Embed Size (px)
Citation preview

1
Transformation of E. coli
&
Plasmid Isolations
All transformations look great – congratulations!
The colonies you see on your plates represent transformation events… ~1 in 100,000. If only 1 transformant in 105 cells, how does one find it?
SELECTION!

2
Selection:
1. Antibiotic or heavy metal resistance: cell cannot grow without resistance geneexamples: growth on kanamycin, tetracycline, rifampicin, mercury
2. Auxotrophies:
a. Anabolic mutation; cell cannot grow without either exogenous nutrient or complementary geneexamples: growth on tryptophan, histidine (usually fungi/yeast)
b. Catabolic: mutant cell cannot break down a nutrient source without a complementary geneexamples: growth on sucrose, toluene
In the miniature ecosystem of the test tube, Nature selects for plasmid maintenance!
What if the genetically engineered organism does not grow neatly in a Petri dish forever? Antibiotic resistance at first, then expression of marker genes: e.g. cells turn color or have other visible property examples: β-glucoronidase, GFP, luciferase (plants); GFP (mammals)
pTYB1 confers the selective advantage of ampicillin resistance to its host bacterial strain.
Selection pressure for plasmid maintenance isachieved by adding ampicillin to the growth medium
Resistance is conferred by expression of bla, whichencodes a β-lactamase enzyme.
β-lactam ring broken open by β-lactamases
β-lactam antibiotics bind to and inhibit enzymes needed for the synthesis of peptidoglycan. They have little effect on resting bacteria, but are lethal to dividing bacteria. Defective walls cannot protect the organism from bursting in hypotonic surroundings.

3
Cell envelopes of Bacteria: Gram -
Few layers of PG
Two membranes:
Inner: phospholipid bilayer
Outer: attached to LPS
Alexander Fleming served as a doctor in WWI, where he noticed that gas gangrene and tetanus were caused by bacteria.
They grew deep in wounds (anaerobic) and couldn’t be reached by antiseptics applied to the surface. He became curious about “chemotherapy” for microbial infections. After the war, as a researcher, he continued this interest.
After a bad cold, he curiously spread his nasal secretions on a petri plate… and discovered “lysozyme”. This is an animal protein found in secretion that serves as a major defense against Bacteria.
We now use lysozyme to help break cells open, for example, in DNA extractions.

