Embed Size (px)
Citation preview

urt.b .ett
THE CLONING AND PRELIMINARYCHARACTERIZATION OF THE CTEA GENE
FROM AspergiZ l¿ts n íd¿t Zøns -
Celia E- A- Dowzer- El-sc-(Hons-)
Department of Genetics, University of Adelaide, South Australia.
Thesis submitted to the University of Adelaide for the degree of
Doctor of Philosophy in March, 1991.
by

T.ABLE OF CONTENTS.
Abstract
Decì arati on
Acknowl edgements
Pubì i cati ons
Abbrevi ati ons
List of tables
List of figures
CHAPTER ONE : I NTRODUCT ION
1.1 MOLECULAR MECHANISMS OF GENE REGULATION IN EUKARYOTES
1.1.1 DNA binding motifs
1.1.2 Activation domains
1.1.3 Negative control sYstems
1.1.4 The reguìation of synthesis and activity of
transcription factors
1.2 CARB0N CATABOLITE REPRESSION IN Escåerichío coli
1.3 CARB0N CATABOLITE REPRESSI0N IN Socchøronyces
1.3.1 Reguìation of the galactose reguìon of
S. cerevísioe
1.3.2 Sucrose utilization in S. cerevísioe
1.4 GLOBAL GENE REGULATION IN Á. nidulons
1.5 CARBON CATABOLITE REPRESSION IN Á. N|dUIONS
1.5.1 Selection of mutations affecting carbon
PAGE
I
IIIIV
v
VI
VII
VIII
1-
3
4
7
I
T2
13
13
18
22
26
30

catabolite repression in Á. nídulons
I.5.2 The phenotype of creA mutants
1.5.3 The phenotype of creB and creC mutants
1.5.4 Proposed roles for creA, creB, creC and cre-34
gene products
1.5.5 Molecular studies of regulation by carbon
catabolite repression in 4. nidulons
1.6 AIMS OF THIS STUDY
CHAPTER TWO: MATERIALS AND
METHODS
2.1 MATERIALS
2.2 BUTFERS AND STOCK SOLUTIONS
2.2.I I X Reaction buffers
2.2.2 Stock solutions
2.3 MEDIA
2.3.L Bacterial and yeast growth media
2.3.2 Aspergil Zus growth medi a
2.4 E. coli STRAINS AND BACTERI0PHAGE
2.5 FUNGAL STRAINS
2.6 PLASMIDS
2.7 METHODS
2.7.L Genetic manipulation and growth testing of
Aspergíllus
2.7.2 Transformation of Aspergíllus
2.7.3 Isolation of nucìeic acids
2.7.4 General reconbinant DNA methods
2.7 .5 Gel electroPhoresis
PAGE
32
35
38
41
42
45
4747
48
48
49
50
50
51
52
53
53
53
53
56
56
59
60

2.7 .6 32P Label ì ing of DNA
2.7.7 Nucleic acid blotting and hybridization
condi ti ons
2.7.8 Quantification of DNA copy number and mRNA
I evel s
2.7.9 DNA sequencing
2.7.I0 Primer extension analYsis
CHAPTER THREE: THE CLONING OF
c re A FROM Aspe rg ¿ L Z ¿ts rt id¿t Z arzs
3.1 THE CLONING OF CrCA
3.1.1 The construction of a wiìdtype genomic ìibrary
from ,4 . nidulons
3.1.2 The identification of a non-revertable creA-
allele
3.1.3 Complementation of the cre\204 mutation
3.1.4 Rescue of transforming sequences
3.1.5 The localization of cre| within pANCI
3.2 ANALYSIS 0F CreA+ TRANSFORMANTS
3.3 EXPRESSI0N 0F creL IN Á. nídulons
3.4 SUMMARY AND CONCLUSIONS
CHAPTER FOUR: ANALYS I S OF
cTeA.d_3O AND THE PHENOTYPE
OF A creA NULL ALLELE4.1 MOLECULAR LOCALIZATION OF THE INVERSION BREAKPOINT IN
creAd-30
4.2 CONSTRUCTIOiI 0F A creA DELETION STRAIN
68
PAGE
61
62
63
63
66
6768
68
73
76
79
81
88
95
97
97
103

4.2.I The isolation of a larger creA genomic clone
4.2.2 The construction of a creA replacement vector
4.2.3 The creation of a creA gene replacement strain
of A. nidulons
4.2.4 Vegetative versus conidial 'lethal ity of the creA
nul I phenotype
4.2.5 Rescuing the ability of TC6330 to form haploids
containing chromosome I having the gene
repì acement
4.3 SEQUENCES H0M0L0G0US T0 creA FROM Á. nidulons
4.4 SUMMARY AND CONCLUSIONS
CHAPTER FIVE: THE PHYSICALCHARACTERIZATION OF THE
Asperg¿iZtts níd¿tZarzs creA GENE
5.1 THE PHYSICAL ANALYSIS OF CTEA
5.1.1 Nucleotide sequence analysis of creA and
characterization of the coding region
5.L.2 The 5' region of the creA gene
5.1.3 The 3' region of the creA gene
5.2 ANALYSIS 0F THE PUTATIVE CreA PR0TEIN
5.2.1. The zinc finger motif
5.2.2 An alanine rich region
5.2.3 The S/TPXX motif
5.2.4 PEST sequences
5.2.5 Other general features of the CreA protein
5.3 SUMMARY
PAGE
103
104
106
115
tL7
119
119
L22t22
122
126
133
133
134
141
t49
149
150
151

CHAPTER SIX: CONCLUDINGDISCUSSION
RE F ERENCES
PAGE
155
L64

I
ABSTRACT -
Previous gentical ana'lyses have led to the suggestion that the
product of the creA gene from Aspergillus nidulons acts as a
negatively act'ing reguìatory protejn in carbon cataboìite repression'
This suggestion was based on the finding that corflrnon crel. mutant
alleles lead to the derepression of synthesis, in the presence of D-
g'lucose, of many enzymes for the ultilization of alternative carbon
sources which are repressed during growth on D-glucose in wildtype
A. nídulons, and these mutant alleles are recessive to the wildtype
allele in diploids.
The creA region has been cloned by complementation of a mutant
allele by transformation with a genom'ic library followed by marker
rescue techniques. The creA gene encodes a transcript of
approxìmately 2kb in ìength which is constitutively expressed' The
nucleotide sequence of the creA genom'ic region and number of cDNA
clones has been determjned. The 390 amjno acid sequence of the
putat'ive crel protein has a number of characteristic features found
jn regulatory and DNA binding proteins including two Cr-H" zinc
finger motifs and several alanine rich regions. Although no overall
sequence simiìarities have been found to other proteins of known
function, regions of simi ìarity, possibìy represent'ing functional
domains, have been identified and their sign'ificance js discussed'
Aìl creA mutant alleles analysed produce a cre| transcript' One
creA alle1e, creAd-30 contains a translocation breakpoint within the

II.
creA gene and has two transcripts. Thus none of the in vívo isolated
mutants tested were nulì alleles at the level of nRNA. A plasmid
construct containing the ríbo} gene from ,4. nidulons fìanked by
sequences up and downstream of the creA gene was used to replace one
of the wììdtype creA genes in a dipìoid strain. Haploidization
analysis of this diploid demonstrates that total ìoss of cre[
function is lethal in vegetative cells.

