Embed Size (px)
Citation preview

A RT I C L E S
NATURE IMMUNOLOGY VOLUME 4 NUMBER 11 NOVEMBER 2003 1093
Differentiation and clonal expansion of TH precursor cells into T effec-tor populations is important in the adaptive immune response andprovides protection against intracellular viruses and pathogenic bacte-ria. However, unrestrained activation of TH effector cells also underliesmany inflammatory disorders. TH1 effector cells are associated withthe pathogenesis of rheumatoid arthritis, inflammatory bowel diseaseand other autoimmune disorders, including type I diabetes and multi-ple sclerosis1,2, as well as allograft rejection3,4. In contrast, TH2 cellactivation is essential in the pathogenesis of allergic asthma5 and hasbeen linked to the acquisition of transplant tolerance3,4. The extent ofT cell activation and the mode of differentiation is determined mainlyby the duration and strength of T cell receptor (TCR)-mediated stimu-lation6. In addition, many costimulatory and accessory molecules,including tumor necrosis factor (TNF) receptor7 and immunoglobu-lin (Ig) superfamily members8, as well as cytokines such as interleukin2 (IL-2), regulate the extent of clonal expansion, deletion and/oranergy induction9. However, whereas many of the cellular and molec-ular mechanisms that regulate naive T cell activation are understood,the molecules that determine the fate of effector T cell subpopulationsremain to be elucidated.
The immunoglobulin superfamily member Tim-3 is a transmem-brane protein preferentially expressed on differentiated TH1 cells10. Ina model of experimental allergic encephalomyelitis, a TH1-mediated
autoimmune disease, in vivo administration of Tim-3 monoclonalantibodies (mAbs) led to more severe inflammatory events within thebrain and more severe clinical disease. Based on these observations,Tim-3 was proposed as a negative regulator of tissue-destructiveimmune responses in experimental allergic encephalomyelitis10.However, it remains uncertain whether these data reflected inhibitionof a negative signal provided by Tim-3 or, conversely, whether Tim-3crosslinking in vivo with the mAb exerted a positive signal to induce Tcell activation and disease exacerbation. Based on results reportedhere, we conclude that Tim-3 engagement by its putative ligand pro-vides an inhibitory signal to dampen inflammatory responses in vivo.Tim-3 pathway blockade through treatment with a Tim-3–Ig fusionprotein accelerated diabetes onset in nonobese diabetic (NOD) miceand abolished the capacity of costimulatory blockade, with either acytotoxic T lymphocyte antigen 4 chimeric fusion protein (CTLA4-Ig)or combined treatment with donor-specific transfusion (DST) plusantibody to CD154 (anti-CD154; also known as CD40 ligand, orCD40L)11–13, to induce tolerance to major histocompatibility complex(MHC)-mismatched allografts. Although the precise mechanismsinvolved remain to be fully elucidated, we propose that Tim-3 regu-lates the outcome of auto- and alloimmune responses at least in partby modulating the capacity of regulatory T cells to dampen inflamma-tory responses.
1Division of Immunology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, Massachusetts 02215, USA. 2Inflammation Department,Experimental Medicine Division, Millennium Pharmaceuticals, Cambridge, Massachusetts 02116, USA. 3Brigham and Women’s Hospital, Harvard Medical School,Boston, Massachusetts 02215, USA. 4Department of Haematology, Division of Investigative Sciences, Faculty of Medicine, Imperial College London, HammersmithHospital, Du Cane Road, London W12 0NN, UK. 5Deutsches Rheumaforschungs Zentrum, Berlin D-10117, Germany. Correspondence should be addressed to A.J.C.([email protected]) or T.B.S. ([email protected]).
Published online 12 October 2003; doi:10.1038/ni987
Tim-3 inhibits T helper type 1–mediated auto- and alloimmune responses and promotesimmunological toleranceAlberto Sánchez-Fueyo1, Jane Tian2, Dominic Picarella2, Christoph Domenig1, Xin Xiao Zheng1, Catherine A Sabatos3, Natasha Manlongat4, Orissa Bender5, Thomas Kamradt5, Vijay K Kuchroo3, José-Carlos Gutiérrez-Ramos2, Anthony J Coyle2 & Terry B Strom1
Although T helper (TH) cell–mediated immunity is required to effectively eliminate pathogens, unrestrained TH activity alsocontributes to tissue injury in many inflammatory and autoimmune diseases. We report here that the TH type 1 (TH1)-specificTim-3 (T cell immunoglobulin domain, mucin domain) protein functions to inhibit aggressive TH1-mediated auto- andalloimmune responses. Tim-3 pathway blockade accelerated diabetes in nonobese diabetic mice and prevented acquisition oftransplantation tolerance induced by costimulation blockade. These effects were mediated, at least in part, by dampening of theantigen-specific immunosuppressive function of CD4+CD25+ regulatory T cell populations. Our data indicate that the Tim-3pathway provides an important mechanism to down-regulate TH1-dependent immune responses and to facilitate the developmentof immunological tolerance.
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
reim
mu
no
log
y

A RT I C L E S
1094 VOLUME 4 NUMBER 11 NOVEMBER 2003 NATURE IMMUNOLOGY
RESULTSTim-3 is preferentially expressed on TH1 effector cellsTo identify genes that may specifically regulate TH effector function,we generated a TH1-versus-TH2 subtractive library from activatedmouse TH1 and TH2 clones, as described14. The full-length clone ofone of the cDNAs represented in the TH1 but not in the TH2 libraryencoded the type 1 membrane protein Tim-3 (ref. 10). We confirmedthe identification of Tim-3 with RNA blot analysis and staining of acti-vated TH1 but not TH2 cells with a Tim-3 specific mAb (data notshown). We first determined the kinetics of Tim-3 expression in vitrowith antigen-specific TCR transgenic CD4+ T cells undergoing activa-tion and polarization into either TH1 or TH2 effector pathways. Tim-3was not expressed during the initial response to antigen, but after res-timulation approximately 25% of CD4+ T cells expressed Tim-3,whether IL-12 plus anti-IL-4 or IL-4 plus anti-IL-12 were used to driveTH1 or TH2 effector cell differentiation, respectively (Fig. 1a).However, repetitive restimulation of these subpopulations with anti-gen-pulsed antigen-presenting cells (APCs) in the presence of IL-12induced most TH1 cells to express Tim-3. In contrast, T cells restimu-lated in the presence of IL-4 had only low expression of Tim-3 after thefourth round of antigenic stimulation (Fig. 1a).
We next studied Tim-3 expression in vivo by means of a graft-versus-host disease–like model in which C57BL/6 (H-2b) lymphocyteslabeled with 5-carboxyfluorescein diacetate succinimidyl ester weretransferred into irradiated allogeneic DBA/2 (H-2d) hosts15. AlthoughTim-3 was not readily detected on resting T cells (data not shown), onday 3 after transfer 30–35% of CD4+ and CD8+ T cells expressed thiscell surface protein (data not shown). The induction of Tim-3 expres-sion on alloactivated T cells increased with each round of prolifera-tion, reaching a plateau at the sixth to the seventh cell cycle division(35–40% of CD4+ or CD8+ T cells positive for Tim-3; Fig. 1b). Theup-regulation of Tim-3 expression in vivo at the sixth to the seventhcell division was closely associated with the production of interferon-γ(IFN-γ) by both CD4+ and CD8+ T cells, as assessed by intracellularcytokine staining (data not shown). Hence, expression of Tim-3 andthe TH1 phenotype are closely linked.
