Embed Size (px)
Citation preview

Journal of NeurochemistryLippincott—Raven Publishers, Philadelphia© 1998 International Society for Neurochemistry
Trimethyltin Stimulates Protein Kinase C TranslocationThrough Receptor-Mediated Phospholipase C
Activation in PC12 Cells
Michael D. Kane, Ching-Wen Yang, Palur G. Gunasekar, and Gary E. Isom
Department of Medicinal Chemistry and Molecular Pharmacology, Purdue University, West Lafayette, Indiana, U.S.A.
Abstract: Trimethyltin (TMT) is a potent neurotoxic com-pound that initiates a delayed neuronal cell death. Previouslywe have shown that TMT-induced cytotoxicity is associatedwith protein kinaseC (PKC) translocation and activation. Thepresent study investigates the mechanism underlying TMT-stimulated PKC translocation in PCi 2 cells. TMT exposureled to a rapid increase in intracellular levels of inositol 1,4,5-trisphosphate (1P3), a product of phospholipase C (PLC).This was significantly decreased by pretreating cells withantagonists to efther the cholinergic muscarinic receptor (at-ropine) or the glutamatergic metabotropic receptor [(+)-a-methyl-4-carboxyphenylglycine; (+ )-MCPG]. Furthermore,the rise in 1P3 level was blocked by pretreating cells with aPLC inhibitor (U-73122) orby a combination of atropine and(+)-MCPG. This pretreatment also significantly decreasedTMT-stimulated PKC translocation, indicating that TMT-me-diated PKC translocation was related to PLC activation, pre-sumably through formation of diacylglycerol, an endogenousactivator of PKC and product of PLC. It is interesting thatatropine and (+ )-MCPG did not provide protection againstTMT-induced cytotoxicity in these cells. However, these datasuggest that TMT causes the release of cellular constituentsthat activate G protein-coupled receptors, ultimately leadingto PKC translocation. Key Words: Tnmethyftin—Proteinkinase C—Inositol 1,4,5-trisphosphate— 1 -[6- [(1 7fi)-3-Methoxyestra-1 3,5(1 0)-trien- 1 7-yl]amino]hexyl]-1H-pyrrole-2,5-dione—U-731 22—Atropine—(+)-a-Methyl-4-carboxyphenylglycine.J. Neurochem. 70, 509—514 (1998).
degeneration occurs predominantly in the hippocampalpyramidal band and fascia dentata accompaniedby cellloss in the amygdala, pyriform cortex, and neocortex(Brown et a!., 1979). The cellular, biochemical, andhistological changes that follow TMT exposure havebeen extensively studied, but the mechanism of neuro-degeneration is poorly understood.
Exposure to TMT causes translocation and activationof protein kinase C (PKC) in the differentiated PCi2cell model (Pavlakovié et al., 1995b). PKC activationwas found to play a significant role in TMT-mediatedneurotoxicity because pretreatment with an inhibitor ofPKC or prior down-regulation of PKC afforded protec-tion against cytotoxicity (Pavlakovié et al., 1995b). Asustained activation of PKC has been shown to play arole in neuronal cell injury in models of excitotoxicity(Favaron et al., 1990), ischemia (Hara et aL, 1990),and hypoxia (Pavlakovié et al., 1995a). The biochemicalevents activated by PKC that lead to cellular injury anddeath remain to be characterized. PKC can be activatedby increases in levels of free intracellular calcium and!or intracellular diacylglycerol, with the latter being aproduct of phospholipase C (PLC) activation (Tanakaand Nishizuka, 1994).
The PC12 cell is a well characterized neuronalmodel originally isolated from a catecholamine-secre-ting tumor (pheochromocytoma) from the rat (Greeneand Tischler, 1976). These cells synthesize and secrete
Trimethyltin (TMT) is a potent neurotoxic organo-tin compound that has been used to study selectiveneurodegeneration and delayed neuronal cell death(Aschner and Aschner, 1992; O’Connell et al.,1994a,b). Dosing of rats and mice with a sublethal,neurotoxic dose of TMT results in significant behav-ioral changes and cognitive deficits described as theTMT syndrome (Dyer et al., 1982). The pattern ofneurodegeneration in the brain and the overall sensitiv-ity to TMT are dependent on the dose regimen andspecies under investigation (Perretta et al., 1993). Inmice a time- and dose-dependent pattern of neuronal
Received August 11, 1997; revised manuscript received Septem-ber 16, 1997; accepted September 17, 1997.
