Therefore, straightforward routes to building blocks containing enantiopure cyclopropanes is of great interest.
Cyclopropylboronates are attractive structures due to the multiple transformations allowed by the carbon-boron bond. Despite the progress in the synthesis of cyclopropane derivatives, there are only a few strategies for the preparation of optically active cyclopropylboronates reported in the literature,[3] none of them including fluorinated groups, although fluorine atoms and fluoroalkyl groups are common motifs in pharmaceutical products.
We have focused on the development of metal-catalyzed protocols, via carbene transfer from a diazo compound, for the stereoselective conversion of alkenylboronates in versatile cyclopropanes. We have recently achieved the enantioselective cyclopropanation of 1-alkenylboronates with ethyl diazoacetate,[4] and in this contribution we will present our most recent results for the stereoselective preparation of versatile trifluoromethyl substituted borocyclopropanes (Scheme 1).
Scheme 1.
References[1] (a) J. Pietruszka Chem. Rev. 2003, 103, 1051-1070; (b) D. Y.-K. Chen, R. H. Pouwer, J.-A. Richard Chem. Soc. Rev. 2012, 41, 4631-4642.[2] T. T. Talele, J. Med. Chem. 2016, 59, 8712−8756.[3] L. Amenós, L. Trulli, L. Nóvoa, A. Parra, M. Tortosa, Angew. Chem. Int. Ed. 2019, 58, 3188–3192 and references cited therein.[4] J. Carreras, A. Caballero, P. J. Pérez Angew. Chem. Int. Ed. 2018, 57, 2334-2338.
PP18

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 81
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Outher-Sphere Alkoxylation of Olefins promoted by Inner-Sphere Oxygen Activation
Cristina Tejel1, Paula Abril1, M- Pilar del Río1, Agustí Lledós2, José A. López1
Miguel A. Ciriano1
1Dpto. de Química Inorganica, Instituto de Síntesis Química y Catalisis Homogénea,(CSIC-UZ), Pedro Cerbuna 12, 50009-Zaragoza (Spain)
E-mail: [email protected] 2Dpto. de Química, Universitat Autònoma de Barcelona, Cerdanyola del Vallès, 08193-Barcelona (Spain)
The electrophilic activation of alkenes by coordination to transition metals is a key for the linkage of a wide range of functional groups to a hydrocarbon skeleton. In particular, the nucleophilic attack of oxygen-based nucleophiles to metal-coordinated olefins is the central step in the palladium-catalyzed Wacker process.[1] It is also relevant in the hydration of olefins to produce alcohols or in hydroalkoxylation reactions leading to ethers.[2] While dominated by palladium, iridium complexes have been poorly explored in this field with very few examples reported in the literature.[3]
In this communication we showcase the easy alkoxylation and hydroxylation reactions of 1,5-cyclooctadiene (cod) in an iridium complex with alcohols and water promoted by the reduction of oxygen to hydrogen peroxide.[4] The exo configuration of the OH/OR groups in the products agrees with the nucleophilic attack at the external face of the olefin as the key step (see figure). The reactions also require the presence of a coordinating protic acid (such as picolinic acid, Hpic) and involve the participation of a cationic diolefin IrIII complex, [Ir(pic)2(cod)]+, which has been isolated.
DFT studies on the mechanism of the reaction show a low-energy proton-coupled electron transfer (PCET) step connecting a superoxide-iridium(II) complex with hydroperoxide-iridium(III) intermediates rather than peroxide complexes, which is essential for the regioselectivity observed.
In addition, the different reactivity of isolated peroxide-complexes will be also discussed.[5]
References[1] R. Jira, Angew. Chem. Int. Ed. 2009, 48, 9034-9037.[2] G. Dong, P. Teo, Z. K. Wickens, R. H. Grubbs, Science 2011, 333, 1609-1612; A. Mifleur, D. S. Mérel, A. Mortreux, I. Suisse, F. Capet, X. Trivelli, M. Sauthier, S. A. Macgregor, ACS Catal. 2017, 7, 6915-6923.[3] B. E. Hauger, J. C. Huffman, K. G. Caulton, Organometallics 1996, 15, 1856-1864.[4] P. Abril, M. P. del Río, J. A. López, A. Lledós, M. A. Ciriano, C. Tejel, Chem. Sci. (SC-EDG-06-2019-003073)[5] M. P. del Río, P.Abril, J. A. López, M. Sodupe, A. Lledós, M. A. Ciriano, C. Tejel, Angew. Chem. Int. Ed. 2019, 58, 3037–3041.
PP19

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 82
Uranium complexes with bulky tetraaryloxide ligands
Rory P. Kelly1, Tatsumi Ochiai1, Francis Y. T. Lam1, Megan L. Seymour1, Jordann A. L. Wells1
Laurent Maron2 and Polly L. Arnold1
1University of Edinburgh, Joseph Black Building, David Brewster Rd, Edinburgh, EH9 3FJ E-mail: [email protected]
2INSA, 135 avenue de Rangueil, 31077 Toulouse, CEDEX 4, France
There is a current resurgence of interest in uranium complexes due to its large size, wide range of accessible oxidation states and the availability of d- and f-orbitals for bonding. These factors make uranium a tantalising prospect for opening up new avenues in reactivity and catalysis [1].
Monometallic uranium complexes are capable of binding and even reducing inert small molecules, e.g. N2 [2], but the presence of two uranium centres can lead to enhanced reactivity, e.g. the reduction of N2 and the coupling of CO by two eq. of [U(OAr)3] (Ar = 2,4,6-tBu3-C6H2) [3]. Ideally, a ligand system capable of binding two uranium centres in close proximity would prime them for cooperative activation of small molecules and provide a more robust system than the combination of two monometallic units. We have explored the use of bulky tetraaryloxide ligands [4] to prepare uranium(IV) complexes with a ‘letterbox’ motif (Fig. 1) that is suitably sized to allow small molecules to enter the pocket and bind to the U centres, setting them up for reductive functionalisation. Two benzylic C–H units point towards the U centres, potentially enabling them to participate in reactivity.
Fig. 1. Bimetallic uranium tetraaryloxide complexes (R = Me, 1U; R = tBu, 1Ut).
References[1] A. R. Fox, S. C. Bart, K. Meyer and C. C. Cummins, Nature, 2008, 455, 341–349.[2] W. J. Evans, S. A. Kozimor and J. W. Ziller, J. Am. Chem. Soc., 2003, 125, 14264–14265.[3] S. M. Mansell, N. Kaltsoyannis,, and P. L. Arnold, J. Am. Chem. Soc., 2011, 133, 9036–9051.[4] J. A. L. Wells, M. L. Seymour, M. Suvova and P. L. Arnold, Dalton Trans., 2016, 45, 16026–16032.
PP20

