Embed Size (px)
Citation preview

ORIGINAL PAPER
The use of a prodrug approach to minimize potential CNSexposure of next generation quinoline methanolswhile maintaining efficacy in in vivo animal models
Jason C. Sousa • Erin Milner • Dustin Carroll • William McCalmont • Sean Gardner • Jay Moon •
Jacob D. Johnson • Patricia Lee • Jennifer Auschwitz • Norma Roncal • Diana Caridha • Anchalee Tungteung •
Qiang Zeng • Sean Reyes • Bryan Smith • Qigui Li • Michael P. Kozar • Victor Melendez • Geoffrey Dow
Received: 31 January 2013 / Accepted: 4 December 2013
� Springer-Verlag France (outside the USA) 2013
Abstract The use of mefloquine (MQ) for antimalarial
treatment and prophylaxis has diminished largely in
response to concerns about its neurologic side effects. An
analog campaign designed to maintain the efficacy of MQ
while minimizing blood–brain barrier (BBB) penetration
has resulted in the synthesis of a prodrug with comparable-
to-superior in vivo efficacy versus mefloquine in a P.
berghei mouse model while exhibiting a sixfold reduction
in CNS drug levels. The prodrug, WR319670, performed
poorly compared to MQ in in vitro efficacy assays, but had
promising in vitro permeability in an MDCK–MDR1 cell
line BBB permeability screen. Its metabolite, WR308245,
exhibited high predicted BBB penetration with excellent
in vitro efficacy. Both WR319670 and WR308245 cured
5/5 animals in separate in vivo efficacy studies. The in vivo
efficacy of WR319670 was thought to be due to the for-
mation of a more active metabolite, specifically
WR308245. This was supported by pharmacokinetics
studies in non-infected mice, which showed that both IV
and oral administration of WR319670 produced essentially
identical levels of WR319670 and WR308245 in both
plasma and brain samples at all time points. In these
studies, the levels of WR308245 in the brain were 1/4 and
1/6 that of MQ in similar IV and oral studies, respectively.
These data show that the use of WR319670 as an anti-
malarial prodrug was able to maintain efficacy in in vivo
efficacy screens, while significantly lowering overall pen-
etration of drug and metabolites across the BBB.
Keywords Malaria � Mefloquine � Quinoline methanol �Blood–brain barrier � Central nervous system � Prodrug
1 Introduction
According to current Centers for Disease Control and
Prevention guidelines (Arguin 2012), mefloquine (MQ)
(Fig. 1a) is arguably the most widely applicable drug for
malaria chemoprophylaxis for non-immune individuals
traveling to endemic countries. Like chloroquine (CQ),
MQ can be administered on a weekly basis, a characteristic
that enhances the likelihood of compliance and is more
forgiving in cases of late or missed dosages. Further, both
drugs are deemed safe for children of all ages/sizes and for
This manuscript was reviewed by the Walter Reed Army Institute of
Research and the US Army Medical Research and Materiel Command,
and there is no objection to its publication or dissemination. The
opinions expressed herein are those of the authors and do not
necessarily reflect the views or opinions of the Department of the Army
and the Department of Defense. All animal experiments were
conducted in compliance with the Animal Welfare Act and other
federal statutes and regulations relating to animals and experiments
involving animals and adhere to the principles stated in the Guide for
the Care and Use of Laboratory Animals (National Academy Press,
1996).
J. C. Sousa (&) � E. Milner � D. Carroll � W. McCalmont �S. Gardner � J. Moon � J. D. Johnson � P. Lee � J. Auschwitz �N. Roncal � D. Caridha � Q. Zeng � S. Reyes � B. Smith �Q. Li � M. P. Kozar � V. Melendez � G. Dow
Walter Reed Army Institute of Research, Silver Spring,
MD 20910, USA
e-mail: [email protected]
G. Dow
e-mail: [email protected]
A. Tungteung
United States Army Medical Component, Armed Forces
Research Institute of Medical Sciences, Bangkok, Thailand
G. Dow
United States Army Medical Materiel Development Activity,
1430 Veterans Drive, Frederick, MD 21702, USA
123
Eur J Drug Metab Pharmacokinet
DOI 10.1007/s13318-013-0162-9

women who are pregnant or breastfeeding. Unfortunately,
the widespread development of CQ-resistant strains of
malaria parasites precludes its use in all but a few select
malaria-endemic regions.
