Embed Size (px)
Citation preview

The Neuropeptide Catestatin Acts As a Novel AngiogenicCytokine via a Basic Fibroblast Growth
Factor–Dependent MechanismMarkus Theurl,* Wilfried Schgoer,* Karin Albrecht, Johannes Jeschke, Margot Egger, Arno G.E. Beer,
Danijela Vasiljevic, Song Rong, Anna Maria Wolf, Ferdinand H. Bahlmann, Josef R. Patsch,Dominik Wolf, Peter Schratzberger, Sushil K. Mahata, Rudolf Kirchmair
Rationale: The neuropeptide catestatin is an endogenous nicotinic cholinergic antagonist that acts as apleiotropic hormone.
Objective: Catestatin shares several functions with angiogenic factors. We therefore reasoned that catestatininduces growth of new blood vessels.
Methods and Results: Catestatin induced migration, proliferation, and antiapoptosis in endothelial cells andexerted capillary tube formation in vitro in a Matrigel assay, and such effects were mediated via G protein,mitogen-activated protein kinase, and Akt. Catestatin-induced endothelial cell functions are further mediated bybasic fibroblast growth factor, as shown by blockade of effects by a neutralizing fibroblast growth factorantibody. Furthermore, catestatin released basic fibroblast growth factor from endothelial cells and stimulatedfibroblast growth factor signaling. In addition to its function on endothelial cells, catestatin also exerted effectson endothelial progenitor cells and vascular smooth muscle cells. In vivo, catestatin induced angiogenesis in themouse cornea neovascularization assay and increased blood perfusion and number of capillaries in the hindlimbischemia model. In addition to angiogenesis, catestatin increased density of arterioles/arteries and incorporationof endothelial progenitor cells in the hindlimb ischemia model, indicating induction of arteriogenesis andpostnatal vasculogenesis.
Conclusion: We conclude that catestatin acts as a novel angiogenic cytokine via a basic fibroblast growthfactor–dependent mechanism. (Circ Res. 2010;107:1326-1335.)
Key Words: angiogenesis � blood vessels � endothelium � endothelial progenitor cells
Chromogranin (Cg)A, the index member of the chromo-granin/secretogranin protein family, is the major soluble
protein of catecholamine storage vesicles of sympatheticnerve terminals and the adrenal medulla.1,2 CgA is a propro-tein giving rise to biological active peptides like the dysgly-cemic hormone pancreastatin,3 the vasodilator vasostatin,4
and catestatin5 (CST) (CgA352–372), which inhibits catechol-amine release by acting as a nicotinic cholinergic antagonist,resulting in a negative-feedback mechanism. Althoughplasma CgA is high in human essential (hereditary) hyper-tension, the plasma concentration of CST is lower in bothestablished cases and in normotensive subjects with a family
history of the disease,6 suggesting a mechanism wherebydiminished CST might increase the risk for later developmentof hypertension. Consistent with the human findings, highblood pressure has been reported in mice after targetedablation of the Chga gene (Chga knockout), and such highblood pressure can be “rescued” either by replacement withCST or introduction of the human ortholog in the Chgaknockout background.7
Angiogenesis, the growth of new vessels from thepreexisting vasculature, is an important process in manyphysiological conditions including embryonic develop-ment and wound healing. However, defects in the regula-
Original received February 25, 2010; revision received September 27, 2010; accepted September 28, 2010. In August 2010, the average time fromsubmission to first decision for all original research papers submitted to Circulation Research was 13.2 days.
From the Departments of Internal Medicine 1 (M.T., W.S., K.A., M.E., A.G.E.B., D.V., J.R.P., P.S., R.K.), Department of Plastic Surgery (J.J.), andTyrolean Cancer Research Institute and Internal Medicine 5 (A.M.W., D.W.), Medical University of Innsbruck, Austria; Department of Nephrology(S.R.), Hannover Medical School, Germany; Klinik fur Innere Medizin IV (F.H.B.), Universitatsklinikum des Saarlandes, Germany; and HypertensionResearch Unit (S.K.M.), University of California at San Diego, La Jolla.
*Both authors contributed equally to this work.Correspondence to Rudolf Kirchmair, MD, Associate Professor of Medicine Department of Internal Medicine 1, Medical University of Innsbruck,
Anichstr. 35, 6020 Innsbruck, Austria (E-mail [email protected]); or Sushil K. Mahata, PhD, Professor of Medicine, University of California,San Diego, Hypertension Research Unit (0838), 9500 Gilman Dr, La Jolla, CA 92093-0838 (E-mail [email protected]).
© 2010 American Heart Association, Inc.
Circulation Research is available at http://circres.ahajournals.org DOI: 10.1161/CIRCRESAHA.110.219493
1326
by guest on April 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on A
pril 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on April 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on A
pril 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on April 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on A
pril 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on April 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on A
pril 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on April 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on A
pril 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on April 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on A
pril 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on April 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on A
pril 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on April 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on A
pril 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on April 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on A
pril 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on April 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on A
pril 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on April 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on A
pril 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on April 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on A
pril 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on April 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on A
pril 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on April 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on A
pril 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on April 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on A
pril 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on April 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on A
pril 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on April 29, 2018
http://circres.ahajournals.org/D
ownloaded from
by guest on A
pril 29, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on April 29, 2018
http://circres.ahajournals.org/D
ownloaded from

tion of angiogenesis often result in pathological conditionssuch as inflammatory diseases, ischemic heart, peripheralvascular diseases, proliferative retinopathy, and solid tu-mors.8 The most potent angiogenic factors are basicfibroblast growth factor (bFGF) and vascular endothelialgrowth factor (VEGF). Besides bFGF and VEGF, a variety ofneuropeptides like substance P (SP), secretoneurin (SN), andneuropeptide (NP)Y exert effects on endothelial cells (ECs) andinduce angiogenesis.9–11
CST shares several features with SP such as vasodilationby CST11a and SP12 and having comparable pI (CST, 12.16;versus SP, 11.14). In addition, CgA was shown to beupregulated by hypoxia in neuronal cells after transienthypoxia,13 a typical characteristic of angiogenic factors likeVEGF. Based on the above similarities of CST with angio-genic peptides, we reasoned that CST would exert effects onECs in vitro and induce angiogenesis in vivo. The presentreport establishes CST as a novel angiogenic cytokine withthe potential of inducing therapeutic effects in animal modelsof ischemia.
MethodsAn expanded Methods section is available in the Online DataSupplement at http://circres.ahajournals.org.
CST Peptide and AntiserumHuman CST and antiserum have been described previously.14
Endothelial Progenitor Cell IsolationEndothelial progenitor cells (EPCs) were isolated and cultured, andEPC chemotaxis assay was performed as described.15
Tube Formation AssayTube formation assay was performed as described.10 Some Matrigelassays were performed with a mixture of 5000 HUVECs and 3000DiI LDL-labeled EPCs.16 The association of EPC to capillarystructures was quantified using Image J.
Cornea Neovascularization AssayPellets containing 500 ng of CST, scrambled CST, or VEGF wereimplanted in C57/BL/6J mice as described.10 On postoperative day 7,mice received an IV injection of 500 �g of BS1 lectin conjugated toFITC (Vector Laboratories). After euthanasia, enucleated eyes werefixed in 1% paraformaldehyde and corneas dissected and examinedby fluorescence microscopy.
Mouse Hindlimb Ischemia ModelC57BL/6 wild-type mice were subjected to unilateral hindlimbsurgery. Mice were injected with saline or 20 �g of CST into thighand calf muscles after operation and every other day for weeks 1and 2 and 2 times per week for weeks 3 and 4. Limb necrosisstatus (see also the Online Data Supplement) was determined ondays 7, 14, and 28.
Bone Marrow Transplantation ModelBone marrow transplantation (BMT) was performed as de-scribed.15,17 Labeling of functional vessels and of EPCs in BMTmice was performed 3 weeks after induction of ischemia as de-scribed.17 EPCs are expressed as number of double positive cells perhigh-power field (magnification, �100).
For confocal microscopy, a Broadband Confocal Leica TCS SP5microscope (lenses: HCX PL APO CS 20�0.7 [dry] and HCX PLAPO CS 63�1.2 [water]) was used.
Blood Flow MeasurementBlood flow measurements were performed using a laser Dopplerperfusion imaging (LDPI) analyzer (Moor Instruments). Bloodperfusion is expressed as the LDPI index representing the ratio of left(operated, ischemic leg) versus right (not-operated, not-ischemic leg)limb blood flow.
ImmunohistochemistryCD31 and �-smooth muscle actin (SMA) staining were performed asdescribed.10,18 For fluorescent microscopy, appropriate secondaryantibodies (Alexa 488 for SMA and Alexa 594 for CD31; both fromInvitrogen 1:200) were used.
Results
In Vitro Results
CST Effects on Migration, Proliferation, and Apoptosis inECs In VitroCST caused dose-dependent induction of chemotaxis inhuman umbilical vein endothelial cells (HUVECs) with amaximum effect at 1 nmol/L (relative chemotaxis index [CI],1.67�0.06, P�0.01 versus control; Figure 1a) comparableto other angiogenic factors like VEGF or SN.10 Blockadeof CST by a specific neutralizing antibody completelyinhibited CST-mediated EC migration, indicating specific-ity of the observed effect (relative CI: CST 1.74�0.06;
Non-standard Abbreviations and Acronyms
bFGF basic fibroblast growth factor
bFGF-Ab basic fibroblast growth factor antibody
BMT bone marrow transplantation
BrdUrd bromodeoxyuridine
CgA chromogranin A
CI chemotaxis index
CST catestatin
Ctr control
EC endothelial cell
EPC endothelial progenitor cell
ERK extracellular signal-regulated kinase
FGFR fibroblast growth factor receptor
GFP green fluorescence protein
HUVEC human umbilical vein endothelial cell
LDPI laser Doppler perfusion imaging
MAPK mitogen-activated protein kinase
NPY neuropeptide Y
PD PD 98059
PDGF platelet-derived growth factor
PI3-kinase phosphatidyl 3-kinase
PTX pertussis toxin
SMA smooth muscle actin
SMC smooth muscle cell
SN secretoneurin
SP substance P
VEGF vascular endothelial growth factor
WM wortmannin
Theurl et al Catestatin and Angiogenesis 1327
by guest on April 29, 2018
http://circres.ahajournals.org/D
ownloaded from