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 83
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Magnetic Bistability in Coordination Nanohoops
María José Heras Ojea1, Lewis C. H. Maddock1, Benjamin M. Day, Fu-Sheng Guo2, Jeff M. Van Raden2, Daniel Pividori3, Jordi Cirera4, Ramesh Jasti2, Karsten Meyer3,
Eliseo Ruiz4, Richard A. Layfield*1
1 Department of Chemistry, University of Sussex, Brighton BN1 9QJ, U.K., E-mail: [email protected]
2 Department of Chemistry and Biochemistry and Materials Science Institute, University of Oregon, Eugene, Oregon 97403, United States
3 Departament de Química Inorgànica i Orgànica and Institut de Recerca de Química Teòrica i Computacional, Universitat de Barcelona, Diagonal 645, 08028 Barcelona, Spain
4 Department of Chemistry and Pharmacy, Inorganic Chemistry, Friedrich-Alexander University Erlangen-Nürn-berg (FAU), Egerlandstrasse 1, D-91058 Erlangen, Germany.
Molecular materials that show magnetic bistability are of interest due to their potential applications as switches, qubits and nanoscale information storage devices.[1,2,3] Single-molecule magnets (SMMs) and spin-crossover (SCO) compounds are materials that provide basic blueprints for targeting magnetic bistability, and we are interested in developing routes to both types using coordination nanohoops, a hugely under-exploited cycloparaphenylene class of ligand.[1,2,4]
In collaboration with the Jasti Group we are exploring the reactivity of a novel family of bipy−embedded polycyclic aromatic ligands, commonly called bipy[n]CPPs, in the presence of different metallo−organic precursors.[4,5] Their unique structures provide an array of attractive photophysical and redox properties, making them ideal candidates for our research goals. Here we present the synthesis and physical characterisation of the first reported 3d and 4f bipy[n]CPPs compounds (n = 8, 9).
Figure 1. Design and physical properties of 3d/4f−nanohoop systems.
References[1] D. Gatteschi, R. Sessoli and J. Villain, Molecular Nanomagnets, OUP, Oxford, 2006.[2] G. Molnár, L. Salmon, W. Nicolazzi, F. Terkib, A. Bousseksou, J. Mater. Chem. C, 2014, 2, 1360-1366.[3] G. Aromí, D. Aguilà, P. Gamez, F. Luis, O. Roubeau, Chem. Soc. Rev., 2012, 41, 537-546.[4] M. R. Golder, R. Jasti, Acc. Chem. Res., 2015, 48 (3), 557-566.[5] J. M. Van Raden, S. Louie, L. N. Zakharov, R. Jasti, J. Am. Chem. Soc., 2017, 139, 2936-2939.
PP21

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 84
Graphene-Supported Gold Nanoparticles Capped with NHC Ligands: A New Method to Synthesize an Active and Recyclable Catalysts for
the Hydration of AlkynesDavid Ventura-Espinosa1, Santiago Martín2, Jose A. Mata1.
1Institute of Advanced Materials (INAM), Av. Sos Baynat s/n, 12071 Castellón de la Plana, E-mail: [email protected]
2Departamento de Química Física, Facultad de Ciencias, Universidad de Zaragoza, C/Pedro Cerbuna 12, 50009 Zaragoza
Although it was initially thought as an inert metal in catalysis, since the seminal works by Haruta [1] and Hutchings [2] during the 1980s gold has drawn a lot of attention from the scientific community.
Specifically, nanoparticulate gold catalysts present unique catalytic activities when they are supported onto base metal oxides or carbonaceous materials. One of the common issues that these catalysts suffer is the Ostwald ripening that gives raise to larger nanoparticles, inactive in catalysis. A usual approach to tackle this problem is coating gold nanoparticles (AuNps) with ligands to increase their stability. However, the presence of ligands on the surface of gold nanoparticles may block the active sites, preventing its contact with the substrates. Therefore, the synthesis of active catalysts based on metal nanoparticles requires a careful design of the coating ligands.
In this communication we present a new methodology to prepare graphene supported gold nanoparticles coated with N-heterocyclic carbenes (NHCs) ligands. The new hybrid catalyst is directly prepared from the interaction between a discrete Au(I)-NHC complex and the graphene without the need of any reducing agent (figure 1). This new platform efficiently catalyses the hydration of alkynes to obtain ketones. In addition, the system is highly recyclable, does not require any additives and proceeds at low catalyst loadings.
Figure 1. Synthesis of Au-NPs coated with NHCs ligands
References[1] Haruta, M., Kobayashi, T., Sano, H., and Yamada, N., Chem. Lett., 1987 16, 405-408[2] Hutchings, G. J., J. Catal., 1985,96, 292-295
PP22

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 85
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Coordination Versatility of Bidentate COC and Tridentate CNC Ligands Containing 1,2,3-Triazol-5-ylidenes
Gregorio Guisado-Barrios1*, Lewis C. Tolley2, Daniela I. Bezuidenhout2*
1Institute of Advance Materials (INAM), Universitat Jaume I, Avenida Vicente Sos Baynat s/n, 12071 Castellon, Spain E-mail: [email protected]
2Molecular Sciences Institute, School of Chemistry, University of the Witwatersrand, Johannesburg 2050, South Africa
Mesoionic 1,2,3-triazolylidene ligands (MICs) have recently received considerable attention due to their intrinsic mesoionic character provisioning them as strong σ-donors.[1,2] The particular advantage of this class of ligands, is the potential for ligand−metal cooperativity combined with their synthetic versatility, giving access to readily available mono-, bi-, and tridentate ligands. In this context, several metal complexes based on poly(triazolylidene) ligands varying either the number of carbene units as well as the substituents at the N1 and N3 (alkyl or aryl) of the triazolylidene, as well the spacer or scaffold employed (flexible or rigid), have been described in the literature.[1] However, despite the significant catalytic implications demonstrated by related ancillary aliphatic pincer ligands,[3] the number of metal complexes featuring poly-(triazolylidene) ligands linked by a flexible aliphatic spacer bearing none or any additional functionality is still limited. Herein, The synthesis of two readily available bis(1,2,3-triazol-5-ylidene) ligand precursors [H2(CXC)](PF6)2 (with X =NH, O), bridged by an ether or amine functionality (Figure 1), respectively, along with the coordination studies towards different Rh(I) and Ir(I) metal precursors will be presented.[4]
Figure 1. Schematic representation of ligand precursors [H2(CXC)](PF6)2 (with X =NH, O).
References[1] G. Guisado-Barrios, M. Soleilhavoup, G. Bertrand, Acc. Chem. Res. 2018, 51, 3236−3244.[2] A. Vivancos, C. Segarra, M. Albrecht, Chem. Rev. 2018, 118, 9493-9586[3] L. Alig, M. Fritz, S. Schneider, Chem. Rev. 2019, 119, 2681−2751.[4] L. C. Tolley, I. Strydom, W. J. Louw, M. A. Fernandes, D. I. Bezuidenhout, G. Guisado-Barrios, ACS Omega 2019, 4, 6360−6374.
PP23

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 86
β(Z)-vinylsilanes produced by hybrid graphene-based alkyne hydrosilylation NHC-rhodium catalyst
Beatriz Sánchez-Page, M. Victoria Jiménez, Jesús J. Pérez-Torrente, Patricia Álvarez, Javier Blasco
Instituto de Síntesis Química y Catálisis Homogénea ISQCH-Instituto de Ciencia de Materiales de Aragón, Universidad de Zaragoza-CSIC, c/ Pedro Cerbuna 12, 50009, Zaragoza, España.
E-mail: [email protected]
In this work, we describe the synthesis of N-heterocyclic carbene ligands such as 1-phenyl-3-methylimidazol-2-ylidene (L1) and 1,4-diphenyl-3-methyl-1,2,3-triazol-5-ylidene (L2), and the corresponding rhodium (III) complexes of the type [Cp*RhI(L)] (L = L1, L2) featuring and ortho-metallated phenyl ring. The Rh(III) complexes are highly active in the catalytic hydrosilylation of alkynes and afford, in a smooth way, to a complete selectivity to the β-(Z) vinylsilane product with a variety of silanes and alkynes as substrates and CDCl3 or acetone-d6 as solvent (Scheme 1).[1] Catalytic hydrosilylation of alkynes is the most direct, powerful and atom-economical process to produce vinylsilanes, but the control of regio and stereo-selectivity is still a major challenge.
A challenge in homogeneous catalysis is the recycling of the catalyst. In this context, graphene surfaces as supports of homogeneous catalysts have the potential to provide special features to the catalytic systems, allowing for the recycling of the catalyst.[2] With this objective in mind, we have supported the previously described carbene-rhodium catalysts on a graphene oxide surface through C-N covalent bonds between the carbon material wall and the carbene ligand of the complexes. The material has been decorated with 3-methyl-4-phenyl-1,2,3-triazolium (L3) groups followed by a trteatment with [Cp*RhCl2]2 to produce the hybrid catalysts. The structure of the rhodium heterogeneous catalysts, TRGO-[Cp*Rh(L3)]I (5), has been confirmed by EA, XPS, ICP (Rh), Raman, EXAFS and TEM analysis. The hybrid catalyst can be recycled up to seven times without a significant loss of either activity or selectivity in the hydrosilylation of 1-octyne with HSiMePh2. The study has been extended to different substrates achieving outstanding results.
References[1] Pérez-Torrente, J. J.; Modrego, F. J. et al. Organometallics 2016, 35, 2410-2422.[2] Blanco, M.; Álvarez, P. et al. Catal. Sci. Technol. 2016, 6, 5504-5514.
PP24

