Embed Size (px)
Citation preview

Journalf. prakt. Chemie. Band 314, Heft 3-4,1972, S. 657-669 J. A. Barth, Leipzig
uber Mehrparameterkorrelationen der kationischen Polymerisation von Vinylmonomeren
Von GUNTHER HEUBLEIN und HORST DAWCZYNSKI Sektion Chemie der Friedrich-Schiller-UniversitLt Jena
Inhaltsiibersicht Es werden eino Reihe p-substituierter Styrole kationisch mit. SnCl, in verschiedenen
Losungsmitteln polymerisiert. Die dilatometrisch ermittelten Bruttogexhwindigkeiten so- wic die Molekulargewichte der Polymeren konnen als ZielgroSen einer Reaktion mit den EinfIuBgrtiOen auf die Elementarprozesse korreliert werden. Substituentenkonstanten, Lo- sungsmittelparameter und spektroskopische Daten liefern lineare Abhiingigkeiten yon den genannt en Zielgr8Ben.
Einleitung In zunehmendem Mal3e wird versucht, eine mathematische Modellierung or-
ganisch-chemischer Reaktionen zu erreichen. Als ein gangbarer Weg erscheint dabei die Gewinnung von molekiilspezifischen Parametern, die fiir bestimmte Elementarprozesse charakteristisch sind, aus geeigneten Modellsystemen. Zu- niichst mu5 uberpriift werden, inwieweit die sich beeinflussenden Faktoren line- are Abhiingigkeiten ergeben, um in einer zweiten Polge deren mogliche Additivi- t L t hinsichtlich der Mehrparameterkorrelation zu untersuchen. Im Ergebnis konnen qualitative und quantitative Voraussagen iiber den Ablauf der betrach- teten Reaktion erhalten werden.
Die kationische Polymerisation wird von verschiedenen Faktoren beein- fluBt, die wir fur den Zweck der Modellierung in EinfluB-, Steuer- und ZielgroBen einteilen [I]. Auf diesem Wege erhaltene Abhangigkeiten konnen dann in Mehr- parameterkorrelationen iiberfuhrt werden [2].
Ergebnisse und Dbkussion
1. S t a n d a r d i s i e r u n g d e r R e a k t i o n s b e d i n g u n g e n -41s Modellsubstanzen fur die kationische Polymerisation wurden mehrere
in p-Stellung substituierte Styrole ausgewiihlt, und zwar Amino-, Dimethyl- amino-, Methoxy-, Methyl-, Dodecyl-, tert.-Butyl-styrol, Styrol, Chlor-, Brom-, Cyano-, Acetoxy- und Nitro-styrol.
An Losungsmitteln zur Polymerisation der genannten Monomeren wurden Hexan, Toluol, Chloroform, Methylenchlorid und Eitromethan verwendet, die unt.erschiedliche Solvatationsfahigkeiten besitzen. Diese konnen durch ihre ET- Werte (kcal/Mol) [3] charakterisiert werden.
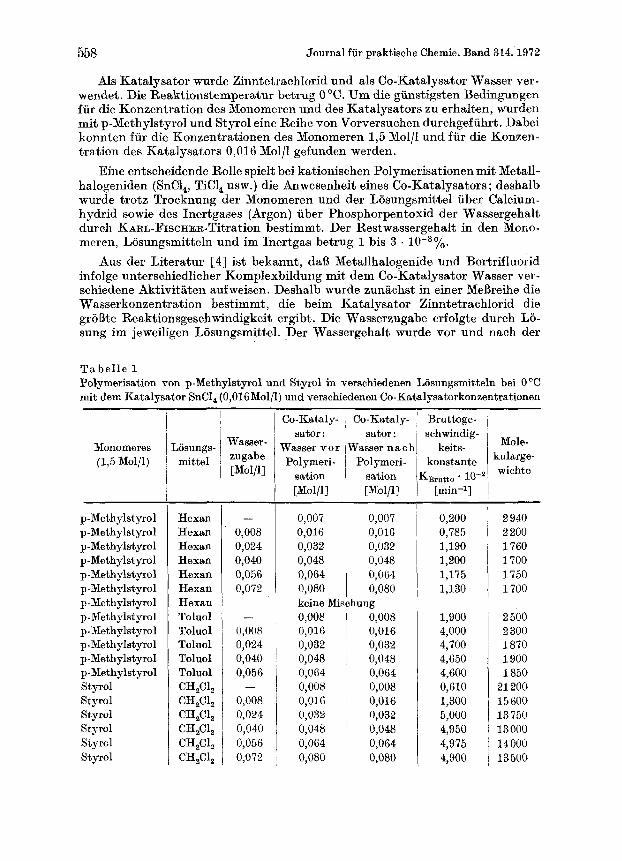
558 Journal fur praktische Chemie. Band 314.1972
~
Co-Kataly- sator:
Wasser vor Polymeri-
Als Katalysator wurde Zinntetrachlorid und als Co-Katalysator Wasser ver- wendet . Die Reaktionstemperatur betrug 0 "C. Um die giinstigsten Bedingungen fur die Konzentration des Monomeren und des Katalysators zu erhalten, wurden mit p-Methylatyrol und Styrol eine Reihe von Vorversuchen durchgefuhrt. Dabei konnten f i i r die Konzentrationen des Monomeren 1,5 Mol/l und fur die Konzen- tration des Katalysators 0,016 Mol/l gefunden werden.