CST�antibody, 1.12�0.06; P�0.01, CST versus CST�Ab;Figure 1b). CST-induced chemotaxis was completely abol-ished by inhibition of G-protein coupled receptors by pertus-sis toxin (PTX) and mitogen-activated protein kinase(MAPK) by PD 98059 (PD), indicating CST signaling via Gprotein–coupled receptors and MAPK pathway (relative CI:CST, 1.74�0.06; CST�PTX, 1.0�0.04; CST�PD, 1.08�0.05; P�0.01, CST versus CST�PTX and CST�PD; Figure1b). Tyrosine kinase inhibitor genistein (1 �mol/L) causedonly partial inhibition of CST-induced EC migration (relativeCI: CST, 1.73�0.05; CST�genistein, 1.41�0.04; P�0.05,CST versus CST�genistein).
CST also induced proliferation of HUVECs (as deter-mined by cell number) with a maximum effect at 1 nmol/L(relative cell number, 1.85�0.11; P�0.01 versus control),and such effect was blocked by pretreatment with CSTantibody (relative cell number, 0.97�0.06; P�0.05 versusCST) and blockade of MAPK by PD (relative cell number,
1.19�0.09; P�0.05 versus CST), indicating specificity ofCST effect and its signaling through MAPK pathway(Figure 1c). CST-induced EC proliferation was also deter-mined by bromodeoxyuridine (BrdUrd) staining (Figure1d). CST, like VEGF, significantly increased BrdUrdincorporation in ECs.
To test the potential effects of CST on EC apoptosis,HUVECs were serum-starved and stained for TUNEL andDAPI. CST caused significant inhibition of apoptosis(percentage of TUNEL-positive apoptotic cells of DAPI-positive cells: control 23.0�0.7%, CST 10.4�0.5%;P�0.01, control versus CST; Figure 1e). Inhibition ofCST-induced antiapoptotic effect by wortmannin (WM)indicates that PI-3 kinase/Akt pathway mediates this effect(Figure 1e).
Inhibition of CST-mediated effects by PD and WM indi-cates involvement of MAPK and PI3-kinase/Akt pathways.CST signaling was evaluated by Western blotting, which
Figure 1. Effects of CST on ECs invitro. a, CST induces EC migration.EC chemotaxis is expressed as che-motactic index relative to control (Ctr)cells without treatment. Cells weretreated with VEGF (50 ng/mL), SN (1nmol/L), or CST with a concentrationof 10�7 to 10�15 mol/L. Results aremeans of 3 to 5 experiments and showthat CST induces EC chemotaxis witha maximum effect at 10�9 mol/L.*P�0.05; **P�0.01. b, CST-inducedEC migration is blocked by CST anti-body, PTX, and inhibition of MAPK byPD. ECs were treated with 1 nmol/LCST with or without preincubation withCST-neutralizing antibody, PTX, or PD.Results (n�3) show that CST-inducedmigration is blocked by treatment withrespective substances, whereas anti-body, PTX, or PD did not affect basalmigration. **P�0.01 CST vs control;§P�0.05 vs CST. c, CST induces ECproliferation. ECs were treated with 1nmol/L SN or different concentrationsof CST; cell number was counted andcompared with untreated cells. A con-centration of 1 nmol/L CST exerted themost potent effect on EC proliferation,and this effect was blocked by treat-ment with a neutralizing CST antibodyor PD. *P�0.05; **P�0.01 vs control;§P�0.05 vs CST; n�3. d, CST inducesEC proliferation/BrdUrd assay. CST-induced EC proliferation was alsodetermined by BrdUrd assay. VEGF
(50 ng/mL) and CST (1 nmol/L) significantly increased BrdUrd incorporation, as expressedas relative light units per second. *P�0.05 vs control; n�3. e, CST inhibits EC apoptosis.ECs were starved and stained for TUNEL and DAPI. CST reduced EC apoptosis (percent-age of TUNEL-positive of DAPI-positive cells) at 1 nmol/L, whereas coincubation with WM(10 nmol/L) inhibited CST-induced protective effect. *P�0.05 vs control; §P�0.05 vs CST;n�3. Representative images of TUNEL and DAPI stains are shown on the right; arrow-heads are examples of double positive cells; arrow in control DAPI, cell positive for DAPIand negative for TUNEL. f, CST- and VEGF-induced ERK and Akt activation. ECs werestimulated with CST at 1 nmol/L and 300 nmol/L, with VEGF (50 ng/mL) (all 10 minutes)with the combination of VEGF and CST 300 nmol/L or with 20% FBS, and extracts wereanalyzed for ERK or Akt activation by Western blotting. CST induced ERK and Akt phos-phorylation at low and even more at high concentrations and did not block VEGF-inducedstimulation of these pathways. Actin and tubulin stains were used as loading controls.
1328 Circulation Research November 26, 2010
by guest on April 29, 2018
http://circres.ahajournals.org/D
ownloaded from
revealed activation of extracellular signal-regulated kinase(ERK) and Akt, as judged by phosphorylation of ERK andAkt (Figure 1f). CST was able to stimulate ERK and Aktphosphorylation at 1 and 300 nmol/L; VEGF was used aspositive control. In contrast to the N-terminal CgA peptidevasostatin,19 CST did not inhibit VEGF-induced ERK acti-vation in ECs.
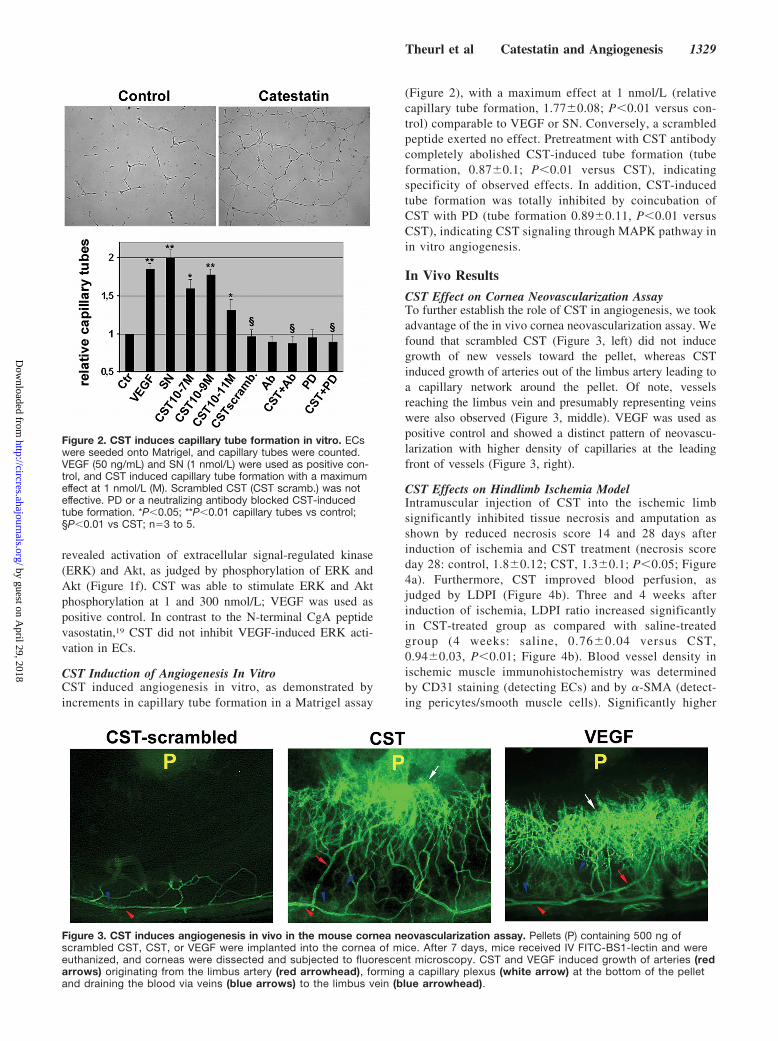
CST Induction of Angiogenesis In VitroCST induced angiogenesis in vitro, as demonstrated byincrements in capillary tube formation in a Matrigel assay
(Figure 2), with a maximum effect at 1 nmol/L (relativecapillary tube formation, 1.77�0.08; P�0.01 versus con-trol) comparable to VEGF or SN. Conversely, a scrambledpeptide exerted no effect. Pretreatment with CST antibodycompletely abolished CST-induced tube formation (tubeformation, 0.87�0.1; P�0.01 versus CST), indicatingspecificity of observed effects. In addition, CST-inducedtube formation was totally inhibited by coincubation ofCST with PD (tube formation 0.89�0.11, P�0.01 versusCST), indicating CST signaling through MAPK pathway inin vitro angiogenesis.
In Vivo Results
CST Effect on Cornea Neovascularization AssayTo further establish the role of CST in angiogenesis, we tookadvantage of the in vivo cornea neovascularization assay. Wefound that scrambled CST (Figure 3, left) did not inducegrowth of new vessels toward the pellet, whereas CSTinduced growth of arteries out of the limbus artery leading toa capillary network around the pellet. Of note, vesselsreaching the limbus vein and presumably representing veinswere also observed (Figure 3, middle). VEGF was used aspositive control and showed a distinct pattern of neovascu-larization with higher density of capillaries at the leadingfront of vessels (Figure 3, right).
CST Effects on Hindlimb Ischemia ModelIntramuscular injection of CST into the ischemic limbsignificantly inhibited tissue necrosis and amputation asshown by reduced necrosis score 14 and 28 days afterinduction of ischemia and CST treatment (necrosis scoreday 28: control, 1.8�0.12; CST, 1.3�0.1; P�0.05; Figure4a). Furthermore, CST improved blood perfusion, asjudged by LDPI (Figure 4b). Three and 4 weeks afterinduction of ischemia, LDPI ratio increased significantlyin CST-treated group as compared with saline-treatedgroup (4 weeks: saline, 0.76�0.04 versus CST,0.94�0.03, P�0.01; Figure 4b). Blood vessel density inischemic muscle immunohistochemistry was determinedby CD31 staining (detecting ECs) and by �-SMA (detect-ing pericytes/smooth muscle cells). Significantly higher
Figure 2. CST induces capillary tube formation in vitro. ECswere seeded onto Matrigel, and capillary tubes were counted.VEGF (50 ng/mL) and SN (1 nmol/L) were used as positive con-trol, and CST induced capillary tube formation with a maximumeffect at 1 nmol/L (M). Scrambled CST (CST scramb.) was noteffective. PD or a neutralizing antibody blocked CST-inducedtube formation. *P�0.05; **P�0.01 capillary tubes vs control;§P�0.01 vs CST; n�3 to 5.
Figure 3. CST induces angiogenesis in vivo in the mouse cornea neovascularization assay. Pellets (P) containing 500 ng ofscrambled CST, CST, or VEGF were implanted into the cornea of mice. After 7 days, mice received IV FITC-BS1-lectin and wereeuthanized, and corneas were dissected and subjected to fluorescent microscopy. CST and VEGF induced growth of arteries (redarrows) originating from the limbus artery (red arrowhead), forming a capillary plexus (white arrow) at the bottom of the pelletand draining the blood via veins (blue arrows) to the limbus vein (blue arrowhead).
Theurl et al Catestatin and Angiogenesis 1329
by guest on April 29, 2018
http://circres.ahajournals.org/D
ownloaded from

