Embed Size (px)
Citation preview

DOI: 10.1002/minf.201300054
Towards the Next Generation of ComputationalChemogenomics ToolsD. Rognan*[a]
Keywords: Chemogenomics · Ligand space · Target space · Profiling
A decade after the term ‘chemogenomics’[1] was introducedto the scientific literature, the herein special issue of Molec-ular Informatics is an excellent opportunity to look at thereally significant advances in the field. There are many pos-sible definitions of chemogenomics.[2,3] We will here definedit as any attempt to match target and ligand spaces inorder to ultimately identify all possible ligands of all possi-ble targets. This vast matrix cannot be obviously addressedonly by experimental data. Computational tools have there-fore a decisive role to fill the gaps and prioritize experimen-tal validation. This review will try to answer to basic ques-tions: (i) Where are we coming from? (ii) Where are wegoing to? Of course, it only reflects the personal opinion ofan academic scientist. As we will see, industrial (pharma,biotech) and academic settings may offer substantially dif-ferent approaches to the same problem, the main differen-ces lying in available resources and (sometimes) in experi-mental data quality. Thanks to the ever increasing availabili-ty of public bioactivity data,[4] these differences have beenminored but still do exist.
Where Are We Coming From?
The basic concept underlying the chemogenomic approachis that ‘similar ligands bind to similar targets.’ Identifyingnovel target-ligand pairs is therefore possible at three con-ditions: (i) availability of bioactivity data (target-annotatedligand sets, ligand-annotated target sets, protein and pro-tein-ligand X-ray structures), (ii) usage of adequate molecu-lar descriptors, (iii) development of a metric to estimate thepair-wise similarity of molecules (ligands, proteins, bindingsites, protein-ligand complexes) of interest.
Although the general applicability domain of chemoge-nomics is the full target-ligand space, the emerging disci-pline has been initially restricted to target family-basedligand design (protein kinases,[1,5,6] G protein-coupled recep-tors,[7] nuclear hormone receptors,[8] proteases[9]). In otherwords, ligands for novel targets were principally inferredfrom existing knowledge on neighboring proteins still fromthe same family.[10] There were two main reasons for thisobservation: (i) pharmaceutical companies have been pio-neering these novel computational methods thanks to thewealth of high-quality binding data accumulated over years
for these main target families, (ii) target comparisons weremuch easier if restricted to a single family (e.g. similarity ofbinding site-lining amino acid sequences[11]).
Despite these limitations, remarkable achievements havebeen noticed using either ligand-centric or target-centricapproaches in numerous fields: (i) classifying target spaceby binding site[11] and ligand[12,13] diversity, (ii) predictingmain,[14] off-,[15] and anti-targets[16] for known drugs and bio-active compounds, (ii) designing clinical candidates witha desired polypharmacological profile,[17] (iii) guiding recep-tor deorphanization.[18]
At this point, one could consider that the ever increasingavailability of structural[19] and bioactivity data[4] will sufficeto enlarge the applicability domain of existing computa-tional chemogenomics. Since the launch of the ChEMBL da-tabase,[20] the number of bioactivity data has grown from2.4 million (Jan 2010) to 10.5 million (Jan 2013). According-ly, the number of distinct ligands increased from 520 000 to1.2 million. After looking at 10 years of evolution, there isnevertheless an obvious need for novel descriptors, meth-ods and strategies in order to (i) improve the predictivity ofchemogenomic approaches, (ii) switch from qualitative toquantitative predictions, (iii) enlarge the target-ligand spaceto yet uncharted regions.
Where Are We Going To? Focusing on Reliableand Information-Rich Data
In its early days, computational chemogenomics was sodata-hungry that quantitative models were rare and ap-plied to a tiny target-ligand pace.[21] For example, com-pounds were assigned to ‘activity classes’ (or targets)[22] ir-respective of their binding affinity and function (receptoragonist/antagonist/inverse agonist, enzyme competitive orallosteric inhibitors).[23] Target-ligand associations were
Specia
lIssu
eC
hem
ogenom
ics
[a] D. RognanUMR 7200 CNRS-Universit� de Strasbourg, MEDALIS DrugDiscovery Center74 route du Rhin, 67400, Illkirch, France*e-mail : [email protected]
Supporting Information for this article is available on the WWWunder http://dx.doi.org/10.1002/minf.201300054.