Eine entscheidende Rolle spielt bei kationischen Polymerisationen mit Metall- halogeniden (SnCl,, TiC1, usw.) die Anwesenheit ekes Co-Katalysators ; deshalb wurde trotz Trocknung der Monomeren und der Losungsmittel iiber Calcium- hydrid sowie des Inertgases (Argon) iiber Phosphorpentoxid der Wassergehalt durch KARL-FIscHER-Titration bestimmt. Der Restwassergehalt in den Mono- meren, Losungsmitteln und im Inertgas betrug 1 bis 3 .
Aus der Literatur [4] ist bekannt, da13 Metallhalogenide und Bortrifluorid infolge unterschiedlicher Komplexbildung mit dem Co-Katalysator Wasser ver- schiedene Aktivitaten aufweisen. Deshalb wurde zuniichst in einer MeBreihe die Wasserkonzentration bestimmt, die beim Katalysator Zinntetrachlorid die groDte Reaktionsgeschwindigkeit ergibt. Die Wasserzugabe erfolgte durch Lo- sung im jeweiligen Liisungsmittel. Der Wassergehalt wurde vor und nach der
~~~~ ~ ~
Co-Kataly- 1 Bruttoge- sator: schwindig-
Polymeri- konstante Wasser nach keits-
T e b e l l e 1 Polymerisation von p-Methylstyrol und Styrol in verschiedenen Losungsmitteln bei 0 "C mit dern Katalysator SnCI, (0,016 Mol/l) und verschiedenon Co-Katalysatorkonzentrationen
sation I sation [MOl/ll I IMol/ll
i
Monomeres (1,5 nIol/l)
K~~~~~~ * 1 0 - 5 [min-l]
p-Methglstyrol p-Methylstyrol p-Methylstyrol p-Methylstyrol p-Methylstyrol p-Methylstyrol p-Mcthylstyrol p-Methylstyrol p-Methylstyrol p-Methylstyrol p-Nethylstyrol p-Methylstyrol Styrol Styrol Styrol Styrol Styrcl Styrol
Hexan Hexan Hexan Hexan Hexan Hexan Hexan Toluol Toluol Toluol Toluol Toluol CH,Cl, CH,Cl, CH,Cl, CHzC1, CH,Clz CH,Cl,
- 0,008 0,024 0,040 0,056 0,072
- 0,008 0,024 0,040 0,056 -
0,008 0,024 0,040 0,056 0,072
0,007 0,016 0,032 0,048 0,064 0,080
0,007
0,032 0,016
0,048 0,064 0,080
keine Mischung
0,016 I 0,016
0,048 ~ 0,048 0,064 0,064
0,016 1 0,016 0,038 , 0,032
0,064 0,064 0,080 I 0,080
0,008 0,008
0,032 0,032
0,008 I O,UO8
0,048 i 0,048
0,200
1,190 1,200
0,785
1,175 ~ 1,130
1,900 4,000 4,700 4,650 4,600 0,610 1,300 5,000 4,950 4,975 4,900
Mole- kularge- wichte
2 940 2 200 1760 1700 1750 1700
2 600 2 300 1870 1900 1850
21 200 15600 13760 13000 14 000 13500

G. HEUBLEIN u. H. DAWCZYNSKI, Mehrparameterkorrelationen 559
Polymerisation bestimmt. Die Polymerisation und die Bestimmung der Poly- merisationsgeschwindigkeit wurden im Dilatometer durchgefuhrt. Die Ergeb- nisse sind in Tab. 1 zusammengefafit und in Abb. 1 graphisch dargestellt.
k
t 3
7
Abb. 1. Abhiingigkeit der Bruttogeschwindigkeitskon- stanten von p-Methylstyrol in Hexan und Toluol so- wie Styrol in Met,hylenchlorid von der Wasserkonzen- tration
Man erkennt an den MeDdaten: 1. Der Wassergehalt im Reaktionsgemisch liegt ohne Wasserzugabe vor und
nach der Polymerisation in der GrBflenordnung des Halbhydrats des verwende- ten Katalysators (SnC1,J.
2. Die Polymerisationsgeschwindigkeit steigt mit der Wasserzunahme bis zu einem Wert, der dem Dihydrat des Katalysators entspricht.
3. Bei weiterer Wasserzugabe bis zur Entmischung im entsprechenden LO- sungsmittel bleibt die Bruttogeschwindigkeit nahezu konstant. Offenbar tritt kein als Abbruchreagenz wirkendes ,,freies Wasser" auf, da hohere Hydrate des Katalysators gebildet werden, die katalytisch inaktiv sind.
4. Wlihrend der Polymerisation wird kein Wasser verkraucht. 5. Die Molekulargewichte und die Schmelzpunkte der gebildeten Polymeren
Alle weiteren Untersuchungen wurden stets mit einer Go-Katalysatorkon- nehmen mit zunehmender Bruttogeschwindigkeitskonstante ab.
zentration durchgefiihrt, die dem Dihydrat des Katalysators entspricht.