densities of capillaries (CD31-positive vessels) were no-ticeable in CST-treated mice 4 weeks after surgery (cap-illaries/mm2: CST, 845.5�47.8; saline, 530.4�41.8;P�0.01; Figure 4c). In addition, we observed increaseddensity of �-SMA–positive vessels after CST treatment(�-SMA–positive vessels/mm2: CST 7.5�0.5 and saline3.9�0.7; P�0.01; Figure 4d). These findings of increaseddensity of arterioles/arteries after CST treatment areconsistent with the induction of arteriogenesis by thispeptide.
We injected CST in green fluorescence protein (GFP)BMT mice subjected to hindlimb ischemia for 3 weeks toevaluate CST-induced vasculogenesis in vivo. CST in-duced incorporation of EPCs, as shown by increasednumber of cells double positive for BS1-lectin (injected IVstaining host vessels) and for GFP (bone marrow– derivedcells after transplantation). Examination of double positivecells per high-powered field (�100) revealed the follow-ing: control, 9.6�2.1; CST, 21.7�2; P�0.001 (Figure 5a).When we examined peripheral blood of these mice, weobserved increased numbers of flk-1�/GFP� cells (per-centage of GFP� cells) in CST-treated mice (saline:
1.2�0.25; CST: 3.1�0.4; P�0.05). We also used confocalmicroscopy to improve colocalization of DAPI-, GFP-, andrhodamine lectin–positive cells. As shown in Figure 5b(right bottom image) cells positive for DAPI, GFP, andlectin can be detected (arrowheads).
CST-Induced Effects Are Mediated by bFGFTo assess effects of VEGF or bFGF on CST-mediated in vitroangiogenesis, CST was incubated with neutralizing VEGFand bFGF antibodies. Inhibition of VEGF did not influenceCST-induced tube formation, but bFGF antibody completelyinhibited CST-mediated effects (tube formation CST,2.05�0.08; CST�VEGF-Ab, 1.98�0.12; CST�bFGF-Ab,1.3�0.06; P�0.05, CST versus CST�bFGF-Ab; Figure 6a).Additionally, bFGF antibody inhibited CST-induced MAPKactivation in ECs. When we performed a time course ofCST-induced MAPK activation in HUVECs, a biphasicactivation of MAPK was observed with a maximum at 10 and50 minutes (Figure 6b). Whereas early activation of MAPKwas not influenced by bFGF antibody, late activation ofMAPK was blocked by bFGF inhibition, indicating thatCST-mediated late activation of MAPK is mediated by bFGF.
Figure 4. CST induces therapeutic angiogenesis and arteriogenesis in the mouse hindlimb ischemia model. a, CST reducesnecrosis score. After hindlimb ischemia, mice (n�10 each group) were treated IM with CST or saline and evaluated for necrosis/amputation at days 7, 14, and 28. Necrosis score was significantly reduced by CST 14 and 28 days after induction of ischemia.Representative images are shown. b, CST improves blood perfusion. Limb perfusion was measured by LDPI before and afteroperation and weekly thereafter for 4 weeks. Results are expressed as LDPI ratio of the operated vs the not-operated leg. CSTimproved perfusion compared with saline 3 and 4 weeks after operation (n�10). Representative pictures are shown. c, CSTincreases capillary density. Sections from ischemic muscles (after 4 weeks) were stained for capillary density by CD31. Values areexpressed as CD31� capillaries/mm2. CST significantly increased capillary density (n�10). d, CST increases density of arteries/arterioles. Sections were also stained for �-SMA. Values are expressed as SMA-positive arteries or arterioles/mm2 (n�10).*P�0.05; **P�0.01, saline vs CST (for a through d). e, Immunofluorescence staining. Adjacent sections of CST-treated ischemiclimb stained for CD31 and �-SMA. Merging of images shows CD31-positive endothelial cells lining the inner surface of the ves-sels, which are surrounded by �-SMA–positive smooth muscle cells.
1330 Circulation Research November 26, 2010
by guest on April 29, 2018
http://circres.ahajournals.org/D
ownloaded from

Because bFGF mRNA was not upregulated by CST over abroad range of concentrations and time points (data notshown and Online Data Supplement), we evaluated whetherCST releases bFGF protein from ECs. We found that ECsstimulated by 1 nmol/L CST released bFGF into cell super-natant from 30 minutes to 6 hours, with a maximum at 1 hour(pg/mL: control, 28.6�2.2; CST, 53.8�3.4; P�0.01; Figure
6c). Concentration of bFGF was not changed in heparin washsolutions or cell lysates after CST treatment (data not shown).To evaluate whether CST stimulates bFGF signaling, weperformed studies on FGF receptor (FGFR)1 activation. Weobserved that CST (after 60 minutes), like bFGF (after 30minutes), stimulated phosphorylation of FGFR1 (Figure 6d).To investigate the mechanism of bFGF release, cells were
Figure 5. CST induces postnatal vasculogenesis. Sections of ischemic muscles (a) were analyzed for bone marrow–derived cells(GFP-positive) and merged with lectin positive host vessels (rhodamine positive). Double positive cells were considered as EPCs andcounted per high-powered field (�100). CST treatment significantly increased number of incorporated EPCs. *P�0.001; n�7 eachgroup. Additionally, confocal microscopy was performed (b) and sections were analyzed for DAPI staining (left), rhodamine-labeledBS1 lectin (second from left), and for GFP� cells (third from left). Merged image (right) shows EPCs positive for DAPI, lectin, andGFP (yellow cells, arrowheads).
Theurl et al Catestatin and Angiogenesis 1331
by guest on April 29, 2018
http://circres.ahajournals.org/D
ownloaded from

incubated with substances inhibiting different signaling trans-duction pathway. We observed that inhibition of MAPK byPD blocked CST-induced bFGF release, whereas inhibitionof PI3-kinase (by WM), of protein kinase C (by GFX), or ofRho kinase (by Y26732) had no effect (Figure 6e). We alsofound that PD and FGF antibody inhibited CST-inducedFGFR1 activation indicating that CST transactivates FGFreceptor by release of bFGF (Figure 6f).
In vivo, inhibition of bFGF by a neutralizing antibodyinhibited CST-induced effects on angiogenesis, as shownby increased necrosis score (Figure 6g) and impaired bloodperfusion in LDPI measurements (day 28: CST�IgG,0.75�0.3; CST�bFGF-Ab, 0.46�0.04; n�7; P�0.01;Figure 6h; control day 28: 0.55�0.08; P�0.05 versusCST�IgG, n�3, data not shown). Additionally, CST-induced vasculogenesis in the GFP BMT mouse wasinhibited by blockade of bFGF, as demonstrated by re-duced GFP�/lectin� EPCs in ischemic limbs (EPCs perhigh-powered field: control, 8.8�0.8; CST�IgG,15.9�1.2; CST�bFGFAb, 10.8�0.8; P�0.05, CST�IgGversus CST�bFGF-Ab; Figure 6i) and reduced circulatingflk-1�/GFP�cells (percentage of GFP� cells; control,1.2�0.25; CST�IgG, 3.1�0.4; CST�bFGF Ab, 1.5�0.3;P�0.05, CST�IgG versus CST�bFGF-Ab; Figure 6j).
Discussion
OverviewCST was initially described as a potent endogenous nico-tinic cholinergic antagonist inhibiting nicotine-evoked cat-echolamine secretion from PC12 cells and primary culturesof bovine chromaffin cells. Subsequently, CST was estab-lished as a pleiotropic peptide. CST acts as a negativeregulator of hypertension,7 inotropy, and lusitropy.20 CST
Figure 6. CST-induced effects on ECs are mediated bybFGF. a, CST-induced tube formation is blocked by a neutraliz-ing bFGF antibody. CST-induced tube formation was calculatedin the presence or absence of a neutralizing VEGF or bFGF anti-body (Ab). Inhibition of bFGF blocked CST-mediated effect.*P�0.05, **P�0.01 vs control; §P�0.05, VEGF vs VEGF�VEGF-Ab, bFGF vs bFGF�bFGF-Ab, or CST vs CST�bFGF-Ab; n�3.b, CST-induced MAPK activation is blocked by a neutralizingbFGF antibody. CST induced phosphorylation of ERK in abiphasic manner with a maximum after 10 and 50 minutes.Incubation with a bFGF antibody inhibited late but not early ERKactivation induced by CST. c, CST induces bFGF release fromECs. ECs were incubated for different time points with 1 nmol/LCST, and bFGF of supernatants was analyzed by ELISA. CSTinduces bFGF release with a maximum at 1 hour. **P�0.01,*P�0.05 vs control; n�3. d, CST induces FGFR1 activation.HUVECs were treated with bFGF (20 ng/mL) for 30, 60, and 90
minutes and with CST (1 nmol/L) for 30, 60, 90, and 180 min-utes. FGFR1 activation was studied by immunoprecipitationusing FGFR1 antibody and subsequent immunoblotting forphospho-tyrosine and FGFR1. CST, like bFGF, induces activa-tion of this receptor. e, CST-induced bFGF release is mediatedby MAPK. PD blocked CST-induced bFGF release in HUVECs,whereas inhibitors of PI3-kinase, protein kinase C, and Rhokinase could not inhibit significant release of bFGF by CST.*P�0.05 vs control; §P�0.05 vs CST; n�3. f, CST-inducedFGFR1 activation is blocked by bFGF antibody and inhibition ofMAPK. After 60 minutes of incubation with CST (1 nmol/L),FGFR1 activation was observed by immunoprecipitation. Theeffect was blocked by PD (10 �mol/L) and bFGF-Ab (1:1000). g,CST-induced effects in vivo are blocked by inhibition of bFGF:necrosis score. After hindlimb ischemia and CST therapybFGF-Ab impaired necrosis score on day 7, 14, and 28 com-pared with IgG. P�0.05; n�7. h, CST-induced effects in vivoare blocked by inhibition of bFGF: LDPI. After hindlimb ischemiaand CST therapy, blood perfusion was significantly reduced inbFGF-Ab–treated mice compared with IgG. *P�0.05, **P�0.01bFGF-Ab vs IgG; n�7. i, CST-induced vasculogenesis is inhib-ited by inhibition of bFGF: EPCs in ischemic muscle. EPCs werecounted in sections of muscles of GFP BMT mice (GFP�/lectin�
cells). Inhibition of bFGF blocked CST-induced increase ofEPCs. *P�0.05 IgG vs bFGF-Ab; n�7. j, CST-induced vasculo-genesis is inhibited by inhibition of bFGF: circulating flk-1�/GFP� cells. Flk-1�/GFP� cells 3 weeks after hindlimb ischemiaand CST IM treatment were significantly inhibited by bFGF-Ab.*P�0.05 IgG vs bFGF-Ab; n�7.
1332 Circulation Research November 26, 2010
by guest on April 29, 2018
http://circres.ahajournals.org/D
ownloaded from