Mol. Inf. 0000, 00, 1 – 6 � 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim &1&
These are not the final page numbers! ��

often predicted in a binary manner[24] without any clueabout the strength of the association. Although many ma-chine learning algorithms are supposed to tolerate noisydata, more precise predictions about the binding affinityand functional outcome of target-ligand pairs should bereached. Pairing ligands to targets is necessary but nomore sufficient. In addition, a fine specificity profile towardshighly related targets (e.g. receptor subtypes) should beavailable, as well as the function of the ligand. Fortunately,we are now in position to be much more stringent on dataselection and only pick the most relevant information forlearning. For example, inhibition constants (Ki) should bepreferred to half maximal inhibitory concentration (IC50)values when both data are available for a target-ligand pair.Filtering bioactivity databases for data consistency andligand properties dramatically reduces the chemogenomicspace (Table 1) but increases the predictivity of models fora better defined target-ligand space.
For example, it is very tempting to select any trustableavailable binding data to train affinity prediction QSARmodels. Our experience (Figure 1) however suggests thatexhaustivity does not guarantee accurate modeling. Theexact applicability domain[25] of chemogenomic modelswith respect to both ligand and target spaces should besystematically investigated and explicitly defined to warrantuseful predictions.
Exploring Novel Ligand and Target Spaces
Current chemogenomic modeling tools, whatever the un-derlying principle (ligand-centric,[27] target-centric,[28] pro-teochemometric[29]) are surprisingly accurate to predicttarget-ligand associations or even binding affinity datawithin a conventional target space (GPCRs, kinases, pro-teases, nuclear hormone receptors) and for drug-like li-gands. The main reason for that observation is that thepharmaceutical industry has gathered enough high-qualitydata on this traditional target-ligand space. Being able topredict a novel protein kinase ATP-competitive inhibitordoes not mean that the corresponding model is in positionto identify the next allosteric inhibitor for this target. First,bioactivity databases should be properly annotated to de-scribe the true functional effect (full or partial agonist, in-
verse agonist, neutral antagonist, competitive or allostericinhibitor) of registered ligands, which demands very accu-rate text-mining techniques. Second, we should not restrictthe available ligand space to conventional definitions.[30]
Small peptides or peptidometics (e.g stapled peptides[31]),macrocyles,[32] natural products[33] may combine oral bio-availability and exquisite target selectivity; they are howev-er still largely under-represented in bioactivity databases.But how to rationally design novel chemotypes matchingyet undisclosed but relevant biological space? Advances inthe design of synthetically available compound librarieshave been noticed recently,[34,35] usually focusing on novelscaffolds or chemotypes. A recent survey of high through-put screening (HTS) data at Novartis indicate that ca. 46 %of their 1.8 million compounds have never been active inany of 230 screening assays.[36] Instead of focusing onchemical diversity, library design should rather be orientedtowards maximum biological diversity (target coverage).HTS fingerprints (HTS-FP) obtained from a very large HTSdata matrix (1.8 million compounds and >230 assays) havejust been described to outperform standard ligand-centricfingerprints in virtual screening for maximum hit rate, scaf-fold rate and biodiverse plate selection.[37] This kind of in-formation is still unavailable to most academic settings, butcomputational surrogates aimed at predicting target-ligandinteractomes do exist[23,15,38–40] and could be used to guidevirtual library design while optimizing potential target di-versity.
Enlarging the target space to which chemogenomicsmethods may apply is of course another topic of interest.
Specia
lIs
sue
Chem
ogenom
ics
Table 1. Filtering the ChEMBL database (release 12) for data con-sistency.
Raw data Filtered data
Ligands 1 076 486[a] 243 625Targets 8703 2337Affinity data 6 186 504[b] 454 611
[a] Ligand filtering: �1 carbon atom; no cycle with more than 25atoms; no organo-metallic structures; <3 peptidic bonds), <13unsubstituted carbon atoms. [b] Data filtering: Keeping the mostrecent pKi value if n = 2; median pKi when n>2 and pKi,max–pKi,min<1; remove otherwise.