2. Losungsmi t te le inf luf l Nachdem die Standardbedingungen (Monomerkonzentration : 1,5 Mol/l,
Katalysator : 0,016 Mol/l, Co-Kafalysator : 0,032 Moljl, Temperatur : 0 "C) fur die kationische Polymerisation der p-substituierten Styrole festgelegt waren, wurde an Hand der Monomeren Styrol und p-Methylstyrol der EinfluB des Losungs-

560 Journal fur praktische Chemie. Band 314.1972
mittels auf die Reaktionsgeschwindigkeit, die Ordnung und das Molekularge- wicht untersucht. Die Polymerisationen wurden in einem von uns entwickelten Dilatometer durchgefuhrt (vgl. experim. Teil) und aus den erhaltenen MeBdaten fur eine Reakt,ionslaufzeit ron jeweils 10 Minuten die Reaktionsordnung und die Polymerisat,ionsgeschwindigkeit (brutto) berechnet [5]. Die Auswertung der Dilatometerdaten ergab, daR die Polymerisationen von Styrol und p-Methyl- styrol in allen Losungsmitteln erster Ordnung in bezug auf das Monomere ver- laufen. Die Ergebnisse der Wasserbestimmung vor und nach der Polymerisation, die Geschwjndigkeitskonstanten und die Molekulargewichte sind in Tab. 2 ZU-
sammengefaat..
d $ -2 4
J 1 , I , 1 1 1 1 1 1 1 1 1 , ,
32 34 36 30 40 42 44 46 €7 - Werf e -
Abb. 2. Korrelation der Geschwindigkeitskonstanten des p-Methglstyrols und Styrols mit den ET-Werten verschiedener Losungsmittel
Stellt man den Logarithmus der Geschwindigkei$skonstanten gegen die ET- Werte der Losungsmittel graphisch dar, so erhalt man fur die Polymerisationen von Styrol und p-Methylstysol in den genannten Losungsmitteln jeweils gerade Linien, die zueinander parallel verschoben sind. Die Ursache der Verschiebung liegt in der unterschiedlichen Reaktivitdt der beiden Monomeren. Die Berech- nung der Regressionsgeraden, der Korrelationskoeffizienten (r) und der Stan- dardabweichungen (s) aus den MeBdaten der Tab. 2 ergibt fur die Polymerisa- tion von Styrol
Ig k,, = 0,1103 E T - 5,8674, r = 0,996, E = f 0,043
und fur Methylstyrol lg kBr = 0,1033 E, - 5,0028; r = 0,986, EI = & 0,12
Das nicht ganz idealeverhalten von p-Methylstyrol in Toluol kann moglicher- weise auf Nebenreaktionen mit dem Losungsmittel beruhen.
Die Auftragung des Logarithmus der Geschwindigkeitskonstanten gegen die DK-Werte des Losungsmittels ergibt erwartungsgem85 keine Geraden, da die DK-Werte die spezifische Solvatation der Losungsmittel nicht beriicksichtigen.
Damit konnte zunachst eine Monolinearitat zwischen dem Losungsmittel- parameter E T und der Bruttogeschwindigkeitskonstante festgestellt werden. Ferner erkennt man aus Tab. 2, da13 der Co-Katalysator Wasser auch bei diesen Polymerisationen nicht verbraucht wird. Die Molekulargewichte von p-Methyl- styrol steigen mit Zunahme der ET-Werte der Losungsmittel an, die des Sty- rols dagegen fallen.

? x
Y
M 9
z 3 T
abcl
le 2
i2
T Po
lym
eris
atio
n vo
n St
yrol
und
p-M
et,h
yl3t
yrol
in v
ersc
hied
enen
Liis
ungs
mitt
eln
bei 0
°C m
it d
eni K
atal
ysat
or S
nCI,
(O,O
lG~M
ol/l)
und
dem
B . Y
Co-
Kat
alys
ator
Was
ser
(0,0
32 M
ol/l)
u
a,
‘d
F
2
0
f:
I ;
w
CL
P
I
Yon
omer
es
1
Mol
,l)
~ L
ij~t
ingm
iitt
el
,
Styr
ol
Styr
ol
Styr
ol
Styr
ol
Styr
ol
p-M
ethy
lsty
rol
p -M
ethy
lst y
rol
p-M
ethy
lsty
rol
p-M
ethy
lst y
rol
p -M
ethy
lsty
rol
Hex
an
Tolu
ol
Chl
orof
orm
M
et hy
lenc
hlor
id
Nit
rom
etha
n H
exan
To
luol
C
hlor
ofor
m
Mct
hyle
nchl
orid
N
itro
met
han
ET-W
erhe
[K
oal/M
ol]
30,9
33
,9
39,l
41
,l
46,3
30
,9
33,9
39
,l 41
,l
46,3
Kat
alys
ator
: Sn
C1,
[Mol
/lI
0,01
6 0,
016
0,01
6 0,
OlG
0,01
6 0,
016
0,01
6 0,
016
0,01
6 0,
016
Zo-
Kat
alys
ator
W
asse
r vo
r Po
lym
eris
stio
n [M
ol/lI
0,03
2 0,
032
0,03
2 0,
032
0,03
2 0,
032
0,03
2 0,
032
0,03
2 0,
032
Co-
Kat
alys
ator
: W
asse
r n
a c h
Po
lym
eris
atio
n [M
ol/lI
0,03
2 0,
032
0,03
2 0,
032
0,03
2 0,
032
0,03
2 0,
032
0,03
2 0,
032
Bru
tto-
ge
sahw
indi
g-
kcits
kons
tant
c K
Bru
tto
10-2
[m
in-1
1
0,33
0,
67
2,95
5,
00
17,8
0 1,
19
4,70
10
,50
17,4
0 59
,lO
p F
Mol
ekul
ar-
gew
icht
e 2 * z j
21 50
0 !?