induces histamine release in vivo in rat21 and in vitro frommast cells.22 Furthermore, CST induces directed migrationof human blood monocytes,14 which qualifies CST to bean inflammatory cytokine.14 Recently, CST has beenshown to induce vasodilation in human dorsal hand vein11a
The present findings establish CST as an angiogenicpeptide.
CST Induces Effects in ECsInflammation and hypoxia are usually accompanied or fol-lowed by increased generation of blood vessels.8 CgA, theprecursor of CST, has been shown to be upregulated in brainin response to hypoxia,13 as has been reported previously forangiogenic factors like VEGF. Here, we found that CSTexerts several effects on ECs including EC migration andproliferation, and consistent with previous findings in well-established angiogenic peptides VEGF and bFGF,23 CST alsosignals through MAPK pathway to induce these effects.Furthermore, inhibition of CST-induced chemotaxis of ECsby PTX indicates CST signaling through G proteins. PI3-kinase/Akt pathway plays a pivotal role in EC survival andwe show that CST stimulates Akt phosphorylation, that CSTinhibits EC apoptosis, and that inhibition of PI3-kinase byWM abrogates CST-mediated effect. These data suggest thatCST signals through the PI3-kinase/Akt pathway to inhibitHUVEC apoptosis.
CST Effects on Other Vascular Cells (EPCs,SMCs), Vasculogenesis, and ArteriogenesisBesides its effects on ECs, CST also induced chemotaxison other vascular cells like EPCs or SMCs (see the OnlineData Supplement). CST also exerted incorporation ofEPCs into vascular structures in vitro, and inhibition ofbFGF blocked this effect, indicating that bFGF, a factorreported to attract EPCs,24 mediates CST-induced effects(see below). These findings indicate that this peptide mayalso induce postnatal vasculogenesis. Consistent with this,we found that CST increases incorporation of EPCs inischemic hindlimbs in GFP BMT mice, which is a well-characterized model to study these cells,16,17 findings thatwere also confirmed by confocal microscopy. We alsofound increased numbers of arteries/arterioles in CSTtreated ischemic hindlimbs, consistent with induction ofarteriogenesis. Upregulation of PDGF-B in ECs mightindicate that CST induces maturation of capillaries.25 Incontrast, MCP-1, an important regulator of maturation ofpreexisting collaterals, was not increased by CST.26
CST Effects on Angiogenesis In VivoConsistent with angiogenic peptides, CST induces angio-genesis in vivo in the mouse cornea neovascularizationassay. This in vivo finding further strengthened the abilityof CST to induce angiogenesis in the hindlimb ischemiamodel. Serial measurements of blood perfusion point outthat CST increases perfusion to levels before ligation ofthe femoral artery, yielding a significant better valuecompared with saline-injected animals. These observationsestablish CST as a novel angiogenic cytokine. In thiscontext, it is also interesting that the CST precursor CgA
was found in motor nerve endplates of skeletal muscles.27
Future studies will have to reveal whether CST plays a rolein physiological processes like nerve–muscle signal trans-mission or in the pathophysiology of injured skeletalmuscle cells, in addition to its ability to increase bloodperfusion to these cells.
CST Effects Are Mediated by bFGFIndirect angiogenic factors like sonic hedgehog inducetheir effects by upregulation of other, direct angiogeniccytokines like VEGF.28 Furthermore, effects of factors likePDGF-BB or prostaglandin E on vascular cells or angio-genesis have been shown to be mediated by bFGF.29,30 Wetherefore tested whether CST-mediated effects on ECsdepend on VEGF or bFGF and found that inhibition ofbFGF by a neutralizing antibody indeed blocked CST-mediated in vitro angiogenesis and late MAPK activation.Additionally, we found that CST releases bFGF from ECswith a maximum as early as 1 hour and stimulates FGFR1activation. Release of bFGF was blocked by inhibition ofMAPK, indicating that this signaling pathway is necessaryfor CST-induced bFGF release. Furthermore, inhibition ofbFGF and MAPK blocked CST-induced FGFR1 activa-tion. This finding, as well as the observation that CST-induced FGFR1 activation was delayed compared withbFGF, indicates that CST transactivates FGFR1 by releaseof bFGF. Therefore, we propose a model for CST action inwhich bFGF is released from ECs by CST via a MAPK-dependent mechanism, stimulates FGFR1, leading to asecond, late activation of MAPK after 50 minutes, inaddition to a first, direct CST-mediated MAPK activationindependent of bFGF after 10 minutes. This prolongedMAPK stimulation seems to be necessary for CST-mediated EC function, as shown by blockade of CST-induced capillary tube formation by inhibition of bFGF. Asimilar effect of bFGF on PDGF-BB–induced vascularSMC function has been reported previously.29 Addition-ally, we also were able to demonstrate that CST-inducedeffects in vivo depend on bFGF, as shown by increasednecrosis, decreased blood perfusion, reduced EPCs inischemic muscles, and reduced flk-1/GFP� cells in thecirculation in mice treated with a neutralizing bFGF-Ab.These findings might indicate that bFGF mediates CST-induced mobilization and homing to ischemic muscletissue.
It should be pointed out that the N-terminal CgA peptidevasostatin inhibits VEGF-induced angiogenesis. In fact, va-sostatin blocked VEGF-induced MAPK activation in ECs andinhibited angiogenesis exerted by this factor in vivo in theMatrigel assay.19 These findings indicate selective inhibitionof EC function and angiogenesis by the CgA peptides. Futurestudies will determine why 2 peptides (vasostatin and CST)derived from CgA exert opposite effects on angiogenesis. It isyet to be determined whether CgA exhibits differentialprocessing to vasostatin and CST in response to differentphysiological demands. In this context, it is also interestingthat TNF-� exhibits dose-dependent, opposing actions onangiogenesis.31
Theurl et al Catestatin and Angiogenesis 1333
by guest on April 29, 2018
http://circres.ahajournals.org/D
ownloaded from

Of note, SN, a peptide derived from another member of thechromogranin/secretogranin family, secretogranin II, alsoinduces therapeutic angiogenesis.16 Several angiogenic cyto-kines have been used to treat patients experiencing peripheralarterial or coronary heart disease32 and CST, as well as SN, isemerging as a promising novel candidate in the therapy ofthese diseases.
AcknowledgmentsWe thank Ursula Stanzl, Eva Huber, and Lydia Markut for excellenttechnical assistance.
Sources of FundingR.K. was supported by a grant of the Oesterreichische Nationalbank(grant no. 10189). S.K.M. was supported by grants from theDepartment of Veterans Affairs and the NIH grants R01 DA011311and P01 HL58120. M.T. was supported by a grant of the Medizinis-che Forschungsfonds Tirol (grant no. 202). M. Theurl was supportedby the intramural funding program of the Medical UniversityInnsbruck for young scientists MUI-START, project 2010012022.
DisclosuresNone.
References1. O’Connor DT, Frigon RP, Sokoloff RL. Human chromogranin A.
Purification and characterization from catecholamine storage vesiclesof human pheochromocytoma. Hypertension. 1984;6:2–12.
2. Winkler H, Fischer-Colbrie R. The chromogranins A and B: the first 25years and future perspectives. Neuroscience. 1992;49:497–528.
3. Tatemoto K, Efendic S, Mutt V, Makk G, Feistner GJ, Barchas JD.Pancreastatin, a novel pancreatic peptide that inhibits insulin secretion.Nature. 1986;324:476–478.
4. Aardal S, Helle KB, Elsayed S, Reed RK, Serck-Hanssen G. Vasostatins,comprising the N-terminal domain of chromogranin A, suppress tensionin isolated human blood vessel segments. J Neuroendocrinol. 1993;5:405–412.
5. Mahata SK, O’Connor DT, Mahata M, Yoo SH, Taupenot L, Wu H,Gill BM, Parmer RJ. Novel autocrine feedback control of catechol-amine release. A discrete chromogranin a fragment is a noncom-petitive nicotinic cholinergic antagonist. J Clin Invest. 1997;100:1623–1633.
6. O’Connor DT, Kailasam MT, Kennedy BP, Ziegler MG, Yanaihara N,Parmer RJ. Early decline in the catecholamine release-inhibitory peptidecatestatin in humans at genetic risk of hypertension. J Hypertens. 2002;20:1335–1345.
7. Mahapatra NR, O’Connor DT, Vaingankar SM, Hikim AP, Mahata M,Ray S, Staite E, Wu H, Gu Y, Dalton N, Kennedy BP, Ziegler MG, RossJ, Mahata SK. Hypertension from targeted ablation of chromogranin Acan be rescued by the human ortholog. J Clin Invest. 2005;115:1942–1952.
8. Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med.2000;6:389–395.
9. Ziche M, Morbidelli L, Masini E, Amerini S, Granger HJ, Maggi CA,Geppetti P, Ledda F. Nitric oxide mediates angiogenesis in vivo andendothelial cell growth and migration in vitro promoted by substance P.J Clin Invest. 1994;94:2036–2044.
10. Kirchmair R, Gander R, Egger M, Hanley A, Silver M, Ritsch A,Murayama T, Kaneider N, Sturm W, Kearny M, Fischer-Colbrie R,Kircher B, Gaenzer H, Wiedermann CJ, Ropper AH, Losordo DW,Patsch JR, Schratzberger P. The neuropeptide secretoneurin acts as adirect angiogenic cytokine in vitro and in vivo. Circulation. 2004;109:777–783.
11a.Fung MM, Salem RM, Mehtani P, Thomas B, Lu CF, Perez B, Rao F,Stridsberg M, Ziegler MG, Mahata SK, O’Connor DT. Direct vasoactiveeffects of the chromogranin A (CHGA) peptide catestatin in humans invivo. Clin Exp Hypertens. 2010;32:278–287.
11. Zukowska-Grojec Z, Karwatowska-Prokopczuk E, Rose W, Rone J,Movafagh S, Ji H, Yeh Y, Chen WT, Kleinman HK, Grouzmann E, GrantDS. Neuropeptide Y: a novel angiogenic factor from the sympatheticnerves and endothelium. Circ Res. 1998;83:187–195.
12. Lembeck F, Holzer P. Substance P as neurogenic mediator of antidromicvasodilation and neurogenic plasma extravasation. Naunyn Schmie-debergs Arch Pharmacol. 1979;310:175–183.
13. Marti E, Ferrer I, Blasi J. Differential regulation of chromogranin A,chromogranin B and secretoneurin protein expression after transientforebrain ischemia in the gerbil. Acta Neuropathol (Berl). 2001;101:159–166.
14. Egger M, Beer AG, Theurl M, Schgoer W, Hotter B, Tatarczyk T,Vasiljevic D, Frauscher S, Marksteiner J, Patsch JR, Schratzberger P,Djanani AM, Mahata SK, Kirchmair R. Monocyte migration: a noveleffect and signaling pathways of catestatin. Eur J Pharmacol. 2008;598:104–111.
15. Kirchmair R, Egger M, Walter DH, Eisterer W, Niederwanger A,Woell E, Nagl M, Pedrini M, Murayama T, Frauscher S, Hanley A,Silver M, Brodmann M, Sturm W, Fischer-Colbrie R, Losordo DW,Patsch JR, Schratzberger P. Secretoneurin, an angiogenic neuro-peptide, induces postnatal vasculogenesis. Circulation. 2004;110:1121–1127.
16. Schgoer W, Theurl M, Jeschke J, Beer AG, Albrecht K, Gander R,Rong S, Vasiljevic D, Egger M, Wolf AM, Frauscher S, Koller B,Tancevski I, Patsch JR, Schratzberger P, Piza-Katzer H, Ritsch A,Bahlmann FH, Fischer-Colbrie R, Wolf D, Kirchmair R. Gene therapywith the angiogenic cytokine secretoneurin induces therapeutic angio-genesis by a nitric oxide-dependent mechanism. Circ Res. 2009;105:994 –1002.
17. Asai J, Takenaka H, Kusano KF, Ii M, Luedemann C, Curry C, EatonE, Iwakura A, Tsutsumi Y, Hamada H, Kishimoto S, Thorne T,Kishore R, Losordo DW. Topical sonic hedgehog gene therapy accel-erates wound healing in diabetes by enhancing endothelial progenitorcell-mediated microvascular remodeling. Circulation. 2006;113:2413–2424.
18. Tsurumi Y, Takeshita S, Chen D, Kearney M, Rossow ST, Passeri J,Horowitz JR, Symes JF. Direct intramuscular gene transfer of naked DNAencoding vascular endothelial growth factor augments collateral devel-opment and tissue perfusion. Circulation. 1996;94:3281–3290.
19. Belloni D, Scabini S, Foglieni C, Veschini L, Giazzon A, Colombo B,Fulgenzi A, Helle KB, Ferrero ME, Corti A, Ferrero E. The vasostatin-Ifragment of chromogranin A inhibits VEGF-induced endothelial cellproliferation and migration. FASEB J. 2007;21:3052–3062.
20. Angelone T, Quintieri AM, Brar BK, Limchaiyawat PT, Tota B, MahataSK, Cerra MC. The antihypertensive chromogranin a peptide catestatinacts as a novel endocrine/paracrine modulator of cardiac inotropism andlusitropism. Endocrinology. 2008;149:4780–4793.
21. Kennedy BP, Mahata SK, O’Connor DT, Ziegler MG. Mechanism ofcardiovascular actions of the chromogranin A fragment catestatin in vivo.Peptides. 1998;19:1241–1248.
22. Kruger PG, Mahata SK, Helle KB. Catestatin (CgA344-364) stim-ulates rat mast cell release of histamine in a manner comparable tomastoparan and other cationic charged neuropeptides. Regul Pept.2003;114:29 –35.
23. D’Angelo G, Struman I, Martial J, Weiner RI. Activation of mitogen-ac-tivated protein kinases by vascular endothelial growth factor and basicfibroblast growth factor in capillary endothelial cells is inhibited by theantiangiogenic factor 16-kDa N-terminal fragment of prolactin. Proc NatlAcad Sci U S A. 1995;92:6374–6378.
24. Murayama T, Tepper O, Silver M, Ma H, Losordo D, Isner J, Asahara T,Kalka C. Determination of bone marrow-derived endothelial progenitorcell significance in angiogenic growth factor-induced neovascularizationin vivo. Exp Hematol. 2002;30:967.
25. Ramsauer M, D’Amore PA. Getting Tie(2)d up in angiogenesis. J ClinInvest. 2002;110:1615–1617.
26. Schaper W. Collateral circulation: past and present. Basic Res Cardiol.2009;104:5–21.
27. Li JY, Leitner B, Lovisetti-Scamihorn P, Winkler H, Dahlstrom A.Proteolytic processing, axonal transport and differential distribution ofchromogranins A and B, and secretogranin II (secretoneurin) in rat sciaticnerve and spinal cord. Eur J Neurosci. 1999;11:528–544.
28. Pola R, Ling LE, Silver M, Corbley MJ, Taylor FR, Baker DP, AsaharaT, Isner JM. The morphogen sonic hedgehog is an indirect angiogenicagent upregulating two families of angiogenic growth factors. Nat Med.2001;7:706–711.
29. Millette E, Rauch BH, Defawe O, Kenagy RD, Daum G, Clowes AW.Platelet-derived growth factor-BB-induced human smooth muscle cell
1334 Circulation Research November 26, 2010
by guest on April 29, 2018
http://circres.ahajournals.org/D
ownloaded from