Address correspondence and reprint requests to Dr. G. E. Isom atDepartment of Medicinal Chemistry and Molecular Pharmacology,School of Pharmacy and Pharmacal Sciences, Purdue University,West Lafayette, IN 47907-1334, U.S.A.
The present addressof Dr. M. D. Kane is Department ofNeurolog-ical and Psychiatric Diseases, Parke-Davis Pharmaceuticals, Warner-Lambert Co., Ann Arbor, MI 48105, U.S.A.
Abbreviations used: 1P3, inositol 1 ,4,5-trisphosphate; (+ )-MCPG,(+ )-a-methyl-4-carboxyphenylglycine; PBS, phosphate-bufferedsaline; PKC,protein kinase C; PLC, phospholipase C; TMT, trimeth-yltin; U-73 122, 1- [6- [(17/3 )-3-methoxyestra-1 ,3,5 (10) -trien-17-yl] amino] hexyl] -1H-pyrrole-2,5-dione.
509

510 M. D. KANE ET AL.
acetylcholine, norepinephrine, and dopamine and con-tain several plasma membrane receptors coupled toPLC that result in inositol 1,4,5-trisphosphate (1P3)formation on activation (for review, see Shafer andAtchison, 1991). The cholinergic muscarinic and glu-tamate metabotropic receptors have been described inthese cells, and their activation increases intracellularlevels of 1P3 (Cross et al., 1984; Horowitz, 1989; Ta-kashima and Kenimer, 1989; Kurozumi et al., 1990).Because of these neurochemica! properties, the PC 12cell line was used in this investigation.
MATERIALS AND METHODS
ChemicalsTMT was purchased from Aldrich Chemical Co. (Milwau-
kee, WI, U.S.A.). myo-[2-3H(N)]Inositol (10—20 mCi!
mmol; in sterile water) and [3HI 1P3 were purchased from
American Radiolabeled Chemicals (St. Louis, MO, U.S.A.).EcoLite liquid scintillation cocktail was obtained from ICNBiomedicals (Irvine, CA, U.S.A.). myo-Inositol was fromFluka Chemical Corp. (Ronkonkoma, NY, U.S.A.). (+)-a-
Methyl-4-carboxyphenylglycine [(+ )-MCPG] was pur-chased from Tocris Neuramin Ltd. (Bristol, U.K.). The PLCinhibitor 1- [6- [(17/3 )-3-methoxyestra-1 ,3,5 (10) -trien-17-ylI amino] hexyl] - 1H-pyrrole-2,5-dione (U-73 122) was pur-chased from Research Biochemicals International (Natick,MA, U.S.A.). Mouse monoclonal anti-PKC antibody MC5(recognizing a and /3 isoforms) was purchased from Amer-sham (Arlington Heights, IL, U.S.A.), and the peroxidase-conjugated anti-mouse secondary antibody was from Bio-Rad (Richmond, CA, U.S.A.).
PC12 cell culturePC12 cells were obtained from American Type Culture
Collection (Rockville, MD, U.S.A.) and propagated by serialpassage. Cells were grown as a loosely adhering monolayeron plastic culture flasks in RPMI 1640 medium supple-mented with 10% heat-inactivated horse serum, 5% fetalbovine serum, and 1% L-glutamine at 37°C.Forphosphoino-sitide experiments, cells were harvested at confluency,washed with loading medium, and resuspended in the load-ing medium at a cell count of 12 x 106 cells/ml.
For PKC translocation experiments, PC12 cells wereplated on collagen-coated cell culture plates with RPMI me-dium supplemented with 10% heat-inactivated horse serum,5% fetal calf serum, and 2.5 mM glutamine. PC12 cell differ-entiation was carried out using RPMI growth medium sup-plemented with 100 ng/ml nerve growth factor (SigmaChemical Co., St. Louis) The cells were grown in six- or12-well collagen-coated plates at density of 5—8>< io~cells!ml of media for 7—10 days before experiments.