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 87
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
An Organometallic Approach to Magnesium Aluminate Complexes for Rechargeable Battery Electrolytes
Stuart D. Robertson, Scott A. BrownWestCHEM, Department of Pure and Applied Chemistry, University of Strathclyde, Glasgow, G1 1XL, UK.
E-mail: [email protected]
As demand for rechargeable batteries increases, magnesium represents a promising alternative to currently favoured lithium, as a consequence of its massive natural abundance advantage and consequent low cost. However, for the rechargeable magnesium battery revolution to be realised, there remains a number of challenges which first must be overcome.[1] One key challenge is the development of suitable electrolytes. One class of magnesium compound which is attracting a lot of attention is magnesium aluminate complexes (of general formula [MgxCl2x-1]
+[AlCl3R]–), since the pairing with another earth-abundant, low cost and low toxicity metal maintains their long-term viability. However, conflicting reports exist on the role of halides within the structure with their corrosivity being presented as a reason to omit them [2] and their facilitation of Mg absorption at the electrode being presented as a reason to include them.[3] Consequently, we have been interested in the development of ‘halide limited’ complexes, that is complexes which maintain the halide in the cationic magnesium moiety but without any halide present in the aluminate anion. As an extension of this approach we are also pursuing an understanding of how to control the aggregation state of the cation (i.e. x = 1, 2, 3)[4] and what aggregation states are best suited as electrolytes. Our final target is developing novel methods to characterize these organometallic species, particularly in solution, since they do not possess any particularly indicative NMR handles. We now present a summary of our most recent and relevant results in the area.
References[1] J. Muldoon, C.B. Bucur and T. Gregory, Angew. Chem. Int. Ed., 2017, 56, 12064-12084.[2] J. Muldoon, C.B. Bucur, A.G. Oliver, J. Zajicek, G.D. Allred and W.C. Boggess, Energy Environ. Sci., 2013, 6, 482-487.[3] K.A. See, K.W. Chapman, L. Zhu, K.M. Wiaderek, O.J. Borkiewicz, C.J. Barile, P.J. Chupas and A.A. Gewirth, J. Am. Chem. Soc., 2016, 138, 328-337.[4] E.V. Brouillet, A.R. Kennedy, K. Koszinowski, R. McLellan, R.E. Mulvey and S.D. Robertson, Dalton Trans., 2016, 45, 5590-5597.
PP25

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 88
Green large-scale production of piezoelectric bioplastics
Valentina Sessini, Marta E. G. MosqueraDepartamento de Química Orgánica y Química Inorgánica, Instituto de Investigación en Química “Andrés
M. del Río” (IQAR), Universidad de Alcalá, Campus Universitario, 28871 Alcalá de Henares, Spain, E-mail: [email protected]
Mechanical energy harvesting is a process by which vibration, kinetic energy, or deformation energy is converted to electrical energy. Piezoelectric energy harvesters (PEHs) are regarded as promising independent renewable power sources for low-power electronic devices such as wireless sensors, portable devices, stretchable electronics and medical implants (biosensor) [1-2]. Piezoelectric materials play a crucial role in the field of PEHs and piezoelectric biobased polymers. Green piezoelectric biopolymers optically active, such as poly(L-lactic acid) (PLLA) [3] and Poly-b-hydroxybutirate (PHB), could represent an interesting alternative that have benefits regarding carbon footprint, thus decreasing the environmental impact of PEHs devices. On the other hand, an attractive synthetic pathway for the production of aliphatic polyesters is the ring-opening polymerization (ROP) of cyclic esters. This process has the advantage of allowing an effective control over the properties of the produced polymers. ROP continues to be the most versatile method of synthesis of major groups of biopolymers, particularly when they are required in big amount [4]. Metallic complexes exhibit a great capability to control the polymer microstructure, molecular weight, and PDI. Aluminium complexes have received considerable attention for their high activity and ability of controlling ROP as catalysts.
This work aims to design and synthetize, by catalytic processes, new biopolymers with piezoelectric properties for possible energy harvesting applications, developing innovative cost-efficient and scalable solutions in the field of smart energy.
Acknowledge This project has received funding from the European Union’s Horizon 2020 research and innovation program under the GET Cofund Marie Skłodowska-Curie grant agreement No 754382.
References[1] F. K. Shaikh and S. Zeadally, Renew. Sust. Energ. Rev. 2016, 55, 1041-1054.[2] R. Hinchet and K. Sang-Woo, ACS nano. 2015, 9.8, 7742-7745.[3] T. Furukawa. IEEE Trans. Electr. Insul., 1989, 24.3: 375-394.[4] F. M. García-Valle, et al. Organometallics, 2018, 37.6: 837-840.
PP26

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 89
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Imidazolium-2-Amidinates:Role as Ligands for Main Group Metals and in C-Cl Bond Activation
ProcessesDavid Sánchez-Roa1, Marta E. G. Mosquera1,*, Juan Cámpora2,*, Tomás Cuenca1,
María Fernández-Millán1
1Departamento de Química Orgánica y Química Inorgánica, Instituto de Investigación en Química Andrés M. del Río, Universidad de Alcalá, Alcalá de Henares
E-mail: [email protected] 2Instituto de Investigaciones Químicas, CSIC-Universidad de Sevilla
Imidazolium-2-amidinates are a promising class of ligands whose full potential is still unveiled. These adducts of N-heterocyclic carbenes and carbodiimides (NHC-CDI) are zwitterionic molecules that exhibit a strong basicity through the amidinate group which endows them with a rich coordination chemistry, and the possibility of tuning the electronic and steric properties through the modification of the aryl groups.1
NHC-CDI adducts have shown a versatile behaviour, being able to stabilize nanoparticles and to form a wide range of organometallic complexes.2 Furthermore, recently we have discovered their capacity to activate C-Cl bonds via a metal free pathway, broadening the scope of research to organocatalysis.3
In this work, we have explored the coordination of ICyCDI(p-Tol) (L) to main group metals such as Zn, Al and Mg. The formation of the adducts [ZnEt2L] and [MgBu2L] was achieved by the direct reaction of the alkyl metal with the ligand. For aluminium, the analogous derivative [AlMe2L]+ was generated via the reaction with the protonated ligand (L·HBPh4) and the alkyl aluminium.
As well, the complex [ZnL3](BPh4)2 was prepared using a combination of these strategies. The generation of the same cation [ZnL3]
2+ had also been achieved by the direct reaction between L and ZnCl2.3 In this case the compound [ZnL3][ZnCl4] was formed, and in comparison to [ZnL3](BPh4)2 an important difference in the coordination modes of the ligand in the inner sphere is observed.
Figure 1. [ZnL3](BPh4)2
References[1] F. T. Edelmann, Adv. Organomet. Chem., 2013, 61, 55-374; A. Baishya, L. Kumar, M. K. Barman, T. Peddarao and S. Nembenna, ChemistrySelect, 2016, 1, 498-503[2] A. Márquez, E. Avila, C. Urbaneja, E. Alvarez, P. Palma and J. Cámpora, Inorg. Chem., 2015, 54, 11007-11017; L. M. Martínez-Prieto, C. Urbaneja, P. Palma, J. Cámpora, K. Philippot and B. Chaudret, Chem. Commun., 2015, 51, 4647-4650; H. S. Biswal, S. Nembenna, et al, Inorg. Chem., 2017, 56, 9535-9546[3] D. Sánchez-Roa, T. G. Santiago, M. Fernández-Millán, T. Cuenca, P. Palma, J. Cámpora and M. E. G. Mosquera, Chem. Commun., 2018, 54, 12586-12589
PP27