III.
DECLARAT I ON .
I declare that this thesis contains no material which has been
accepted for the award of any other degree or diploma in any
University, and to the best of my knowìedge and belief contains no
material previously published or written by another person, except
where due reference is made in the text of the thesis.
I give my consent for this thesis to be made available for
photocopying and loan.
Celia E. A. Dowzer.

IV.
AC KNOW L EDG EMENTS .
I am extremeìy gratefuì for the excellent supervisjon given by
Dr. Joan Ke]ìy during my candidature in this department. I wouìd like
to thank Joan on a personal basis for her friendship, along with the
encouragement and enthusiasm she has shown for this proiect.
I appreciate the vaìuable discussions, conments and suggestions
on written and/or experimental work prov'ided by Dr. Robin Lockington,
Dr. Meryì Davis and Prof. Michael Hynes and the interest they have
shown in the progress of this work
I am indebted to Dr. Matthew 0'Conne'll for his friendship and
the unfailing encouragement he has given. I also appreciate very much
his advice on experimental techniques and written work and the time
he has spent helping with conputer analyses.
Dr. Dara }{hisson provided many suggestions on experìmental
methods and has aìso been a good friend over the past few years' I
would also like to thank Angela Binns and Michelle Adamou for their
technical help in lab109. I wish to thank Doug Anderson and Ulrik
John for their time and effort in proof-reading this thesis.
I acknowledge the financial support of a Conmonwealth
postgraduate Researôh Award and an Australian Postgraduate Priority
Research Award.

V
#,t,f
PUBLICATIONS -
The work presented in this thesis has been submitted for
pubìication or published under the foì1wìng titles:
Dowzer, C.E.A. and Keìly, J.M. 1989. Cloning of the creA gene from
Aspergillus nidulonsz a gene involved in carbon catabol ite
repressi on. Curr. Genet. 15: 457-459.
Dowzer, C.E.A. and Kelly, J.M. 1991. Physicaì ana'lysis of the creA
gene of Aspergillus nídu|øns, and determination of the phenotype
of a deletion. Mol. CeLl. BioI. submitted-
Arst, H.N. , Toì lervey, D. , Dowzer, C.E.A. and Keì ly, J 'M'
inversion truncating the creL gene of Aspergillus
resuìts in carbon catabolite derepression. Mol. Micro.
854.
1990. An
niduløns
4: 851-
rI

VI.
üI
ABBREV I AT I ONS .
A: Adenosine; bp: base pajr(s); BSA: bovine serum albumin; c:
cytidine; cDNA: DNA comp'lementary to RNA; Ci: curie; dATP: 2'-deoxy-
adenos i ne-5 ' -tri phosphate; dCTP: 2 ' -deoxy-cyt ì d i ne-5 ' -tri phosphate;
ddATP z 2',3' -dideoxy-adenosine-s' -trìphosphate; ddCTP : 2',3' -dideoxy-
cytidine-S'-triphosphate; ddGTPz 2',3'-dideoxy-guanosine-5'-
triphosphate; ddNTP 2 2',3' -dideoxy-nucleoside-5' -triphosphate; ddTTP:
2',3'-dideoxy-thymidine-5' -triphosphate; dGTP: 2' -deoxy-guanosìne-5' -
tr.iphosphate; dITP: 2'-deoxy-jnosine-5'-triphosphate; DNA:
deoxyri bonuc I ei c ac i d; dNTP: 2 ' -deoxy-nuc I eos i de-S ' -tri phosphate;
dTTP: 2'-deoxy-thymidine-5'-triphosphate; DTT: dithiothrejtol ; EDTA:
(ethy'lenedinitrilo)tetraacetìc acid; G: guanosine; g: gram/force of
gravity; kb: kilobase(s); mg: milligram; ml: millilitre; mRNA
messenger RNA; NAD: ß -nicotinamjde-adenjne dinucleotide; NADP: ß -
nicotinanide-adenine dinucleotide phosphate; p: plasmid; RNA:
ribonucleic acid; r.p.m: revolutions per mjnute; SDS: sodium dodecyl
sulphate; T: thymidine; Tris: 2-amino-2-(hydroxymethyl)-1,3-
propanediol; u: uridine; uci: microcurie; ug: microgram; uì:
microlitre; vfvz volume per volume; w/v: weight per volume.
II
,
3
I

VI I.
TABLE
2.5.t
2.6.r
2.7.1
3.1 .1
3.2.r
3.3.1
3.3.2
4.1.1
5.1.1
LIST OF TABLES.
Genotypes of Aspergillus strains.
Plasmids used in this study.
dNTP solutions for DNA sequenc'ing.
Spontaneous and induced "reversion" of creAd-I
and creA204.
creA copy number in CreA+ transformants.
Relative levels of expression of creA in
wildtype A. nidulons and strain 664.
Relative levels of expression of cre[ in crel
mutant strains and CreA+ transformants.
Expected bands of hybridization of pANC4 to
wildtype A. nídulsns D1{4.
Codon usage in the creA gene of A- níduløns-
PAGE
54
55
65
100
r27
N¡i{ü
I
72
85
92
92
I{i
r
t
i

VIII.
FIGURE
3.1. 1
3.1.2
3.1.3
3.1.4
3.1.5
3.1.6
3.r.7
3.1.8
3.2.r
3.3. I
3.3.2
3.3.3
3.3.4
LIST OF FIGURES.
Spontaneous and induced "reversion" of cred-I
and creA204.
Growth testing of T41a, T41b and T41c'
Complementation of C43 with pANCl.
Partial restriction maP of PANCl.
Southern blot analysis of wi'ldtype A' nidulons
and T41.
Partial restriction map of the pANC4 insert'
Southern blot analysis of wildtype A' nidulons
DNA probed with PANC3.
Southern blot analysis of creA mutant alleles'
Southern blot analysis of Á. nidulons pANC4
transfonnants.
Northern blot analysis of wildtype A' nidulons
and cre\204 Probed with PANC4.
Northern blot analysis of wildtype A' nídulons
probed with pANCl.
Northern blot analysis of creA nutant strains'
Northern blot analYsis of CreA+
transformants.
Southern blot analysis of creAd-30'
PAGE
70
75
77
78
rIIïI
I
80
82
83
84
87
90
91
94
96
99
*
4.1.1

