Embed Size (px)
Citation preview

Pergamon
Geochimica et Cosmochimica Acta, Vol. 61, No. 14, pp. 3013-3018, 1997 Copyright 0 1997 Elsevier Science Ltd Printed in the USA. All rights reserved
0016-7037/97 $17.00 + .OO
PI1 SOO16-7037( 97)00122-l
SCIENTIFIC COMMENT
The surface chemistry and structure of acid-leached albite: New insights on the dissolution mechanism of the alkali feldspars
ROBERT GOUT, I ERIC H. OELKERS, ’ JACQUES SCHOIT, ’ and ANTOINE ZWICK’ ‘Laboratoire de GBochimie, CNRS UMR 5563, UniversitC Paul Sabatier, 31400 Toulouse, France
‘Laboratoire de Physique du Solide, CNRS-UniversitB Paul Sabatier, 31062 Toulouse Cedex, France
(Received September 25, 1996; accepted in revised form March 10, 1997)
Abstract-The surfaces of fresh albite crystals, and of albite crystals following their two hour leaching in an aqueous 0.3 molal HCl solution at a temperature of 2OO”C, were analyzed by Raman spectroscopy, scanning electron microscopy, and Energy Dispersive Spectroscopy (EDS ) . Scanning electron micros- copy reveals that dissolution is nonuniform and reacted surfaces exhibit a range from fresh to extremely altered regions, the latter characterized by the presence of etch pits and crater-like structures. Energy dispersive spectroscopy indicates that the chemical composition of the near surface is heterogeneous; the most altered regions have significantly lower Na/Si and Al/Si ratios compared to both unreacted albite and the fresher appearing regions of reacted albite. This result demonstrates that ( 1) initial dissolution produces spatially discontinuous altered layers, and (2) the depletion of both alkali cations and aluminum from the near surface region is associated with locally enhanced alkali-feldspar dissolution. This latter observation is strong evidence that the inverse relationship observed between far from equilib- rium constant temperature/pH alkali feldspar dissolution rates and aqueous alkali cation and/or aluminum concentration stems from a reaction mechanism involving the selective removal of these elements from the mineral structure. The Raman spectra of increasingly altered surface regions confirm that alkali feldspar dissolution is the result the sequential breaking of bonds in the mineral structure. A close correspondence is apparent between the Raman spectra of amorphous silica and the reacted albite’s most altered regions. As altered layers on albite surfaces are discontinuous, the accurate determination of their thickness requires techniques which can distinguish chemical differences on a micron scale. Copyright 0 1997 Elsevier Science Ltd
1. INTRODUCTION
The dissolution mechanism of alkali feldspars is of particular interest because, as they constitute nearly one half of the volume of the crust, their dissolution plays a major role in surficial weathering processes, diagenesis of sedimentary rocks, and mass transfer in hydrothermal systems. Much recent controversy, however, surrounds this mechanism and the identity of the rate controlling surface complex (cf. Blum, 1994; Blum and Stillings, 1995). Blum and Lasaga ( 1988, 1991) suggested that protonated surface aluminum hydrolyls are the rate limiting precursor complexes of feld- spar dissolution. Brady and Walther ( 1989, 1992) concluded that albite dissolution rates are controlled by the protonation of surficial aluminum sites at acidic conditions, but by the deprotonation of surticial silicon sites at basic conditions. Oelkers and Schott (1992, 1995a,b), Oelkers et al. (1994), and Gautier et al. ( 1994) described alkali feldspar dissolu- tion as a three step process consisting of ( 1) the relatively rapid exchange of hydrogen and alkali ions near the mineral surface, (2) an exchange reaction between three hydrogen atoms in solution with one aluminum atom in the mineral structure resulting in the breaking of Al-O bonds, coupled with the formation of a rate controlling Si rich precursor
complex, and (3) the hydrolysis of Si-0 bonds releasing the precursor complexes into solution. In contrast, Brantley and Stillings ( 1994, 1996) proposed that an exchange of hydro- gen for alkali cations forms the rate controlling surface com- plex.