CD4+ T cells express a putative Tim-3 ligandThe structure of Tim-3 is somewhat reminiscent of that of mucosaladdressin cell adhesion molecule-1, which contains two immunoglobu-lin domains and a mucin-rich region10. To assess the distribution ofputative Tim-3 ligands (Tim-3Ls), we constructed human IgG1-derived
Tim
-3-p
ositi
ve c
ells
(%
)C
ell c
ount
s
Tim
-3-p
ositi
ve c
ells
(%
)
Tim
-3
CD8+ T cells
CD4+ T cells CD4+ T cells
CD8+ T cells
CD4+ CD8+ B220+ CD11c+ CD11b+
CD4+CD25– CD4+CD25+
Resting conditions
24 h in vitroactivation
48 h in vitro activation
a
c
b
d
TH1
TH2
Cel
l cou
nts
PE
PE
Figure 1 Expression of Tim-3 and Tim-3L. (a) Tim-3 mAb 8H7 preferentiallybinds to activated TH1, but not TH2, clones after repeated restimulation (RS;below horizontal axis) in vitro. 1° Stim, primary stimulation. (b) Tim-3 isexpressed in vivo by both CD4+ and CD8+ proliferating C57BL/6 T cells 3 dafter adoptive transfer into DBA/2 allogeneic, irradiated recipients. CFSE, 5-carboxyfluorescein diacetate succinimidyl ester. (c) Both flTim-3–Ig andthe truncated sTim-3–Ig fusion protein bind to resting CD4+ T cells and a portion of CD11c+ but not CD11b+, B220+ or CD8+ T cells. Filledhistograms, isotype control staining; open histograms, binding with flTim-3-Fc (dotted lines) and sTim-3–Ig (solid lines). (d) CD4+CD25– but not regulatory CD4+C25+ T cells down-regulate Tim-3L after in vitrostimulation. Both CD4+CD25– and CD4+CD25+ T cells bind sTim-3–Ig inresting conditions (top). Although there are no changes in Tim-3L expressionafter 24 h of in vitro stimulation with anti-CD3, anti-CD28 and rIL-2(middle), at 48 h Tim-3L is detected only on CD4+CD25+ T cells (bottom).Filled histograms, isotype control staining; open histograms, binding with sTim-3–Ig. Data are representative of three independent experiments.
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
reim
mu
no
log
y

A RT I C L E S
NATURE IMMUNOLOGY VOLUME 4 NUMBER 11 NOVEMBER 2003 1095
Fc fusion proteins incorporating the extracellular domain of mouseTim-3 (full-length; flTim-3–Ig), or a truncated, soluble, immunoglobu-lin domain only (sTim-3–Ig)16. Both flTim-3–Ig and sTim-3–Ig boundto resting CD4+ T cells, but not CD8+, B220+ or CD11b+ cells (Fig. 1c),with sTim-3–Ig binding more intensely. We also noted some binding onsplenic CD11c+ dendritic cells (Fig. 1c), but the pattern of staining wasfar less consistent than that of CD4+ T cells. Data reported in an accom-panying paper16 indicate that terminally differentiated resting, but notactivated, TH1 and TH2 CD4+ T cell clones bind Tim-3–Ig fusion pro-teins. Hence the putative Tim-3L seems to be expressed mainly on CD4+
T cells. TH1-TH2 polarization does not affect Tim-3L expression.However, in vitro activation of these clones induced Tim-3L down-regulation. Given the amplified staining pattern seen with sTim-3–Ig,compared with that of flTim-3–Ig, the immunoglobulin domain ofTim-3 seems to be responsible for interactions with Tim-3L.
CD4+CD25+ T cells maintain Tim-3L after activationIn resting conditions, Tim-3-related fusion proteins bound similarly toboth effector CD4+CD25– and regulatory CD4+CD25+ T cell subpopu-lations (Fig. 1d, top). To identify potential mechanisms through whichTim-3 might selectively target the function of either regulatory or effec-tor CD4+ T cells, we studied the kinetics of Tim-3L expression, assessedby the binding of sTim-3–Ig, on CD4+CD25– and CD4+CD25+ T cellsafter activation in vitro. Stimulation with anti-CD3 and anti-CD28 didnot alter Tim-3L expression throughout the first 24 h of culture (Fig. 1d,middle). In contrast, Tim-3L expression was down-regulated on acti-vated CD4+CD25– T cells at 48 h of culture, whereas Tim-3L expressionpersisted on CD4+CD25+ regulatory T cells (Fig. 1d, bottom).
We also found down-regulation of Tim-3L expression onCD4+CD25– T cells after in vitro activation with concanavalin A orlipopolysaccharide (data not shown). The preferential expression ofthe putative Tim-3L on activated regulatory T cells was mirrored byTim-3 expression on activated TH1 cells. Hence, we investigated thehypothesis that Tim-3–Tim-3L interactions are crucial to the acquisi-tion and/or maintenance of immunoregulation and tolerance in TH1-mediated immune responses.
Tim-3 blockade accelerates diabetes in NOD miceTo address the function of Tim-3 in TH1-dependent autoimmuneresponses, we studied the effects of administration of anti-Tim-3,flTim-3–Ig or control immunoglobulin to NOD mice. These micespontaneously develop insulin-dependent diabetes as a result of selec-tive T cell–dependent destruction of the insulin-producing β-cells,
and the cytokine profile of islet-infiltrating lymphocytes is consistentwith a TH1-type inflammatory response17. In our test model, diabeteswas induced in NOD–severe combined immunodeficient (SCID)recipient mice through the adoptive transfer of T cell–enrichedsplenocyte populations from overtly diabetic donor NOD mice18.Treatment of recipient mice with a mAb specific for Tim-3 hastenedthe onset of diabetes, as almost 80% of the mice treated with anti-Tim-3 were diabetic by week 6, versus 0% in the group treated with controlantibody (Fig. 2a). We obtained similar results when we blocked Tim-3–Tim-3L pathway by administering flTim-3–Ig (Fig. 2b). Becausetargeting the Tim-3 pathway with either a mAb or with the flTim-3–Igprotein accelerated autoimmunity, it is likely that Tim-3 triggered anegative signal on TH1 cells and/or amplified immunoregulatorypathways that dampen autoimmune responses. The magnitude of thelymphocytic infiltrate surrounding and/or invading islets was similarin both treated and control mice, indicating that in this model Tim-3blockade did not increase proliferation and/or migration of islet-spe-cific TH1 cells, but instead enhanced their capacity to mediate tissueinjury (Fig. 2c,d).
