Embed Size (px)
Citation preview

Eur. J. Biochem. 66, 25-36 (1976)
The Tentative Identification in Escherichia coli of a Multienzyme Complex with Glycolytic Activity John MOWBRAY and Vivian MOSES
Department of Chemical Biodynamics and Lawrence Radiation Laboratory, University of California, Berkeley
(Received October 20, 1975/February 20, 1976)
Penicillin spheroplasts of Escherichia coli were ruptured osmotically, by freezing and thawing, or mechanically. Differential centrifugation sedimented 20 - 30 % of the glycolytic enzymes without increasing their specific activities. There was, however, evidence of distinct groups of sedimenting enzymes ; growth on different carbon sources could influence the distribution. Sucrose gradient studies gave no evidence of enzyme association but provided estimations of the molecular weight of each enzyme which were close to those subsequently observed on gel filtration. Using the determined molecular weight and a literature value for specific activity, the measured activity ratio of the enzymes was compared with that expected from an equimolar mixture. All values agreed within a factor of five, except for hexokinase. The relative roles of hexokinase and phosphotransferase in E. coli are briefly considered.
An equimolar multienzyme aggregate of all the enzymes of glycolysis would have a molecular weight of about 1.6 x lo6. Chromatography on a Biogel column yielded one fraction, corresponding to a molecular weight of 1.6 x lo6, which contained a proportion of all the glycolytic enzyme studied; the remaining portion of each enzyme activity was eluted from the column at the position expected from its individual molecular weight. The fraction of mol. wt 1600000 was tested for complete glycolysis pathway activity and found not to be different from a reconcentrated mixture of the separat- ed enzymes. Both the eluted and the reconstructed systems showed unexpected activity changes at different protein concentrations. The specific radioactivity of pyruvate formed by these systems from ['4C]glucose 6-phosphate was reduced by the presence of unlabelled 3-phosphoglycerate, but by less than would have been expected had the latter been able to participate fully in glycolytic activity. This result indicates that these preparations were capable of selectivity compartmenting glycolytic intermediates.
Electron microscope investigation of both systems showed large numbers of regular 30 nm dia- meter particles which, on disruption, appeared to be composed of smaller units: it is possible that these particles may have been aggregates containing glycolytic enzymes. The possible advantages of a glycolytic multienzyme complex are briefly discussed.
The demonstration that well-known soluble meta- bolic pathways, such as fatty acid synthesis [1] or the
Enzymes. Hexokinase (EC 2.7.1 .I); hexosephosphate isomerase (EC 5.3.1.9); fructose-6-phosphate kinase, phosphofructokinase (EC 2.7.1.1 1); glyceraldehyde-3-phosphate dehydrogenase (EC 1.2.1.12); pyruvate kinase (EC 2.7.1.40); fructose-bisphosphate aldolase or aldoldse (EC 4.1.2.13); triosephosphate isomerase (EC 5.3.1.1); glycerol-3-phosphate dehydrogenase (EC 1.1.1.8); eno- lase(EC4.2.1.11); 3-phosphoglycerate kinase (EC2.7.2.3); phospho- glyceromutase (EC2.7.5.3); catalase (EC 1.11.1.6); lactate dehydro- genase(EC1.1.1.27); glucose-6-phosphatedehydrogenase(EC 1.1.1. 49) ; deoxyribonuclease (EC 3.1.4.5) ; glucosephosphate isomerase or phosphoglucose isomerase (EC 5.3.1.9) ; alanine aminotransferase (EC 2.6.1.2).
decarboxylation of pyruvate [2], are catalysed by a specific aggregation of the component enzymes has lent impetus to the idea that a significant degree of organisation may exist among apparently soluble enzyme systems. In particular, mounting evidence sug- gests that in some cells glycolytic enzymes are not freely dispersed. It has long been known that myogen, a press juice preparation from muscie containing mostly glycolytic enzymes, forms a single slow-moving peak in boundary electrophoresis [3] and that a mixed enzyme fraction from this, myogen A, crystallises readily and is monomigratory in the ultracentrifuge [4]. Histochemical staining of muscle sections showed that

26 Glycolytic Multienzyme Complexes in Escherichiu coli
lactate dehydrogenase and glyceraldehyde 3-phos- phate dehydrogenase activities were confined to the isotropic zones [5,6]. Subsequent work in Pette's laboratory has shown that several other glycolytic and related enzymes (though not hexokinase) could be located in muscle I-band [7] and that the major I-band protein, F actin [S], can bind aldolase, pyruvate kinase and glyceraldehyde-3-phosphate dehydrogenase. Lac- tate dehydrogenase and phosphoglycerate kinase are bound by F actin at low ionic strength to a lesser degree [9,10]. Myosin was found to bind the enzymes much less tightly [9]. In an electrophoretic study under cellular conditions of ionic strength, myosin and troponiyosin polymer (delta protein) formed com- plexes with some glycolytic enzymes provided the protein concentration was high [ll]. Under similar conditions, several glycolytic enzymes in a myogen preparation co-sedimented in the ultracentrifuge and their association was aided by the presence of myosin [12]. Aldolase also binds to troponin-tropomyosin complex (present in I-band too) at low ionic strength
The suggestion of an association between glycolytic enzymes and structural components is not confined to muscle. Evidence has been presented that many of these enzymes may be bound to cell membrane in red cell and yeast [14], and in retina [15]. Moreover, the internal surface of human red cell membrane appears to have high affinity binding sites for glycer- aldehyde-3-phosphate dehydrogenase and these show species specificity [16].
Such evidence of physical association is comple- mented by observations of functional compartmen- tation of glycolysis. In a variety of tissues measurement of the specific radioactivity in one or other glycolytic intermediate has yielded a value much at variance with those of its presumed metabolic precursor and product, suggesting that distinct pools of those inter- mediates exist in the cell [17-211. Lyophilised and extracted yeast has been found to respire glucose, hexose phosphates and pyruvate and to ferment glucose, fructose and mannose but not hexose phos- phates, triose phosphates or pyruvate [22]. This ob- servation suggests that the glycolytic pathway used for fermentation is distinct from that used for respi- ration. Similarly, we have found evidence in Eschr- richia coli for distinct coexisting glycolytic pathways, which contribute unequally to cell's carbon pool and are selective in their source of hexose [23,24].
It thus seemed likely that the glycolytic enzymes, present in many tissues at near equimolar amounts [25,26], might exist as a multienzyme complex. We set out to isolate such a complex, choosing E.coli, since this organism is apparently devoid of internal structures which might bind the complex or adsorb the enzymes and because it is possible to rupture the cells relatively gently if the coat is first removed.
t131.
MATERIALS AND METHODS
Cells
E. coli, Cavalli strain (met-, thy-) was obtained from Aleen Simmons (Department of Molecular Biology, University of California, Berkeley) and main- tained on minimal medium 63 [27], containing 0.9 %, glucose and supplemented with methionine 50 pg/ml and thymine 2 pg/ml.
