Embed Size (px)
Citation preview

14Life and Death of Trypanosoma brucei: New Perspectivesfor Drug DevelopmentTorsten Barth, Jasmin Stein, Stefan Mogk, Caroline Sch€onfeld,Bruno K. Kubata, and Michael Duszenko�
AbstractCell death is a life-long companion for any cell and molecular processes evolved inorder to deal with life-threatening situations like starvation or poisoning. It is alsoknown that in metazoa different pathways exist by which a cell undergoes suicidefor the benefit of the whole organism, collectively known as apoptosis. Althoughprotozoa generally lack caspases, a formerly thought indispensable prerequisitefor apoptosis, an inducible caspase-independent form of apoptosis as a mecha-nism of cell density regulation has been described in African trypanosomes. Here,we review the current status of cell death mechanisms in Trypanosoma bruceiincluding necrosis, autophagy, and apoptosis. We will discuss which of theseevents are necessary to cope with stressful situations, and which are probablyneeded to ensure continuance of infection and transmittance to the insect vector.Since autophagy and apoptosis are essential for the parasite’s survival, we will alsodiscuss possible pathway junctions suitable for drug development.
Necrosis
Any cell will undergo necrosis if changes of the environmental conditions arecontradictory to cell integrity. This might be extensive temperature shifts, highpressure, non-isotonic conditions, or the appearance of substances that eitherdissolve the membrane integrity or lead to a breakdown of the energy metabolism.In any case, this is an accidental cell death and mostly a rather rapid process.Detection of necrosis is usually performed by light or electron microscopy, orby uptake of specific dyes that are unable to cross intact membranes. Propidiumiodide is often used in this respect, because it is excluded from viable cells, butpenetrates damaged membranes, intercalates into DNA (one molecule of dyebinds to about 5 bp), and is then readily detectable by fluorescence-activated cellsorting measurements (Figure 14.1). However, if conditional changes appeargradually, a cell would react accordingly to cope with the respective stress
� Corresponding Author
Trypanosomatid Diseases: Molecular Routes to Drug Discovery, First edition. Edited by T. Jäger, O. Koch, and L. Flohé.# 2013 Wiley-VCH Verlag GmbH & Co. KGaA. Published 2013 by Wiley-VCH Verlag GmbH & Co. KGaA.
j261

condition. In this case, signs of autophagic cell death or apoptosis may also bedetected as a consequence of the cellular response [1].Necrosis in bloodstream-form trypanosomes is easily detected by incubating the
parasites in, for example, glucose-free media or using sufficient concentrations oftrypanocidal drugs (Figure 14.1). Although induction of necrosis in trypanosomesmight appear as a perfect alternative to remove parasites from the host, severe side-effects may appear during this process that could threaten the host’s life. First, drugsintervening with the parasites metabolism in a way to cause necrosis will probablyalso interact with at least some of the host’s cells. For example, depletion of glucosewould kill the trypanosomes, but would also lead to anemia, to coma, and finally todeath caused by a hypoglycemic shock. The reason is that most, if not all, of thecentral metabolic pathways are evolutionarily highly conserved. Here again, glycoly-sis is a very good example: trypanosomes as well as the other flagellates of the orderKinetoplastida possess an organelle called a glycosome that contains most of theglycolytic enzymes [2,3]. Thus, although the organization of this pathway as well as
Figure 14.1 Necrosis in African trypanosomesafter H2O2 treatment. (a) Scanning electronmicrograph of trypanosomes incubated for 3 hin culture medium containing 40 mMH2O2.(b) Flow cytometry double staining withpropidium iodide (PE-A) and Annexin-V-FLUOS(FITC-A) to detect necrosis and apoptosis. Leftpanel: control trypanosomes incubated in
culture medium; right panel: trypanosomesincubated for 3 h in culture medium containing40mMH2O2. Quadrants: dots in the lower-leftquadrant represent viable cells; dots in thelower-right quadrant represent apoptotic cells(Annexin positive); dots in the upper-rightquadrant represent necrotic cells (Annexin andpropidium iodide positive).
262j 14 Life and Death of Trypanosoma brucei: New Perspectives for Drug Development

the structure of several glycolytic enzymes show differences and peculiarities ascompared with their mammalian counterpart, so far no specific inhibition ofglycolysis in trypanosomes could be established, although crystal structures ofmany glycolytic enzymes exist of both mammalian and trypanosomal origin [4–6].Secondly, mass destruction of parasites would lead to a massive inflammationreaction that, especially in local areas like the meningeal compartment, may havedeleterious effects.
Autophagic Cell Death
Autophagy in general is a survival mechanism of a cell to cope with different stresssituations. The most plausible and best-analyzed scenario is starvation. In this casepart of the cytosol including organelles is engulfed by a double membrane that isformed elsewhere within the cell and delivered to the lysosome for degradation.Following fusion of the outer membrane of the autophagosome with the lysosome,the inner autophagosomal membrane as well as its contents are degraded bylysosomal hydrolases and the remaining substrates (e.g., nucleosides, amino acids,carbohydrates, and lipids) are released into the cytosol for biosynthesis of essentialbiopolymers (nucleic acids, proteins, etc.). In this way, a cell can survive for sometime by eating its less important constituents to form urgently needed molecules. Ifnutrition does not return to a normal supply, the cell will enter autophagic cell death,most obviously characterized by a massive increase of lysosomes and vacuolization.In contrast to the constitutively occurring engulfment of parts of the cytosol by thelysosome itself (microautophagy), the process has been called macroautophagy.Interestingly, this is not the only cellular event wheremacroautophagy is involved. Infact, it became increasingly clear during the last decade that autophagy is alsoinvolved during cell differentiation and generally during cellular remodeling as anadaptation to environmental changes [7]. In addition, it plays significant roles indisease [8] and infection [9]. We will briefly describe the general molecularmachinery involved in the autophagic process, before we concentrate on trypano-somes. Macroautophagy (further referred to as autophagy) starts with the formationof a phagophore (also called an isolation membrane) at a certain place within the cellthat has been called the “phagophore assembly site” or “pre-autophagosomalstructure” (PAS). It is a long-standing question where the PAS originates. Datahave been presented that, on the one hand, it is formed as an elongation of theendoplasmic reticulum [10] or, on the other hand, that the outer mitochondrialmembrane contributes to PAS formation [11]. Although autophagy occurs consti-tutively at a low basal level, the PAS is massively formed in response to a definedsignal (e.g., in the absence of amino acids). The central regulator is the serine/-threonine kinase TOR, an enzyme that was named due to its inhibition byrapamycin, hence its name “target of rapamycin.” Rapamycin, a macrolide, wasoriginally isolated from Streptomyces hygroscopicus and used as an anti-fungal drug,before it was discovered that it has a pronounced immunosuppressive effect byinhibiting the intracellular response of B- and T-cells to interleukin-2 [12]. As we
Autophagic Cell Death j263