4
Seven years later, returning from vacation, he noted inhibition of Gram positive bacteria (Micrococcus luteus) on petriplates in the sink that had become contaminated with fungi.
He investigated further and realized that this was due to a water-soluble substance secreted by the fungus Penicillium(although not by other fungi) and which was antibacterial.
Comparing peptidoglycan of gram-positive and gram-negative bacteria
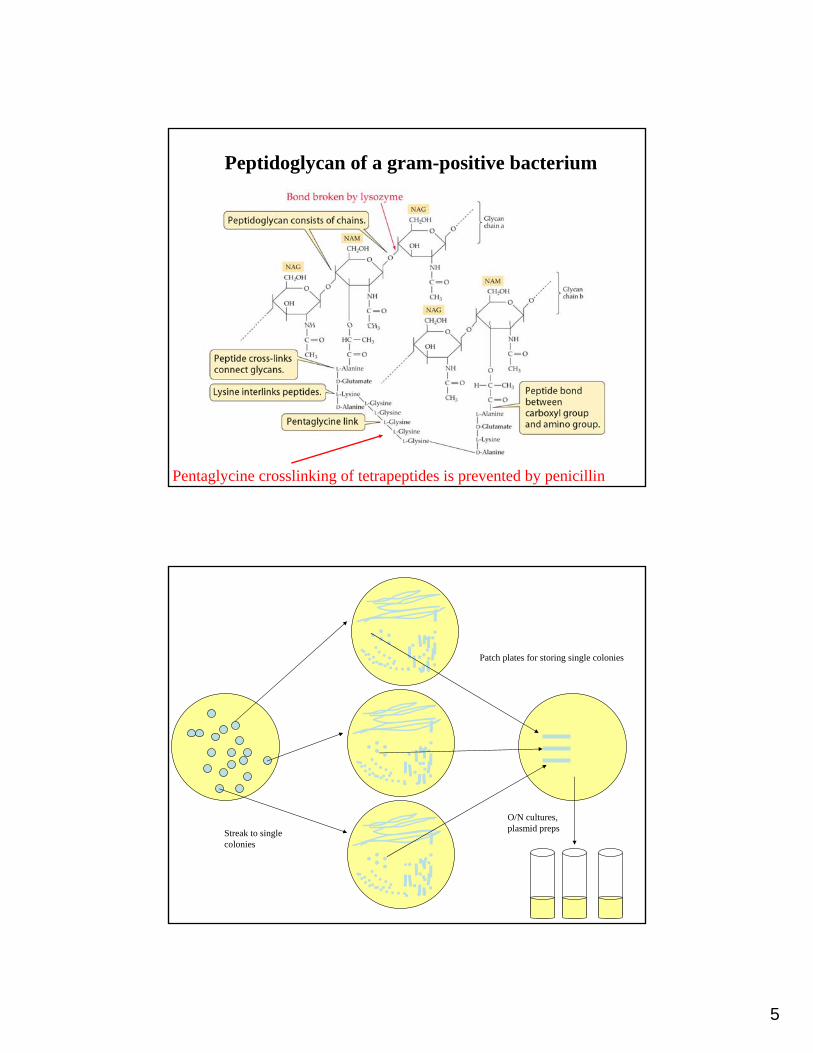
5
Peptidoglycan of a gram-positive bacterium
Pentaglycine crosslinking of tetrapeptides is prevented by penicillin
O/N cultures, plasmid prepsStreak to single
colonies
Patch plates for storing single colonies

6
“Sibling” or satellite colonies:
β-lactamase is a secreted enzyme. A zone in the vicinity of resistant colonies will be free of ampicillin, and therefore “friendly” to the reproduction of non-plasmid containing cells:
1. Offspring of transformants that lost plasmid through unequal plasmid partitioning (cloning vectors don’t usually have par locus)
2. Contaminants
Beware of tiny colonies that grow around the original colonies later on!!
*Ampicillin is heat-sensitive. Adding to hot medium will inactivate it and destroy your selection.
Not all competent cells take up plasmid DNA. Transformation efficiency determinations tell you how many cells actually do take up plasmid DNA.
Transformation efficiency = # colonies/μg DNA
These can range from 103 to 108. Which would you use if you were transforming bacteria with a library of rRNAs harvested from an undersea volcano that cost $50K per day to sample?
If the transformation efficiency of a batch of competent cells was 103, would you amplify 1 ng of plasmid DNA using these cells?

7
Plasmid Isolations
Alkaline SDS Lysis
Want to get rid of lipids, proteins, sugars, and most nucleic acids, but selectively retain plasmid DNA. How??

8
Alkaline SDS Lysis
1. Cell resuspension buffer (P1): Isoosmotic buffer, usually 10 mM Tris buffer (pH 8) and glucose (a.k.a. dextrose).
2. SDS/NaOH solution (P2):NaOH denatures double-stranded (ds) DNA to single-stranded (ss) DNA, denatures proteins,
and hydolyzes RNA. SDS (sodium dodecyl sulfate) is a detergent and breaks up the lipid cell membrane. It also complexes proteins and denatures them. Lengthy incubation in NaOH/SDS will permanently denature and damage DNA.
3. Potassium acetate solution (N3):Potassium acetate neutralizes the alkali, allowing renaturation of the DNA. The potassium
ions replace the sodium ions in SDS, creating insoluble precipitates of KDS/cellular components. Large fragments of DNA are caught in the precipitated cell debris. Small DNA molecules stay in solution.
Contains guanidine HCl, a chaotropic salt which denatures proteins. DNase is biggest concern.
Alkaline SDS Lysis
4. Optional wash in PBAdditional wash step in PB washes away DNase with guanidine HCl
5. Wash buffer (PE)Ethanol in PE buffer washes off buffer.or isopropanol precipitate DNA and RNA from the supernatant.
6. Elution buffer (EB)TE buffer contains Tris and EDTA. Tris is a buffer, EDTA chelates (=binds) cations which
nucleases need to function. This disables such DNA degrading enzymes so they won’t chew up your DNA!

