Embed Size (px)
Citation preview

Proc. Natl. Acad. Sci. USAVol. 92, pp. 5266-5272, June 1995
Review
Gene targeting approaches to complex genetic diseases:Atherosclerosis and essential hypertension
(animal models/gene disruption/gene duplication/quantitative traits)
Oliver Smithies and Nobuyo MaedaDepartment of Pathology, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599-7525
ABSTRACT Gene targeting allowsprecise, predetermined changes to bemade in a chosen gene in the mousegenome. To date, targeting has been usedmost often for generation of animals com-pletely lacking the product of a gene ofinterest. The resulting "knockout" micehave confirmed some hypotheses, haveupset others, but have rarely been unin-formative. Models of several human ge-netic diseases have been produced by tar-geting-including Gaucher disease, cysticfibrosis, and the fragile X syndrome.These diseases are primarily determinedby defects in single genes, and their modesof inheritance are well understood. Whenthe disease under study has a complexetiology with multiple genetic and envi-ronmental components, the generation ofanimal models becomes more difficult butno less valuable. The problems associatedwith dissecting out the individual geneticfactors also increase substantially and thedistinction between causation and corre-lation is often difficult. To prove causa-tion in a complex system requires rigor-ous adherence to the principle that theexperiments must allow detection of theeffects of changing only a single variableat one time. Gene targeting experiments,when properly designed, can test the ef-fects of a precise genetic change com-pletely free from the effects of differencesin any other genes (linked or unlinked tothe test gene). They therefore allow proofsof causation.
Atherosclerosis and essential hyperten-sion are two common diseases that ac-count for a major portion of morbidity anddeath in the United States. Although pop-ulation studies have long suggested ge-netic components in these complex dis-eases, the details of their inheritance arenot well understood, in part because mul-tiple factors are involved, both genetic andenvironmental. A reasonable working hy-pothesis is that these diseases in manypatients are the consequence of variouscombinations of genetic defects that indi-vidually may have only modest effects.Gene targeting in mice provides a meansof testing this hypothesis. Individual genescan be modified and their effects on thedevelopment of atherosclerosis or on
blood pressures can be evaluated in a
manner that excludes the effects of anyother genes (linked or unlinked to thetarget gene). The identification of com-pensatory mechanisms induced by the pri-mary gene modification can point to othergenes that antagonize the effects of thetarget gene. Combinations of geneticchanges can then be made that allow thedetection of synergistic or antagonisticinteractions. In addition, at any stage ofthe genetic investigations, the effects ofenvironmental factors such as high cho-lesterol and fat or salt in the diet can beevaluated, so that the interactions be-tween environment and genotype can beassessed. In what follows, we describe ourexperiences while using gene targeting tostudy the genetics of atherosclerosis andhypertension, and some of the challengesencountered, in order to illustrate howsuch experiments can generate useful an-imal models for further study and developan increased understanding of complexgenetic diseases.
Atherosclerosis
Atherosclerosis in humans is character-ized by the development of intraarteriallesions that may eventually become occlu-sive (1). The earliest recognizable stage isthe preatherosclerotic "fatty streak" inwhich an aggregation of lipid-rich macro-phages can be found in the intima, be-neath the endothelial lining of the vessels.Fatty streaks progress to intermediate le-sions that have smooth muscle prolifera-tion as well as fatty components. Maturelesions typically have fibrous caps and anacellular lipid core and may develop re-gions of calcification. Plaque fissuring fol-lowed by thrombosis may eventually oc-cur. Of numerous risk factors that havebeen associated with atherosclerosis,plasma lipoprotein levels are the best stud-ied. Elevated levels of low density lipopro-tein (LDL) and reduced levels of highdensity lipoprotein (HDL) in plasma arecorrelated with a higher risk for athero-sclerosis in humans (2).No mouse strains are known in which
atherosclerosis occurs spontaneouslywhen they are fed regular mouse chow(4.5% fat/0.02% cholesterol). However,
5266
experiments by Paigen and her collabora-tors (3) have shown that some inbredstrains develop vascular lesions when fed ahigh fat (15.8%)/high cholesterol (1.25%)diet. But the lesions, even in a susceptiblestrain such as C57BL/6J (B6), rarelyprogress beyond the fatty streak stage.Mice naturally have high plasma levels ofHDL and low levels of LDL, conditionsthat in humans are protective against ath-erosclerosis. Mice also lack lipoprotein(a),an atherogenic lipoprotein, and their lev-els of cholesteryl ester transfer protein areextremely low; both these proteins arepresent in humans. Despite these differ-ences, the general pathways of lipid trans-port and metabolism in mice and humansare otherwise so similar that one couldreasonably expect derangements in spe-cific parts of the pathways to have similareffects in the two species. Nevertheless,when we began our gene targeting exper-iments in mice, we expected that fullydeveloped atherosclerosis might not beachievable without combining several de-fects.A series of targets were initially cho-
sen. The first gene (Apoe) codes forapolipoprotein E (apoE), which plays acentral role in hepatic clearance from thecirculation of chylomicron remnants andvery low density lipoprotein remnants.The second gene (Apoal) codes for apo-lipoprotein A-I (apoA-I), which is themajor protein component of HDL. Dis-ruption of theApoe gene was expected toabolish apoE-mediated clearance andtherefore lead to an increase in the tran-sit times of lipid in the circulation and tohypercholesterolemia. Removal ofapoA-I was expected to cause a reduc-tion in plasma HDL. Because of ouruncertainty that atherosclerosis wouldoccur consequent to these primarychanges in mice, targeted changes inseveral other genes were also initiated,including those coding for apoB, apo-CIII, and hepatic lipase.
Abbreviations: ACE, angiotensin-convertingenzyme; AGT, angiotensinogen; apo, apoli-poprotein; HDL, high density lipoprotein;LDL, low density lipoprotein; ANP, atrial na-triuretic peptide; ES cell, embryonic stem cell.