know now, rapamycin associates with the soluble protein FK-binding protein 12(FKBP12) before it directly binds to the TOR complex 1 (TORC1) to inhibit its kinaseactivity. TORC1 and TORC2 are two distinct cytosolic complexes consisting ofseveral proteins with TOR as the catalytic center of both complexes [13]. TORC1senses many different cellular signals like the amino acid concentration, theavailability of growth factors, the energy status of the cell, and stress. Sufficientsupply of these signals and the absence of stress lead the cell to an anabolic ratherthan catabolic metabolism, inducing cell growth, cell cycle progression, and cellproliferation. On the contrary, deficiency of these signals, an increase in stress, orbinding of rapamycin–FKBP12 leads to the opposite effects and induces autophagy(Figure 14.2a) [13]. Usually, TORC2 does not bind rapamycin–FKBP12 and does notinduce autophagy, but is involved in cytoskeleton organization and cell volumecontrol [14]. Inhibition of TORC1 blocks phosphorylation of the downstream signal
Figure 14.2 Schematic description ofautophagy. (a) Regulation of the induction ofautophagy; (b) vesicle nucleation andgeneration of membranes; (c) autophagosome
formation by the Atg8 and Atg12 ubiquitin-likepathways; and (d) fusion of a lysosome andautophagosome to an autophagolysosome andvesicle breakdown. (See text for more details.).
264j 14 Life and Death of Trypanosoma brucei: New Perspectives for Drug Development

molecules and induces autophagy. Key players in autophagy induction are autoph-agy-related proteins (Atg) which are encoded in autophagy-related genes (ATG).TORC1 phosphorylates Atg13 to inhibit autophagy. Under starvation conditions,TORC1 becomes inactive and Atg13 is readily dephosphorylated, forming a complexwith Atg1 and Atg17. The activated complex induces formation of the PAS byactivating a protein complex consisting beside others (e.g., Atg6, Atg14, Vps15) ofclass III phosphatidylinositol-3 kinase (Vps34). This Vps34 (Vps¼ vacuolar proteinsorting) leads to formation of phosphatidylinositol-3-phosphate that is enriched inthe inner bilayer of the autophagosome membrane thereby attracting additional Atgproteins like the Atg18–Atg2 complex to increase the membrane’s size(Figure 14.2b). The next step (i.e., formation of a vesicular structure) includesbending of the double membrane, which is induced by binding of Atg8 tophosphatidylethanolamine (PE). Atg8, a ubiquitin-like protein, reacts with thecysteine protease Atg4, which removes amino acids until a glycine residue isexposed at the C-terminus. It is then conjugated to PE by the E1-like activatingenzyme Atg7 and the E2-like conjugating enzyme Atg3. Conjugation of Atg8 issupported by Atg12, Atg5, and Atg16 – a protein complex that works as an E3-likeenzyme and needs activity of Atg7 and Atg10 for its own formation. Finally, Atg8 isreleased from PE by Atg4 (Figure 14.2c) [15]. The newly formed autophagosomescarry a cargo of engulfed cytoplasmic materials including cytosol with all solutes,macromolecules like ribosomes, and organelles. The outer membrane of theautophagosome fuses with a lysosome, thereby forming an autophagolysosome.Now the lysosomal hydrolases will degrade the inner autophagosomal membraneand all constituents of the cargo to release the degradation products (amino acids,nucleosides, sugars, lipids) into the cytosol (Figure 14.2d).Wewill now describe differences in the autophagymechanisms in trypanosomes
in order to discuss possible drug targets. There is no doubt that trypanosomesundergo autophagy, which is clearly visible in electron micrographs (Figure 14.3).Obviously, autophagy is already visible under control conditions (Figure 14.3a), butis especially induced during starvation or incubation with nanoparticles(Figure 14.3b and c). Since a bioinformatics survey had revealed that a considerablylower number ofATG genes seem to be present in Kinetoplastida, it was speculatedthat autophagy in these parasites may be more simply organized than in highereukaryotes [16]. However, as we know now, the major principles are very muchcomparable, although some peculiarities exist that could indeed serve as targets fordrug development. It already starts with the TORC complexes. As described above,TORC1 is usually the target of rapamycin and its inhibition leads to the induction ofautophagy. In African trypanosomes, both TORC1 and TORC2 are present andbuild from two different TOR kinases (TbTOR1 and TbTOR2) and different adapterproteins, including TbTOR-like 1 and TbTOR-like 2 [17]. As in the mammaliansystem, both complexes are involved in cell cycle progression and cell division, butin contrast, TORC1 is not affected by rapamycin in the nanomolar range intrypanosomes. Instead, rapamycin inhibits TORC2, leading to a disruption ofcytokinesis and subsequently cell death [17]. Since TOR and TOR-like proteins areconsiderably different to their mammalian counterparts and rapamycin inhibits
Autophagic Cell Death j265