9
protein
SDS:-Coats lipids in a micelle of SDS-Coats hydrophobic patches on proteins,
denaturing and linearizing and solubilizing them
-Does NOT interact with DNA (SO4 and PO4 charges repel one another)
Alkaline SDS Lysis
Chromosomal DNA stays with pellet because it is attached to cell membrane.
Violent mixing will shear chromosomal DNA and get it into the supernatant with your plasmid.
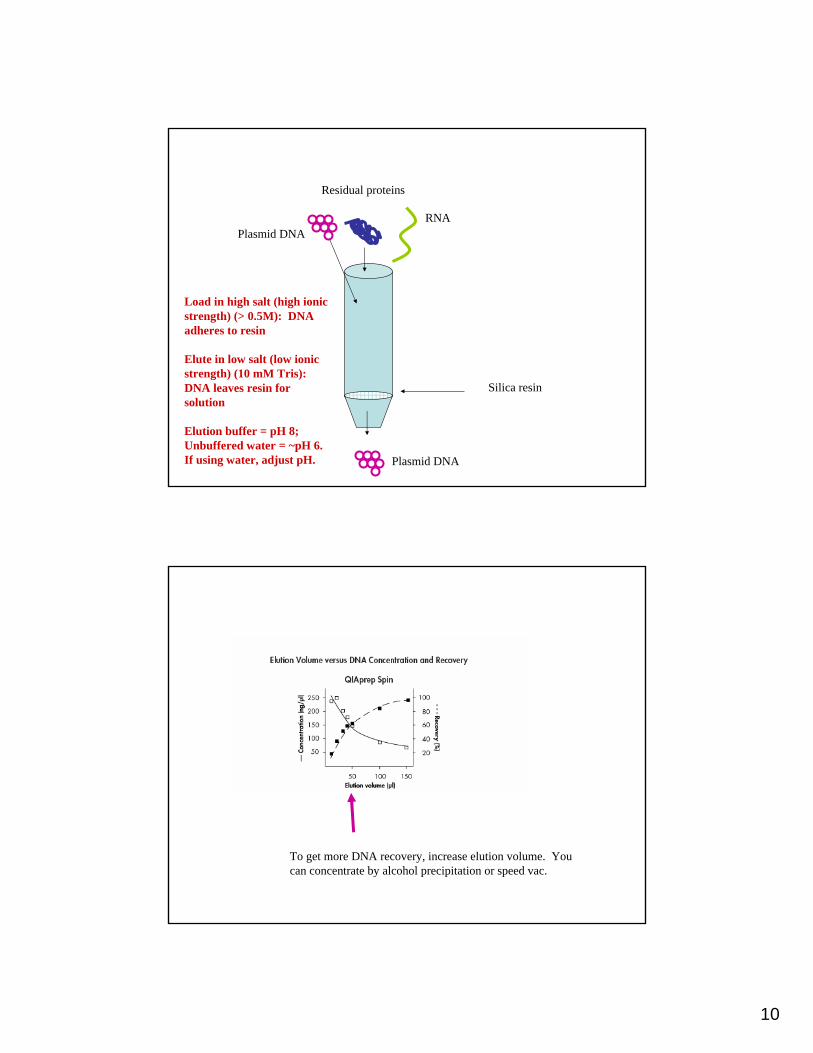
10
Silica resin
Load in high salt (high ionic strength) (> 0.5M): DNA adheres to resin
Elute in low salt (low ionic strength) (10 mM Tris): DNA leaves resin for solution
Elution buffer = pH 8; Unbuffered water = ~pH 6. If using water, adjust pH.
Plasmid DNA
Residual proteins
RNA
Plasmid DNA
To get more DNA recovery, increase elution volume. You can concentrate by alcohol precipitation or speed vac.