Sp heroplust Format ion
Cells were grown overnight at 37°C in medium as described above. A portion (0.1 ml) of this station- ary suspension was transferred to 500 ml of the same medium and the culture shaken at 37 "C for 14 h. The whole 500-ml culture was then added to 4.5 1 of a nutrient medium containing (g/l) glucose, 0.9 ; NaCl, 3.15; KzHP04, 3.31; KH2P04, 1.19; Bacto yeast extract, 1.35; Bacto Peptone, 2.25; and Bacto Nutri- ent broth, 3.6. The culture was oxygenated continu- ously and stirred vigorously. When the absorbance at 650 nm reached 1.0, 2.55 1 of pre-warmed sucrose nutrient broth (composition as above and containing, in addition, 685 g/1 sucrose) was added to the culture, followed by 15.7 g of MgC12 and 14000 units of penicillin [28]. The absorbance at 650 nm normally dropped to 0.2-0.3 and then increased during the next 100-150min to about 0.7, by which time spheroplast formation (followed microscopically, magnification SO0 x ) was essentially complete. The spheroplasts were harvested at 4°C either by cen- trifuging the culture for 20 min at 9000 rev./min in the GS-3 head of a Sorvall RC-2B centrifuge and washing once in 0.85 M sucrose, or by using a Sharples separator at 35000 rev./min with a flow rate of about SO ml per minute.
Spheroplast Disruption
Osmotic. The packed spheroplasts were dispersed in an equal volume of 100 mM potassium phosphate buffer, pH 7.0, containing 5 mM MgC12, 2 mM cys- teine, 2.5 mM KCI and chloramphenicol 0.1 mg/ml, and either stored for 4- 15 h at 4 "C or cooled to - 198°C and rewarmed to 37°C three times. DNA was hydrolysed by the addition of a crystal of deoxy- ribonuclease and incubation for 5 min at 37°C. The pH was tested during the above procedures and re- aqjusted to 7 if necessary.
Mechanical. The cell sludge, cooled in ice, was homogenised for 45 s at 32000 rev./min in a Virtis 45 homogeniser, a crystal of deoxyribonuclease was added and the preparation incubated at 25°C for 5 min. The homogenisation and incubation were repeated twice more and the mixture was finally centrifuged at 13000 rev./min for 20 min in the SS-34 head of a Sorvall RC2B at 4 "C.

J. Mowbray and V. Moses 27
Differential Centr ifugut ion
To remove debris the broken cell mixture was centrifuged for 20 min at 11 000 rev./min in the GSA rotor of a Sorvall-RC2B at 4 “C. All further centrifu- gation was carried out at 4°C in a Spinco model L2-65B. The slightly milky 20000 x g supernatant was spun at 33000 rev./min in a type 50 rotor for 30 min, and the resulting 86000 x g pellet washed once in the buffer used to suspend the cells. The wash was retained. The supernatant was then subjected to 50000 rev./min in the same rotor for 90 min, yielding a translucent pellet and a final 198000 x g supernatant. As with the 86000 x g pellet, this final pellet was resuspended in the solution in which the cells were ruptured, and recentrifuged.
Density Gradient Analysis
Linear 10 - 30 ”/, sucrose gradients [29] in 100 mM potassium phosphate, 5 mM MgS04, pH 7.0, were poured at 4°C to a final volume of 13 ml. Centrifu- gation was for 15.5 h at 40000 rev./min in a Beckman Spinco L2-65B with an SW41 rotor at 4 “C. Fractions of 5 drops each were collected.
Enzyme Assays
Hexokinase was assayed by the method of Joshi and Jagannathan [30], except that 10 % of the recom- mended ATP concentration was found to be sufficient. Hexose isomerase activity was estimated with the hexokinase assay system, replacing both glucose and ATP with 3.7 mM fructose 6-phosphate. Fructose 6-phosphate kinase was assayed using system A de- scribed by Mansour [31]. Glyceraldehyde-3-phos- phate dehydrogenase activity was estimated by the method of Krebs (321, except that pyrophosphate buffer (25 mM in assay mixture, pH 8.5) was used, pyruvate kinase was assayed according to Biicher and Pfleiderer [33], while aldolase was measured using the method of Rutter et a/. [34]. Triose phosphate iso- merase activity was determined by adding 100 pl of 25 mM glyceraldehyde 3-phosphate to 2.9 ml enzyme solution containing a final concentration of 0.05 M glycylglycine buffer (pH 7.5), 0.015 M cysteine, 0.163 mM NADH and 50 pg glycerol-3-phosphate dehydrogenase. Enolase was estimated using the pyruvate kinase assay conditions in the absence of phosphoenolpyruvate and containing in addition 1 I.U. pyruvate kinase. The reaction was started by adding 100 pl of 60 mM 2-phosphoglycerate. Phospho- glyceromutase was assayed by a further extension of the enolase method: enolase (5 I.U.) was added to the reaction mixture together with enough 2,3-bis- phosphoglycerate to reach 0.04 mM ; 3-phosphoglyc- erate (60 mM) replaced 2-phosphoglycerate as the
substrate. 3-Phosphoglycerate kinase activity was determined by the method of Bucher [35], and catalase by the method of Martin and Ames [36].
Total Pathway Activity
This was studied by incubating glucose or one of the glycolytic intermediates labelled with 14C together with an appropriate quantity of enzyme preparation plus coenzymes. The pyruvate formed was transam- inated through the use of glutamic acid and alanine aminotransferase, the residual glutamic acid and the alanine formed being separated by paper chromatog- raphy. The incubation was carried out at 37 ’C in a solution containing 2.5mM KC1, 10mM ADP, 10mM potassium phosphate, 2 mM cysteine, 10 mM NAD+, 0.2 mM ATP, 5 mM MgC12,0.04 mM 2,3-bisphospho- glyceric acid, 30 mM glutamic acid, and alanine aminotransferase 2.5 units/ml, adjusted to pH 7.0 with KOH. The reaction was terminated by mixing a sample of the mixture with an equal volume of ethanol. A portion (50 pl) of the ethanol mixture was applied to Ederol No, 202 filter paper (J. C. Binzer Paperfabrik, 3559 Hatzfeld/Eder, West Germany) and chromatographed using the two-dimensional system of Bassham and Calvin [37]. Autoradiography was used to locate the spots of labelled substances which were then excised and counted by immersing them in a toluene-based scintillator solution (0.382 PPO and 0.0080/, POPOP) using a Packard Liquid Scin- tillation spectrometer model 3375. The statistical counting error was always less than 2 ”/,.
Column Chromatography
Column chromatography was performed at 0 “C using 3 cm diameter jacketed columns and an LKB refrigerated fraction collector. The flow rate was maintained at between 0.2 and 0.4 ml per minute and the 100mM potassium phosphate buffer, pH 7.2, contained 5 mM MgC12, 100 mM 2-mercaptoethanol and chloramphenicol 100 pg/ml. Fractions (20 drops ; 1.04 ml) were collected. Amino acids and protein were measured by the methods of Moore and Stein [38] and Lowry et al. [39], respectively. Haemoglobin was assayed by its absorption at 405 nm and dextran by the difference in absorbance at between 600 nm and 475 nm.
Electron Microscopy
Electron Microscopy was performed with a Siemens Elmiskop I. Fractions were placed on formvar-covered grids and negatively stained with 2 % ammonium molybdate or phosphotungstic acid.