Figure 1. Averaged cross-validated correlation coefficient (meanQ2) for affinity interpolation models[26] from ChEMBL binding data(release 12) and different target spaces. A) any human target withmore than 5 different ligands (2087 targets), B) any human targetwith more than 25 different ligands (1473 targets),C) identical tomodel B but with an homogenous distribution of pKi values in 3 in-tervals ([4–6], [6–8], and [8–10] and at least 15 % of all data in eachinterval (271 targets), D) any human target with more than 100 dif-ferent ligands (767 targets).
&2& www.molinf.com � 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Mol. Inf. 0000, 00, 1 – 6
�� These are not the final page numbers!
Communication D. Rognan
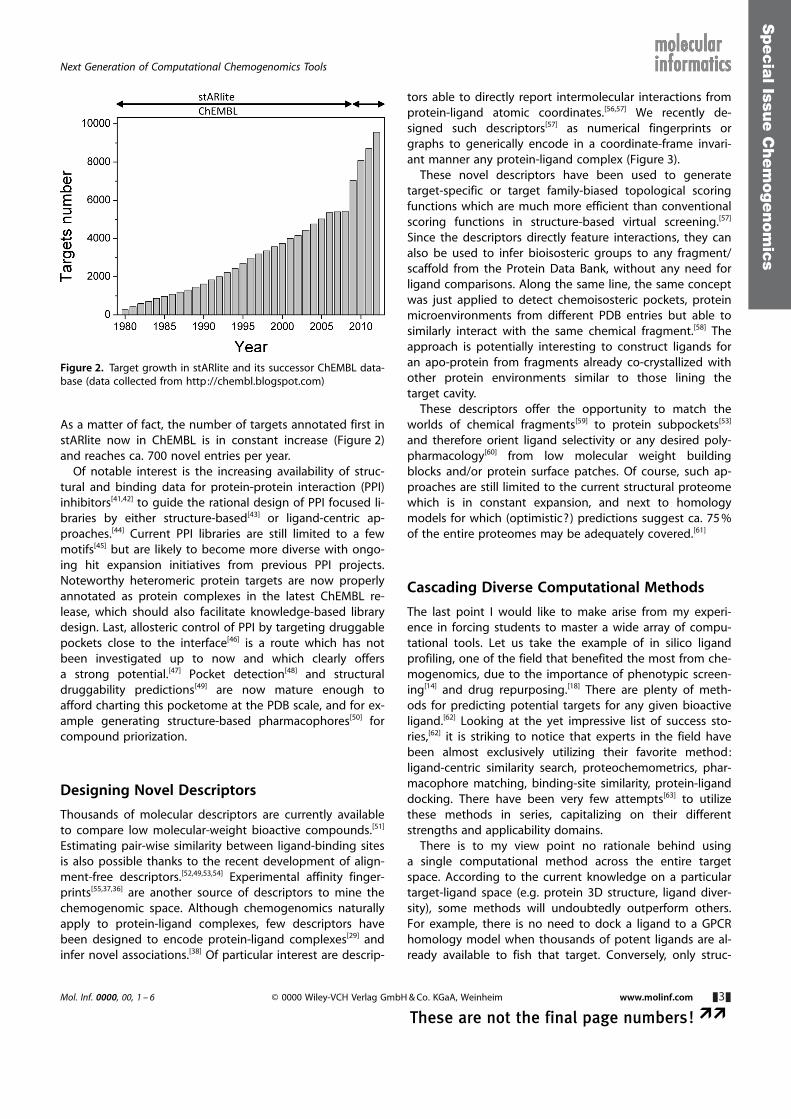
As a matter of fact, the number of targets annotated first instARlite now in ChEMBL is in constant increase (Figure 2)and reaches ca. 700 novel entries per year.