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 90
Titanium(IV)-modified silica@magnetic nanocomposites. Synthesis and application as efficient nanocatalysts for sulfide oxidation with H2O2
Joan Martín Vinueza, Gerardo Jiménez, Vanessa TaberneroDpto Química Orgánica y Química Inorgánica Universidad de Alcalá, Instituto de Investigación Química
Andrés M. del Río. E-mail: [email protected]
One of the biggest challenges in catalysis is the development of heterogenous catalysts by anchoring active homogeneous catalysts on a suitable support with the aim of bringing together the advantages of both kind of catalysts. In this context, metal-silsesquioxane complexes are regarded as well-defined soluble models of heterogeneous catalysts based on metal species supported on silica.[1]
We have reported the first example of soluble oxidation catalyst based on titanasilsesquioxane species (Ti-POSS) suitable for efficiently oxidizing sulfides and olefins using aqueous solutions of hydrogen peroxide as primary oxidant, and under mild conditions.[2] Such complexes have been prepared via the reaction of complexes [Ti(h5-C5H4SiMe3-nCln)Cl3] (n = 1, 1; 2, 2; 3, 3) with different incompletely condensed silsesquioxanes.[3] This approach has allowed to prepare a new family of titanium complexes containing an innovative cyclopentadienyl-silsesquioxane ligand (CpPOSS), which arises as a consequence of the union of both ligands through the Cp-silicon atom.
Following such a strategy, within this work we describe the synthesis of highly efficient and magnetically retrievable nanocatalysts [4] based on cyclopentadienyltitanium (IV) compounds. Such species have been synthesized by covalent anchoring of organometallics compounds 1, 2 and 3 onto silica coated magnetic nanoparticles by reaction of the Si-Cl and Ti-Cl bonds with the surface silanol groups. These nanocatalysts show excellent behavior as sulfoxidation catalyst.
References[1] Severn, J. R.; Chadwick, J. C.; Duchateau, R.; Friederichs, N. Chem. Rev. 2005, 105, 4073.[2] Ventura, M., Mosquera, M.E.G., Cuenca, T., Royo, B., Jiménez, G. Inorg. Chem. 2012, 51, 6345.[3] Ventura, M., Tabernero, V., Cuenca. T., Royo, B., Jiménez, G. Eur. J. Inorg. Chem. 2016, 2843.[4] Z. Zhang, J. of Colloid and Interface Science 2011, 360, 189.
PP28

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 91
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
New heterometallic aluminium alkali metal compounds with oximate ligands
Jesús Damián Burgoa, Tomás Cuenca, Marta E.G. Mosquera*Departamento de Química Orgánica y Química Inorgánica. Instituto de Investigación en Química
“Andrés M. del Rio”. Universidad de Alcalá. Alcalá de Henares, Madrid. E-mail: [email protected]
Aluminium is the most abundant metal on Earth’s crust and displays a very attractive chemical behavior.[1] Aluminates are a very interesting type of species where the aluminium atom is combined with a more electropositive metal, such as an alkaline metal, frequently through a bridging ligand. A type of ligand that has been rarely explored for the preparation of these heterometallic species is the oximate, in fact there is only another example reported for aluminates.[2] In this communication, we present the synthesis of new aluminate compounds with oximates acting as bridging ligands.
Using the methodology frequently employed in our group,[3] we initially treated the oxime with an alkali metal base and an insoluble solid precipitate. The addition of AlMe3 to this solid leads to the formation of the new oximate derivatives 1 and 2 that present unprecedented solid-state structures. A different outcome is observed for the sodium and potassium compounds. In the first case, the derivative [NaAlMe3(OR)]2 (1) is formed as the only product of the reaction (figure 1). However, when potassium is the alkali metal, the final species isolated 2 is an aggregate of the derivatives [KAlMe2(OR)2]2 and [KAlMe3(OR)]2 that are combined generating a chain (figure 2). These chains are arranged in layers via Me···M interactions in the crystal packing.
Figure 1 [NaAlMe3(OR)]2 Figure 2 [KAlMe2(OR)2]2·[KAlMe3(OR)]2
References[1] P.P. Power, Chem. Rev., 1999, 99, 3463. S. Woodward, S. Dagorne, Modern Organoaluminum Reagents, Springer, 2012; H. Sinn, W. Kaminsky, Adv. Organomet. Chem. 1980, 18, 99; E. Y.-X. Chen, T. J. Marks, Chem. Rev. 2000, 100, 1391.[2] Fernandez-Millan, M.; Temprado, M.; Cano, J.; Cuenca, T.; Mosquera, M. E. G. Dalton Trans. 2016, 45, 10514−10518.[3] M. T. Muñoz, C. Urbaneja, M. Temprado, M. E. G. Mosquera, T. Cuenca, Chem. Commun. 2011, 47, 11757; M. T. Muñoz, T. Cuenca, M. E. G. Mosquera, Dalton Trans. 2014, 43, 14377. F. M. García-Valle, V. Tabernero, T. Cuenca, J. Cano, M. E. G. Mosquera, Dalton Trans., 2018, 47, 6499.
PP29
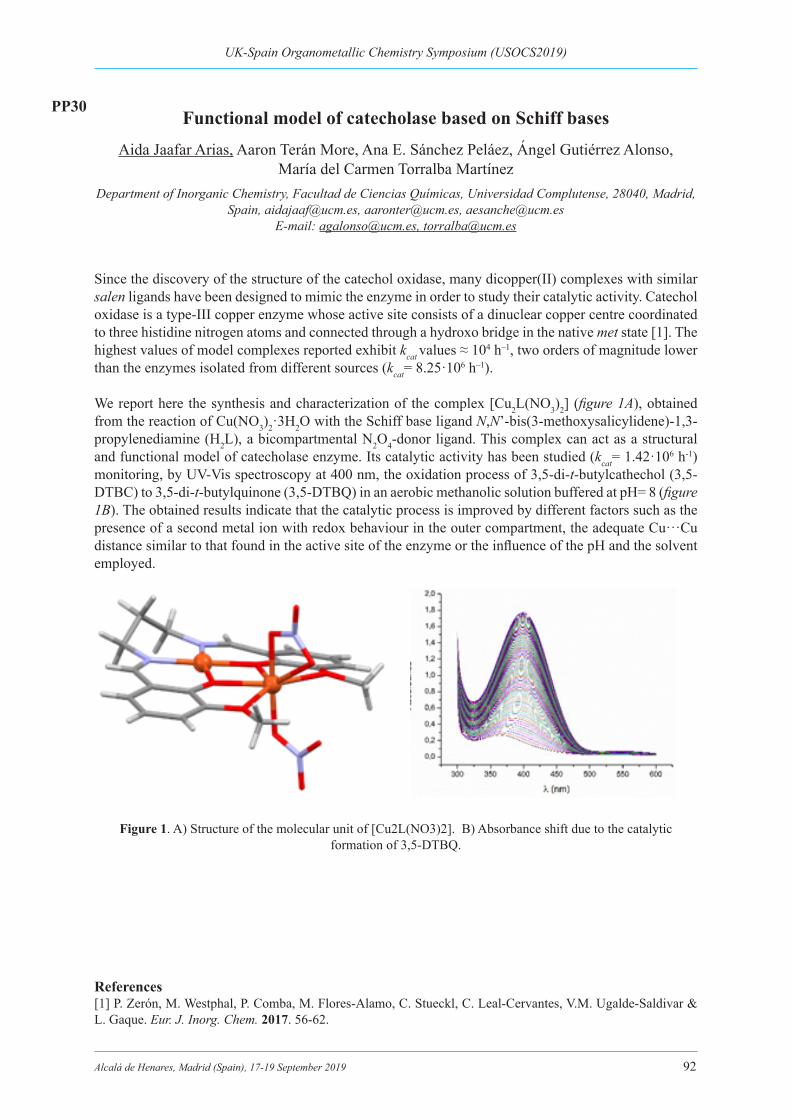
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 92
PP30Functional model of catecholase based on Schiff bases
Aida Jaafar Arias, Aaron Terán More, Ana E. Sánchez Peláez, Ángel Gutiérrez Alonso, María del Carmen Torralba Martínez
Department of Inorganic Chemistry, Facultad de Ciencias Químicas, Universidad Complutense, 28040, Madrid, Spain, [email protected], [email protected], [email protected]
E-mail: [email protected], [email protected]
Since the discovery of the structure of the catechol oxidase, many dicopper(II) complexes with similar salen ligands have been designed to mimic the enzyme in order to study their catalytic activity. Catechol oxidase is a type-III copper enzyme whose active site consists of a dinuclear copper centre coordinated to three histidine nitrogen atoms and connected through a hydroxo bridge in the native met state [1]. The highest values of model complexes reported exhibit kcat values ≈ 104 h–1, two orders of magnitude lower than the enzymes isolated from different sources (kcat= 8.25·106 h–1).
We report here the synthesis and characterization of the complex [Cu2L(NO3)2] (figure 1A), obtained from the reaction of Cu(NO3)2·3H2O with the Schiff base ligand N,N’-bis(3-methoxysalicylidene)-1,3-propylenediamine (H2L), a bicompartmental N2O4-donor ligand. This complex can act as a structural and functional model of catecholase enzyme. Its catalytic activity has been studied (kcat= 1.42·106 h-1) monitoring, by UV-Vis spectroscopy at 400 nm, the oxidation process of 3,5-di-t-butylcathechol (3,5-DTBC) to 3,5-di-t-butylquinone (3,5-DTBQ) in an aerobic methanolic solution buffered at pH= 8 (figure 1B). The obtained results indicate that the catalytic process is improved by different factors such as the presence of a second metal ion with redox behaviour in the outer compartment, the adequate Cu···Cu distance similar to that found in the active site of the enzyme or the influence of the pH and the solvent employed.
Figure 1. A) Structure of the molecular unit of [Cu2L(NO3)2]. B) Absorbance shift due to the catalytic formation of 3,5-DTBQ.
References[1] P. Zerón, M. Westphal, P. Comba, M. Flores-Alamo, C. Stueckl, C. Leal-Cervantes, V.M. Ugalde-Saldivar & L. Gaque. Eur. J. Inorg. Chem. 2017. 56-62.