specifically TORC2 instead of TORC1 as in the host’s system, the parasitic TOR andTOR-like proteins could indeed be valuable targets for drug development, espe-cially as they are involved in cell cycle control and progression. This is furthersupported by the finding that the TOR-like 1 kinase seems specifically involved information of acidocalcisomes – unique and essential organelles in Kinetoplastida[18]. The next step, namely formation of the autophagosome bilayer, dependsprimarily on Vps34. The respective ortholog of this protein has been investigated intrypanosomes, and seems especially involved in Golgi segregation as well as inreceptor-mediated endocytosis and in exocytosis [19]. Its involvement in autophagywas not explored in this study, but RNA interference-induced knockdown mutantsshowed severe growth defects, indicating that TbVps34 is needed for a correctcytokinesis. It might thus be advisable to investigate more about PAS membraneformation in trypanosomes in order to gainmore information about this step and toanalyze its suitability for drug development. Finally, autophagosome formation andprogress of autophagy has been investigated in several laboratories [9].
Figure 14.3 Electron micrographs showing thedevelopment of autophagy in T. brucei. (a)Untreated control cells showing a very low butvisible rate of autophagy; (b) autophagyinduced by nanoparticles labeled with bovineserum albumin (BSA) for uptake bytrypanosomes; and (c) final stage of autophagyclose to autophagic cell death induced by amino
acid starvation for 24 h. AP, autophagosome;APL, autophagolysosome; APP,autophagophore; fp, flagellar pocket; G,glycosome; Gol, Golgi apparatus; kDNA,kinetoplast DNA; L, lysosome; M,mitochondrion; PAS, phagophore assemblingsite; N, nucleus.
266j 14 Life and Death of Trypanosoma brucei: New Perspectives for Drug Development

Trypanosomes contain all orthologs for the Atg8 pathway and possess threepossible ATG8 genes, named ATG8.1, ATG8.2, and ATG8.3 [9]. The respectiveprotein Atg8.3 contains an insertion of 16 amino acids and seems not to be involvedin autophagosome formation. The function of Atg8.1 is confusing. Since it is oneamino acid shorter and ends with a glycine residue on its C-terminal end, it will notbe processed by Atg4. It is, however, expressed (as judged from our unpublishedreverse transcription-polymerase chain reaction data) and is probably responsiblefor the constitutively occurring basal autophagy observed at any time in thisparasite independent of autophagy induction (see control cells, Figure 14.3a). Incontrast, Atg8.2 contains a single amino acid extension on the C-terminal end. Wehave solved the three-dimensional structure of TbAtg8.1/2 and could show byhomology modeling with Atg4 from rats that it fits perfectly with its ubiquitin foldto this protease, extending its C-terminus into the catalytic site (Figure 14.4) [20].Obviously, Atg8.2 represents the true ortholog of LC3, the human counterpart, andis responsible for execution of induced autophagy. In this case it is activated uponlimited proteolysis by Atg4 and binds to PE. Accordingly, immunofluorescenceimages of trypanosomes starved by amino acid removal from the media show apunctuated (i.e., membrane-bound) localization of this protein, while it has a clearcytosolic distribution in non-starved control cells [21]. So far it remains to bedemonstrated thatT. brucei contains a functioningAtg12–Atg5 conjugation system.While a bioinformatics survey did not reveal clear homologs in trypanosomes,experimental evidence was presented showing that the respective proteins areactive in Leishmania [22].
There are several ways to induce autophagy in trypanosomes, even though the useof rapamycin is critical because it inhibits TORC2 rather than TORC1. The mostobvious way is amino acid starvation. In this case, we see at the level of electronmicroscopy clear morphological signs of autophagy, including formation of PASs,
Figure 14.4 Homology modeling docking the crystal structure of TbAtg8 to Atg4 from rat. Note:the C-terminal amino acid (cysteine) of Atg8 fits perfectly into the active center of Atg4.
Autophagic Cell Death j267

autophagosomes, and autophagolysosomes. If starvation proceeds, trypanosomesshow an increasing number of lysosomes, vacuolization of the cell, and eventuallycell death (Figure 14.3c).Interestingly, we see the same signs of autophagy when we treat trypanosomes
with different nanoparticles. Owing to their small size, in the range of 1–1000 nm,they offer new properties and new functions as compared to larger particles. Theycan be subdivided into different classes, such as liposomes, emulsions, ceramicnanoparticles, metallic nanoparticles, gold shell nanoparticles, and quantum dots[23,24]. These particles can be made up of different materials like TiO2, ZnO2, SiO2,
semiconductor material with unique fluorescent properties, or supermagnetic ironoxide [23]. Beyond their size, one main advantage of these particles is the surfacevariability, which allows a wide range of use. Different coats can be attached to thesurface of the particles. These coats can be fluorophores to trace the routes theparticles take within the cell and to study the mode of action, but also proteins orantibodies to target specific cells. In addition, nanoparticles can be filled withdifferent drugs to utilize them as a drug carrier system. In our study we used eitherhuman transferrin (Tf)- or bovine serum albumin (BSA)-coated Fe2O3 nanopar-ticles, both of a size of around 10 nm. The latter coat is known to be taken up by fluid-phase endocytosis [25,26], while transferrin is incorporated due to receptor-medi-ated endocytosis by trypanosomes [25]. Treatment of bloodstream-form T. bruceiwith these two nanoparticle formulations causes massive endocytosis and readilyinduced autophagy detectable via electron microscopy. We detected PASs, autopha-gosomes, and autophagolysosomes after nanoparticle treatment (Figure 14.3b).
Apoptosis in Protozoan Parasites
Apoptosis, formerly called programmed cell death, was originally described as a wayfor metazoan organisms to get rid of unwanted cells that are either no longernecessary, like in embryology [27,28], or even perilous for the survival of the wholeorganism [29]. Two mechanisms have been described: (i) the respective cell gets asignal from the organism via so-called death receptors to kill itself [30] or (ii) vital cellfunctions are damaged (e.g., by radiation or viral infection) forcing this cell todisappear [31,32]. In both cases, caspases, a defined class of cysteine proteases, areactivated by limited proteolysis to induce a controlled cell death avoiding inflam-mation [33]. The necessity of this form of cell death is most obvious in the case ofimmune cells, where myriads of “wrong” cells (i.e., cells expressing an antibody or aT-cell receptor with deleterious effects on the organism itself) have to die in a silentway. Either way of classical apoptosis is characterized by three indispensableprerequisites: the function of caspases as starting points of self-destruction, theavoidance of inflammatory reactions, and the benefit of the organism as a drivingforce during evolution of this induced cell death. Cellular self-destruction induces aset of morphological and biochemical changes that are used to define apoptosis on amolecular basis, and to separate it from necrosis and autophagic cell death. Amongothers, the most prominent changes are: shrinkage of the cell, blebbing,
268j 14 Life and Death of Trypanosoma brucei: New Perspectives for Drug Development

