Embed Size (px)
Citation preview

THE EFFECT OF HIGH HYDROSTATIC PRESSURE ON STABILITY OF PYRUVATE
OXIDASE FROM AEROCCOCUS SPECIES
by
Luke Wallace
(Under the Direction of José Reyes I. De Corcuera)
ABSTRACT
Pyruvate oxidase (POX) catalyzes the oxidative decarboxylation of pyruvate to
acetylphosphate in the presence of oxygen, a divalent cation, and its cofactors thiamine
pyrophosphate and flavin adenine dinucleotide. High hydrostatic pressure (HHP) was applied at
selected temperatures to elucidate its effect on the thermostability of POX. High pressure
generally destabilizes POX. Rate of inactivation was slowed slightly between atmospheric
pressure and 50 MPa at 35 °C, where kinact decreased from 0.004 ± 0.003 min-1 to 0.001 ± 0.001
min-1. Activation volume halved to 29 ± 13 cm3/mol at 45 °C from 68 ± 43 cm/mol at 35 °C and
71 ± 12 cm/mol at 25 °C. Data from non-denaturing gel electrophoresis and ultraviolet
spectroscopy suggest that POX remains in a native conformation before dissociating and
precipitating with the application of HHP. Low density polyethylene reduced POX activity,
though the specific reason for this affect is unclear.
INDEX WORDS: Pyruvate oxidase; High hydrostatic pressure; Enzyme kinetics; Stabilization

THE EFFECT OF HIGH HYDROSTATIC PRESSURE ON STABILITY OF PYRUVATE
OXIDASE FROM AEROCCOCUS SPECIES
by
LUKE WALLACE
B.S.A., The University of Georgia, 2013
A Thesis Submitted to the Graduate Faculty of the University of Georgia in Partial Fulfillment of
the Requirements for the Degree
MASTER OF SCIENCE
ATHENS, GEORGIA
2017

© 2017
Luke Wallace
All Rights Reserved

THE EFFECT OF HIGH HYDROSTATIC PRESSURE ON STABILITY OF PYRUVATE
OXIDASE FROM AEROCCOCUS SPECIES
by
Luke Smith Wallace
Major Professor: José I. Reyes De Corcuera
Committee: Fanbin Kong
Derek Dee
Electronic Version Approved:
Suzanne Barbour
Dean of the Graduate School
The University of Georgia
August 2017

ACKNOWLEDGEMENTS
I would like to thank my major advisor, Dr. Reyes, for taking me on during a challenging
transition and for his constant support and enthusiasm throughout my project. I acknowledge and
appreciate the funding and financial assistance provided to me by Dr. Reyes, the department of
Food Science and Technology, and the USDA-NIFA-AFRI grant #2014-67021-21604, without
which my graduate education would not have been possible. To my advisory committee, Dr.
Kong and Dr. Dee, I thank you for your assistance and feedback in developing my project and
for your service in my committee. Lastly, I would especially like to thank my lab members, both
current and former, and my friends Dr. Garcia, Ali, Martina, Victoria, Daoyuan, Tristin, Brittnee,
Vivian, Hanna, Erica, Jess and many others, both for their help in my project, and for their
relentless support in helping me survive graduate school.
iv

v
TABLE OF CONTENTS
Page
ACKNOWLEDGEMENTS.……………………………………………………...………………iv
LIST OF TABLES………………………………………………………………………………..vi
LIST OF FIGURES…………………………………………………………………………...…vii
CHAPTER
1. LITERATURE REVIEW……………………………………………………………..1
Introduction……………………………………………………………………………1
Characteristics of Pyruvate Oxidase…………………………………………………..2
Detection of Pyruvate………………………………………………………………....4
Applications for Pyruvate Oxidase in Biosensors…………………………………….6
High Hydrostatic Pressure Stabilization of Enzymes………………………………..13
Gap of Knowledge…………………………………………………………………...17
Hypothesis……………………………………………………………………………17
Objectives……………………………………………………………………………17
2. THE EFFECT OF HIGH HYDROSTATIC PRESSURE ON STABILITY OF
PYRUVATE OXIDASE FROM AEROCOCCUS SPECIES……………………….19
Introduction…………………………………………………………………………..19
Materials and Methods……………………………………………………………….20
Results and Discussion………………………………………………………………34
3. FINAL COMMENTS………………………………………………………………..49
REFERENCES…………………………………………………………………………………..51
APPENDIX……………………………………………………………………………………....57

vi
LIST OF TABLES
Page
Table 1.1. Comparison of biosensors based on pyruvate oxidase………………………….……10
Table 1.2. Pyruvate oxidase biosensor stabilization technique comparison……...………….…..12
Table 2.1. Reaction cocktail composition for pyruvate oxidase activity assay....……………….25
Table 2.2. Residual activity for pyruvate oxidase treated using original pouch design at 100
MPa at 5 °C relative to both 0 s treatment time and fresh working enzyme solution
(n=3)…………………………………………………………………………………...…30
Table 2.3. Residual pyruvate oxidase activity for samples sealed in new pouches relative to
fresh enzyme activity over time………………………………………………..………...31
Table 2.4. First order rate of pyruvate oxidase inactivation ± standard error determined by the
linear regression of residual activity versus treatment time….…………………………..39
Table 2.5. Residual activity data for confirmation experiments using new pyruvate oxidase
batch compared to residual activity for the old batch calculated based on kinact.………...42
Table 2.6 Apparent activation energy of inactivation ± standard error determined by the linear
regression of ln(k) versus 1/T……………...…………...……………………………….43
Table 2.7 . Activation volume ± standard error determined by the linear regression for each
temperature………………………………………...…………………………………….45

vii
LIST OF FIGURES
Page
Figure 1.1. Typical elliptical pressure-temperature diagram with active and inactive enzyme
regions……………………………………………………………………………….…..15
Figure 1.2. Monomeric subunit of pyruvate oxidase (PDB ID: 1VF5) with its cofactors FAD
(1) and TPP (2), three largest cavities (3-5), and active site, GLU54 (6), labeled.….…..16
Figure 2.1. High hydrostatic pressure system...……………………………………………….....22
Figure 2.2. Pressure (○) and temperature (□) profiles for a sample treated at 35 °C at 100 MPa
for 300 s and pressure (●) and temperature (■) profiles for a sample treated at 35 °C
at 100 MPa for 0 s ……………………………………………………….………………24
Figure 2.3. Comparison of original (left) and newer (right) pouch design to scale………...……29
Figure 2.4. Residual activity relative to fresh enzyme over time for pyruvate oxidase in low-
density polyethylene pouch ( ), high density polypropylene tube ( ), and amber glass
vial ( )..……….…….…………………………………………………………………....32
Figure 2.5. Corrected absorbance measurements for individual replicates of fresh pyruvate
oxidase (black) compared with samples treated at 0.1 MPa at 35 °C for 0 (dark grey)
or 600 s (light grey)…………………………………………………………………........34
Figure 2.6. Residual activity for t0 treatments relative to fresh enzyme at 25 ( ), 35 ( ), or
45 °C ( )…………………………….……………………………………………………35
Figure 2.7. Reactor (open symbol) versus syringe (closed symbol) temperature at 25 (light grey),
35 (dark grey), or 45 °C (black)……………………………………………………….....36

viii
Figure 2.8. Residual activity for POX at 25 (A), 35 (B), and 45 °C (C) at 0.1 ( ), 50 ( ),
100 ( ) or 150 ( ) MPa…………………………………………………………………..37
Figure 2.9. Logarithm of residual activity at 25 (A), 35 (B), and 45 °C (C) at 0.1 ( ), 50 ( ),
100 ( ) and 150 ( ) MPa………………………...……………………………………….40
Figure 2.10. Rate constant of inactivation versus pressure at 25 ( ), 35 ( ), and 45 °C ( )….….45
Figure 2.11. Relative distance traveled through 4-20 % polyacrylamide gel by native and
inactivated pyruvate oxidase samples ………………………………………………..….47
Figure 2.12. Ultraviolet spectra for Native (black), Inactivated (grey), and blank (dotted)
samples …………………………………………………………………………………..47
Figure A.1. Pressure (closed symbol) and temperature (open symbol) for samples one
(dark grey) and two (light gray) for supplemental sample treated at 0.1 MPa at 25 °C
for 3000 s………………………………………………………………………………...57
Figure A.2. Pressure (closed symbol) and temperature (open symbol) for samples one
(dark grey) and two (light gray) for supplemental sample treated at 50 MPa at 25 °C
for 0 s ……………………………………………………………………………….…...58
Figure A.3. Pressure (closed symbol) and temperature (open symbol) for samples one
(dark grey) and two (light gray) for supplemental sample treated at 50 MPa at 25 °C
for 3000 s……………………………………………………………...…………………58
Figure A.4. Pressure (closed symbol) and temperature (open symbol) for samples one
(dark grey) and two (light gray) for supplemental sample treated at 0.1 MPa at 35 °C
for 0 s ………………………………………………………………………………...….58

ix
Figure A.5. Pressure (closed symbol) and temperature (open symbol) for samples one
(dark grey) and two (light gray) for supplemental sample treated at 0.1 MPa at 35 °C
for 3000 s …………………………………………………………………………...…...59
Figure A.6. Pressure (closed symbol) and temperature (open symbol) for samples one
(dark grey) and two (light gray) for supplemental sample treated at 50 MPa at 35 °C
for 0 s …………………………………………………………………………………....59
Figure A.7. Pressure (closed symbol) and temperature (open symbol) for samples one
(dark grey) and two (light gray) for supplemental sample treated at 50 MPa at 35 °C
for 3000 s …………………………………………………………………………...…...59
Figure A.8. Pressure (closed symbol) and temperature (open symbol) for samples one
(dark grey) and two (light gray) for supplemental sample treated at 0.1 MPa at 45 °C
for 0 s ………………………………………………………………………………...….60
Figure A.9. Pressure (closed symbol) and temperature (open symbol) for samples one
(dark grey) and two (light gray) for supplemental sample treated at 0.1 MPa at 45 °C
for 1000 s ……………………………………………………………………….…...…..60
Figure A.10. Pressure (closed symbol) and temperature (open symbol) for samples one
(dark grey) and two (light gray) for supplemental sample treated at 50 MPa at 45 °C
for 0 s ……………………………………………………………………...………….…60

1
CHAPTER 1
LITERATURE REVIEW
Introduction
Pyruvate oxidase (POX) catalyzes the oxidative decarboxylation of pyruvate into
acetylphosphate and hydrogen peroxide in the presence of oxygen and phosphate. Pyruvate
oxidase is a homotetrameric flavoprotein that is integral to the process of microbial aerobic
respiration. The high substrate specificity of POX enables it to be utilized in electrochemical
biosensors used in the quantitative analysis of both pyruvate and phosphate.
Biosensors are useful tools for detecting or quantifying the concentration of specific
chemicals or biological materials. Because they offer high sensitivity and specificity while
maintaining short response times, simple analysis methods, and relatively low costs, biosensors
are often preferable to other analytical methods. Biosensors using POX have been constructed
both for the detection of pyruvate and phosphate. In the case of phosphate detection, biosensors
are preferable to conventional spectrophotometric methods due to faster measurement speed,
reusability, higher specificity, and the potential for miniaturization and transportability [1, 2]. In
the case of pyruvate, biosensors can achieve a lower limit of detection, potentially at a lower cost
than many conventional detection methods [3, 4].
Pyruvate oxidase biosensors have potentially important roles in the food industry, the
medical field, and in environmental science. However, due to the poor stability of POX, current

2
biosensors lack the longevity to make them practical tools for most applications. Table 1.1
compares existing POX biosensors with respect to their operational and storage stability.
High hydrostatic pressure (HHP) has a stabilizing effect on many enzymes, and in certain
cases has been shown to enhance enzymatic activity [5]. It is not yet clear if HHP alone can be
used to enhance the practicality of enzymatic biosensors, but it could play a synergistic role
when paired with other stabilization techniques such as chemical, physical, and/or genetic
modification towards creating a more stable biosensor.
This chapter will review the current knowledge of POX activity, stability, and stabilization
within the context of biosensor fabrication.
Characteristics of Pyruvate Oxidase
Pyruvate oxidase is a homotetrameric flavoprotein in which each unit (65.5 kDa) binds
one FAD and one thiamine pyrophosphate (TPP) in the presence of a divalent cation, most
commonly Mn2+ or Mg2+. Pyruvate oxidase catalyzes the oxidative decarboxylation of pyruvate
according to the equation:
𝐶𝐻3𝐶𝑂𝐶𝑂𝑂− + 𝑂2 + 𝐻𝑃𝑂4
−2 + 2𝐻+ → 𝐶𝑂2 + 𝐻2𝑂2 + 𝐶𝐻3𝐶𝑂𝑂𝑃𝑂3𝐻−
The catalytic activity of POX can be broken down into five steps. The first is the deprotonation
of the C2-H of TPP, as with all TPP-dependent enzymes. Secondly, pyruvate is bound to the C2
atom of enzyme-bound TPP. Next is the decarboxylation of pyruvate to form hydroxyethyl-TPP,
followed by the oxidation of hydroxyethyl-TPP by FAD, and finally the reoxidation of FAD by
oxygen [6].
The optimum temperature for POX from Aerococcus sp. is 25 °C [7], 38 °C from
Lactobacillus plantarum [8], and 42 °C from Escherichia coli [9]. The optimum pH for POX
from Aerococcus is 7 [1] and 5.6 from Lactobacillus plantarum [8].