Of notable interest is the increasing availability of struc-tural and binding data for protein-protein interaction (PPI)inhibitors[41,42] to guide the rational design of PPI focused li-braries by either structure-based[43] or ligand-centric ap-proaches.[44] Current PPI libraries are still limited to a fewmotifs[45] but are likely to become more diverse with ongo-ing hit expansion initiatives from previous PPI projects.Noteworthy heteromeric protein targets are now properlyannotated as protein complexes in the latest ChEMBL re-lease, which should also facilitate knowledge-based librarydesign. Last, allosteric control of PPI by targeting druggablepockets close to the interface[46] is a route which has notbeen investigated up to now and which clearly offersa strong potential.[47] Pocket detection[48] and structuraldruggability predictions[49] are now mature enough toafford charting this pocketome at the PDB scale, and for ex-ample generating structure-based pharmacophores[50] forcompound priorization.
Designing Novel Descriptors
Thousands of molecular descriptors are currently availableto compare low molecular-weight bioactive compounds.[51]
Estimating pair-wise similarity between ligand-binding sitesis also possible thanks to the recent development of align-ment-free descriptors.[52,49,53,54] Experimental affinity finger-prints[55,37,36] are another source of descriptors to mine thechemogenomic space. Although chemogenomics naturallyapply to protein-ligand complexes, few descriptors havebeen designed to encode protein-ligand complexes[29] andinfer novel associations.[38] Of particular interest are descrip-
tors able to directly report intermolecular interactions fromprotein-ligand atomic coordinates.[56,57] We recently de-signed such descriptors[57] as numerical fingerprints orgraphs to generically encode in a coordinate-frame invari-ant manner any protein-ligand complex (Figure 3).
These novel descriptors have been used to generatetarget-specific or target family-biased topological scoringfunctions which are much more efficient than conventionalscoring functions in structure-based virtual screening.[57]
Since the descriptors directly feature interactions, they canalso be used to infer bioisosteric groups to any fragment/scaffold from the Protein Data Bank, without any need forligand comparisons. Along the same line, the same conceptwas just applied to detect chemoisosteric pockets, proteinmicroenvironments from different PDB entries but able tosimilarly interact with the same chemical fragment.[58] Theapproach is potentially interesting to construct ligands foran apo-protein from fragments already co-crystallized withother protein environments similar to those lining thetarget cavity.
These descriptors offer the opportunity to match theworlds of chemical fragments[59] to protein subpockets[53]
and therefore orient ligand selectivity or any desired poly-pharmacology[60] from low molecular weight buildingblocks and/or protein surface patches. Of course, such ap-proaches are still limited to the current structural proteomewhich is in constant expansion, and next to homologymodels for which (optimistic?) predictions suggest ca. 75 %of the entire proteomes may be adequately covered.[61]
Cascading Diverse Computational Methods
The last point I would like to make arise from my experi-ence in forcing students to master a wide array of compu-tational tools. Let us take the example of in silico ligandprofiling, one of the field that benefited the most from che-mogenomics, due to the importance of phenotypic screen-ing[14] and drug repurposing.[18] There are plenty of meth-ods for predicting potential targets for any given bioactiveligand.[62] Looking at the yet impressive list of success sto-ries,[62] it is striking to notice that experts in the field havebeen almost exclusively utilizing their favorite method:ligand-centric similarity search, proteochemometrics, phar-macophore matching, binding-site similarity, protein-liganddocking. There have been very few attempts[63] to utilizethese methods in series, capitalizing on their differentstrengths and applicability domains.
There is to my view point no rationale behind usinga single computational method across the entire targetspace. According to the current knowledge on a particulartarget-ligand space (e.g. protein 3D structure, ligand diver-sity), some methods will undoubtedly outperform others.For example, there is no need to dock a ligand to a GPCRhomology model when thousands of potent ligands are al-ready available to fish that target. Conversely, only struc-
Specia
lIssu
eC
hem
ogenom
ics
Figure 2. Target growth in stARlite and its successor ChEMBL data-base (data collected from http://chembl.blogspot.com)
Mol. Inf. 0000, 00, 1 – 6 � 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.molinf.com &3&
These are not the final page numbers! ��
Next Generation of Computational Chemogenomics Tools

ture-based approaches will permit, whenever possible, todeorphanize in silico still orphan GPCRs far different fromany existing liganded receptor.[64] A pragmatic but logicalapproach to enhance the coverage and accuracy of in silicoprofiling methods would thus be: (i) to set the rules permit-ting to fix the exact applicability domain of each method,(ii) to apply these methods to each of the target sub-spacein which they are predicted to be efficient. Preliminarytrials from our group to set these rules[40] and design a com-posite profiling workflow clearly show that all these meth-ods are indeed complementary and contribute -albeit todifferent extents- to successfully profile bioactive com-pounds.