segmentation of the nucleus, single-strand DNA breaks, exposure of phosphati-dylserine in the outer leaflet of the plasma membrane, and loss of potential of theinner mitochondrial membrane [34,35]. To detect these changes, usually light orelectron microscopic techniques together with FACS analyses are used. The formerinclude the TUNEL test (terminal deoxynucleotidyl transferase dUTP nick end-labeling) to detect DNA strand breaks, the latter Annexin staining (to measurephosphatidylserine exposure) and tetramethylrhodamine ethyl ester staining tomeasure loss of the inner membrane potential (DY).Considering apoptosis in trypanosomes poses two serious problems: (i) protozoa
do not possess caspases and (ii) is it hard to believe that a single-cell organism maydevelop a molecular mechanism for its own death during evolution. Nevertheless,investigating cell death in protozoa showed that at least some of the hallmarks ofapoptosis (see above) can also be found in single-cell organisms [36,37], includingtrypanosomes [38,39].In this situation, the discovery of metacaspases in plants, fungi, and protists as a
class of proteins closely related to caspases in animals seemed like the missinglink. However, although metacaspases seem to be involved in cell death in plantsand fungi, there is no conclusive evidence for their participation in protozoaapoptosis. In fact, T. brucei contains five metacaspases, but they are not involved inapoptosis here, as we [40] and others [41] could show. This is also true forLeishmania [42,43]. The next step to solve the dilemma of apoptosis in protozoanparasites was the observation that a caspase-independent apoptosis exists inmetazoa [44]. In this case, a small redox enzyme located in the intermembranespace of mitochondria, the so-called apoptosis-inducing factor (AIF), is releasedand translocates to the nucleus to induce chromatin condensation and DNAdegradation [45,46]. Unfortunately, no ortholog of AIF has so far been detected inprotozoan parasites, but endonuclease G (Endo G) seems to perform the samesort of action if released from the intermembrane space during induction of celldeath [47]. We have analyzed cell death in trypanosomes upon prostaglandin (PG)D2 (PGD2) application (Figure 14.5). The reason to investigate PGD2 effects on theparasite was based on our discovery that trypanosomes produce prostaglandins[48], which was especially interesting because the PGD2 concentration is remark-ably high in the cerebrospinal fluid (CSF) of late-stage sleeping sickness patients[49]. We originally considered that PGD2 may induce differentiation from slenderto stumpy parasites, but readily observed cell death of the stumpy population.Interestingly, we detected nearly all classical hallmarks of apoptosis, but found noindication of necrosis or autophagic cell death [50,51]. These results clearly showthe parallel appearance of different populations that respond differently tosignaling molecules. In terms of parasite survival within the host this seemsto have major consequences. Cell density regulation is a major problem for aparasite to survive. Considering continuous proliferation with a generationdoubling time of about 8 h (as in trypanosomes), parasite density would exceedan acceptable level in blood, thus killing a human in less than 2 weeks.Trypanosomes deal with this problem by expressing a dense surface coatconsisting of 107 identical protein molecules, the so-called variant surface
Apoptosis in Protozoan Parasites j269
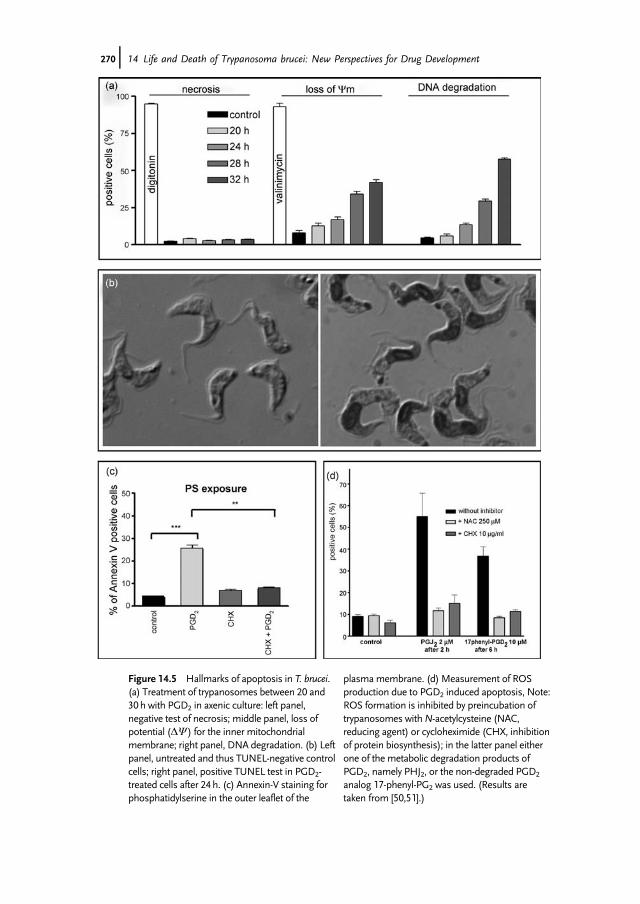
Figure 14.5 Hallmarks of apoptosis in T. brucei.(a) Treatment of trypanosomes between 20 and30h with PGD2 in axenic culture: left panel,negative test of necrosis; middle panel, loss ofpotential (DY) for the inner mitochondrialmembrane; right panel, DNA degradation. (b) Leftpanel, untreated and thus TUNEL-negative controlcells; right panel, positive TUNEL test in PGD2-treated cells after 24h. (c) Annexin-V staining forphosphatidylserine in the outer leaflet of the
plasmamembrane. (d) Measurement of ROSproduction due to PGD2 induced apoptosis, Note:ROS formation is inhibited by preincubation oftrypanosomes withN-acetylcysteine (NAC,reducing agent) or cycloheximide (CHX, inhibitionof protein biosynthesis); in the latter panel eitherone of the metabolic degradation products ofPGD2, namely PHJ2, or the non-degraded PGD2
analog 17-phenyl-PG2 was used. (Results aretaken from [50,51].)
270j 14 Life and Death of Trypanosoma brucei: New Perspectives for Drug Development