Dow
nloa
ded
by g
uest
on
May
28,
202
0

Proc. Natl. Acad. Sci. USA 92 (1995) 5267
ApoE-Deficient Mice Develop Athero-sclerosis Spontaneously. Disruption ofthe Apoe gene by us (4) and by Plump andhis collaborators at the Rockefeller Uni-versity (5), in practice, proved by itself tobe sufficient to generate a mouse strain inwhich mature atherosclerotic lesions de-velop spontaneously. Homozygotes forthe disrupted Apoe gene (-/-) have se-
vere hypercholesterolemia (4-5 times nor-
mal levels); they develop atherosclerosison normal mouse chow, which is low in fatand cholesterol; and their lesions progressto a complex form (Fig. 1) that is essen-tially identical to that seen in human pa-tients (6, 7).The regularity with which advanced ath-
erosclerosis develops spontaneously in theapoE-deficient mice raises at least twoquestions. First, is apoE deficiency as suchimportant in the etiology of the humandisease? Second, is the mouse model ofthe disease made by this single mutationrelevant to the disease in humans, whichhas a complex genetic etiology? An an-
swer to the first question is that it is veryunlikely that an appreciable proportion ofpatients have atherosclerosis because of a
complete absence of apoE, since onlythree families have been described withsuch an absence (8-10). However, thephenotype in mice does not depend on
complete absence of apoE. Thus, whenmice heterozygous (+/-) for the Apoegene disruption were fed the high fat/highcholesterol diet for 3 months, 9/9 devel-oped lesions. Most of the lesions werefatty streaks, but fibrous cap formationwas seen in the heterozygote that had thelargest lesion. Only 3/9 wild-type (Apoe+/+) animals fed the diet had lesions,which averaged -1/10th the size of thosein the heterozygous animals; none werecomplex (11). van Ree et al (12) have alsostudied heterozygous mice (Apoe +/-)
and have demonstrated diet-induced hy-percholesterolemia and atherosclerosis inthem. A hypothesis stemming from thesefindings is that genetic reduction in apoElevels in humans may constitute a riskfactor for atherogenesis, particularly un-
der dietary stress.The second question, concerning the
relevance of the mouse condition gener-
ated by mutation of a single gene to thehuman disease, which is genetically com-
plex, can be addressed by comparing thelesions seen under various circumstances.As described above, the complex lesions inthe apoE-deficient mice are very like thecomplex lesions seen in humans. Similarly,atherosclerotic lesions that occur sponta-neously in pigs having one particular poly-morphic form of the apolipoprotein con-
stituent of LDL (apoB) (13) are also verysimilar in histology to human lesions.Likewise, the lesions that occur in Wa-tanabe heritable hyperlipidemic rabbits asa consequence of a defect in the genecoding for the LDL receptor (14) are
comparable to the human lesions. Finally,in mice homozygous for a disrupted LDLreceptor gene (15) the lesions are again ofthe same type, although a high fat diet isrequired for their development (16). TheapoE-deficient mice consequently providea source of animals that, on a normal diet,rapidly and regularly develop atheroscle-rotic lesions of an advanced and appar-ently general type that is common to manyspecies (including humans) independentof the specific genetic etiology. This highlyreproducible phenotype is now being usedto investigate the effects of other geneticmodifications, drug treatments, dietaryfactors, and the like.Some differences in the detailed effects
of a targeted mutation in mice and of a
comparable mutation in humans are nev-
ertheless to be expected and may be very
*S~~~~~~~~~~~~~~~~. _47-
FIG. 1. A complex atherosclerotic lesion in the aorta of a 60-week-oldApoe -/- mouse fedregular mouse chow. a, Calcified region; b, fibrous cap; c, acellular lipid core with cholesterolcrystal clefts; d, smooth muscle cells infiltrated with lipid. (x60.)
informative. For example, vascular occlu-sions in human patients with advancedatherosclerosis are frequently the conse-quence of thrombosis initiated by plaquefissuring, which exposes extracellular de-bris to the clotting system. No evidence ofthrombotic incidents or sequelae has beenreported in the ApoE-deficient mice. Thissuggests the presence in the mouse ofprotective factors or the absence of dele-terious factors present in humans. Identi-fication of such factors would be of con-siderable interest.
Effects of Other Gene Modifications onAtherogenesis. Thanks to the pioneeringwork of Brown and Goldstein on familialhypercholesterolemia (17), defects in thegene coding for the LDL receptor are thebest documented genetic causes of prema-ture atherosclerosis and coronary heartdisease in humans. It was therefore tosome degree unexpected that mice lackingthe LDL receptor, generated by Ishibashiet al (15), had only mild pathologicalmanifestations when fed normal mousechow, with only about twice normal levelsof plasma cholesterol. Severe lesions were,however, seen when the mice were fed anatherogenic diet (16). This result is incontrast to humans with familial hyper-cholesterolemia due to lack of the LDLreceptor; homozygous humans with thiscondition can have >5 times normalplasma cholesterol and develop severeatherosclerosis even in childhood.Although they are contributing substan-
tially to our understanding of factors thatmodulate lipid metabolism, none of theother single gene defects so far tested havehad such a dramatic effect on atherogen-esis as the apoE deficiency or absence ofthe LDL receptor. For example, as we hadexpected, gene-targeted mice lackingapoA-I have reduced HDL cholesterollevels (about one-fourth normal). In hu-mans, lower levels ofHDL cholesterol areassociated with a higher risk of athero-sclerosis. We were therefore somewhatsurprised to find that mice having reducedlevels of HDL cholesterol caused byapoA-I deficiency do not develop athero-sclerotic lesions on normal chow and areno more susceptible than wild-type ani-mals to diet-induced atherosclerosis (18).However, this finding agrees with reportsthat some human individuals with muta-tions in theAPOAl gene that cause severereductions in HDL cholesterol do notappear to have an increased risk of coro-nary artery disease (19). Thus, reductionof HDL cholesterol is most likely notsufficient to cause atherosclerosis by itself.Combining Mutations. The success of
Apoe gene disruption in generating severeatherosclerosis in the mouse is allowingnew ways of studying the genetic complex-ity of atherogenesis. Thus, we and othershave begun to determine how mutationsin second or third genes modify the inci-dence and progression of the disease gen-
Review: Smithies and Maeda
Dow
nloa
ded
by g
uest
on
May
28,
202
0
5268 Review: Smithies and Maeda
erated by the Apoe gene mutation. Pub-lished accounts of this type of experimentnow include the protective effects on thedisease of genetically increasing the levelsof apoA-I (20, 21). The results show thatincreased levels of apoA-I synthesis me-diated by a human APOA1 transgenecause a 2- to 3-fold increase in HDL anda highly significant reduction in the inci-dence and size of the atherosclerotic le-sions. In a similar fashion, ApoE-deficientmice are allowing evaluation of the rolesof various other genetic factors involved inthe detailed pathology of atherosclerosisat the level of the arterial wall, such asfactors regulating smooth muscle prolifer-ation and inflammation.The combination in mice of two genetic
manipulations, one producing a seriousand complex disease and the other at-tempting to ameliorate its severity, islikely to be a powerful tool for investigat-ing potential modes of gene therapy.When a human disease is the consequenceof a known defect in a specific gene, thefirst choice for gene therapy is likely to bereplacement of the defective gene func-tion. When the disease is caused by defectsin more than one gene, as is probably thecase in human patients with atherosclero-sis or essential hypertension, gene therapyby replacement is less practical. The pos-sibility that effective therapy can beachieved by increasing the product of agene (such asApoal) that is not itself thecause of the disease is therefore exciting.The apoE-deficient mouse provides agood model on which to test this type of"generic" therapy. Conceivably, therapiesaimed at simultaneously increasing or de-creasing production of several proteinsmight also be used in the future for thetreatment of complex multifactorial disease.
