Embed Size (px)
Citation preview

Chemical Physics Letters 602 (2014) 16–21
Contents lists available at ScienceDirect
Chemical Physics Letters
journal homepage: www.elsevier .com/locate /cplet t
Tuning the C–X. . .p interaction of benzene–chloroacetylene complexesby aromatic substitutions
http://dx.doi.org/10.1016/j.cplett.2014.04.0100009-2614/� 2014 Elsevier B.V. All rights reserved.
⇑ Corresponding author.E-mail address: [email protected] (J.Y. Lee).
S. Karthikeyan, Jin Yong Lee ⇑Department of Chemistry, Sungkyunkwan University, Suwon 440-746, Republic of Korea
a r t i c l e i n f o
Article history:Received 22 January 2014In final form 7 April 2014Available online 13 April 2014
a b s t r a c t
The interaction between chloroacetylene (C2HCl) and substituted benzenes has been investigated using densityfunctional theory (DFT), MP2, and CCSD(T)/CBS methods. The results derived from these calculations revealedpredominant non-covalent p. . .ClC2H interactions in all cases. The predicted interaction energies for substitutedbenzene/ClC2H complexes span a narrow range from �1.61 to �3.96 kcal/mol, indicating that the p. . .ClC2Hinteraction is comparable in strength to well-documented C–H. . .p interactions. The trend for interaction energieswas found to be hexafluorobenzene–ClC2H < sym-tetrafluorobenzene–ClC2H < sym-trifluorobenzene–ClC2-H < sym-difluorobenzene–ClC2H < benzene–ClC2H < sym-dimethylbenzene–ClC2H < sym-trimethylben-zene–ClC2H < sym-tetramethylbenzene–ClC2H < hexamethylbenzene–ClC2H. We have shown thatelectron-withdrawing groups weaken the complex, whereas electron-donating groups strengthen theinteraction energy of the complex.
� 2014 Elsevier B.V. All rights reserved.
1. Introduction
In recent years, non-covalent interactions between organic hal-ogen compounds and aromatic rings are become of pivotal impor-tance in the design of novel drugs, materials, crystal engineering,and in the study of biological systems [1–44]. Of these interactions,C–X. . .p interactions can be compared with C–H. . .p interactions.The interaction between a halogen atom and an electron donorwas first reported by Guthrie in 1863 [1], but the key role of thehalogen bond in molecular recognition, crystal engineering, andbio-molecular systems has only recently been revealed. Since boththe halogen atom and the halogen-bonded electron donor are con-sidered to be negatively charged, the very existence of a halogenbond is surprising, an inconsistency first raised by Politzer et al.[2–4]. The importance of the halogen interaction in crystal struc-tures was noticed by Hassel et al. from X-ray structures of a greatnumber of inclusion compounds [5–7]. Subsequently, Murray-Rustand co-workers [8–10] surveyed the Cambridge Structural Data-base (CSD) and found that a large number of fragments were sur-prisingly involved in non-covalent interactions between arylcomponents and halogens. In addition, it was reported that halo-gen. . .p complexes are key intermediates in electrophilic halogen-ations of organic molecules [11–13].
A halogen bond can be considered to be the electrostatic attrac-tion between the positive potential of the halogen and the negativesite on the counterpart. The region of positive potential on thehalogen surface is often described as a positive r-hole [3,4,14–17] and has been termed an electropositive crown [2] as exempli-fied by the electrostatic potential surface of chloroacetyleneshown in Figure 1.