Despite its favorable properties, MQ also has drawbacks
that prohibit more universal application for malaria chemo-
prophylaxis and treatment. First, while much less prevalent
than CQ resistance, MQ-resistant strains of malaria parasites
have begun to appear in Southeast Asia. Perhaps more sig-
nificant are MQ’s reported adverse reactions. In addition to
gastrointestinal upset, MQ has been implicated in more
serious neuropathic side effects, including dizziness, vivid
dreams, hallucinations, paranoia, and schizophrenia (CDC
2009; Yelmo et al. 2010). While more common at larger
treatment doses, these effects have been noted at lower
prophylactic doses as well. Further, these symptoms have
been known to persist well beyond the completion of the
normal dosing regimen. As a result, MQ is not recommended
for use in patients with active symptoms of, or a history of,
depression or other psychiatric disorders (Yelmo et al. 2010).
Among the recent drug development programs at the
Walter Reed Army Institute of Research (WRAIR) is an
effort to produce an analog of MQ that is less capable of
penetrating the blood–brain barrier (BBB), while main-
taining levels of efficacy that are equal to or greater than
MQ. Because the precise target responsible for MQ’s
neurological effects is not known (Toovey 2009), the pre-
vailing strategy in this effort has been to prevent overall
drug levels in the brain. To that end, a library of MQ
analogs has been synthesized to explore the relationship
between a range of physiochemical properties and efficacy/
BBB permeability (Milner et al. 2010a, b).
NH
OH
N
F
FF
FF
F
OH
N
F
FF
FF
F
NHH3C
(A) (B)
OH
N
F
FF
FF
F
NH3C NH2
(C)
Fig. 1 a Mefloquine
(WR142490); b WR308245;
and c WR319670
Eur J Drug Metab Pharmacokinet
123

One of the compounds from this library, WR308245
(Fig. 1b), initially generated substantial interest due to its
promising results in in vitro efficacy and cytotoxicity
assays. However, its apparent permeability (Papp) in
in vitro studies using the MDCK cell line transfected with
human MDR1 suggested high BBB penetration. In vivo
mouse PK studies confirmed this finding with drug levels in
the brain approximately six times that of MQ at the Cmax
after IV dosing.
Another drug in this class of analogs, WR319670
(Fig. 1c), was found to have in vivo activity against P.
berghei in a mouse model of antimalarial efficacy while
exhibiting poor activity levels in [3H] hypoxanthine
(Chulay et al. 1983; Milhous et al. 1985) and SYBR Green
(Smilkstein et al. 2004; Johnson et al. 2007) in vitro effi-
cacy screens. These data suggested that the activity of
WR319670 in the in vivo model could be due to drug
metabolism, i.e., that WR319670 acted as a prodrug. Based
on the structure of WR319670 and common Phase I bio-
transformations, it was postulated that one of the metabo-
lites would be WR308245. In vivo mouse PK confirmed
the formation of WR308245 upon IV administration of
WR319670. In addition, drug brain levels for both
WR319670 and WR308245 achieved a Cmax of approxi-
mately 1/3 that of MQ.
While the use of prodrugs in the treatment of malaria is
not new (Chung et al. 2008), we believe that this is the first
reported use of the prodrug approach to minimize overall
penetration of the BBB and thereby reduce complications
due to adverse CNS effects.
2 Materials and methods
2.1 MDCK–MDR1 permeability
All BBB permeability screens using the MDCK cell line
transfected with human MDR1 were performed under
contract by absorption systems (Exton, PA).