1330
0 3
8 800
!i
1870
k
13 75
0 3 3
6 300
17
60
1660
21
50
2 440
L?. 8 B

562
3. Subs t i t uen tene in f luB Auf der Grundlage der vorher beschriebenen Standardbedingungen wurde die
kationische Polymerisation von 10 verschiedenen p-substituierten Styrolen in Methylenchlorid durchgefiihrt und der EinfluB der Monomerreaktivitat auf die Reaktionsordnung, die Reaktionsgeschwindigkeit und das Molekulargewicht untersucht. Aus der Berechnung der Dilatometerwerte ergab sich, da5 die Poly- merisationen von p-Dimethylamino-, p-Methoxy-, p-Dodecyl, p-tert.-Butyl, p-Methyl-styrol, Styrol, p-Chlor-, p-Brom- und p-Cyano-styrol einem Geschwin- digkeitsgesetz erster Ordnung entsprechen ; p-Nitrostyrol und p-Acetoxy- Ftyrol [17] lieBen sich unter den angewendeten Reaktionsbedingungen nicht polymerisieren. Die Ergebnisse der Co-Katalysatorbestimmung vor und nach der Polymerisation, die Geschwindigkeitskonskanten und die Molekulargewichte fur die p-substitnierten Styrole sind in Tab. 3 zusammengefafib.
Journal fur praktisehe Chemie. Band 314.1972
6-
-4bb. 3. Karrelation zwischen den HAMDIETT- when a-Konstanten und den Bruttoge- schwindigkeiten der p-substituierten Styrole
Man erkennt : 1. daR kein Verbrauch drs Co-Katalysators wahrend der Poly- merisation auftritt, 2. daR die Polymerisationsgeschwindigkeit stark von der Polaritat des Substituenten abhiingt, 3. daR die Molekulargewichte der Polyme- ren in Abhangigkeit vom SubstituenteneinfluB unterschiedliche Trends zeigen (vgl. Abb. 4).
Als Mal3, inwieweit ein gegebener Substituent die Elektronendichte am Reak- tionszentrum verandert, kann die HAMMETT-Konstante cr dienen [6]. Unter der Voraussetzung hoher Polymerisationsgrade und der Gultigkeit des Stationari- tatsprinzipes ist das Verhiiltnis der Initiierungs- zur Abbruchgeschwindigkeit gegenuber dcr Wachstumsgeschwindigkeit vernachlassigbar klein. Fur diesen Fall kann die Bruttogeschwindigkeitskonstante als Ausdruck der Monomerak- tivitat bei der Wachstumsreaktion gelten. Es wurden deshalb die dekadischen Logarithmer, der Bruttopolymerisationsgeschwindigkeiten der p-substituierten Styrole gegen ihre o-Werte aufgetragen, wobei dabei annahernd eine Gerade erhalten wurde (Abb. 3).
Bei der Berechnung der Regressionsgeraden, des Korrelationskoeffizienten (r) und der Standardabweichung (s) aus den Meljdaten der Tab. 3 wurde der Wert fiir p-Dimethylaminostyrol nicht berdcksichtigt. da seine Bruttogeschwindig- keitskonstante wesentlich kleiner als die zu erwartende war und sich so eine Verfdsehung der HAMMETT-Reziehung f iir die iibrigen Monomeren ergeben wurde.
Die Regressionsgerade lautet lg li,, = -3,36 appitra -1,24; r = -0,995, s = 10,104.

9
x 9
W
Q,
M
F 1
Tab
elle
3
Poly
mer
ieat
ion
vers
ohie
dene
r p-s
ubst
ituie
rter
Sty
role
in M
ethy
lenc
hlor
id b
ei 0
“C m
it de
m K
atal
ysat
or S
nC1,
(0,0
16 M
oll])
und
dem
Co-
P
Kat
alys
ator
Was
ssr
(0,0
32 M
ol/l)
Mon
omer
es
[I ,5
Mol
/l]
p-di
met
hyla
min
osty
ro
p-B
enzy
loxy
styr
ol
p-M
etho
xyst
yrol
p-
Dod
ecyl
styr
ol
p -M
ethy
lsty
rol
p-C
hlor
styr
ol
p-B
rom
styr
ol
p-Jo
dsty
rol
p-C
yano
styr
ol
p-N
itros
tyro
l
p-t0
d.-B
Ut$
styr
Ol
Styr
ol
HA
MM
ET
T-
Kon
stan
te
upar
e
-0,6
00
-0,4
15
-0,2
68
-0,2
56
-0,1
97
-0,1
70
0,00
0 +0
,227
+0
,232
+0
,276
+0
,628
$0
,778
Co-
Kat
alye
ator
: W
asse
r vo
r Po
lym
eris
a tio
n “O
l/ll
0,03
2 0,
032
0,03
2 0,
032
0,03
2 0,
032
0,03
2 0,
032
0,03
2 0,
032
0,03
2 0,
032
Co-
Kat
alys
ator
: W
asse
r n a
c h
Poly
mer
isat
ion
[Mol
/lI
0,03
2 0,
032
0,03
2 0,
032
0,03
2 0,
032
0,03
2 0,
032
0,03
2 0,
032
0,03
2 0,
032
Bru
ttoge
schw
indi
g ke
itsko
nsta
nte
[min
-l]
KB
rutt
o * lo
-’
2,10
0 79
,400
69
,800
61
,000
25
,200
17
,400
5,
000
0,91
0 0,
860
0,63
1 0,
052
Mol
ekul
arge
- w
icht
e [D
IG]
4000
33
600
1900
0 4 7
00
7 600
2 1
50
1375
0 14
700
15 00
0 15
900
19 10
0 -
3,60
21
E
3,88
08
%
4,13
83
8 8
4,52
63
~ k
4,27
88
$ 3,
6721
9
3,33
24
P? m,
4,16
73
4,17
61
~
4,20
14
B 4,
2810

564 Journal fur praktische Chemie. Rand 314.1972
Die verringerte Bruttopolymerisationsgeschwindigkeit von p-Dimethyl- aminostyrol in Methylenchlorid kann nur so erklart werden, daB das freie Elek- tronenpaar am Stickstoffatom der Dimethylaminogruppe mit der LEWIS-Skure SnCl, einen N-Donator-Akzeptorkomplex bildet und damit einen Teil des wirk- samen Katalysators bindet sowie die Basizitat der Vinylgruppe im p-Dimethyl- aminostyrol verringert. Fur p-Dodecylstyrol wurde in der Literatur keine cr-Kon- stante gefunden. Sie wurde aus der Beziehung opara/vCn, bestimmt und ergab -0,256. Dieser Wert ist groBenordnungsma8ig und bezuglich des Vorzeichens vertretbar, wenn man den 0-Wert mit iihnlichen G-Konstanten vergleicht.