The halogen center may act as a Lewis acid as well as a Lewisbase. For example, nucleophiles approach head-on in the polarpositive region, whereas electrophiles tend to attack side-on inthe equatorial negative region. Thus, a halogen bond can be formedwith lone-pair-possessing atoms or p systems. Numerous theoret-ical studies of halogen bonding (Csp3–X. . .p; where Csp3 is sp3
hybridized carbon) have been reported in recent years [18–28].To the best of our knowledge, no systematic study has been
reported to account for the influence of substituents on theCsp–X. . .p interaction (where Csp is an sp hybridized carbon). Inthe present work, the benzene–chloroacetylene (Bz. . .ClC2H) dimerwas chosen as a reference dimer. Fluorine and methyl substituentswere appropriately selected to analyze their influence on theC–X. . .p interaction. While the fluorine atom is an electron-with-drawing group (the most electronegative atom in the periodictable), the methyl substituent is an electron-donating group.Substitutions were made such that the total dipole moment ofthe aromatic molecule would remain zero. The following aro-matic moieties were chosen: sym-difluorobenzene (2FBz), sym-trifluorobenzene (3FBz), sym-tetrafluorobenzene (4FBz), and

Figure 1. Electrostatic potential surface of choloracethylene.
S. Karthikeyan, J.Y. Lee / Chemical Physics Letters 602 (2014) 16–21 17
hexafluorobenzene (6FBz). Methyl substitutions were made in asimilar fashion. In addition, the influences of some other key sub-stituents (NO2, CN, COOH, Br, Cl, OH, and NH2) were also investi-gated. This study provides useful information on the nature andmagnitude of halogen. . .p interactions and contributes to a betterunderstanding of electrophilic aromatic substitution reactions.
Figure 2. MP2/cc-pVTZ optimized structures of multiple fluo
2. Computational methods
M05-2X, M06-2X, B97-D, wB97X-D, and CCSD(T) calculationswere performed using the GAUSSIAN 09 suite of programs [45], andBLYP-D3 and B3LYP-D3 calculations were carried out using theORCA package [46]. Computations with DFT methods were per-formed with the aug-cc-pVTZ basis set. The geometries of all com-plexes were obtained using counterpoise-corrected gradientoptimization at the MP2/cc-pVTZ level of theory. Hobza and co-workers [47] showed that this method yields geometries in goodagreement with those from the CCSD(T)/CBS method. Subse-quently, single-point energy calculations were performed at theMP2/aug-cc-pVDZ, MP2/aug-cc-pVTZ, and CCSD(T)/CBS levels.The core electrons were