Much of the effort to characterize the alkali feldspar hy- drolysis mechanism has focused on the nature of the surface during and after dissolution. Techniques commonly used to illuminate the nature of this mineral surface include titration (Wollast and Chou, 1992), the stoichiometry of element release during initial dissolution (Schweda, 1989)) X-ray photoelectron spectroscopy (XPS ) (Bemer and Holdren, 1979), secondary ion mass spectrometry (SIMS; Nesbitt and Muir, 1988), and ion beam techniques (Petit et al., 1989). The bulk of these techniques indicate the presence of alkali cation and/or Al depleted layers at both acidic and basic conditions (Nesbitt and Muir, 1988; Casey et al., 1988, 1989a,b; Petit et al., 1989; Muir and Nesbitt, 1991; Nesbitt et al., 1991; Hellmann et al., 1990; Hellmann, 1994). One major limitation of these analytical techniques is that they provide little information on the degree of spatial heteroge- neity of the dissolution process which may be significant (Bemer and Holdren, 1979; Bemer et al., 1985; Schott and
3013

3014 R. Gout et al.
Petit, 1987; Hochella et al., 1988). Consequently the inter- pretation of these data commonly relies on the assumption that changes of the near surface structure and composition due to mineral-solution interaction is spatially homogeneous.
In the present study Raman spectroscopy, scanning elec- tron microscopy, and Energy Dispersive Spectroscopy (EDS) are used to spatially characterize the structure and composition of alkali feldspar surfaces during its initial dis- solution. Results obtained from these analyses are used to assess the homogeneity of surface processes and to establish a direct link between the depletion of elements in the albite near surface and its dissolution. The purpose of this commu- nication is to present the results of these analyses and to apply them to the better understanding of the alkali feldspar dissolution mechanism.
2. EXPERIMENTAL METHODS
Natural albite crystals originating from Amelia Courthouse, Vir- ginia, USA, of an average length of 1.5 cm were obtained from Wards Natural Science. Four crystals were placed in a titanium mixed flow reactor and dissolved for 2 h in an aqueous 0.3 m HCl solution flowing through the reactor at 10 g/min at a temperature of 2OO”C, assuring a reactive solution extremely undersaturated with respect to all possible solid phases. The reactive fluid consisted of deionized HZ0 and Merck reagent grade HCl. The reactor system was identical to that used for the dissolution of albite and K-feldspar described by Oelkers et al. (1994) and Gautier et al. ( 1994). Pump- ing of the inlet fluid through the system began immediately after the crystals were placed in the reactor. The reactor attained a temperature of 200°C within 20 min, and was maintained at this temperature for 2 h. The reactor was then cooled in less than 10 min to 25°C and the albite grains subsequently removed from the liquid. These crys- tals were dried immediately by blowing nitrogen gas over the sur- faces, then they were placed in an oven kept at 60°C overnight. Although these conditions are not typical of natural processes, they were chosen to generate albite surfaces exhibiting structural features corresponding to the complete range of initial leached layer forma- tion.
Microscopic and elemental analyses of fresh and altered albite surfaces were performed using a Jeol JSM840a scanning electron microscope equipped with a Tracer Energy Dispersive Spectrometer (EDS) This spectrometer is equipped with a lithium drifted silicon detector protected by a beryllium window. The electron source for spectroscopy was a tungsten filament, the scattering voltage was 20 kV, and the current was 0.1 nA. This low current was selected in an attempt to minimize possible sodium ion mobility during the EDS analyses. The working distance is 34 mm. The beam spot size is estimated to be 50 A; in analytic mode it allows a spatial and depth resolution of -2 pm (Maurice, 1978; Eberhart, 1989). The sample surface was metallized with Au. To overcome potential ambiguities due to irregular surfaces on elemental analyses, results are presented as elemental ratios. The effect of irregular surfaces on measured chemical compositions was assessed by tilting the sample to various orientations relative to the beam. Tilting changed measured elemen- tal ratios by less than 10%. Consequently, the overall uncertainty in the elemental ratios reported in the present study is estimated to be approximately t 10%. The degree of sodium ion mobility during the EDS analyses was evaluated by measuring Na/Si elemental ratios of several characteristic sample points as a function of beam interac- tion time and of scattering voltage from 15 to 20 kV. No significant change in Na/Si elemental ratios were observed by varying these parameters.