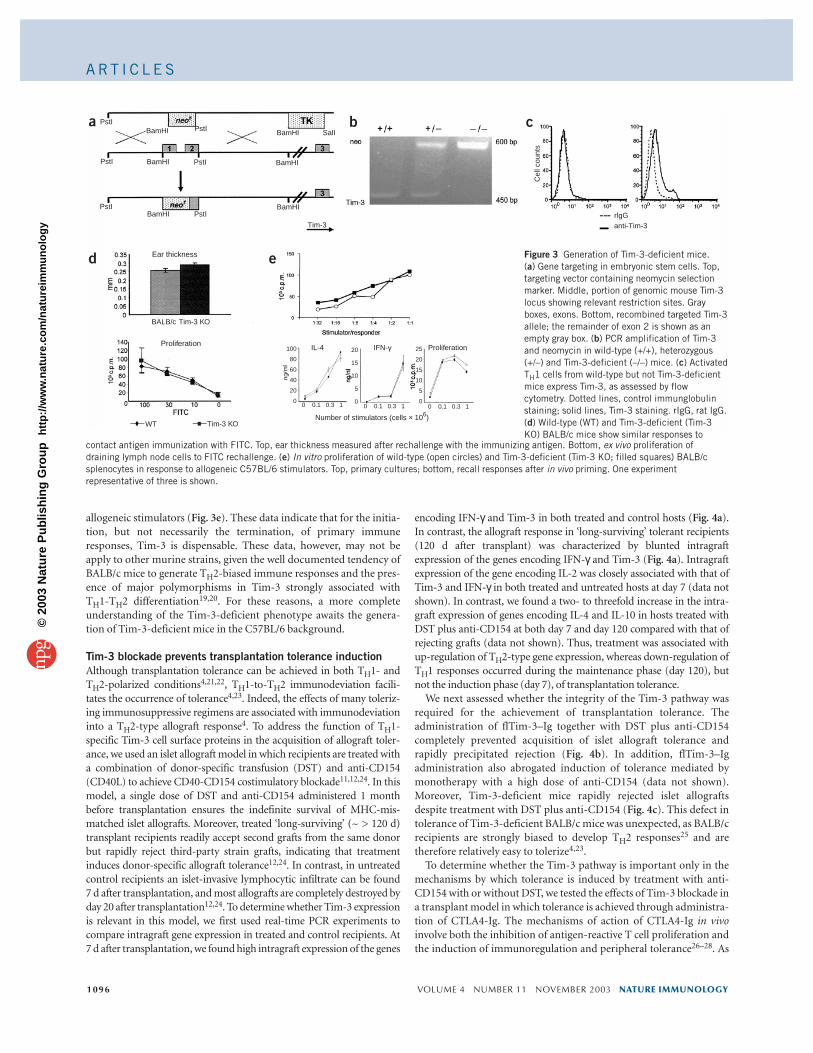
Generation of Tim-3-deficient miceTo further address the function of Tim-3 in the negative regulation ofimmune responses, we generated Tim-3-deficient mice by targeteddeletion of the first and part of the second exon of the gene encodingTim-3 (official gene name, Havcr2; Fig. 3a,b). In CD4+ T cells fromTim-3-deficient mice, we did not find Tim-3 expression by flowcytometry with three rounds of anti-CD3 and anti-CD28 stimulationin TH1-polarizing conditions (IL-12 plus anti-IL-4; Fig. 3c). We firstcreated chimeric mice from Tim-3-deficient embryonic stem cellclones, and then crossed these mice with BALB/c females for six toeight generations. Tim-3-deficient BALB/c mice were viable andshowed normal thymic development, with numbers and ratios ofCD4+ and CD8+ T cells in the periphery that were not different fromthose of wild-type mice. There was no evidence of autoimmune orlymphoproliferative traits (data not shown). Serum antibody concen-trations were similar in deficient and wild-type mice after immuniza-tion with a thymic-dependent antigen, indicating that T cell helperfunction was not impaired (data not shown). Contact hypersensitivityafter immunization with fluorescein isothiocyanate (FITC) was unim-paired in Tim-3-deficient mice (Fig. 3d). Finally, although in primarymixed-lymphocyte reactions Tim-3-deficient lymphocytes showedslightly increased basal proliferation, we found no substantial differ-ences between the response of wild-type and Tim-3-deficient cells to
Figure 2 Treatment with both Tim-3 mAb and flTim-3–Ig augments autoimmune diabetes in an adoptive transfer NOD model. NOD-SCID recipient micereceived T cell–enriched splenocyte populations from spontaneously diabetic NOD mice and were subsequently treated with Tim-3 mAb (a), flTim-3–Ig fusion protein (b) or isotype-matched mAbs. Both Tim-3 mAb and flTim-3–Ig treatments accelerate the occurrence of diabetes in the adoptively transferredrecipients compared with that of mice treated with control immunoglobulin. (c) Pancreatic islet sections from adoptively transferred NOD-SCID mice(paraffin sections stained with hematoxylin and eosin). (d) Serial histological examination of insulitis in adoptively transferred NOD-SCID mice.
a b c d
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
reim
mu
no
log
y
A RT I C L E S
1096 VOLUME 4 NUMBER 11 NOVEMBER 2003 NATURE IMMUNOLOGY
allogeneic stimulators (Fig. 3e). These data indicate that for the initia-tion, but not necessarily the termination, of primary immuneresponses, Tim-3 is dispensable. These data, however, may not beapply to other murine strains, given the well documented tendency ofBALB/c mice to generate TH2-biased immune responses and the pres-ence of major polymorphisms in Tim-3 strongly associated with TH1-TH2 differentiation19,20. For these reasons, a more completeunderstanding of the Tim-3-deficient phenotype awaits the genera-tion of Tim-3-deficient mice in the C57BL/6 background.
Tim-3 blockade prevents transplantation tolerance inductionAlthough transplantation tolerance can be achieved in both TH1- andTH2-polarized conditions4,21,22, TH1-to-TH2 immunodeviation facili-tates the occurrence of tolerance4,23. Indeed, the effects of many toleriz-ing immunosuppressive regimens are associated with immunodeviationinto a TH2-type allograft response4. To address the function of TH1-specific Tim-3 cell surface proteins in the acquisition of allograft toler-ance, we used an islet allograft model in which recipients are treated witha combination of donor-specific transfusion (DST) and anti-CD154(CD40L) to achieve CD40-CD154 costimulatory blockade11,12,24. In thismodel, a single dose of DST and anti-CD154 administered 1 monthbefore transplantation ensures the indefinite survival of MHC-mis-matched islet allografts. Moreover, treated ‘long-surviving’ (~ > 120 d)transplant recipients readily accept second grafts from the same donorbut rapidly reject third-party strain grafts, indicating that treatmentinduces donor-specific allograft tolerance12,24. In contrast, in untreatedcontrol recipients an islet-invasive lymphocytic infiltrate can be found 7 d after transplantation, and most allografts are completely destroyed byday 20 after transplantation12,24. To determine whether Tim-3 expressionis relevant in this model, we first used real-time PCR experiments tocompare intragraft gene expression in treated and control recipients. At 7 d after transplantation, we found high intragraft expression of the genes
encoding IFN-γ and Tim-3 in both treated and control hosts (Fig. 4a). In contrast, the allograft response in ‘long-surviving’ tolerant recipients(120 d after transplant) was characterized by blunted intragraft expression of the genes encoding IFN-γ and Tim-3 (Fig. 4a). Intragraftexpression of the gene encoding IL-2 was closely associated with that ofTim-3 and IFN-γ in both treated and untreated hosts at day 7 (data notshown). In contrast, we found a two- to threefold increase in the intra-graft expression of genes encoding IL-4 and IL-10 in hosts treated withDST plus anti-CD154 at both day 7 and day 120 compared with that ofrejecting grafts (data not shown). Thus, treatment was associated withup-regulation of TH2-type gene expression, whereas down-regulation ofTH1 responses occurred during the maintenance phase (day 120), butnot the induction phase (day 7), of transplantation tolerance.
We next assessed whether the integrity of the Tim-3 pathway wasrequired for the achievement of transplantation tolerance. Theadministration of flTim-3–Ig together with DST plus anti-CD154completely prevented acquisition of islet allograft tolerance andrapidly precipitated rejection (Fig. 4b). In addition, flTim-3–Igadministration also abrogated induction of tolerance mediated bymonotherapy with a high dose of anti-CD154 (data not shown).Moreover, Tim-3-deficient mice rapidly rejected islet allograftsdespite treatment with DST plus anti-CD154 (Fig. 4c). This defect intolerance of Tim-3-deficient BALB/c mice was unexpected, as BALB/crecipients are strongly biased to develop TH2 responses25 and aretherefore relatively easy to tolerize4,23.
To determine whether the Tim-3 pathway is important only in themechanisms by which tolerance is induced by treatment with anti-CD154 with or without DST, we tested the effects of Tim-3 blockade ina transplant model in which tolerance is achieved through administra-tion of CTLA4-Ig. The mechanisms of action of CTLA4-Ig in vivoinvolve both the inhibition of antigen-reactive T cell proliferation andthe induction of immunoregulation and peripheral tolerance26–28. As
BALB/c
WT Tim-3 KO
Tim-3 KO
Number of stimulators (cells × 106)
IL-4 IFN-γ Proliferation
Cel
l cou
nts
rlgGanti-Tim-3
0 0.1 0.3 1 0 0.1 0.3 1 0 0.1 0.3 1
25
20
15
10
5
0
20
15
10
5
0
100
80
60
40
20
0
PstI
PstI
PstI
BamHI
BamHI
BamHI
Tim-3
BamHI
BamHI
BamHI PstI
PstI
PstI
SalIa b c
d e
Proliferation
Ear thickness Figure 3 Generation of Tim-3-deficient mice. (a) Gene targeting in embryonic stem cells. Top,targeting vector containing neomycin selectionmarker. Middle, portion of genomic mouse Tim-3locus showing relevant restriction sites. Grayboxes, exons. Bottom, recombined targeted Tim-3allele; the remainder of exon 2 is shown as anempty gray box. (b) PCR amplification of Tim-3and neomycin in wild-type (+/+), heterozygous(+/–) and Tim-3-deficient (–/–) mice. (c) ActivatedTH1 cells from wild-type but not Tim-3-deficientmice express Tim-3, as assessed by flowcytometry. Dotted lines, control immunglobulinstaining; solid lines, Tim-3 staining. rIgG, rat IgG.(d) Wild-type (WT) and Tim-3-deficient (Tim-3KO) BALB/c mice show similar responses to
contact antigen immunization with FITC. Top, ear thickness measured after rechallenge with the immunizing antigen. Bottom, ex vivo proliferation ofdraining lymph node cells to FITC rechallenge. (e) In vitro proliferation of wild-type (open circles) and Tim-3-deficient (Tim-3 KO; filled squares) BALB/csplenocytes in response to allogeneic C57BL/6 stimulators. Top, primary cultures; bottom, recall responses after in vivo priming. One experimentrepresentative of three is shown.