considered static in all of the calculations.The interaction energies for the dimers were computed using thesuper-molecule approach. All of the interaction energies were cor-rected for basis set superposition error (BSSE) using the counter-poise-correction (CP) method. MP2/CBS energy values weredetermined by exploiting the two-point extrapolation scheme pro-posed by Helgaker et al. [48]. Furthermore, the estimated CCSD(T)/CBS energy was computed using the following formula:
DECCSDðTÞ=CBS ¼ DEMP2=CBS þ ðDECCSDðTÞ=aVDZ � DEMP2=aVDZÞ
ro- and methyl-substituted benzene–ClC2H complexes.
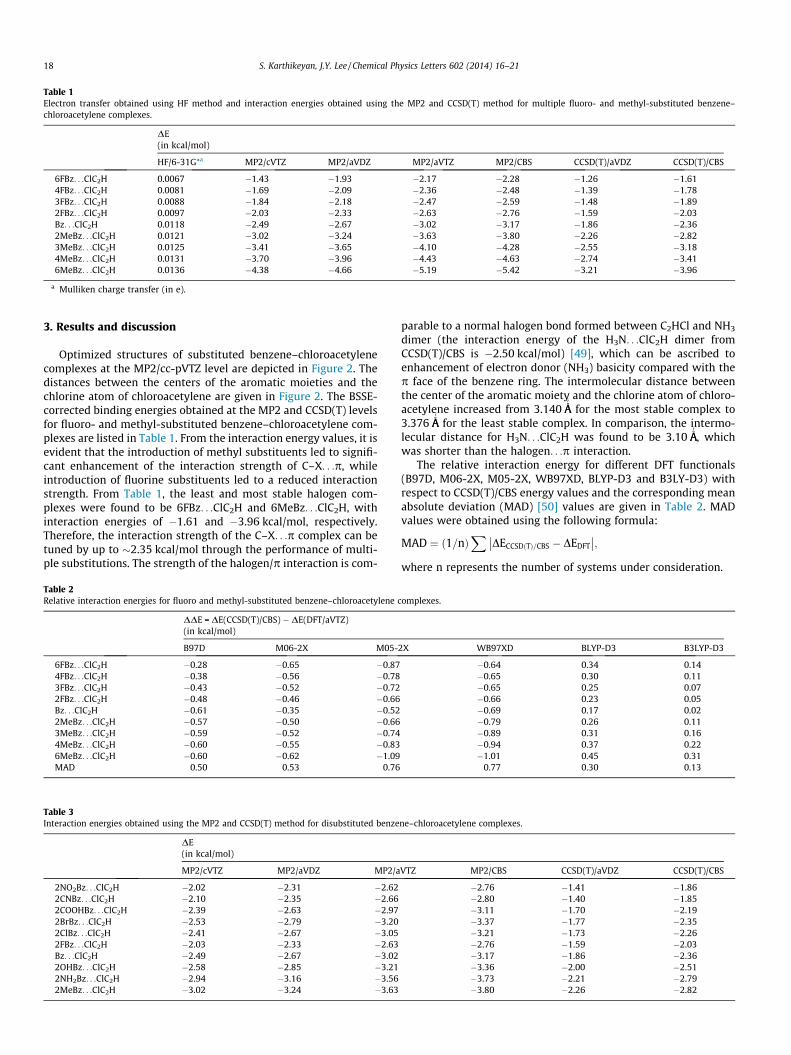
Table 1Electron transfer obtained using HF method and interaction energies obtained using the MP2 and CCSD(T) method for multiple fluoro- and methyl-substituted benzene–chloroacetylene complexes.
DE(in kcal/mol)
HF/6-31G⁄a MP2/cVTZ MP2/aVDZ MP2/aVTZ MP2/CBS CCSD(T)/aVDZ CCSD(T)/CBS
6FBz. . .ClC2H 0.0067 �1.43 �1.93 �2.17 �2.28 �1.26 �1.614FBz. . .ClC2H 0.0081 �1.69 �2.09 �2.36 �2.48 �1.39 �1.783FBz. . .ClC2H 0.0088 �1.84 �2.18 �2.47 �2.59 �1.48 �1.892FBz. . .ClC2H 0.0097 �2.03 �2.33 �2.63 �2.76 �1.59 �2.03Bz. . .ClC2H 0.0118 �2.49 �2.67 �3.02 �3.17 �1.86 �2.362MeBz. . .ClC2H 0.0121 �3.02 �3.24 �3.63 �3.80 �2.26 �2.823MeBz. . .ClC2H 0.0125 �3.41 �3.65 �4.10 �4.28 �2.55 �3.184MeBz. . .ClC2H 0.0131 �3.70 �3.96 �4.43 �4.63 �2.74 �3.416MeBz. . .ClC2H 0.0136 �4.38 �4.66 �5.19 �5.42 �3.21 �3.96
a Mulliken charge transfer (in e).
18 S. Karthikeyan, J.Y. Lee / Chemical Physics Letters 602 (2014) 16–21
3. Results and discussion
Optimized structures of substituted benzene–chloroacetylenecomplexes at the MP2/cc-pVTZ level are depicted in Figure 2. Thedistances between the centers of the aromatic moieties and thechlorine atom of chloroacetylene are given in Figure 2. The BSSE-corrected binding energies obtained at the MP2 and CCSD(T) levelsfor fluoro- and methyl-substituted benzene–chloroacetylene com-plexes are listed in Table 1. From the interaction energy values, it isevident that the introduction of methyl substituents led to signifi-cant enhancement of the interaction strength of C–X. . .p, whileintroduction of fluorine substituents led to a reduced interactionstrength. From Table 1, the least and most stable halogen com-plexes were found to be 6FBz. . .ClC2H and 6MeBz. . .ClC2H, withinteraction energies of �1.61 and �3.96 kcal/mol, respectively.Therefore, the interaction strength of the C–X. . .p complex can betuned by up to �2.35 kcal/mol through the performance of multi-ple substitutions. The strength of the halogen/p interaction is com-