Raman spectroscopy is used in the present study to characterize the near surface structure of albite. Owing to the spatial resolution, ease of data collection, and sensitivity of the spectral pattern to slight differences in crystal structure (McMillan and Hofmeister, 1988; Malezieux, 1990) this method is ideally suited for the characteriza-
tion of altering mineral surfaces. Raman spectra were obtained using a Dilor XY spectrometer equipped with a double monochromator and a multichannel photodiode array detector. The 457.9 nm line of an Argon laser oriented at a 90” angle with the mineral surface was used as the excitation source. The sample was analyzed through a 100X objective microscope. A diaphragm located in the microscope image plane limits the volume of analyzed sample. The analyzed sample area is approximately 5 pm in diameter, and the sample depth is approximately 5 pm. All analyses were performed at a temperature of 20 5 2°C. Spectra were acquired in the backscattering mode over the range from 150 to 4000 cm-‘. Due to fluorescence, interpretation of the altered albite spectra at Raman shifts greater than 1700 cm-’ was not possible. Fresh albite only exhibited Raman bands below - 1100 cm-‘. Because its optics are inferior to those of the scanning electron microscope, the locations of the Raman spectrographic analyses are different from the EDS analyses.
3. RESULTS AND DISCUSSION
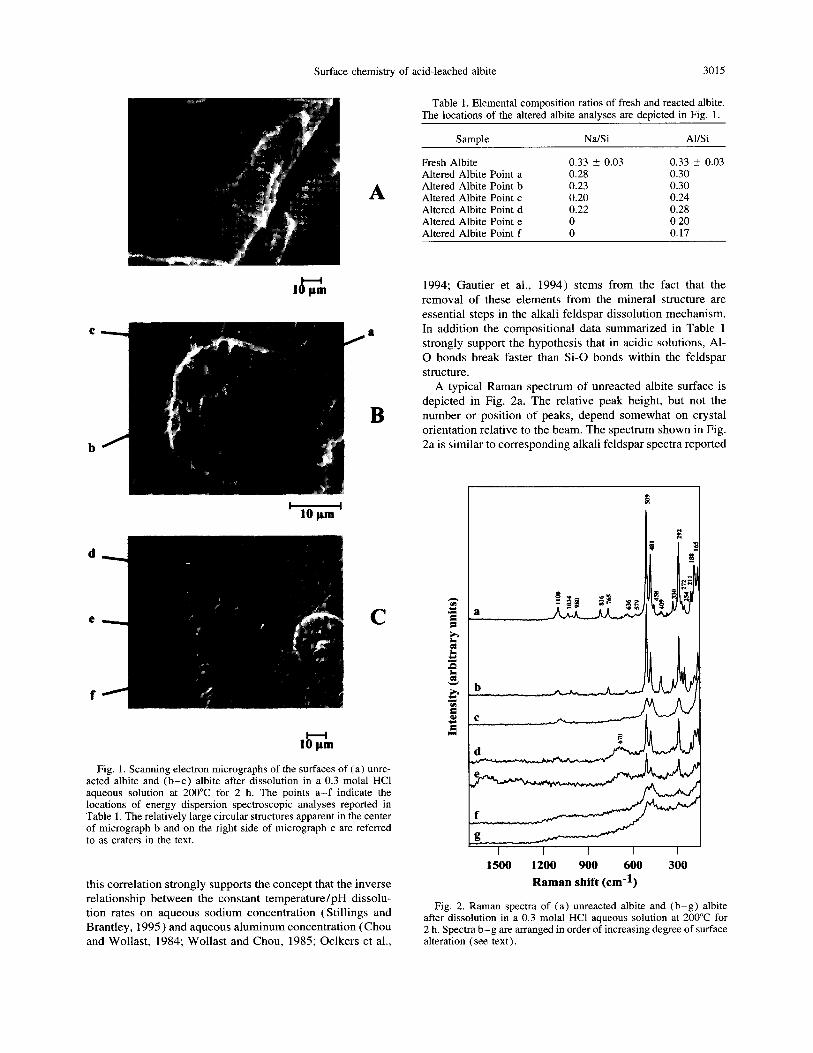
Scanning electron micrographs of the surfaces of both fresh and reacted albite are presented in Fig. 1. As can be seen in this figure the reacted albite surfaces exhibit a large variation in surface features. Some sections of reacted albite appear to be relatively fresh, while others display features of aggressive mineral dissolution including etch pits and dissolution craters (see Fig. 1). No evidence is seen for precipitation of any secondary phase.