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 93
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
PP31Multifunctional molecular materials combining optical and liquid
crystal propertiesBrais González1, Cristián Cuerva1,2, José A. Campo1, Mercedes Cano1, Carlos Lodeiro 2,3
1Departamento de Química Inorgánica, Facultad de Ciencias Químicas, Universidad Complutense, 28040-Madrid (Spain).
2BIOSCOPE Research Group, LAQV@REQUIMTE, Chemistry Department, Faculty of Science and Technology, University NOVA of Lisbon, 2829-516 Caparica (Portugal)
3PROTEOMASS Scientific Society, Rua dos Inventores, Madam Parque, Caparica Campus, 2829-516 Caparica E-mail: [email protected]
Our interest in the development of Eu(III) nanoclusters has been established, on the one hand, on the basis of their relevant optical properties and, on the other hand, by considering the potential ability of these systems to achieve the supramolecular ordering in the liquid crystal mesophases through an adequate design of Eu(III)/ligand complexes. With this approach, diketonate compounds as multidentate ligands with great versatility for their coordination and bearing long-chained substituents are good candidates to obtain single-size nanoscale polymetallic clusters, because they could form the metallic-cluster periphery, so giving rise a nanoshell structure.
On the other hand, taking into account the applications of the multifunctional molecular materials, it has been considered that the inclusion of the inorganic cluster in a polymeric organic matrix improves the properties, so avoiding the formation of aggregates.
We present in this work the synthesis, and both the optical and the liquid crystal properties of a series of Eu(III) complexes containing the b-diketonate ligands HL4N(n) (Fig. 1), which are established as tetranuclear clusters by bridged ligands and OH groups (Fig. 2).
AcknowledgementsMINECO/FEDER (CTQ2015-63858-P); FCT/MEC (UID/QUI/50006/2013); PROTEOMASS Scientific Society
References[1] P.C. Andrews, T. Beck, B. Fraser, P.C. Junk, M. Massi, B. Moubaraki, M. Silberstein, Polyhedron, 2009, 28, 2123-2130.[2] V. Baskar, P.W. Roesky, Z. Anorg. Allg. Chem., 2005, 631, 2782-2785.

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 94
PP32Monocatenar and dicatenar b-diketonylpyridinium salts:
ionic liquid crystalsM. Eugenia Aznárez1, Arturo Pareja1, Cristián Cuerva 1,2, José A. Campo 1, Mercedes Cano1,
Carlos Lodeiro2,3
1Departamento de Química Inorgánica, Facultad de Ciencias Químicas, Universidad Complutense, 28040-Madrid (Spain)
2BIOSCOPE Research Group, LAQV@REQUIMTE, Chemistry Department, Faculty of Science and Technology, University NOVA of Lisbon, 2829-516 Caparica (Portugal)
3PROTEOMASS Scientific Society, Rua dos Inventores, Madam Parque, Caparica Campus, 2829-516 Caparica E-mail: [email protected]
We have extended our previous studies related on new salts based on monocatenar long-chained b-diketonyl-pyridinium cations [1,2] to those containing dicatenar ligands by using inorganic anions such as Cl-, BF4
-, PF6-, ReO4
-, or metallocomplexes of the type MCl42- (M = Zn, Cu) as counterions. We
have also studied the influence of the position of the nitrogen atom at the pyridine substituent, at the 2-position or the 4-position (Fig. 1). Most of the compounds do not exhibit mesomorphism, and melt at relatively high temperatures. By contrast, ZnCl4
2- salts show the typical pseudo-focal conic textures of columnar mesophases. We have also solved the crystal structure of a chloride derivative, which shows a layer distribution of columns defined by hydrogen bonds (Fig. 2). The protonation of the nitrogen atom at the pyridine substituent has been followed by photophysical studies.
Figure 1. Crystal structure of [Ru2Cl(Aux1)(DPhF)3]. Hydrogen atoms have been omitted for clarity
AcknowledgementsMINECO/FEDER (CTQ2015-63858-P); FCT/MEC (UID/QUI/50006/2013); PROTEOMASS Scientific Society
References[1] M.J. Pastor, C. Cuerva, J.A. Campo, R. Schmidt, M. Cano, Materials, 2016, 9, 360.[2] M. J. Mayoral, P. Ovejero, J.A. Campo, J.V. Heras, M.R. Torres, M. Cano, Inorg. Chem. Commun., 2009, 12, 214-218.