proliferation depends on basic FGF release and FGFR-1 activation. CircRes. 2005;96:172–179.
30. Finetti F, Donnini S, Giachetti A, Morbidelli L, Ziche M. ProstaglandinE(2) primes the angiogenic switch via a synergic interaction with thefibroblast growth factor-2 pathway. Circ Res. 2009;105:657–666.
31. Fajardo LF, Kwan HH, Kowalski J, Prionas SD, Allison AC. Dual role oftumor necrosis factor-alpha in angiogenesis. Am J Pathol. 1992;140:539–544.
32. Gupta R, Tongers J, Losordo DW. Human studies of angiogenic genetherapy. Circ Res. 2009;105:724–736.
Novelty and Significance
What Is Known?
● Neuropeptides such as substance P, neuropeptide Y, or secretoneurininduce angiogenesis.
● Catestatin (human chromogranin A352–372; bovine chromograninA344–364) was initially identified as a catecholamine release–in-hibitory peptide inhibiting catecholamine secretion in an auto-crine/paracrine manner by acting as a nicotinic cholinergicantagonist.
● Subsequent studies established catestatin as a pleiotropic peptidethat lowers blood pressure. It induces vasodilation, is a negativeinotrope, and has antimicrobial activity. It also stimulates mono-cyte chemotaxis.
What New Information Does This Article Contribute?
● Catestatin induces migration and proliferation of endothelial cells(ECs) and stimulates chemotaxis in vascular smooth muscle cellsor endothelial progenitor cells in vitro. These effects are mediatedby the activation of Akt and mitogen-activated protein kinase(MAPK) in ECs.
● Catestatin acts as an angiogenic factor in vivo, specifically in thecornea neovascularization model and the hindlimb ischemiamodel. It stimulates the incorporation of endothelial progenitorcells into ischemia hindlimbs.
● Both the in vitro and in vivo effects of catestatin were blocked by aneutralizing basic fibroblast growth factor (bFGF) antibody, impli-
cating the induction of bFGF-mediated signaling by catestatin viastimulation of bFGF release from ECs and activation of FGFreceptor-1.
We hypothesized that the neuropeptide catestatin induces theformation of new blood vessels because of its reported effectson vasodilation coupled with hypoxia-induced upregulation ofthe catestatin precursor chromogranin A. We found that cates-tatin induces chemotactic, proliferative, and antiapoptotic ef-fects on ECs in vitro by activating Akt and MAPK. Catestatininduced angiogenesis in the mouse cornea neovascularizationassay. In the hindlimb ischemia model, catestatin therapyimproved blood flow and reduced necrosis. Immunofluorescentstudies revealed increased density of capillaries and arteries andincorporation of endothelial progenitor cells by catestatin, im-plying that the peptide induces angiogenesis, arteriogenesis,and postnatal vasculogenesis. The in vitro and in vivo effects ofcatestatin were inhibited by a neutralizing bFGF antibody,indicating that bFGF mediates the action of catestatin. Of note,catestatin releases bFGF from ECs and stimulates FGFreceptor-1 in these cells. These findings indicate that catestatininduces therapeutic angiogenesis in the hindlimb ischemiamodel. Additional studies are required to evaluate the therapeu-tic potential of this peptide in other ischemic conditions such asischemic heart disease.
Theurl et al Catestatin and Angiogenesis 1335
by guest on April 29, 2018
http://circres.ahajournals.org/D
ownloaded from

Patsch, Dominik Wolf, Peter Schratzberger, Sushil K. Mahata and Rudolf KirchmairBeer, Danijela Vasiljevic, Song Rong, Anna Maria Wolf, Ferdinand H. Bahlmann, Josef R.
Markus Theurl, Wilfried Schgoer, Karin Albrecht, Johannes Jeschke, Margot Egger, Arno G.E.Dependent Mechanism−Growth Factor
The Neuropeptide Catestatin Acts As a Novel Angiogenic Cytokine via a Basic Fibroblast
Print ISSN: 0009-7330. Online ISSN: 1524-4571 Copyright © 2010 American Heart Association, Inc. All rights reserved.is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation Research
doi: 10.1161/CIRCRESAHA.110.2194932010;107:1326-1335; originally published online October 7, 2010;Circ Res.
http://circres.ahajournals.org/content/107/11/1326World Wide Web at:
The online version of this article, along with updated information and services, is located on the
/content/110/9/e69.full.pdfAn erratum has been published regarding this article. Please see the attached page for:
http://circres.ahajournals.org/content/suppl/2010/10/07/CIRCRESAHA.110.219493.DC1Data Supplement (unedited) at:
http://circres.ahajournals.org//subscriptions/
is online at: Circulation Research Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer about this process is available in the
located, click Request Permissions in the middle column of the Web page under Services. Further informationEditorial Office. Once the online version of the published article for which permission is being requested is
can be obtained via RightsLink, a service of the Copyright Clearance Center, not theCirculation Researchin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on April 29, 2018
http://circres.ahajournals.org/D
ownloaded from

Correction
The Neuropeptide Catestatin Acts As a Novel Angiogenic Cytokine via a Basic Fibroblast GrowthFactor–Dependent Mechanism: Correction
In the article that appears on page 1326 of the November 26, 2010 issue, the following sentenceshould have been included in the Sources of Funding section:
M. Theurl was supported by the intramural funding program of the Medical UniversityInnsbruck for young scientists MUI-START, project 2010012022.
The authors apologize for this error, and the error has been noted and corrected in the onlineversion of the article, which is available at http://circres.ahajournals.org/content/107/11/1326.full.
DOI: 10.1161/RES.0b013e3182571591
(Circ Res. 2012;110:e69.)© 2012 American Heart Association, Inc.
Circulation Research is available at http://circres.ahajournals.org
e69

Supplement Material.
Materials and Methods
Materials
Neutralizing basic fibroblast growth factor (bFGF) and vascular endothelial growth
factor (VEGF) antibodies, PD 98059, pertussis toxin, genistein and DAPI were purchased
from Sigma (Germany). Anti-phospho tyrosine antibody was from Millipore, antibodies
against FGF Receptor 1 and 3 were from Santa Cruz. Scrambled CST peptide was from
PiChem, Austria. SDF-1 was from Bachem.
Catestatin peptide and antiserum
Human catestatin CGA352–372 (SSMKLSFRARGYGFRGPGPQL) was synthesized by
the solid-phase method, using 9-fluorenylmethoxycarbonyl protection chemistry, as described
previously 1. Peptides were purified to 95% homogeneity by preparative reversed-phase high-
performance liquid chromatography on C-18 silica columns. Authenticity and purity of
peptides were further verified by analytical chromatography (reversed phase high-
performance liquid chromatography), and electrospray-ionization or matrix-assisted laser
desorption ionization mass spectrometry. Polyclonal rabbit antisera recognizing human
catestatin were developed by a modification of protocols previously described for other
chromogranin peptides.
Endothelial cell (EC) and smooth muscle cell (SMC) chemotaxis assay:
The migratory response of human umbilical vein endothelial cells (HUVEC) and
human umbilical artery smooth muscle cells (HUASMC) (both from Promo Cell, Germany)

2
was measured using a modified 48-well Boyden chemotaxis chamber in which a 8 µm pore
sized cellulose nitrate filter (Sartorius, Göttingen, Germany) separated the upper and the
lower chamber. Cells were detached from the tissue flask with 0.05% trypsin and EDTA
(Gibco) and re-suspended at a density of 3x105 cells/well in chemotaxis medium [EBM-2
(Clonetics) for HUVEC or smooth muscle cell medium with 0.75% bovine serum albumin;
PAA] before being placed in the upper wells of the chemotaxis chamber (Neuroprobe,
Bethesda, Maryland).
Cells were allowed to migrate toward different soluble concentrations of catestatin in a
humidified atmosphere (37°C, 5% CO2) for 8 hours (HUASMC) and 16 hours (for HUVEC).
After the incubation time the nitrocellulose filters were dehydrated, fixed and stained with
haematoxilin-eosin. Migration into the filter was quantified by measuring microscopically the
distance from the surface of the filter to the leading front of the cells. Data are expressed as
relative chemotaxis index, which is the ratio between the distance of migration towards test
attractants and that toward control medium into the filters. Basal migration of cells typically
was 20-30 µm.
To investigate the involvement of mitogen activated protein kinases, G-protein
coupled receptors and tyrosine kinases-receptors HUVECs were pre-treated for 90 minutes
with PD 98059 (Sigma) 10µmol/L, pertussis toxin (Sigma) 1µg/ml or genistein (Sigma),10-
6M before the chemotaxis assay. For some experiments cells were incubated with CST
antibody (1:1000).
EC proliferation assays:
To investigate a possible effect of catestatin on endothelial cell proliferation, cells
(30.000/well) were seeded in multiwell plates (24 well) and incubated for 36 hours in EBM-2

3
(Clonetics) containing 3% FBS (PAA) and solvent, different concentrations of catestatin or
10 ng/ml secretoneurin (Sigma). Subsequently, cells were stained with DAPI (Sigma) and cell
numbers were counted using ImageJ 1.4g. Three randomly selected fields were counted per
well and each condition was performed in quadruplicates. Three independent experiments
were performed. Additionally, EC proliferation was determined by BrdU Assay (Cell
Proliferation ELISA BrdU Kit from Roche) as determined by the manufacturer. Briefly, cells
were incubated with dilution medium (EBM2+1.5% FBS), VEGF (50 ng/ml) or CST
(1nmol/L) for 20 hours. Afterwards cells were labeled with BrdU for 4 hours and processed as
suggested by the manufacturer. Signals were analyzed using an Anthos Lucy 1 luminometer.
Apoptosis Assay:
For apoptosis assay cells were incubated with control medium or CST 1 nmol/L in
serum free EBM-2 for 18 hours. To investigate the effect of inhibition of phosphatidyl-
inositol-3 (PI-3) kinase/Akt pathway on CST mediated effects on EC apoptosis ECs were
treated with wortmannin (WM, from Sigma) at a concentration of 10 nM alone or in
combination with CST. TUNEL assay was performed according to the manufacturer’s
instructions (Roche) and cells positive fur TUNEL staining and for DAPI staining were
counted and results are expressed as % of TUNEL positive cells of all DAPI stained cells.
Western Blotting:
All primary antibodies were purchased from Cell Signaling. HUVECs were plated on
60 mm tissue culture dishes coated with gelatine 0,2 % (Sigma), starved for 12 hours in serum
free EBM-2 (Clonetics), treated with catestatin or VEGF (50 ng/ml), and processed for
western blotting, as suggested by the manufacturer. Serum free medium served as negative
control and 20% FBS (PAA) as positive control. For some experiments cells were co-
incubated with CST and bFGF Antibody 1:1000.