3
Size-exclusion HPLC and analytical ultracentrifugation show that the holoenzyme retains
its tetrameric state down to 20 µg/mL. The apoenzyme, however, shows a stepwise tetramer-
dimer-monomer dissociation at a protein concentration of 20 µg/mL. The quaternary structure is
stabilized in the presence of the cofactors, TPP, FAD, and a divalent cation. In the presence of
divalent cations, FAD, and TPP bind to the apoenzyme, forming the binary complex. Both TPP
and FAD affect the association of the quaternary structure by shifting the equilibrium towards a
dimer or tetramer structure. High FAD concentrations elicit a strong stabilizing effect against
urea and heat denaturation, while excess TPP has no effect [10].
POX activity requires the presence of a divalent metal cation. Blake, et al. [11] compared
the activity of pyruvate oxidase in the presence of Mg2+, Ca2+, Zn2+, Mn2+, Ba2+, Ni2+, Co2+,
Cu2+, and Cr3+. The results showed that while the most consistent steady-state kinetics were
observed with Mg2+ and Mn2+, the enzyme lacked specificity for the metal ion required for
catalysis. These findings suggest that the divalent metal cation is not a true cofactor of POX, but
that the cation instead binds to TPP, forming a metal-TPP complex that is the true enzymatic
cofactor.
To elucidate the conditions for thermal inactivation of the enzyme, POX from Bacillus
stearothermophilus was isolated along with other enzymes such as cytochrome oxidase, malic
dehydrogenase, and adolase from the cell granule using lysozyme. Unlike all other enzymes
isolated from the thermophile in this study, POX was quickly inactivated at the optimal growing
temperature range for the bacteria (60-65 °C). After one hour at 60 °C, the activity of POX
decreased to only 12%. The presence of pyruvate, Mg2+, and oxygen all increased thermal
stability. Pyruvate oxidase left within unlysed cells showed stability at 60 °C up to an hour, at
which point the system began to run out of oxygen and substrate [12].

4
Chang and Ruan [13] explored the pressure-induced dissociation of pyruvate oxidase
from E. coli using a fluorescence spectrophotometer attached to a pressurized chamber to
monitor the enzyme’s dissociation from 0.1 to 300 MPa. Results indicated that the native enzyme
dissociated completely at 220 MPa, but that the dissociation was likely reversible. FAD had a
stabilizing effect against pressure. When FAD was removed from the native enzyme, the
resulting apoenzyme had a half-dissociation pressure of only 98 MPa as opposed to 130 MPa for
the native enzyme. Based on the fluorometric data, the dissociation of the apo-pyruvate oxidase
occurred in a single step, the dissociation of the four subunits. In contrast, the native enzyme
showed an extra step wherein FAD was removed before the full dissociation of the subunits
occurred. This, along with calculated changes in molar volume, led to the author’s theory that
FAD induces a conformation change resulting in better matching of the subunit interface within
POX. Lastly, as enzyme dissociation was compared at pH values of 7.5, 8.5, or 9.5, results
showed that stronger alkalinity promoted POX dissociation. This may be due to a decrease in
subunit affinity caused by a change in ionization of amino acid sidechains. In the case of acidic
pH values, measurements were impossible due to enzyme precipitation under high pressure.
Detection of Pyruvate
Pyruvate is an important part of many metabolic pathways, perhaps most notably as a
product of glycolysis and as a starting point for the Krebs cycle, oxidative phosphorylation, and
amino acid synthesis. The detection of pyruvate is important to the food industry, the medical
field, and in environmental science.
The detection of pyruvate is of importance to the process of food fermentation [14].
During the production of wine and beer, the presence of pyruvic acid and other keto acids in the
beer wort or grape must is associated with important flavor compound production by yeast in the

5
finished product. During fermentation, specific amino acids (valine, leucine, isoleucine,
methionine, and phenylalinine) are metabolized by Saccharomyces cerevisiae into keto acids that
cannot be used in primary carbon metabolism. These acids are redirected into the Ehrlich
pathway where they are converted into fusel alcohols, which unlike ethanol contain more than
two carbons. In high concentrations, fusel alcohols contribute undesirable off-flavor. However,
in lower, controlled concentrations, these products help to contribute to the distinct flavor
profiles of many products [15, 16].
In onions, pyruvate serves as an important quality indicator of pungency [17].
Schwimmer and Weston [18] suggested that pungency arises from an interaction of L-cysteine
derivatives and allinase enzymes when the onion tissue is damaged, resulting in the production
of pyruvic acid. Biosensors made from immobilized pyruvate oxidase have differentiated
between low and high-pungency onion by measuring pyruvate concentrations [4]. Conventional
pyruvate measurements for alliums can be time consuming and expensive. With low-pungency
onions growing in popularity, means of accurately measuring pyruvate are becoming more
important [19].
Pyruvate oxidase biosensors have potential applications in the medical field. The
monitoring of L-lactate dehydrogenase (LDH) in human blood serum is important for medical
analysis. It catalyzes the reaction:
𝐿 − 𝑙𝑎𝑐𝑡𝑎𝑡𝑒 + 𝑁𝐴𝐷+𝐿𝐷𝐻→ 𝑝𝑦𝑟𝑢𝑣𝑎𝑡𝑒 + 𝑁𝐴𝐷𝐻 + 𝐻+
The conversion of NAD+ to NADH can be measured spectrophotometrically, but this is
difficult to do in colored samples, such as blood serum. Pyruvate oxidase presents a potential
alternative to the rapid detection of LDH in serum [20]. Additionally, the monitoring of
pyruvate in blood serum as fluctuations can be indicative of diabetic acidosis or vitamin B1

6
deficiency [14]. Similarly to LDH, pyruvate in serum can be monitored spectrophotometrically,
but problems arise due to the turbidity of blood samples.
Perhaps the main application for pyruvate oxidase-based biosensors is the monitoring of
phosphorous in aquatic environments. Phosphorous is an essential component to metabolism in
all known life forms, serving as an essential building block for nucleic acids and various
intermediate metabolites [21]. Monitoring phosphorous levels in lakes and rivers is a major
environmental concern due to eutrophication. Eutrophication occurs when waters become over-
enriched with nutrients, with phosphorous being the most common cause of eutrophication in
freshwater. During eutrophication, the over-enrichment of nutrients in water increases the
production of autotrophs such as algae and cyanobacteria. The accompanying increase in cellular
respiration causes hypoxia or anoxia, resulting in the death of aquatic animals and a detriment to
biodiversity. The detection of phosphorous is particularly important, because unlike nitrogen and
carbon which can be acquired in the atmosphere, phosphorous is usually found in more limiting
amounts, most commonly from surface water runoff. Human activities, mainly agriculture, are
the main contributors to phosphorous in surface runoff [22].
Applications for Pyruvate Oxidase for Biosensors
Existing alternatives for Pyruvate and Phosphate Detection
A biosensor is an integrated receptor-transducer device that converts a biologically-
induced recognition event, such as enzyme activity, into a detectable signal via a transducer and
processor. The signal can be used to depict the presence and/or concentration of the analyte of
interest using a display. Electrochemical biosensors are based on the monitoring of either the
production or consumption of electroactive species. Transduction occurs under a variety of
methods that fall under either potentiometry or amperometry. Potentiometric biosensors monitor

7
the electric potential of an electrochemical cell in a working electrode relative to a reference
electrode with zero current flow. Measuring the concentration of the target analyte can be
achieved by measuring ions or gases that are either generated or consumed during enzymatic
activity. Amperometric biosensors tend to be more common, both for single-use and multi-
measurement devices. Unlike potentiometric devices, amperometric biosensors utilize a constant
potential applied between a working and a reference electrode. The resulting net current flow
from the imposed potential is proportional to the concentration of the electroactive species in
solution [23].
Akyilmaz and Yorganci [24], developed a biosensor to investigate the effect of thiamine
on pyruvate oxidase. The biosensor was prepared by covalently immobilizing pyruvate oxidase
onto a dissolved oxygen probe using gelatin and glutaraldehyde as a cross-linking agent. During
the enzymatic reaction, the dissolved oxygen layer in the interval space decreased in relation to
concentration of added substrate. The difference in dissolved oxygen (ΔDO) was measured
between before and after the addition of substrate to the reaction medium. The principle of the
biosensor relied on detecting this ΔDO in relation to thiamine concentration to construct a
standard curve for the determination of thiamine. All measurements were done at 35°C in a
thermostatic reaction cell filled with oxygen-saturated phosphate buffer (50 mM, pH 7.0). The
biosensor could detect a linear range of thiamine over a constant concentration of pyruvate.
As previously mentioned, while phosphorus is an essential element to all life, elevated
levels caused by surface runoff from agricultural or other human activities can cause an
excessive growth of algae or cyanobacteria, leading to eutrophication [25]. Pyruvate oxidase is
the most commonly-used enzyme for multi-enzymatic phosphate biosensors. These are generally
considered to be simpler to construct and operate than other multi-enzyme phosphate biosensors

8
[26]. Gavalas and Chaniotakis [27] created a phosphate biosensor using recombinant pyruvate
oxidase from Lactobacillus plantarum stabilized by a polyelectrolyte on a porous carbon rod.
Two polyelectrolytes were tested, diethylamonoethyl-dextran (DEAE-Dextran) at concentrations
0.10, 0.25, and 0.50 (%w/v) and DNA at 0.10 and 0.25 (%w/v) the 0.10 [DEAE-Dextran} and
0.25 [DNA] were chosen for further analysis, as they had the greatest sensitivity and stability of
those polyelectrolyte concentrations tested. The DEAE-Dextran showed the greatest stability,
with 67% remaining activity after 220 hours, compared with 56% for DNA, and 50% for the
control with no polyelectrolyte. Ogabiela, et al. [28] developed a highly-sensitive (limit of
detection o.1 µM) phosphate biosensor for use in freshwater sources. Using a highly-ordered
gold nanowire array (AuNWA) that integrated the enzyme’s cofactors, the concentration of POX
required could be reduced around 100-fold when compared to similar biosensors. The AuNWA
biosensor was resistant to interference from common freshwater interferants such as chloride,
sulfate, fluoride, nitrite, and nitrate ions. The device showed no loss in activity after two weeks
of daily measurements, and with a recovery of 96.6 ± 4.9%, making it one of the most stable and
most sensitive POX-based biosensors designed for detecting phosphate in water sources.
Pyruvate oxidase biosensors have low operational and storage stability compared to other
enzymatic biosensors. This is due in most part to the poor stability of POX [29]. Currently, many
techniques have been explored aiming to stabilize POX for use in biosensors. Table 1.2
compares different POX stabilization techniques in biosensors. The stability of POX biosensors
has seen only moderate improvement since one of the earliest designs by Zapatabacri and
Burstein [14] in 1987. However, the sensitivity of biosensors based on POX has improved over
the past few decades. One of the most recent devices, the previously-mentioned design by
Akyilmaz and Yorganci [30] in 2007 using POX from Aerococcus sp., has produced the most

9
sensitive pyruvate biosensor to date, capable of detecting pyruvate linearly between 0.0025 –
0.05 µM, while still contending with other designs in terms of stability.