Essential Hypertension
Essential hypertension (elevated bloodpressure not associated with a recogniz-able predisposing condition) like athero-sclerosis has a complex etiology, with ge-netic factors being responsible for abouttwo-thirds of the familial aggregation ofblood pressures and with transmission ofcultural factors being responsible for theremaining one-third (22). Normal bloodpressures are maintained by several inter-acting systems that control cardiac output,peripheral resistance, blood volume, renalfunction, and sodium balance. The renin/angiotensin system is one of these systems,and it is intimately concerned with thecontrol of systemic and intrarenal bloodpressures and sodium retention. Its rele-vance to essential hypertension is exem-plified by the use of inhibitors of elementsof the system to treat hypertension. Cap-topril and other orally active inhibitors ofthe angiotensin-converting enzyme(ACE), which converts the inactive pep-tide angiotensin I to the hypertensive pep-
tide angiotensin II, are among the earliestand most successful. Losartan (DuP753),an orally active inhibitor of type 1 recep-tors for angiotensin II, is one of the mostrecent to undergo clinical trial. Virtuallyall the genes coding for elements in therenin/angiotensin system are conse-quently a priori candidates for involve-ment in hypertension.Recent advances in the mapping of
genes that affect complex phenotypes pro-vide a second reason for selecting therenin/angiotensin system as a source ofcandidate genes. For example, analyses ofcrosses between spontaneously hyperten-sive stroke-prone rats and wild-type Wist-ar-Kyoto rats have shown that a geneticdeterminant of the blood pressure of an-imals fed a high-salt diet segregates with achromosomal region known to include thegene coding forACE (23, 24). Similarly, inhumans, a specific variant (M235T) of thegene coding for angiotensinogen (AGT),which is the protein precursor of angio-tensin I, has been found to segregate withhypertension in a sibling pair study (25).The variant was also associated with mod-estly elevated (120% normal) plasmaAGT levels.
Linkage experiments must, however, beinterpreted with caution. Although theydemonstrate a genetic correlation (link-age) between the phenotype and thescored genetic variant(s), they do not inthemselves prove a causative relationshipbetween the two. In particular, they do notexclude the possibility that the significantfactor is a difference in some other genelinked to the scored variant, which itselfmay be a neutral marker.Gene Titration. Some of the problems