Quantification of 1P3IF3 levels were determined based on the method developed
by Berridge (1983) with minor modifications. In brief, cellswere loaded with myo-[
3H]inositol in loading medium(Dul-becco’s modified Eagle’s medium, 5% fetal bovine serum,and 1% L-glUtamate) with 5 ~Ci/ml myo-[3H]inositol for36 h, washed twice with loading medium (without myo-[3HIinositol) to remove unincorporated myo-[3HIinositol,and then incubated in loading medium (containing 10 mMLiC1) containing atropine, (+ ) -MCPG, or U-73 122, when
appropriate, for 15 mm at 37°C.Immediately following TMTtreatment, 1 ml of ice-cold 12% HC1O
4 was added to termi-nate the response, and inositol polyphosphates were sepa-rated on Accell QMA Sep-Pak cartridges (Waters, Milford,MA, U.S.A.) with triethylammonium bicarbonate solutionunder vacuum. [
3H]IF3 levels were quantified by liquid scin-
tillation. Results represent three separate experiments carriedout in triplicate. The statistical significance was assessedusing a Tukey post hoc multiple range t test. Values of p<0.05 were considered statistically significant.
PKC western blottingRPMI medium was substituted with Krebs—Ringerbuffer
(125 mM NaCI, 5 mM KC1, 1.2 mM KH2PO4, 5 mMNaHCO3, 25 mM HEPES, 6 mM glucose, 1.2 mM MgSO4,and 1 mM CaC12, pH 7.4) 15 mm before initiation of theexperiments. Differentiated PC12 cells were then treated byexposure to TMT for 30 rain, followed by cell lysis. For apositive control of PKC translocation, cells were treated with100 nM phorbol 12-myristate 13-acetate for 30 mm. Krebs—Ringer buffer was aspirated, cells were washed once withice-cold phosphate-buffered saline (PBS) containing 5 mMEGTA, and the cells were lysed by addition of 0.5 ml perwell of ice-coldlysis buffer(20mM Tris-HC1, 2mM EDTA,5 mM EGTA, 2 mM dithiothreitol, 1 mM phenylmethylsul-fonyl fluoride, 1 mg/ml bacitracin, 10 ~zg/mlleupeptin, and10 j.tg/ml aprotinin). The particulate (membrane) fractionsof the lysates were separated by centrifugation at 40,000 gfor 40 mm at 4°C.Pellets were resuspended in lysis bufferwith 1% Triton X-100. A 50-j.il aliquot of each sample wasremoved for protein content determination and frozen at—80°C until analysis. Protein concentration of the sampleswasdetermined using theBio-Rad protein assay kit. Sampleswere normalized for total protein and separated by sodiumdodecyl sulfate—polyaciylamide gel electrophoresis.
Western blot analysis with the enhanced chemilumines-cence detection method was performed as described bySchneppenheim et al. (1991). Before protein separation bypolyacrylamide gel electrophoresis, samples were mixedwith a denaturation buffer (50 mM Tris-HC1, 2% sodiumdodecyl sulfate, 4% glycerol, and 2% 2-mercaptoethanol) inthe ratio of 1:2. Samples were boiled in a water bath for 90s and then sonicated for 10 s to break up aggregated nucleicacids and proteins. Equivalent amounts of sample proteins(~0,ug per lane) were separated using a 7.5% (wt/vol)polyacrylamide separatinggelunder reducing conditions andtransferred to a PVDF membrane. Following the transfer,membranes were washed in PBS for 10 mm and incubatedin 3% bovine serumalbumin in PBS with 0.5% Triton X-100for 60 mm. Next, PKC wasimmunolabeled with monoclonalantibody MC5 (Amersham), which recognizes PKCa andPKC~3isoforms (dilution 1:1,000 in PBS). Following incu-bation with the primary antibody, membranes were washedin PBS (three times for 5 mm each) and then incubated for2 h with peroxidase-conjugated secondary antibody. Mem-branes were washed in PBS (three times for 10 mm each),and antibody-complexed antigens were visualized using theenhancedchemiluminescence detection system (Amersham)and autoradiography film. For quantitative analysis, the pro-tein band intensity was determined using a model GS300transmittance!reflectance densitometer (Hoeffer Scientific,Sacramento, CA, U.S.A.). PKC translocation experimentswere repeated four times.