4
bFGF-ELISA:
ELISA was purchased from R&D. HUVEC were starved for 6 hours and treated with
1nmol/L CST for 30 minutes to 6 hours. Supernatants were removed from cells, cells washed
with PBS and a heparin solution and finally lysed as described 2. bFGF ELISA was performed
from supernatants, heparin washes and cell lysates.
FGF-Receptor-1 and -3 activation
HUVEC were stimulated with bFGF (20 ng/mL) for 30, 60 and 90 minutes or CST (1
nmol/L) for 30, 60, 90 and 180 minutes. For some experiments CST was co-incubated with
PD 98059 (10 µM) or a bFGF antibody (1:1000). Afterwards cells were processed for FGF-
Receptor-1 phosphorylation by immunoprecipitation for FGF-Receptor-1 and subsequent
phosphor-tyrosine immunoblotting and immunoblotting for FGF-Receptor-1 (antibodies from
Santa Cruz, USA) as described 3 4. For FGF-Receptor-3 activation similar experiments using
FGF-Receptor-3 antibody instead of FGF-Receptor-1 antibody were performed for control
and 60 minutes stimulation with bFGF and CST as described above.
PCR:
PCR was performed as described 5 to detect mRNA expression of respective proteins
and RNA of cells was extracted (Qiagen, RNeasy Mini Kit, Vienna, Austria). To eliminate
genomic DNA, samples were digested with DNase I (desoxyribonuclease I, Amplification
Grade, Invitrogen, Vienna, Austria), then subjected to reverse transcription (Superscript TM
First-Strand Synthesis System for RT-PCR, Invitrogen). cDNA was finally used as template
for PCR reactions using Biorad C 1000 cycler with CFX96 optical reaction module. As
mastermix MESA GREEN qPCR Mastermix Plus for SYBR Assay (Eurogentec) was used.
Omitting reverse transcriptase enzyme or cDNA template was used as negative control.

5
Following primers were used:
human MCP-1 (256 bp)
fwd CACCAATAGGAAGATCTCAGTGC
rev AACAGGGTGTCTGGGGAAAG
Following primers were used according to published literature 6
human SDF-1 (77 bp):
fwd GAAGCGAAAAAATCAGTGAATAAACC
rev TGGAACCTGAAACCCTGCTG
human PDGF-B (111 bp):
fwd CATTCCCGAGGAGCTTTATGAG
rev TCCAACTCGGCCCCATCT
human VEGF-A (72 bp):
fwd GTGCCCACTGAGGAGTCCA
rev TCCTATGTGCTGGCCTTGGT
human FGF2 (85 bp):
fwd AGCGACCCTCACATCAAGCTA
rev CCAGGTAACGGTTAGCACACACT
Endothelial progenitor cell (EPC) isolation
EPCs were isolated and cultured and EPC chemotaxis assay was performed as we
described before 7, 8. Cultured cell were analyzed by FACS for P1H12 and CD31 7 and by
PCR for KDR and CD31 8.

6
FACS analysis
FACS analysis of blood samples was performed in GFP bone marrow transplanted
mice 3 weeks after induction of hindlimb ischemia and treatment with CST or saline i.m. For
some experiments mice also received bFGF-neutralizing antibody or IgG (see above). 200 µl
of blood was withdrawn by puncture of the retro-orbital plexus. Cells positive for GFP were
analyzed also for VEGF-receptor 2 (flk-1) by incubating samples with PE-conjugated
antibody against flk-1(BD Pharmingen) for 15 min in the dark. Appropriate
fluorochrome-conjugated isotype control was used (BD Pharmingen). After incubation,
erythrocytes were lysed using FACS lysing solution (BD Pharmingen) and washed with PBS
before analysis. Cells positive for flk-1 and GFP are expressed as percentage of all GFP+
cells. Cells were analyzed using a FacsCalibur (Beckton-Dickinson) and CellQuest
software (Beckton-Dickinson).
Tube formation assay:
For tube formation assays a growth factor reduced matrigel was used (Chemicon). 96-
well plates were coated with matrigel and HUVECs (5000/well) were seeded in serum free
EBM-2 with or without test substances (SN, CST, CST+PD 98059; CST+Cat Antibody
1:1000; CST+bFGF Antibody 1:1000; CST+VEGF Antibody 1:1000) and incubated for 8 to
12 hours at 37°C and 5% CO2. Images of tube formation were obtained using an inverted light
microscope (Olympus CK30) and the degree of tube formation was quantified by measuring
the intersection points in a grid (Plugin „Analyze-Grid Image J 1.4g).
Some matrigel assays were performed with a mixture of 5000 HUVECs and 3000 DiI LDL-
labelled EPCs as described 9, 10. All EPCs per high power field were counted using Image J
1.4g (Plugin-Analyze-Cell counter) and cells associated with tubes (in the same field) were
expressed as percentage of total EPCs. For more detailed studies of EPC –EC interaction ECs

7
were labelled with PKH67 green fluorescent cell linker kit (Sigma) as described by the
manufacturer.
Animal models
All protocols were approved by the Animal Care and Use Committee of the Medical
University Innsbruck and Hannover. Mice were purchased from Charles River Laboratories,
Kisslegg, Germany. Anesthesia was performed with 2,2,2-tribromoethanol (880 mmol/kg
body wt i.p.; Sigma-Aldrich).
Cornea Neovascularisation Assay:
Pellets containing 500 ng scrambled CST, CST or VEGF were implanted in C57/BL/6J
mice (age of 8 weeks) as described 11. On postoperative day 7 mice received an intravenous
injection of 500 µg BS1 lectin conjugated to FITC (Vector Laboratories). After euthanasia,
enucleated eyes were fixed in 1% paraformaldehyde, corneas dissected and examined by
fluorescence microscopy.
Mouse hindlimb ischemia model
Male C57BL/6 wild-type mice at the age between 12 and 15 months were subjected to
unilateral hind-limb surgery as described 12. Briefly, the left femoral artery was exposed,
ligated with 5-0 silk ligatures, and excised. Mice were injected with saline or 20 µg catestatin
(n=10 mice/group) into thigh and calf muscles after operation and every other day for weeks 1
and 2 and 2 times per week for weeks 3 and 4 according to previously published work 13. All
further measurements and analyses were performed by investigators blinded to the treatment
of the animals. For some experiments mice were treated with a mouse monoclonal
neutralizing bFGF antibody or respective IgG control (both from Abcam) via subcutaneously

8
implanted osmotic minipumps (Alzet) to allow continuous release of a total of 10 µg antibody
over 4 weeks. In some mice hindlimb muscles were homogenized 3 days after ischemia and
CST content of ischemic and control hindlimbs was determined by CST ELISA (Bachem). In
another group of mice CST was injected into hindlimbs (20 µg) and CST content in muscles
and serum was analyzed by ELISA after different time-points.
Limb necrosis score was determined as described previously in detail 14. Briefly, mice
were investigated at day 7, 14 and 28 and scored with 1 point if no necrosis or defect was
observed, with 2 points if skin necrosis or below ankle amputation was present and with 3
points if above ankle amputation was present. Scores were averaged for catestatin or saline
group.
Bone marrow transplantation model
Bone marrow transplantation (BMT) was performed as described 8 15 10. Briefly, 8
weeks old wild-type C56BL/6J recipient mice were lethally irradiated. ß-actin/GFP mice
(C56BL/6J background) that ubiquitously express GFP were used as bone marrow donors.
After irradiation, the recipient mice received bone marrow cells (5 x 106) from the ß-
actin/GFP mice by tail vein injection. After 3 weeks reconstitution of transplanted marrow
was confirmed by FACS analysis of peripheral leukocytes and >98% of cells were GFP
positive (data not shown). 6 weeks after transplantation, hind-limb ischemia surgery and
injection of catestatin or saline was performed. Mice were injected with saline or 20 µg
catestatin into thigh and calf muscles after operation and every other day for weeks 1 and 2
and 2 times per week for week 3. For some experiments mice were treated with a mouse
monoclonal neutralizing bFGF antibody or respective IgG control (both from Abcam) via
osmotic minipumps (Alzet) to allow continuous release of a total of 10 µg antibody over 3
weeks. Labelling of functional vessels and of EPCs in BMT-mice was performed 3 weeks
after induction of ischemia as described 15. Briefly, host-derived vessels were labelled by

9
intra-venous (i.v.) injection of 100 µL of rhodamine-conjugated BS-1 lectin 20 minutes before
euthanasia. Hind-limb muscles were embedded in OCT compound (TISSUE-TEK®, Sakura
Finetek) and snap-frozen in liquid nitrogen. 5 µm-thick frozen sections were analysed for red
(rhodamine) labelled host derived vessels and for green (GFP) labelled graft bone marrow
derived cells. EPCs are considered as double positive stained cells (red for incorporation into
the host vasculature and green for bone-marrow origin). EPCs are expressed as number of
double positive cells per high-power field (magnification x100). Each muscle was divided
into 2 parts and EPCs were counted and averaged from 5 sections of each part. A group of
mice was also treated with bFGF neutralizing monoclonal antibody or IgG as described
above.
For confocal microscopy a Broadband Confocal Leica TCS SP5 microscope (lenses:
HCX PL APO CS 20x0.7 (dry) and HCX PL APO CS 63x1.2 (Water)) was used.
Blood flow measurement
Blood flow measurements were performed using a laser Doppler perfusion image
(LDPI) analyzer (Moor Instruments, USA) as described 12. To minimize data variables
attributable to ambient light and temperature animals were kept on a heating plate at 37°C
before measurement and blood perfusion is expressed as the LDPI index representing the ratio
of left (=operated, ischemic leg) versus right (=not-operated, not-ischemic leg) limb blood
flow. A ratio of 1 before operation indicates equal blood perfusion of both legs, whereas after
femoral artery excision this ratio drops to values between 0.15 and 0.26, indicating severe
attenuation of leg blood supply in the operated leg.
Immunofluorescence

10
For tissue staining, mice were sacrificed and ischemic limb tissues were retrieved after
4 weeks. Specimens were fixed in 10% (v/v) buffered formaldehyde, dehydrated with graded
ethanol series and embedded in paraffin. Alternatively, fresh tissue was embedded in OCT
compound (TISSUE-TEK®, Sakura Finetek) and snap-frozen in liquid nitrogen. Tissues were
sliced into 5-µm sections. Vascular endothelial cells were identified by CD-31 (Pharmingen)
or FITC-isolectin-B4 (Vector, dilution 1:100) staining and for assessment of artery/arteriole
density sections were stained with a mouse monoclonal alpha-smooth muscle actin (SMA)
antibody (Pharmingen) as described 11. For fluorescent microscopy appropriate secondary
antibodies (Alexa 488 or 594 for SMA and Alexa 594 for CD31; both from Invitrogen 1:200)
were used. Adductor muscle samples of each leg were divided into 2 parts and capillaries
(CD31 positive) and arteries (alpha-smooth muscle actin positive) were counted in five
sections of each part and are expressed as capillary and arteriole density per mm2.
For staining of myelo-monocytic cells a CD11b antibody was used (clone M1/70 from
Bioscience) and detected by a goat-anti-rat Alexa 647 secondary antibody (Invitrogen).
For cell apoptosis assays sections of muscles subjected to hind-limb ischemia and
treated by CST or saline after 7 days were stained using the TUNEL assay from Roche (In-
Situ Cell Death Detection Kit, Fluorescein) as described by the manufacturer. Sections were
additionally stained for DAPI and CD-31.
Statistical Analysis
All results are expressed as mean ± SEM. Statistical comparisons between 2 groups
were performed by Student-t test. Multiple groups were analysed by 1-way ANOVA test
followed by appropriate post hoc tests to determine statistical significance. Probability values
<0.05 were considered statistically significant. All experiments were repeated at least in
triplicate.