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 95
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
PP33Phytohormones as bridging ligands in diruthenium complexesIsabel Coloma, Miguel Cortijo, Inés Fernández-Sánchez, Santiago Herrero,
Reyes Jiménez-Aparicio, José L. PriegoDepartament of Inorganic Chemistry, Facultad de Ciencias Químicas, Universidad Complutense,
28040, Madrid, Spain E-mail: [email protected]
Several diruthenium compounds have been described showing a paddlewheel structure. The most usual oxidation state for these complexes is Ru2
5+, where metal-metal bond order is 2.5 [1]. Due to the strong electronic delocalization between the two Ru atoms, these formally mixed-valence complexes are, actually, of average valence. Similar compounds have been reported to interact with biological systems such as RNA [2]. In addition to the applications in biochemistry, one of the main interests in these diruthenium compounds is their great variety of magnetic behaviours given by the different energy of π* and δ* orbitals that can lead to three different configurations: Q(π*δ*)3 (high spin), Qπ*3 or Qδ*2π* (both low spin) in which Q denotes the underlying σ2π4δ2 core [3].
Auxins are phytohormones involved in the life cycle of plants. In this work, we have prepared diruthenium compounds with auxins as bridging ligands to study its controlled release in plants. Thus, we report the synthesis, characterization and study of the magnetic properties of a family of [Ru2Cl(Aux)(DPhF)3] compounds (Aux1 = indol 3-acetate, Aux2 = 2,4-dichlorophenoxyacetate, Aux3 = naphthaleneacetate, DPhF = N,N’-diphenylformamidinate) (Figure 1).
Figure 1. Crystal structure of [Ru2Cl(Aux1)(DPhF)3]. Hydrogen atoms have been omitted for clarity.
References[1] F. A. Cotton, C. A. Murillo, R. A. Walton. Multiple bonds between metal atoms. Third ed., Springer Science and Business Media Inc., New York, USA. 2005.[2] G. Lozano, R. Jiménez-Aparicio, S. Herrero, E. Martínez-Salas, RNA, 2016, 22(3), 330-338.[3] M. C. Barral, T. Gallo, S. Herrero, R. Jiménez-Aparicio, M. R. Torres, F. A. Urbanos, Chem. Eur. J., 2007, 13(36), 10088-10095.

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 96
PP34New insights in the electronic structure of formamidinate-supported Ru2
5+ compounds by Raman resonant spectroscopy
A. Inchausti1, A. Lobato1, S. Herrero2, R. González-Prieto2, R. Jiménez-Aparicio2, M. Taravillo1, V. G. Baonza1,3
1MALTA-Consolider Team, Dpto. de Química Física, Facultad de Ciencias Químicas, Universidad Complutense, 28040 Madrid, Spain
2Dpto. de Química Inorgánica, Facultad de Ciencias Químicas, Universidad Complutense, 28040 Madrid, Spain 3Instituto de Geociencias IGEO (CSIC-UCM), 28040 Madrid, Spain
E-mail: [email protected]
Paddlewheel mixed-valence diruthenium metal-metal bonded species have attracted much interest during the years for their multiple applications, the main ones inferring from their magnetic and electronic properties. These interesting features derive from the energy gap between the π* and δ* orbitals in the Ru-Ru bond. This means that a change in the energy of these orbitals and, in consequence, in the metal-metal bond force constant, will provide information about the systems properties. So far, a diatomic molecule model to estimate the Ru-Ru bond distance from the fundamental frequency was introduced [1]. Nevertheless, this model cannot be assumed, as the π* and δ* contributions change for each case. In this work, a frequency vs. distance tendency has been studied, and it was observed that it is very different from the diatomic molecule. This clearly shows the importance of the description of a new empirical model, and we suggest Raman spectroscopy in order to obtain frequency data for this propose.
Also, it is worth to mention that these molecules absorb light in the visible region. This means that the laser used in the Raman experiments might match with an electronic transition, giving rise to resonance phenomena and an increase in intensity of several normal modes. This technique gives not only vibrational information, but also electronic, and it allows the assignment of such transitions. For high spin complexes, with a 4A2 ground state, two configurations are possible, (π*2δ*) and (δ*π*2), which lead to two different electronic spectra profiles [2]. We present the resonance Raman spectroscopy as a useful tool to identify the electronic transitions for each profile, and to take a step further in the understanding of the dependence of the energy gap between the antibonding orbitals involved in the metal-metal bond, in order to be able to carry out a selective design of diruthenium complexes.
References[1] C. D. Tait, J. M. Garner, J. P. Collman, A. P. Sattelberger, W. H. Woodruff. J. Am. Chem. Soc. 1989, 111, 7806-7811.[2] M. C. Barral, T. Gallo, S. Herrero, R. Jiménez-Aparicio, M. R. Torres, F. A. Urbanos. Chem. Eur. J. 2007, 13, 10088-10095.

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 97
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
PP35[Ru2Cl(DArF)x(O2CMe)4-x] compounds as starting materials for
macromolecules and RNA probesA. de Marcos Galán1, A. Inchausti2, A. Manchado-Parra1, R. González-Prieto1, S. Herrero1,
R. Jiménez-Aparicio1
1 Departamento de Química Inorgánica, Facultad de Ciencias Químicas, Universidad Complutense de Madrid, Ciudad Universitaria, 28040 Madrid, Spain
2 MALTA-Consolider Team, Departamento de Química Física, Facultad de Ciencias Químicas, Universidad Complutense de Madrid, Ciudad Universitaria, 28040 Madrid, Spain
E-mail: [email protected]
Due to their electronic and magnetic properties, diruthenium paddlewheel structured complexes are among the most investigated multiple metal-metal bonding dinuclear compounds. Inside this family, the Ru2
5+ core complexes are the most stable ones, as a result of their half-filled highest occupied molecular orbitals. Moreover, compounds with the general formula [Ru2Cl(DArF)x(O2CMe)4-x] (x = 1-4; DArF = N,N’-diarylformamidinate) have been used as paramagnetic building blocks for supramolecular complexes, coordination polymers or porous metal-organic frameworks [1]. In addition, the interaction between [Ru2Cl2(DPhF)3] (DPhF = N,N’-diphenylformamidinate) and RNA can be employed to recognise RNA secondary structural motifs [2,3]. Thus, a new research aim is directed to see how small changes, as the substitution of the para H atoms in the ligand DPhF by fluorine, can affect the electronic and steric properties of the Ru2
5+ complexes and its effect on the ruthenium-RNA interaction. [Ru2Cl2(DPhF)3] is obtained from [Ru2Cl(DPhF)3(O2CMe)]. Therefore, we have studied the substitution reaction of acetate ligands by Dp-FPhF (N,N’-bis(p-fluorophenyl)formamidinate) in [Ru2Cl(O2CMe)4] to isolate the intermediate compounds [Ru2Cl(Dp-FPhF)x(O2CMe)4-x], to be used as starting materials to build inorganic macromolecules and RNA probes. A plot of the crystalline structure of one of these intermediate compounds obtained is shown in the Figure 1.
Figure 1. Crystal structure of [Ru2Cl(Dp-FPhF)2(O2CMe)2]. Hydrogen atoms have been omitted for clarity.
References[1] Y. Kataoka et al. Polyhedron, 2017, 136, 87-92.[2] G. Lozano et al. RNA, 2016, 22, 330-338. [3] S. Herrero et al. Compuestos de dirrutenio con estructura de rueda de paletas abierta (open-paddlewheel) y su interacción con ácidos nucleicos, 2018, patent number: ES 2603257 B2.