J. Neurochem., Vol. 70, No. 2, 1998
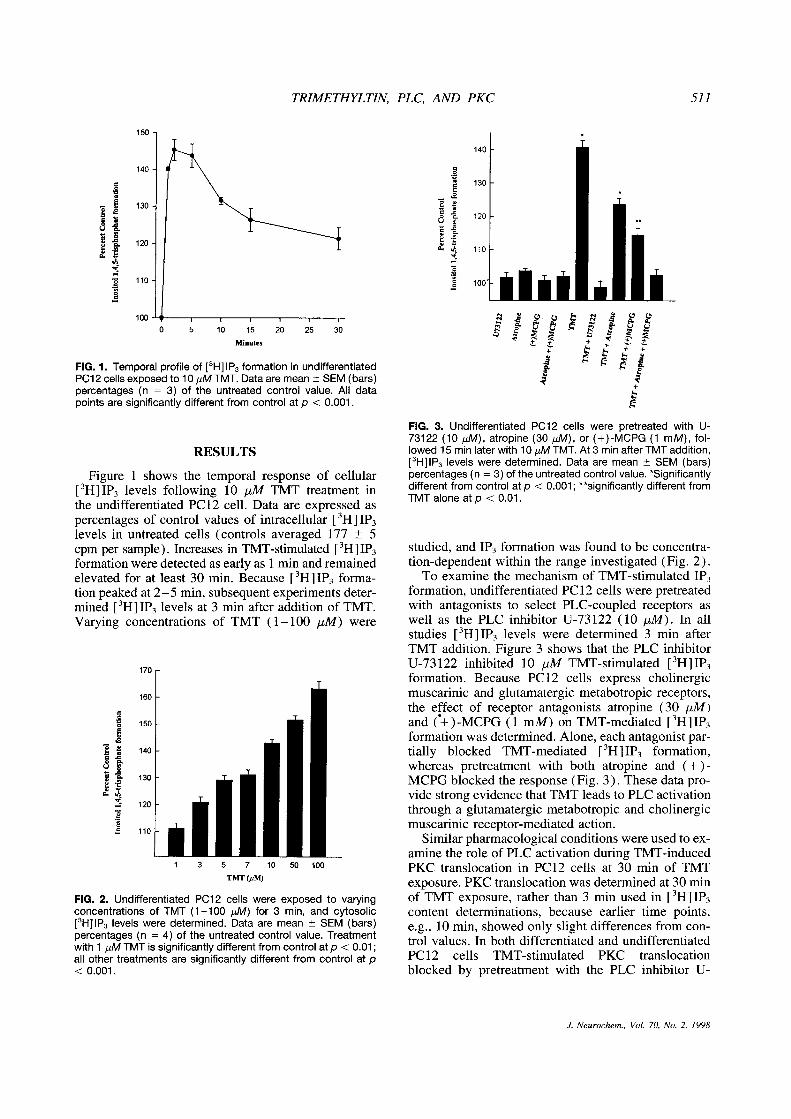
TRIMETHYLTIN, PLC, AND PKC 511
FIG. 1. Temporal profile of [3H]1P3formation in undifferentiated
PC12 cells exposed to 10 1iM TMT. Data are mean ±SEM (bars)percentages (n = 3) of the untreated control value. All datapoints are significantly different from control at p < 0.001.
RESULTS
Figure 1 shows the temporal response of cellular[3H]IP
3 levels following 10 ,iM TMT treatment inthe undifferentiated PC12 cell. Data are expressed aspercentages of control values of intracellular {
3H] 1P3
levels in untreated cells (controls averaged 177 ±5cpm per sample). Increases in TIVIT-stimulated [
3H]1P3
formation were detected as earlyas 1 mm and remainedelevated for at least 30 mm. Because [
3H]1P3 forma-
tion peaked at 2—5 mm, subsequent experiments deter-mined [
3H]1P3 levels at 3 mm after addition of TMT.
Varying concentrations of TMT (1—100 ~.tM) were
FIG. 2. Undifferentiated PC12 cells were exposed to varyingconcentrations of TMT (1—100 ~tM) for 3 mm, and cytosolic[3HJIP
3levels were determined. Data are mean ±SEM (bars)percentages (n = 4) of the untreated control value. Treatmentwith 1 p~MTMT is significantly different from control at p <0.01;all other treatments are significantly different from control at p<0.001.