associated with proving causation can beillustrated by describing gene targetingexperiments initially aimed at determin-ing whether the AGT variant M235T cancause hypertension in the mouse. Inspec-tion of the published amino acid se-quences of human AGT and mouse AGTquickly showed that position 235 is in apoorly conserved region of the protein. Asimple equivalent of the human M235T
150
E 100ECu
50oc0o
substitution cannot therefore be made inthe mouse. In its place, we decided to testthe broader hypothesis advanced by Jeune-maitre et al (25)-namely, that the in-creased level of plasma AGT associatedwith the variant allele was the true causeof the hypertension.To test whether variants of the mouse
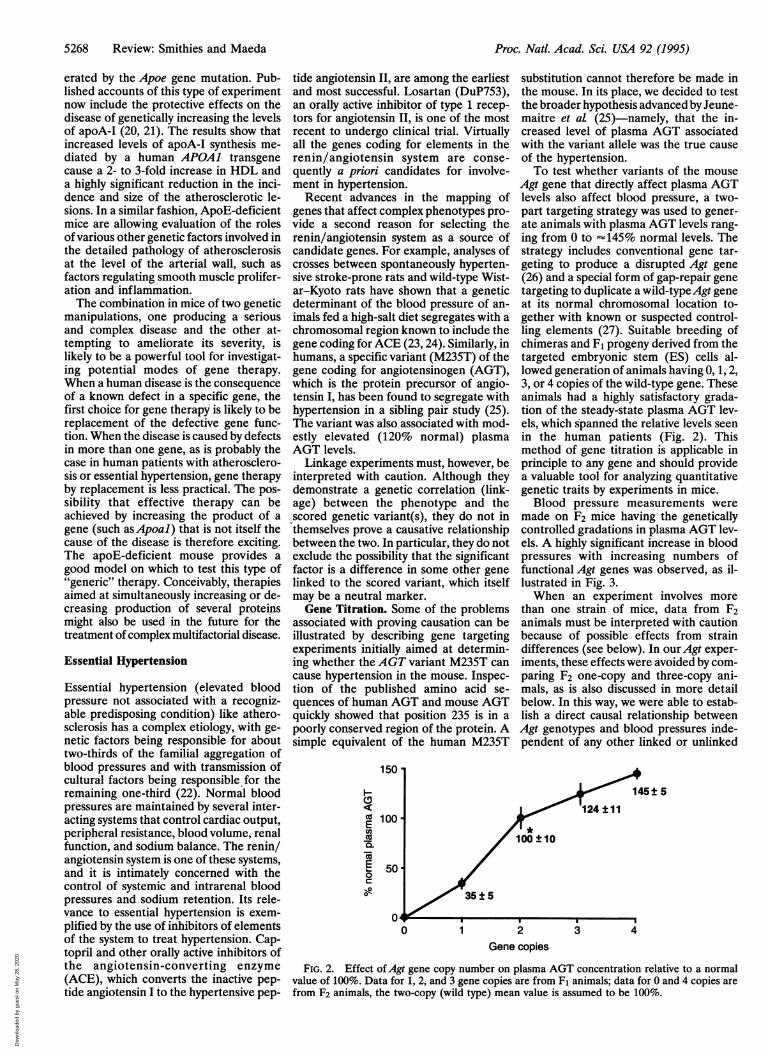
Agt gene that directly affect plasma AGTlevels also affect blood pressure, a two-part targeting strategy was used to gener-ate animals with plasma AGT levels rang-ing from 0 to "145% normal levels. Thestrategy includes conventional gene tar-geting to produce a disrupted Agt gene(26) and a special form of gap-repair genetargeting to duplicate a wild-typeAgt geneat its normal chromosomal location to-gether with known or suspected control-ling elements (27). Suitable breeding ofchimeras and F1 progeny derived from thetargeted embryonic stem (ES) cells al-lowed generation of animals having 0, 1, 2,3, or 4 copies of the wild-type gene. Theseanimals had a highly satisfactory grada-tion of the steady-state plasma AGT lev-els, which spanned the relative levels seenin the human patients (Fig. 2). Thismethod of gene titration is applicable inprinciple to any gene and should providea valuable tool for analyzing quantitativegenetic traits by experiments in mice.Blood pressure measurements were
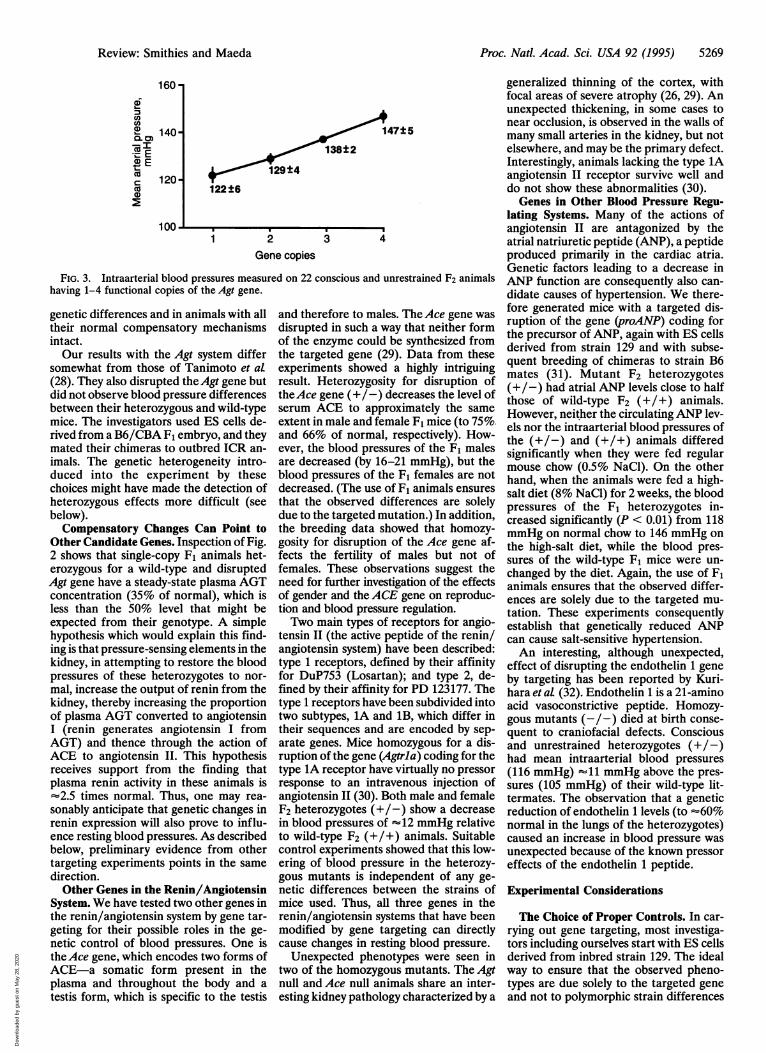
made on F2 mice having the geneticallycontrolled gradations in plasma AGT lev-els. A highly significant increase in bloodpressures with increasing numbers offunctional Agt genes was observed, as il-lustrated in Fig. 3.When an experiment involves more
than one strain of mice, data from F2animals must be interpreted with cautionbecause of possible effects from straindifferences (see below). In ourAgt exper-iments, these effects were avoided by com-paring F2 one-copy and three-copy ani-mals, as is also discussed in more detailbelow. In this way, we were able to estab-lish a direct causal relationship betweenAgt genotypes and blood pressures inde-pendent of any other linked or unlinked
145±5124 +11
0 1 2 3 4Gene copies
FIG. 2. Effect ofAgt gene copy number on plasma AGT concentration relative to a normalvalue of 100%. Data for 1, 2, and 3 gene copies are from F1 animals; data for 0 and 4 copies arefrom F2 animals, the two-copy (wild type) mean value is assumed to be 100%.