I
Abb. 4. Korrelation zwischen den HAXMETT schen a-Konstanten der p-substituierten Styrole und den Molekulargewichten der erhaltenen Polymeren
In Abb. 4 wurde der dekadische Logarithmus der Molekulargewichte der Polymeren gegen die HAMMETT-Konstante aufgetragen, um daraus einen Riick- schluB auf die Reaktionsfahigkeit der wachsenden Kationen wibhrend der Poly- merisation zu erhalten. Die Berechnung der Regressionsgeraden, der Korrela- tionskoeffizienten (r) und der Standardabweichungen (9) fur die Beziehung lg MG/apma der MeBdaten von Tab. 3 ergab fur die Polymerisation der p-sub- stituierten Styrole mit elektronenanziehenden Substituenten
IgMG = 0 , 2 3 5 ~ ~ ~ ~ ~ + 4,1286; r = 0,975, s = &0,012
und mit elektronenabgebenden Substituenten
lg MG = -4,23 aFara + 2,8341; r = -0,85, s = &0,291.
Nach Abb. 4 verhalten sich nucleophile und elektrophile Substituenten in Relation zum unsubstituierten Styrol gegenliiufig. Die Reaktionsfiihigkeit der wachsenden Kationen im Polymeren wird urn so groBer, je starker die elektro- nenanziehende Natur des Substituenten ist. I n diesem MaBe wird die Elektro- philie der Kettentriiger erhoht . Fur elektronenliefernde SubstiOuenten ergibt sich ein umgekehrter Sachverhalt . Eine gegenlaufige Tendenz der Monomerreaktivi- tiit (Abb. 3) und der Reaktivitiit der elektrophilen Kettentrager im Wachstums- schritt (Abb. 4) wurde bereits in anderem Zusammenhang festgestellt [7]. Da die Wachstumsgeschwindigkeit jedoch in starkem MaBe von auBeren Faktoren ab- hangt [l], setzt die Verallgemeinerung dieser SchluBfolgerung eine konkrete Analyse des jeweiligen Systems voraus*).
*) So konnten wir in neueren Untersuchungen bereits beim Wechsel des Katalysators (z. B. TiCI,, FeCI,) eine Anordnung beider Substituentengruppen auf einer Geraden fest- stellen, mit Ausnahme der Punkte fur Dodecyl- bxw. tert.-Butylstyrol,

G. HEUBLEIN u. H. DAWCZYNSKI, Mehrparameterkorrelationen 565
Die hier gezeigten Monolinearitiiten zwischen der Monomerreaktivittit bzw. der Reaktionsfahigkeit der Kettentriiger und der Bruttopolymerisationsge- schwindigkeit sowie den Molekulargewichten der erhaltenen Polymeren konnen im folgenden Abschnitt um sogenannte ,,statkche" Parameter aus spektrosko- pischen Daten erweitert werden.
4. Spek t roskop i sche D a t e n Einem Vergleich der IR-Spektren ist zu entnehmen, dafi mit Veranderung
der Reaktivitiit der Monomeren sich auch die fiir die Vinylgruppe charakteristi- schen Banden verschieben. Am starksten traten solche Verschiebungen fur die CH,- wagging-Schwingung auf. Die Lage dieser Bande bei den einzelnen Mono- meren ist in Tab. 4 dargestellt.
Eine iihnliche Verschiebung der liingstwelligen UV-Absorptionsbande war beim Vergleich der UV-Spektren festzustellen. Die einzelnen Ergebnisse sind gleichfalls in Tab. 4 enthalten.