10
Table 1.1. Comparison of biosensors based on pyruvate oxidase
Substrate
Detected
POX
Source
Immobilization
Technique
Working
Electrode
Detection
Limit
Linear
Range
Operatio
nal
Stability
Storage
Stability
Ref Notes
Pyruvate Pediococ
cus spp.
Physical adsorption
with medolas blue
dye
Screen-
printed,
medolas
blue
mediated
electrode
1-2
µmol/g
fresh
weight
1-4
µmol/g
fresh
weight
[4] Disposable
biosensor to
detect pyruvate
in onions
Pyruvate Aerococc
us sp.
Cross-linked with
gelatin using
gluteraldehyde
YSI type
DO probe
covered
with
Teflon
membrane
0.0025
-
0.05µM
86% after
9 hours
[30]
Pyruvate Aerococc
us sp.
Cross-linked with
gelatin using
gluteraldehyde
YSI type
DO probe
covered
with
Teflon
membrane
0.0025
-0.05
µM
90% after
20 days
[24] Investigating the
effect of
thiamine on
POX
Pyruvate Electropolymerizati
on, conductive
redox polymer
Glassy
carbon
electrode
1 µM –
1.8 mM
[31]
Pyruvate Recombi
nant POX
from
Lactobac
illus
plantaru
m
Co-immobilized
with horseradish
peroxidase in a
carbon paste using
methylene green
Modified
carbon
paste
electrodes
0.1-3
mM
[32]

11
Pyruvate Microorg
anisms
Modified pyrrole
monomer with
thiophene
Platinized
glassy-
carbon
electrode
0.0 –
1.0 mM
[29] Attempting to
develop
biosensor
capable of
detecting in O2-
free samples
Phosphate Recombi
nant POX
from
Lactobac
illus
plantaru
m
Genetic
modification and
physical adsorption
onto the carbon
tube
Porous
carbon
soaked in
polyelectr
olyte
solution
<0.3 mM 0.05 –
1.0 mM
67% after
220 hours
49% after
24 weeks
[27] Optimized to
measure
phosphate ion
activity in serum
Pyruvate 34 µM 90 –
600 µM
[33] Measured
pyruvate in
onions and garlic
Pyruvate Chemically bound
using methylene
green
Modified
carbon
electrode
0.38 –
1.03
mM
[34]
Pyruvate Chemically bound
to olyazetidine
prepolymer, nylon
membrane,
Modified
carbon
electrode
1-10
mM
[35]
Phosphate Aerococc
us sp.
Immobilized on
polypyrrole
Polished
platinum
3 µM 15-400
µM
[2]
Pyruvate Covalent
attachment to
polytyramine
Glassy
carbon
electrode
0.05 µM 0.1 –
3.0 mM
74% after
50 days
[3]
Pyruvate Cross-linked with
gelatin using
gluteraldehyde
pO2 meter Up to 2
mM
“several
months”,
~250
assays
[14]

12
Table 1.2. Pyruvate oxidase biosensor stabilization technique comparison
Method Most Stable Version Measure of Stability Ref.
POX immobilized with gelatin and
insoluble film using glutaraldehyde
(GA) crosslinked with glucose
35 °C, pH 7.0, 50
mM phosphate buffer
>85% activity
remaining after 9
hours at 35 °C
[30]
Electropolymerization of
mercaptohydroquinone in the presence
of POX
0.2 mM TPP, 10 µM
MgSO4 at 20 °C
5% loss after 7 days
stored at 5 °C
[31]
POX immobilized using GA on a self-
assembled monolayer made of 3-
mercaptopropionic acid and 6-
aminocaproic acid
4-hour assembly of
self-assembled
monolayer
90.5% activity after
15 assays, 26% loss
after 10 days of
refrigerated storage
[36]
Recombinant POX from Lactobacillus
plantarum co-immobilized with
horseradish peroxidase in organic carbon
paste
Addition of raffinose
and protective
dialysis membrane
89% after 3 months,
66% after 6 months
with storage in
desiccator at 4 °C
[32]
POX from Aerococcus sp. crosslinked
on Pt/Au alloy nanowires along with
cofactors
40%Pt + 60% Au,
150 mM TPP, 5 µM
FAD, 10 U/L POX
48% loss after 2
weeks, stored in 0.1
M citrate buffer, pH
7.0 at 4 °C
[37]
POX from L. plantarum covalently
immobilized on copolymer poly (5-
hydroxy-1,4-napthoquinone-1,4-
nathoquinone acid)
pH 7.5 40% loss after 3 uses,
50% loss after 7 days
in storage at 4 °C
[38]
Potentiostatic redox film with
polypyrrole film to prevent POX
leaching
5mM, oxygen-
saturated phosphate
buffer on pure
polythiophene film
14% remaining after
10 days stored in pH
6.5 phosphate buffer
along with cofactors
at 4 °C
[29]
Recombinant POX from L. plantarum
stabilized with polyelectrolyte DEAE-
dextran or DNA
0.10% w/v DEAE-
dextran with 20-hour
polarization
67% after 220h, 49%
after 24 weeks under
dry storage at -20 °C
[27]
POX from Aerococcus viridans
immobilized by nafion matrix covered in
poly (caromoyl) sulfunate hydrogel
Use of both nafion
and PCS hydrogel
>85% after 12 hours
of continuous
operation
[1]
POX from Aerococus sp. crosslinked
with bovine serum albumin and
glutaraldehyde along with cofactors on
gold nanowire array
Crosslinkage with
both BSA and GA
100% activity after 2
weeks of repeated
use, stored in 50Mm
citrate buffer in
refrigerator
[28]

13
High Hydrostatic Pressure Stabilization of Enzymes
The application of high hydrostatic pressure (HPP) is effective in inactivating certain
deteriorative enzymes in foods [39, 40]. Pressure has often been viewed as a protein denaturant,
as it disrupts the multimeric structure of many proteins. There exists, however, examples of
pressure-enhanced stability in proteins, due mainly to pressure’s effect on hydrophobic, van der
Waals, and electrostatic interactions [41]. Generally, the effect of pressure on physiochemical
processes at equilibrium are governed by Eyring’s Equation [42]:
(𝛿 ln 𝑘
𝛿𝑝)𝑇 = −(
Δ𝑉≠
𝑅𝑇)
where k is the rate constant, p is pressure, T is absolute temperature, R is the ideal gas constant,
and ΔV≠ is the activation volume that represents the influence of pressure on the reaction rate.
This equation can be integrated and rearranged to produce the equation:
ln 𝑘 = −∆𝑉≠
𝑅𝑇×𝑃 + ln 𝑘𝑝0
where k is the rate constant at a reference pressure, p0. This means that a reaction with a
negative volume change will be shifted towards the more compact state under pressure, while
one with a positive volume change will be slowed.
For changes in protein structure, the precise magnitude and sign of ΔV≠ is dependent
upon the specific molecular interactions [43].. Furthermore, unlike chemical catalysis, calculating
ΔV≠ can be difficult to calculate for enzymes under pressure, as it is dependent on changes in
enzyme conformation, enzyme solvation (including interactions with media and other proteins),
specific chemical equilibrium, and interactions with other proteins. Values for ΔV≠ typically
have a magnitude of less than 30 cm-3mol-3, but values can range from -70 to +60 cm-3mol-3 [44].

14
Noncovalent interactions are the primary molecular force altered through pressure. The
solvation of charged groups is associated with a reduction in volume, whereas the formation of
coulombic interactions accompanying the dehydration of charged atoms results in an increase in
volume. The formation of hydrophobic interactions between aliphatic groups (associated with a
positive ΔV≠) is destabilized under pressure, whereas the stacking of aromatic rings is favored by
increased pressure, as it is associated with a small decrease in volume. Hydrogen bonding is
associated with virtually no change in volume, and is therefore largely independent of pressure
[45, 46]. Under a high-pressure simulation of bovine pancreatic trypsin inhibitor (BPTI),
Kitchen, et al. [47] found that the hydration shell around the molecule was more ordered under
high pressure as compared to the low pressure simulation. This effect was especially pronounced
around more nonpolar surface groups.
In studying the stabilizing effect of pressure on α-chymotrypsin against thermal
inactivation, Mozhaev, et al. [48] suggested that the stabilizing effect of pressure may be due to
the opposing effects of temperature and pressure on the ability of protein functional groups to
interact with water. This was supported by Taniguchi and Suzuki [49], who stated that while
increased heat is accompanied by protein unfolding, the resulting unfolding exposes more
hydrophobic residues to the surrounding water. Under high-pressure conditions, the hydration of
these newly-exposed functional groups is associated with a decrease in molar volume of water
with the increase in ordering of the hydration shell [50]. It is likely that the promotion of
nonpolar functional groups interacting with water under high pressure is key to the stabilizing
effect of pressure against thermal inactivation. Hawley [51] devised an elliptical model (Figure
1.1) to describe the effects of pressure and temperature on enzyme conformation. The active
conformation is represented by the darker, inner region, surrounded by the lighter region

15
representing the point of reversible denaturation, with all areas outside the ellipse representing
complete denaturation.
Figure 1.1. Typical elliptical pressure-temperature diagram with active and inactive enzyme
regions
More recent studies have examined the role that hydrophobic cavities play in the
behavior of proteins under HHP. The decrease in volume associated with protein unfolding under
pressure has been attributed to the difference between the density of bulk water and water
associated with the protein, the loss of internal cavities, or some combination of the two. Roche,
et al. [52] studied the structural and energetic details of unfolding in several staphylococcal
nuclease mutants with varying cavity sizes using simulations based on high-pressure NMR
spectroscopy. They determined that greater internal void volume increased the magnitude of the
volume change associated with unfolding, and that internal cavities were the primary
determinants of pressure unfolding in proteins. While the hydrophobic effect would predict that
the transfer of nonpolar hydrocarbons from the interior of the protein into water would be
expected to be unfavorable [53], Collins, et al. [54] found that, at high pressures, the filling of
these hydrophobic cavities became so favorable that new cavities would grow to accommodate
water molecules. In another homotetrameric oxidoreductase, urate oxidase, the high-pressure
Pressure
Active/Native
Inactive/Denatured
Reversibly Denatured
Tem
pera
ture

16
perturbation of a large hydrophobic cavity near the active site led to the unfolding of that enzyme
until quaternary structure damage caused irreversible aggregation [55]. In POX, there are three
cavities large enough to accommodate at least one water molecule (≥ 30 Å3) located directly
beside the active site, around GLU54 in each of its four subunits. Figure 1.2 shows a rendering of
one subunit of POX from Aerococcus viridans generated from crystallographic data from Juan,
et al. [56], with its three largest cavities and active site labeled.
Figure 1.2. Monomeric subunit of pyruvate oxidase (PDB ID: 1VF5) with its cofactors FAD (1)
and TPP (2), three largest cavities (3-5), and active site, GLU54 (6), labeled.
1
2
3
4 5
6

17
Gap of Knowledge
Many studies have utilized HHP to stabilize enzymes. While the exact mechanisms by
which HHP increases thermal stability in enzymes are unknown, it is thought that the effects of
HHP and temperature counteract each other in certain cases. Specifically, greater pressure
increases order, thereby decreasing entropy, while increased temperature decreases order, thus
increasing entropy [57]. Newer studies have suggested that the size and location of internal
cavities in the enzyme play an important role in how that protein behaves under HHP [52, 54,
55]. There exists no current mathematical model for predicting the effect of HHP on a given
enzyme based on molecular structure. Empirical models must be constructed for each enzyme to
observe the effects of HHP. At this point, no such empirical model exists for POX.
In our laboratory, we recently studied the effect of HHP on glucose oxidase[58], xanthine
oxidase [59], and alcohol oxidase [60]. These enzymes were stabilized under HHP, but the effect
of HHP on POX from Aeroccus sp. is unknown. Though HHP has been used to stabilize
enzymes, stability does not persist after depressurization. If there is a rapid loss in stability for
the presumptive HHP-stabilized POX, techniques would need to be developed to capture the
stabilized form onto the working electrode. This could potentially mean constructing the POX-
based biosensor under high pressure conditions. Further experiments would be required to
determine if the immobilized HHP-stabilized POX biosensor offered improvements in stability,
sensitivity, and reproducibility over currently POX biosensor models.