IX.
4.r.2
4.2.r
4.2.2
4.2.3
4.2.4
4.2.5
4.2.6
4.4.r
5.1.1
5.t.2
5.1.3
5.2.1
5.2.2
5.2.3
5.2.4
5.2.5
5.2.6
5.2.7
5.2.8
5.2.9
5.2.t0
5.2.rL
5.2.t2
Northern blot analysis of creAd-30.
Partial restriction map of the insert in pANC6.
Partiaì restriction map of the insert in pANC8.
Northern blot analysis using pANCS as a probe.
Southern blot of pANCS transfonnants of C62.
Strategy used for deleting the creA gene fron
A. nidulons.
Southern analyses of TC6330 and TC6332.
Southern blot of DNA from various fungi probed
with the creA clone.
creA sequencing strategy.
Nucleotide sequence of creA.
Primer extension analysis.
Aìignment of zinc finger domains.
The finger motifs proposed for zinc fingers I
and II in CreA. 136
Comparison of CreA and MIG1 zinc fingers- 138
Comparison of H-C links in zinc finger proteins' 138
Plotstructure of Peptidestructure for CreA. 140
Comparison of zinc finger loop regions in zinc
finger proteins. L42
Comparison of CreA and Kruppeì amino acid sequences. 144
Comparison of CreA and Engrailed amino acid sequences. 145
Comparison of CreA and Evenskjpped amino acid
PAGE
r02
105
r07
108
110
111
113
I20
r23
125
r29
135
sequences.
Comparison of CreA and CycS amino acid sequences'
Chou and Fasman CreA secondary structure.
GOR CreA secondary structure.
146
148
L52
153

CHAPTER ONE
I NTRODUCT I ON .
Carbon cataboljte repressìon, also referred to as glucose
repressì on, i s a wi de domai n regul atory system whi ch al I ows the
regulation of a large number of genes in response to the carbon
source available. It functions to repress the expression of a large
number of genes involved in the utilization of alternative carbon
sources when a readiìy utilizable carbon source such as glucose or
sucrose i s al so present, thus al I owi ng more effi ci ent use of
resources. The metabolism of aìternative carbon sources requires the
induction and/or derepression of express'ion of subsets of genes for
the enzymes required for a specific metabolic pathway, in response
to parti cul ar envi ronmental stimul i .
The mechanism of carbon cataboì ite repressjon is fairly wel I
understood jn E. coli. However, the molecular basis for this process
in eukaryotes appears to be more complex and has not been well
characterized. Considerable interest has been generated over the past
few years for elucidating the basis for carbon catabolite repression
at the molecular level. The study of carbon catabolite repression
provides a means by which major systems of gene reguìation may be
understood from both a pure research perspective and also in the
application of this knowledge to the control of gene express'ion in
industrially important organìsms. A large number of nutants showjng
an altered response to carbon catabol ite repression have been

2
isolated in various s'imple eukaryotes. For example, at least a dozen
putative reguìatory genes invoìved in gìucose repression in
Socchoromyces cerevisíoe have been identified by genetic anaìysis
(reviewed by Entian 1986, Gancedo and Gancedo 1986). Aìthough many of
these have been geneticalìy characterized, extensive studies at the
moìecular level are required to determine the sequence of events
involved jn the regu'lation of gene expression by carbon cataboìite
repress i on .
This thesis outlines a study of cre| which is a wide domain
regu'l atory gene i nvol ved j n carbon catabol i te repress i on i n
Aspergillus nídulons. This provides a model system for understanding
some properties of maior mechanisms of gene regulation in simple
eukaryotes. There are many advantages to studying complex systems of
gene regulation in A. nidulons as both classical genetic analyses and
the molecular characterization of genes involved in specific
regulatory pathways can be readily undertaken.
The filamentous ascomycete fungus, A. nídulons, is metabolica'lly
versatile and can grow on defined solid and liquid media. It has a
hapìoid conrpìement of eight chromosomes (Elliot 1960) and a large
number of mutants have been mapped to the eight linkage groups
(reviewed by Clutterbuck 1984) providing an extensive genetic map of
this species. The existence of both sexuaì and parasexual stages in
i ts ì i fe cycì e al l ows both mei oti c, mi toti c and hap'loi di zati on
ana'lyses. These are desirabìe features to cornpìement molecular
techniques for comprehensive studies into gene function and
regulation. A. nidulqns has a relatively small genome of about 3 X
10?bp (timberìake 1978, Brody and Carbon 1989), which facilitates the
identificatjon and isolation of particular sequences of interest

3
from genomic libraries.
A number of transformatjon systems have been deveìoped for A'
nídulons and related species using seìectable markers ìn either
pl asmi d or cosmi d vectors for homol ogous and non-homol ogous
'i ntegrati on into the host genome. These i ncl ude both the
complementation of recessjve auxotrophic mutations with cloned genes
and the use of dominant selectable markers (reviewed by Gurr et ol'
1987, Fincham 1989), âllowing both the complementation analysis of
cloned sequences and the analysis of mutations by reverse genetics' A
further .important feature of AspergíItus species is that celluìar
differentiation during sporolation provides a system to study the
mechanisms by which a large number of genes are regulated both
spacially and temporaìly (reviewed by Timberlake and Marshall 1988).
Therefore , A. nidulons provides an amenable nodel for investigating
the molecular mechanjsms of wide domain regulatory systems in
eukaryotes.
As a background to the study of the creA gene from A ' nidulons '
it is important to review some of the maior mechanisms by which
eukaryot'ic reguìatory genes exert their effect at the molecular
level. In addition some of the major observations and interpretations
of the more wideìy studied genes ìnvolved in carbon catabolite
repression in both prokaryotes and eukaryotes are reviewed.
1.1 MOLECULAR MECHANISMS OF GENE REGULATION IN EUKARYOTES:
The major control of gene regulation in eukaryotic cells is at
the level of transcription (Darneìt 1982, reviewed by Latchman 1990).
This transcript'ionaì regulation of eukaryotic genes is mediated by

4
the interaction of cis-acting DNA sequences with trons-acting factors
(reviewed by Dynan and Tjian 1985). As many of the reguìatory genes
characterized from A. nídulons, including creR (this thesis) code
for proteins having similar structural domains to known DNA binding
proteins and have been shown to bind DNA, it is relevant to review
the najor characteristics of eukaryotic transcription factors.
1.1.1 DNA bindin moti fs:
Analyses of the stucture and derived amino acid sequences of
eukaryotìc transcription factors have demonstrated that there are
several motifs in these proteins which enable them to bind to DNA in
a sequence specific manner (reviewed by Latchman 1990, reviewed by
Nussinov 1990).
X-ray crystal lography of the bacteriophage Cro protein
(Anderson et ø1. 1981) the E. coZ¿ CRP (McKay and Steitz 1981) and
the Cl (Dodd and Egan 1987) proteins revealed the helix-turn-helix
motif responsible for the ability of these proteins to bjnd in a
sequence specific manner to DNA. This motif is present as the
essential DNA binding domain in a number of regulatory proteins,
inc'luding the proteins encoded by the honeotic genes of Drosophilø
nelonogoster, referred to as homeodomains (Mihara and Kaiser 1988,
Mulìer et ol. 1988). Anaìysis of the 3-dimensional structure of the
helix-turn-helix motif has shown that it consists of an a-helix
followed by a turn followed by another o-helix in the protein' When
interacting w'ith DNA, the first a-heìix lies across the maior groove
of the DNA target sequence whi I e the second cr -heì i x ì i es part'ly
within the major groove and makes the sequence specific contact with