FIG. 3. Undifferentiated PC12 cells were pretreated with U-73122 (10 1zM), atropine (30 bLM), or (+)-MCPG (1 mM), fol-lowed 15 mm later with 10 j.tM TMT. At 3 mm after TMT addition,[3H]1P
3levels were determined. Data are mean ±SEM (bars)percentages (n = 3) of the untreated control value. °Significantlydifferent from control at p < 0.001; **significantly different fromTMT alone at p < 0.01.
studied, and 1P3 formation was found to be concentra-tion-dependent within the range investigated (Fig. 2).
To examine the mechanism of TMT-stimulated IP3formation, undifferentiated PC12 cells were pretreatedwith antagonists to select PLC-coupled receptors aswell as the PLC inhibitor U-73122 (10 pM). In allstudies [
3H]IF3 levels were determined 3 mm after
TMT addition. Figure 3 shows that the PLC inhibitorU-73 122 inhibited 10 ,iM TMT-stimulated [
3H]1P3
formation. Because PC12 cells express cholinergicmuscarinic and glutamatergic metabotropic receptors,the effect of receptor antagonists atropine (30 tiM)and (+)-MCPG (1 mM) on TMT-mediated [
3H11P3
formation was determined. Alone, each antagonist par-tially blocked TMT-mediated [
3HI 1P3 formation,
whereas pretreatment with both atropine and (+ ) -
MCPG blocked the response (Fig. 3). These data pro-vide strong evidence that TMT leads to PLC activationthrough a glutamatergic metabotropic and cholinergicmuscarinic receptor-mediated action.
Similar pharmacological conditions were used toex-amine the role of PLC activation during TMT-inducedPKC translocation in PC12 cells at 30 mm of TMTexposure. PKC translocation was determined at 30 mmof TMT exposure, rather than 3 mm used in [
3H]IF3
content determinations, because earlier time points,e.g., 10 mm, showed only slight differences from con-trol values. In both differentiated and undifferentiatedPC 12 cells TMT-stimulated PKC translocationblocked by pretreatment with the PLC inhibitor U-
J. Neurochem., Vol. 70, No. 2, 1998

512 M D. KANE ET AL.
FIG. 4. PC12 cells pretreated for 15 mm with U-73122 (10 tiM)followed by a 30-mm exposure to 10 ~MTMT. Cells were lysed,and the extent of PKC translocation to cellular membranes wasdetermined: (A) undifferentiated PC12 cell membrane fractionand (B) differentiated P012 cell membrane fraction (40 /ig oftotal protein per lane). Lane assignments: 1, control; 2, TMT(alone); 3, U-73122 (alone); 4, U-73122 + TMT.
73122 (Fig. 4). Similarly, pretreating cells with thecombination of atropine (30 jiM) and (+ )-MCPG (1mM) blocked TMT-induced PKC translocation inbothdifferentiated and undifferentiated PC12 cells (Fig. 5).These data suggest that TMT-stimulated PKC translo-cation is linked to PLC activation, presumably owingto diacylglycerol (a PLC product), which is an endog-enous activatorof PKC. It is interesting that the combi-nation of atropine and (+ ) -MCPG did not provideprotection against TMT-induced cytotoxicity in differ-entiated PC 12 cells (data not shown), yet PKC inhibi-tion has been shown to be protective against TMT inthis model (Pavlakovié et a)., 1995b). Note that TMT-induced cytotoxicity assays were performed at 48 h ofTMT exposure (Pavlakovid et al., l995b), and similarassays carriedout at 30 mm of TMT exposure revealedno significant cell death.
DISCUSSION
The present study shows TMT- stimulated PKCtranslocation to be dependent on PLC activation in thePC 12 cell, presumably through diacylglycerol forma-tion, because a selective inhibitor of PLC significantlyblocked PKC translocation. Furthermore, PLC activa-tion occurs through the activation of the cholinergicmuscarinic and glutamatergic metabotropic receptorsin the PC 12 cell. Receptor-mediated activation of PLCoccurs through a G protein mechanism, which resultsin the formation of IP3 and diacylglycerol, with thelatter being an endogenous activator of PKC (Tanakaand Nishizuka, 1994). TMT-mediated PKC activationhas been shown to contribute to TMT-mediated cyto-toxicity in these cells (Pavlakovié et al., 1995b). Thisstudy shows that receptor-mediated PLC activation andsubsequent IF3 and diacyiglycerol formation underlieTMT-mediated PKC translocation.