18
To characterize the effect of HHP on POX and determine if there is an optimal pressure-
temperature combination for maximal stability.
Hypothesis:
High hydrostatic pressure will stabilize pyruvate oxidase
Overall Objective:

19
CHAPTER 2
THE INFLUENCE OF HIGH HYDROSTATIC PRESSURE ON STABILITY OF PYRUVATE
OXIDASE
Introduction
The rapid and accurate detection and quantification of pyruvate and phosphorous is
important to the food industry, and for the fields of medical and environmental science. For
flavor and quality purposes, it is important to monitor the starting pyruvate levels in fermented
products such as beer wort or wine must [15]. Pyruvate levels in onions are a predictor of
pungency, which is an important indicator of quality for that crop [4, 19]. Detection of pyruvate
in blood serum can assist in clinical diagnoses, as fluctuations in pyruvate levels in blood serum
can be indicative of a variety of medical issues [14]. Accurate on-site monitoring of phosphorous
levels in freshwater is vital for predicting and controlling the environmentally-destructive
process of eutrophication [22]. Current analytical methods mainly involve spectroscopy, which
currently lack the transportability and specificity needed for on-site environmental measurements
of phosphorous, the specificity and ease of use for pyruvate in food samples, and may not be able
to measure turbid blood samples [1, 4, 20].
Pyruvate oxidase (POX) is a homotetrameric flavoprotein with a molecular weight of 262
kDa. It catalyzes the oxidative decarboxylation of pyruvate to acetylphosphate (Equation 2.1) in
the presence of oxygen. Current designs for POX-based biosensors offer higher specificity and

20
sensitivity than other analytical methods for pyruvate and phosphorous, but are limited due to the
poor thermal stability of the enzyme.
Originally, high hydrostatic pressure (HHP) was used as a nonthermal means of
inactivating deleterious enzymes in food, but more recently the utility of HHP has expanded.
Under certain combinations of pressure and temperature, many enzymes have shown enhanced
stability [5]. Presently, there exist no mathematical models for predicting the effect of pressure
on enzymes based on molecular structure, so we currently must rely on empirical data built on a
case-by-case basis for each enzyme.
Before POX biosensors can be effectively utilized, better enzyme stabilization techniques
need to be developed. Currently, POX stabilization has largely only been studied in the context
of constructing biosensors. The most common method of stabilization is by crosslinking the
enzyme, usually using glutaraldehyde [14, 24, 30]. To the best of our knowledge, no previous
study has examined the effect of HHP on the stability of POX. The objective of this research was
to examine the effect of HHP on POX at selected temperatures, and to determine if there is an
optimal pressure-temperature combination for promoting stability.
Materials and Methods
Materials and Equipment
Pyruvate oxidase from Aerococcus sp. (product number P4105-100UN), peroxidase from
horseradish (POD, product number P8250-25KU), flavin adenine dinucleotide (FAD), sodium
(2.1)

21
pyruvate, and thiamine pyrophosphate (TPP), N-Ethyl-N-(2-hydroxy-3-sulfopropyl)-m-toluidine
(EHSPT), ethylenediaminetetraacetic acid (EDTA), magnesium sulfate, and 4-aminoantipyrine
were all purchased from Sigma-Aldrich (St. Louis, MO, USA) for use in the enzymatic activity
assay for POX. Enzyme buffers were made from potassium phosphate disodium salt from Fisher
Scientific (Pittsburg, PA, USA). Activity was assayed spectrophotometrically using a BioTek
Synergy™ HTX Multi-Mode Microplate Reader (VT, USA) using a xenon flash lamp and
monochromator at 550 nm. Data was collected using BioTek’s Gen5 Data Analysis Software.
For treatment of POX samples, the HHP system (Figure 2.1), was composed of a
micropump (model MP5), an 8.5-mL high pressure reactor (model U111), and a pump controller
(MP5 micropump control unit), from Unipress Equipment (Warsaw, Poland). Reactor
temperature was controlled by water baths, Isotemp 3016D (5 °C) and Isotemp 6200 R28 (25 -
45 °C) from Fischer Scientific (Pittsburg, PA, USA), which fed water through the jacket of the
high-pressure reactor. Selection of flow from each water bath was controlled by a pair of Sirai
Z110A solenoid pinch valves (Busseri, Italy). Temperature was monitored by a type K
thermocouple inserted through the bottom of the reactor with the tip flush to the bottom of the
vessel. A program written in LabVIEW controlled process time, pressure, and the activation of
solenoid valves with a data acquisition board (NI cDAQ 9174) from National Instruments
(Austin Texas). Pressure, time, and temperature were recorded by the LabVIEW program.
Enzyme samples were treated in modified 0.5 mL high-density polypropylene syringes (Medi-
Dose, Ivyland, PA, USA) added to the reaction vessel with Sil 180 oil bath liquid (Thermo
Scientific, Rockford, IL, USA). This was the same system used by Eisenmenger and Reyes-De-
Corcuera [57], apart from an additional BK Precision 1666 DC regulated power supply (B&K
Precision, Yorba Linda, CA, USA) used to isolate on of the DAQ modules to rectify a ground

22
loop issue that was discovered to cause inaccurate temperature readings during preliminary
measurements.
Figure 2.1. High hydrostatic pressure system.
The native polyacrylamide gel electrophoresis (PAGE) system consisted of 4-20%
polyacrylamide pre-cast gels (product number 456-1094) placed inside a Mini-PROTEAN®
Tetra cell, powered by a Power Pac 3000 power supply, all purchased form Bio-Rad Industries
(Hercules, CA, USA). Tris/glycine running buffer, native running buffer (product number 161-
0738), and Coomassie G-250 stain were also purchased from Bio-Rad. Staining occurred on a
Red-Rotor PR70-115V shaker platform (Hoefer Scientific, Holliston, MA, USA).
Sample spectra for native and inactivate POX samples were recorded using a
NanoDrop™ One Microvolume UV-Vis spectrophotometer (Thermo Fisher Scientific, Waltham,
MA, USA).

23
High Hydrostatic Pressure Processing
Syringes were modified by removing the plunger and truncating the body of the syringe
at the 0.15 mL mark before replacing and trimming the plunger. A 100 µL sample of POX was
slowly pipetted into a modified syringe while simultaneously withdrawing plunger to eliminate
as much headspace as possible before replacing the cap. The sample was submerged in the
reactor, which was pre-filled with silicon oil and held at 5 °C. The reactor was sealed with its
threaded cap before initiating pressurization. Once the set point for pressure was reached, water
flow was switched to the second, warmer water bath maintaining the preset incubation
temperature. Process time began once the reactor reached 95% of the temperature set point.
Figure 2.2 shows the pressure and temperature profile for a sample treated at 35 °C at 100 MPa
for 0 and 300 s. To determine 100% residual activity for a time of 0 s (t0), the reactor
immediately began cooling once 95% of the temperature set point was reached. Once the reactor
reached 15 °C depressurization was initiated as cooling continued to 5 °C. Samples were
immediately removed and analyzed for activity within two minutes of treatment.
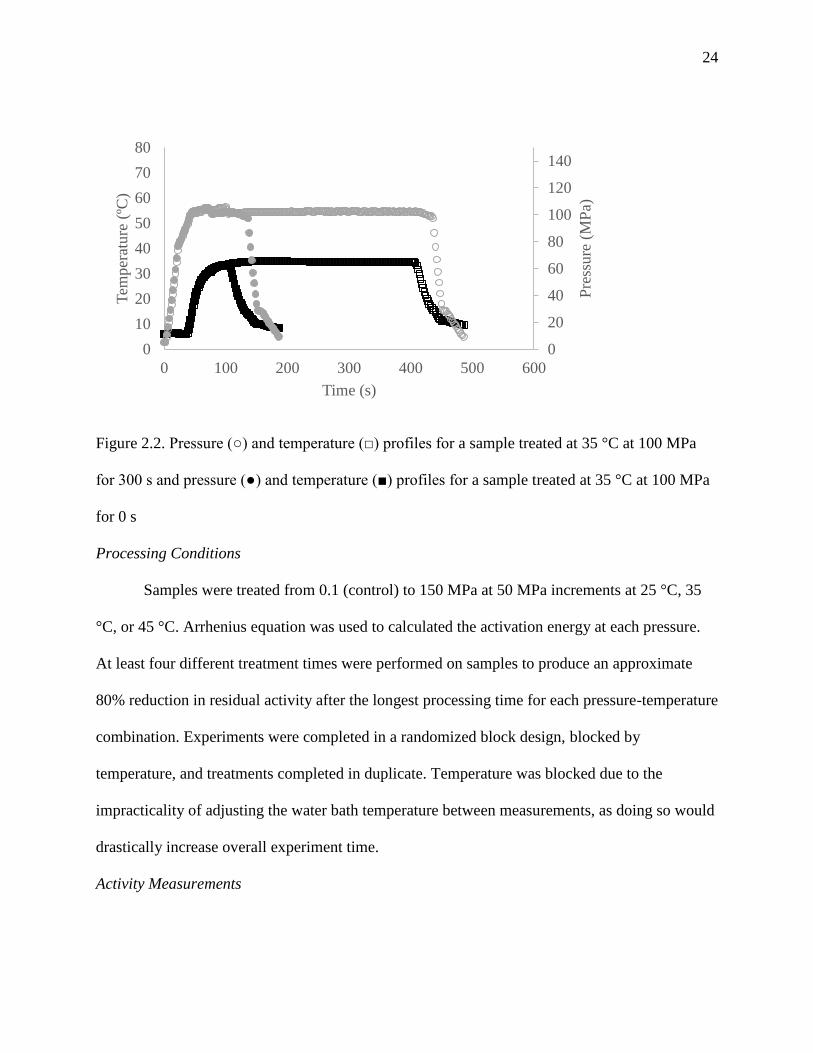
24
Figure 2.2. Pressure (○) and temperature (□) profiles for a sample treated at 35 °C at 100 MPa
for 300 s and pressure (●) and temperature (■) profiles for a sample treated at 35 °C at 100 MPa
for 0 s
Processing Conditions
Samples were treated from 0.1 (control) to 150 MPa at 50 MPa increments at 25 °C, 35
°C, or 45 °C. Arrhenius equation was used to calculated the activation energy at each pressure.
At least four different treatment times were performed on samples to produce an approximate
80% reduction in residual activity after the longest processing time for each pressure-temperature
combination. Experiments were completed in a randomized block design, blocked by
temperature, and treatments completed in duplicate. Temperature was blocked due to the
impracticality of adjusting the water bath temperature between measurements, as doing so would
drastically increase overall experiment time.
Activity Measurements
0
20
40
60
80
100
120
140
0
10
20
30
40
50
60
70
80
0 100 200 300 400 500 600
Pre
ssure
(M
Pa)
Tem
per
ature
(ºC
)
Time (s)

25
A bi-enzymatic assay adapted to a 96-well microplate was used to measure the activity of
POX spectrophotometrically at 37 °C [61]. H2O2 produced by the POX-catalyzed reaction
(equation 2.2) was monitored at 550 nm by the POD-catalyzed conversion of EHSPT to
quinoneimine dye. (equation 2.3).
𝑃𝑦𝑟𝑢𝑣𝑎𝑡𝑒 + 𝑂2 + 𝑃𝑖𝑃𝑂𝑋→ 𝐴𝑐𝑒𝑡𝑦𝑙𝑝ℎ𝑜𝑠𝑝ℎ𝑎𝑡𝑒 + 𝐶𝑂2 + 𝐻2𝑂2 (2.2)
2𝐻2𝑂2 + 4 − 𝐴𝑚𝑖𝑛𝑜𝑎𝑛𝑡𝑖𝑝𝑦𝑟𝑖𝑛𝑒 + 𝐸𝐻𝑆𝑃𝑇𝑃𝑂𝐷→ 𝑄𝑢𝑖𝑛𝑜𝑛𝑒𝑖𝑚𝑖𝑛𝑒 𝑑𝑦𝑒 + 4𝐻2𝑂 (2.3)
A stock solution of approximately 8 units/mL POX was prepared and stored in an amber glass
bottle at 5 °C. Each day, a 0.8-unit/mL working solution was prepared by diluting 0.1 mL of
stock with 0.9 mL 50 mM potassium phosphate buffer, pH 5.7 (enzyme diluent) in a 8-mL amber
glass vial. New stock solution was prepared approximately every three weeks during
measurement, once the activity of the fresh enzyme began to decrease. A reaction cocktail was
prepared according to Table 2.1, adapted from Sigma [61]. Peroxidase, FAD, and TPP were
prepared fresh each day. All other solutions, including pyruvate and buffers were prepared
beforehand. When not in use, all reagents were held at 4 °C and stored on ice during
experiments.
Table 2.1. Reaction cocktail composition for pyruvate oxidase activity assay
Reagent Volume (mL)
150 mM Potassium phosphate buffer, pH 5.9 2.0
7.4 mM 4-Aminoantipyrine 0.4
0.3% (w/v) EHSPT 0.4
3 mM TPP 0.4
0.15 mM FAD 0.4
15 mM EDTA 0.4
150 mM MgSO4 0.4
50 units/mL POD 0.6
Total 5.0