glycoprotein (VSG). This coat protects the parasite from the cellular immuneresponse as well as from complement. It is, however, immunogenic and leads toformation of specific antibodies. All parasites carrying the same coat are thenopsonized and killed by the immune system. Due to antigenic variation, newpopulations appear spontaneously carrying a different coat [52–54]. A trypano-somal infection is thus characterized by an oscillating population density in blood.In addition to this host-induced density regulation, trypanosomes differentiate inblood from a dividing slender to a non-dividing stumpy form that is preadapted tosurvive in the insect vector [55–57]. This differentiation depends on the slenderpopulation density [58] and is induced by a secreted low-molecular-weight factor(stumpy inducing factor (SIF) [58]) of so far unknown chemical identity thatenriches in conditioned culture media [59]. Most likely SIF works like anautocrine mediator that is released from slender cells and induces slender cellsto differentiate to the stumpy form.The presence of two different populations offers the basis for a parasite-induced
density regulation. In fact, trypanosomes do not behave like single self-containedindividual organisms, but as members of populations, talking to each other viaquorum sensing [9,60]. In this way, the dividing slender population grows untilthe local population density reaches a point where the concentration of thesecreted SIF is high enough to induce cell differentiation in its neighbor cells.Consequently, the number of slender cells decreases while the stumpy populationincreases. Stumpy trypanosomes do not divide anymore and do not undergoantigenic variation [61]. In addition, they are vulnerable to PGD2 that inducesformation of reactive oxygen species (ROS) which then leads to changes of themitochondrion membrane integrity and the release of Endo G [50,51]. Endo Gtranslocates to the nucleus and leads by activation of its nuclease activity to theonset of this caspase-independent form of apoptosis [62]. Considering thisconcept, it seems like a mechanistic strategy of the parasite to control its celldensity during the course of infection (Figure 14.6). The advantages are: (i) theparasitemia will usually not reach a density that would kill the host just by the purenumber of parasites (e.g., because of its high glucose consumption), (ii) theslender population cannot become completely extinct, since the SIF concentrationwould decrease with a decrease of the slender population and the movement of theparasite, (iii) an apoptotic cell death of the stumpy population would not induceinflammation, and (iv) this mechanism is independent of VSG-specific antibodiesand works also in immunologically privileged organs like the brain. The latterpoint seems especially important during the late stage of sleeping sickness. Takentogether, apoptosis of the stumpy population has indeed an altruistic quality as itenables the dividing slender form to keep the infection going and increases thechances of the parasite to be transmitted to the insect vector.Analyzing the brain stage of sleeping sickness, two possible entry gates exists: the
blood–brain barrier and the blood–CSF barrier. In the literature, data have beenpresented to support either the first way [63,64] or the second way [65]. We haveinvestigated the early brain infection stage in rodents (i.e., between 20 and 35 dayspost-infection) using electron microscopy, and detected parasites within the stroma
Apoptosis in Protozoan Parasites j271

of the choroid plexus, in the ventricles, and in the meningeal space, especially in apial and subpial location outside the glia limitans [66]. This space just outside thebrain parenchyma seems an ideal area for the parasite to reside. Here they are notattacked by immune cells or VSG-specific antibodies and they can move back intolymph (via the nasal lymphatic vessels [67]) or blood (via the arachnoid villi)to produce the observed relapses after killing of the blood parasites, such as bysuramin – a drug that does not penetrate into the brain. On the other hand, PGD2 ofparasite origin would be needed to control parasite density in this very limited spaceand could also be distributed via CSF to induce the well-known sleep disturbances[68]. Thus, apoptosis of stumpy cells seems to be an inevitable prerequisite to allowthe survival of trypanosomes for a prolonged time in the mammalian host.Considering apoptosis in trypanosomes as a possible drug target, it seems obvious
that neither induction of apoptosis or inhibition of apoptosis would offer a suitablestrategy. In the first case, only non-dividing stumpy forms would disappear with littleeffect on the ongoing infection, while in the latter case an unhampered parasitemiawould result with a disastrous effect on the host. However, the addition of the stumpyinduction factor would be very effective. From our study we know that this factor isfairly small (molecular weight below 500 Da), hydrophilic, and relatively unstable.Especially the latter point renders handling during preparation difficult and thus thechemical identity of SIF is still elusive. However, due to its expected functionality,
Figure 14.6 Mechanism of cell densityregulation in T. brucei. Schematic representationof SIF-induced differentiation from slender tostumpy parasites and PGD2-induced apoptosis
that leads to cell-cycle-arrested trypanosomesand finally cell death. Scanning electronmicrographs of cells obtained from infected ratsor from PGD2-treated parasites in cell culture.
272j 14 Life and Death of Trypanosoma brucei: New Perspectives for Drug Development