The need to bridge bio- and chemoinformatics methodsis obviously even higher to go beyond just a list of poten-tial targets and infer potential side effects[65,66] as well as se-lectively regulate specific metabolic pathways.[13] Integrated
systems biology databases[67,68] are now in place to enablefurther progress in this exciting arena.
Conclusions
Despite being still in its infancy, the field of computationalchemogenomics has undergone a remarkable evolutionboth in accuracy and applicability domain. Initial qualitativepredictions focusing on a small set of target families havebeen replaced by remarkably accurate affinity estimates ona large array of diverse targets. Driving forces to this evolu-tion have been first the pioneering work of a few pharma-ceutical companies and second the public release of bioac-tivity data that brought chemogenomic methods to thedesktop of any computational chemist. It is of prime impor-tance that the field does not remain in the sole hands of
Specia
lIs
sue
Chem
ogenom
ics
Figure 3. Encoding protein-ligand complexes by fingerprints or graphs. From atomic coordinates of the protein-ligand complex , interac-tions are detected on the fly and described by interaction pseudoatoms (IPAs) featuring 7 interaction types (Hydrophobic, H, green balls ;Aromatic, Ar, yellow balls; H-bond donor, HBD, blue balls ; H-bond acceptor, HBA, red balls ; positive ionizable, PI, not featured here; nega-tive ionizable, NI, not featured here; metal complexation, Me, dark (see Supporting Information for color)). The set of IPAs can be represent-ed by a disconnected graph (Step 3). All possible triplets (3 properties, 3 distances) of IPAs are generated and matched to a triplet list(Step 4). The count of each triplet type is encoded through a fingerprint of 12508 integers that is further pruned to 210 integers (Step 5) tofeature the most frequently occurring triplets.[57]
&4& www.molinf.com � 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Mol. Inf. 0000, 00, 1 – 6
�� These are not the final page numbers!
Communication D. Rognan

experts but lives from collaborative efforts with medicinalchemists, structural biologists, pharmacologists, toxicolo-gists and many more. I am very much confident that thediscipline will continue its evolution and pursue remarkableachievements in the next decade.
Acknowledgements
I warmly acknowledge all current and former members ofthe Structural Chemogenomics Laboratory (UMR 7200 CNRS-Universit� de Strasbourg) for their invaluable help all overthe years and their team spirit. My thanks also go to theIPHC grid (Strasbourg) and CC-IN2P3 (Villeurbanne) for pro-viding first-class computational resources and support.
References
[1] P. R. Caron, M. D. Mullican, R. D. Mashal, K. P. Wilson, M. S. Su,M. A. Murcko, Curr. Opin. Chem. Biol. 2001, 5, 464 – 470.
[2] M. Bredel, E. Jacoby, Nat. Rev. Genet. 2004, 5, 262 – 275.[3] D. Rognan, Br. J. Pharmacol. 2007, 152, 38 – 52.[4] G. Nicola, T. Liu, M. K. Gilson, J. Med. Chem. 2012, 55, 6987 –
7002.[5] Z. Deng, C. Chuaqui, J. Singh, J. Med. Chem. 2004, 47, 337 –
344.[6] M. Vieth, J. J. Sutherland, D. H. Robertson, R. M. Campbell,
Drug Discov. Today 2005, 10, 839 – 846.[7] T. Klabunde, G. Hessler, Chembiochem 2002, 3, 928 – 944.[8] J. Mestres, L. Martin-Couce, E. Gregori-Puigjane, M. Cases, S.