26
Four microwels were used for each assay. First, 250 µL of reaction cocktail and 50 µL of
300 mM pyruvate solution were added to each of the wells. The pre-heated microplate mixed the
plate by swirling for 30 s, then monitored the absorbance for 5 min to ensure constant
absorbance before addition of enzyme. The plate was then ejected, 10 µL of enzyme diluent were
added to the first two wells to serve as a blank and 10 µL of enzyme sample were added to the
second two. The absorbance was then monitored for 20 min, with measurements occurring
approximately every 10 s, for a total of 121 measurements for each well. After each assay, the
data from the four cells was transferred into an Excel spreadsheet where the average of the two
blank wells was subtracted from the average of the two test wells to account for any non-
enzymatic oxidation of the substrate. Once this corrected absorbance was calculated for each
replication, the slope of absorbance versus time was calculated every 10 successive data points.
The activity (abs/time) was calculated as the maximum slope from each replicate. Residual
activity for a given replicate was calculated by comparing the activity of that replicate to the
average activity of the two replicates at time zero of the same treatment. The activity of fresh
enzyme was recorded daily to ensure no loss of activity of stock enzyme between comparable
treatments. All absorbance measurements were corrected for path length. Actual enzyme activity
(units/mL) was calculated according to equation 2.4.
𝑈𝑛𝑖𝑡𝑠 𝑚𝐿 𝐸𝑛𝑧𝑦𝑚𝑒⁄ = (∆𝐴550 𝑇𝑒𝑠𝑡−∆𝐴550 𝐵𝑙𝑎𝑛𝑘)×0.31
(36.88)(0.01)(2.4)
where 0.31 is the total volume in milliliters of the assay, 36.88 is the millimolar extinction
coefficient of quinonimine dye, 0.01 is the milliliter volume of enzyme used in the assay, and
one unit of POX is defined as producing 1.0 µmole of H2O2 per minute under assay conditions
(pH 5.7, 37 °C) [61].

27
Based on the good fit of the linear regression of natural logarithm of residual activity
versus time (figure 2.9), first-order kinetics were used to calculate the rate constant of
inactivation (k) at each selected temperature and pressure.
The Arrhenius equation (equation 2.5) was used to calculate the activation energy at each
pressure from the first-order rate constants of inactivation.
ln(𝑘) = (−𝐸𝑎𝑖
𝑅×1
𝑇) + ln (𝑘𝑇0) (2.5)
Where k is the rate constant, T is the absolute temperature, R is the ideal gas constant (8.314 J
mol-1 K-1), Eai is the activation energy, and 𝑘𝑇0 is the rate constant at a reference absolute
temperature. The “i” was added to the symbol Eai to denote that it is the activation energy of
inactivation as opposed to the activation energy of activation.
Activation volume of POX was calculated using Eyring’s equation (Equation 2.6) at each
temperature tested with the rates of inactivation:
ln(𝑘) = (∆𝑉≠
𝑅𝑇×𝑃) + ln (𝑘𝑃0) (2.6)
where k is the rate constant, ΔV≠ is the activation volume, P is pressure, 𝑘𝑃0is the rate constant at
reference pressure P0.
Standard error of the linear regression was reported for inactivation rates, activation energies,
and activation volumes.
Original Design with Plastic Pouches
The initial methods of this study were based on similar studies on the protective effects of
HHP on the stability of alcohol oxidase [60], xanthine oxidase, and glucose oxidase [58]. As
with enzyme samples in the aforementioned studies, POX samples were originally contained in
plastic pouches. These pouches were fabricated from low-density polyethylene zip-top bags,
using a heat sealer to divide the bags into 1.9 cm2 portions, which were cut into individual

28
squares so that the top edge of each square was left unsealed. A 100 µL aliquot of POX working
solution was added to each pouch slowly to allow for space between the bulk of the solution and
the edge of the bag, which would be heat sealed before the sample was added to the HHP system.
Initial results were inconsistent and poorly reproducible, with severe activity loss
occurring between fresh POX and unheated pressurized samples. Aside from pressure, heat from
the sealer and pouch material were considered as possible factors for loss in activity.
To account for potential loss from the heat sealer, pouch design and handling methods
were adjusted. The pouch design was modified to allow for more space between the heat sealer
and enzyme solution. In this new, narrower design, the pouches were cut into 1.3 x 3.8 cm
rectangles, with the open end on one of the narrower edges. Figure 2.3 shows a comparison of
the two pouch designs. A smaller volume (50 µL) of POX working solution was added to these
pouches to allow for a greater space between the heating element of the sealer and the enzyme
solution. Additionally, the pouches were kept on ice constantly from filling to addition to the HP
reactor, other than when they were being sealed at which point they were handled with tweezers
to avoid any temperature change from body heat. To examine the loss in activity due to heat
from the heat sealer, environment, and manual handling, the activity of POX sealed in the new
pouches and handled to minimize external heat was compared with fresh enzyme solution. These
samples were all pipetted into the pouches and sealed, then their activity was recorded.

29
Unsealed Edge
Figure 2.3. Comparison of original (left) and newer (right) pouch design to scale
After only modest improvements in enzyme activity retention from adjustments to pouch
design and handling, the possible effect of sample container material was considered. Enzyme
solution was added to an amber glass vial, one of the plastic pouches, and a high-density
polypropylene centrifuge tube, all stored in a covered ice bucket. The plastic pouch was left
unsealed so that any inactivating effects would be due to container material and not incidental
heating. The activity of the POX solution was recorded from each container at 0, 25, and 50 min.
Unlike the original activity assay protocol that involved two test wells and two blank wells, each
sample was added to a single well, along with a single blank well so that all samples could be
measured at the same time.
Unsealed Edge
Sample
(100 µL)
Sample
(50 µL)

30
Pouch Study Results
Residual activity measurements using the original pouch showed highly inconsistent
results. Table 2.2 details residual activity for POX treated with 100 MPa without raising
temperature using the square pouches. On average, virtually no activity was lost over the short
treatment time of 75 s, however, the standard deviation (n = 3) between the triplicates was
extremely high. Relative to the fresh POX working solution, which remained in an amber glass
vial, the loss for POX treated in the original pouches was severe (75 – 80%).
Table 2.2. Residual activity for pyruvate oxidase treated using original pouch design at 100 MPa
at 5 °C relative to both 0 s treatment time and fresh working enzyme solution (n=3)
Residual Activity (%)
Time (s) Relative to 0 s Relative to Fresh
0 100 ± 44 % 21 ± 9 %
75 116 ± 68 % 25 ± 14 %
Based on the results from experiments using the original pouch design, it was considered
that enzyme activity was being affected by sources of heat outside of the reactor, namely heat
from the sealer, but possibly also atmospheric and body temperature. However, new pouch
design and handling methods resulted only in slight improvements in activity loss. Table 2.3
outlines residual activity values for these unpressurized samples relative to fresh POX solution.

31
Table 2.3. Residual pyruvate oxidase activity for samples sealed in new pouches relative to fresh
enzyme activity over time
Sample 1 Sample 2 Sample 3
Residual Activity (%)
Time (min)
61%
0
49%
30
12%
60
At best, there was still at least a 60% loss in activity using the new pouch design and handling
technique. It is important to note that all three samples were prepared at the same time, and each
remained on ice while the previous sample’s activity was recorded, meaning there was
approximately 30 – 35 min between activity assays. Results indicated that POX loses activity
over time as it remained in the pouch, as the activity of Sample 1 was recorded immediately after
being sealed in the pouch, Sample 2 remained in the pouch for approximately 30 min, and
Sample 3 for at least 60 min.
The comparison of activity over time for POX contained in a plastic pouch, glass vial,
and hard plastic tube (Figure 2.4) revealed that, over the span of 50 min, the activity of POX was
reduced by more than half when stored in the plastic pouches, while the samples stored in glass
or hard plastic decreased from 88% to 70% and 79% to 61% respectively. Data for the POX
stored in the vial at 25 min was excluded due to pipetting error into the single microwell. This
experiment was not replicated.

32
Figure 2.4. Residual activity relative to fresh enzyme over time for pyruvate oxidase in low-
density polyethylene pouch ( ), high density polypropylene tube ( ), and amber glass vial ( )
In a study that compared the retention of eight radiolabeled proteins in containers made
of different plastic and glass materials commonly found in lab settings, Goebel-Stengel, et al.
[62] concluded that selecting the proper can improve protein retention dramatically. In their
comparison of polypropylene and polystyrene, the two most commonly-used plastics in
laboratories, retention for polypropylene was as good or better than polystyrene. In the case of
cholecystokinin, the difference in retention was 86 vs 16% for the two plastics respectively.
Specific protein-plastic interactions are difficult to predict due to the variability in peptide
structure between different proteins. Based on data from these preliminary studies, all future
experiments in this study were completed using high-density polypropylene syringes.
Native and Inactivated Enzyme Comparison
Native polyacrylamide gel electrophoresis (PAGE) was performed to attempt to
determine whether the tetrameric structure of pyruvate oxidase broke into subunits (dimers or
monomers) once completely inactivated with pressure. The gels were submerged in a 25-mM
Tris, 192 mM glycine running buffer. To prepare the inactivated sample, 100 µL of POX stock
0%
20%
40%
60%
80%
100%
120%
0 25 50
Res
idual
Act
ivit
y R
elat
ive
to F
resh
Enzy
me
Time (min)