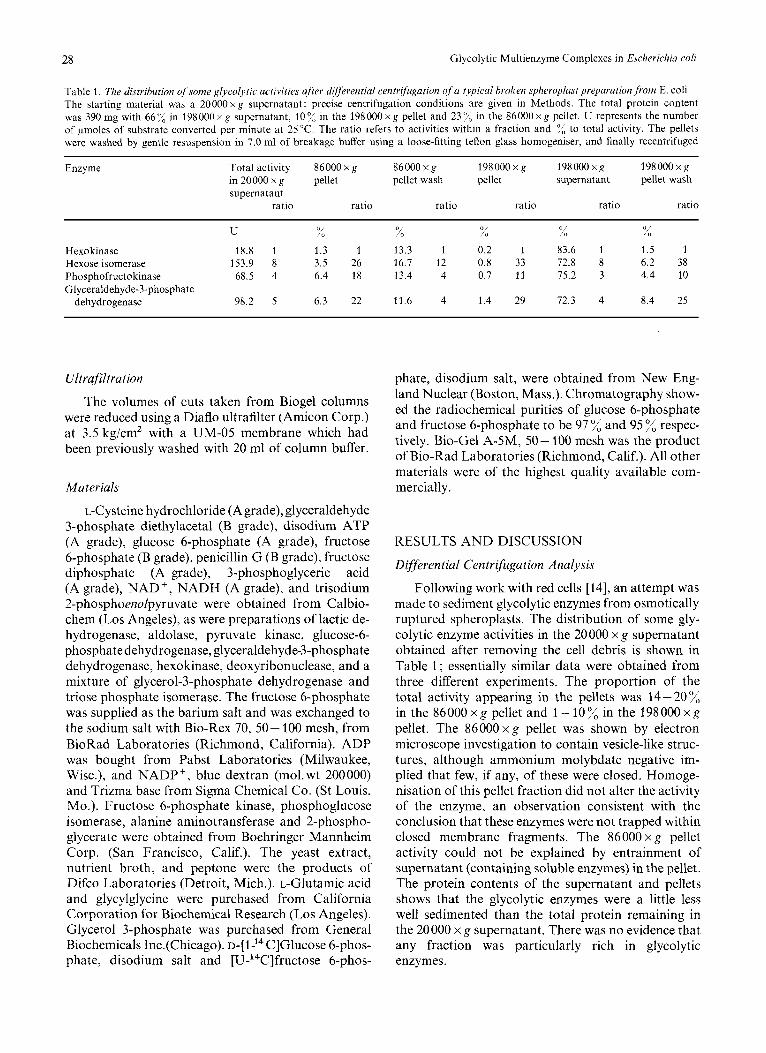
28 Glycolytic Multienzyme Complexes in Escherichia coli
Table 1. The distribution of some glycolytic activities after differential centrjfugation o j a typical broken spheroplast preparation from E. cob The starting material was a 20000 x g supernatant: precise centrifugation conditions are given in Methods. The total protein content was 390 mg with 66 % in 198 000 x g supernatant, I0 % in the 198 000 x g pellet and 23 ;< in the 86000 x g pellet. U represents the number of pmoles of substrate converted per minute at 25°C. The ratio refers to activities within a fraction and "/, to total activity. The pellets were washed by gentle resuspension in 7.0 ml of breakage buffer using a loose-fitting teflon glass homogeniser, and finally recentrifuged
Enzyme 198000 x g Total activity 86000 x g 86000 x g 198 000 x g 198000 x g in 20000 x g pellet pellet wash pellet supernatant pellet wash supernatant
ratio ratio ratio ratio ratio ratio
U % % % % % 1.5 1 Hexokinase 18.8 1 1.3 1 13.3 I 0.2 1 83.6 1
Hexose isomerase 153.9 8 3.5 26 16.7 12 0.8 33 72.8 8 6.2 38
Glyceraldehyde-3-phosphate Phosphofructokinase 68.5 4 6.4 18 13.4 4 0.7 11 75.2 3 4.4 10
8.4 25 dehydrogenase 98.2 5 6.3 22 11.6 4 1.4 29 72.3 4
Ultrafiltration
The volumes of cuts taken from Biogel columns were reduced using a Diaflo ultrafilter (Amicon Corp.) at 3.5 kg/cm2 with a UM-05 membrane which had been previously washed with 20 ml of column buffer.
Materials
L-Cysteine hydrochloride (A grade), glyceraldehyde 3-phosphate diethylacetal (B grade), disodium ATP (A grade), glucose 6-phosphate (A grade), fructose 6-phosphate (B grade), penicillin G (B grade), fructose diphosphate (A grade), 3-phosphoglyceric acid (A grade), NAD', NADH (A grade), and trisodium 2-phosphoenolpyruvate were obtained from Calbio- chem (Los Angeles), as were preparations of lactic de- hydrogenase, aldolase, pyruvate kinase, glucose-6- phosphate dehydrogenase, glyceraldehyde-3-phosphate dehydrogenase, hexokinase, deoxyribonuclease, and a mixture of glycerol-3-phosphate dehydrogenase and triose phosphate isomerase. The fructose 6-phosphate was supplied as the barium salt and was exchanged to the sodium salt with Bio-Rex 70, 50- 100 mesh, from BioRad Laboratories (Richmond, California). ADP was bought from Pabst Laboratories (Milwaukee, Wisc.), and NADP', blue dextran (mol. wt 200000) and Trizma base from Sigma Chemical Co. (St Louis, Mo.). Fructose 6-phosphate kinase, phosphoglucose isomerase, alanine aminotransferase and 2-phospho- glycerate were obtained from Boehringer Mannheim Corp. (San Francisco, Calif.). The yeast extract, nutrient broth, and peptone were the products of Difco Laboratories (Detroit, Mich.). L-Glutamic acid and glycylglycine were purchased from California Corporation for Biochemical Research (Los Angeles). Glycerol 3-phosphate was purchased from General Biochemicals Inc.(Chicago). ~ - [ l - ' ~ CIGlucose 6-phos- phate, disodium salt and [U-'4C]fructose 6-phos-
phate, disodium salt, were obtained from New Eng- land Nuclear (Boston, Mass.). Chromatography show- ed the radiochemical purities of glucose 6-phosphate and fructose 6-phosphate to be 97 % and 95 % respec- tively. Bio-Gel A-5M, 50- 100 mesh was the product of Bio-Rad Laboratories (Richmond, Calif.). All other materials were of the highest quality available com- mercially.
RESULTS AND DISCUSSION
D ifferen t ial Cen tr fuga t ion A n d y sis
Following work with red cells [14], an attempt was made to sediment glycolytic enzymes from osmotically ruptured spheroplasts. The distribution of some gly- colytic enzyme activities in the 20000 x g supernatant obtained after removing the cell debris is shown in Table 1 ; essentially similar data were obtained from three different experiments. The proportion of the total activity appearing in the pellets was 14-20% in the 86000 x g pellet and 1 - 10 % in the 198 000 x g pellet. The 86000 x g pellet was shown by electron microscope investigation to contain vesicle-like struc- tures, although ammonium molybdate negative im- plied that few, if any, of these were closed. Homoge- nisation of this pellet fraction did not alter the activity of the enzyme, an observation consistent with the conclusion that these enzymes were not trapped within closed membrane fragments. The 86000 x g pellet activity could not be explained by entrainment of supernatant (containing soluble enzymes) in the pellet. The protein contents of the supernatant and pellets shows that the glycolytic enzymes were a little less well sedimented than the total protein remaining in the 20000 x g supernatant. There was no evidence that any fraction was particularly rich in glycolytic enzymes.