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
reim
mu
no
log
y

A RT I C L E S
NATURE IMMUNOLOGY VOLUME 4 NUMBER 11 NOVEMBER 2003 1097
expected, treatment with CTLA4-Ig produced indefinite engraftmentof islet allografts in seven of nine treated recipients. In contrast, coad-ministration of flTim-3–Ig with CTLA4-Ig resulted in the rejection ofmost of the allografts (Fig. 4d). Together with results reported in theaccompanying paper16, these data indicate that Tim-3 is a critical reg-ulator of immunological tolerance.
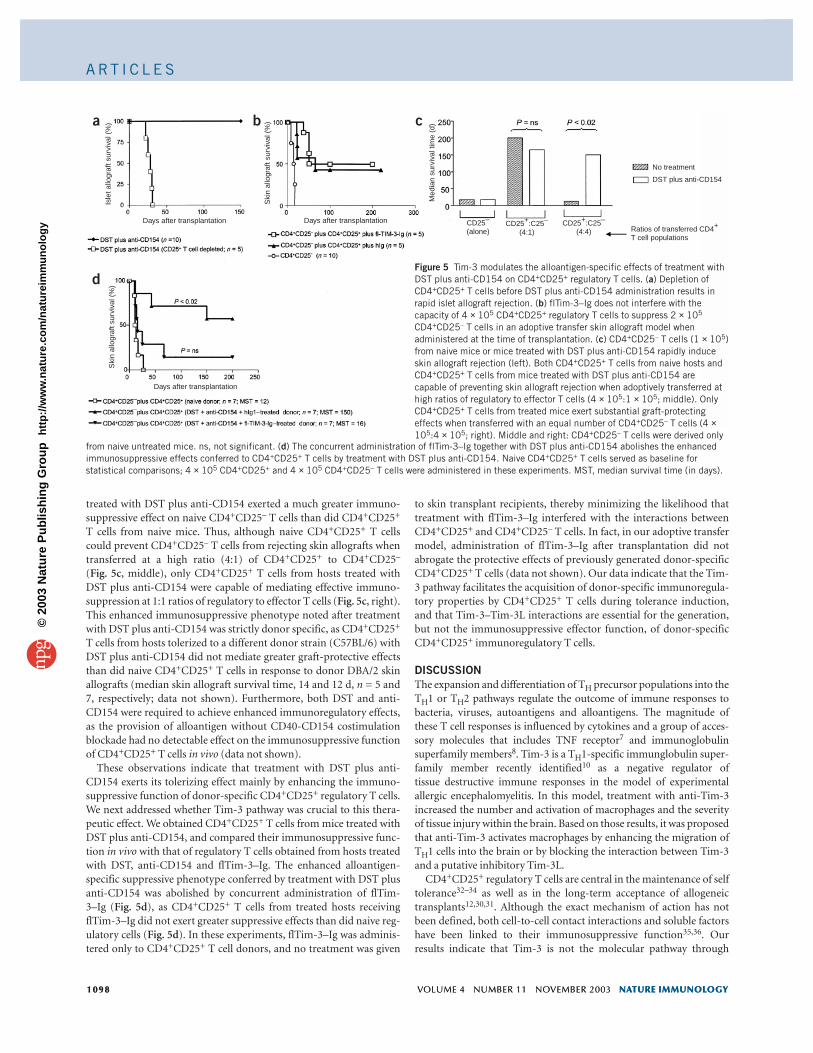
Naive CD4+CD25+ T cells block rejectionCD4+CD25+ regulatory T cells are involved in the maintenance of thetolerant state induced with DST plus anti-CD154 (ref. 12). We testedwhether this regulatory T cell subset was also required for the induc-tion phase of transplant tolerance. The consequences of CD4+CD25+
T cell depletion were similar to the effects induced by flTim-3–Igadministration, and we found rapid rejection of the islet allografts inhosts treated with anti-CD25 (Fig. 5a).
Based on these results, we hypothesized that engagement of Tim-3expressed on TH1 effector cells by Tim-3L-bearing CD4+CD25+ regula-tory T cells present in naive hosts might be a prerequisite for theirimmunosuppressive function. To determine whether regulatory T cells from naive mice can exert their immunosuppressive function inthe absence of Tim-3 engagement, we did adoptive transfer experi-ments assessing the capacity of naive CD4+CD25+ T cells to preventallograft rejection mediated by CD4+CD25– T cells in the presence ofTim-3 blockade. In this model, the transfer of as few as 1 × 105
CD4+CD25– or CD8+ T cells into immunodeficient MHC-mismatchedskin allograft recipients results in rapid skin allograft rejection, whereastransferred naive CD4+CD25+ T cells do not induce rejection and pre-vent CD4+CD25– effector T cell populations from destroying thegrafts29,30. Administration of flTim-3–Ig to C3H/He SCID recipients ofDBA/2 skin allografts did not inhibit the capacity of transferred naiveCD4+CD25+ T cells to prevent cotransferred CD4+CD25– T cells frommounting allograft rejection (Fig. 5b). We obtained similar results invitro when we cultured naive CD4+CD25+ T cells with either naiveCD4+CD25– T cells or TH1-polarized CD4+ T cells and stimulatedthem with soluble anti-CD3 in the presence of flTim-3–Ig (data not
shown). In addition, the transfer of 1 × 105 CD4+CD25– T cells aloneresulted in equally rapid skin graft rejection regardless of whetherflTim-3–Ig or a control human IgG was administered (data notshown). In agreement with these results with flTim-3–Ig, we found asimilar tempo of skin allograft rejection after transfer of wild-type orTim-3-deficient CD4+CD25– T cells (data not shown). Moreover, wild-type naive CD4+CD25+ T cells were equally capable of suppressing thecapacity of Tim-3-deficient or wild-type CD4+CD25– T cells to mountskin allograft rejection as well as the proliferative response of wild-typeand Tim-3-deficient antigen-activated TH1 cells to anti-CD3 in vitro(data not shown). Finally, CD4+CD25+ T cells from naive wild-typeand Tim-deficient BALB/c mice showed similar immunosuppressiveproperties in response to MHC-mismatched alloantigens in vivo in theadoptive-transfer skin transplant model (data not shown).
These results indicate that CD4+CD25+ regulatory T cells fromnaive alloantigen-inexperienced mice can suppress TH1-dependentcytopathic responses in the absence of Tim-3 engagement. Hence,Tim-3 blockade does not abolish the immunoregulatory effectorfunction of alloantigen-inexperienced CD4+CD25+ T cells, nor doesit substantially enhance the capacity of CD4+CD25– T cells to rejectskin allografts.