33
solution (6 units mL-1), prepared in 50 mM, pH 5.7 potassium phosphate buffer, was added to a
syringe and treated at 45 °C at 150 MPa for 30 min. This treatment was determined based on
prior data to achieve complete inactivation of the enzyme. Before loading onto the gels, 50 µL of
fresh enzyme stock solution, inactivated solution, and potassium phosphate buffer were each
separately mixed with an equal volume of native running buffer. For each sample, 40 µL of
solution, containing approximately 4 µg of protein, was added to two wells. A 200-V potential
was applied to the system for 60 min. Afterwards, the gel was removed and stained with
Coomassie blue for 30 min on a shaker platform, then rinsed with water for 30 min on the same
platform.
To further understand the effect of the inactivation treatment on the protein, the
absorbance spectrum between 240 – 350 nm was recorded using a NanoDrop™ One
Microvolume UV-Vis spectrophotometer for both inactivated and native enzyme samples, as
well as a sample of 50 mM phosphate buffer treated at 150 MPa at 45 °C for 30 min to detect
any potential contaminants from the plastic syringe that may absorb within the spectrum. For
each of the three samples, 2 µL was pipetted directly onto the loading stage and the optical piece
was lowered such that the sample droplet rested underneath.
34
Results and Discussion
Activity Assay
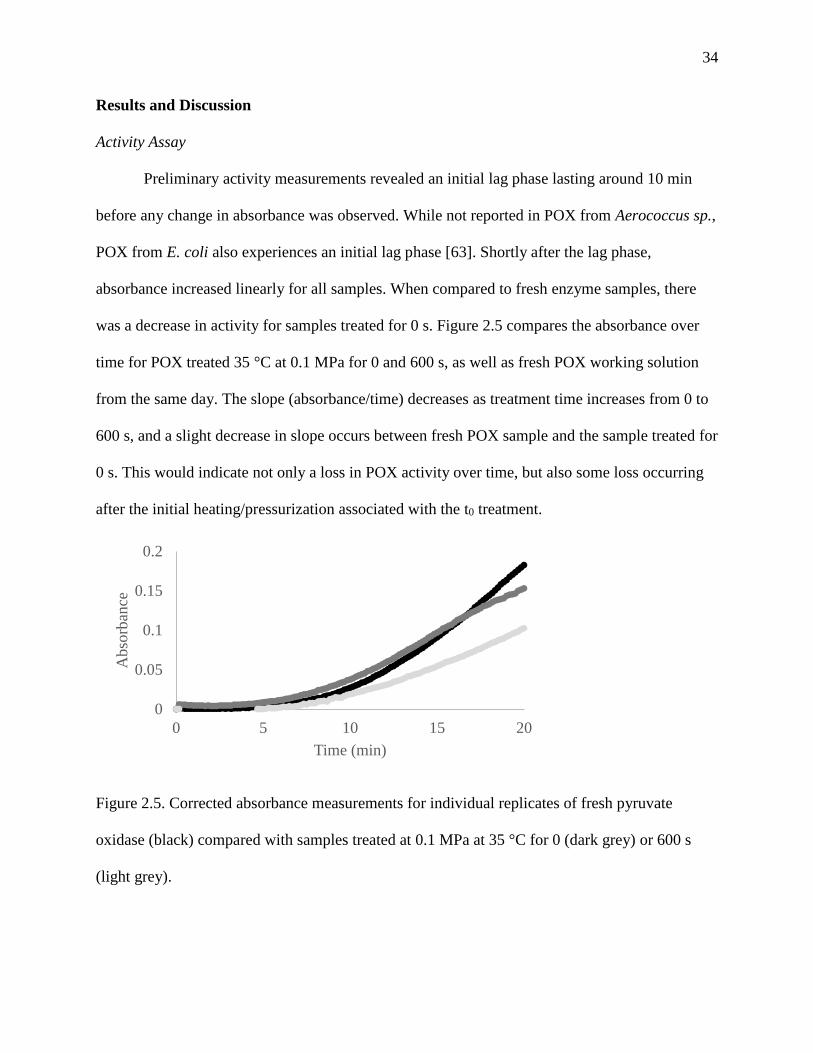
Preliminary activity measurements revealed an initial lag phase lasting around 10 min
before any change in absorbance was observed. While not reported in POX from Aerococcus sp.,
POX from E. coli also experiences an initial lag phase [63]. Shortly after the lag phase,
absorbance increased linearly for all samples. When compared to fresh enzyme samples, there
was a decrease in activity for samples treated for 0 s. Figure 2.5 compares the absorbance over
time for POX treated 35 °C at 0.1 MPa for 0 and 600 s, as well as fresh POX working solution
from the same day. The slope (absorbance/time) decreases as treatment time increases from 0 to
600 s, and a slight decrease in slope occurs between fresh POX sample and the sample treated for
0 s. This would indicate not only a loss in POX activity over time, but also some loss occurring
after the initial heating/pressurization associated with the t0 treatment.
Figure 2.5. Corrected absorbance measurements for individual replicates of fresh pyruvate
oxidase (black) compared with samples treated at 0.1 MPa at 35 °C for 0 (dark grey) or 600 s
(light grey).
0
0.05
0.1
0.15
0.2
0 5 10 15 20
Abso
rban
ce
Time (min)

35
The loss in activity due to the t0 treatment at each pressure and temperature combination relative
to fresh enzyme stock solution is shown in Figure 2.6. The process of bringing samples to the
temperature and pressure setpoint reduced activity relative to fresh in all cases. From 0.1 to 100
MPa, there was a consistent reduction in activity of about 20% at all temperatures, with an
increase in reduction at 150 MPa. Higher reduction in enzyme activity between fresh enzyme
and those treated for 0 s occurred at lower temperatures at 150 MPa.
Figure 2.6. Residual activity for t0 treatments relative to fresh enzyme at 25 ( ), 35 ( ), or
45 °C ( )
Additionally, there is a delay between the heating of the reaction vessel and the interior of
the syringes. By comparing the two temperatures (Figure 2.7), it is evident that the syringes take
approximately an additional two minutes to reach the temperature setpoint after the reactor.
0%
20%
40%
60%
80%
100%
120%
0 50 100 150
Res
idual
Act
ivit
y R
elat
ive
to
Fre
sh (
%)
Pressure (MPa)

36
Figure 2.7. Reactor (open symbol) versus syringe (closed symbol) temperature at 25 (light grey),
35 (dark grey), or 45 °C (black)
Effect of HHP on Stability
Pyruvate oxidase residual activity decreased over time at all pressure-temperature
combinations. Figure 2.8 shows the loss in residual activity for samples treated at 0.1, 50, 100,
and 150 MPa at all three temperatures. Residual activity generally decreased as pressure
increased.
0
10
20
30
40
50
0 50 100 150 200 250 300
Tem
per
ature
(°C
)
Time (s)

37
Figure 2.8. Residual activity for POX at 25 (A), 35 (B), or 45 °C (C) at 0.1 ( ), 50 ( ), 100 ( )
or 150 ( ) MPa
First order models were used to describe the relationship between residual activity of
POX and treatment time. First order kinetics are, to our knowledge, the most commonly used
model for the rate of thermal and pressure inactivation of enzymes, as in the case of glucose
oxidase [58], pectin methylesterase [64], and alcohol oxidase [60]. Such models provided a good
-30%
20%
70%
120%
0 1000 2000 3000 4000 5000 6000 7000 8000
Res
idual
Act
ivit
y (
%)
Time (s)
0.0%
50.0%
100.0%
150.0%
0 500 1000 1500 2000 2500 3000 3500 4000 4500 5000 5500
Res
idual
Act
ivit
y (
%)
Time (s)
0.0%
50.0%
100.0%
150.0%
0 500 1000 1500 2000
Res
idual
Act
ivit
y (
%)
Time (s)
A
B
C

38
fit for residual activity as a function of treatment time and allowed for meaningful comparison to
other data from the literature
First order rate constants of inactivation (kinact) are reported in Table 2.4. At 25 °C, the
rate of inactivation increased with pressure at pressures above 50 MPa, with there being no
statistically significant change between atmospheric pressure and 50 MPa. The rate of
inactivation for 50 MPa at 35 °C was significantly smaller than 0.1 and 100 MPa, while there
was no significant difference between 0.1 and 100 MPa. The poor coefficient of determination of
determination (R2) for the 50 MPa treatment at 35 °C can be partially explained by the very
small slope for the rate of inactivation, as seen in Figure 2.9.

39
Table 2.4. First order rate of pyruvate oxidase inactivation ± standard error determined by the
linear regression of residual activity versus treatment time.
Pressure Temperature (°C)
25 R2 35 R2 45 R2
(MPa) kinact (min-1)
0.1 0.003a ± 0.001 0.42 0.004a ± 0.003 0.22 0.089a ± 0.007 0.95
50 0.004a ± 0.002 0.50 0.001* ± 0.001 0.08 0.065a ± 0.021 0.49
100 0.027 ± 0.002 0.95 0.006a* ± 0.002 0.57 0.138 ± 0.013 0.90
150 0.182 ± 0.035 0.72 0.177 ± 0.019 0.90 0.433 ± 0.045 0.91
a Represents statistically similar treatments as determined by ± standard error calculated from the
linear regression for each temperature.
* Indicates treatments completed with a newer batch of enzyme
At 45 °C, the rate of inactivation generally increased with pressure, except for a small
decrease from 0.1 to 50 MPa, but this decrease was not statistically significant. There was a large
increase in rate of inactivation between 100 and 150 MPa at all temperatures. While pressure-
induced inactivation of POX from Aeroccocus has not been explored in the current literature,
fluorescence data shows that in POX from E. coli, the cofactors FAD and TPP dissociate from
the enzyme before unfolding occurs as pressure approaches 220 MPa [13].

40
Figure 2.9. Logarithm of residual activity at 25 (A), 35 (B), or 45 °C (C) at 0.1 ( ), 50 ( ),
100 ( ) or 150 ( ) MPa
To investigate a possible discrepancy between enzyme batches, a series of experiments
using the new batch were completed comparing the residual activity overtime between 0.1 and
50 MPa at all three temperatures. The results from these supplemental experiments (Table 2.5)
show a similar response between enzyme batches. The residual activity values for the new batch
1.00
2.00
3.00
4.00
5.00
0 1000 2000 3000 4000 5000 6000 7000 8000ln(R
esid
ual
Act
ivit
y,
%)
Time (s)
1.000
2.000
3.000
4.000
5.000
0 1000 2000 3000 4000 5000 6000ln(R
esid
ual
Act
ivit
y,
%)
Time (s)
1.000
2.000
3.000
4.000
5.000
0 200 400 600 800 1000 1200 1400 1600ln(R
esid
ual
Act
ivit
y,
%)
Time (s)
A
B
C

41
was with those calculated based on kinact values for the old data at the same treatment times. The
difference between batches was mostly on the same order as the variability between replicates in
the same batch, but variability increased at 45 °C. The pressure/temperature treatment profiles
for these supplemental experiments (Figures A.1 – A.10) show that the treatments are both
consistent and reproducible.

42
Table 2.5. Residual activity data for confirmation experiments using new pyruvate oxidase batch
compared to residual activity for the old batch calculated based on kinact.
Treatment Time (s) Rep 1 Rep 2 Average
Old Batch
(Calculated)
Difference
(New Batch -
Old)
0.1 MPa 25 °C
0 98.6% 101.4% 100.0%
3000 94.2% 88.9% 91.5% 94.9% -3.4%
50 MPa 25 °C
0 98.9% 101.1% 100.0%
3000 100.2% 86.6% 93.4% 92.4% 1.0%
0.1 MPa 35 °C
0 93.4% 106.6% 100.0%
3000 102.8% 107.7% 105.3% 92.2% 13.1%
50 MPa 35 °C
0 94.2% 105.8% 100.0%
3000 98.0% 124.6% 111.3% Same Batch
0.1 MPa 45 °C
0 103.9% 96.1% 100.0%
1000 56.8% 34.2% 45.5% 70.0% -24.5%
50 MPa 45 °C
0 103.5% 96.5% 100.0%
1000 87.9% 113.3% 100.6% 77.0% 23.6%
Based on the data for this batch-to-batch comparison, there was insufficient evidence to discard
residual activity values based on new batch POX from future calculations.
Activation Energy of Inactivation
The apparent activation energy of inactivation (Eai) was calculated using the Arrhenius
equation. Table 2.6 reports Eai for 0.1 to 150 MPa at increments of 50 MPa. With every increase
in pressure of 50 MPa, the apparent Eai decreased by slightly less than a half, meaning that the
enzyme’s sensitivity to temperature increases with pressure over the observed range of pressure.

43
Table 2.6. Apparent activation energy of inactivation ± standard error determined by the linear
regression of ln(k) versus 1/T
Pressure Eai R2
(MPa) (kJ mol-1)
0.1 135 ± 66 0.81
50 100 ± 15 0.33
100 62 ± 113 0.24
150 33 ± 21 0.71
Activation energy of inactivation for POX decreased by around 75% as pressure was
increased from 0.1 to 150 MPa. The decrease in Eai for POX was more dramatic over a smaller
pressure range than many other enzymes from similar studies. Comparatively, Eai for glucose
oxidase dropped from 378.1 to 281.0 kJ/mol between 0.1 – 300 MPa [58] and xanthine oxidase
saw a decrease from 181.7 to 97.0 kJ/mol over 0.1 – 300 MPa [59]. In alcohol oxidase, Eai for
the thermoresistant fraction of the enzyme saw no significant change from 105.1 to 101.3 MPa
over a pressure range of 40 – 200 MPa. Even for some enzymes inactivacted by pressure, unlike
those previously mentioned, the degree of change in Eai was often less than that observed for
POX. The activation energy for pectinmethylesterase from navel oranges decreased from 177 to
95 kJ/mol over range of 100-750 MPa [64] while Eai for plum polyphenoloxidase just halved
from 130.6 to 62.7 over a dramatic pressure increase from 0.1 to 900 MPa [65]. Compared to the
plum polyphenol oxidase and pectinmethyesterase from navel orange, which saw their Eai halved
over a pressure range of 650 and 900 MPa respectively, the Eai of POX was reduced by 75% over
a range of only 150 MPa. This rate in reduction of Eai in POX as pressure increases is nearly an
order of magnitude greater, demonstrating its remarkable sensitivity to thermal inactivation as
pressure increases.