2.2 In vitro activity
In vitro activities against several strains of Plasmodium
falciparum were evaluated using both the [3H] hypoxan-
thine method first described by Desjardins et al. (1979) and
later modified by Milhous et al. (1985), and an SYBR
Green I fluorescence assay introduced by Smilkstein et al.
(2004) and later modified by Johnson et al. (2007). In brief,
the [3H] hypoxanthine assay measures the amount of radio-
labeled hypoxanthine incorporated by P. falciparum para-
sites cultured in erythrocytes in response to exposure to a
range of drug concentrations. Plotting radioactivity (as
counts per minute) of the incorporated isotope versus drug
concentration can be used to determine the IC90 for the test
compound. The SYBR Green I assay involves the culturing
of P. falciparum parasites in erythrocytes while exposed to
a range of drug concentrations, followed by cell lysis and
exposure to the nucleic acid-intercalating dye SYBR Green
I. Fluorescence is plotted versus drug concentration to
determine the IC90 of the test compound.
These analyses were repeated using several strains of P.
falciparum, including W2 (CQ resistant, MQ sensitive), D6
(CQ sensitive, less susceptible to MQ), and C235 (resistant
to CQ and MQ).
2.3 In vitro cytotoxicity
In vitro cytotoxicity assays were performed according to
the method described by Caridha et al. (2008). In brief, the
assay measures a compound’s ability to disrupt calcium
homeostasis in rodent macrophage (RAW-264.7) cells
using a 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazo-
lium bromide (MTT) indicator of cell viability. Plots of
absorbance versus concentration of test compound are used
to determine LC50.
2.4 In vivo efficacy
In vivo efficacy against malaria parasites was evaluated
using a modified Thompson test (Dow et al. 2006),
whereby male ICR mice were infected on Day 0 with P.
berghei (KBG-173 strain) and dosed once orally with test
compound, on Day 3. Five mice were used in each dosing
group. Animals were monitored daily through Day 31 for
clinical signs of infection and periodic malaria smears were
taken. Efficacy was expressed as mean survival time where
cures were defined as survival at 31 days post-treatment.
Typically untreated control mice are usually euthanized
humanely on days 6–10 once parasitemia levels become
too high or symptoms of malaria infection are observed.
Dosages were either 160 or 320 mg/kg and dosing vehicle
was either HECT (0.5 % hydroxyethylcellulose in 0.2 %
Tween 80) or 5 % ethanol/5 % Cremophor EL/90 %
HECT.
2.5 Pharmacokinetic studies
Pharmacokinetic studies were performed using both IV and
PO administration. For each time point to be acquired, four
male FVB mice, aged 5–6 weeks, were dosed at 5 mg/kg
(IV) or 160 mg/kg (PO). Drug vehicle for IV studies was
0.5/5/5 % v/v Cremophor EL/dimethylsulfoxide/glucose
solution in 20 mM citrate buffer (pH 3.0), administered at
50 lL/20 g. For PO dosing, drug vehicle was 5 % ethanol/
5 % Cremophor EL/90 % HECT, administered at 150 lL/
20 g.
Eur J Drug Metab Pharmacokinet
123

At each time point, plasma, whole blood, brain, and
liver samples were collected. Whole blood was collected
by cardiac puncture. Following the separation of appro-
priate aliquots, plasma was obtained from the remaining
whole blood via centrifugation. All liquid and tissue sam-
ples were immediately preserved at -80� C and stored
until analytical work was performed.
2.6 Pharmacokinetics
2.6.1 Extraction
Liquid samples were extracted by adding two volumes of
acetonitrile to one volume of blood/plasma, followed by
vigorous vortexing for 10–15 s. Tissue samples were
weighed and 5 mL of deionized water were added for each
gram of tissue, followed by homogenization using a sonic
dismembrator. From this homogenate, aliquots were
extracted in a manner identical to the liquid samples. All
samples were centrifuged at 13,000 rpm for 10 min
immediately after extraction, and the supernatant liquid
was removed for analysis by LC–MS/MS.