Tim-3 regulates allospecific CD4+CD25+ tolerant T cell actionSome therapeutic regimens that cause transplant tolerance are knownto bolster the potency of alloantigen-specific CD4+CD25+ regulatoryT cells29–31. To determine the effect of treatment with DST plus anti-CD154 on the generation of alloantigen-specific CD4+CD25+ T cells,we did additional adoptive transfer experiments to compare theimmunosuppressive capacity of CD4+CD25+ T cells from naive hostsor from mice treated with DST plus anti-CD154. CD4+CD25– orCD8+ T cells from treated or untreated naive mice did not differ intheir capacity to promote acute skin allograft rejection (Fig. 5c,CD4+CD25– T cells), indicating that in this model, treatment withDST plus anti-CD154 does not have a notable effect on effector T cellpopulations. In contrast, CD4+CD25+ T cells sorted from hosts
Figure 4 Tim-3 contributes to the tolerizingeffects of treatment with DST plus anti-CD154 in an MHC-mismatched islet allograft model. (a) Transcripts of genes encoding Tim-3 and IFN-γare up-regulated in rejecting but not in long-termsurviving islet allografts. Data are expressed asrelative fold difference between target samplesand a calibrator (isolated islets), and representthe mean ± s.e.m. obtained from fourindependent experiments (b) Concurrent
administration of flTim-3–Ig together with DST plus anti-CD154 prevents the induction of transplantation tolerance to islet allografts. (c) Tim-3-deficientmice rapidly reject islet allografts despite treatment with DST plus anti-CD154. (d) Administration of flTim-3–Ig significantly decreases the tolerance-promoting capacity of CTLA4-Ig. MST, median survival time (in days).
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
reim
mu
no
log
y
A RT I C L E S
1098 VOLUME 4 NUMBER 11 NOVEMBER 2003 NATURE IMMUNOLOGY
treated with DST plus anti-CD154 exerted a much greater immuno-suppressive effect on naive CD4+CD25– T cells than did CD4+CD25+
T cells from naive mice. Thus, although naive CD4+CD25+ T cellscould prevent CD4+CD25– T cells from rejecting skin allografts whentransferred at a high ratio (4:1) of CD4+CD25+ to CD4+CD25–
(Fig. 5c, middle), only CD4+CD25+ T cells from hosts treated withDST plus anti-CD154 were capable of mediating effective immuno-suppression at 1:1 ratios of regulatory to effector T cells (Fig. 5c, right).This enhanced immunosuppressive phenotype noted after treatmentwith DST plus anti-CD154 was strictly donor specific, as CD4+CD25+
T cells from hosts tolerized to a different donor strain (C57BL/6) withDST plus anti-CD154 did not mediate greater graft-protective effectsthan did naive CD4+CD25+ T cells in response to donor DBA/2 skinallografts (median skin allograft survival time, 14 and 12 d, n = 5 and7, respectively; data not shown). Furthermore, both DST and anti-CD154 were required to achieve enhanced immunoregulatory effects,as the provision of alloantigen without CD40-CD154 costimulationblockade had no detectable effect on the immunosuppressive functionof CD4+CD25+ T cells in vivo (data not shown).
These observations indicate that treatment with DST plus anti-CD154 exerts its tolerizing effect mainly by enhancing the immuno-suppressive function of donor-specific CD4+CD25+ regulatory T cells.We next addressed whether Tim-3 pathway was crucial to this thera-peutic effect. We obtained CD4+CD25+ T cells from mice treated withDST plus anti-CD154, and compared their immunosuppressive func-tion in vivo with that of regulatory T cells obtained from hosts treatedwith DST, anti-CD154 and flTim-3–Ig. The enhanced alloantigen-specific suppressive phenotype conferred by treatment with DST plusanti-CD154 was abolished by concurrent administration of flTim-3–Ig (Fig. 5d), as CD4+CD25+ T cells from treated hosts receivingflTim-3–Ig did not exert greater suppressive effects than did naive reg-ulatory cells (Fig. 5d). In these experiments, flTim-3–Ig was adminis-tered only to CD4+CD25+ T cell donors, and no treatment was given
to skin transplant recipients, thereby minimizing the likelihood thattreatment with flTim-3–Ig interfered with the interactions betweenCD4+CD25+ and CD4+CD25– T cells. In fact, in our adoptive transfermodel, administration of flTim-3–Ig after transplantation did notabrogate the protective effects of previously generated donor-specificCD4+CD25+ T cells (data not shown). Our data indicate that the Tim-3 pathway facilitates the acquisition of donor-specific immunoregula-tory properties by CD4+CD25+ T cells during tolerance induction,and that Tim-3–Tim-3L interactions are essential for the generation,but not the immunosuppressive effector function, of donor-specificCD4+CD25+ immunoregulatory T cells.
DISCUSSIONThe expansion and differentiation of TH precursor populations into theTH1 or TH2 pathways regulate the outcome of immune responses tobacteria, viruses, autoantigens and alloantigens. The magnitude ofthese T cell responses is influenced by cytokines and a group of acces-sory molecules that includes TNF receptor7 and immunoglobulinsuperfamily members8. Tim-3 is a TH1-specific immunglobulin super-family member recently identified10 as a negative regulator of tissue destructive immune responses in the model of experimentalallergic encephalomyelitis. In this model, treatment with anti-Tim-3increased the number and activation of macrophages and the severityof tissue injury within the brain. Based on those results, it was proposedthat anti-Tim-3 activates macrophages by enhancing the migration ofTH1 cells into the brain or by blocking the interaction between Tim-3and a putative inhibitory Tim-3L.
CD4+CD25+ regulatory T cells are central in the maintenance of selftolerance32–34 as well as in the long-term acceptance of allogeneictransplants12,30,31. Although the exact mechanism of action has notbeen defined, both cell-to-cell contact interactions and soluble factorshave been linked to their immunosuppressive function35,36. Ourresults indicate that Tim-3 is not the molecular pathway through
1098 VOLUME 4 NUMBER 11 NOVEMBER 2003 NATURE IMMUNOLOGY
CD25–
(alone)CD25+:C25–
(4:1)CD25+:C25–
(4:4)
Med
ian
surv
ival
tim
e (d
)
No treatment
DST plus anti-CD154
Ratios of transferred CD4+ T cell populations
Days after transplantation
Ski
n al
logr
aft s
urvi
val (
%)
Days after transplantation
Isle
t allo
graf
t sur
viva
l (%
)S
kin
allo
graf
t sur
viva
l (%
)
Days after transplantation
a b c
dFigure 5 Tim-3 modulates the alloantigen-specific effects of treatment withDST plus anti-CD154 on CD4+CD25+ regulatory T cells. (a) Depletion ofCD4+CD25+ T cells before DST plus anti-CD154 administration results inrapid islet allograft rejection. (b) flTim-3–Ig does not interfere with thecapacity of 4 × 105 CD4+CD25+ regulatory T cells to suppress 2 × 105
CD4+CD25– T cells in an adoptive transfer skin allograft model whenadministered at the time of transplantation. (c) CD4+CD25– T cells (1 × 105)from naive mice or mice treated with DST plus anti-CD154 rapidly induceskin allograft rejection (left). Both CD4+CD25+ T cells from naive hosts andCD4+CD25+ T cells from mice treated with DST plus anti-CD154 arecapable of preventing skin allograft rejection when adoptively transferred athigh ratios of regulatory to effector T cells (4 × 105:1 × 105; middle). OnlyCD4+CD25+ T cells from treated mice exert substantial graft-protectingeffects when transferred with an equal number of CD4+CD25– T cells (4 ×105:4 × 105; right). Middle and right: CD4+CD25– T cells were derived only
from naive untreated mice. ns, not significant. (d) The concurrent administration of flTim-3–Ig together with DST plus anti-CD154 abolishes the enhancedimmunosuppressive effects conferred to CD4+CD25+ T cells by treatment with DST plus anti-CD154. Naive CD4+CD25+ T cells served as baseline forstatistical comparisons; 4 × 105 CD4+CD25+ and 4 × 105 CD4+CD25– T cells were administered in these experiments. MST, median survival time (in days).
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
reim
mu
no
log
y

A RT I C L E S
NATURE IMMUNOLOGY VOLUME 4 NUMBER 11 NOVEMBER 2003 1099
which these CD4+CD25+ T cells deliver their immunosuppressiveeffects, as CD4+CD25+ T cells from naive or tolerant hosts canstrongly suppress CD4+CD25– T cells lacking effective expression ofTim-3 both in vivo and in vitro. However, a Tim-3–Tim-3L–sensitivepathway is responsible for the functional generation of donor-specificCD4+CD25+ regulatory T cells that emerge after the administration oftolerizing treatments such as DST plus anti-CD154. Hence, both Tim-3-dependent and Tim-3-independent pathways are involved inCD4+CD25+ T cell–dependent immunoregulation.