isolation and chemical identification is an ongoing project in our group. Another bigproblem to deal with in sleeping sickness is caused by the infiltration of the brainduring the late stage. Asmentioned above, trypanosomes enter the choroid plexus andtravel via CSF to the meningeal space to settle within the pia mater [66]. In order toreach the CSF, parasites have to cross two barriers – the fenestrated endothelium toenter the stroma of the plexus andfinally the epitheliumcell wall to enter the ventricle.Both cell layers are placed on a basal membrane, which has also to be opened by theparasite. So far, themolecular mechanism of how trypanosomes open the gates is notwell understood. We have observed that at least some parasites within the stroma losttheir VSG coat prior to their uptake and digestion by phagocytes. We consider it mostlikely that trypanosomes, similar to cancer cell migration, need the action of secretedmetalloproteases to gain access through the cell barrier. Trypanosomes encode a zinc-dependent protease (MSP-B) that is especially expressed and secreted during trans-formation from blood forms to the procyclic insect form to remove the VSG coat [69].This protease seems also to be a very good candidate to facilitate entry into the stromaand maybe furthermore into the ventricle. In order to block the parasite’s entry intoCSF, one may consider developing inhibitors for this pathway, such as an effectiveprotease inhibitor.However, thiswould onlywork prior to the late stage as a preventivedrug. Instead, it seems more promising to treat late-stage disease by administeringtrypanocidal drugs intrathecally by lumbar injection. In this case it does not have tocross the blood–brain barrier or the blood–CSF barrier and should have access totrypanosomes in the pial compartment. It should be noted that this form ofapplication is used in brain cancer chemotherapy with considerable success [70].In this case, the drug should be soluble and stable in CSF, and be able to movebetween the pial cell layers. At the moment it seems worthwhile to explore this routeof drug administration in more detail, because it could probably be performed underfield conditions.
Outlook
Trypanosomes, as any other cell, possess a spectrum of measures to resist cell death.The most obvious scenario is autophagy, but this is also true for the onset ofnecrosis. In addition, the stumpy form undergoes PGD2-induced apoptosis andother forms of apoptosis may exist [38]. It is certainly too early to announce drugcandidates that interfere with the parasite’s ability to induce and control cell deathmechanisms. However, induction of an irrepressible autophagy (e.g., by nano-particles) seems possible and should be explored in detail. This would be especiallypromising using nanoparticles that carry trypanocidal drugs inside to be releasedafter uptake by the parasite. Another promising aspect seems to us to be theintrathecal application of trypanocidal drugs to attack trypanosomes residing withinthe pia mater. Apart from that, the ability of trypanosomes to handle life-threateningsituations and their disposition to use cell death mechanisms for epidemiologicallyeffective infections should be analyzed in molecular detail to explore additiontoeholds to fight trypanosomiasis.
Outlook j273

References
1 Maiuri, M.C., Zalckvar, E., Kimchi, A., andKroemer, G. (2007) Self-eating and self-killing: crosstalk between autophagy andapoptosis. Nat. Rev. Mol. Cell Biol., 8,741–752.
2 Opperdoes, F.R. and Borst, P. (1977)Localization of nine glycolytic enzymes in amicrobody-like organelle in Trypanosomabrucei: the glycosome. FEBS Lett., 80,360–364.
3 Coley, A.F., Dodson, H.C., Morris, M.T.,and Morris, J.C. (2011) Glycolysis in theAfrican trypanosome: targeting enzymesand their subcellular compartments fortherapeutic development.Mol. Biol. Int.,2011, 123702.
4 Verlinde, C.L., Hannaert, V., Blonski, C.,Willson, M., Perie, J.J. et al. (2001)Glycolysis as a target for the design of newanti-trypanosome drugs. Drug Resist.Updat., 4, 50–65.
5 Wierenga, R.K., Noble, M.E., Postma,J.P., Groendijk, H., Kalk, K.H. et al.(1991) The crystal structure of the“open” and the “closed” conformation ofthe flexible loop of trypanosomaltriosephosphate isomerase. Proteins, 10,33–49.
6 Sirover, M.A. (1999) New insights into anold protein: the functional diversity ofmammalian glyceraldehyde-3-phosphatedehydrogenase. Biochim. Biophys. Acta,1432, 159–184.
7 He, C. and Klionsky, D.J. (2009) Regulationmechanisms and signaling pathways ofautophagy. Annu. Rev. Genet., 43, 67–93.
8 Mehrpour, M., Esclatine, A., Beau, I., andCodogno, P. (2010) Autophagy in healthand disease. 1. Regulation and significanceof autophagy: an overview. Am. J. Physiol.,298, C776–C785.
9 Duszenko, M., Ginger, M.L., Brennand, A.,Gualdron-Lopez, M., Colombo, M.I. et al.(2011) Autophagy in protists. Autophagy, 7,127–158.
10 Hayashi-Nishino, M., Fujita, N., Noda, T.,Yamaguchi, A., Yoshimori, T. et al. (2010)Electron tomography reveals theendoplasmic reticulum as a membranesource for autophagosome formation.Autophagy, 6, 301–303.
11 Hailey, D.W., Rambold, A.S., Satpute-Krishnan, P., Mitra, K., Sougrat, R. et al.(2010) Mitochondria supply membranesfor autophagosome biogenesis duringstarvation. Cell, 141, 656–667.
12 Calne, R.Y., Collier, D.S., Lim, S., Pollard,S.G., Samaan, A. et al. (1989) Rapamycinfor immunosuppression in organallografting. Lancet, 2, 227.
13 Foster, K.G. and Fingar, D.C. (2010)Mammalian target of rapamycin (mTOR):conducting the cellular signalingsymphony. J. Biol. Chem., 285,14071–14077.
14 Sarbassov, D.D., Ali, S.M., Kim, D.H.,Guertin, D.A., Latek, R.R. et al. (2004)Rictor, a novel binding partner ofmTOR, defines a rapamycin-insensitiveand raptor-independent pathway thatregulates the cytoskeleton. Curr. Biol.,14, 1296–1302.
15 Geng, J. and Klionsky, D.J. (2008) The Atg8and Atg12 ubiquitin-like conjugationsystems in macroautophagy. ‘Proteinmodifications: beyond the usual suspects’review series. EMBO Rep., 9, 859–864.
16 Herman, M., Gillies, S., Michels, P.A., andRigden, D.J. (2006) Autophagy and relatedprocesses in trypanosomatids: insightsfrom genomic and bioinformatic analyses.Autophagy, 2, 107–118.
17 Barquilla, A., Crespo, J.L., and Navarro, M.(2008) Rapamycin inhibits trypanosomecell growth by preventing TOR complex 2formation. Proc. Natl. Acad. Sci. USA, 105,14579–14584.
18 de Jesus, T.C.L., Tonelli, R.R., Nardelli,S.C., Augusto, L.D., Motta, M.C.M. et al.(2010) Target of rapamycin (TOR)-like 1kinase is involved in the control ofpolyphosphate levels and acidocalcisomemaintenance in Trypanosoma brucei. J. Biol.Chem., 285, 24131–24140.
19 Hall, B.S., Gabernet-Castello, C., Voak, A.,Goulding, D., Natesan, S.K. et al. (2006)TbVps34, the trypanosome orthologue ofVps34, is required for Golgi complexsegregation. J. Biol. Chem., 281,27600–27612.
20 Koopmann, R., Muhammad, K., Perbandt,M., Betzel, C., and Duszenko, M. (2009)
274j 14 Life and Death of Trypanosoma brucei: New Perspectives for Drug Development