2.6.2 LC–MS/MS
Chromatography was performed using a surveyor pump
(Thermo Scientific, Waltham, MA) with Waters XTerra
MS C18 50 mm 9 2.1 mm id, 3.5 lm particle size col-
umns (Waters Corp., Milford, MA). Mobile phase con-
sisted of a water/0.1 % formic acid (Solvent A)/
acetonitrile/0.1 % formic acid (Solvent B) gradient. The
gradient began at 2 % B, rose to 98 % B from 1 to 3.5 min,
held steady for 2 min, then returned immediately to its
starting composition and allowed to equilibrate for
1.5 min. Flow rate was 300 lL/min. Samples were injected
using an HTC PAL autosampler (LEAP Technologies,
Carrboro, NC). Tandem mass spectrometry was performed
using a TSQ Quantum AM (Thermo Scientific). Monitored
transitions were: WR319670 (380.11–321.08, ESI?);
WR308245 (339.08–270.03, ESI?); and mefloquine
(379.02–361.42, ESI?).
3 Results
In vitro permeability with MDR1-transfected MDCK cells
was used to rank order potential MQ analogs in terms of
decreasing likelihood to penetrate the BBB. Table 1 sum-
marizes the permeability, in vitro activity, in vitro cyto-
toxicity and in vivo efficacy results for WR308245 and
WR319670 along with that of MQ, used as a baseline
comparison for rank ordering purposes. While the in vitro
permeability data suggested greater BBB penetration than
MQ, WR308245 generated sufficient interest due to its
comparable in vitro efficacy coupled with its superior
in vitro toxicity results. WR319670, while suggesting a
promising decrease in BBB permeability and a comparable
cytotoxicity profile, did not produce impressive in vitro
efficacy results. However, in the in vivo efficacy model
using P. berghei, WR319670 cured 5/5 mice after a single
oral dose, compared to 4/5 for MQ and 5/5 for WR308245
after a 3-dose regimen.
Figure 2 shows the brain (solid line) and plasma (dashed
line) drug levels for WR308245 and MQ in an IV PK
studies for both drugs using non-infected mice. As sug-
gested by the in vitro screen, WR308245 has much higher
BBB permeability than MQ, peaking at about six times the
concentration at Cmax. In Fig. 3, a comparable PK study
with IV dosing of WR319670 confirms the in vitro pre-
diction of lower BBB penetration (about 1/4 that of MQ in
Fig. 2).
In an effort to explain the discrepancy between
WR319670’s in vitro and in vivo efficacy, the PK samples
from WR319670 IV dosing were also monitored for the
formation of WR308245, which is a potential metabolite of
WR319670 based on common Phase I biotransformations.
These data are also included in Fig. 3. Throughout the
course of the study, WR308245 and WR319670 were
present in similar concentrations in both plasma and brain.
Figure 4 summarizes the total brain concentration of
MQ, WR319670, and WR308245 after IV dosing of 5
mg/kg, as well as the appearance of WR308245 in the brain
after the dosing of WR319670.