Proc. Natl. Acad. Sci. USA 92 (1995)
Dow
nloa
ded
by g
uest
on
May
28,
202
0
Proc. Natl. Acad. Sci. USA 92 (1995) 5269
100I **
147±5
138±2
4129±4
122±6
1 2 3 4
Gene copies
FIG. 3. Intraarterial blood pressures measured on 22 conscious and unrestrained F2 animalshaving 1-4 functional copies of the Agt gene.
genetic differences and in animals with alltheir normal compensatory mechanismsintact.Our results with the Agt system differ
somewhat from those of Tanimoto et at
(28). They also disrupted theAgt gene butdid not observe blood pressure differencesbetween their heterozygous and wild-typemice. The investigators used ES cells de-rived from a B6/CBA F1 embryo, and theymated their chimeras to outbred ICR an-imals. The genetic heterogeneity intro-duced into the experiment by thesechoices might have made the detection ofheterozygous effects more difficult (seebelow).Compensatory Changes Can Point to
Other Candidate Genes. Inspection of Fig.2 shows that single-copy F1 animals het-erozygous for a wild-type and disruptedAgt gene have a steady-state plasma AGTconcentration (35% of normal), which isless than the 50% level that might beexpected from their genotype. A simplehypothesis which would explain this find-ing is that pressure-sensing elements in thekidney, in attempting to restore the bloodpressures of these heterozygotes to nor-mal, increase the output of renin from thekidney, thereby increasing the proportionof plasma AGT converted to angiotensinI (renin generates angiotensin I fromAGT) and thence through the action ofACE to angiotensin II. This hypothesisreceives support from the finding thatplasma renin activity in these animals is-2.5 times normal. Thus, one may rea-sonably anticipate that genetic changes inrenin expression will also prove to influ-ence resting blood pressures. As describedbelow, preliminary evidence from othertargeting experiments points in the samedirection.Other Genes in the Renin/Angiotensin
System. We have tested two other genes inthe renin/angiotensin system by gene tar-geting for their possible roles in the ge-netic control of blood pressures. One istheAce gene, which encodes two forms ofACE-a somatic form present in theplasma and throughout the body and a
testis form, which is specific to the testis
and therefore to males. The Ace gene wasdisrupted in such a way that neither formof the enzyme could be synthesized fromthe targeted gene (29). Data from theseexperiments showed a highly intriguingresult. Heterozygosity for disruption oftheAce gene (+/-) decreases the level ofserum ACE to approximately the sameextent in male and female F1 mice (to 75%.and 66% of normal, respectively). How-ever, the blood pressures of the F1 malesare decreased (by 16-21 mmHg), but theblood pressures of the F1 females are notdecreased. (The use of F1 animals ensuresthat the observed differences are solelydue to the targeted mutation.) In addition,the breeding data showed that homozy-gosity for disruption of the Ace gene af-fects the fertility of males but not offemales. These observations suggest theneed for further investigation of the effectsof gender and the ACE gene on reproduc-tion and blood pressure regulation.Two main types of receptors for angio-
tensin II (the active peptide of the renin/angiotensin system) have been described:type 1 receptors, defined by their affinityfor DuP753 (Losartan); and type 2, de-fined by their affinity for PD 123177. Thetype 1 receptors have been subdivided intotwo subtypes, lA and iB, which differ intheir sequences and are encoded by sep-arate genes. Mice homozygous for a dis-ruption of the gene (Agtrla) coding for thetype lA receptor have virtually no pressorresponse to an intravenous injection ofangiotensin II (30). Both male and femaleF2 heterozygotes (+/-) show a decreasein blood pressures of -12 mmHg relativeto wild-type F2 (+/+) animals. Suitablecontrol experiments showed that this low-ering of blood pressure in the heterozy-gous mutants is independent of any ge-netic differences between the strains ofmice used. Thus, all three genes in therenin/angiotensin systems that have beenmodified by gene targeting can directlycause changes in resting blood pressure.Unexpected phenotypes were seen in
two of the homozygous mutants. The Agtnull and Ace null animals share an inter-esting kidney pathology characterized by a
generalized thinning of the cortex, withfocal areas of severe atrophy (26, 29). Anunexpected thickening, in some cases tonear occlusion, is observed in the walls ofmany small arteries in the kidney, but notelsewhere, and may be the primary defect.Interestingly, animals lacking the type lAangiotensin II receptor survive well anddo not show these abnormalities (30).Genes in Other Blood Pressure Regu-
lating Systems. Many of the actions ofangiotensin II are antagonized by theatrial natriuretic peptide (ANP), a peptideproduced primarily in the cardiac atria.Genetic factors leading to a decrease inANP function are consequently also can-didate causes of hypertension. We there-fore generated mice with a targeted dis-ruption of the gene (proANP) coding forthe precursor of ANP, again with ES cellsderived from strain 129 and with subse-quent breeding of chimeras to strain B6mates (31). Mutant F2 heterozygotes(+/-) had atrial ANP levels close to halfthose of wild-type F2 (+/+) animals.However, neither the circulating ANP lev-els nor the intraarterial blood pressures ofthe (+/-) and (+/+) animals differedsignificantly when they were fed regularmouse chow (0.5% NaCl). On the otherhand, when the animals were fed a high-salt diet (8% NaCl) for 2 weeks, the bloodpressures of the F, heterozygotes in-creased significantly (P < 0.01) from 118mmHg on normal chow to 146 mmHg onthe high-salt diet, while the blood pres-sures of the wild-type F1 mice were un-changed by the diet. Again, the use of F,animals ensures that the observed differ-ences are solely due to the targeted mu-tation. These experiments consequentlyestablish that genetically reduced ANPcan cause salt-sensitive hypertension.An interesting, although unexpected,
effect of disrupting the endothelin 1 geneby targeting has been reported by Kuri-hara et at (32). Endothelin 1 is a 21-aminoacid vasoconstrictive peptide. Homozy-gous mutants (-/-) died at birth conse-quent to craniofacial defects. Consciousand unrestrained heterozygotes (+/-)had mean intraarterial blood pressures(116 mmHg) -11 mmHg above the pres-sures (105 mmHg) of their wild-type lit-termates. The observation that a geneticreduction of endothelin 1 levels (to -60%normal in the lungs of the heterozygotes)caused an increase in blood pressure wasunexpected because of the known pressoreffects of the endothelin 1 peptide.