Although the precise cellular and molecular interactions involvingTim-3 and its ligand(s) remain to be fully elucidated, the expressionpatterns noted for Tim-3 and Tim-3L indicate that a direct interactionbetween Tim-3-positive TH1 effector cells and Tim-3L-positive regula-tory T cells might constitute a mechanism through which CD4+CD25+
T cells acquire enhanced immunosuppressive function. Alternatively,given that Tim-3L expression is also found on some dendritic cells, theeffects of Tim-3 on regulatory T cells could be exerted indirectlythrough the intervention of APCs. This last interpretation would beconsistent with observations concerning the function of dendritic cellphenotype in the modulation of CD4+CD25+ T cell–mediatedimmunoregulation37,38. Finally, although Tim-3 might also exert adirect inhibitory signal on TH1 cells themselves, the absence of anincrease in the frequency of IL-2- and IFN-producing effector T cells inthe spleens of transplant recipients treated with flTim-3–Ig (data notshown) indicates that in transplantation, Tim-3 has a more potenteffect on regulatory than on effector T cell populations. These resultsare also consistent with our finding that the initiation of effectorimmune responses is almost unimpaired in Tim-3-deficient mice.
Our findings offer a new insight into the mechanisms throughwhich treatment with DST plus anti-CD154 promotes toleranceinduction in transplantation. The current understanding states thatthe provision of alloantigen to host T cells in the context of an anti-CD154-induced immunosuppressive environment directly inactivatesalloreactive CD4+ and CD8+ T lymphocytes by anergy and/or apopto-sis, and subsequently promotes the expansion of regulatory T cellscapable of self-perpetuating the tolerant state11–13,24,39. We have pro-vided evidence indicating that in polyclonal systems, DST plus anti-CD154 acts mainly by increasing the immunosuppressive func-tion of CD4+CD25+ T cells in an alloantigen-specific way, whereas theeffects of treatment on the capacity of effector T cell populations toreject allografts are much less potent.
Hypotheses about the mechanisms through which prior encounterwith alloantigen in the absence of CD40-CD154 costimulation inducethese Tim-3-sensitive immunoregulatory networks are speculative.Regulatory T cell clones capable of recognizing allogeneic peptidesmight undergo selective expansion40,41 and/or acquire a more efficientimmunosuppressive function, thereby strengthening donor-specificimmunoregulatory ‘circuits’. Indeed, the lack of an effect of DSTmonotherapy in our model indicates that CD154 blockade, known topromote effector T cell apoptosis and anergy13,39, spares or evenenhances the function and survival of regulatory T cells. Alternatively,CD40-CD154 costimulation blockade might modulate the phenotypeof resident APCs, resulting in the recruitment into the immunoregula-tory compartment of naive donor-reactive T cells undergoing activa-tion. Lack of CD40 expression on APCs promotes antigen-specifictolerance42–45, in some cases through the generation of CD4+ regula-tory T cells45. Hence, given our findings, CD40 and Tim-3 may haveopposing effects and their balance may serve as a checkpoint in the‘decision’ between tolerance and immunity.
The importance of Tim-3–Tim-3L pathway in facilitating immuno-logical tolerance is not exclusively restricted to the mechanisms
through which treatment with DST plus anti-CD154 achieves trans-plantation tolerance. Indeed, based on the experiments reported in theaccompanying article16 on the inability to induce high-dose tolerancein Tim-3-deficient mice, and our own observations that flTim-3–Igabrogates tolerance induction after treatment with CTLA4-Ig, we canconclude that Tim-3 is fundamental in regulating TH1-mediatedimmune responses and facilitating the generation of immunologicaltolerance.
Thus, we have shown that the immunoglobulin superfamily memberTim-3 functions to inhibit aggressive TH1 mediated auto- and alloim-mune responses. These effects seem to be mediated, at least in part, bythe regulation of the immunosuppressive potency of CD4+CD25+ reg-ulatory T cells. Hence, expression of Tim-3 on TH1 cells provides a keycheckpoint that serves to dampen proinflammatory TH1-dependent Tcell responses and limit the associated tissue injury.
METHODSMice. All mice were obtained from The Jackson Laboratories and maintained inspecific pathogen–free conditions in conventional animal facilities atMillennium Pharmaceuticals, Beth Israel Deaconess Medical Center (Boston,Massachusetts) or Deutsches Rheumaforschungs Zentrum (Berlin, Germany).Animal experiments were approved by all three institutional committees.
Identification and cloning of mouse Tim-3. A TH1-versus-TH2 subtractivelibrary from activated mouse TH1 and TH2 clones was generated as described14.One of the clones in the TH1 library consisted of an 857-base-pair cDNA frag-ment and was used to obtain the full-length clone with a cDNA library frommurine TH1 cells. The human homolog was subsequently identified andsequenced from a human spleen cDNA library. Mouse antigen–specific TH1(AE7 and Dorris) and TH2 (D10.G4, DAX, CDC25) clones were stimulatedevery 10–14 d with peptide, mitomycin C–treated APCs and IL-2 (100 U/ml).Cells were activated with anti-CD3 (2C11; Pharmingen) and RNA was isolated.Differential expression of Tim-3 cDNA from resting and anti-CD3 activatedclones was subsequently confirmed by RNA blot.
Generation of Tim-3 mAb and Tim-3 fusion proteins. A DNA sequence con-taining the extracellular domain of Tim-3 was amplified by PCR and clonedinto a vector containing the CD5 signal sequence and the human IgG1 constantregion. COS cells were transfected and the recombinant protein was purifiedover a protein A column. Wky rats were immunized with purified murine Tim-3 fusion protein (100 µg) in CFA and boosted intraperitoneally and subcuta-neously. Splenocytes were fused with SP/2 myeloma cells, and the resultingclones were screened for binding on Tim-3-transfected CHO cells. One of theseclones, 8H7, was selected based on specific binding to Tim-3 but not inducibleT cell costimulatory transfectants. Tim-3 mAb 8H7 was isotyped as a rat IgG1with specific antibodies (BD Pharmingen). Both flTim-3–Ig and sTim-3–Igwere constructed as human IgG1 Fc tail fusion proteins and expressed in NS.1cells as described16. Biotinylated Tim-3-related fusion proteins or human IgG1together with fluorochrome-conjugated streptavidin (BD Pharmingen) wereused for Tim-3L staining experiments.
Expression of Tim-3 on TH1 and TH2 effector cells. Resting transgenic CD4+ Tcells obtained from mice expressing the transgene encoding the DO11.10 αβTCR were cultured with a peptide consisting of ovalbumin amino acids323–339 (10 µg/ml) and mitomycin C–treated splenocytes in TH1- or TH2-polarizing conditions. For TH1 phenotype development, recombinant murineIL-12 (10 ng/ml; Genetics Institute) and neutralizing mAb to IL-4 (11B11) wereadded, and for TH2 development, recombinant murine IL-4 (BioSourceInternational) and neutralizing anti-IL-12 (C17.8) were used. After every 7 d,cells were restimulated with fresh APCs and maintained in culture in appropri-ate polarizing cytokines. Expression of Tim-3 was determined by flow cytome-try every second day with Tim-3 mAb (8H7).
NOD adoptive transfer model of insulin-dependent diabetes mellitus. FemaleNOD mice with blood glucose concentrations in excess of 250 mg/dl for 3 con-secutive days were considered diabetic and were used as donors of T cell–enriched
NATURE IMMUNOLOGY VOLUME 4 NUMBER 11 NOVEMBER 2003 1099
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
reim
mu
no
log
y

A RT I C L E S
1100 VOLUME 4 NUMBER 11 NOVEMBER 2003 NATURE IMMUNOLOGY
splenocytes populations, prepared as described23. T cell–enriched splenocytepopulations (5 × 106 cells) were then injected into the tail veins of 4- to 6-week-old NOD-SCID recipients. Mice were treated with either Tim-3 mAb (8H7) or arat isotype control mAb (A110-1; BD Pharmingen). The mAbs (0.5 mg each)were administered intraperitoneally on day 0 (6 h before injection of cells) fol-lowed by administration of 100 µg mAb twice a week for the duration of theexperiment (6–8 weeks). Alternatively, NOD-SCID recipients received eitherflTim-3–Ig or a human isotype control IgG1 mAb (0.5 mg each; Sigma) on day 0followed by 100 µg twice a week. Histological examination of pancreatic islets wasdone at different time points on three randomly selected adoptively transferredmice (at least 20 islets per mice examined). Insulitis was assigned histologicalscores according a published procedure46.