J . Mowbray and V. Moses 29
Washing the pellets in buffer released a consider- able portion of their enzymic activity to the super- natant (Table 1). If an organised multienzyme complex were involved the ratio of activities in any fraction might be expected to be constant. In the case of those enzymes assayed, the activity ratios fell into two distinct groups, with the pellets and 198000 x g pellet wash in one group, and the soluble fraction and the 860OOxg pellet wash in the other. The data do not fit a simple adsorption or partition model, and further investigation of this possibility by washing the pellets repeatedly only reinforced this conclusion. However, interpretation of the results is complicated by the possibility that the observed binding may change with handling and environment. The total enzyme activities recovered in the various fractions, compared with that present in the 20000 x g supernatant were: hexokinase, 90 %; hexose isomerase, 42 ”/, ; phosphofructokinase, 298 %, ; and glyceraldehyde-3-phosphate dehydroge- nase, 125%.
Another set of observations suggests that the distribution of enzymes between fractions reflects real intracellular organisation. Cells were grown in medium in which glucose was replaced as carbon source by equal carbon quantities of either lactose or maltose. The glucose and lactose cultures were virtu- ally identical in their doubling time (60 min) and yield of cells. Maltose growth was slower (doubling time of 70 min) and yielded a cell mass only 70”/;: of the other substrates. The specific activities of both hexo- kinase and phosphofructokinase were identical in the case of the lactose and glucose-grown cells, and more than double that in the maltose culture. Further, whereas glucose and lactose yielded a sedimentation distribution very close to that in Table 1 for hexo- kinase, hexose isomerase and phosphofructokinase (the only the enzymes assayed in this experiment), in the maltose-grown cells 67 x, 67 %, and 58 %, respec- tively, of these enzymes were in the soluble fraction and a correspondingly higher proportion in the pellets (36 average compared with 26 :d in the glucose and lactose cultures). If this represents a true division of some sets of glycolytic enzymes between membrane and soluble phases, it might go some way towards explaining the observed metabolic compartmentation of hexose derived from a monosaccharide or di- saccharide source [23,24]. One possible difference be- tween the metabolism of monosaccharide and di- saccharides is that hexose may be released intracellu- larly as the 6-phosphate ester if it is derived from free hexose transport mainly by the phosphotransferase system [40,41], whereas one hexose unit from a di- saccharide will presumably require phosphorylation by hexokinase after intracellular hydrolysis. In this context it is interesting to note that the membrane fractions were relatively poor in hexokinase activity (Table 1).
Sucrose Gradient Studies
Since this technique has been successfully used in isolating multienzyme complexes from bacteria [42] and from fungi [43], it was used to investigate the aggregation of glycolytic enzymes in the 20000 x g supernatant and in the fractions obtained by differ- ential centrifugation. Far from demonstrating any co-migration, the various activities peaked separately and in identical places irrespective of the fraction applied to the gradient. Using added catalase (mol. wt 250000) and haemoglobin (mol. wt 67000) as markers, it was possible [36] to estimate the approximate molecular weight of all the enzymes (Table 2) except pyruvate kinase, which was inactived in the sucrose buffer. Table 2 also presents molecular weight esti- mates obtained using the same markers, together with blue dextran (mol. wt 2000000), on a Biogel-5M column (see below). The estimates agree very well with one another, except in the cases of phospho- fructokinase and 3-phosphoglyceric acid kinase, where the Biogel form is clearly a dimer of the species mi- grating in the sucrose gradient. Species of phospho- fructokinase with an apparent molecular weight of 205 000 were also observed. Although specific activity data are not available in the literature for these E. coli glycolytic enzymes, values from a range of other species [44] have been used in conjunction with the molecular weight estimates to predict an activity ratio for an equimolar aggregate of the enzymes [26]. These values, relative to phosphofructokinase, are set beside the values determined by assay of the fractions ob- tained by sucrose density gradient fractionation. With the exception of hexokinase, the values agree within a factor of five. The conclusion are, of course, very tentative, but considering the degree of uncertainty in these data, it is surprising that the agreement i3 so close. For hexokinase, however, the ratio is between one and two orders of magnitude lower than predicted, but because glucose transport in E. coli phosphorylates glucose at the 6-position at the expense of phospho- enolpyruvate [40,41], hexokinase might best be re- garded as a non-glycolytic enzyme in this cell. These results suggest that in E. coli the glycolytic enzymes are present in the cell at near equimolar amounts. The molecular weight of a multienzyme complex containing this ratio of enzymes would be about 1 600000.