Experimental Considerations
The Choice of Proper Controls. In car-rying out gene targeting, most investiga-tors including ourselves start with ES cellsderived from inbred strain 129. The idealway to ensure that the observed pheno-types are due solely to the targeted geneand not to polymorphic strain differences
160 -ci~32? 140-0.0),_ E
C 120-CoCu
Review: Smithies and Maeda
Dow
nloa
ded
by g
uest
on
May
28,
202
0

5270 Review: Smithies and Maeda
between the animals under study is tobreed the chimeras to mates of the sameinbred strain 129. We therefore regularlydo this to ensure the future availability ofthe targeted mutation in the inbred strain129 background. Unfortunately, confiningall subsequent breeding to strain 129 isimpractical if many animals are requiredbecause of the poor breeding characteris-tics of the strain and its susceptibility toinfection. These problems point to theneed for easily targetable ES cells frominbred strains that are more fecund andhardy than strain 129.
In practice, as described above, we nor-mally mate the chimeras generated frominbred strain 129-targeted ES cells to an-other inbred strain, typically B6, to obtainfirst generation progeny (F1). (Heterozy-gotes obtained from breeding the chime-ras to strain 129 mates can be used for thispurpose when chimeras cease to be avail-able.) These hybrid F1 animals are vigor-ous and fecund, and the animals of eachsex are genetically identical except for thegene (wild type or targeted) they receivefrom the strain 129-derived ES cell. Com-parisons between wild-type (+ chromo-some from B6/+ chromosome from 129)and heterozygous (+ chromosome fromB6/targeted chromosome from 129) F1animals therefore provide a test of theeffects of targeted modifications on thephenotype of interest completely freefrom any genetic heterogeneity intro-duced by the use of two strains (Fig. 4).
Effect of Genes Unlinked to the Tar-geted Locus. Proceeding to the F2 gener-ation by intercrossing F1 +/- animalsmakes it necessary to consider the effectson the phenotype of differences betweenthe two parental strains at loci other thanthe target locus. Any genes that differbetween the two strains and are not linkedto the target locus will in general segre-gate randomly within F2 animals. If any ofthese unlinked differences affect bloodpressure, for example, they will increasethe spread of blood pressures observed indifferent animals, even though they haveidentical genotypes at the target locus.Differences in unlinked genes conse-quently make it more difficult to detectsubtle effects of the targeted gene, al-though they do not affect the averagemagnitude of the effect.
Experiments with the proANP locus il-lustrate how this spread of the phenotypein F2 animals can be used in an advanta-geous way to identify other genes likely tobe important. Some strains of mice (in-cluding B6) naturally have one copy perhaploid genome of a gene that codes forrenin; other strains (including strain 129)naturally have two renin genes per haploidgenome (33, 34). The renin locus Ren isnot linked to the proANP locus. Typinganimals in the proANP targeting experi-ment for Ren genotypes showed that thoseF2 animals that by chance received both
p
Fl
Proc. Natl. Acad. Sci. USA 92 (1995)
Strain 2 Strain I(e.g. B6 mate) (e.g.129 chimera)
Wild Type A Heterozygote B
18180AxB
F2
C D E C D F G C F H
F2 Wildtypes: C, D, E Heterozygous mutants: F, G Homozygous mutant: H
FIG. 4. Cosegregation of strain-specific chromosomal regions and the target locus influencesexperimental design and the choice of controls when two inbred strains are used in the breeding.Comparison of F1 animals, wild-type A and mutant heterozygote B, allows assessment of theeffects of the targeted mutation free from biases due to any genetic differences between the twostrains. Note that there are three classes of F2 wild-type animals (C, D, and E) and two classesof F2 heterozygotes (F and G), which can all be distinguished in ways that are discussed in thetext. Comparison of the three classes of F2 wild-type animals allows assessment of the effects onthe trait of interest of any naturally occurring strain differences at the target locus or in any geneslinked to it. Wild-type animal D is an excellent control for the mutant heterozygote F, as iswild-type animal E for the mutant heterozygote G and for the mutant homozygote H. Effects ofstrain differences in linked genes could be avoided in future generations if only class E, G, andH animals are mated. P, parental animals: usually a chimera derived from strain 1-targeted EScells (typically strain 129) and a strain 2 mate (typically B6). F1, first generation offspring. F2, threesets of second generation offspring that can be derived from intercrossing F1 animals. Strain1-derived chromosomes carrying the targeted (-) or the wild-type (+) gene at the target locusare shown in white. Homologous chromosomes from the second strain are shown in black; all carrya wild-type gene (+) at the target locus. F2 progeny chromosomes are shown without centromeresand telomeres to signify that crossovers reduce the extent of cosegregation to linked regions ratherthan whole chromosomes.
their Ren loci from strain 129 (and there-fore have four copies of a renin gene) havehigher blood pressures when fed 2% saltthan animals of the same proANP geno-type that received three copies. These inturn have pressures higher than the two-copy animals that received both Ren locifrom B6 (John H. Krege and Simon W. M.John, personal communication). Thesepreliminary experiments do not eliminatethe possibility that in this case the spreadsin phenotype are not in part or completelydue to strain differences in genes linked tothe Ren locus, but they do show how theeffects of segregation of a second chro-mosomal region unlinked to the targetlocus can be detected and how it caninfluence the phenotype resulting fromthe planned change at the target locus.
Effect of Genes Linked to the TargetLocus. Polymorphic differences in geneslinked to the target locus can cause moreserious complications. The frequency withwhich related genes are linked to targetloci is much greater than would be ex-pected by chance because related genesoften occur as linked clusters in the mam-malian genome. For example, threemouse apolipoprotein genes-Apoal,
Apoc3, andApoa4-are all within a 20-kbstretch of DNA (unpublished data). Sim-ilarly, two mouse natriuretic peptide pre-cursor genes-proANP and proBNP-arewithin 20 kb (Mark E. Steinhelper, per-sonal communication).Polymorphism in a related linked gene
is unimportant if the polymorphism has noeffect on the phenotype of interest. Butthis will no longer be true if the linkedpolymorphic allele affects the phenotype.Imagine, for example, that strain 129 hasa linked polymorphic allele that affects thephenotype under study positively whencompared to the corresponding allele instrain B6. The effects of any negativemutations at the target locus will in thiscase be underestimated (or even reversed)in a conventional comparison of F2 prog-eny derived from intercrossing F1 het-erozygotes. This is because all the F2progeny that are homozygous mutant(-/-) at the target locus will also behomozygous for the strain 129-derived(more positive) allele at the linked locus(Fig. 4). Likewise all the F2 progeny fromthis intercross that are wild type (+/+) atthe target locus will also be homozygousfor the B6-derived (less positive) allele at
Dow
nloa
ded
by g
uest
on
May
28,
202
0

Proc. Natl. Acad. Sci. USA 92 (1995) 5271
the linked locus. Thus, polymorphisms atlinked phenotype-related loci can alter theapparent magnitude of the effects of thetargeting if only conventional F2 +/+,+/-, and -/- progeny derived fromintercrossing F1 +/- animals are com-pared. Conceivably, they could even re-verse its sign, or give an apparent effectfrom the targeting when none is there.Linked genes will not, however, changethe spread of values within a specific tar-get locus genotype.