11
Plasmid copy number will affect yield
Depends on origin of replication
Replication also depends on medium (adding NaCl can increase yield), culture age, host strain (you won’t always use E. coli), and how much you used for inoculum.

12
How do you determine your plasmid yield?
-DNA-binding agents such as EtBr: add to your sample and to a standard curve of known concentrations
-Run on agarose gel and compare to known mass of DNA standards using ImageJ
-Measure UV absorbance via spectrophotometry
DNA quantification via spectrophotometry
DNA absorbs in the UV range around 260 nm. Proteins do not absorb much in this region.
1. Purines have an absorbance maximum slightly below 260 nm.
2. Pyrimidines have a maximum slightly above 260 nm.
Purines have a higher molar absorptivity than pyrimidines. Therefore, the absorbance maximum and absorptivity of a segment of DNA depends on its base composition.

13
DNA quantification via spectrophotometry
Proteins have two absorbance peaks in the UV region:
1. Between 215-230 nm (peptide bonds)
Measurements are generally not performed at this wavelength because commonly used buffers and solvents, such as Tris, also absorb at these wavelengths.
2. At about 280 (aromatic amino acids, e.g. tyrosine, tryptophan and phenylalanine).
Nucleic acids also have some absorbance at this wavelength.
Both proteins and nucleic acids absorb light at 280 nm. Therefore, if nucleic acids and proteins are mixed in the same sample, their spectra interfere (overlap) with one another.
DNA quantification via spectrophotometry
Absorbance at 270 nm? Probably due to phenol contamination.
Use 320 nm as a “control” – there should be no absorbance from proteins or DNA here.
DNAProtein (BSA)
DNA/Protein mix
200 nm300 nm

14
Using absorbance values to obtain quantitative measurements of DNA and protein:
1. Construct a calibration curve using standards of known concentration.
The linear range for UV absorbance of DNA ranges from about 5-50 µg/mL. UV analysis of proteins at 280 nm has a linear range from about 0.1 - 5 mg/mL.
You may need to dilute your samples to get accurate readings.
2. Use Beer’s law to determine “average” absorptivity constant for nucleic acid
A = Ecd
Where:
A = absorbance of solute in solution (nm at λ max)E =extinction coefficientC = concentration of solute (moles/L)d = distance (cm) the light must travel through solution
Approximate relationship of absorbance and nucleic acid concentration using Beer’s Law and the conversions that some nice person already did for you…
1 absorbance unit at 260 nm = approximately 50 µg/mL of pure ds DNA.1 absorbance unit at 260 nm = approximately 33 µg/mL of pure ss DNA. 1 absorbance unit at 260 nm = approximately 40 µg/mL of pure ss RNA.
Thus:
# of Units of Abs260 x 50μg/mL x Dilution factor = μg/mL of DNA
Values for proteins vary by amino acid composition. A very rough rule is that if a sample containing pure protein has an absorbance of 1 at 280 nm, then it contains approximately 1 mg/mL of protein.

15
Estimation of the Purity of a Nucleic Acid Preparation
You can also use UV spectrophotometry to estimate the purity of preparation of nucleic acids.
Measure absorbance of the solution at 260 nm and 280 nm, and calculate the Abs260/Abs280ratio:
A ratio of 2.0 is characteristic of pure RNA. A ratio of 1.8 is characteristic of pure DNA. A ratio of about 0.6 is characteristic of pure protein
Therefore, a ratio of > 1.8 is desired when purifying nucleic acids.
A ratio less than 1.8 means there is probably a contaminant in the solution (usually either protein or phenol).





