Column Chromatography
Provided it did not dissociate when diluted, a multi- enzyme complex with a molecular weight over 1 x lo6 would be expected to separate easily from the indi- vidual glycolytic enzymes by molecular sieve tech- niques. An attempt was therefore made to separate all species with molecular weights over 0.5 x lo6 by
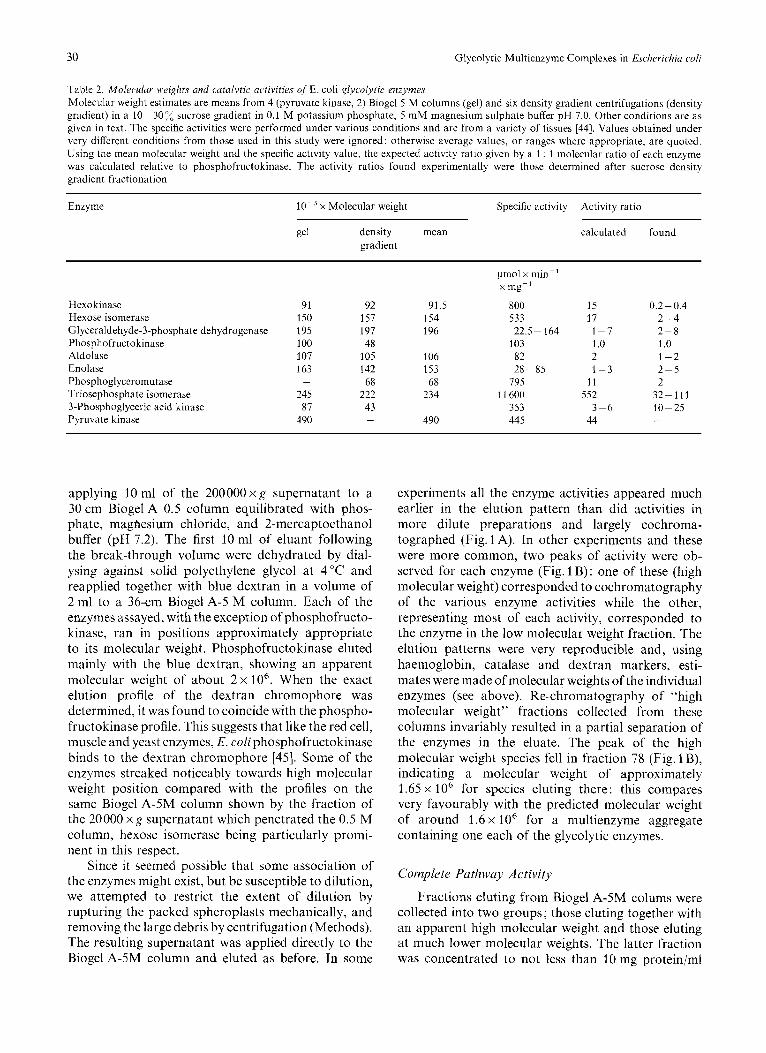
30 Glycolytic Multienzyme Complexes in Escherichia co/i
Table 2. Molecdur weights and cutatytic activities of E. coli glycolytic enzymes Molecular weight estimates are means from 4 (pyruvate kinase, 2) Biogel 5 M columns (gel) and six density gradient centrifugations (density gradient) in a 10-30”/, sucrose gradient in 0.1 M potassium phosphate, 5 mM magnesium sulphate buffer pH 7.0. Other conditions are as given in text. The specific activities were performed under various conditions and are from a variety of tissues 1441. Values obtained under very different conditions from those used in this study were ignored; otherwise average values, o r ranges where appropriate, are quoted. Using the mean molecular weight and the specific activity value, the expected activity ratio given by a 1 : 1 molecular ratio of each enzyme was calculated relative to phosphofructokinase. The activity ratios found experimentally were those determined after sucrose density gradient fractionation
Enzyme x Molecular weight Specific activity Activity ratio
gel density mean gradient
calculated found
Hexokinase Hexose isomerase Glyceraldehyde-3-phosphate dehydrogenase Phosphofructo kinase Aldolase Enolase Phosphogl yceromutase Triosephosphate isomerase 3-Phosphoglyceric acid kinase Pyruvate kinase
91 150 195 100 107 163
245 87 490
-
92 157 197 48 105 142 68 222 43 -
91.5 154 196
106 153 68 234
490
pmol x min-’ x mg-‘
800 533 22.5- 164 103 82
795 1 1 600
353 445
28-85
15 17 1-7 1.0 2 1-3
1 1 552
44 3-6
0.2-0.4 2-4 2-8 1 .o 1-2 2-5 2 32-111 10-25 -
applying 10 ml of the 200000 x g supernatant to a 30 cm Biogel A 0.5 column equilibrated with phos- phate, magnesium chloride, and 2-mercaptoethanol buffer (pH 7.2). The first 10 ml of eluant following the break-through volume were dehydrated by dial- ysing against solid polyethylene glycol at 4 “C and reapplied together with blue dextran in a volume of 2 ml to a 36-cm Biogel A-5 M column. Each of the enzymes assayed, with the exception of phosphofructo- kinase, ran in positions approximately appropriate to its molecular weight. Phosphofructokinase eluted mainly with the blue dextran, showing an apparent molecular weight of about 2 x lo6. When the exact elution profile of the dextran chromophore was determined, it was found to coincide with the phospho- fructokinase profile. This suggests that like the red cell, muscle and yeast enzymes, E. coli phosphofructokinase binds to the dextran chromophore [45]. Some of the enzymes streaked noticeably towards high molecular weight position compared with the profiles on the same Biogel A-5M column shown by the fraction of the 20000 x g supernatant which penetrated the 0.5 M column, hexose isomerase being particularly promi- nent in this respect.
Since it seemed possible that some association of the enzymes might exist, but be susceptible to dilution, we attempted to restrict the extent of dilution by rupturing the packed spheroplasts mechanically, and removing the large debris by centrifugation (Methods). The resulting supernatant was applied directly to the Biogel A-5M column and eluted as before. In some
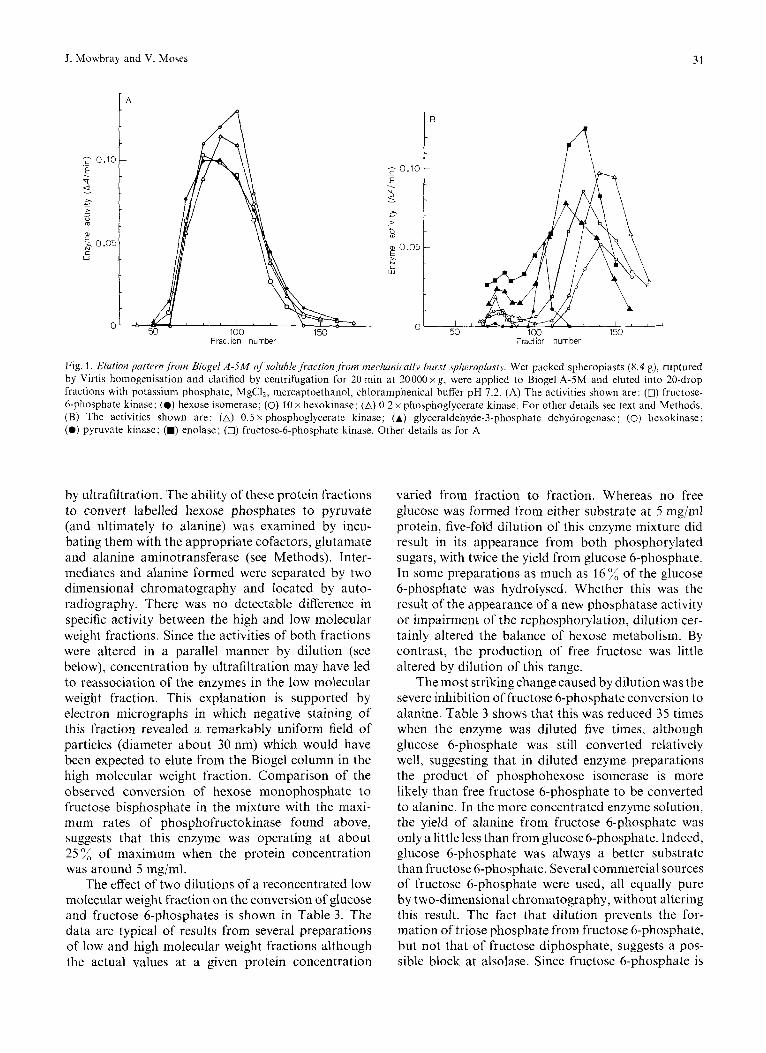
experiments all the enzyme activities appeared much earlier in the elution pattern than did activities in more dilute preparations and largely cochroma- tographed (Fig. 1 A). In other experiments and these were more common, two peaks of activity were ob- served for each enzyme (Fig. 1 B) : one of these (high molecular weight) corresponded to cochromatography of the various enzyme activities while the other, representing most of each activity, corresponded to the enzyme in the low molecular weight fraction. The elution patterns were very reproducible and, using haemoglobin, catalase and dextran markers, esti- mates were made of molecular weights of the individual enzymes (see above). Re-chromatography of “high molecular weight” fractions collected from these columns invariably resulted in a partial separation of the enzymes in the eluate. The peak of the high molecular weight species fell in fraction 78 (Fig. 1 B), indicating a molecular weight of approximately 1.65 x lo6 for species eluting there: this compares very favourably with the predicted molecular weight of around 1.6 x lo6 for a multienzyme aggregate containing one each of the glycolytic enzymes.