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 98
PP36NMR studies of diruthenium-protein compounds
Aarón Terán More1, Jose Manuel Pérez-Cañadillas2, Santiago Herrero1, Reyes Jiménez-Aparicio1
1Departament of Inorganic Chemistry, Facultad de Ciencias Químicas, Universidad Complutense, 28040, Madrid, Spain
2Deparment of Biologial Physical Chemistry, Instituto de Química-Física Rocasolano, Consejo Superior de Investigaciones científica, Madrid, Spain
E-mail: [email protected]
Diruthenium complexes with paddlewheel or open-paddlewheel type structure (Figure 1) have attracted scientific interest due to their interaction with biological species such as RNA[1] or proteins[2]. They usually are formed by Ru2
5+ units, which are mixed valence Ru(II)-Ru(III) species with an intermediate oxidation state, a 2.5 bond order and present three unpaired electrons (paramagnetic behaviour).
Figure 1. Schematic representation fo a diruthenium compound with paddlewheel (A) or open-paddlewheel (B)
type structure. Reactive positions indicate with an asterisk.
The interaction of diruthenium compounds with biological species offer a new way of studying structure and activity. The application of paramagnetic agents has also been greatly developed in recent years due to the additional information that can be provided through techniques such as Nuclear Magnetic Resonance (NMR) when structural and dynamic studies are carried out in biomolecules. The introduction of “paramagnetic-binding tags” improves the sensitivity of NMR experiments, resonance assignments, studies of conformational heterogeneity or resolving heavily overlapped protein NMR spectra[3].
Four diruthenium compounds have been essayed in the present study: [Ru2Cl(µ-O2CCH3)4] (A), [Ru2Cl(µ-DPhF)3(µ-O2CCH3)]·2THF (B), [Ru2Cl2(µ-DPhF)3] (C) and K3[Ru2(µ-CO3)4]·4H2O (D). It has been proved their interaction with RNA binding proteins (RBP) such as Nrd1 or HRB1 proteins by NMR spectroscopy using 1H-15N HSQC spectra (Figure 2) and it has been analysed their affinity for certain positions.
Figure 2. Intensity variation of signals in 1H-15N HSQC spectra in Hrb1-A sample
References[1] G. Lozano, R. Jimenez-Aparicio, S. Herrero, and E. Martinez-Salas, RNA, 2016, 22, 330–338. [2] L. Messori, T. Marzo, R. N. F. Sanches, Hanif-Ur-Rehman, D. de Oliveira Silva, and A. Merlino, Angew. Chem. Int. Ed., 2014, 53, 6172–6175.[3] X.-C. Su and G. Otting, J. Biomol. NMR, 2010, 46, 101–112.

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 99
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
PP37Coordination capacity of thymine-1-acetate towards diruthenium
complexes with open-paddlewheel structureMiguel Cortijo, Inés Fernández-Sánchez, Santiago Herrero, Reyes Jiménez-Aparicio,
Aarón TeránDepartament of Inorganic Chemistry, Facultad de Ciencias Químicas, Universidad Complutense,
28040, Madrid, Spain E-mail:[email protected]
Our research group has recently started to explore the bioinorganic applications of diruthenium formamidinato complexes showing that [Ru2Cl2(DPhF)3(dmso)] (N,N´-diphenylformamidinate) (OPW-Ru), with open paddlewheel structure (Figure 1, left), can be useful as a probe to obtain information regarding the secondary structure of RNA. Particularly, this complex interacts with nucleosides placed on the junctions of the RNA or near of them giving complementary information to that obtained with other chemical agents such as N-methylisatoic anhydride that interacts in loops and bulges of RNA [1]. For this reason, the study of the reactions of OPW-Ru with nucleobases, nucleosides and nucleotides is of special interest in order to gain more insight on the interaction of this compound with nucleic acids. Thus, we reported the study of the reactions of this compound with cytosine, adenine, cytidine, adenosine, cytidine 2′,3′-cyclic monophosphate sodium salt, and adenosine 3′,5′-cyclic monophosphate in a recent publication [2].
In this work, we have employed thymine-1-acetate (O2CCH2Ty) as a simplified model of a nucleotide in order to gain knowledge about the potential interaction of OPW-Ru with thymine derivatives. Several reactions conditions have been tested giving rise to compounds with different structures (Figure 1, right and center) that have been characterized by IR and electronic spectroscopies, elemental analysis, mass spectrometry and X-ray diffraction.
Figure 1. Crystal structure of OPW-Ru (left), [Ru2Cl(DPhF)3(O2CCH2Ty)] (center) and [{Ru2Cl(DPhF)3}2(O2CCH2Ty)] (right).
References[1] G. Lozano, R. Jiménez-Aparicio, S. Herrero, E. Martínez-Salas, RNA, 2016, 22, 330–338.[2] A. Valentín-Pérez, J. Perles, S. Herrero, R. Jiménez-Aparicio, J. Inorg. Biochem., 2018, 187, 109–115.

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 100
PP38Remote Functionalized Bidentate Phosphine-based Ruthenium (II) Catalysts for Ethanol/Methanol Upgrading to Advanced Biofuels
Folasade J. Sama,1 Richard L. Wingad,1 and Duncan F. Wass1
1School of Chemistry, Cardiff University, Park Place, Cardiff, CF10 3AT, UK E-mail:[email protected]
Ruthenium complexes with bidentate phosphines show good catalytic performance in Guerbet-type reactions [1, 2], where alcohols such as methanol and ethanol can be upgraded to butanols, which find application as advanced biofuels. We are interested in further developing ligands for this application in which remote groups can be used for heterogenization or to increase solubility. Such bidentate diphosphines can be functionalization through phosphination [3], nucleophilic addition [4] and lithiation [5]. Also, the functionalization of these ligand types can be carried out before or after complexation, with the complex itself acting as a protecting group. The presence of remote functional groups (alkoxylsilyl group) in our catalysts offers a route for anchoring them onto support materials. The synthesis of these materials, catalytic results and preliminary heterogenization experiments are reported..
References[1] G. R. M. Dowson, M. F. Haddow, J. Lee, R. L. Wingad and D. F. Wass, Angew. Chem. Int. Ed. 2013, 52, 9005-9008.[2] K. J. Pellow, R. L. Wingad and D. F. Wass, Catal. Sci. Technol., 2017, 7, 5128-5134.[3] P. Braunstein, H. P. Kormann, W. Meyer‐Zaika, R. Pugin and G. Schmid, Chem. Eur. J., 2000, 6, 4637-4646.[4] S. J. Higgins, M. K. McCart, M. McElhinney, D. C. Nugent and T. J. Pounds, Chem. Commun., 1995, 2129-2130.[5] S. Kabehie, M. Xue, A. Z. Stieg, M. Liong, K. L. Wang and J. I. Zink, J. Am. Chem. Soc., 2010, 132, 15987-15996.
EtOH OH0.1 mol% [Ru]
200 mol% NaOMe180oC, 2-20h
[Ru] =
RuPPh2
Ph2P
Ph2PPPh2
Cl
ClNN Si(OMe)3(MeO)3Si
+ 2H2O
RuPPh2
Ph2P
Ph2PPPh2
Cl
ClNH
NH Si(OEt)3(EtO)3Si
2 MeOH+

Index of authors

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 102
INDEX OF AUTHORS
A
ABENGÓZAR, Alberto: 61ABRIL, Paula: 81AGUILAR, Enrique: 78ALBÉNIZ, Ana Carmen: 23, 60, 70ALDRIDGE, Simon: 45ALMENARA, N.: 67ALTAREJOS, Julia: 80ÁLVAREZ, Patricia: 86ÁLVAREZ-RUIZ, Elena: 73ANTIÑOLO, Antonio: 64ARIAS-PÉREZ, María-Selma: 58ARNOLD, Polly L.: 48, 82ASENSIO-BARTOLOMÉ, Miryam: 50
B
BATSANOV, Andrei S.: 44BAUCHEREL, Xavier: 71BEAUMONT, Simon: 71BERRYMAN, Victoria: 48BEUCHER, Hélène: 65BEZUIDENHOUT, Daniela I.: 85BIOSCA, Maria: 39BLASCO, Javier: 86BOCHMANN, Manfred: 38BRODIE, Claire N.: 44BROWN, Scott A.: 87BURGOA, Jesús Damián: 91BUSTOS, Itxaso: 68
C
CABALLERO, Ana: 80CÁMPORA, Juan: 89CAMPOS, Jesús: 33, 74CAÑADAS, Purificación: 66CAPRIATI, Vito: 43CARMONA, Ernesto: 74CARRASCO, Ana C.: 59CARRASCOSA, José L.: 59CARRERAS, Javier: 80CARRILLO-HERMOSILLA, Fernando: 64CATIVIELA, Carlos: 75, 76