5
the DNA (Ptashne 1986).
Another motif is the leucine zipper (Landschulz et ol. 1988a).
This element has been shown to be present in a number of proteìns
including the marnnalian enhancer binding protein C/EBP (Johnson et
qI. 1987), Gcn4 from yeast, the proto-oncogene proteins Myc, Fos and
Jun and the Drosophílo daughterless protein (Landschulz et ø1. 1988a,
reviewed by Prendergast and Ziff 1989). The leucine zipper is
characterized by a region containing four leucine residues positioned
at intervals of seven amino acids in an cl-heìix, whereby the leucine
residues occur every two turns on the same side of the helix. This
structure is required for dimerization of the protein essential for
subsequent DNA bindìng. The leucine zipper is therefore involved in
protein-proteìn interactions and is not a DNA binding motìf itself.
Dimerization of the protein (or the formation of heterodimers) is
medjated by the interlocking of two leucine containing helices on
different molecules and results in the correct conformation for
binding in the adjacent DNA binding region of the protein or protein
complex (Johnson et ol 1987, Landschulz et ol. 1988a, b).
Zinc finger motifs were originalìy identified as DNA binding
structures in the RNA polymerase III transcription factor TFIIIA
which binds to the internal control region of the 5S RNA gene in
Xenopus (Miller et ol. 1985). Subsequently, ôt least two types of
zinc fingers have been found in DNA binding factors involved in
transcription mediated by RNA poìymerase II. TFIIIA-like zinc fingers
have been identified in a large number of protein sequences (reviewed
by Berg 1990) including the mammalian transcription factor SpI
(Kadonga et ol. 1987), the DrosophúZø kruppel protein (Redmann et ø1.
1988) and the yeast AdrI protein (Parraga et ol. 1988). The zinc

6
finger consensus sequence'is Tyr/Phe-X-Cys-Xr, o-Cys-X.-Phe-Xu-Leu-Xr-
His-X.,o-His, where X is a variable amino acid (Mi11er et o1.1985).
This sequence contains two invariant pairs of cysteine and histidine
residues that stabilize the domain by tetrahedralìy co-ordìnating a
7n2+ ion. Protein modelìing predicts that this would result in a
finger-ìike structure where the conserved pheny'laìanine and leucine
residues in the finger project from the surface of the protein. The
tips of these fingers directly contact the major groove of DNA in the
protein's target site where alternate fingers bind on opposite sides
of the helix (K'lug and Rhodes 1987). TFIIIA has nine tandemly
arranged zjnc finger motifs (Mill er et ol. 1985). The solution
structure of a number of these zinc finger proteins have been
identified using nuclear magnetic resonance data and confirm the
major characteristics of the previousìy predicted models for these
proteins involved in metal binding (Lee et ø1. 1989, Klevit ef sL-
1990, Omichinshi et øl 1990).
A second class of zinc finger proteins are found in some
steroid/thyroid hormone receptors which activate or repress gene
expression by binding to specific DNA sequences in their target genes
(Evans 1988, Beato 1989) and have also been observed in a number of
other proteins such as the yeast transcription factor Gal4 (Giniger
et sl . 1985) . The bi ndi ng regi on of these f i ngers was ori g'inal 'ly
thought to consist of four cysteine residues which co-ordinate the
7n2+ ion (Holtenberg et ol. 1987). However, more recent studies of
the metal binding domain of Gal4 which consists of a Cys-Xr-Cys-Xu-
Cys-Xu-Cys-Xr-Cys-Xu-Cys motif have suggested that instead of forming
a zinc finger structure, the six cysteine residues form a binuclear
metal ion cluster. Pan and Coleman (1990) have demonstrated that this

7
motif in Gal4 binds two metaì ions which are coordinated by the six
cyste'ines of the motif where two of these form connecting ligands
between the jons. This alternative model for the structure of crc,
zinc fingers is yet to be demonstrated for other transcrjption
factors containing this motif, however the sequence conservation of
such domains suggests that they are ìikeìy to form similar binuclear
cl usters .
I.I.2 Activation domains:
Following the binding of transcription factors to specific DNA
sequences in the 5' region(s) of their target genes, these proteins
must interact with other transcniption factors or with RNA po]ymerase
in order to infìuence transcription either in a positive way by
activation (reviewed by Guarente 1988, reviewed by Ptashne and Gann
1990) or negatively by repressìon (reviewed by Levine and Manley
1989, reviewed by Renkawitz 1990).
In a number of cases specific acidic domains, distinct from
those that mediate DNA binding, are required for transcriptionaì
activation by positive'ly actìng transcription factors (Hope and
Struhl 1986, Holìenberg and Evans 1988). These activation domains
probabìy interact with other proteins such as the TATA box binding
factor TFIID (Sawadago and Roeder 19B5) rather than by a direct
interactjon with RNA polymerase. For example, the binding of the
yeast transcription factor Gaì4 to its target site has been shown to
alter the conformation of already bound TFIID, such that TFIID
interacts with both the TATA box and the start point of
transcription, rather than with the TATA box alone. The altered

8
binding of TFIID facilitates the binding of other transcription
factors and RNA polymerase into a transcription compìex and allows
transcription to be initiated (Horikoshi et ol. 1988)'
1.1.3 Neqative control sYstems:
Negativeìy acting transcription factors have been found to be
less cormon in eukaryotic cells, however some such as the yeast Gal80
protein have been well characterized. Levine and Manley (1989)
some of the mechanisms by which negative regulation may be achieved.
The simp'lest mechanism of repress'ion occurs where positively acting
transcript'ion factors are prevented from binding to their target
sites by the binding of a repressor molecule. This is also comnon in
bacteria, for example the ZøcI gene product blocks transcription of
the Lac-operon (reviewed by Busby 1986). Similarly, a trons-acting
protein may bind positive regulatory molecules preventing their
binding to the transcription complex. A negatively acting factor may
also interfere with the activation of transcription by an already
bound transcription factor. For example, the negatively acting Gal80
protein from yeast prevents activation mediated by Gal4 of the
structural genes for galactose utilization by binding to and masking
the activation domain of Gal4 (Johnston et ol. 1987). The interaction
between Gal4 and Gal80 is discussed further in section 1.3.
Studies of eukaryotic gene reguìation have focused mainly on
positive regulatory sequences and positively acting transcription
factors. However, a number of regulatory systens involving regulation
by negatively acting factors (repressors) have been the subject of
extensive analyses.