It is apparent that TMT treatment stimulates PLCby activating the cholinergic muscarinic and gluta-matergic metabotropic receptors in these cells. Themechanism of cholinergic muscarinic and gluta-matergic metabotropic receptor activation following
TMT treatment is not known but may involve TMT-stimulated release of acetylcholine and glutamate fromthe cells, which subsequently activate their respectivereceptors. Release of these constituents may lead to acascade of intracellular events involving receptor acti-vation, PLC activation, IF3 formation, PKC activation,and eventual cell death. Because the PC12 cell synthe-sizes, stores, and secretes acetylcholine, its release inresponse to TMT would be expected to activate thecholmnergic receptor that is expressed in this cell line(Cross et al., 1984; Horowitz, 1989; Takashima andKenimer, 1989). Alterations in the cholinergic musca-rinic receptor system have been shown to be involvedin the mechanism of TMT-mediated hippocampal neu-rotoxicity in vivo (O’Connell et al., 1994a,b). Numer-ous studieshave described perturbations in neurotrans-mitter systems and neurotransmitter release followingTMT exposure. These include the cholinergic (O’Con-nell et al., 1994a,b), glutamatergic (Patel et al., 1990),GABAergic (Brodie et al., 1990), dopaminergic(Mailman et al., 1983), and serotoninergic (Earleyet al., 1992) neurotransmitter systems. However, themechanism of TMT-stimulated neurotransmitter re-lease and its contribution to TMT-mediated cytotoxic-ity have not been clearly established.
TMT also interacts with the glutamate neurotrans-mitter system. TMT-mediated glutamate release hasbeen shown in the partially depolarized hippocampalslice (Patel et al., 1990) and in the hippocampus invivo (Brodie et al., 1990). TMT causes the release ofglutamate from secretory vesicles by decreasing theATP-dependent membrane potential of the vesicle(Shioi et al., 1989). For the present study, it is im-portant to note that the PC 12 cell has not been shownto contain glutamate as a neurotransmitter. BecauseTMT exposure leads to activation of the PC 12 cellglutamate metabotropic receptors, this suggests thatTMT treatment causes cytosolic glutamate release, per-haps by reversing the glutamate uptake mechanism.The possibility that TMT causes the release of gluta-thate through reversal of the glutamate transporter, orby exocytosis, may indicate a role of endogenous gluta-mate release in the development of the hippocampallesion in vivo. Excessive glutamate signaling has been
FIG. 5. P012 cells were treated for 15 mm with atropine (30~M) and (+)-MCPG (1 mM), followed by a 30-mm exposure to10 ~M TMT. Cells were lysed, and the extent of PKC transloca-tion was determined: (A) undifferentiated P012 cell membranefraction and (B) differentiated P012 cell membrane fraction (40~.tgof total protein per lane). Lane assignments: 1, control; 2,TMT (alone); 3, (+)-MCPG + atropine; 4,(+)-MCPG + atropine+ TMT; 5, phorbol 12-myristate 13-acetate (positive control).
J. Neurochem., Vol. 70, No. 2, 1998

TRIMETHYLTIN, PLC, AND PKC 513
proposed to underlie TMT-mediated hippocampal le-sions in vivo by causing hyperexcitation of neuronalcells and excitotoxic cell death (Chang, 1986). Thehippocampus is sensitive to excitotoxicity in rodentmodels of brain ischemia and hypoxia (for review,see Schmidt-Kastner and Freund, 1991). The role ofendogenous glutamate release in the development ofTMT-mediated neurotoxicity remains to be deter-mined.