Boyer, J. Chem. Inf. Model. 2006, 46, 2725 – 2736.[9] H. Matter, W. Schwab, J. Med. Chem. 1999, 42, 4506 – 4523.
[10] R. E. Martin, L. G. Green, W. Guba, N. Kratochwil, A. Christ, J.Med. Chem. 2007, 50, 6291 – 6294.
[11] J. S. Surgand, J. Rodrigo, E. Kellenberger, D. Rognan, Proteins2006, 62, 509 – 538.
[12] G. V. Paolini, R. H. Shapland, W. P. van Hoorn, J. S. Mason, A. L.Hopkins, Nat. Biotechnol. 2006, 24, 805 – 815.
[13] H. Lin, M. F. Sassano, B. L. Roth, B. K. Shoichet, Nat. Methods2013, 10, 140 – 146.
[14] C. Laggner, D. Kokel, V. Setola, A. Tolia, H. Lin, J. J. Irwin, M. J.Keiser, C. Y. Cheung, D. L. Minor, Jr. , B. L. Roth, R. T. Peterson,B. K. Shoichet, Nat. Chem. Biol. 2011, 8, 144 – 146.
[15] M. J. Keiser, V. Setola, J. J. Irwin, C. Laggner, A. I. Abbas, S. J.Hufeisen, N. H. Jensen, M. B. Kuijer, R. C. Matos, T. B. Tran, R.Whaley, R. A. Glennon, J. Hert, K. L. Thomas, D. D. Edwards,B. K. Shoichet, B. L. Roth, Nature 2009, 462, 175 – 181.
[16] E. Lounkine, M. J. Keiser, S. Whitebread, D. Mikhailov, J.Hamon, J. L. Jenkins, P. Lavan, E. Weber, A. K. Doak, S. Cote,B. K. Shoichet, L. Urban, Nature 2012, 486, 361 – 367.
[17] J. Besnard, G. F. Ruda, V. Setola, K. Abecassis, R. M. Rodriguiz,X. P. Huang, S. Norval, M. F. Sassano, A. I. Shin, L. A. Webster,F. R. Simeons, L. Stojanovski, A. Prat, N. G. Seidah, D. B. Con-stam, G. R. Bickerton, K. D. Read, W. C. Wetsel, I. H. Gilbert, B. L.Roth, A. L. Hopkins, Nature 2012, 492, 215 – 220.
[18] D. E. Gloriam, P. Wellendorph, L. D. Johansen, A. R. Thomsen,K. Phonekeo, D. S. Pedersen, H. Brauner-Osborne, Chem. Biol.2011, 18, 1489 – 1498.
[19] There are currently over 85 000 structures deposited in theProtein Data Bank; http://www.rcsb.org/pdb/statistics/hol-dings.do, accessed March, 2013.
[20] A. Gaulton, L. J. Bellis, A. P. Bento, J. Chambers, M. Davies, A.Hersey, Y. Light, S. McGlinchey, D. Michalovich, B. Al-Lazikani,J. P. Overington, Nucleic Acids Res. 2011, 40, D1100 – D1107.
[21] J. R. Bock, D. A. Gough, J. Chem. Inf. Model. 2005, 45, 1402 –1414.
[22] A. Schuffenhauer, J. Zimmermann, R. Stoop, J. J. van der Vyver,S. Lecchini, E. Jacoby, J. Chem. Inf. Comput. Sci. 2002, 42, 947 –955.
[23] Nidhi, M. Glick, J. W. Davies, J. L. Jenkins, J. Chem. Inf. Model.2006, 46, 1124 – 1133.
[24] N. Weill, D. Rognan, J. Chem. Inf. Model. 2009, 49, 1049 – 1062.[25] A. Varnek, I. Baskin, J. Chem. Inf. Model. 2012, 52, 1413 – 1437.[26] D. Vidal, J. Mestres, Mol. Inf. 2010, 29, 543 – 551.[27] M. J. Keiser, J. J. Irwin, B. K. Shoichet, Biochemistry 2010, 49,
10267 – 1076.[28] D. Rognan, Mol. Inf. 2010, 29, 176 – 187.[29] G. J. P. van Westen, J. K. Wegner, A. P. Ijzerman, H. W. T. van Vlij-
men, A. Bender, Med. Chem. Commun. 2011, 2, 16 – 30.[30] P. Kirkpatrick, Nat. Rev. Drug. Discov. 2012, 11, 900 – 901.[31] G. L. Verdine, G. J. Hilinski, Methods Enzymol. 2012, 503, 3 – 33.[32] H. R. Hoveyda, E. Marsault, R. Gagnon, A. P. Mathieu, M.