11
Supplemental Results
1. Effects of CST on EPCs in-vitro.
To determine potential effects of CST on EPCs we performed cell migration
experiments. CST induced migration of EPCs with the maximum effect at 1 nmol/L (relative
CI CST 1.64±0.04; P<0.05; Online Figure Ia). SDF-1 and SN were used as positive controls.
CST also caused significant increase in percentage of EPCs associated with capillary tubes in
the co-culture matrigel assay of HUVEC with DiI-labelled EPCs and this effect was blocked
by a bFGF antibody (Ctr 57.6±2.7% of EPCs associated with capillary tubes; CST 1 nmol/L
78.8±2.7%; CST+bFGF Ab 53.4±2.6%; P<0.01 CST vs. Ctr; and CST+bFGF Ab; Online
Figure Ib). These findings indicate that CST-mediated EPC-EC interaction in-vitro is
mediated by bFGF like other effects of CST on ECs in vitro and on angiogenesis and
vasculogenesis in-vivo (Fig. 6).
For more detailed studies of EPC –EC interaction in co-culture EPCs were labelled with DiI-
acetylated LDL and ECs with PKH67. Online Figure Ic shows close interaction of EPC (red)
and EC (green) (arrowheads), some EPCs are also without contact to tubes (arrow).
2. CST effects on smooth muscle cells (SMCs)
Since formation of arteries or arteriogenesis, requires migration of SMCs, we tested
whether catestatin could induce chemotaxis of SMCs. Like the well-established SMC
chemotactic agent platelet-derived growth factor (PDGF)-BB, CST also induced chemotaxis
of SMCs with a maximum effect at 10-9 mol/L (CI 1.64±0.05, P<0.05 vs. Ctr; Online Figure
IIa). We additionally evaluated if CST induces release of bFGF from SMCs and found
significant release after 1 hour but not at later time-points (pg bFGF/ml: Ctr. 21.9±2.4, CST 1
hour: 35.1±1.7; P<0.05, n=3). Addition of PD but not of bFGF antibody inhibited CST-

12
induced SMC chemotaxis (Online Figure IIa) indicating that SMC chemotaxis by CST is
independent of bFGF like also shown for PDGF-BB 2. We also investigated if CST stimulates
MAPK in SMCs and found activation of ERK with a maximum after 10 minutes (Online
Figure IIb).
3. Regulation of angiogenic factors in ECs by CST
To evaluate if CST up-regulates angiogenic factors in ECs cells were incubated with
CST in different concentrations (1 nmol/L, 10 nmol/L) and time-points (3, 6 and 12 hours)
and respective m-RNAs were measured by real-time PCR. We found up-regulation of PDGF-
B after 6 hours of stimulation with 1 nmol/L CST (rel. m-RNA/18S: 1.62±0.14; P<0.05 vs.
Ctr). We did not detect regulation of other factors including VEGF, bFGF, MCP-1 or SDF-1
(Online Figure III).
4. Isolectin staining of capillaries in ischemic muscles
Beside CD31 we also used isolectin to identify ECs in ischemic muscles treated with
saline or catestatin. Co-staining with α-SMA was used to identify arterioles/arteries. We
found that catestatin increased density of capillaries and arteries using isolectin staining
(Online Figure IV).
5. Characterisation of GFP-positive cells after BMT
To evaluate CST-induced post-natal vasculogenesis we used the GFP-bone marrow
transplanted mouse. As described above in the Methods section (bone marrow transplantation
model) mice were subjected to hindlimb ischemia operation and received i.v. injection of
rhodamine-labelled BS-1 lectin before euthanasia. Sections of ischemic muscles were
analyzed for GFP+ cells (bone marrow origin) and rhodamine-lectin positive cells. Double

13
positive cells were considered as EPCs. To additionally characterize cells for monocyte origin
we stained cells for CD11b. As shown in Online Figure V we found cells positive for CD11b
and GFP (arrows) and, however distinct cells, positive for GFP and BS-1 lectin (arrowheads).
This finding indicates that EPCs and CD11b positive monocytes are different cells.
6. CST concentration in ischemic limbs, CST tissue half life
After 3 days of ischemia hindlimb muscles of ischemic and control side were homogenized
and CST content analyzed by ELISA. In the non-ischemic limb CST level was below the
detection limit of the assay. In the ischemic limb CST concentration was 1.04±0.34 ng/1g wet
weight (n=5). This corresponds to an approximately 0.5 nmol/L concentration.
To evaluate half life of GST in the limb tissue 20 µg CST was injected i.m. and CST content
was measured in tissue homogenates 1, 2, 4 and 6 hours after injection as well as 48 hours
after injection (the time-point when the next injection was performed). From the slope of the
curve a half-life of CST of approximately 12 hours can be calculated (Online Figure VI).
7. FGF Receptor 3 activation
FGF Receptor 3 (FGFR3) activation was studied by immunoprecpitation as described above.
We found that bFGF and CST activate FGFR3 (Online Figure VII).
8. Catestatin effects on p38 and JNK activation.
Possible activation of p38 and JNK by CST was evaluated in HUVEC by western blotting.
Only a slight activation of p38 after 10 and 20 minutes was observed whereas JNK was not
activated by CST (Online Figure VIII).
9. Akt-activation of catestatin is not blocked by bFGF antibody.

14
HUVEC were treated with CST in the presence or absence of a neutralizing bFGF antibody
for different time-points and evaluated for Akt activation by western blotting. Akt activation
by CST was not blocked by the bFGF-Ab (Online Figure IX).
10. Catestatin inhibits apoptosis in ischemic hindlimbs
Sections of ischemic hindlimbs 7 days after induction of ischemia were stained with DAPI
and TUNEL. Calculations revealed significantly decreased apoptotic cells by CST treatment
(%TUNEL-positive cells of DAPI-positive cells: Ctr. 2.0±0.1, CST 1.46±0.1; P<0.05; n=4,
Online Figure X).

15
References 1. Kennedy BP, Mahata SK, O'Connor DT, Ziegler MG. Mechanism of cardiovascular
actions of the chromogranin A fragment catestatin in vivo. Peptides. 1998;19:1241-
1248.
2. Millette E, Rauch BH, Defawe O, Kenagy RD, Daum G, Clowes AW. Platelet-derived
growth factor-BB-induced human smooth muscle cell proliferation depends on basic
FGF release and FGFR-1 activation. Circ Res. 2005;96:172-179.
3. Finetti F, Solito R, Morbidelli L, Giachetti A, Ziche M, Donnini S. Prostaglandin E2
regulates angiogenesis via activation of fibroblast growth factor receptor-1. J Biol
Chem. 2008;283:2139-2146.
4. Finetti F, Donnini S, Giachetti A, Morbidelli L, Ziche M. Prostaglandin E(2) primes
the angiogenic switch via a synergic interaction with the fibroblast growth factor-2
pathway. Circ Res. 2009;105:657-666.
5. Egger M, Schgoer W, Beer AG, Jeschke J, Leierer J, Theurl M, Frauscher S, Tepper
OM, Niederwanger A, Ritsch A, Kearney M, Wanschitz J, Gurtner GC, Fischer-
Colbrie R, Weiss G, Piza-Katzer H, Losordo DW, Patsch JR, Schratzberger P,
Kirchmair R. Hypoxia up-regulates the angiogenic cytokine secretoneurin via an HIF-
1alpha- and basic FGF-dependent pathway in muscle cells. Faseb J. 2007;21:2906-
2917.
6. Cho HJ, Lee N, Lee JY, Choi YJ, Ii M, Wecker A, Jeong JO, Curry C, Qin G, Yoon
YS. Role of host tissues for sustained humoral effects after endothelial progenitor cell
transplantation into the ischemic heart. J Exp Med. 2007;204:3257-3269.
7. Walter DH, Rittig K, Bahlmann FH, Kirchmair R, Silver M, Murayama T, Nishimura
H, Losordo DW, Asahara T, Isner JM. Statin therapy accelerates reendothelialization:

16
a novel effect involving mobilization and incorporation of bone marrow-derived
endothelial progenitor cells. Circulation. 2002;105:3017-3024.
8. Kirchmair R, Egger M, Walter DH, Eisterer W, Niederwanger A, Woell E, Nagl M,
Pedrini M, Murayama T, Frauscher S, Hanley A, Silver M, Brodmann M, Sturm W,
Fischer-Colbrie R, Losordo DW, Patsch JR, Schratzberger P. Secretoneurin, an
angiogenic neuropeptide, induces postnatal vasculogenesis. Circulation.
2004;110:1121-1127.
9. Shibata R, Skurk C, Ouchi N, Galasso G, Kondo K, Ohashi T, Shimano M, Kihara S,
Murohara T, Walsh K. Adiponectin promotes endothelial progenitor cell number and
function. FEBS Lett. 2008;582:1607-1612.
10. Schgoer W, Theurl M, Jeschke J, Beer AG, Albrecht K, Gander R, Rong S, Vasiljevic
D, Egger M, Wolf AM, Frauscher S, Koller B, Tancevski I, Patsch JR, Schratzberger
P, Piza-Katzer H, Ritsch A, Bahlmann FH, Fischer-Colbrie R, Wolf D, Kirchmair R.
Gene therapy with the angiogenic cytokine secretoneurin induces therapeutic
angiogenesis by a nitric oxide-dependent mechanism. Circ Res. 2009;105:994-1002.
11. Kirchmair R, Gander R, Egger M, Hanley A, Silver M, Ritsch A, Murayama T,
Kaneider N, Sturm W, Kearny M, Fischer-Colbrie R, Kircher B, Gaenzer H,
Wiedermann CJ, Ropper AH, Losordo DW, Patsch JR, Schratzberger P. The
neuropeptide secretoneurin acts as a direct angiogenic cytokine in vitro and in vivo.
Circulation. 2004;109:777-783.
12. Couffinhal T, Silver M, Zheng LP, Kearney M, Witzenbichler B, Isner JM. Mouse
model of angiogenesis. Am J Pathol. 1998;152:1667-1679.
13. Emanueli C, Salis MB, Pinna A, Graiani G, Manni L, Madeddu P. Nerve growth
factor promotes angiogenesis and arteriogenesis in ischemic hindlimbs. Circulation.
2002;106:2257-2262.