Table 2Relative interaction energies for fluoro and methyl-substituted benzene–chloroacetylene c
DDE = DE(CCSD(T)/CBS) � DE(DFT/aVTZ)(in kcal/mol)
B97D M06-2X M05-
6FBz. . .ClC2H �0.28 �0.65 �0.874FBz. . .ClC2H �0.38 �0.56 �0.783FBz. . .ClC2H �0.43 �0.52 �0.722FBz. . .ClC2H �0.48 �0.46 �0.66Bz. . .ClC2H �0.61 �0.35 �0.522MeBz. . .ClC2H �0.57 �0.50 �0.663MeBz. . .ClC2H �0.59 �0.52 �0.744MeBz. . .ClC2H �0.60 �0.55 �0.836MeBz. . .ClC2H �0.60 �0.62 �1.09MAD 0.50 0.53 0.76
Table 3Interaction energies obtained using the MP2 and CCSD(T) method for disubstituted benze
DE(in kcal/mol)
MP2/cVTZ MP2/aVDZ MP2/a
2NO2Bz. . .ClC2H �2.02 �2.31 �2.622CNBz. . .ClC2H �2.10 �2.35 �2.662COOHBz. . .ClC2H �2.39 �2.63 �2.972BrBz. . .ClC2H �2.53 �2.79 �3.202ClBz. . .ClC2H �2.41 �2.67 �3.052FBz. . .ClC2H �2.03 �2.33 �2.63Bz. . .ClC2H �2.49 �2.67 �3.022OHBz. . .ClC2H �2.58 �2.85 �3.212NH2Bz. . .ClC2H �2.94 �3.16 �3.562MeBz. . .ClC2H �3.02 �3.24 �3.63
parable to a normal halogen bond formed between C2HCl and NH3
dimer (the interaction energy of the H3N. . .ClC2H dimer fromCCSD(T)/CBS is �2.50 kcal/mol) [49], which can be ascribed toenhancement of electron donor (NH3) basicity compared with thep face of the benzene ring. The intermolecular distance betweenthe center of the aromatic moiety and the chlorine atom of chloro-acetylene increased from 3.140 ÅA
0
for the most stable complex to3.376 ÅA
0
for the least stable complex. In comparison, the intermo-lecular distance for H3N. . .ClC2H was found to be 3.10 ÅA
0
, whichwas shorter than the halogen. . .p interaction.
The relative interaction energy for different DFT functionals(B97D, M06-2X, M05-2X, WB97XD, BLYP-D3 and B3LY-D3) withrespect to CCSD(T)/CBS energy values and the corresponding meanabsolute deviation (MAD) [50] values are given in Table 2. MADvalues were obtained using the following formula:
MAD ¼ ð1=nÞX
DECCSDðTÞ=CBS � DEDFT
�� ��;
where n represents the number of systems under consideration.
omplexes.
2X WB97XD BLYP-D3 B3LYP-D3
�0.64 0.34 0.14�0.65 0.30 0.11�0.65 0.25 0.07�0.66 0.23 0.05�0.69 0.17 0.02�0.79 0.26 0.11�0.89 0.31 0.16�0.94 0.37 0.22�1.01 0.45 0.31
0.77 0.30 0.13
ne–chloroacetylene complexes.
VTZ MP2/CBS CCSD(T)/aVDZ CCSD(T)/CBS
�2.76 �1.41 �1.86�2.80 �1.40 �1.85�3.11 �1.70 �2.19�3.37 �1.77 �2.35�3.21 �1.73 �2.26�2.76 �1.59 �2.03�3.17 �1.86 �2.36�3.36 �2.00 �2.51�3.73 �2.21 �2.79�3.80 �2.26 �2.82

Table 4Relative interaction energies for disubstituted benzene–chloroacetylene complexes.