Energy dispersive spectrometry was used to characterize the near surface composition of the albite on the different surface regions indicated in Fig. 1. The results of these mea- surements, given in Table 1, reveal that the Na/Si ratio varies depending on the surface region. Fresher appearing sections of the altered albite surfaces have a Na/Si ratio similar to the 0.33 Na/Si ratio of fresh stoichiometric albite. Corre- sponding analyses of the near surface albite compositions in a dissolution etch pit (points e and f) indicate total sodium removal. A similar trend appears in the Al/Si ratio of the near surface albite. The fresher appearing regions of the albite surfaces have an Al/Si ratio of 0.25 or greater, which approaches that of pure albite, but this ratio decreases to as low as 0.17 in dissolution etch pits. Analyses performed of the surfaces of other crystallographic faces show a similar depletion of Na and Al relative to Si in dissolution etch pits and craters. Although the magnitudes of measured elemental ratios may correspond to an average between a completely depleted surface and relatively unaltered albite at depth, the results presented in Table 1 demonstrate that alteration of the surface is nonuniform in both apparent reactability and chemical composition. It follows that adoption of the as- sumption that the release of cations from a dissolving mineral surface is spatially homogeneous can lead to serious inter- pretative ambiguities when estimating depleted surface layer depths from spatially insensitive analytical techniques such as XPS, SIMS, and ion beam techniques.
The data listed in Table 1 illustrate a correlation between the selective removal of both Na and Al from this surface and the location of dissolution etch pits. Because etch pits are sites of locally enhanced dissolution, this correlation im- plies that the lower the Na/Si and AI/Si ratio of a given surface region, the faster the local albite dissolution rate. Although the position of these enhanced dissolution regions may be controlled by twin boundaries or surface defects,
Surface chemistry of acid-leached albite 3015
B
C
Fig. 1. Scanning electron micrographs of the surfaces of (a) unre- acted albite and (b-c) albite after dissolution in a 0.3 molal HCl aqueous solution at 200°C for 2 h. The points a-f indicate the locations of energy dispersion spectroscopic analyses reported in Table 1. The relatively large circular structures apparent in the center of micrograph b and on the right side of micrograph c are referred to as craters in the text.
this correlation strongly supports the concept that the inverse relationship between the constant temperature/pH dissolu- tion rates on aqueous sodium concentration (Stillings and Brantley, 1995 ) and aqueous aluminum concentration (Chou and Wollast, 1984; Wollast and Chou, 1985; Oelkers et al.,
Table 1. Elemental composition ratios of fresh and reacted albite. The locations of the altered albite analvses are depicted in Fig. 1.
Samnle Na/Si Al/Si
Fresh Albite 0.33 r 0.03 0.33 z 0.03 Altered Albite Point a 0.28 0.30 Altered Albite Point b 0.23 0.30 Altered Albite Point c 0.20 0.24 Altered Albite Point d 0.22 0.28 Altered Albite Point e 0 0.20 Altered Albite Point f 0 0.17
1994; Gautier et al., 1994) stems from the fact that the removal of these elements from the mineral structure are essential steps in the alkali feldspar dissolution mechanism. In addition the compositional data summarized in Table 1 strongly support the hypothesis that in acidic solutions, Al- 0 bonds break faster than S-0 bonds within the feldspar structure.
A typical Raman spectrum of unreacted albite surface is depicted in Fig. 2a. The relative peak height, but not the number or position of peaks, depend somewhat on crystal orientation relative to the beam. The spectrum shown in Fig. 2a is similar to corresponding alkali feldspar spectra reported
A
1500 1200 900 600 300
Raman shift (cm-l)
Fig. 2. Raman spectra of (a) unreacted albite and (b-g) albite after dissolution in a 0.3 molal HCl aqueous solution at 200°C for 2 h. Spectra b-g are arranged in order of increasing degree of surface alteration (see text).
3016 R. Gout et al.
in the literature (von Stengel, 1977; Matson et al., 1986). This spectrum can be compared with the corresponding spec- tra of increasingly altered regions of the reacted albite, which are provided in Fig. 2b-g. A comparison of the different spectra shown in Fig. 2 confirms the fact that the surface of reacted albite is heterogeneous. Four different types of spec- tra are apparent on the surface of the reacted feldspar.
3.1. Type I
Sections of the altered albite exhibit spectra that are essen- tially identical to that of unreacted albite. A representative example of these spectra is given in Fig. 2b. The differences between the relative peak heights of spectra 2a and 2b is due to crystal orientation differences.