I
The homeodomains of D. nelonogosúer homeobox proteins mediate
the bìnding of these proteins to specific DNA target sequences to
regulate the transcription from target promoters. Most of these genes
control 1 ing ear'ly embryonic development in Drosophilo function
positively to activate transcription of their target genes. However,
a number of these have also been shown to be transcriptionaì
repressors, while others have repressor as well as activator function
where the mode of activity appears to depend on the promoter they are
reguìating (reviewed by Hayashi and Scott 1990). Induction of the
segment poìarity gene engroíted by the homeobox proteins zerknullt-
related, fushi tarazu and paired is inhibited by the evenskípped and
engrøiled gene products (Han eú ø2. 1989) which recognize similar
DNA target sequences (reviewed by Levine and Manley 1989). It has
therefore been suggested that these proteins compete with positiveìy
acting factors for target sites to mediate their repressor function.
0ther studies have suggested that evenskipped may act as a repressor
by d'irectìy interfering with the activities of general transcription
factors, such as TFIID, which form the basaì transcription complex
(Ohkuma et ol. 1990), rather than preventing an activator protein
fron bind'ing to the same DNA sequence element. In vitro transcription
of the uttrobithorøx gene is similarly inhibited by evenskipped and
it has been demonstrated that this repression is dependent on the
interaction of evenskipped with specific sequences downstream of the
uttrobithorox start point of transcription. Furthermore, these sites
can also mediate repression by evenskìpped when pìaced upstream of a
heterologous promoter (giggin and Tiian 1989). Evidence that
repression by evenskipped is mediated by interference with the
transcriptjon compìex, rather than by the competitive binding of
transcription factors, comes from the finding that the binding of the

10.
zerknul lt homeobox protein to these sites does not alter
evenskipped's repression of ultrobíthorox transcription (Aiggin and
Tj'ian 1989). Domajns impìicated ìn repressor activity by some of the
D. nelanogoster homeobox proteins are discussed further in Chapter 5.
In Neurosporo crosso the expression of the structuraì genes
involved in the utilization of nitrogen sources is regulated by the
positiveìy acting nít-2 gene (Fu and Marzluf 1987) as welì as a
negatively acting gene nnr (Premakumar et ol. 1980, Dunn-Coleman et
ø1. 1981). The nít-Z gene encodes a protein containing a zinc finger
motif and is presumed to function as a DNA binding factor which
activates transcription of the structural genes invoìved jn nitrogen
assimilation (Fu and Marzluf 1990). Mutatjons in the nmr gene lead to
the constitutive expression of nitrogen related structural genes
(Dunn-Coìeman eú ø2. 1981) and therefore probab'ly acts as a repressor
of the structural genes of the nitrogen circuit. nmr is expressed
constitutively at low levels in l1f. crosso and does not appear to
regulate nit-Z expression (Fu et øt. 1988) . nnr encodes a protein
which has regions sirnilar to the product of the AGRII gene from
Søcchøromyces cerevisioe, although unìike AgrII it does not contain
DNA binding motifs, suggesting that Nmr is not a DNA binding protein
which mediates transcriptionaì repression (Young et ol. 1990, Jarai
and Marzluf 1990). Alternative models for nrnr's role in nitrogen
repression propose that Nmr may bind to the Nit-2 protein which
interferes with the activation of transcription by Nit-2, oF the DNA
bind'ing domains of Nit-2 (Jarai and Marzluf 1990). The manner in
which the Nit-2 and Nmr proteins interact remain to be elucidated'
The utilization of quinìc ac'id as a carbon source in lV. crosso'is
control I ed by a cl uster of fi ve structural genes and two the

11.
regulatory genes qo-lF and gø-lS (Giles et ol. 1985). The genes of
this cìuster are induced by quinic acid and subiect to gìucose
repression (patel et ol. 1981). Mutations in qø-lF lead to a non-
inducabìe phenotype and are recessive (Case and Giles 1975). qo-lF
has been shown to encode an activator which is positively required
for transcri pti on of al I the qo genes i ncì udi ng i tsel f duri ng
induction (reveiwed by Geever et ol. 1989) which is consistent with
the phenotype of qo-IF- mutants. The qa-lF protein activates
transcript'ion by binding to target sites which are present upstream
of each gø gene in the cluster (Baum et ø1. 1987). Two classes of
mutations in qo-15 have been identified and lead to either a dominant
non-inducable phenotype (qo-15- ) or a recessive constitutive
phenotype (qø-15") (Case and Giles 1975). These authors implied from
the properties of the mutants that go-I.S codes for a repressor that
interferes with the activation of transcriptìon by Qa-lF and that the
repressor itself is inhibited by quìnic acid. The ana'lyses of
wiìdtype and mutant qo-15 nucleotide sequences are consistent with
the hypothesis that gø-IS encodes a repressor. The protein encoded by
qo-15 does not contain any motifs characteristic of DNA bìnding
proteins (Huiet and Giles 1986) and therefore its target is more
like'ly to be Qa-lF where it may abolish the ability of Qa-lF to bind
to its target sequences or its abiìity to activate transcription at
these promoters.
The gø cluster of fr/. crosss corresponds to the qut cluster which
encodes six genes involved in quinic acid utilization in A. nidulons
(reveiwed by Grant et ol. 1988). Regulation of the qut cluster is
mediated simiìarly by a positively acting regu'latory protein within
the cluster, qutl, and a negative'ly acting regulator encoded by the

12.
c'losely linked guúR gene (Beri et ø1. t987, Grant et ol. 1988). These
are the ,4 . nidulons equivalents of qo-1F and qo-15 respectively.
Like qo-7F, qut| is presumed to activate transcription of the qut
structural genes by binding to target sites upstream of these genes.
potential QutA target sites have been identified and await further
anaìyses (Hawkins et o1.1988). Mutations in the guúR locus result in
induced levels of expression of the structural genes, but expression
remains subject to carbon catabolite repression in guúR- strains. The
mechanisms by which Qa-lS and QutR act as repressor at the molecular
ìevel remain to be determined and their targets identified.
1 . 1 .4 The requl ati on of svnthesis and activity of transcriPtion
factors:
In order for specific gene regulation mediated by transcription
factors to occur, the factors themselves must respond to specific
environmental stimul i. A number of mechanisms exist which may
regul ate the speci fic activiti es of transcri ption factors ' The
expression of genes encoding transcription factors may be regulated
themselves so that the synthesis of the encoded proteins on'ly occurs
under conditions for which they are required or in specific cell
types. This is very conmon, for example, in cell differentiation in
higher eukaryotes (reviewed by Mitchell and Tiian 1989). The
synthesis of eukaryotic transcription factors is often reguìated
post-transcriptionally. For exanp'le, the synthesis of Gcn4 which
activates genes encoding enzymes for the biosynthesis of amino acids
in yeast cells occurs in response to amino acid starvation. Synthesis
of Gcn4 is mediated via increased translation of its mRNA. Regulation
is made possible by the inhibition of translation initiation at AUG