44
Activation Volume
Eyring’s equation was used to calculate activation volume (ΔV≠). Table 2.7 reports
activation volume for 25, 35, or 45 °C. For 25 and 35 °C, ΔV≠ was very similar. However, at 45
°C the activation volume fell by over 50% relative to the other two treatments, suggesting that
pyruvate oxidase is far more sensitive to pressure at 45 °C than the two lower temperatures.
Different trends in ΔV≠ could be observed in previously-studied oxidases. For xanthine oxidase,
ΔV≠ roughly tripled from 9.5 to 28.9 cm3/mol as temperature increased from 55.0- 70 °C [59]. In
glucose oxidase, ΔV≠ changed from 22.8 to 45.8 cm3/mol over a temperature range of 58.8 – 80.0
°C, with the greatest ΔV≠ of 57.0 cm3/mol occurring at 74.5 °C [58]. Additionally, polyphenol
oxidase from strawberry (Fragraria ananassa) saw an increase in ΔV≠ from -53.41 to -2.81
cm3/mol between 45 and 65 °C [66]. As with each of these enzymes, an increase in ΔV≠ is
associated with higher stability, indicating that more pressure is needed to achieve inactivation as
stability increases. In the case of POX, the decrease in ΔV≠ at 45 °C from 25 and 35 °C is within
reason, given the much higher rates of inactivation at 45 °C than those at the two lower
temperatures.

45
Table 2.7 . Activation volume ± standard error determined by the linear regression for each
temperature
Temperature ∆V≠ R2
(°C) (cm3 mol-1)
25 71 ± 12 0.95
35 68 ± 43 0.56
45 29 ± 13 0.72
* Indicates values calculated excluding new batch data, for which standard error and R2 could not
be calculated
When comparing the rate constant of inactivation versus pressure at each temperature (Figure
2.10) the plots form a gentle curve at the bottom of which sit 50 MPa. Based on the projected
shape of these plots, it is unlikely that a significantly lower inactivation rate could be achieved
just above or below 50 MPa.
Figure 2.10. Rate constant of inactivation versus pressure at 25 ( ), 35 ( ), and 45 °C ( )
0.000E+00
1.000E-03
2.000E-03
3.000E-03
4.000E-03
5.000E-03
6.000E-03
7.000E-03
8.000E-03
0 50 100 150 200
k (
s-1)
Pressure (MPa)

46
Native and Inactivated Enzyme Comparison
The stained polyacrylamide gel (Figure 2.11) showed that both the native and a portion of
the inactivated enzyme traveled the same distance, signifying that they had the same mass/charge
ratio. The bands for the inactivated enzyme were much fainter than those for the native, and there
was protein that remained in the top of the wells for the inactivated samples. This suggests that a
portion of the protein may have formed an aggregate too large to enter the gel, while the
remaining portion has returned to a native conformation upon depressurization. In a study on
another homotetrameric enzyme, urate oxidase, Girard, et al. [55] observed the enzyme structure
under high pressure and measured activity before and after pressurization. They observed that
above 150 MPa urate oxidase approaches a threshold of irreversible damage to the quaternary
structure, wherein a portion of the enzyme aggregates while the rest of the still-soluble enzyme
remains in a native, tetrameric state. Evidence of unfolding in pyruvate oxidase can be seen
spectrophotometrically (Figure 2.12). The “peak broadening” as absorbance increases, especially
below 270 nm, could be a result of cofactors being released as the enzyme unfolds. Thiamine
pyrophosphate exhibits a strong peak around 240-250 nm at neutral pH [67]. The release of
buried TPP from POX could explain the increase in absorbance on the lower end of the UV
spectra. The lack of absorbance in the blank sample would imply no contamination from the
syringe during processing.

47
Figure 2.11. Relative distance traveled through 4-20 % polyacrylamide gel by native and
inactivated pyruvate oxidase samples.
Figure 2.12. Ultraviolet spectra for Native (black), Inactivated (grey), and blank (dotted) samples
-0.2
0
0.2
0.4
0.6
0.8
1
1.2
220 240 260 280 300 320 340 360
Abso
rban
ce
Wavelength (nm)
Native Inactivated Blank

48
Conclusion
In conclusion, both HHP and temperature largely had an additive inactivating effect on
pyruvate oxidase from Aerococcus species, with pressure offering little to no protection against
thermal inactivation except for a small protective effect at 50 MPa at 35°C. Given the findings of
this study and the current literature on POX stability, HHP is not a viable technique for
stabilizing POX from Aerococcus sp. for the fabrication of biosensors.

49
Chapter Three
Final Comments
Overview
Data showed that material from the low-density polyethylene containers diminished
enzyme activity. This could either be through POX adsorbing to the plastic surface or due to the
plastic having an inactivating effect on the enzyme. Either way, these results further highlighting
the general importance of selecting appropriate containers when working with enzymes. While
the high-density polypropylene syringes did not have the same inactivating effect of POX, there
was a delay between the reactor temperature that was recorded and that of the temperature of the
samples.
Future Work
Further work is required to determine if lower pressures (around 50 MPa) can be utilized
in conjunction with other techniques to increase the feasibility of biosensors based on pyruvate
oxidase. Results from this study show some potential for pressures beneath what is usually
considered high pressure (100-700 MPa), perhaps in combination with other stabilization
techniques. Given the slight stabilization seen at 50 MPa in both the original and new batch data,
it would be useful to examine the effect of pressure between 15 – 75 MPa at a similar
temperature range to this study.
Based on data from native PAGE and UV-spectroscopy, it may be possible that POX,
like urate oxidase, maintains its native conformation until approaches a threshold of irreversible

50
aggregation. More sophisticated methods in which pyruvate oxidase is directly observed under
high pressure are needed to confirm the accuracy of this model, and to confirm if the internal
cavities play a role in the process of unfolding and aggregation. This could be achieved using
protein simulations informed by x-ray crystallography and/or fluorescence spectroscopy under
high pressure.

51
REFERENCES
[1] R.C.H. Kwan, H.F. Leung, P.Y.T. Hon, J.P. Barford, R. Renneberg, A screen-printed
biosensor using pyruvate oxidase for rapid determination of phosphate in synthetic wastewater,
Appl. Microbiol. Biotechnol. 66(4) (2005) 377-383.
[2] E. Ogabiela, S.B. Adeloju, A potentiometric phosphate biosensor based on entrapment of
pyruvate oxidase in a polypyrrole film, Anal. Methods 6(14) (2014) 5290-5297.
[3] M. Situmorang, J.J. Gooding, D.B. Hibbert, D. Barnett, The development of a pyruvate
biosensor using electrodeposited polytyramine, Electroanalysis 14(1) (2002) 17-21.
[4] L.A. Abayomi, L.A. Terry, S.F. White, P.J. Warner, Development of a disposable pyruvate
biosensor to determine pungency in onions (Allium cepa L.), Biosens. Bioelectron. 21(11)
(2006) 2176-2179.
[5] M.J. Eisenmenger, J.I. Reyes-De-Corcuera, High pressure enhancement of enzymes: A
review, Enzyme Microb. Technol. 45(5) (2009) 331-347.
[6] K. Tittmann, D. Proske, M. Spinka, S. Ghisla, R. Rudolph, G. Hubner, G. Kern, Activation of
thiamin diphosphate and FAD in the phosphate-dependent pyruvate oxidase from Lactobacillus
plantarum, J. Biol. Chem. 273(21) (1998) 12929-12934.
[7] L. Gilbert, A.T. Jenkins, S. Browning, J.P. Hart, Development of an amperometric assay for
phosphate ions in urine based on a chemically modified screen-printed carbon electrode, Anal
Biochem 393(2) (2009) 242-247.
[8] B. Sedewitz, K.H. Schleifer, F. Gotz, Purification and biochemical-characterization of
pyruvate oxidase from lactobacillus-plantarum, J. Bacteriol. 160(1) (1984) 273-278.
[9] A. Tomar, M. Eiteman, E. Altman, The effect of acetate pathway mutations on the production
of pyruvate in Escherichia coli, Appl. Microbiol. Biotechnol. 62(1) (2003) 76-82.
[10] B. Risse, G. Stempfer, R. Rudolph, H. Möllering, R. Jaenicke, Stability and reconstitution of
pyruvate oxidase from Lactobacillus plantarum: dissection of the stabilizing effects of coenzyme
binding and subunit interaction, Protein Sci. 1(12) (1992) 1699-1709.
[11] R. Blake, T.A. Obrien, R.B. Gennis, L.P. Hager, Role of the divalent metal cation in the
pyruvate oxidase reaction, J. Biol. Chem. 257(16) (1982) 9605-9611.

52
[12] W. Militzer, L. Burns, Thermal enzymes. VI. Heat stability of pyruvic oxidase, Arch.
Biochem. Biophys. 52(1) (1954) 66-73.
[13] T.F. Chang, K.C. Ruan, Exploration of pressure-induced dissociation of pyruvate oxidase,
Cell. Mol. Biol. 50(4) (2004) 323-328.
[14] A.M. Zapatabacri, C. Burstein, Enzyme electrode composed of the pyruvate oxidase from
pediococcus species coupled to an oxygen-electrode for measurement of pyruvate in biological
media, Biosensors 3(4) (1987) 227-237.
[15] L.A. Hazelwood, J.M. Daran, A.J.A. van Maris, J.T. Pronk, J.R. Dickinson, The ehrlich
pathway for fusel alcohol production: a century of research on Saccharomyces cerevisiae
metabolism, Appl. Environ. Microbiol. 74(8) (2008) 2259-2266.
[16] S. Sentheshanmuganathan, Mechanism of the formation of higher alcohols from amino
acids by Saccharomyces cerecisiae, Biochem. J. 74 (1960) 568-576.
[17] M.M. Wall, J.N. Corgan, Relationship between pyruvate analysis and flavor and flavor
perception for onion pungency determination, Hortscience 27(9) (1992) 1029-1030.
[18] S. Schwimmer, W.J. Weston, Onion flavor and odor- enzymatic development of pyruvic
acid in onion as a measure of pungency, J. Agric. Food Chem. 9(4) (1961) 301-304.
[19] K.S. Yoo, L.M. Pike, Determination of background pyruvic acid concentrations in onions,
Allium species, and other vegetables, Sci. Hortic. 89(4) (2001) 249-256.
[20] N. Minoura, S. Yamada, I. Karube, I. Kubo, S. Suzuki, Determination of lactate-
dehydrogenase in serum by using a pyruvate sensor, Anal. Chim. Acta 135(2) (1982) 355-357.
[21] D.L. Correll, The role of phosphorus in the eutrophication of receiving waters: A review, J.
Environ. Qual. 27(2) (1998) 261-266.
[22] G.E. Likens, A.F. Bartsch, G.H. Lauff, J.E. Hobbie, Nutrients and eutrophication, Science
172(3985) (1971) 873-874.
[23] L.A. Terry, S.F. White, L.J. Tigwell, The application of biosensors to fresh produce and the
wider food industry, J. Agric. Food Chem. 53(5) (2005) 1309-1316.
[24] E. Akyilmaz, E. Yorganci, A novel biosensor based on activation effect of thiamine on the
activity of pyruvate oxidase, Biosens. Bioelectron. 23(12) (2008) 1874-1877.
[25] S.R. Carpenter, N.F. Caraco, D.L. Correll, R.W. Howarth, A.N. Sharpley, V.H. Smith,
Nonpoint pollution of surface waters with phosphorus and nitrogen, Ecol. Appl. 8(3) (1998) 559-
568.