3.2. Type II
Sections of the altered albite are characterized by broad bands situated in the same position as unreacted albite. An example of this type of spectrum is shown in Fig. 2c. Band broadening may correspond to disorganization of an intact albite structure, perhaps due to exchange of hydrogen for sodium ions in the near surface.
3.3. Type III
Examples of Raman spectra obtained from slightly more altered parts of the albite surface (Fig. 2d and e) exhibit a broad peak at -670 cm-‘. Although such a peak is not found in the Raman spectra of fresh alkali feldspars, a peak at this position is observed in the spectra of cyclosilicates, inosili- cates (amphiboles and pyroxenes), and phyllosilicates (White, 1975; Sharma et al., 1983; Tlili et al., 1989; Smith and Boyer, 1985, 1987) and has been attributed to symmetri- cal stretching plus bending of bridging oxygens in T-O-T. It thus seems likely that this spectra corresponds to the exis- tence of a partially broken aluminosilicate framework. Such a structure may be an intermediate step in the formation of extensive leached layers on the albite surface.
3.4. Type IV
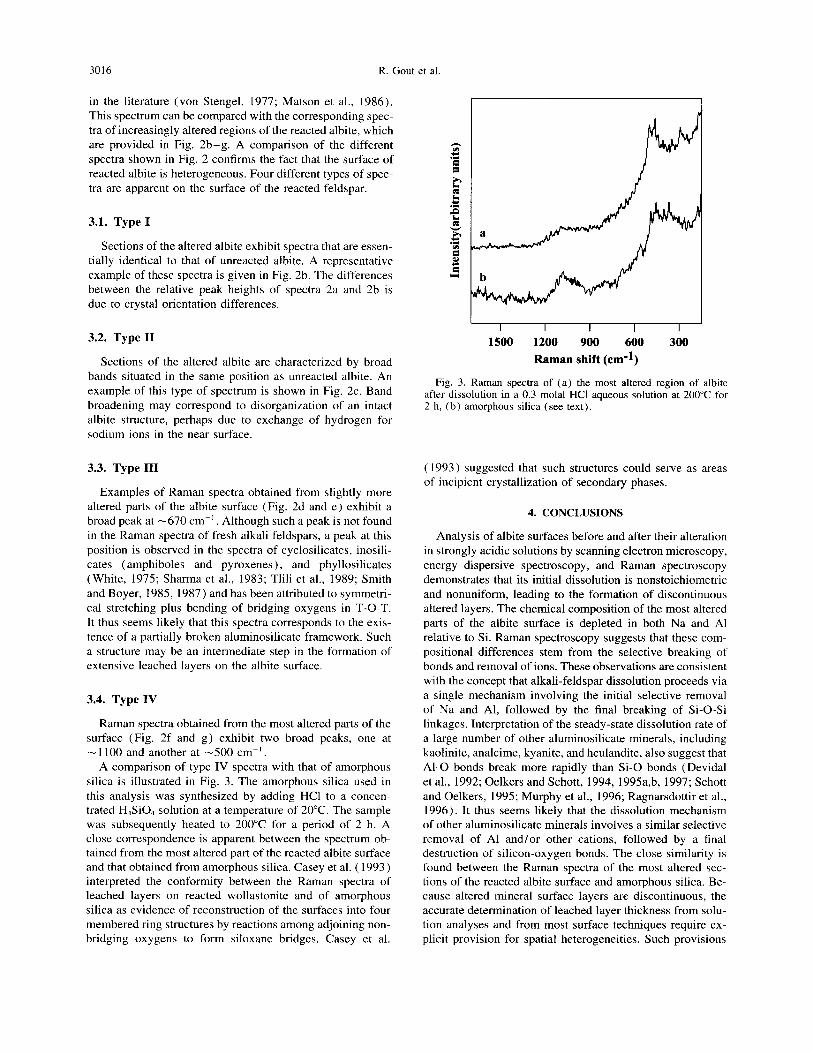
Raman spectra obtained from the most altered parts of the surface (Fig. 2f and g) exhibit two broad peaks, one at - 1100 and another at -500 cm-‘.
A comparison of type IV spectra with that of amorphous silica is illustrated in Fig. 3. The amorphous silica used in this analysis was synthesized by adding HCl to a concen- trated H,SiO, solution at a temperature of 20°C. The sample was subsequently heated to 200°C for a period of 2 h. A close correspondence is apparent between the spectrum ob- tained from the most altered part of the reacted albite surface and that obtained from amorphous silica. Casey et al. ( 1993) interpreted the conformity between the Raman spectra of leached layers on reacted wollastonite and of amorphous silica as evidence of reconstruction of the surfaces into four membered ring structures by reactions among adjoining non- bridging oxygens to form siloxane bridges. Casey et al.