In gene knockout experiments in whichthe effects of absence of the target geneproduct are extreme and overriding, com-plications from linked strain differencesare usually of little concern (as in the caseof Apoe -/- mice). However, when thephenotype is a quantitative variable andthe effects may be subtle, rigorous con-trols in which the targeted mutation is thesole variable become important. Fig. 4illustrates some of the complexities intro-duced by the use of two strains, even whenboth are inbred. For example, the figureillustrates that there are three classes (C,D, and E) rather than one class of F2wild-type animals. All are +/+ at thetarget locus, but they differ in the sourcesof their wild-type alleles. Likewise, thereare two classes (F and G) of mutant F2heterozygote (+/-). Choice of rigorouscontrols requires careful consideration ofthese classes, as indicated in the legend toFig. 4. The different classes of F2 animalscan readily be distinguished if a naturalpolymorphism can be found between thetwo strains at the target locus. Otherwise,it may be necessary to use simple sequencelength polymorphisms linked to the targetlocus in order to identify them-a strategythat was used in a related situation todistinguish three-copy and four-copy F2animals in our Agt gene experiments (26).There are other ways of avoiding the
effects of strain differences in genes linkedto the target locus. For example, in the genetitration experiment carried out at the Agtlocus, we compared the one-copy and three-copy animals (26). The one-copy animalsand the three-copy animals both have awild-type Agt allele and nearby DNA de-rived from strain B6, and both have a tar-geted Agt allele and nearby DNA derivedfrom strain 129; they consequently differsystematically only at the targeted locus.
This technical discussion is intended toemphasize that, even in a complex geneticsystem, gene targeting experiments canwith careful attention to details be de-signed to prove that a specific phenotypeis caused by the targeted change and notby any other differences between the par-ticipating strains. This is the elusive proofof causation referred to previously. Oncethis proof has been secured, it may not benecessary to continue to preserve the ge-netic "purity" of a mutant strain. Alter-natively, by backcrossing to an inbredstrain (such as B6) the genetic purity of a
targeted mutant can be recovered. Theresulting mutant strain will be inbred B6except for being homozygous for a smallregion derived from the ES cell strain thatincludes and surrounds the target locus.The complications associated with poly-morphisms in genes closely linked to thetarget loci therefore still remain, unlessbackcrossing is also carried out to secureanimals having the ES cell-derived wild-type gene at the target locus, but thecomplications associated with polymor-phisms elsewhere in the genome are elim-inated. Consequently, when the effects ofcombining targeted mutations at severalloci are to be investigated, animals back-crossed to B6 (or any other chosen inbredstrain) are very valuable.
Conclusions
How are gene targeting experiments inmice helping to develop a better under-standing of the genetic etiologies of ath-erosclerosis and essential hypertension,and what can be expected from them inthe future?At present, the most significant contri-
bution toward this understanding for ath-erosclerosis is probably the generation ofprecisely defined mouse models of thedisease: mice with disruption in the genescoding for apoE or the LDL receptor.These models are allowing assessment ofthe effects of other genes on atherogenesisby using the powerful genetic tools avail-able in the mouse. Carefully controlledinvestigations of the effects of diet or ofdrugs are also facilitated by these animals.At present, the most significant contribu-
tion to understanding the genetic etiology ofessential hypertension is probably the dem-onstration that discrete alterations in theexpression of a variety of different genes canindividually cause changes in the blood pres-sures of mice, even when the mice have alltheir compensatory mechanisms intact.These effects are readily detected in animalshaving moderate decreases in gene functiondue to heterozygosity for gene disruptionsor modest increases due to gene duplica-tions.
Considerable work still remains to bedone in dissecting out in a rigorous man-ner the effects of alterations in singlegenes on the induction or progression ofatherosclerosis and on the control ofblood pressures. Perhaps even more excit-ing is the opportunity now becoming avail-able to breed animals in which the effectsof precise differences in more than onegene can be studied in combination. Itwould be overly optimistic to expect thatall data from the mouse will directly applyto humans. Some species differences areto be expected, but it will be important notto dismiss unexpected findings too easily.Indeed, determining the basis of the un-expected findings with the help of mousegenetics may provide important insights
into the causes and treatment of thesecomplex multifactorial diseases.
We thank Tom Coffman, Beverly Koller,Kathy Sulik, and the members of our laboratoryfor their help in preparing this review. Ourworkis supported by grants from the National Insti-tutes of Health (GM20069, HL42630, andHL49277).
1. Ross, R. (1993) Nature (London) 362,801-809.
2. Gordon, T., Castelli, W. P., Hjortland,M. C., Kannel, W. B. & Dawber, T. R.(1977) Am. J. Med. 62, 707-714.
3. Paigen, B., Morrow, A., Brandon, C.,Mitchell, D. & Holmes, P. (1985) Athero-sclerosis 57, 65-73.
4. Zhang, S. H., Reddick, R. L., Piedrahita,J. A. & Maeda, N. (1992) Science 258,468-471.
5. Plump, A. S., Smith, J. D., Hayek, T.,Aalto-Setala, K., Walsh, A., Verstuyft,J. G., Rubin, E. M. & Breslow, J. L. (1992)Cell 71, 343-353.