Vezina, A. Landry, Z. Wang, K. Benakli, S. Beaubien, C. Saint-Louis, M. Brassard, J. F. Pinault, L. Ouellet, S. Bhat, M. Ramase-shan, X. Peng, L. Foucher, S. Beauchemin, P. Bherer, D. F. Veber,M. L. Peterson, G. L. Fraser, J. Med. Chem. 2011, 54, 8305 –8320.
[33] B. Over, S. Wetzel, C. Grutter, Y. Nakai, S. Renner, D. Rauh, H.Waldmann, Nat. Chem. 2012, 5, 21 – 28.
[34] M. Hartenfeller, H. Zettl, M. Walter, M. Rupp, F. Reisen, E. Pro-schak, S. Weggen, H. Stark, G. Schneider, PLoS Comput. Biol.2012, 8, e1002380.
[35] L. Ruddigkeit, L. C. Blum, J. L. Reymond, J. Chem. Inf. Model.2013, 53, 56 – 65.
[36] P. M. Petrone, A. M. Wassermann, E. Lounkine, P. Kutchukian, B.Simms, J. Jenkins, P. Selzer, M. Glick, Drug Discov. Today 2013,18, 674 – 680.
[37] P. M. Petrone, B. Simms, F. Nigsch, E. Lounkine, P. Kutchukian,A. Cornett, Z. Deng, J. W. Davies, J. L. Jenkins, M. Glick, ACSChem. Biol. 2012, 7, 1399 – 1409.
[38] F. Wang, D. Liu, H. Wang, C. Luo, M. Zheng, H. Liu, W. Zhu, X.Luo, J. Zhang, H. Jiang, J. Chem. Inf. Model. 2011, 51, 2821 –2828.
[39] L. Yang, K. Wang, J. Chen, A. G. Jegga, H. Luo, L. Shi, C. Wan,X. Guo, S. Qin, G. He, G. Feng, L. He, PLoS Comput. Biol. 2011,7, e1002016.
[40] J. Meslamani, J. Li, J. Sutter, A. Stevens, H. O. Bertrand, D.Rognan, J. Chem. Inf. Model. 2012, 52, 943 – 955.
[41] M. J. Basse, S. Betzi, R. Bourgeas, S. Bouzidi, B. Chetrit, V.Hamon, X. Morelli, P. Roche, Nucleic Acids Res. 2012, 41,D824 – D827.
[42] D. M. Kruger, G. Jessen, H. Gohlke, J. Chem. Inf. Model. 2012,52, 2807 – 2811.
[43] D. Fry, K. S. Huang, P. Di Lello, P. Mohr, K. Muller, S. S. So, T.Harada, M. Stahl, B. Vu, H. Mauser, ChemMedChem 2013, 8,726 – 732.
[44] C. Reynes, H. Host, A. C. Camproux, G. Laconde, F. Leroux, A.Mazars, B. Deprez, R. Fahraeus, B. O. Villoutreix, O. Sperandio,PLoS Comput. Biol. 2010, 6, e1000695.
[45] L. R. Whitby, D. L. Boger, Acc. Chem. Res. 2012, 45, 1698 – 1709.[46] M. Gao, J. Skolnick, Proc. Natl. Acad. Sci. USA 2012, 109, 3784 –
379.[47] M. Cazorla, J. Premont, A. Mann, N. Girard, C. Kellendonk, D.
Rognan, J. Clin. Invest. 2011, 121, 1846 – 1857.