I I I I I I
I
1500 1200 900 600 300 Raman shift (cm-l)
Raman spectra of (a) the most altered region of albite after dissolution in a 0.3 molal HCl aqueous solution at 200°C for 2 h, (b) amorphous silica (see text).
( 1993) suggested that such structures could serve as areas of incipient crystallization of secondary phases.
4. CONCLUSIONS
Analysis of albite surfaces before and after their alteration in strongly acidic solutions by scanning electron microscopy, energy dispersive spectroscopy, and Raman spectroscopy demonstrates that its initial dissolution is nonstoichiometric and nonuniform, leading to the formation of discontinuous altered layers. The chemical composition of the most altered parts of the albite surface is depleted in both Na and Al relative to Si. Raman spectroscopy suggests that these com- positional differences stem from the selective breaking of bonds and removal of ions. These observations are consistent with the concept that alkali-feldspar dissolution proceeds via a single mechanism involving the initial selective removal of Na and Al, followed by the final breaking of Si-0-Si linkages. Interpretation of the steady-state dissolution rate of a large number of other aluminosilicate minerals, including kaolinite, analcime, kyanite, and heulandite, also suggest that Al-0 bonds break more rapidly than Si-0 bonds (Devidal et al., 1992; Oelkers and Schott, 1994, 1995a,b, 1997; Schott and Oelkers, 1995; Murphy et al., 1996; Ragnarsdottir et al., 1996). It thus seems likely that the dissolution mechanism of other aluminosilicate minerals involves a similar selective removal of Al and/or other cations, followed by a final destruction of silicon-oxygen bonds. The close similarity is found between the Raman spectra of the most altered sec- tions of the reacted albite surface and amorphous silica. Be- cause altered mineral surface layers are discontinuous, the accurate determination of leached layer thickness from solu- tion analyses and from most surface techniques require ex- plicit provision for spatial heterogeneities. Such provisions

Surface chemistry of acid-leached albite 3017
require reliable analytical techniques that can distinguish
chemical differences on a micron scale.
Acknowledgments-This manuscript is contribution number 43 of the Dynamique et Bilan de la Terre (DBT II) program of the CNRS. We are grateful to Jean-Louis Dandurand, Gilles Berger, Marwan Alkattan, Christophe Monnin, Gleb Pokrovski, Igor Diakonov, Jean- Pal Fortune, Susan Brantley, Patrick Brady, Jean-Marie Gautier, and Stacey Callahan for helpful discussions during the course of this study. Constructive reviews by Bernard Pirou, William M. Murphy, and Roy Wogelius led to significant improvements to the text. We are indebted to Jocelyne Escalier, Richard Etchevery, Jean Claude Harrichoury, Claude Lurde, Roger Sirvin, and Michel Thibaut for technical assistance. Support from Centre National de la Recherche Scientifique is gratefully acknowledged.
Editorial handling: G. Faure
REFERENCES
Bemer R. A. and Holdren G. R. (1979) Mechanism of feldspar weathering II. Observations of feldspars from soils. Geochim. Cosmochim. Actu 43, 1173- 1186.
Berner R. A., Holdren G. R., and Schott J. (1985) Protective surface layers on dissolving silicates. Comment on the paper “Study of the weathering of albite at room temperature and pressure with a fluidized bed reactor” by L. Chou and R. Wollast. Geochim. Cosmochim. Actu 49, 1657-1658.
Blum A. E. (1994) Feldspars in weathering. In Feldspars and Their Reactions (ed. I. Parsons), pp. 595-629. Kluwer Academic Publ.
Blum A. E. and Lasaga A. C. ( 1988) Role of surface speciation in the low temperature dissolution kinetics of minerals. Nature 334, 43 1-433.
Blum A. E. and Lasaga A. C. (1991) Role of surface speciation in the dissolution of albite. Geochim. Cosmochim. Actu 55, 2193- 2201.
Blum A. E. and Stillings L. L. (1995) Feldspar dissolution kinetics. Rev. Miner. 31, 291-351.