Tabe l l e 4 Lage der CH,-wagging-Schwingugen im I R und UV- Maxima bei p-substituierten Styrolen. Die IR-Spektren wurden in CC1,und die UV-Spektren in Hexan aufgenom- men
Monomeres
p-Dimethylaminostyrol p- Aminostyrol p-Methoxyst p o l p-Dodecylstyrol p-tert.-Butylstyrol p-Methylstyrol
p-Chlorstyrol p-Bromstyrol p-Cyanostyrol p-Nitrostpol
Styrol
CH,-wagging [cm-l]
893 896 902 906 908 910 912 916 916 924 926
UV-Maxima [nml
295 280 261 254 253 243 247 254 260 265 292
TrLgt man den dekadischen Logarithmus der Bruttopolymerisationsge- schwindigkeitskonstanten (lg kBr) gegen die Lage der CH,-wagging-Schwin- gung der Monomeren (VCH,) auf, so erhiilt man eine weitere Monolinearitiit zwi-* schen den statischen Monomerparametern (aus IR- und UV-Banden) und der Bruttogeschwindigkeit als ZielgroBe f i i r die kationische Polymerisation (Abb. 5). Die Gleichung der Regressionsgeraden fi.ir lg kBr/YCH,, ihr Korrelationskoeffi- zient (r) und die Standardabweichung (s) sind :
lg kBr = -0,166 vCH, + 140,93; r = -0,985, s = f 0,22
Die Oberpriifung des Additivitiitsverhaltens der gezeigten Monolinearitiiten und ihre Weiterfiihrung zur Mehrparameterkorrelation wird in einer weiteren Mitteilung erortert.

5 66 Journal fur praktische Chemie. Band 314.1972
Y CH2 - Wagghg -Schwingung -
Abb. 5. Korrelation zwischen der Lageder CH,- wagging-Schwingung im IR-Spektrum und den Bruttopol ymerisationsgeschwindig keitskon- stanten verschiedener p-substituierter Styrole in Methylenchlorid bei 0 "C, dem Katalysator SnC1, (0,016 Mol/l) und dem Co-Katalysator Wasser (0,032 Mol/l)
Beschreibung der Versucho
D a r s t e l l u n g d e r Monomeren
p-Aminostyrol [8] wurde aus gaschromatographisch reinem p-Nitrostyrol durch Re- duktion mit Aluminiumamalgam [9] gewonnen. Die Reinigung des Aminostyrols erfolgte nicht wie in der Literatur beschrieben durch Destillation, sondern durch mehrmaliges Um- fgllen uber das stabile Aminostyrolhydrochlorid in peroxidfreiem Ather: IR-Banden vN=, = 3 405 und 3 492 cm-l.
p-Dimethylaminostyrol wurde nach [lo] ausgehend von p-Dimethylaminobenzaldehyd dargestellt. Auf der Stufe der GRIGNARD-Reaktion mu13 mit peroxidfreiem Ather nnd unter Schutzgas gearbeitet werden, um eine Polymerisation des p-Dimethylaminostyls zu ver- hindern. Ausbeute 65% ; Kp. 68"/0,1 Torr; F. 15-16' ; gaschromatographisch rein [Chromo- sorb W (60/80) mit 15% Silikongummi, T, =- 192", t, = 33 min], IR-Bande vC,,* = 1355 cm-l.
p-Nethoxystyrol wurde nach Ell] durch Decarboxylierung aus p-Methoxyzimtsgure ge- wonnen. Ausb. 70%; Kp. 45--66"C/0,5 Torr; gaschromatographisch rein [Chromosorb W (60/80) mit 10% Silikonol DC-710, T, = 160", t, = 1,86 rnin].
p-Methylstyrol wurde nach [l2] durch katalytische Dehydratisierung von p-Methyl- phenylmethylcarbinol dargestellt. Ausb. 70%; Kp. 65-66"/19 Tom; gaschromatographisch rein {Diaphorit mit 10% Athylenglykoladipinsaurepolyester, T, = 121", t, = 2,3 min].
p-Dodecylstyrol wurde nach [13] aus p-Dodecylphenylmethylcarbinol dargestellt. Ausb. 65%; Kp. 155-162 'C/l Torr; gaschromatographisch rein [Porolith (0,2/0,25 mm) mit 9,7% AthylenglykoladipinsBurepolyester, T, = 191", t, = 8,7 min].
p-tert. Butylstyrol wurde narh [13] analog p-Dodecylstyrol dargestellt. Ausbeute 65%, Kp. 52'/1 Torr ; gaschromatographisch rein [Diaphorit mit 10% Athylenglykola,dipinsaure- polyester, T, = 160", t, = 1,0 rnin].
Styrol lag als Handelsprodukt vor (VEB Laborchemie Apolda). Zur Eeinigung und Destabilisierung wurde dieses mit l0proz. Natronlauge mehrmals geschuttelt, mit Waaser bis zum Pu'eutralpunkt gewaschen und iiber Calciumchlorid vorgetrocknet. Danach wurde es unter Argon uber Calciumhydrid i. Vak. destilliert. Gaschromatographisch rein [Chromo- sorb WS 30-60 rnit 15% Silikongummi BE = 30, T, = 91", t, = 1,2 min].
p-Chlorstyrol wurde nach [14] aus p-Chlorphenylmethylcarbinol dargestellt. Busbeute 60%; Kp. 74"/12 Torr; gaschromatographisch rein [Chromosorb. WS 30-60 mit 15% Sili- kongummi SE = 30, T, = 199", t, = 0,7 min].
p-Bromstyrol wurde nach [15] analog dem p-Chlorstyrol gewonnen. Ausb. SOY0, Kp. 87"/13 Torr; gaschromatographisch rein [Chromosorb WS 30-60 mit 15% Silikon- gummi SE = 30, T, = 199", tr = 0,7 min].