Table 1 Summary of results for MQ, WR308245, and WR319670
from in vitro MDCK–MDR1 permeability, in vitro efficacy by both
the [3H] hypoxanthine (HX) and SYBR Green I (SG) methods,
in vitro toxicity (MTT), and in vivo efficacy against P. berghei in a
murine model of malaria infection
MQ WR308245 WR319670
MDCK–MDR1 (910-6
cm/s)
9.3 24 5.0
HX W2 (ng/mL) 6.2 ± 2.8
(532)
17 180
HX D6 (ng/mL) 17 ± 11
(536)
45 290
HX C235 (ng/mL) 52 ± 30
(367)
58 710
SG D6 (ng/mL) 12 ± 2 (6) NT 340
SG C235 (ng/mL) 43 ± 12 (6) NT 700
LC50 MTT (ng/mL) 4,000 31,000 3,800
Cures at 160 mg/kg 91
PO
4/5 5/5 5/5
Numbers in parentheses indicate the number of runs used in deter-
mining the reported values
NT not tested
Eur J Drug Metab Pharmacokinet
123
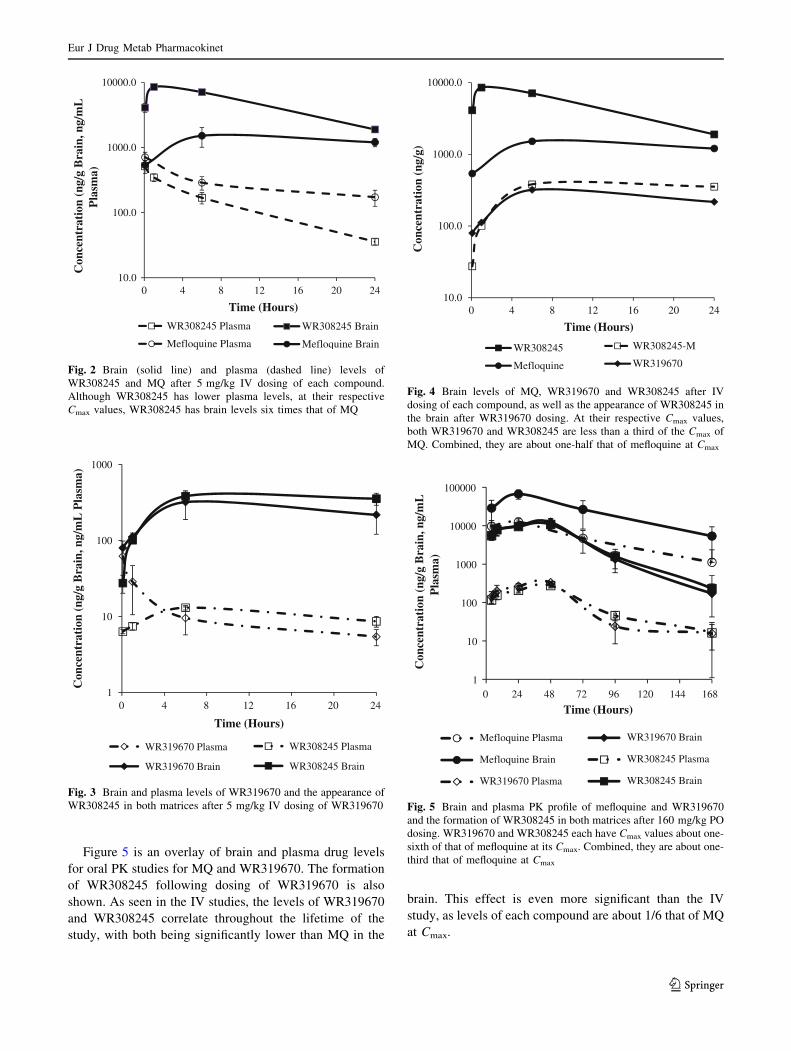
Figure 5 is an overlay of brain and plasma drug levels
for oral PK studies for MQ and WR319670. The formation
of WR308245 following dosing of WR319670 is also
shown. As seen in the IV studies, the levels of WR319670
and WR308245 correlate throughout the lifetime of the
study, with both being significantly lower than MQ in the
brain. This effect is even more significant than the IV
study, as levels of each compound are about 1/6 that of MQ
at Cmax.