13.
codons upstream from the correct initiation site, 'leading to an
increase in translation from the correct start codon ìn response to
amjno acid starvation (Fink 1986).
Other transcription factors may be expressed or synthesized
constitut'ively in cells and may be activated/inactivated at the post-
translational level by tigand binding (for example, the modification
of the yeast AceI protein by copper - Furst eú ø1. 1988) , protein
modi fications vi a gìycosy] ation (Jackson and Ti i an 1988) or
phosphorylation (Sorger and Pelham 1988) or by a mechanism which
disrupts protein-protein interactions such as that seen for the Gal80
and Gal4 reguìatory proteins in response to gaìactose (see section
1.3.1) .
1.2 CARBON CATABOLITE REPRESSION IN fscheríchío coliz
The most extensiveìy studied example of carbon catabol ite
repression in prokaryotic organisms is that of the regulation of the
lactose operon in Escheríchio colí. A great deal is now known about
how the regulatory molecules involved in the utilization of lactose
as a carbon source act at the molecular leveì to actjvate and repress
the expression of the ìactose operon (reviewed by Busby 1986).
The I actose operon i s i nducibl e, and subiect to carbon
cataboìite repression. The operon is negatively regulated by the
product of the ZøcI gene, the Lac repressor protein. In the absence
of an inducer the Lac repressor protein binds to one major and two
auxilìary operator.sites in the Zøc promoter (Reznikoff et ol' L974,
Gilbert et oL. 1976, Fried and Crothers 1981) which hinders the
binding of RNA polymerase and therefore the initi ation of

14.
transciption, resulting in repression of the Zøc operon. Binding to
one of the minor operator sites may aìso prevent the binding of the
cycì ic-3',5' - adenosine monophosphate (cAMP) receptor protein (CRP)'
which is posìtively required for the initiation of transcription of
the lactose operon (Qehler et ø1. 1990). However, when Lac repressor
protein is complexed with an inducer, such as lactose or allolactose,
it can no longer bind to the major operator site to prevent
transcription and derepressed levels of transcription occur.
The expression of the lactose operon is also subject to positive
regulation via CRP, which also regulates the expression of other
operons which are glucose sensitìve and involved in the utilization
of alternative carbon sources. Regulation by carbon catabol ite
repression in E. col¿ acts at the level of transcription, resulting
in decreased synthesi s of loc ¡RNA (Varmus 1970a and 1970b). The
operons which are subject to reguìation by CRP have been co'llectiveìy
named the cAMP - CRP regulon (Magasanik and Neidhardt 1987). Ïfhen E.
coli celìs are grown in the presence of lactose as the carbon source
the intracellular concentration of cAMP rises rapidly and upon the
addition of g'lucose to the mediu¡n this concentration decreases
inrmediateìy (reviewd by Ullmann and Danchin 1983). If the level of
cAMP within ceììs is high, cAMP binds to CRP. This cAMP - CRP complex
assumes a new conformation, which allows it to bind to specific DNA
sequences in or near catabolite sensitive promoters including the
promoter region of the lactose operon (reviewed by de Crornbugghe et
qt.1984). Such binding allows the initiation of transcription of the
lactose operon. During growth of E. coli in rnedium containing glucose
the intracellular level of cAMP drops and cAMP - CRP nediated
transciptìon is suppressed. The CRP molecule is composed of two

15.
identical subunits (Anderson et ol. 1971, Aiba et ol. 1982, Cossart
and Gicque'l-Sanzey 1982) enabling two molecules of cAMP to bind to
this dimeric protein, one molecule per subunit (l{eber et ol' 1982)'
Each CRP subunit is composed of two structural dornains. The C00H
terminal domain contains a DNA binding helix-turn-helix motif and the
NHr-terminal domain contains a single cAMP binding site and a subunit
interaction site (Krakow and Pastan Lg73' Eilen et ol. L978, Aiba and
Krakow 1981, McKay and Steitz 1981). Chemical protection experiments
with the loc promoter indicate that the protein has its major
contacts in two successive major grooves in the target DNA bìnding
site (Simpson 1980). The amino acid residues of CRP which are
directìy involved in making sequence specific contacts with this DNA
have been identified (Ebright et øt. 1984). Comparisons of a number
of cAMp - CRp binding sites has shown that these sites span 22bp with
the consensus sequence being 5' aaaTGTGAtctagaTCACAttt 3', containing
an inverted repeat of an llbp eìenent where the upper case letters
represent the highìy conserved nucleotides (Jayaraman et ol' 1989) '
The distance between the start point of transcription and the CRP
binding site within CRP responsive promoters has been shown not to
be critical and therefore there is probably not direct contact
between the cAMP - CRP compìex and the RNA po'lynerase molecule in
order for the initiation of transcription (Ullmann and Danchin 1983).
when cAMP - CRP binds to its target site, the DNA sequences flanking
thìs site wrap around the CRP which stabilizes the binding (Unger' eÚ
oL 1983) and probably causes the DNA to bend in the E. coli loc
promoter (l,rlu and Crothers 1984). The wildtype løc promoter (P1) is an
inefficient promoter in the absence of CRP and cAMP. A second loc
promoter P2 has been ìdentified in the Zøc operon reguìatory region
which leads to the initiation of transcription at low frequency 22bp

16.
upstream from the major point of transcription (Hawley et ø1. L982,
Reznikoff et ol. 1982). It has therefore been suggested that RNA
polymerase I has a greater affinity for P2 and other promoters in the
absence of cAMP - CRP. However, in the presence of cAMP - CRP, the
binding of thìs complex to Pl inhibits transcription from the less
efficient promoter P2 by bìocking RNA polymerase binding. Under these
conditions Pl no longer competes for RNA polymerase binding and
transcription is initiated at high frequency from the maior start
point (Hawìey et ø1. 1932). Therefore, cAMP - CRP activates Lac
transcription by both increasing the binding rate of RNA polymerase
and its affinity to Pl, and by excluding competing RNA polymerase
binding sites.
Adenylate cyclase has been shown to be the sensor protein which
ìs activated/inactivated in response to whether the carbon source is
gìucose or a poor one and converts ATP to cAMP accordingly.
Adeny'late cyclase is activated/inactivated by a process involving
phosphorylation - dephosphorylation of the sugar phosphotransferase
system, aìthough the precise mechanism is not known (Rosenan and
Meadow 1990).
1.3 CARBON CATABOLITE REPRESSION IN Søcchoronvces ¡
Studies of carbon catabolite repression in simple eukaryotes
have mainly concentrated on the regulation of carbohydrate metabolism
in the yeast Socchoronyces cerevisioe. In the presence of glucose the
synthesis of many enzymes is repressed, particularly those involved
in g'luconeogenesis, the tricarboxylic acid cycle, the glyoxylate
cyc'le and the catabolisn of exogenously supplied sugars such as