Trypanosoma brucei ATG8: structuralinsights into autophagic-like mechanismsin protozoa. Autophagy, 5, 1085–1091.
21 Alvarez, V.E., Kosec, G., Anna, C.S., Turk,V., Cazzulo, J.J. et al. (2008) Autophagy isinvolved in nutritional stress response anddifferentiation in Trypanosoma cruzi. J. Biol.Chem., 283, 3454–3464.
22 Williams, R.A.M., Woods, K.L., Juliano, L.,Mottram, J.C., and Coombs, G.H. (2009)Characterization of unusual families ofATG8-like proteins and ATG12 in theprotozoan parasite Leishmania major.Autophagy, 5, 159–172.
23 Medina, C., Santos-Martinez, M.J.,Radomski, A., Corrigan, O.I., andRadomski, M.W. (2007) Nanoparticles:pharmacological and toxicologicalsignificance. Br. J. Pharmacol., 150,552–558.
24 McNeil, S.E. (2005) Nanotechnology for thebiologist. J. Leukoc. Biol., 78, 585–594.
25 Coppens, I., Opperdoes, F.R., Courtoy, P.J.,and Baudhuin, P. (1987) Receptor-mediated endocytosis in the bloodstreamform of Trypanosoma brucei. J. Protozool.,34, 465–473.
26 Webster, P. (1989) Endocytosis by Africantrypanosomes. I. Three-dimensionalstructure of the endocytic organelles inTrypanosoma brucei and T. congolense. Eur. J.Cell Biol., 49, 295–302.
27 Vogt, C. (1842)Untersuchungen uber dieEntwicklungsgeschichte derGeburtshelferkroete (Alytes obstetricans),Jent & Gassmann, Solothurn.
28 Jacobson, M.D., Weil, M., and Raff, M.C.(1997) Programmed cell death in animaldevelopment. Cell, 88, 347–354.
29 Saunders, J.W.Jr. (1966) Death inembryonic systems. Science, 154,604–612.
30 Denecker, G., Vercammen, D., Declercq,W., and Vandenabeele, P. (2001) Apoptoticand necrotic cell death induced by deathdomain receptors. Cell Mol. Life Sci., 58,356–370.
31 Cosulich, S. and Clarke, P. (1996)Apoptosis: does stress kill? Curr. Biol., 6,1586–1588.
32 Basu, S. and Kolesnick, R. (1998) Stresssignals for apoptosis: ceramide and c-Junkinase. Oncogene, 17, 3277–3285.
33 Hotchkiss, R.S. and Nicholson, D.W.(2006) Apoptosis and caspases regulatedeath and inflammation in sepsis. Nat. Rev.Immunol., 6, 813–822.
34 Van Cruchten, S. and Van den Broeck,W. (2002) Morphological andbiochemical aspects of apoptosis, oncosisand necrosis. Anat. Histol. Embryol., 31,214–223.
35 Welburn, S.C., Macleod, E., Figarella, K.,and Duzensko, M. (2006) Programmed celldeath in African trypanosomes.Parasitology, 132 (Suppl.), S7–S18.
36 Madeo, F., Engelhardt, S., Herker, E.,Lehmann, N., Maldener, C. et al. (2002)Apoptosis in yeast: a new model systemwith applications in cell biology andmedicine. Curr. Genet., 41, 208–216.
37 Madeo, F., Herker, E., Maldener, C.,Wissing, S., Lachelt, S. et al. (2002) Acaspase-related protease regulatesapoptosis in yeast.Mol. Cell, 9, 911–917.
38 Welburn, S.C., Dale, C., Ellis, D., Beecroft,R., and Pearson, T.W. (1996) Apoptosis inprocyclic Trypanosoma brucei rhodesiense invitro. Cell Death Differ., 3, 229–236.
39 Welburn, S.C., Lillico, S., and Murphy,N.B. (1999) Programmed cell death inprocyclic form Trypanosoma bruceirhodesiense – identification of differentiallyexpressed genes during con A induceddeath.Mem. Inst. Oswaldo Cruz, 94,229–234.
40 Szallies, A., Kubata, B.K., and Duszenko,M. (2002) A metacaspase of Trypanosomabrucei causes loss of respirationcompetence and clonal death in the yeastSaccharomyces cerevisiae. FEBS Lett., 517,144–150.
41 Mottram, J.C., Helms, M.J., Coombs, G.H., and Sajid, M. (2003) Clan CD cysteinepeptidases of parasitic protozoa. TrendsParasitol., 19, 182–187.
42 Zangger, H., Mottram, J.C., and Fasel, N.(2002) Cell death in Leishmania induced bystress and differentiation: programmedcell death or necrosis? Cell Death Differ., 9,1126–1139.
43 Ambit, A., Fasel, N., Coombs, G.H., andMottram, J.C. (2008) An essential role forthe Leishmania majormetacaspase in cellcycle progression. Cell Death Differ., 15,113–122.
References j275