DDE = DE(CCSD(T)/CBS) � DE(DFT/aVTZ)(in kcal/mol)
B97D M06-2X M05-2X WB97XD BLYP-D3 B3LYP-D3
2NO2Bz. . .ClC2H �0.65 �0.82 �1.02 �0.86 0.19 0.042CNBz. . .ClC2H �0.68 �0.69 �0.90 �0.85 0.22 0.052COOHBz. . .ClC2H �0.60 �0.72 �0.93 �0.82 0.25 0.082BrBz. . .ClC2H �0.64 �0.66 �0.88 �0.89 0.26 0.052ClBz. . .ClC2H �0.66 �0.62 �0.81 �0.89 0.24 0.042FBz. . .ClC2H �0.48 �0.46 �0.66 �0.66 1.54 0.05Bz. . .ClC2H �0.61 �0.35 �0.52 �0.69 1.40 0.022OHBz. . .ClC2H �0.55 �0.47 �0.64 �0.79 0.27 0.092NH2Bz. . .ClC2H �0.55 �0.49 �0.64 �0.87 0.30 0.112MeBz. . .ClC2H �0.57 �0.50 �0.66 �0.79 0.26 0.11MAD 0.60 0.56 0.75 0.81 0.52 0.06
S. Karthikeyan, J.Y. Lee / Chemical Physics Letters 602 (2014) 16–21 19
The BSSE-corrected interaction energies for various DFT func-tionals are similar to those for the CCSD(T)/CBS method. FromTable 2, we observe that the B3LYP-D3 method yields a lowerMAD value, while the DFT functionals B97-D, M06-2X, M05-2X,and WB97XD overestimate the interaction energies. Among thedifferent DFT functionals used, the B3LYP-D3 method appears tobe best, with a MAD value of 0.13 kcal/mol. The BLYP-D3, B97D,M06-2X, M05-2X, and WB97XD methods gave MAD values of0.30, 0.50, 0.53, 0.76, 0.77 kcal/mol, respectively.
We also investigated the interaction energies of various para di-substituted benzene–C2HCl complexes at the DFT, MP2, and
Figure 3. MP2/cc-pVTZ optimized structures of variou
CCSD(T)/CBS level of theory. The interaction energy values arelisted in Tables 3 and 4, and the corresponding geometries aredepicted in Figure 3. The selected substituents can be classifiedinto two categories: electron-withdrawing groups (NO2, CN, COOH,Br, Cl, and F) and electron-donating groups (OH, NH2, and CH3). Thepresence of electron-withdrawing groups on benzene resulted inan appreciable decrease in the interaction energy, while the intro-duction of electron-donating substituents increased the interactionenergy. For example, NO2 and CN substituents decreased the inter-action energy by approximately 0.5 kcal/mol relative to the unsub-stituted dimer; it is noteworthy that the electron-withdrawing
s para di-substituted benzene–C2HCl complexes.

Figure 4. Electrostatic potential results for substituted benzenes (the most negative electrostatic potential of the aromatic ring (atomic unit) is given in parentheses).
20 S. Karthikeyan, J.Y. Lee / Chemical Physics Letters 602 (2014) 16–21
ability of two nitro (or two CN) groups is comparable to that ofthree fluorine atoms. This can be rationalized by the fact thatNO2 and CN groups exert both �R (resonance) and �I (inductive)effects, whereas F has a +R effect on the benzene ring. While bothNO2 and F groups are electron-withdrawing, they differ in reso-nance effects. The three halogens are less deactivating than theNO2 and CN groups. The OH, NH2, and CH3 groups are known tobe electron-donating groups, and consequently, their complexeshave greater interaction energies compared to the unsubstituteddimer. The magnitude of the stabilization energy can be rational-ized by the increased electron density due to the presence of elec-tron-donating groups on benzene. As seen from Tables 3 and 4, theinteraction energy of the methyl-substituted dimer was higherthan those of the other dimers because the methyl group has botha positive inductive effect and a hyperconjugation effect.
The electrostatic potentials of p systems are shown in Figure 4.The most negative electrostatic potential on the aromatic ring (inatomic units) is given in Figure 4. The introduction of electron-donating groups on benzene increases the negative electrostaticpotential of the p ring, whereas electron-withdrawing groups havethe opposite effect. Interestingly, the negative electrostatic poten-tials of the p electrons correlate well with the interaction energies.