1
10
100
1000
0 4 8 12 16 20 24
Con
cent
rati
on (
ng/g
Bra
in, n
g/m
L P
lasm
a)
Time (Hours)
WR319670 Plasma
WR319670 Brain
WR308245 Plasma
WR308245 Brain
Fig. 3 Brain and plasma levels of WR319670 and the appearance of
WR308245 in both matrices after 5 mg/kg IV dosing of WR319670
10.0
100.0
1000.0
10000.0
0 4 8 12 16 20 24
Con
cent
rati
on (
ng/g
)
Time (Hours)
WR308245
Mefloquine
WR308245-M
WR319670
Fig. 4 Brain levels of MQ, WR319670 and WR308245 after IV
dosing of each compound, as well as the appearance of WR308245 in
the brain after WR319670 dosing. At their respective Cmax values,
both WR319670 and WR308245 are less than a third of the Cmax of
MQ. Combined, they are about one-half that of mefloquine at Cmax
1
10
100
1000
10000
100000
0 24 48 72 96 120 144 168
Con
cent
rati
on (
ng/g
Bra
in, n
g/m
L
Pla
sma)
Time (Hours)
Mefloquine Plasma
Mefloquine Brain
WR319670 Plasma
WR319670 Brain
WR308245 Plasma
WR308245 Brain
Fig. 5 Brain and plasma PK profile of mefloquine and WR319670
and the formation of WR308245 in both matrices after 160 mg/kg PO
dosing. WR319670 and WR308245 each have Cmax values about one-
sixth of that of mefloquine at its Cmax. Combined, they are about one-
third that of mefloquine at Cmax
10.0
100.0
1000.0
10000.0
0 4 8 12 16 20 24
Con
cent
rati
on (
ng/g
Bra
in, n
g/m
L
Pla
sma)
Time (Hours)
WR308245 Plasma
Mefloquine Plasma
WR308245 Brain
Mefloquine Brain
Fig. 2 Brain (solid line) and plasma (dashed line) levels of
WR308245 and MQ after 5 mg/kg IV dosing of each compound.
Although WR308245 has lower plasma levels, at their respective
Cmax values, WR308245 has brain levels six times that of MQ
Eur J Drug Metab Pharmacokinet
123

4 Discussion
The library of compounds from which this data is derived
was designed with the goal of reducing BBB penetration
compared to MQ through the manipulation of a variety of
physicochemical properties (Milner et al. 2010a, b). It was
an unexpected observation in the combined in vivo and
in vitro efficacy data that exposed the prodrug nature of
WR319670. While prodrugs are commonly used to
improve bioavailability, reduce toxicity by directing
metabolism and activation near the target, or to enhance
penetration and transport across biological membranes, the
use of this approach to maintain therapeutic activity while
minimizing drug levels in the CNS is unusual.
In a PK comparison against MQ with IV dosing,
WR308245 exhibited brain concentrations nearly six times
that of MQ at Cmax. This is consistent with the observed
results in the in vitro MDCK–MDR1 assay, which sug-
gested permeability across the BBB 2–3 times that of MQ.
When MQ and WR319670 were each dosed orally, both
WR319670 and its metabolite WR308245 were present in
the brain at about 1/3 the level of MQ at Cmax. Because
WR319670 has little intrinsic activity against the malaria
parasite, as demonstrated in the [3H] hypoxanthine and
SYBR Green I in vitro assays, the drug’s activity in an
in vivo mouse model can most likely be attributed to the
formation of active metabolites, such as WR308245. Fur-
ther, a net 18-fold change in brain levels of the active
WR308245 metabolite was not associated with a demon-
strable change in efficacy, providing vindication of this
prodrug approach. Since the levels of WR308245 were
essentially equal to those of WR319670 after dosing, it is
safe to assume that it is the major metabolite contributing
to antimalarial activity. It is not contended here that the
biotransformation of WR319670 to WR308245 in any way
impacts the mechanism of BBB penetration of the latter
compound. Rather, it is simply shown that the use of
WR319670 as a prodrug maintains antimalarial efficacy
while significantly diminishing the overall drug levels in
the CNS.