Brady P. V. and Walther J. V. (1989) Controls on silicate dissolution in neutral and basic pH conditions at 25°C. Geochim. Cosmochim. Actu 53, 2823-2830.
Brady P. V. and Walther J. V. (1992) Surface chemistry and silicate dissolution at elevated temperatures. Amer. J. Sci. 292, 639-658.
Brantley S. L. and Stillings L. ( 1994) An integrated model for feld- spar dissolution under acid conditions. Miner. Mug. %A, 117- 118.
Brantley S. L. and Stillings L. (1996) Feldspar dissolution at 25°C and low pH. Amer. J. Sci. 296, 101-127.
Casey W. H., Westrich H. R., and Arnold G. W. (1988) Surface chemistry of labradorite feldspar reacted with aqueous solution at pH = 2, 3, and 12. Geochim. Cosmochim. Acta 52, 2795-2807.
Casey W. H., Westrich H. R., Massis T., Banfield J. F., and Arnold G. W. (1989a) The surface chemistry of labradorite feldspar after acid hydrolysis. Chem. Geol. 78, 205-218.
Casey W. H., Westrich H. R., Arnold G. W., and Banfield J. F. ( 1989b) The surface chemistry of dissolving labradorite feldspar. Geochim. Cosmochim. Actu 53, 821-832.
Casey W. H., Westrich H. R., Banfield J. F., Ferruzzi G., and Arnold G. W. (1993) Leaching and reconstruction at the surfaces of dis- solving chain silicate minerals. Nature 366, 253-255.
Chou L. and Wollast R. ( 1984) Study of the weathering of albite at room temperature and pressure with a fluidized bed reactor. Geochim. Cosmochim. Actu 48, 2205-2217.
Devidal J. L., Dandurand J. L., and Schott J. (1992) Dissolution and precipitation kinetics of kaolinite as a function of chemical affinity (T = 150°C pH = 2 and 7.8) In Water Rock Interaction (ed. Y. K. Kharaka and A. S. Maest), pp. 93-96. Balkema.
Eberhart J. P. ( 1989) Anulyse structurule et chimique des mute’riuux. Durod.
Gautier J.-M., Oelkers E. H., and Schott J. (1994) Experimental study of K-feldspar dissolution rates as a function of chemical
affinity at 150°C and pH 9. Geochim. Cosmochim. Acta 58,4549- 4560.
Hellmann R. (1994) A leached layer hydrolysis model: A better way to understanding feldspar dissolution at elevated temperatures and pressures. Miner. Mug. %A, 400-401.
Hellmann R., Eggleston C. M., Hochella M. F., and Crerar D. A. ( 1990) The formation of leached layers on albite surfaces during dissolution under hydrothermal conditions. Geochim. Cosmochim. Actu 54, 1267-1281.
Hochella M. F., Ponader H. B., Turner A. M., and Harris D. W. (1988) The complexity of mineral dissolution as viewed by high resolution scanning Auger microscopy: Laboradorite under hydrothermal conditions. Geochim. Cosmochim. Actu 52, 385- 394.
Malezieu J.-M. ( 1990) Contribution of Raman spectroscopy to min- eral studies. In Absorption Spectroscopy in Mineralogy (ed. A. Mottana and F. Burragato), pp. 40-60. Elsevier.
Matson D. W., Sharma S. K., and Philpotts J. A. (1986) Raman spectra of some tectosilicates and of glasses along the orthoclase- anorthite and nepheline-anorthite joins. Amer. Miner. 71, 694- 704.
Maurice F. (1978) Emissions X. In Microunulyse et Microscopic Electronique ir Bulayuge, pp. 171-214. Ed. Phys.
McMillan P. F. and Hofmeister A. M. (1988) Infrared and Raman spectroscopy. Rev. Mineral. 18, 99- 159.
Muir I. J. and Nesbitt H. W. ( 1991) Effects of aqueous cations on the dissolution of labradorite feldspar. Geochim. Cosmochim. Actu 55, 3181-3189.
Murphy W. M., Pabalan R. T., Prikryl J. D., and Goulet C. J. ( 1996) Reaction kinetics and thermodynamics of aqueous dissolution and growth of analcime and Na-clinoptilolite at 25°C. Amer. J. Sci. 296, 128- 186.
Nesbitt H. W. and Muir I. J. (1988) SIMS depth profiles of weath- ered plagioclase and processes affecting dissolved Al and Si in some acidic soils. Nature 334, 336-338.