Because atropine and (+ )-MCPG were shown toblock PKC translocation during 30 mm of TMT treat-ment, it was thought that these antagonists could pro-vide protection against TMT-mediated cytotoxicity, asinhibition of PKC was shown to protect differentiatedPC 12 cells against TMT-mediated cytotoxicity (Pavla-kovié et al., 1995b). However, pretreatment/treatmentwith these antagonists failed to provide significant pro-tection against TMT-mediated cytotoxicity (data notshown). This may be due to the factthat cytotoxicity isassayed 48 h following TMT exposure, whereas PKCtranslocation was measured at 30 mm following TMTexposure. Because these time points differ dramati-cally, it is possible that during the 48 h of TMT expo-sure, the release of acetyicholine and/or glutamateoverwhelms the antagonist-mediated blockade, therebyactivating PLC-coupled receptors, resulting in PKC ac-tivation. Also, other cellular changes, such as increasesin free cytosolic calcium levels, may develop later dur-ing the 48-h TMT exposure and contribute to PKCactivation and cytotoxicity. Cytotoxicity assays carriedout at 30 mm of TMT exposure revealed no significantcell death at concentrations used in this study (datanot shown).
As intracellular 1P3 can mobilize intracellular poolsof calcium (Fisher et al., 1992), the role of increasedfree cytosolic calcium levels in the mechanism ofTMT-mediated PKC translocation was investigated.TMT exposure does not increase free intracellular cal-cium content within 30 mm in the differentiated andundifferentiated PC 12 cell at TMT concentrations usedin this study (Viviani et al., 1995; M.D.K., unpublisheddata). It is concluded that TMT does not produce im-mediate changes in intracellular calcium homeostasis.However, the role of altered calcium homeostasis inthe PC 12 cell during long-term exposure to TMT re-mains to be determined.
We have recently shown that TMT treatment of dif-ferentiated PC 12 cells results in the translocation andactivation of PKC (Pavlakovié et al., 1 995b). Further-more, pretreatment of differentiated PC12 cells withthe PKC inhibitor chelerythrine produces significantprotection against TMT-mediated cytotoxicity. Thesefindings indicate PKC activation plays a critical role inTMT-mediated cytotoxicity in the differentiated PC 12cell model (Pavlakovié et al., 1 995b). It appears thatthe mechanism of TMT-mediated PKC translocationmay involve the release of acetylcholine and gluta-mate, which then activate their respective plasma
membrane receptors, resulting in PLC activation andsubsequent formation of IP3 and diacylglycerol. Theintracellular events that lead to neurotransmitter re-lease and follow PKC activation remain to be deter-mined.
REFERENCES
Aschner M. and Aschner J. L. (1992) Cellular and molecular effectsof trimethyltin and triethyltin: relevance to organotin neurotox-icity. Neurosci. Behav. Rev. 16, 427—435.
Berridge M. J. (1983) Rapid accumulation of inositol triphosphatereveals that agonists hydrolyze polyphosphoinositides insteadof phosphatidylinositol. Biochem. J. 212, 849—858.
Brodie M. E., Opaka-Juffry J., Peterson D. W., and Brown A. W.(1990) Neurochemical changes in hippocampal and caudatedialysates associated with early trimethyltin neurotoxicity inrats. Neurotoxicology 11, 35—46.
Brown A. W., Aidridge W. N., Street B. W., and Verschoyle R. D.(1979) The behavioral and neuropathologic sequelae of intoxi-cation by trimethyltin compounds in the rat. Am. J. Pathol. 97,59—82.
Chang L. W. (1986) Neuropathology of trimethyltin: a proposedpathogenic mechanism. Fundam. Appi. Toxicol. 6, 217—232.
Cross A. J., Johnson J. A., Frith C., and Taylor G. R. (1984) Musca-rinic cholinergic receptor in a rat pheochromocytoma cell line.Biochem. Biophys. Res. Commun. 119, 163—167.
Dyer R. S., Walsh T. J., Wonderlin W. F., and Bercegeay M. (1982)The trimethyltin syndrome in rats. Neurobehav. Toxicol. Tera-tol. 4, 127—133.
Earley B., Burke M., and Leonard B. E. (1992) Behavioral, bio-chemical, and histological effects of trimethyltin (TMT) in-duced brain damage in the rat. Neurochem. lot. 21, 35 1—366.
Favaron M., Manev H., Siman R., Bertolino M., Szekely A. M.,DeErausquin G., Guidotti A., and Costa E. (1990) Down-regu-lation of protein kinase C protects cerebellar granule neuronsin primary culture from glutamate-induced neuronal death.Proc. Nati. Acad. Sci. USA 87, 1983—1987.