The amounts of electrons transferred from the donor (substi-tuted benzene) to the acceptor (C2HCl) are shown in Table 1. Inall cases, the HF method was combined with the 6-31G⁄ basis
set. Mulliken charge transfer (electron unit) of all complexes aregiven in Table 1. Interestingly, the amount of charge transferincreased linearly with the number of electron-donating groupsin the complexes, whereas it decreased linearly with the numberof electron-withdrawing groups.
4. Conclusion
We investigated the interactions between chloroacetylene andsubstituted benzenes and found that the binding energy of thebenzene–chloroacetylene complexes could be tuned by up to�2.35 kcal/mol by substituting hydrogen atoms of the benzenemolecule with multiple electron-donating or electron-withdraw-ing groups. The introduction of electron-donating groups (CH3,NH2, and OH) on benzene increased the interaction energy,whereas electron-withdrawing groups (NO2, CN, COOH, Br, Cl,and F) had the opposite effect. The dispersion energy is primarilyresponsible for the overall stabilization of the complexes; however,electrostatic interactions are also significant.
Acknowledgments
This work was supported by NRF grants (Nos. 2007-0056343and 2011-0015767) funded by MEST, Republic of Korea. Theauthors would like to acknowledge support from the KISTI

S. Karthikeyan, J.Y. Lee / Chemical Physics Letters 602 (2014) 16–21 21
supercomputing center through the strategic support program forsupercomputing application research [No. KSC-2012-C2-43].
References
[1] F. Guthrie, J. Chem. Soc. 16 (1863) 239.[2] T. Brinck, J.S. Murray, P. Politzer, Int. J. Quantum Chem. Quantum Biol. Symp.
19 (1992) 57–64.[3] P. Politzer, P. Lane, M.C. Concha, Y.G. Ma, J.S. Murray, J. Mol. Model. 13 (2007)
305.[4] T. Clark, M. Hennemann, J.S. Murray, P. Politzer, J. Mol. Model. 13 (2007) 291.[5] O. Hassel, C. Romming, Q. Rev, Chem. Soc. 16 (1962) 1.[6] E. Damm, O. Hassel, C. Romming, Acta Chem. Scand. 19 (1965) 1159.[7] P. Metrangolo, G. Resnati, T. Pilati, S. Biella, in: Halogen Bonding Fundamentals
and Applications, Springer, Berlin/Heidelberg, 2008, p. 105.[8] P. Murray-Rust, W.D.S. Motherwell, J. Am. Chem. Soc. 101 (1979) 4374.[9] P. Murray-Rust, W.C. Stallings, C.T. Monti, R.K. Preston, J.P. Glusker, J. Am.
Chem. Soc. 105 (1983) 3206.[10] N. Ramasubbu, R. Parthasarathy, P. Murray-Rust, J. Am. Chem. Soc. 108 (1986)
4308.[11] A. Legon, Angew. Chem. Int. Ed. 38 (1999) 2686.[12] D. Lenoir, Angew. Chem. Int. Ed. 42 (2003) 854.[13] D. Lenoir, C. Chiappe, Chem. Eur. J. 9 (2003) 1036.[14] K.E. Riley, P. Hobza, J. Chem. Theory Comput. 4 (2008) 232.[15] P. Auffinger, F.A. Hays, E. Westhof, P.S. Ho, Proc. Natl. Acad. Sci. U.S.A. 101
(2004) 16789.[16] E. Bosch, C.L. Barnes, Cryst. Growth Des. 2 (2002) 299.[17] A. Chana, M.A. Concejero, M. de Frutos, M.J. Gonzalez, B. Herradon, Chem. Res.
Toxicol. 15 (2002) 1514.[18] J.W. Zou, Y.J. Jiang, M. Guo, G.X. Hu, B. Zhang, H.Ch. Liu, Q.S. Yu, Chem. Eur. J. 11
(2005) 740.[19] W.Z. Wang, N.B. Wong, W.X. Zheng, W.X. Zhang, A.M. Tian, J. Phys. Chem. A
108 (2004) 1799.[20] A. Karpfen, J. Phys. Chem. A 104 (2000) 6871.[21] P. Romaniello, F. Lelj, J. Phys. Chem. A 106 (2002) 9114.[22] I. Alkorta, J. Rozas, J. Elguero, J. Phys. Chem. A. 102 (1998) 9278.[23] W. Wang, P. Hobza, J. Phys. Chem. A 112 (2008) 4114.[24] Y.X. Lu, J.W. Zou, Y.H. Wang, Q.S. Yu, Chem. Phys. 334 (2007) 1.