Generation of Tim-3-deficient mice. We isolated the mouse gene encodingTim-3 from a 129SVJ library with the full-length (846 base pairs) cDNAprobe. The targeting vector replaced a 5.4-kilobase genomic fragment with aneomycin-resistance cassette in sense orientation relative to Tim-3 tran-scription. After selection, homologous recombinants were identified by PCRand verified by Southern blot analysis. Chimeric mice generated from Tim-3-deficient embryonic stem cell clones were crossed with BALB/c femalemice for six to eight generations to produce Tim-3-deficient mice.
Contact hypersensitivity assays. Tim-3-deficient or wild-type BALB/c micewere immunized on their shaved abdomens with 0.5% FITC (Sigma) in acetoneand dibutylpthalate. Mice were challenged with FITC on both sides of one earat day 7, and rechallenged on day 9. Ear thickness was measured on day 10. Onday 13, draining lymph nodes were collected and single-cell suspensions werestimulated in vitro for 48 h with different concentrations of FITC.
Islet transplantation. DBA/2 (H-2d) into C57BL/6 (H-2b) islet transplanta-tion was done as described47. The tolerizing protocol consisted of intra-venous administration of 1 × 107 DBA/2 splenocytes 28 d beforetransplantation (day –28), and 250 µg hamster anti-mouse CD154 (MR1,IgG2a, ATCC HB11048; American Type Culture Collection) intraperi-toneally on days –28, –26 and –24. Control human IgG1 or flTim-3–Ig wasadministered intraperitoneally at a dose of 250 µg on days –28, –26 and –24.In some recipients, 200 µg rat anti-mouse CD25 (PC61, 5.3, IgG1, ATCCTB222; American Type Culture Collection) was also administered intraperi-toneally on days –40, –38 and –36. At such doses, anti-CD25 eliminates morethan 80% of CD4+CD25+ T cells in secondary lymphoid tissues12. In someexperiments, tolerance was induced by administration of 0.1 µg CTLA4-Igon days 0, 2, 4, 6 and 8 after transplantation.
Real-time PCR. Total RNA was extracted from islet grafts with Trizol andreverse transcription was done with Multiscribed Reverse TranscriptaseEnzyme (PE Applied Biosystems). Real-time PCR was done with the ABI7700 sequence detector system (PE Applied Biosystems) as described10.Glyceraldehyde phosphodehydrogenase, IFN-γ, IL-4 and IL-10 primer-probesets were purchased from Applied Biosystems. Tim-3 primer-probesequences have been described10.
Adoptive cell transfer into skin allograft recipients. Lymph node cells wereobtained from naive C3H/He mice (H-2k), or from C3H/He mice treated withan intravenous injection of 1 × 107 DBA/2 splenocytes (28 d before; day –28),and either anti-CD154 (250 µg on days –28, –26 and –24) or anti-CD154 plusflTim3–Ig (250 µg each on days –28, –26 and –24). Cells were stained with anti-CD25 and anti-CD4 (all from BD Pharmingen) and sorted on a MoFlo High-Performance Cell Sorter (Cytomation). The purity of the CD4+CD25– andCD4+CD25+ preparations was consistently >90%. Varying numbers ofCD4+CD25+ regulatory T cells and CD4+CD25– effector T cells were injectedinto the tail veins of C3H/He SCID mouse hosts undergoing DBA/2 skin trans-plantation 24 h later. In some experiments, three doses of 250 µg flTim-3–Igwere administered on days 0, 2 and 4 after skin transplant. Full-thicknessDBA/2 tail skin allografts were done as described48, and graft survival was mon-itored daily. Rejection was defined as a complete necrosis of the skin grafts.Similar experiments were done with wild-type C57BL/6 mice as donors of T cell populations and C57BL/6 mice deficient in recombination activationgene 2 as recipients of DBA/2 skin allografts.
T cell proliferation assays. In vitro mixed-lymphocyte reactions and in vivo 5-carboxyfluorescein diacetate succinimidyl ester proliferation assays have beendescribed12,15. For in vivo mixed-lymphocyte reactions, wild-type or Tim-3-deficient BALB/c mice were injected in the footpads with 2.5 × 106 mitomycinC–treated C57BL/6 spleen cells. On day 5, draining lymph nodes were removedand cells were cultured together with C57BL/6 stimulators, and on day 7cytokine concentrations measured by enzyme-linked immunosorbent assay(Endogen; Perbio Science). Sorted CD4+CD25+ and CD4+CD25– T cells werestimulated in vitro with 2 µg/ml of anti-CD3, 5 µg/ml of anti-CD28 and 100 U/ml of recombinant IL-2 (all from PharMingen). Cells were collected andstained with biotinylated sTim-3–Ig or control human IgG1 followed by strep-tavidin-cytochrome at 24 and 48 h, and were analyzed by flow cytometry.
ACKNOWLEDGMENTSWe thank Chimerigen (Allston, Massachusetts) for providing purified fusionproteins, and Y. Tian for technical assistance. Supported by the Juvenile DiabetesResearch Foundation (X.X.Z. and A.S.-F.), the National Institute of Allergy andInfectious Diseases (T.B.S.) and the Juvenile Diabetes Research Foundation Centerfor Islet Transplantation at Harvard Medical School (T.B.S.).
COMPETING INTERESTS STATEMENTThe authors declare competing financial interests (see the Nature Immunologywebsite for details).
Received 17 June; accepted 22 September 2003Published online at http://www.nature.com/natureimmunology/
1. Romagnani, S. Lymphokine production by human T cells in disease states. Annu.Rev. Immunol. 12, 227–257 (1994).
2. Kamradt, T. & Mitchison, N.A. Tolerance and autoimmunity. N. Engl. J. Med. 344,655–664 (2001).
3. Strom, T.B. et al. The Th1/Th2 paradigm and the allograft response. Curr. Opin.Immunol. 8, 688–693 (1996).
4. Li, X.C., Zand, M.S., Li, Y., Zheng, X.X. & Strom, T.B. On histocompatibility barriers,Th1 to Th2 immune deviation, and the nature of the allograft responses. J. Immunol.161, 2241–2247 (1998).
5. Anderson, G.P. & Coyle, A.J. TH2 and ‘TH2-like’ cells in allergy and asthma: pharma-cological perspectives. Trends. Pharmacol. Sci. 15, 324–332 (1994).
6. Kundig, T.M. et al. Duration of TCR stimulation determines costimulatory require-ment of T cells. Immunity 5, 41–52 (1996).
7. Locksley, R.M., Killeen, N. & Lenardo, M.J. The TNF and TNF receptor superfamilies:integrating mammalian biology. Cell 104, 487–501 (2001).
8. Salomon, B. & Bluestone, J.A. Complexities of CD28/B7: CTLA-4 costimulatory path-ways in autoimmunity and transplantation. Annu. Rev. Immunol. 19, 225–252 (2001).
9. Refaeli, Y., Van Parijs, L., London, C.A., Tschopp, J. & Abbas, A.K. Biochemical mecha-nisms of IL-2-regulated Fas-mediated T cell apoptosis. Immunity 8, 615–623 (1998).
10. Monney, L. et al. Th1-specific cell surface protein Tim-3 regulates macrophage acti-vation and severity of an autoimmune disease. Nature 415, 536–541 (2002).
11. Parker, D.C. et al. Survival of mouse pancreatic islet allografts in recipients treatedwith allogeneic small lymphocytes and antibody to CD40 ligand. Proc. Natl. Acad.Sci. USA 92, 9560–9564 (1995).
12. Sanchez-Fueyo, A., Weber, M., Domenig, C., Strom, T.B. & Zheng, X.X. Tracking theimmunoregulatory mechanisms active during allograft tolerance. J. Immunol. 168,2274–2281 (2002).