17.
,l
Ë\Ë
,f
I
I
i'
maltose, sucrose and galactose (reviewed by t'lills 1990). There is a
growing amount of genetical and biochemical evidence that the role of
cAMp in carbon catabolite repression in S. cerevísíoe does not have
the same direct role as that found in E. coli (Matsurnoto et ol.
1983a, Eraso and Gancedo 1984). It has been shown that intracellular
levels of cAMP increase transiently in response to glucose in the
growth medium. This puìse of cAMP may be sufficient to start a
cascade effect, bringing about the activation/inactjvation of cAMP
dependent protein kinases, which in turn leads to either repression
or synthesis of enzymes involved in carbon metabolism (Beullens et
o1.1988). A príori, there is no reason to believe that mechanisms of
carbon catabolite repress'ion are conserved between prokaryotes and
eukaryotes.
Different strains of .S. cerevísioe are extremely specialized in
their ability to metaboìize both fermentable and non-fermentable
substrates. This special ization has resulted from the strong
seìection for industrial strains capabìe of metabolizing specific
carbon sources efficiently for the production of various metabolites
such as ethanol. In 5. cerevísíøe, families of genes are involved in
the utilization of various carbon sources. Therefore, different
strains may have fixed allelic differences and gene family loci may
vary between strains (reviewed by Carlson 1986). Not surprisingly,
there are many conflicting reports on the abiìity of various strains
to utilize specific carbon sources and on the specific regulatory
mechanisms involved in the netabolism of such carbon sources.
A ì arge number of mutati ons have been i denti fied in S'
cerevisíoe which lead to an altered response to carbon catabolite
repression. Some of these are allelic despite showing quite different
r

:
18
ill\l;
,i
phenotypic effects (revìewed by Gancedo and Gancedo 1986). l.lith
studies now focusjng on the study of these genes at the molecular
level, the complexit'ies of the reguìation of structural genes by a
number of reguìatory systems are beginning to emerge. The regu'latory
systems can be positìve or negative (reviewed by Guarente 1984) and
encompass a number of specific and global regulatory molecules
effecting a response in a hierarchical manner. The study of carbon
catabolite repression in yeast is further complicated by the fìnding
that the products of some genes are required for normal repression in
the presence of glucose while the products of other genes are
required for the derepression process (reviewed by Gancedo and
Gancedo 1986) . Another compl i cati ng factor i s that whi I e the
synthesis of some enzymes are subiect to carbon catabol ite
repression, these enzymes may also be readily inactivated by the
addition of g'lucose, that is, subject to catabolite inactivation
(reviewed by Holzer 1976).
1.3.1 Requlation of the oalactose requ ìon of S. cerevísioez
The reguìation of the structural genes involved in the
utjìization of galactose as a carbon source has been extensive'ly
studied in S. cerevís¿oe and provides a good example of control by
multiple regul atory factors.
The structural genes GALI, GALT and GALIO, which are requìred
for the utilization of galactose, are subject to galactose regulon
specific induction by both positively and negatively acting
regulatory molecules (revìewed by Johnston 1987). Posit'ive regulation
is effected by the product of the 6,4[4 gene. The Gal4 protein is a¡
ï
i

tl
19.
ß¡
DNA binding prote'in which binds in a cooperative manner to four
upstream activating sequences (UAS) of 17bp in 'length, upstream from
the structural genes (Bram and Kornberg 1985, Gininger et ol. 1985'
Bram eú ol. 1986, Tajima et ø1. 1986) and activates transcription
from these promoters. Gal80 is a negatìve regulator of gal-regulon
transcriptìon. 6Á¿80 transcription is subject to induction by the
presence of gaìactose although the mechanism is not understood. t'lhen
cells are grown in the absence of inducer the Gal80 protein complexes
with Gal4, preventing it from activating transcription of the
structura'l genes. However in the presence of galactose, either
gaìactose itself or a metabolite of galactose, causes this complex to
disassociate freeing Gal4 which can then activate transcription
(reviewed by Oshima 1982, reviewed by Wills 1990). The association
between Gal4 and Gal80 probably occurs while Gal4 is bound to its
target sites, since it has been found to be bound to UAS's in both
induced and uninduced growth conditions (Giniger et ol. 1985' Lohr
and Hopper 1985).
Expression of the GAI genes required for the metaboìism of
gaìactose in S. cerevisioe is stringently regulated by the carbon
source available to the cells (reviewed by Johnston 1987). When
gl ucose i s present Gal 4 di sassoci ates from i ts UAS' s and the
gaìactose regulon is repressed (Matsumoto et ø1. 1983, Selleck and
Majors 1987). However, the mechanisn by which growth on glucose
causes repression of the 6,4t and other genes is not well understood.
A nunber of genes have been identified by mutational analysis to be
involved in carbon catabolite repression of the gaìactose regu'lon.
Mutations in HEXT, encoding hexokinase PII which catalyzes the
phosphorylation of glucose, ìead to the insensitivity of the GAL, SUC
rI
j
ì

20.
,IM'q
I
(sucrose utilization) and MAL (maltose utiìization) gene families to
glucose repression (Zirnmermann and Scheel L977, Entian 1980' Ma and
Botstein 1986). Such pleiotropic effects suggest that HEXZ has a
gìobal regu'latory role in gìucose repression. Mutations in the genes
REGL and 6RR1 also lead to the loss of carbon catabolite repression
of the galactose regulon (Matsumoto et ol. 1983, Baiìey and Woodword
1984, Neigeborn and Carlson 1987) . GAL82 and 6,4[83 have an undefined
role in mediating repression and mutations in these genes lead to the
loss of carbon catabolite repression of the 6,4L genes specificalìy
(Matsumoto et al. 1981, Matsumoto et o1.1983). Along with the genes
required for normal repression, six S/VF genes have been found to be
required for the derepression of genes subject to carbon catabolite
repression (Carlson et ol. 1981, Neigeborn and Carìson 1984, Celenza
and Carlson 1986). SÍl/Fl encodes a protein kinase, further suggesting
that protein phosphorylation has a major roìe in carbon catabolite
repression in .S. cerevisíoe (Ceìenza and Carlson 1986)'
Molecular analysis of the G,4t1 promoter region has identified
two elements which are involved in carbon catabolite repression of
6,411 transcription (Flick and Johnston 1990). The first element UASG,
inhibits the activation of transciption of GAL| by Gal4. The second
eìement URS, of which there are probably multiple sequences, mediates
glucose repression of G,4L1 transcription which is independent of
Gal4. GAL83, REGI, 6RR1, SS/tl6 ønd SNF1 are required for repression at
both of these elements, whì|e GAL82 and HXK2 are required for the
glucose repression independent of GAL4. Therefore, glucose repression
of GALI appears to be conferred by two independent pathways which
respond to growth on glucose (Flick and Johnston 1990). A third
regulatory circuit may involve the competitive interaction between
,¡rl
;(
III
I
!I