G. HIWJBLEIN u. H. DAWCZYNSKI, Mehrparameterkorrelationen 567
p-Cyanostyrol wurde aus p-Cyanoecetophenon durch zehnstiindiges Erhitzen von p- Bromacetophenon mit Cu,(CN), [16] in Dimethylformamid bei 150" erhalten. Nach Auf- arbeitimg des Reaktionsproduktes nnd mehrmaligem Umkristallisieren aus Petroliither (SO-SO "C) wurde gaschromatographisch reines p-Cyanoacetophenon erhalten. F. 56-57' [Chromosorb WS 30-60 mit IS:/, Silikongummi SE = 30, T, = 186", tr = 1,05 min]. Daraus wurde nach [121 und [13] p-Cysnostyrol dargestellt. Ausb. 60%, Kp. 84"/1 Torr; gaschromatographisch rein [Chromosorb WS 30-60 mit 15% Silikongummi, T, = 164, tr = 1,3 min].
p-Nitrostyrol wurde nach {lO] durch Dehydrohalogenierung von p-Nitrophenylathyl- bromid gewonnen. Die Reinigung erfolgte durch mehrmaliges Umkristallisieren aus Petrol- ather bei -40'. Ausb. 85%; F. 21,4"; gaschromatographisch rein [Chromosorb WS 30-60 mit 15% Silikongummi SE = 30, T, = 185", t, = 1,15 min].
Zur Reinigung der Losungsmittel und des Katalysators wurde nach bekannten Metho- den verfahren [IS]. Die Wasserbestimmung in den gereinigten und getrockneten Losungs- mitteln, den Nonomeren und im gefiillten PolymerisationsgefiiB vor und nach der Polymeri- sation erfolgte nach der Methode der visuellen KARL-FISCRER-Titration [19].
Zur Messung d e r Polymer isa t ionsgeachwindigkei t diente ein von uns ent- wickeltes Dilatometer, das zur Tieftemperaturdilatometrie besonders geeignet erscheint und daher kurz charakterisiert werden soll.
-b - - 5 e - - - - _ Abb. 6. Aufban des Dilatometere. 1 Birnenkolben; 2 KPG-Kapillare; 3 DewargefBO; 4 , 5 Einlaufstutzen; 6 LIEBIG-Kiihler ; 7 Magnetriihrer

568 Journal fur praktische Chemie. Band 314.197 2
Das Dilatometer (vgl. Abb. 6) besteht aus einem 10-ml-Birnenkolben (1) rnit NS-14,5 und einer etwa 0,5m 1angenPrazisions-KPG-Kapillare (2) (Innendurchmesser : 0,6--1,6mm), an derem unteren Ende sich ein Kern NS-14,5 und eine schriig nach oben angesetzte NS-5 Hiilse befindet. Am oberen Ende sind eine Hulse NS-14,5, ein seitlicher Ansatz NS-5 und ein Hahnansat.2 angebracht. Die beiden NS-5 Hiilsen werden >-or der Polymerisation mit Silikongummi abgedichtet. Das Dilatometer wird bis zum Kapillaransatz in ein Dewarge- faB (3) mit, abgeflachtem Boden getaucht, das zwei seitliche Ansatze (4 und 5) besitzt, durch die standig die Kiiltelosung (Wasser/Glycerin) eines Kryostaten flieBt. Die Kapillare wird von einem LIEBIG-Kuhler (6) umgeben, durch den ebenfalls die Kaltelosung stromt. Die Reaktionekomponenten werden durch einen Magnetriihrer durchmischt (7).
Zum Fullen dcs Dilatometers wird das Monomere entw-eder im Kolben eingewogen und bei aufgeeetzter Kapillare eingefroren und entgast oder das bereits entga8t.e Monomere wird mittels Injektionsspritze durch den Silikongummi des unteren seitlichen NS-5 Schliffes inji- ziert. Nach dem Evakuieren wird das Losungsmittel aus einem VorratsgefaB uber die Kapil- lare in den Dilatometerkolben gegeben. Das Volumen des Dilatometers wird entweder durch Auslitern mit Quecksilber vorher bestimmt oder das Losungsmittel wird mit einer Burette gemessen, die zwischen Kapillare und VorratsgefiiB geschaltet wird. Eventuell vorhandene Gasblasen konnen uber den unteren mit Silikongummi verschlossenen NS-5 mit einer diinnen Kanule entfernt werden. AnschlieBend wird das Dilatometer zum Temperieren in die Bad- flussigkeit gebracht. Nach Temperaturausgleich wird das LosungsmittelvorratsgefLB unter Argon entfernt und mit einer langen Kanule im Argonstrom aus der Kapillare so vie1 Lo- sungsmittel entnommen, daB nach Injektion des Katalysators der Meniskus im Anfangsbe- reich der KapilIarenska.la, liegt. Danach wird der lhgnetruhrer in Betrieb genommen und nach 3 bis 4 Minuten der Katalysator mit einer Mikroliterspritze iiber den unteren seitlichen Ansatz injiziert. Die Volumenkontraktion wird visuell abgelesen; ohne erheblichen appara- tiven Aufwand ist auch eine a,utomatische Rcgistrierung moglich. Das beschriebene Dilato- meter ist fur die ionische, speziell die kationische Polymerisation geeignet. Die Verwendbar- keit wurde im Temperaturbereich von -78 bis 1-50 "C nachgewiesen. Voraussetzung ist eine Temperaturstabilisierung von 0,Ol "C.