Complete Pathway Activity
Fractions eluting from Biogel A-5M colums were collected into two groups; those eluting together with an apparent high molecular weight and those eluting at much lower molecular weights. The latter fraction was concentrated to not less than 10 mg protein/ml
J. Mowbray and V. Moses
I A
31
B A
Fig. 1. Elution paitri.n,frotn Biogel A-5M nf.solublefraction,from mcchanicaily burst sphrropbsts. Wet packed spheroplasts (8.4 g), ruptured by Virtis homogenisation and clarified by centrifugation for 20 min at 20000 xg, were applied to Biogel A-5M and eluted into 20-drop fractions with potassium phosphate, MgCIZ, mercaptoelhanol, chlordmphenical buffer pH 7.2. (A) The activities shown are: (0) fructose- 6-phosphate kinase; (0) hexose isomerase; (0) 10 x hexokinase; (A) 0.2 x phoaphoglycerate kinase. For other details see text and Methods. (B) The activities shown are: (A) 0.5 x phosphoglycerate kinase; (A) glyceraldehyde-3-phosphate dehydrogenase; (0) hexokinase; (0) pyruvate kinase; (M) enolase; (0) fructose-6-phosphate kinase. Other details as for A
by ultrafiltration. The ability of these protein fractions to convert labelled hexose phosphates to pyruvate (and ultimately to alanine) was examined by incu- bating them with the appropriate cofactors, glutamate and alanine aminotransferase (see Methods). Inter- mediates and alanine formed were separated by two dimensional chromatography and located by auto- radiography. There was no detectable difference in specific activity between the high and low molecular weight fractions. Since the activities of both fractions were altered in a parallel manner by dilution (see below), concentration by ultrafiltration may have led to reassociation of the enzymes in the low molecular weight fraction. This explanation is supported by electron micrographs in which negative staining of this fraction revealed a remarkably uniform field of particles (diameter about 30 nm) which would have been expected to elute from the Biogel column in the high molecular weight fraction. Comparison of the observed conversion of hexose monophosphate to fructose bisphosphate in the mixture with the maxi- mum rates of phosphofructokinase found above, suggests that this enzyme was operating at about 25% of maximum when the protein concentration was around 5 mg/ml.
The effect of two dilutions of a reconcentrated low molecular weight fraction on the conversion of glucose and fructose 6-phosphates is shown in Table 3. The data are typical of results from several preparations of low and high molecular weight fractions although the actual values at a given protein concentration
varied from fraction to fraction. Whereas no free glucose was formed from either substrate at 5 mg/ml protein, five-fold dilution of this enzyme mixture did result in its appearance from both phosphorylated sugars, with twice the yield from glucose 6-phosphate. In some preparations as much as 16 of the glucose 6-phosphate was hydrolysed. Whether this was the result of the appearance of a new phosphatase activity or impairment of the rephosphorylation, dilution cer- tainly altered the balance of hexose metabolism. By contrast, the production of free fructose was little altered by dilution of this range.
The most striking change caused by dilution was the severe inhibition of fructose 6-phosphate conversion to alanine. Table 3 shows that this was reduced 35 times when the enzyme was diluted five times, although glucose 6-phosphate was still converted relatively well, suggesting that in diluted enzyme preparations the product of phosphohexose isomerase is more likely than free fructose 6-phosphate to be converted to alanine. In the more concentrated enzyme solution, the yield of alanine from fructose 6-phosphate was only a little less than from glucose6-phosphate. Indeed, glucose 6-phosphate was always a better substrate than fructose 6-phosphate. Several commercial sources of fructose 6-phosphate were used, all equally pure by two-dimensional chromatography, without altering this result. The fact that dilution prevents the for- mation of triose phosphate from fructose 6-phosphate, but not that of fructose diphosphate, suggests a pos- sible block at alsolase. Since fructose 6-phosphate is

32 Glycolytic Multienzyme Complexes in Escherichia coli
Table 3. Conversion of labelled substrate by ajraction from a Biogel column containing all the glycolytic enzymes Low molecular weight fractions from a Biogel A-5M column were collected and concentrated by ultrafiltration (Methods). A portion (500 ~ 1 ) of this solution in chromatography buffer, containing the amount of protein shown, was added to 500 pl of a 2 x concentrated buffer mixture containing glycolytic co-factors together with 30 mM glutamate, alanine amino transferase and 20 mM l4C-1abelled substrate (0.025 Ci per mol). The reaction was stopped with ethanol after 1 h and the intermediates separated, identified and 14C measured. For additional details see Methods. A sample (200 pl) was taken at zero time as a blank. The 14C content of each sample is presented as a percentage of the 14C initially present in the hexose monophosphate used as substrate
Substrate Amount l4C content of of substrate
glucose fructose hexose mono- fructose triose alanine phosphates bisphosphate phosphate (glucose 6-phosphate and fructose 6-phosphate combined)
mgprotein/ % reaction mixture
Glucose 6-phosphate 5.0 0 1.5 37.9 26.5 6.3 9.1 Fructose 6-phosphate 5.0 0 2.4 29.1 28.8 6.8 7.3 Glucose 6-phosphate 1 .0 6.8 1.5 36.5 28.5 3.1 1.2 Fructose 6-phosphate 1 .0 3.7 0.9 70.8 28.2 0 0.2
known to be a competitive inhibitor of class I1 al- dolases (Ki for yeast 4 mM, pH 7.5, 30 "C) [34], this might explain the apparent preference for glucose phosphate over fructose phosphate. However, it seems less likely that a modest change in enzyme protein concentration from 5 mg/ml to 1 mg/ml would so alter the effective concentration of the 20mM fructose 6-phosphate that there would be a change from almost no inhibition compared with glucose 6-phosphate at 5 mg/ml to virtually complete inhibition (1 mg/ml, Table 3). Thus, if aldolase inhibition is a realistic mechanism in the dilute enzyme solution the data suggest that this enzyme is being protected by means other than inhibitor sequestration at the higher con- centration. Taken together, these data emphasise how a change in protein concentration may alter the balance of metabolism of hexose phosphates. Although the implication is that protein-protein interactions are important in specifying the overall activity, the data do not necessarily imply the existence of an organised multienzyme aggregate.
The last point was investigated by measuring the complete pathway activity using 20 mM ['4C]glucose 6-phosphate as substrate, with or without added 3-phosphoglyceric acid (20 mM) as competitor, and concentrated low molecular weight fraction from a Biogel A5M column as enzyme source. Samples were taken at zero time and during a three-hour incubation period, and enzymic activity was terminated by adding an equal volume of ethanol, as before. Alanine and glucose 6-phosphate were isolated chromatographi- cally from each sample, assayed for 14C content, and the chemical amounts present determined with nin-
hydrin and phosphomolybdate [46], respectively. Although the yield of alanine varied widely with different preparations (from 0.05-0.2 pmol x mg protein-' x hour-'), it was produced linearly over the whole time of incubation and there was no signifi- cant difference between incubations in the presence and absence of phosphoglycerate. Pyruvate kinase may be rate-limiting in these conditions, although added purified rabbit muscle pyruvate kinase did not enhance alanine production. In the presence of phos- phoglycerate there was some exchange of carbon be- tween the hexose phosphate and triose phosphate pools but this did not reduce the specific activity of hexose phosphate by more than about 10% after 1 h. In four experiments in the absence of phospho- glyceric acid, the specific radioactivity of the [1-14C]- glucose 6-phosphate supplied was 110000 dis. x min- ' xpmol-', and that of the alanine formed after 1 h incubation was 46000 & 5000 dis. x min-' x pmol-'. In parallel experiments containing 20 mM phospho- glycerate the specific activity of alanine was reduced to 10000 f 600 dis, x min-' x pmol-l, only 22% of the earlier value, showing that a considerable amount of unlabelled triose had been converted to alanine. However, at the time those samples were taken, the specific activity of phosphoglycerate in the mixtures which had been made 20mM in this substance was 2800 f 300 dis. x min-' x pmol-', nearly 20 times lower than its specific activity (54000 & 6000 dis. x min-' x pmol-') in the absence of added unlabelled phosphoglycerate. Moreover, the specific activity of the phosphoglycerate after 1 h of incubation, being lower than that of glucose 6-phosphate, its precursor,

J. Mowbray and V. Moses 33
and therefore a function increasing with time, must have been even lower during the course of the experiment than it was at the end of 1 h. Thus, the specific activity of alanine produced was several times higher than that of its apparent precursor. An obvious possible explanation is that the phosphoglycerate pool was not homogeneous and that part of it had a much higher specific activity than the average value. These data therefore demonstrate that alanine arose preferentially from that phosphoglycerate deriving from hexose phosphate. For this to have occurred in a stirred cell-free solution implies that there must have been selective transfer of metabolic intermediates between pathway enzymes. So the enzyme mixture used in this experiment appears capable of catalysing some sequential steps in glycolysis without allowing equilibration of substrate or product with the bulk phase. This is exactly the property to be expected of an organised multienzyme complex, and suggests that the observed aggregation of glycolytic enzymes is unlikely to be random and in all probability represents the state in vivo of this series of enzymes.