Fisher S. K., Heacock A. M., and Agranoff B. W. (1992) Inositollipids and signal transduction in the nervous system: an update.J. Neurochem. 58, 18—38.
Greene L. A. and Tischler A. 5. (1976) Establishment of a noradren-ergic clonal line of rat adrenal pheochromocytoma cells whichrespond to nerve growth factor. Proc. Nati. Acad. Sci. USA 73,2424—2428.
Hara H., Onodera H., Yoshidomi M., Matsuda Y., and Kogure K.(1990) Staurosporine, a novel protein kinase C inhibitor, pre-ver~tspostischemic neuronal damage in the gerbil and rat. J.Cereb. Blood Flow Metab. 10, 646—653.
Horowitz J. (1989) Muscarinic receptor stimulation increases inosi-tol-phospholipid metabolism and inhibits cyclic AMP accumu-lation in PC12 cells. J. Neurochem. 53, 197—204.
Kurozumi K., Murayama T., and Nomura Y. (1990) Generation ofinositol phosphates, cytosolic Ca
2~and secretion of noradrena-line in PC12 cells treated with glutamate. FEBS Lett. 270, 225—228.
Mailman R. B., Krigman M. R., Frye G. D., and Hanin I. (1983)Effects of postnatal trimethyltin or triethyltin treatment on CNScatecholamine, GABA, and acetylcholine systems in the rat. J.Neurochem. 40, 1423—1429.
O’Connell A., Earley B., and Leonard B. E. (l994a) Changes inmuscarinic (Ml and M2 subtypes) and phencyclidine receptordensity in the rat brain following trimethyltin intoxication. Neu-
rochem. mt. 25, 243—252.O’Connell A., Earley B., and Leonard B. E. (l994b) The neuropro-
tective effects of tacrine on trimethyltin induced memory andmuscarinic receptor dysfunction in the rat. Neurochem. mt. 25,555 —566.
Patel M. N., Ardelt B. K., Yim G. K. W., and lsom G. E. (1990)
J. Neurochem., Vol. 70, No. 2, 1998

514 M. D. KANE ET AL.
Interaction of trimethyltin with hippocampal glutamate. Neuro-toxicology 11, 601—608.
Pavlakovié G., Eyer C. L., and Isom G. E. (1995a) Neuroprotectiveeffects of PKC inhibition against chemical hypoxia. Brain Res.676, 205—211.
Pavlakovié G., Kane M. D., Eyer C. L., Kanthasamy A., and IsomG. E. (1995b) Activation of protein kinase C by trimethyltin:relevance to neurotoxicity. J. Neurochem. 65, 2338—2343.
Perretta G., Righi F. R., and Gozzo S. (1993) Neuropathologicaland behavioral toxicology of trimethyltin exposure. Ann. 1st.Super. Sanita 29, 167—174.
Schmidt-Kastner R. and Freund T. F. (1991) Selective vulnerabilityof the hippocampus in brain ischemia. Neuroscience 40, 599—636.
Schneppenheim R., Budde U., Dahlmann N., and Rautenberg P.(1991) Luminography—a new, highly sensitive visualizationmethod for electrophoresis. Electrophoresis 12, 367—372.
Shafer T. J. and Atchison W. D. (1991) Transmitter, ion channeland receptor properties of pheochromocytoma (PC12) cells: amodel for neurotoxicological studies. Neurotoxicology 12,473—492.
Shioi J., Naito S., and Ueda T. (1989) Glutamate uptake into synap-tic vesicles of bovine cerebral cortex and electrochemical poten-tial difference of proton across the membrane. Biochem. J. 258,499—504.
Takashima A. and Kenimer J. G. (1989) Muscarinic-stimulated nor-epinephrine release and phosphoinositide hydrolysis in PCI2cells are independent events. J. Biol. Chem. 264, 10654—10659.
Tanaka C. and Nishizuka Y. (1994) The protein kinase C familyfor neuronal signaling. Annu. Rev. Neurosci. 17, 551—567.
Viviani B., Rossi A. D., Chow S. C., and Nicotera P. (1995) Organo-tin compounds induce calcium overload and apoptosis in PC12cells. Neurotoxicology 16, 19—25.
J. Neurochem., Vol. 70, No. 2. 1998





