Zur D u r c h f u h r u n g d e r P o l y m e r i s a t i o n wird das Monomere in das Dilatometer- kolbchen eingewogen, das Dilatometer mehrmals sekuriert und das Losungsmittel uber die Kapillare eingefullt. Durch den seitlichen Ansatz an der Kapillare werden der Co-Kataly- sator Wasser, der Katalysator und zur Beendigung der Polymerisation die Abbruchreagen- zien durch eine mit einer 0,06-mm-Kanule versehene Mikroliter- oder Tnjektionsspritze in den Kolben eingespritzt. Nach Beendigung der Polymerisation werden durch den seitlichen Ansatz an der Kapillare jeweils drei Proben zii je einem ml zur Bestimmung des Wasser- und dea Restmonomerengehaltes gezogen. Die verbliebene Polymerlosung wird in eine Methanollosung getropft und das Polymere ausgefiillt. Durch Losung in Benzol und Flillung rnit Methanol wird das Polymere drei- bis viermal umgefallt und schlieBlich sieben Tage bei 40" im Vakuumtrockenschrank getrocknet. Polymerlosungen, die beim Vcrsetzen mit Metha- nol infolge zu niedriger Molekulargewichte keine Fiillung ergaben, wurden im Rotationsum- laufverdampfer eingeengt.
Der Restmonomergehalt in der Polymerlosung wird mit Hilfe der Kaliumbromid-Bro- mattitration [ Z O ] bestimmt. Die Methode wurde zuvor mit allen Monomeren getestet und ergab eine Genauigkeit von & 2%, mit Ausnahme der Monomeren p-Amino- und p-Dimethyl- aminostyrol. Fur diese beiden Monomeren konnte diese Methode Z U ~ Umsetzbe$timmung nicht verwendet werden. Bei der Polymerisation von p-Dimethylaminostyro1 wurde der Umsatz durch Auswagen des Polymeren bestimmt .

G. HEUBLEIN u. H. DAWCZYNSRI, Mehrparameterkorrelationen 569
Die Bestimmung der Molekulargewichte erfolgte in Benzol bei 45" mit einem Dampf- druckosmometer nach KNAUER.
Die UV-Spektren der Monomeren wurden niit dem Uvispek Photoelectric Spectro- photometer H 700,30 von Hilger & Watts aufgenommen. Zur Aufnahme der IR-Spektren diente das Spektralphotometer UR 10 des VEB Carl Zeiss Jena.
Literaturverzeichnis [1] G. H E U B L E ~ , Plaste und Kautschuk 19, 177 (1972). [2] V. A. PALM, Grundlagen der quantitativen Theorie organiseher Reaktionen, Akade-
[3] CHR. REICHARDT u. K. DIMROTH, Fortschr. chem. Forsch. 11/1, 1 (1968). [4] P. GIUSTI, E'. ANDRVZZI, P. CERRAI u. G. L. POSSANZINI, Makromolekulare Chemie
136, 97 (1970). [5] A. A. FROST 11. R. G . PEARSON, Kinetik und Mechanismen homogener chemisoher
Reaktionen, S. 36 und 37, Verlag Chemie, Weinheim 1964. [6] H. H. JAFFE, Chem. Reviews 63, 191 (1953). [7] Y. IMANISHI, A. MIzOTE, T. HIGASLIIMURA, u. S. OKABTURA, Chem. High Polymers
[8] J. H. BOYER u. H. ALIEL, J. Amer. chem. SOC. 81, 2137 (1959). [9] A. VOGEL, A Textbook of Practical Organic Chemistry, Longmans, Green & Go., Lon-
[lo] R. w. STRASSBTJRQ, R. A. GREGG u. CH. WALLING, J. Amer. rhem. soc. 69, 2141
1111 W. J. DALE u. H. F. HENNES, J. Amer. chem. Soc. 81, 2143 (1959). [12] D. T. MOWRY, M. RENOLL u. W. F. HUBER, J. Amer. chem. SOC. 68, 1105 (1946). [13] C. B. MERBERGER u. J. H. SAUNDERS, Org. Synth. 28, 31 (1948). [14] C. S. MARWEL u. 0. L. SCHERTZ, J. Amer. chem. SOC. 65, 2056 (1943). [15] W. KERN u. D. BRAVN, Makromolekulare Chemie 27, 23 (1958). [16] L. FRIEDMAN u. H. SHECHTER, J. org. Chemistry 26, 2522 (1961). [17] B. B. CARSON, J. o g . Chemistry 23, 547 (1958). [18] W. BUKGE in HOUBEN-WEYL, Methoden der Organischen Chemie, Bd. 1/2, S. 770ff.,
[19] E. EBERIUS, KARL-FISCHER-L6SUng, Verlag Chemie, Weinheim/BergstraBe 1958. [20] HOUBEN-WEYL, Methoden der Organischen Chemie, Bd. 14j1, S. 358, Georg Thieme
mie-Verlag, Berlin 1971.
[Tokyo] 20, 213 (1963).
don 1956, p. 198.
(1947).
Georg Thieme Verlag, Stuttgart 1959.
Verlag, Stuttgart 1961.
Bei der Redaktion eingegangen am 26. Oktober 1971.
Anschr. d. Verf.: Prof. Dr. G. HEUBLEIN und Dr. H. DAWCZYNSKI, Sektion Chemie der Universitgt Jena, DDR- 69 Jena., Humboldtstr. 10.





