17
14. Bosch-Marce M, Okuyama H, Wesley JB, Sarkar K, Kimura H, Liu YV, Zhang H,
Strazza M, Rey S, Savino L, Zhou YF, McDonald KR, Na Y, Vandiver S, Rabi A,
Shaked Y, Kerbel R, Lavallee T, Semenza GL. Effects of aging and hypoxia-inducible
factor-1 activity on angiogenic cell mobilization and recovery of perfusion after limb
ischemia. Circ Res. 2007;101:1310-1318.
15. Asai J, Takenaka H, Kusano KF, Ii M, Luedemann C, Curry C, Eaton E, Iwakura A,
Tsutsumi Y, Hamada H, Kishimoto S, Thorne T, Kishore R, Losordo DW. Topical
sonic hedgehog gene therapy accelerates wound healing in diabetes by enhancing
endothelial progenitor cell-mediated microvascular remodeling. Circulation.
2006;113:2413-2424.

18

19
Online Figure I. Catestatin exerts effects on EPCs.
I.a. Catestatin induces EPC migration. Results are expressed as chemotactic index relative
to untreated cells. Catestatin induces chemotaxis of EPCs with a maximum effect at 1 nmol/L.
*P<0.05, **P<0.01 vs. Ctr.; n=3
I.b. Catestatin increases EPC incorporation into capillary tubes. ECs were seeded onto
matrigel together with acylated LDL-DiI labeled EPCs. EPCs incorporated and associated
with capillary tubes per high power field were counted and compared to total EPCs in the
same field. Catestatin (10-7 mol/L, 10-9 mol/L increased numbers of EPCs associated with
capillary tubes and effect was blocked by bFGF antibody. * P<0.05; ** P<0.01 vs. Ctr.; §
P<0.05 CST vs. CST+bFGF-Ab; n=3.
EC and EPC co-culture: double fluorescent labelling (right lower picture). EPCs were
labelled with DiI-acetylated LDL and ECs with PKH67. After co-culture picture reveals close
interaction of both cell types (arrowheads), some EPCs are located between tubes (arrow).

20
Online Figure II. Catestatin induces effects on vascular smooth muscle cells.
II.a. Catestatin induces SMC migration. Migration was evaluated for PDGF-BB (10 ng/ml)
and different concentrations of catestatin. Catestatin induced migration of these cells with a

21
maximum effect at a concentration of 10-9mol/L. Effect was blocked by PD but not by bFGF-
Ab.*P<0.05 vs. Ctr.; § P<0.05 CST vs. CST+PD; n=3
II.b. Catestatin induces MAPK activation in SMC. CST at a concentration of 1 nmol/L
stimulated ERK activation in SMCs with a maximum effect at 10 and 20 minutes.

22
Online Figure III: Regulation of angiogenic factors in ECs by CST.
ECs were serum starved and treated with 1 nmol/L CST for 6 hours. Real time PCR was used
to detect respective m-RNAs. * P<0.05 PDGF-B vs. Ctr, n=3.

23
Online Figure IV: Catestatin increases lectin-positive capillaries and arteries.
Sections of ischemic hindlimb muscles treated by saline or catestatin were stained by isolectin
(FITC labelled, left panels) or SMA antibody (labelled by a red fluorescent secondary
ALEXA antibody, middle panels); merged pictures: right panels. Catestatin increases lectin-
positive vessels.

24
Online Figure V: Characterisation of GFP positive cells in the GFP-bone marrow
transplanted mouse.
After BMT mice were subjected to hindlimb ischemia operation and treated with CST. Before
sacrifice host blood vessels were stained with i.v. injection of rhodamine labelled BS1-lectin.
Sections of ischemic hindlimbs were stained with DAPI and CD11b antibody and analysed
for GFP+ cells and rhodamine BS1 lectin positive vessels. Merging of pictures (right lower
picture) revealed GFP+/CD11b+ cells (arrows) and, distinct from them, GFP+/lectin+ cells
(arrowheads).

25
Online Figure VI: CST tissue half live after i.m. injection
CST levels in hindlimbs were determined by ELISA in tissue homogenates at different time-
points after injection of 20 µg CST. From the slope of the curve a half-life of approximately
12 hours can be calculated.
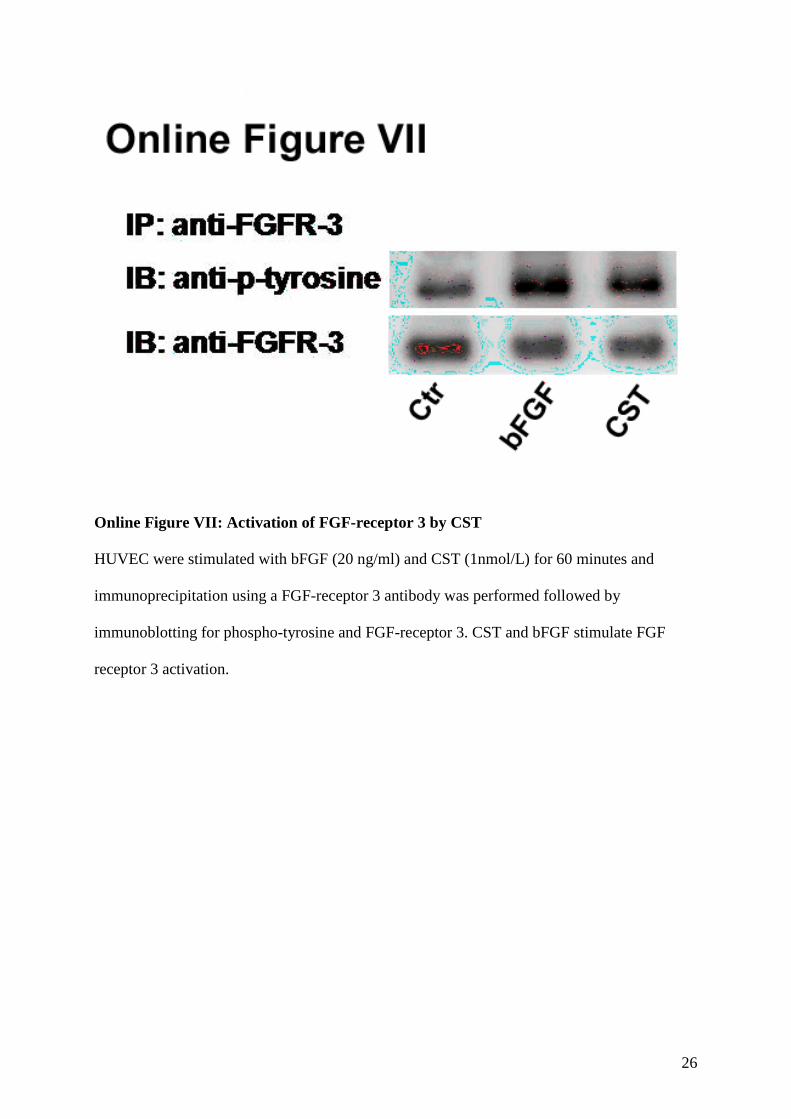
26
Online Figure VII: Activation of FGF-receptor 3 by CST
HUVEC were stimulated with bFGF (20 ng/ml) and CST (1nmol/L) for 60 minutes and
immunoprecipitation using a FGF-receptor 3 antibody was performed followed by
immunoblotting for phospho-tyrosine and FGF-receptor 3. CST and bFGF stimulate FGF
receptor 3 activation.

27
Online Figure VIII: Catestatin and p38 and JNK
HUVEC were stimulated with 1 nmol/L CST for different time-points and evaluated for
activation of p38 and JNK by western blotting. A slight activation of p38 but no activation of
JNK was observed.
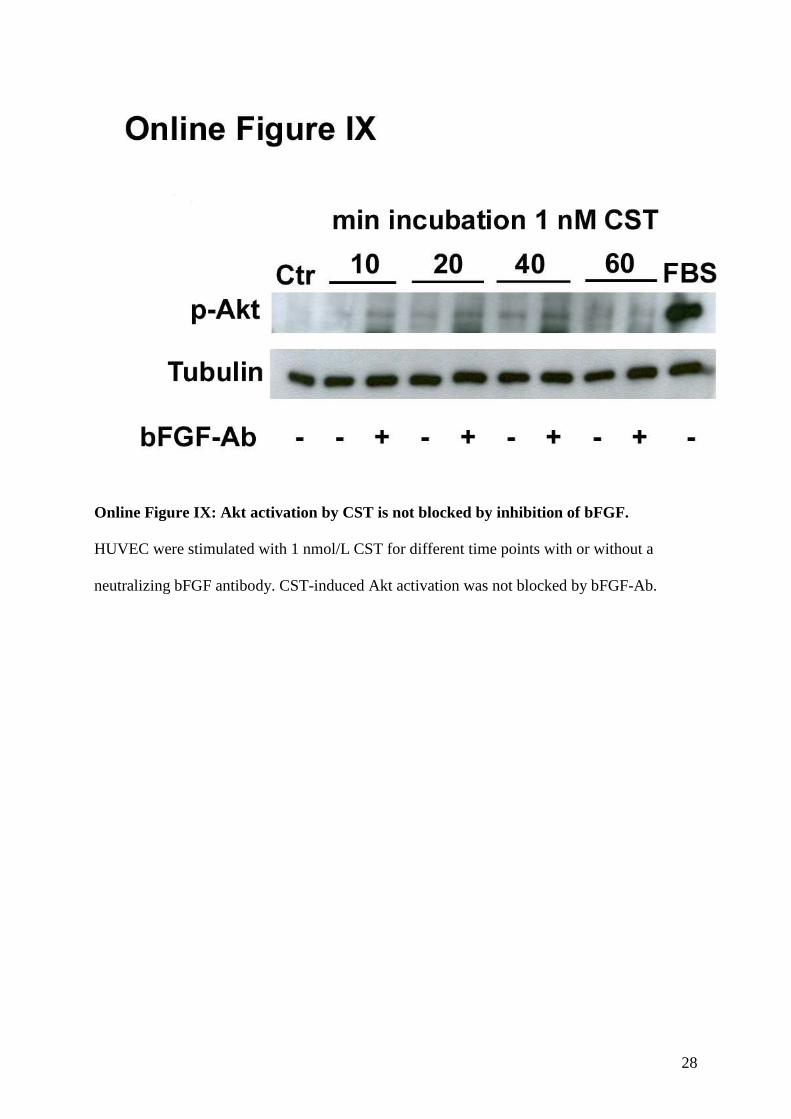
28
Online Figure IX: Akt activation by CST is not blocked by inhibition of bFGF.
HUVEC were stimulated with 1 nmol/L CST for different time points with or without a
neutralizing bFGF antibody. CST-induced Akt activation was not blocked by bFGF-Ab.

29
Online Figure X: Catestatin inhibits apoptosis in-vivo.
7 days after induction of ischemia and treatment with CST or saline sections of ischemic
muscles were analyzed for apoptotic cells by TUNEL staining. Fewer cells were TUNEL-
positive after CST treatment.

Correction
The Neuropeptide Catestatin Acts As a Novel Angiogenic Cytokine via a Basic Fibroblast GrowthFactor–Dependent Mechanism: Correction
In the article that appears on page 1326 of the November 26, 2010 issue, the following sentenceshould have been included in the Sources of Funding section:
M. Theurl was supported by the intramural funding program of the Medical UniversityInnsbruck for young scientists MUI-START, project 2010012022.
The authors apologize for this error, and the error has been noted and corrected in the onlineversion of the article, which is available at http://circres.ahajournals.org/content/107/11/1326.full.
DOI: 10.1161/RES.0b013e3182571591
(Circ Res. 2012;110:00.)© 2012 American Heart Association, Inc.
Circulation Research is available at http://circres.ahajournals.org
1





