13. Quezada, S.A. et al. Mechanisms of donor specific transfusion tolerance: pre-emp-tive induction of clonal T cell exhaustion via indirect presentation. Blood1920–1926 (2003).
14. Coyle, A.J. et al. The CD28-related molecule ICOS is required for effective T cell-dependent immune responses. Immunity 13, 95–105 (2000).
15. Wells, A.D., Gudmundsdottir, H. & Turka, L.A. Following the fate of individual T cellsthroughout activation and clonal expansion. Signals from T cell receptor and CD28differentially regulate the induction and duration of a proliferative response. J. Clin.Invest. 100, 3173–3183 (1997).
16. Sabatos, C.A. et al. Tim-3/Tim-3-Ligand interaction regulates TH1 responses andinduction of peripheral tolerance. Nat. Immunol. advance online publication 12October 2003; doi:10.1038/ni988.
17. Delovitch, T.L. & Singh, B. The nonobese diabetic mouse as a model of autoimmunediabetes: immune dysregulation gets the NOD. Immunity 7, 727–738 (1997).
18. Christianson, S.W., Shultz, L.D. & Leiter, E.H. Adoptive transfer of diabetes into immun-odeficient NOD-scid/scid mice. Relative contributions of CD4+ and CD8+ T-cells fromdiabetic versus prediabetic NOD.NON-Thy-1a donors. Diabetes 42, 44–55 (1993).
19. McIntire, J.J. et al. Identification of Tapr (an airway hyperreactivity regulatory locus)and the linked Tim gene family. Nat. Immunol. 2, 1109–1116 (2001).
20. Kuchroo, V.K., Umetsu, D.T., DeKruyff, R.H. & Freeman, G.J. The TIM gene family:emerging roles in immunity and disease. Nat. Rev. Immunol. 3, 454–462 (2003).
21. Li, X.C. et al. IL-2 and IL-4 double knockout mice reject islet allografts: a role fornovel T cell growth factors in allograft rejection. J. Immunol. 161, 890–896 (1998).
22. Kishimoto, K. et al. The role of CD154-CD40 versus CD28-B7 costimulatory path-ways in regulating allogeneic Th1 and Th2 responses in vivo. J. Clin. Invest. 106,
1100 VOLUME 4 NUMBER 11 NOVEMBER 2003 NATURE IMMUNOLOGY
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
reim
mu
no
log
y

A RT I C L E S
NATURE IMMUNOLOGY VOLUME 4 NUMBER 11 NOVEMBER 2003 1101
63–72 (2000).23. Sho, M. et al. Physiological mechanisms of regulating alloimmunity: cytokines,
CTLA-4, CD25+ cells, and the alloreactive T cell clone size. J. Immunol. 169,3744–3751 (2002).
24. Zheng, X.X. et al. CTLA4 signals are required to optimally induce allograft tolerancewith combined donor-specific transfusion and anti-CD154 monoclonal antibodytreatment. J. Immunol. 162, 4983–4990 (1999).
25. DeKruyff, R.H., Fang, Y. & Umetsu, D.T. IL-4 synthesis by in vivo primed keyholelimpet hemocyanin specific CD4+ T cells. I. Influence of antigen concentration andantigen presenting cell type. J. Immunol. 149, 3468–3476 (1992).
26. Judge, T.A. et al. The in vivo mechanism of action of CTLA4Ig. J. Immunol. 156,2294–2299 (1996).
27. Waaga, A.M. et al. Regulatory functions of self-restricted MHC class II allopeptide-specific Th2 clones in vivo. J. Clin. Invest. 107, 909–916 (2001).
28. Lee, R.S. et al. CTLA4Ig-induced linked regulation of allogeneic T cell responses. J. Immunol. 166, 1572–1582 (2001).
29. Maurik Av, A., Herber, M., Wood, K.J. & Jones, N.D. Cutting edge: CD4+CD25+
alloantigen-specific immunoregulatory cells that can prevent CD8+ T cell-mediatedgraft rejection: implications for anti-CD154 immunotherapy. J. Immunol. 169,5401–5404 (2002).
30. Kingsley, C.I., Karim, M., Bushell, A.R. & Wood, K.J. CD25+CD4+ regulatory T cellsprevent graft rejection: CTLA-4- and IL-10- dependent immunoregulation of allore-sponses. J. Immunol. 168, 1080–1086 (2002).
31. Graca, L. et al. Both CD4+CD25+ and CD4+CD25– regulatory cells mediate dominanttransplantation tolerance. J. Immunol. 168, 5558–5565 (2002).
32. Sakaguchi, S. & Sakaguchi, N. Thymus and autoimmunity: capacity of the normalthymus to produce pathogenic self-reactive T cells and conditions required for theirinduction of autoimmune disease. J. Exp. Med. 172, 537–545 (1990).
33. Sakaguchi, S., Sakaguchi, N., Asano, M., Itoh, M. & Toda, M. Immunologic self-toler-ance maintained by activated T cells expressing IL-2 receptor α-chains (CD25).Breakdown of a single mechanism of self-tolerance causes various autoimmune dis-eases. J. Immunol. 155, 1151–1164 (1995).
34. Salomon, B. et al. B7/CD28 costimulation is essential for the homeostasis of the
CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity12, 431–440 (2000).
35. Wood, K.J. & Sakaguchi, S. Regulatory lymphocytes: regulatory T cells in transplanta-tion tolerance. Nat. Rev. Immunol. 3, 199–210 (2003).
36. Shevach, E.M. CD4+CD25+ suppressor T cells: more questions than answers. Nat.Rev. Immunol. 2, 389–400 (2002).
37. Caramalho, I. et al. Regulatory T cells selectively express Toll-like receptors and areactivated by lipopolysaccharide. J. Exp. Med. 197, 403–411 (2003).
38. Pasare, C. & Medzhitov, R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science 299, 1033–1036 (2003).
39. Iwakoshi, N.N. et al. Treatment of allograft recipients with donor-specific transfusionand anti-CD154 antibody leads to deletion of alloreactive CD8+ T cells and prolongedgraft survival in a CTLA4-dependent manner. J. Immunol. 164, 512–521 (2000).
40. Yamazaki, S. et al. Direct expansion of functional CD25+CD4+ regulatory T cells byantigen-processing dendritic cells. J. Exp. Med. 198, 235–247 (2003).
41. Walker, L.S., Chodos, A., Eggena, M., Dooms, H. & Abbas, A.K. Antigen-dependent pro-liferation of CD4+CD25+ regulatory T cells in vivo. J. Exp. Med. 198, 249–258 (2003).
42. Buhlmann, J.E. et al. In the absence of a CD40 signal, B cells are tolerogenic.Immunity 2, 645–653 (1995).
43. Foy, T.M., Aruffo, A., Bajorath, J., Buhlmann, J.E. & Noelle, R.J. Immune regulationby CD40 and its ligand GP39. Annu. Rev. Immunol. 14, 591–617 (1996).
44. Hollander, G.A. et al. Induction of alloantigen-specific tolerance by B cells fromCD40-deficient mice. Proc. Natl. Acad. Sci. USA 93, 4994–4998 (1996).
45. Martin, E., O’Sullivan, B., Low, P. & Thomas, R. Antigen-specific suppression of aprimed immune response by dendritic cells mediated by regulatory T cells secretinginterleukin-10. Immunity 18, 155–167 (2003).
46. Yoon, J.W. et al. Control of autoimmune diabetes in NOD mice by GAD expression orsuppression in beta cells. Science 284, 1183–1187 (1999).
47. Steiger, J., Nickerson, P.W., Steurer, W., Moscovitch-Lopatin, M. & Strom, T.B. IL-2knockout recipient mice reject islet cell allografts. J. Immunol. 155, 489–498 (1995).
48. Li, Y. et al. Blocking both signal 1 and signal 2 of T-cell activation prevents apoptosisof alloreactive T cells and induction of peripheral allograft tolerance. Nat. Med. 5,1298–1302 (1999).
NATURE IMMUNOLOGY VOLUME 4 NUMBER 11 NOVEMBER 2003 1101
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
reim
mu
no
log
y





