53
[26] L.S.B. Upadhyay, N. Verma, Recent advances in phosphate biosensors, Biotechnol. Lett.
37(7) (2015) 1335-1345.
[27] V.G. Gavalas, N.A. Chaniotakis, Phosphate biosensor based on polyelectrolyte-stabilized
pyruvate oxidase, Anal. Chim. Acta 427(2) (2001) 271-277.
[28] E. Ogabiela, S.B. Adeloju, J.W. Cui, Y.C. Wu, W. Chen, A novel ultrasensitive phosphate
amperometric nanobiosensor based on the integration of pyruvate oxidase with highly ordered
gold nanowires array, Biosens. Bioelectron. 71 (2015) 278-285.
[29] N. Gajovic, K. Habermuller, A. Warsinke, W. Schuhmann, F.W. Scheller, A pyruvate
oxidase electrode based on an electrochemically deposited redox polymer, Electroanalysis
11(18) (1999) 1377-1383.
[30] E. Akyilmaz, E. Yorganci, Construction of an amperometric pyruvate oxidase enzyme
electrode for determination of pyruvate and phosphate, Electrochim. Acta 52(28) (2007) 7972-
7977.
[31] G. Arai, T. Noma, H. Habu, I. Yasumori, Pyruvate sensor based on pyruvate oxidase
immobilized in a poly (mercapto-p-benzoquinone) film, Journal of electroanalytical chemistry
464(2) (1999) 143-148.
[32] W. Bergmann, R. Rudolph, U. Spohn, A bienzyme modified carbon paste electrode for
amperometric detection of pyruvate, Anal. Chim. Acta 394(2-3) (1999) 233-241.
[33] M.E. Ghica, C.M.A. Brett, Development of novel glucose and pyruvate biosensors at
poly(neutral red) modified carbon film electrodes. Application to natural samples,
Electroanalysis 18(8) (2006) 748-756.
[34] J. Kulys, L. Wang, N. Daugvilaite, Amperometric methylene green-mediated pyruvate
electrode based on pyruvate oxidase entrapped in carbon paste, Anal. Chim. Acta 265(1) (1992)
15-20.
[35] M. Mascini, F. Mazzei, Amperometric sensor for pyruvate with immobilized pyruvate
oxidase, Anal. Chim. Acta 192 (1987) 9-16.
[36] E. Bayram, E. Akyilmaz, A new pyruvate oxidase biosensor based on 3-mercaptopropionic
acid/6-aminocaproic acid modified gold electrode, Artif. Cell. Nanomed. Biotechnol. 42(6)
(2014) 418-422.
[37] J.W. Cui, E.E. Ogabiela, J.N. Hui, Y. Wang, Y. Zhang, L. Tong, J.F. Zhang, S.B. Adeloju,
X.Y. Zhang, Y.C. Wu, Electrochemical biosensor based on Pt/Au alloy nanowire arrays for
phosphate detection, J. Electrochem. Soc. 162(3) (2015) B62-B67.
[38] L.A. Dang, J. Haccoun, B. Piro, M.C. Pham, Reagentless recycling of pyruvate oxidase on a
conducting polymer-modified electrode, Electrochim. Acta 51(19) (2006) 3934-3943.

54
[39] J.A. Guerrero-Beltran, G. Barbosa-Canovas, B.G. Swanson, High hydrostatic pressure
processing of fruit and vegetable products, Food Rev. Int. 21(4) (2005) 411-425.
[40] J. Raso, G.V. Barbosa-Canovas, Nonthermal preservation of foods using combined
processing techniques, Crit. Rev. Food Sci. Nutr. 43(3) (2003) 265-285.
[41] B.B. Boonyaratanakornkit, C.B. Park, D.S. Clark, Pressure effects on intra- and
intermolecular interactions within proteins, Biochim. Biophys. Acta-Protein Struct. Molec.
Enzym. 1595(1-2) (2002) 235-249.
[42] H. Eyring, J.L. Magee, Application of the theory of absolute reaction rates to bacterial
luminescence, J. Cell. Comp. Physiol. 20(2) (1942) 169-177.
[43] C.R. Robinson, S.G. Sligar, [18] Hydrostatic and osmotic pressure as tools to study
macromolecular recognition, Methods in enzymology 259 (1995) 395-427.
[44] P.C. Michels, D.S. Clark, Pressure-enhanced activity and stability of a hyperthermophilic
protease from a deep-sea methanogen, Appl. Environ. Microbiol. 63(10) (1997) 3985-3991.
[45] M. Gross, R. Jaenicke, Proteins under pressure – the influence of high hydrostatic-pressure
on struture, function and assembly of proteins and protein complexes, Eur. J. Biochem. 221(2)
(1994) 617-630.
[46] K. Heremans, High-pressure effects on proteins and other biomolecules, Annual Review of
Biophysics and Bioengineering, Annual Review of Biophysics and Bioengineering 11 (1982) 1-
21.
[47] D.B. Kitchen, L.H. Reed, R.M. Levy, Molecular-dynamics simulations of solvated protein
at high-pressure, Biochemistry 31(41) (1992) 10083-10093.
[48] V.V. Mozhaev, R. Lange, E.V. Kudryashova, C. Balny, Application of high hydrostatic
pressure for increasing activity and stability of enzymes, Biotechnol. Bioeng. 52(2) (1996) 320-
331.
[49] Y. Taniguchi, K. Suzuki, Studies of polymer effects under pressure .7. pressure inactivation
of alpha-chymostrypsin, J. Phys. Chem. 87(25) (1983) 5185-5193.
[50] V.V. Mozhaev, K. Heremans, J. Frank, P. Masson, C. Balny, High pressure effects on
protein structure and function, Proteins 24(1) (1996) 81-91.
[51] S.A. Hawley, Reversible pressure-temperature denaturation of chymotrypsinogen,
Biochemistry 10(13) (1971) 2436-2442.

55
[52] J. Roche, J.A. Caro, D.R. Norberto, P. Barthe, C. Roumestand, J.L. Schlessman, A.E.
Garcia, B. Garcia-Moreno, C.A. Royer, Cavities determine the pressure unfolding of proteins,
Proc. Natl. Acad. Sci. U. S. A. 109(18) (2012) 6945-6950.
[53] W. Kauzmann, Protein stabilization – thermodynamics of unfolding, Nature 325(6107)
(1987) 763-764.
[54] M.D. Collins, G. Hummer, M.L. Quillin, B.W. Matthews, S.M. Gruner, Cooperative water
filling of a nonpolar protein cavity observed by high-pressure crystallography and simulation,
Proc. Natl. Acad. Sci. U. S. A. 102(46) (2005) 16668-16671.
[55] E. Girard, S. Marchal, J. Perez, S. Finet, R. Kahn, R. Fourme, G. Marassio, A.C. Dhaussy,
T. Prange, M. Giffard, F. Dulin, F. Bonnete, R. Lange, J.H. Abraini, M. Mezouar, N. Colloc'h,
Structure-function perturbation and dissociation of tetrameric urate oxidase by high hydrostatic
pressure, Biophys. J. 98(10) (2010) 2365-2373.
[56] E.C. Juan, M.M. Hoque, M.T. Hossain, T. Yamamoto, S. Imamura, K. Suzuki, T.
Sekiguchi, A. Takenaka, The structures of pyruvate oxidase from Aerococcus viridans with
cofactors and with a reaction intermediate reveal the flexibility of the active-site tunnel for
catalysis, Acta crystallographica. Section F, Structural biology and crystallization
communications 63(Pt 11) (2007) 900-907.
[57] M.J. Eisenmenger, J.I. Reyes-De-Corcuera, High hydrostatic pressure increased stability
and activity of immobilized lipase in hexane, Enzyme Microb. Technol. 45(2) (2009) 118-125.
[58] A. Halalipour, M.R. Duff, E.E. Howell, J.I. Reyes-De-Corcuera, Glucose oxidase
stabilization against thermal inactivation using high hydrostatic pressure and hydrophobic
modification, Biotechnol. Bioeng. 114(3) (2017) 516-525.
[59] A. Halalipour, M.R. Duff, E.E. Howell, J.I. Reyes-De-Corcuera, Effects of high hydrostatic
pressure or hydrophobic modification on thermal stability of xanthine oxidase, Enzyme Microb.
Technol. 103 (2017) 18-24.
[60] M.I. Buchholz, Increase stability and activity of alcohol oxidase under high hydrostatic
pressure, Food Science and Technology, University of Georgia, 2016.
[61] Sigma-Aldrich, Assay procedure for pyruvate oxidase bacterial.
http://www.sigmaaldrich.com/technical-documents/protocols/biology/assay-procedure-for-
pyruvate-oxidase-bacterial.html. (Accessed April 1 2016).
[62] M. Goebel-Stengel, A. Stengel, Y. Taché, J.R. Reeve, The importance of using the optimal
plastic and glassware in studies involving peptides, Anal. Biochem. 414(1) (2011) 38-46.
[63] M. Mather, R. Gennis, Kinetic studies of the lipid-activated pyruvate oxidase flavoprotein
of Escherichia coli, J. Biol. Chem. 260(30) (1985) 16148-16155.

56
[64] A.C. Polydera, E. Galanou, N.G. Stoforos, P.S. Taoukis, Inactivation kinetics of pectin
methylesterase of greek Navel orange juice as a function of high hydrostatic pressure and
temperature process conditions, J. Food Eng. 62(3) (2004) 291-298.
[65] C. Weemaes, L. Ludikhuyze, I. Van den Broeck, M. Hendrickx, High pressure inactivation
of polyphenoloxidases, J. Food Sci. 63(5) (1998) 873-877.
[66] I. Dalmadi, G. Rapeanu, A.v. Loey, C. Smout, M. Hendrickx, Characterization and
inactivation by thermal and pressure processing of strawberry (Fragaria ananassa) polyphenol
oxidase: a kinetic study, Journal of Food Biochemistry 30(1) (2006) 56-76.
[67] J.L. Melnick, Ultraviolet absorption spectra of cocarboxylase, thiamine, and their reduction
products, J. Biol. Chem. 131 (1939) 615-620.

57
APPENDIX
SUPPLEMENTAL EXPERIMENT TEMPERATURE AND PRESSURE PROFILES
The following figures compare the temperature and pressure profiles of samples from the
supplemental experiments using the new batch of POX, to show consistency in treatment
between replicates. Profiles for samples treated a 0.1 MPa at 25 °C for 0s have been omitted, as
the combination of low pressure, short time, and low pressure erroneously trigger the LabView
program to cease recording as treatment continues. The file for the second replication treated at
50 MPa at 45 °C for 1000 s was mistakenly overwritten.
Figure A.1. Pressure (closed symbol) and temperature (open symbol) for samples one (dark grey)
and two (light gray) for supplemental sample treated at 0.1 MPa at 25 °C for 3000 s

58
Figure A.2. Pressure (closed symbol) and temperature (open symbol) for samples one (dark grey)
and two (light gray) for supplemental sample treated at 50 MPa at 25 °C for 0 s
Figure A.3. Pressure (closed symbol) and temperature (open symbol) for samples one (dark grey)
and two (light gray) for supplemental sample treated at 50 MPa at 25 °C for 3000 s
Figure A.4. Pressure (closed symbol) and temperature (open symbol) for samples one (dark grey)
and two (light gray) for supplemental sample treated at 0.1 MPa at 35 °C for 0 s

59
Figure A.5. Pressure (closed symbol) and temperature (open symbol) for samples one (dark grey)
and two (light gray) for supplemental sample treated at 0.1 MPa at 35 °C for 3000 s
Figure A.6. Pressure (closed symbol) and temperature (open symbol) for samples one (dark grey)
and two (light gray) for supplemental sample treated at 50 MPa at 35 °C for 0 s
Figure A.7. Pressure (closed symbol) and temperature (open symbol) for samples one (dark grey)
and two (light gray) for supplemental sample treated at 50 MPa at 35 °C for 3000 s

60
Figure A.8. Pressure (closed symbol) and temperature (open symbol) for samples one (dark grey)
and two (light gray) for supplemental sample treated at 0.1 MPa at 45 °C for 0 s
Figure A.9. Pressure (closed symbol) and temperature (open symbol) for samples one (dark grey)
and two (light gray) for supplemental sample treated at 0.1 MPa at 45 °C for 1000 s
Figure A.10. Pressure (closed symbol) and temperature (open symbol) for samples one (dark
grey) and two (light gray) for supplemental sample treated at 50 MPa at 45 °C for 0 s





