Nesbitt H. W., Macrae N. D., and Shotyk W. ( 1991) Congruent and incongruent dissolution of labradorite in dilute acidic salt solu- tions. J. Geol. 99, 429-442.
Oelkers E. H. and Schott J. (1992) The dissolution rate of albite as a function of chemical affinity and the stoichiometry of activated complexes in aluminosilicate dissolution reactions. Geol. Sot. Amer. ABSTR. PROGR. 24, A207.
Oelkers E. H. and Schott J. ( 1994) Experimental study of kyanite dissolution rates as a function of aluminum and silicon concentra- tion. Miner. Msg. 58A, 659-660.
Oelkers E. H. and Schott J. (1995a) The dissolution and crystalliza- tion rates of silicate minerals as a function of their structure and composition. In Water-Rock Interaction (ed. Y. K. Kharaka and 0. V. Chudaev), pp. 153-156. Balkema.
Oelkers E. H. and Schott J. (1995b) An experimental study of anor- thite dissolution and the relative mechanism of feldspar hydrolysis. Geochim. Cosmochim. Acta 59, 5039-5053.
Oelkers E. H. and Schott J. ( 1997) An experimental study of kyanite dissolution as a function of chemical affinity and solution compo- sition. Geochim. Cosmochim. Actu (in prep).
Oelkers E. H., Schott J., and Devidal J. L. (1994) The effect of aluminum, pH, and chemical affinity on the rates of aluminosili- cate dissolution reactions. Geochim. Cosmochim. Actu 58, 2011- 2024.
Petit J.-C., Dran J. C., Paccagurlla A., and Della Mea G. (1989) Structural dependence of crystalline silicate hydration during aqueous dissolution. Earth Planet. Sci. Lett. 93, 292-298.
Ragnarsdottir K. V., Graham C. M., and Allen G. C. ( 1996) Surface chemistry of reacted heulandite: A SIMS and XPS study. Chem. Geol. 131, 167-181.
Schott J. and Petit J. C. (1987) New evidence for the mechanisms of dissolution of silicate minerals. In Aquatic Surface Chemistry (ed. W. Stumm), pp. 293-315. Wiley.
Schott J. and Oelkers E. H. (1995) Dissolution and crystallization rates of silicate minerals as a function of chemical affinity. Pure Appl. Chem. 67, 903-910.

3018 R. Gout et al.
Sharma S. K., Simons B., and Yoder H. S., Jr. (1983) Raman study of anorthite, calcium Tschermack’s pyroxene, and gehlenite in crystalline and glassy states. Amer. Min. 68, 1113- 1125.
Schweda P. (1989) Kinetics of alkali feldspar dissolution at low temperature. In Water Rock Interaction (ed. D. L. Miles), pp. 609-612. Balkema.
Smith D. C. and Boyer H. (1985) Raman microprobe fingerprinting of ordered and disordered pyroxenes in the system diopside-om- phacite-jadeite. Terra Cog&a 5, 429.
Smith D. C. and Boyer H. ( 1987) An exploration of the Raman spectra of several natural high-pressure amphiboles. Terra Cog&a 7, 429.
Stillings L. L. and Brantley S. L. (1995) Feldspar dissolution at 25°C and pH 3: Reaction stoichiometry and the effect of cations. Geochim. Cosmochim. Acta 59. 1483- 1496.
Tlili A., Smith D. C., Beny J.-M., and Boyer H. ( 1989) A Raman microprobe study of natural micas. Mineral. Msg. 53, 165- 179.
von Stengel M. 0. ( 1977) Normalschwingungen von alkalifelds- paten. 2. Kristallogr. 146, 1 - 18.
White W. B. ( 1975) Structural interpretation of lunar and terrestrial minerals by Raman spectroscopy. In Infrared and Raman Spec- troscopy of Lunar and Terrestrial Materials (ed. M. Karr), pp. 325-358. Academic Press.
Wollast R. and Chou L. (1985) Kinetic study of the dissolution of albite with a continuous flow through reactor. In The Chemistry of Weathering (ed. J. Drever), pp. 75-96. Redel.
Wollast R. and Chou L. (1992) Surface reactions during the early stages of weathering of albite. Geochim. Cosmochim. Acta 56, 3113-3122.





