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 103
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
CHIRILA, Paula G.: 40CICCO, Luciana: 43CID, M. B.: 72CIRERA, Jordi: 83CIRIANO, Miguel A.: 79, 81CLOETENS, Peter: 59COLLINS, Richard: 63CONAGHAN, P. J.: 38CONEJO-RODRÍGUEZ, Verónica: 53CONESA, José Javier: 59COSTAS, Miquel: 29, 46CREDINGTON, D.: 38CRIMMIN, Mark R.: 47CUÉLLAR, Elena: 50CUENCA, Tomás: 42, 89, 91CUEVA-ALIQUE, Isabel de la: 58
D
DALMAU, David: 76DAY, Benjamin M.: 77, 83DÍAZ-GARCÍA, Diana: 51DÍAZ-SÁNCHEZ, Miguel: 51DIÉGUEZ, Montserrat: 39DIEZ-VARGA, Alberto: 50DIRK BOEGE, Matthias: 57DURRANT, James: 77DYER, Philip W.: 44, 71
E
ESPINET, Pablo : 53
F
FERNÁNDEZ-BAEZA, Juan: 56FERNÁNDEZ-GALÁN, Rafael: 64FERNÁNDEZ-GONZÁLEZ, Miguel Ángel: 61FERNÁNDEZ-MILLÁN, María: 89FOX, Thomas: 65FRANCO FERNÁNDEZ, M. : 72FRUTOS, Luis Manuel: 61FUENTES, M. Ángeles: 45
G
GARCÉS, Andrés: 56GARCÍA-ÁLVAREZ, Joaquín: 43GARCÍA-GARCÍA, Patricia: 61, 78

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 104
GARCÍA-HERBOSA, Gabriel: 50GARCÍA-VIVÓ, Daniel: 64GARÇON, Martí: 47GARRALDA, M. A.: 67GARRALDA, María Ángeles: 68GARRE, M. Soledad: 78GEER, Ana M.: 79GENOUX, Alexandre: 65GIMÉNEZ, María: 31GIMENO, M. Concepción: 26, 57GOICOCHEA, José: 24GÓMEZ, Manuel: 73GÓMEZ-RUIZ, Santiago: 51GONZÁLEZ-SABÍN, Javier: 43GUDE, Lourdes: 58GUISADO-BARRIOS, Gregorio: 85GUO, Fu-Sheng: 83
H
HAMILTON, Alex: 40HANTON, Martin J.: 44HECK, Juergen: 57HERAS OJEA, María José: 83HERNÁNDEZ-PRIETO, Cristina: 73HERNÁN-GÓMEZ, Alberto: 46HEVIA, Eva: 28HORNO, Estefanía del: 69HUERGO, Estefanía: 64HUERTOS, M. A.: 67
I
INFANTE-TADEO, Sonia: 41
J
JASTI, Ramesh: 83JIMÉNEZ, Gerardo: 90JIMÉNEZ, M. Victoria: 86JIMÉNEZ-APARICIO, Reyes: 69, 95, 96, 97, 98, 99JOHNSON, Alice: 55
K
KALTSOYANNIS, Nikolas: 48KAYS, Debbie: 32KELLY, Rory P.: 82

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 105
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
L
LAM, Francis Y. T.: 82LARA-SÁNCHEZ, Agustín: 56LAYFIELD, Richard A.: 30, 54, 63, 77, 83LINNOLAHTI, M.: 38LLEDÓS, Agustí: 81LÓPEZ, José A.: 81
M
MACGREGOR, Stuart A.: 49MADDOCK, Lewis C. H.: 83MARON, Laurent: 82MARTÍN VINUEZA, Joan: 90MARTÍN, Avelino: 73MARTÍN, Santiago: 84MARTÍN-ALVAREZ, José M.: 50MARTÍNEZ-MARTÍNEZ, Antonio J.: 49, 55MASERAS, Feliu: 39MATA, José A.: 84MENA, Miguel: 69, 73MENA-PALOMO, Irene: 51MENDICUTE-FIERRO, Claudio: 68MERILLAS, Beatriz: 50MERINO, Estíbaliz: 65, 78MEYER, Karsten: 83MIGUEL, Daniel: 50MIRANDA, J. I.: 67MIRANDA-PIZARRO, Juan: 74MOSQUERA, Marta E. G. : 88, 89, 91MULVEY, Robert: 22MUÑOZ, M. Teresa: 42
N
N. PEÑAS-DEFRUTOS, Marconi: 53NAVARRO, Marta: 56NEVADO, Cristina: 65
O
OCHIAI, Tatsumi: 48, 82ORO, Luis: 20
P
PALENZUELA, Miguel: 42PÀMIES, Oscar: 39

UK-Spain Organometallic Chemistry Symposium (USOCS2019)
Alcalá de Henares, Madrid (Spain), 17-19 September 2019 106
PARELLA, Teodor: 46PARSONS, Simon: 48PEREIRO, Eva: 59PÉREZ, Julio: 66PÉREZ, Pedro J.: 80PÉREZ-REDONDO, Adrián: 69PÉREZ-TORRENTE, Jesús J.: 86PERICÀS, Miquel, A.: 39PINILLA, Cintya: 70PIVIDORI, Daniel: 83PIZARRO, Ana M.: 41POP, Alexandra: 75, 76PRASHAR, Sanjiv: 51PRICE, Amy N.: 48PRIEGO, José Luis: 69, 95
R
RAMOS, Alberto : 64RIERA, Lucía: 66RÍO, M- Pilar del: 81RÍOS-LOMBARDÍA, Nicolas: 43ROBERTSON, Stuart D.: 87RODRÍGUEZ, Mónica: 46RODRÍGUEZ-ÁLVAREZ, María Jesús: 43RODRÍGUEZ-DIÉGUEZ, Antonio: 64, 67RODRÍGUEZ-FANJUL, Vanessa: 59ROMANOV, A. S.: 38ROYO, Eva: 58RUIZ, Eliseo: 83
S
SALAMANCA, Vanesa: 70SALGADO, Antonio: 61SAMA, J. Folasade: 100SÁNCHEZ, Pablo: 75SÁNCHEZ-BARBA, Luis F.: 56SÁNCHEZ-PAGE, Beatriz: 86SÁNCHEZ-ROA, David: 89SANTAMARÍA, Cristina: 73SERRANO-PINDADO, Álvaro: 51SESSINI, Valentina: 88SEYMOUR, Megan L.: 82SHEPHARD, Jacob, J.: 48SIERRA, Sonia: 76SILVESTRU, Cristian: 75, 76SMITH, Alana: 71SUCUNZA, David: 61, 78, 80

Alcalá de Henares, Madrid (Spain), 17-19 September 2019 107
UK-Spain Organometallic Chemistry Symposium (USOCS2019)
T
TABERNERO, Vanessa: 107TANG, Jinkui : 63TEJEL, Cristina: 79, 81TOLLEY, Lewis C.: 85TORRE-RUBIO, Elena de la: 58TORROBA, Tomás: 50TORTOSA, M.: 72
U
URRIOLABEITIA, Esteban P.: 75, 76
V
VALENCIA, Isabel: 61VAN DE WATER, Leon: 71VAN RADEN, Jeff M.: 83VAQUERO, Juan J.: 78, 80VARELA, Victor: 79VENTURA-ESPINOSA, David: 84VILLAFAÑE, Fernando: 50VILLALBA, Francisco: 60, 70
W
WELLER, Andrew S.: 49, 55WELLS, Jordann A. L. : 82WHITE, Andrew J. P.: 47WHITEOAK, Christopher J.: 40WILLANS, Charlotte: 34WRIGHT, Dominic S.: 25
Y
YANG, Yang: 59YÉLAMOS, Carlos: 69

www.usocs2019.com





