[25] D.J.R. Duarte, M.M. De Las Vallejos, N.M.J. Peruchena, Mol. Model. 16 (2010)737.
[26] H.G. Wallnoefer, T. Fox, K.R. Liedl, C.S. Tautermann, Phys. Chem. Chem. Phys.12 (2010) 14941.
[27] A. Forni, S. Rendine, S. Pieraccini, M. Sironi, ChemPhysChem 13 (2012) 4224.[28] A. Forni, S. Pieraccini, S. Rendine, M. Sironi, J. Comp. Chem. 35 (2014) 386.[29] K.S. Kim, P. Tarakeshwar, J.Y. Lee, Chem. Rev. 100 (11) (2000) 4145.[30] P. Tarakeshwar, H.S. Choi, K.S. Kim, J. Am. Chem. Soc. 123 (14) (2001) 3323.[31] E.C. Lee et al., J. Am. Chem. Soc. 127 (12) (2005) 4530.[32] S. Vaupel, B. Brutschy, P. Tarakeshwar, K.S. Kim, J. Am. Chem. Soc. 128 (16)
(2006) 5416.[33] P. Tarakeshwar, S.J. Lee, J.Y. Lee, K.S. Kim, J. Chem. Phys. 108 (17) (1998) 7217.[34] P. Tarakeshwar et al., J. Chem. Phys. 111 (13) (1999) 5838.[35] N.J. Singh, S.K. Min, D.Y. Kim, K.S. Kim, J. Chem. Theory Comput. 5 (3) (2009)
515.[36] E.C. Lee, D. Kim, P. Jurecka, P. Tarakeshwar, P. Hobza, K.S. Kim, J. Phys. Chem. A
111 (18) (2007) 3446.[37] K.S. Kim, S. Karthikeyan, N.J. Singh, J. Chem. Theory Comput. 7 (11) (2011)
3471.[38] I. Geronimo, E.C. Lee, N.J. Singh, K.S. Kim, J. Chem. Theory Comput. 6 (7) (2010)
1931.[39] S.K. Min, E.C. Lee, H.M. Lee, D.Y. Kim, D. Kim, K.S. Kim, J. Comput. Chem. 29 (8)
(2007) 1208.[40] S. Karthikeyan, S. Nagase, J. Phys. Chem. A 116 (2012) 1694.[41] B.K. Mishra, S. Karthikeyan, V. Ramanathan, J. Chem. Theory Comput. 8 (6)
(2012) 1935.[42] M. Guin, G.N. Patwari, S. Karthikeyan, K.S. Kim, Phys. Chem. Chem. Phys. 13
(2011) 5514.[43] S. Maity, A. Dey, G.N. Patwari, S. Karthikeyan, K.S. Kim, J. Phys. Chem. A 114
(42) (2010) 11347.[44] S. Karthikeyan, H.M. Lee, K.S. Kim, J. Comput. Chem. 6 (10) (2010) 3190.[45] M.J. Frisch et al., GAUSSIAN 09, Revision B.01, Gaussian Inc., Wallingford, CT,
2010.[46] F. Neese et al., Orca Version 3, Program package, 2011.[47] I. Dabkowska, P. Jurecka, P. Hobza, J. Chem. Phys. 122 (2005) 204322.[48] T. Helgaker, W. Klopper, H. Koch, J. Noga, J. Chem. Phys. 106 (1997) 9639.[49] S. Karthikeyan, R. Sedlak, P. Hobza, J. Phys. Chem. A. 115 (2011) 9422.[50] S. Karthikeyan, V. Ramanathan, B.K. Mishra, J. Phys. Chem. A. 117 (2013) 6687.





