Because the exact target causing adverse CNS side
effects with MQ is not known, the next generation quino-
line methanol project team has set as a goal a fivefold
decrease in overall brain concentration for any potential
lead candidates. As WR319670 does not provide an ade-
quate window under this criterion, it is not a likely can-
didate for further development and a fuller characterization
of its metabolic profile has not been deemed a priority.
However, the discovery of this prodrug strategy as a means
of maintaining efficacy in in vivo models while minimizing
CNS drug levels may prove to be an important tool in the
future design of quinoline methanol analogs.
References
Arguin PM, Mali S (2012) Malaria. CDC Yellow Book. http://
wwwnc.cdc.gov/travel/yellowbook/2012/chapter-3-infectious-
diseases-related-to-travel/malaria.htm. Accessed 12 Dec 2012
Caridha D, Yourick D, Cabezas M, Wolf L, Hudson TH, Dow GS
(2008) Mefloquine-induced disruption of calcium homeostasis in
mammalian cells is similar to that induced by ionomycin.
Antimicrob Agents Chemother 52:684–693
Chulay JD, Haynes JD, Diggs CL (1983) Plasmodium falciparum—
assessment of in vitro growth by [H-3]-labeled hypoxanthine
incorporation. Exp Parasitol 55:138–146
Chung MC, Ferreira EI et al (2008) Prodrugs for the treatment of
neglected diseases. Molecules 13:616–677
Desjardins RE, Canfield CJ, Haynes JD, Chulay JD (1979) Quanti-
tative assessment of antimalarial activity in vitro by a semiau-
tomated microdilution technique. Antimicrob Agents Chemother
16:710–718
Dow GS, Heady TN et al (2006) Utility of alkylaminoquinolinyl
methanols as new antimalarial drugs. Antimicrob Agents Che-
mother 50:4132–4143
Dow GS, Magill AJ, Ohrt C (2008) Clinical development of new
prophylactic antimalarial drugs after the 5th Amendment to the
Declaration of Helsinki. Ther Clin Risk Manag 4:803–819
Johnson JD, Dennull RA, Gerena L, Lopez-Sanchez M, Roncal NE,
Waters NC (2007) Assessment and continued validation of the
malaria SYBR green I-based fluorescence assay for use in
malaria drug screening. Antimicrob Agents Chemother 51:
1926–1933
Milhous WK, Weatherly NF, Bowdre JH, Desjardins RE (1985)
In vitro activities of and mechanisms of resistance to antifol
antimalarial drugs. Antimicrob Agents Chemother 27:525–530
Milner E, McCalmont W et al (2010a) Structure-activity relationships
amongst 4-position quinoline methanol antimalarials that inhibit
the growth of drug sensitive and resistant strains of Plasmodium
falciparum. Bioorg Med Chem Lett 20:1347–1351
Milner E, McCalmont W et al (2010b) Anti-malarial activity of a non-
piperidine library of next-generation quinoline methanols. Malar
J 9:51–60
Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P, Riscoe M
(2004) Simple and inexpensive fluorescence-based technique for
high-throughput antimalarial drug screening. Antimicrob Agents
Chemother 48:1803–1806
Toovey S (2009) Mefloquine neurotoxicity: a literature review. Travel
Med Infect Dis 7:2–6
Yelmo S, Morera-Fumero AL, Henry M, Renshaw A, Gracia-Marco
R (2010) Mania Associated With Mefloquine Prophylaxis. J Clin
Psychopharmacol 30:339–341
Eur J Drug Metab Pharmacokinet
123



![Synthesis of highly functionalized benzo[h]quinoline and ...shodhganga.inflibnet.ac.in/bitstream/10603/39020/17/17...quinoline ) (16 ) and tetracyclic quinoline (3-(epimin omethano)](https://img.dokumen.tips/doc/110x75/606a70077d4f6141007ad728/synthesis-of-highly-functionalized-benzohquinoline-and-quinoline-16.jpg)

