6. Reddick, R. L., Zhang, S. H. & Maeda, N.(1994) Arterioscler. Thromb. 14, 141-147.
7. Nakashima, Y., Plump, A. S., Raines,E. W., Breslow, J. L. & Ross, R. (1994)Arterioscler. Thromb. 14, 133-140.
8. Ghiselli, G. E., Schaefer, E. J., Gascon, P.& Brewer, H. B. (1981) Science 214,1239-1241.
9. Mabuchi, H., Itoh, H., Takeda, M., Kaji-nami, K., Wakasugi, T., Koizumi, J.,Takeda, R. & Asagami, C. (1989) Metab-olism 38, 115-119.
10. Kurosaka, D., Teramoto, T., Matsushima,T., Yokoyama, T., Yamada, A., Aikawa,T., Miyamoto, Y. & Kurokawa, K. (1991)Atherosclerosis 88, 15-20.
11. Zhang, S. H., Reddick, R. L., Burkey, B.& Maeda, N. (1994) J. Clin. Invest. 94,937-945.
12. van Ree, J. H., van den Broek, W. J. A. A.,Dahlmans, V. E. H., Groot, P. H. E., Vid-geon-Hart, M., Frants, R. R., Wieringa,B., Havekes, L. M. & Hofker, M. H.(1994) Atherosclerosis 111, 25-37.
13. Rapacz, J., Hasler-Rapacz, J., Taylor,K. M., Checovich, W. J. & Attie, A. D.(1986) Science 234, 1573-1577.
14. Buja, L. M., Kita, T., Goldstein, J. L., Wa-tanabe, Y. & Brown, M. S. (1983)Arterio-sclerosis 3, 87-101.
15. Ishibashi, S., Brown, M. S., Goldstein,J. L., Gerard, R. D., Hammer, R. E. &Herz, J. (1993)J. Clin. Invest. 92, 883-893.
16. Ishibashi, S., Goldstein, J. L., Brown,M. S., Herz, J. & Burns, D. K. 91994) J.Clin. Invest. 93, 1885-1893.
17. Brown, M. S. & Goldstein, J. L. (1986)Science 232, 34-47.
18. Li, H., Reddick, R. L. & Maeda, N. (1993)Arterioscler. Thromb. 13, 1814-1821.
19. Assmann, G., von Eckardstein, A. &Funke, H. (1993) Circulation 87, Suppi.III, 28-34.
20. Paszty, C., Maeda, N., Verstuyft, J. & Ru-bin, E. M. (1994)J. Clin. Invest. 94,899-903.
21. Plump, A. S., Scott, C. J. & Breslow, J. L.(1994) Proc. Natl. Acad. Sci. USA 91,9607-9611.
22. Ward, R. (1990) in Hypertension: Patho-physiology, Diagnosis, and Management,ed. Laragh, J. H. & Brenner, B. M.(Raven, New York), pp. 81-100.
Review: Smithies and Maeda
Dow
nloa
ded
by g
uest
on
May
28,
202
0

5272 Review: Smithies and Maeda Proc. Natl. Acad. Sci. USA 92 (1995)
23. Hilbert, P., Lindpaintner, K., Beckmann,J. S., Serikawa, T., Soubrier, F., Dubay, C.,Cartwright, P., De Gouyon, B., Julier, C.,Takahasi, S., Vincent, M., Ganten, D.,Georges, M. & Lathrop, G. M. (1991)Nature (London) 353, 521-529.
24. Jacob, H. J., Lindpaintner, K, Lincoln,S. E., Kusumi, K., Bunker, R. K., Mao,Y.-P., Ganten, D., Dzau, V. J. & Lander,E. S. (1991) Cell 67, 213-224.
25. Jeunemaitre, X., Soubrier, F., Kotelevt-sev, Y. V., Lifton, R. P., Williams, C. S.,Charru, A., Hunt, S. C., Hopkins, P. N.,Williams, R. R., Lalouel, J.-M. & Corvol,P. (1992) Cell 71, 169-180.
26. Kim, H.-S., Krege, J. H., Kluckman,K. D., Hagaman, J. H., Hodgin, J. B.,
Best, C. F., Jennette, J. C., Coffman,T. M., Maeda, N. & Smithies, 0. (1995)Proc. Natl. Acad. Sci. USA 92, 2735-2739.
27. Smithies, 0. & Kim, H.-S. (1994) Proc.Natl. Acad. Sci. USA 91, 3612-3615.
28. Tanimoto, K., Sugiyama, F., Goto, Y.,Ishida, J., Takimoto, E., Yagami, K,Fukamizu, A. & Murakami, K. (1994) J.Biol. Chem. 269, 31334-31337.
29. Krege, J. H., John, S. W. M., Langenbach,L. L., Hodgin, J. B., Hagaman, J. R.,Bachman, E. S., Jennette, J. C., O'Brien,D. A. & Smithies, 0. (1995) Nature (Lon-don), in press.
30. Ito, M., Oliverio, M. I., Mannon, P. J.,Best, C. F., Maeda, N., Smithies, 0. &
Coffman, T. M. (1995) Proc. Natl. Acad.Sci. USA 92, 3521-3525.
31. John, S. W. M., Krege, J. H., Oliver, P.,Hagaman, J. R., Hodgin, J. B., Pang, S. C.,Flynn, T. G. & Smithies, 0. (1995) Science267, 679-681.
32. Kurihara, Y., Kurihara, H., Suzuki, H., Ko-dama, T., Maemura, K, Nagai, R., Oda, H.,Kuwaki, T., Cao, W.-H., Kamada, N.,Jishage, K, Ouchi, Y., Azuma, S., Toyoda,Y., Ishikawa, T., Kumada, M. & Yazaki, Y.(1994) Nature (London) 368, 703-710.
33. Piccini, N., Knopf, J. L. & Gross, K. W.(1982) Cell 30, 205-213.
34. Panthier, J.-J., Holm, I. & Rougeon, F.(1982) EMBO J. 1, 1417-1421.
Dow
nloa
ded
by g
uest
on
May
28,
202
0





