Electron Microscopy
Fractions obtained from Biogel columns were investigated by electron microscopy after negative staining with ammonium molybdate or phospho- tungstic acid. Fractions collected from the high mo- lecular weight eluate showed large numbers of par- ticles of diameter 30 - 40 nm. A very few larger species with the appearance of small vesicle fragments could also be seen. Rather surprisingly the collected low molecular weight fractions concentrated by ultra- filtration showed uniform fields containing par- ticles with a diameter about 30 nm (Fig. 2) almost indistinguishable from those in the high molecular weight eluate. The uniform appearance of these par- ticles strongly suggests that they were not random aggregates. Dilution leads to dissociation of the aggre- gates and since no particle of this size would be ex- pected to penetrate the column as easily as a single, free glycolytic enzyme, it is possible that the particles reform after concentration by ultrafiltration of the dilute low molecular weight fraction. It further seems likely, in view of the channelling of the complete pathway activity observed in this reconcentrated frac- tion, that the glycolytic enzymes also aggregate after ultrafiltration. Whether the particles represent aggre- gates of the glycolytic enzymes is not known. Attempts to investigate further the structure of the particles by washing the formvar grid after application of the enzyme solution, but prior to fixing with phospho- tungstic acid, demonstrated that the aggregates may be composed of between seven and thirteen smaller units (Fig. 2).
General Discussion
The data presented show that the glycolytic en- zymes from E. coli can self-aggregate. Although ap- proximately equimolar amounts of the glycolytic enzymes, apart from hexokinase, are present in the cells, there seems no apriori reason for expecting a random association with a molecular weight close to that predicted for an equimolar complex. The obser- vation ofjust such an aggregation in the elution pattern from molecular seive columns and in electron micro- graphs suggests that the association is specific. Whether a structural protein is additionally required for complex formation, as it appears to be in muscle [10,12], is not known. Further, the overall catalytic properties of a fraction containing all the enzymes of glycolysis and showing aggregation are appreciably altered by changing the protein concentration, and can exhibit selective compartmentation of substrate. Taken together, these findings imply that glycolytic activity in E. coli K12 is catalysed by a complex of the enzymes in equimolar proportions. The association of such complexes with different sources of glucose 6-phosphate (e.g. from hexokinase or from the phos- photransferase system) could form a basis for ex- plaining the compartmentation of glycolytic substrates produced from glucose derived intracellularly as op- posed to glucose supplied free in the external medium [23,24]. The observation that growth on different sugars can lead to a redistribution of some enzyme activities among fractions, as revealed by differential centrifugation, may reflect differences in the pro- portions of complexes associated with the different phosphorylation systems in response to changes in carbon source.
It seems reasonable to ask what advantages a gl ycolytic multienzyme complex might confer because, at first sight, the vast differences in turnover number between different enzymes (Table 2) suggest that equi- molar quantities of all the enzymes are unnecessary : a molar ratio of triose phosphate isomerase to dehy- drogenase of 100: 1 , for example, would seem ade- quate. The advantages inherent to all multienzyme complexes of increased efficiency and control potential have been pointed out [47]. More recently Atkinson [48] has made a case that the solvent capacity of the cell may be so limited that it would not be possible to reconcile the capacity to solvate all “soluble” enzymes at a concentration for each enzyme allowing sufficient catalytic activity. Such a situation would lead to competition for solvent between small mole- cules and proteins, and the consequent necessity of maintaining the former in as low a concentration as feasible to prevent “salting out” of enzymes. Indeed, Sols and Marco [49] have shown, by consideration of binding constants and available binding sites that, in eukaryotic cells, many metabolites (and notably

34 Glycolytic Multienzyme Complexes in Escherichiu coli
Fig. 2. Fraction containing glycol-vtic enzvrncs negatively stained with phosphotungstic acid. The sample shown was from fractions 105 - 180, Fig. I B, collected and concentrated by ultrafiltration (see Methods). Following application of enzyme solution, the grid was washed once with water to damage the particle organisation prior to negative staining. Some disintegrated particles can he seen: in other areas (not shown) extensive disintegration took place. The distance bar represents 100 nm, magnification 256000
several glycolytic intermediates) are probably mainly orders of magnitude smaller than for the same sub- protein-bound. This phenomenon is presumably stances in aqueous solution [50- 541. Thus, the choice largely responsible for the finding that diffusion coef- of multienzyme aggregates with restricted diffusion ficients for small molecules within the cell are several paths may be forced on the cell by the need to achieve

J. Mowbray and V. Moses 35
efficient function ar very low free metabolic concen- trations.
The glycolytic pathway plays a major role in both energy production and biosynthesis. The latter role can be very extensive, glycolytic intermediates being the source of all other hexoses, pentoses, amino-sugars (and hence polysaccharides), glycero-lipids, serine, cysteine, glycine, phenylalanine, tyrosine, tryptophan, alanine, valine, isoleucine and leucine. These functions may conflict in certain conditions, and it may be advantageous, by localising some complexes within the cell, to earmark some glycolytic capacity for a given function. An example where this seems obviously beneficial is the siting of glycolytic enzymes within the isotropic zone of skeletal muscle [7] where they can be maximally responsive to the need for con- traction energy. Since large changes in glycolytic rate occur in muscle depending on energy demand, such sequestration may allow inhibition of the major por- tion of these enzymes in resting conditions without impairing the tissue’s biosynthetic function. In ad- dition, muscle glycolysis has to be able to show vast changes in rate over a very short time. To achieve such a complete activity change without any appreciable accumulation of intermediary products [SS] may re- quire the restricted environment and coordinated con- trol offered by a multienzyme complex.
The work reported here was sponsored by the United States Atomic Energy Commission. We are grateful to the Carnegie Trust for a Senior Scholarship for J . M. and to Dr Melvin Calvin for suppoi-t and encouragement. For advice and help with electron micrographs we arc indebted to Miss Ann M. Hughes and Dr R. B. Park of this laboratory; to Dr Robley C. Williams of the Virus Laboratory and Department of Molecular Biology, University of California, Berkeley ; and to Miss Carol Ilavie of the Biochcmistry Department, University Collcge London.
REFERENCES 1. Lynen. J., Oesterhelt, D., Schweiaer, E. & Willeke. K. (1968)
in Cellular ConipurtniPntira/iori and Control of Fair], Arid Mriaholism (Gran, F. C., ed.) pp. 1-24, Academic Press, London and New York.