Specia
lIssu
eC
hem
ogenom
ics
Mol. Inf. 0000, 00, 1 – 6 � 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.molinf.com &5&
These are not the final page numbers! ��
Next Generation of Computational Chemogenomics Tools

[48] A. Volkamer, D. Kuhn, T. Grombacher, F. Rippmann, M. Rarey, J.Chem. Inf. Model. 2011, 52, 360 – 372.
[49] J. Desaphy, K. Azdimousa, E. Kellenberger, D. Rognan, J. Chem.Inf. Model. 2012, 52, 2287 – 2299.
[50] D. R. Koes, C. J. Camacho, Nucleic Acids Res. 2012, 40, W387 –W392.
[51] R. Todeschini, V. Consonni, Handbook of Molecular Descriptors,Wiley-VCH, 2008.
[52] N. Weill, D. Rognan, J. Chem. Inf. Model. 2010, 50, 123 – 135.[53] T. Kalliokoski, T. S. Olsson, A. Vulpetti, J. Chem. Inf. Model.
2013, 53, 131 – 141.[54] M. M. von Behren, A. Volkamer, A. M. Henzler, K. T. Schomburg,
S. Urbaczek, M. Rarey, J. Chem. Inf. Model. 2013, 53, 411 – 422.[55] M. Bieler, R. Heilker, H. Koppen, G. Schneider, J. Chem. Inf.
Model. 2011, 51, 1897 – 1905.[56] M. Weisel, H. M. Bitter, F. Diederich, W. V. So, R. Kondru, J.
Chem. Inf. Model. 2012, 52, 1450 – 1461.[57] J. Desaphy, E. Raimbaud, P. Ducrot, D. Rognan, J. Chem. Inf.
Model. 2013, 53, 623 – 637.[58] X. Jalencas, J. Mestres, J. Chem. Inf. Model. 2013, 53, 279 – 292.[59] D. Rognan, Top. Curr. Chem. 2012, 317, 201 – 222.[60] R. Morphy, J. Med. Chem. 2010, 53, 1413 – 37.[61] J. Skolnick, H. Zhou, M. Gao, Curr. Opin. Struct. Biol. 2013, 23,
191 – 197.
[62] A. Koutsoukas, B. Simms, J. Kirchmair, P. J. Bond, A. V. Whit-more, S. Zimmer, M. P. Young, J. L. Jenkins, M. Glick, R. C. Glen,A. Bender, J. Proteomics 2011, 74, 2554 – 2574.
[63] J. D. Durrant, R. E. Amaro, L. Xie, M. D. Urbaniak, M. A. Fergu-son, A. Haapalainen, Z. Chen, A. M. Di Guilmi, F. Wunder, P. E.Bourne, J. A. McCammon, PLoS Comput. Biol. 2010, 6,e1000648.
[64] B. L. Roth, ACS Med. Chem. Lett. 2013, 4, 316 – 318.[65] M. Campillos, M. Kuhn, A. C. Gavin, L. J. Jensen, P. Bork, Science
2008, 321, 263 – 266.[66] J. Scheiber, B. Chen, M. Milik, S. C. Sukuru, A. Bender, D. Mi-
khailov, S. Whitebread, J. Hamon, K. Azzaoui, L. Urban, M.Glick, J. W. Davies, J. L. Jenkins, J. Chem. Inf. Model. 2009, 49,308 – 317.
[67] B. Chen, X. Dong, D. Jiao, H. Wang, Q. Zhu, Y. Ding, D. J. Wild,BMC Bioinformatics 2010, 11, 255.
[68] S. Kim Kjaerulff, L. Wich, J. Kringelum, U. P. Jacobsen, I. Kous-koumvekaki, K. Audouze, O. Lund, S. Brunak, T. I. Oprea, O. Ta-boureau, Nucleic Acids Res. 2013, 41, D464 – D469.
Received: March 28, 2013Accepted: June 11, 2013
Published online: && &&, 0000
Specia
lIs
sue
Chem
ogenom
ics
&6& www.molinf.com � 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Mol. Inf. 0000, 00, 1 – 6
�� These are not the final page numbers!
Communication D. Rognan





