44 Susin, S.A., Lorenzo, H.K., Zamzami, N.,Marzo, I., Snow, B.E. et al. (1999)Molecular characterization ofmitochondrial apoptosis-inducing factor.Nature, 397, 441–446.
45 Cande, C., Cecconi, F., Dessen, P., andKroemer, G. (2002) Apoptosis-inducingfactor (AIF): key to the conserved caspase-independent pathways of cell death? J. CellSci., 115, 4727–4734.
46 Joza, N., Pospisilik, J.A., Hangen, E.,Hanada, T., Modjtahedi, N. et al. (2009)AIF: Not just an apoptosis-inducing factor.Ann. NYAcad. Sci., 1171, 2–11.
47 Li, L.Y., Luo, L., and Wang, X.D. (2001)Endonuclease G is an apoptotic DNasewhen released from mitochondria. Nature,412, 95–99.
48 Kubata, B.K., Duszenko, M., Kabututu, Z.,Rawer, M., Szallies, A. et al. (2000)Identification of a novel prostaglandin f(2alpha) synthase in Trypanosoma brucei.J. Exp. Med., 192, 1327–1338.
49 Pentreath, V.W., Rees, K., Owolabi, O.A.,Philip, K.A., and Doua, F. (1990) Thesomnogenic T lymphocyte suppressorprostaglandin D2 is selectively elevated incerebrospinal fluid of advanced sleepingsickness patients. Trans. R. Soc. Trop. Med.Hyg., 84, 795–799.
50 Figarella, K., Rawer, M., Uzcategui, N.L.,Kubata, B.K., Lauber, K. et al. (2005)Prostaglandin D2 induces programmedcell death in Trypanosoma bruceibloodstream form. Cell Death Differ., 12,335–346.
51 Figarella, K., Uzcategui, N.L., Beck, A.,Schoenfeld, C., Kubata, B.K. et al. (2006)Prostaglandin-induced programmed celldeath in Trypanosoma brucei involvesoxidative stress. Cell Death Differ., 13,1802–1814.
52 Vickerman, K. (1969) On the surface coatand flagellar adhesion in trypanosomes. J.Cell Sci., 5, 163–193.
53 Vickerman, K. (1978) Antigenic variationin trypanosomes. Nature, 273, 613–617.
54 Cross, G.A. (1978) Antigenic variation intrypanosomes. Proc. R. Soc. Lond. B, 202,55–72.
55 Matthews, K.R. (1999) Developments inthe differentiation of Trypanosoma brucei.Parasitol. Today, 15, 76–80.
56 Brown, R.C., Evans, D.A., and Vickerman,K. (1973) Changes in oxidative metabolismand ultrastructure accompanyingdifferentiation of the mitochondrion inTrypanosoma brucei. Int. J. Parasitol., 3,691–704.
57 Feagin, J.E., Jasmer, D.P., and Stuart, K.(1986) Differential mitochondrial geneexpression between slender and stumpybloodforms of Trypanosoma brucei.Mol.Biochem. Parasitol., 20, 207–214.
58 Vassella, E., Reuner, B., Yutzy, B., andBoshart, M. (1997) Differentiation ofAfrican trypanosomes is controlled by adensity sensing mechanism which signalscell cycle arrest via the cAMP pathway. J.Cell Sci., 110, 2661–2671.
59 Hamm, B., Schindler, A., Mecke, D., andDuszenko, M. (1990) Differentiation ofTrypanosoma brucei bloodstreamtrypomastigotes from long slender to shortstumpy-like forms in axenic culture.Mol.Biochem. Parasitol., 40, 13–22.
60 van Zandbergen, G., Luder, C.G., Heussler,V., and Duszenko, M. (2010) Programmedcell death in unicellular parasites: aprerequisite for sustained infection? TrendsParasitol., 26, 477–483.
61 Black, S.J., Hewett, R.S., andSendashonga, C.N. (1982) Trypanosomabrucei variable surface antigen isreleased by degenerating parasites butnot by actively dividing parasites.Parasite Immunol., 4, 233–244.
62 Gannavaram, S., Vedvyas, C., andDebrabant, A. (2008) Conservation of thepro-apoptotic nuclease activity ofendonuclease G in unicellulartrypanosomatid parasites. J. Cell Sci., 121,99–109.
63 Mulenga, C., Mhlanga, J.D., Kristensson,K., and Robertson, B. (2001)Trypanosoma brucei brucei crosses theblood–brain barrier while tight junctionproteins are preserved in a rat chronicdisease model. Neuropathol. Appl.Neurobiol., 27, 77–85.
64 Grab, D.J. and Kennedy, P.G. (2008)Traversal of human and animaltrypanosomes across the blood–brainbarrier. J. Neurovirol., 14, 344–351.
65 Schmidt, H. (1983) The pathogenesis oftrypanosomiasis of the CNS. Studies on
276j 14 Life and Death of Trypanosoma brucei: New Perspectives for Drug Development

parasitological and neurohistologicalfindings in Trypanosoma rhodesienseinfected vervet monkeys. Virchows Arch. A,399, 333–343.
66 Wolburg, H., Mogk, S., Acker, S., Frey, C.,Meinert, M. et al. (2012) Late stageinfection in sleeping sickness. PLoS ONE,7, e34304.
67 Johnston, M., Zakharov, A.,Papaiconomou, C., Salmasi, G., andArmstrong, D. (2004) Evidence ofconnections between cerebrospinal fluidand nasal lymphatic vessels in humans,non-human primates and othermammalian species. Cerebrospinal FluidRes., 1, 2.
68 Hayaishi, O., Urade, Y., Eguchi, N., andHuang, Z.L. (2004) Genes forprostaglandin d synthase and receptor aswell as adenosine A2A receptor areinvolved in the homeostatic regulation ofNREM sleep. Arch. Ital. Biol., 142,533–539.
69 de Sousa, K.P., Atouguia, J., and Silva, M.S.(2010) Partial biochemical characterizationof a metalloproteinase from thebloodstream forms of Trypanosoma bruceibrucei parasites. Protein J., 29, 283–289.
70 Cradock, J.C., Kleinman, L.M., andDavignon, J.P. (1977) Intrathecalinjections – a review of pharmaceuticalfactors. Bull. Parent. Drug Assoc., 31, 237–247.
References j277





