2. Reed, L. J. & Oliver, R. M. (1968) Brookhaven S.rmp. Bid . 21,
3. Amberson, W. R., Bauer, A. C., Philpott, D. E. & Raisen, F.
4. Baranowski, T. & Niederland, T. R. (1949) J . Biol. Chem. 180,
5. Fahimi, H. D. & Amardsingham, C. R. (1964) J . Cell Bid. 22,
6. Brandau, H . &Pette, D. (1966) Enzymol. b id . elin. 6 , 123- 156. 7. Siegel, P. & Pette, D. (1969) J . Histoehem. Cytochem. 17,
8. Gergely, J . (1966) Annu. Rev. Biochem. 35, 691 -722. 9. Arnold, H. & Pette, D. (1968) Eur. J . Biochrm. 6 , 163-171.
397-412.
(1964) J . CdI, Comp. Pliysiol. 63, 7-24.
543- 551.
29-48.
225- 237.
10. Arnold, H., Henning, R. & Pette, D. (1971) Eur. J . Biochrm. 22, 121 - 126.
11. Amberson, W. R. & Bauer, A. C. (1971) .I. Cell Physiol. 77.
12. Clarke, F. M. & Masters, C. J. (1974) Binchim. Bio~?hy,s. A m ,
13. Clarke, F. M. , Masters, C. J . & Winzot, D. J . (1974) Biochcwr.
14. Green, D. E., Murer, E., Hultin, H. O., Richardson, S. H. , Salmon, B., Brierley, G. P. & Baum, H. (1965) Arch. Biochrm.
15. Cecchi, X., Canessa-Fischer, M., Maturana, A. & Fischer. S.
16. McDaniel, C. F., Kirtley, M. E. & Tanner, M. J. A. (1974) J .
17. Moses, V., Holm-Hansen, O., Bassham. J . A. & Calvin, M.
18. Landau, B. R . & Sims, E. A. H. (1967) J . B id . Chrm. 242.
19. Kalant, N. & Beitner, R . (1971) . J . Biol. Clrc~w~. 246. 504-507. 20. Mowbray, J. & Ottaway, J . H. (1973) Eur. J . Biochertr. 36,
21. Das, 1. &Chain, E. B. (1973) Biocliem. Soc. Tram. I , 599-601. 22. Rothstein, A,, Jennings, D. H., Dennis, C. & Bruce. M. (1959)
23. McBricn, D. C. H. & Moses, V. (1968) J . Grn. Microhiol. 51.
24. Macnab, R., Moses, V. & Mowbray, J . (1973) Eur. J . Biochenr.
25. Pctte, D., Luk, W. & Bucher, Th. (1962) Bioc,hmi. Biophy.s.
26. Mier, P. D. & Cotton, D. W. K. (1966) Naturt, iLond.) 209,
27. Pardee, A. B. & Prestidge, L. S. (1961) Biochini. Bi(qJhy.7. Actu.
28. Tonomura, B. & Rabinowit;., J . C. (1967) J . Mol. Biol. 24. 177 - 202.
29. NoII, H. (1969) in Techniques in Protein Bio.syn/hc.c.is (Campbell, P. N. & Sargent, J. R. . eds) vol. 2, pp. 101 - 180. Academic Press, London and New York.
30. Joshi, M. D . & Jagannathan, V. (1966) Mcihod.s Enn-jmol. Y.
31. Mansour, T. E. (1963) J . B id . Cli~rn. 238, 2285-2292. 32. Krebs, E. G. (1955) Mrthods Lnzyniol. I , 407-41 1. 33. Bucher. Th. & Pfleiderer, G. (1955) Meihodi Enz.ynzol. I , 435-
440. 34. Ruttcr, W. J., Hunsley. J . R., Groves, W. E., Calder, J . , Ka,i-
kumar, T. V. & Woodfin, B. M. (1966) h4eihod.s Enzym)l .
281 - 300.
358, 193-207.
J. 139, 785- 788.
BlOJ)/JjS. 112, 635- 647.
(1971) Arch. Biochcm. Biopl7ys. 145, 240- 247.
Bid . Chem. 249, 6478 - 6485.
(1959) J . Mol. Bid. I . 21-29.
163 - 172.
369- 379.
Biochrm. J . 71, 99 - 106.
159-172.
34, 15-19.
Res. Commun. 7, 419-424.
1022- 1023.
49, 77-88.
371 -381.
9, 479-491. 35. Bucher, Th. (1955) Methods Enq>mol. I , 415-422. 36. Martin, R. G. & Ames, B. N . (1961) J . Biol. Chefit. 236,
1372- 1379. 37. Bassham, J. A. & Calvin, M. (1957) in Thc Puth o/ Curhon
in Pho/o.rynthesis, Prentice-Hall, Inc., Unglewood Cliffs, N. J. wood Cliffs, N.J.
38. Moore, S. & Stein, W. 13. (1954) J . Biol. Chem. 211, 907-913. 39. Lowry, 0. H., Rosebrough, N. J., Farr, A. L. & Randall, R. J.
40. Roseman, S. (1969) J . G m . Phjsiol. 54, 139-180. 41. Roseman, S. (1972) in Metabolic Pathways (Hokin, L. E., ed.)
vol. 6, pp. 42-89, Academic Press, New York and London. 42. Gaertner, F. H. & DeMoss, J. A. (1969) J . Bid. Chem. 244,
2716- 2125. 43. Berlyn, M. B. & Giles, N. H. (1969) J . Bacteriol. 99, 222-230. 44. Barman, T. E. (1969) Enz-yrne Handbook, vol. 1, 2, Springer-
45. Kopperschlager, G., Diezel, W., Freyer, R., Liebe, S. & Hof-
46. Chen, P. S., Jr, Toribdrd, T. Y. & Warner, H. (1956) Anal.
(1951) J . Biol. Chem. 193, 265-275.
Verlag, Berlin, Heidelberg, New York.
mann, E. (1971) Eur. J . Biochrm. 22, 40-45.
Che~7. 28, 1756- 1758.

36 J. Mowbray and V. Moses: Glycolytic Multienzyme Complexes in Escherichia coli
47. Reed, L. J. & Cox, D. J. (1966) Annu. Rev. Biochem. 35, 57- 84. 48. Atkinson, D. E. (1969) Curr. Top. Cell. Regut. I , 29-42. 49. Sols, A. & Marco, R. (1970) Curr. Top. Cell. Regul. 2,227-273. 50. Harris, E. J. (1957) J. Gen. Physiol. 41, 169-195.
52. Fenichel, I. R. & Horowitz, S. B. (1963) Acta Physiol. Scand.
53. Dick, D. A. T. (1964) J . Theor. Biol. 7, 504-531. 54. Ling, G. N. & Cope, F. W. (1969) Science (Wash. D.C.) 163,
55. Helmreich, E. & Cori, C. F. (1965) Adv. Enzyme Re&. 3,
60, Suppl. 221.
51. Harris, E. J. & Prankerd, T. A. J. (1957) J . Gen. Physiol. 41, 1335- 1336. 197-218.
91 - 107.
J. Mowbray, Department of Biochemistry, University College London, Cower Street, London, Great Britain, WCIE 6BT
V. Moses, Department of Plant Biology and Microbiology, Queen Mary College, London, Great Britain, El 4NS











![Methods for quantification of cerebral glycolytic ...€¦ · Methods for quantification of cerebral glycolytic metabolism using 2-deoxy-2-[18F]fluoroglucose in small animals Silvana](https://img.dokumen.tips/doc/110x75/5ffc7d36312aae2966337c5a/methods-for-quantification-of-cerebral-glycolytic-methods-for-quantification.jpg)

