21.
the effectors glucose and galactose on the Gal80 protein in the
cytopìasrn or by a mechanism which lowers the intracel lular
concentration of ga'l-regulon inducer vja the inhibition of gal-
permease (Matern and Hoìzer 1977). These regulatory circuits have
been incorporated into a model for carbon catabolite repression of
the gaìactose regulon by Matsumoto et ø1. (19S3), but the details of
the mechanisms invoìved renain unclear.
The Lacg protein, which positiveìy regulates the galactose-
lactose reguìon of Kluyveromyces lqctís, can partially substitute for
Gal4 function in 5. cerevísioe (|.|ray et ol. 1987) and vice versa
(Riley et ol. 1987). The proteins have three homologous domains, one
of which is the DNA binding domain, and have similar target sequences
(Wray et ol. 1987). As discussed above, the gal-reguìon of S.
cerevísíoe is subject to glucose repression, however the gal-1ac-
regulon of K. Ioctis is not subject to such severe glucose repression
(Dickson and Markin 1980). Interestingly, compìementation of lsc9
with G,4[4 leads to a glucose repressible gal-lac regulon in K. løctis
(Riìey et ol. 1987) and compìementation of a gol4 defective strain
with Locg leads to glucose repression of the gal-reguìon in S.
cerevís¿oe which is ìess severe than in strains containing GAL4 (Wray
et al. 1987). This suggests that Lacg function may have sone response
to the presence of glucose not previously detected. It has recently
been confirmed that gtucose repression is mediated by Lac9 in strains
having [øc genes subject to glucose repression and that this activity
is separate from its ability to activate transcriptìon of the gal-lac
reguìon of K. loctis (Breunig 1989). Genetic evidence suggested that
an analogous gene to the S. cerevisioe GAL80 gene functions to
negatively reguìate the gal-lac-regulon of K. loctis (designatedI
ì
I

22
LACLO) (Dickson et ol. 1990). It has subsequently been demonstrated
that Lacg can interact with Gal80 (Salmeron et ol. 1989). Molecular
and mutational analyses of a glucose sensitive [AC9 al]ele show that
Lacg is a limiting facfor for ß-galactosidase gene expression and
that a negative reguìatory protein like Gal80, ilôY interact with Lac9
in the presence of gìucose, Since moderate overproduction of this
Lacg protein was found to be sufficient to overcome inhibitory
effects of gìucose (Kuger et ol. 1990).
1.3.2 Sucrose utilization in S. cerevisisez
The SUC2 gene frorn S. cerevisioe encodes a secreted invertase
which is responsible for the extracellular hydrolysis of sucrose
(Carìson and Botstein 1982). The SUC2 gene is particularly useful for
studying carbon catabol ite repression since this is the on'ly
reguìatory mechanism controlling SIICZ expression. SUCL expression is
not subject to regulation by induction by the substrate as is the
case with most of the structural genes involved in the utiìization of
alternative carbon sources (Carlson and Bolstein 1982). However, the
regulation of SIJC2 by carbon catabolite repression remains a complex
system. Carìson (1987) has reviewed the genes which affect glucose
repression of SuC2, of which at least twelve have been identified.
These include SÍr/FI-$ and SflF7 , REGI, CIDI, 55/r/6, TUPL, (reviewed by
Carlson 1987) and RGR1 (Sakai et ol. 1990). Mutations in all of these
genes can aìso lead to altered reguìation of other genes subject to
carbon cataboì ite repression.
Analysis of the SUC2 promoter has identified a region which
confers glucose-repressibìe expression of the St/C2 gene (Sarokin and

23.
Carlson 1984) and mutations in the trans-acting genes S,VF1-5 and SilF7
partially or completely block derepression of SttCZ gene expression
(Carìson et ø2. 1981, Neighborn and Carlson 1984). Further analysis
of SNFZ and SilFS at the molecular level using gene disruptions
suggested that these genes are required for high level expression of
SIJCI, but are not directly involved in regulation (Abrams et ol'
1986). Although S/r/F$ was identified as a gene required for SucZ
expression (Neigeborn an Carlson 1984), further studies suggest that
S/VF5 has a more general role in transcription rather than being
restricted to gìucose repression. For example, SiVFS is also required
for derepression of ac'id phosphatase during phosphate starvation
(Abrams et ol. 1986) and mutations in 5/VF5 aìso lead to increase
express'ion of protease B in stationary phase cells (Moehle and Jones
1990). A role for SilFS in transc¡iption is also supported by the
find'ing that. mutations in the essential gene SPf6 affect
transcription and are abl e to restore regul ati on of i nvertase
expression in snl5 mutations (Neigeborn eú ol. 1986, reviewed by
Laurent et ø1. 1990). S/r/FS has been cloned and sequenced (Laurent eÚ
sl. 1990) and found to encode a g'lutamine and proline rich protein
which has trans-activator function without the capacity to bind to
DNA itself. Other associated proteins such as Snf2 and Snf6 may be
required for the activation function of Snf5. Snf2, Snf5 and Snf6 are
proposed by Laurent et ol. (1990) to form a heteromeric compìex where
Snfs provides a transcriptioal activation domain and Snf2 and Snf6
may provide DNA binding activity. No evidence has yet been reported
that Snf2 or Snf6 are capable of binding upstream of SUCZ.
The S,VF3 gene encodes a high affinity g'lucose transporter
homologous to the mannnalian protein (Celenza et ol. 1988) and exerts

24.
its effect early in the regu'latory circuit Interestingly, disruption
of sl/F3 reduces growth on sucrose and raffinose but has no effect on
SUCZ expression (Nejgeborn et ol. 1986b), while S,VF3 missense
mutations lead to altered regulation of SIJCZ expression (Neigeborn
and Carlson 1984). The role of S,VF3 in carbon catabolite repressìon
js further supported by the fact that there is some evidence that
hìgh affinity glucose transport in S. cerevisiøe is associated with
hexokinase PII, which is probably one of the regulatory proteins
involved in glucose repress'ion (Entian and Frolich 1984). Genetic
studies suggest that SilFl (which encodes a serine/threonine protein
kinase) and silF4 are functionally related genes. Ljke sivFl, S/vF4 is
aìso required for the expression of many gìucose repressible genes'
Both s/vFl and s/vF4 act at the transcri pti onal ì evel of sucz
expression (Carìson and Botstein 1982). More recently Celenza et ol.
(1989) have provided further genetic and biochernicaì evidence which
suggest that snf4 is a positive effector of the protein kinase
encoded by SÍtlFI and that the two proteins are probably phys'ical'ly
associated. Celenza et ol. (1989) propose that either Snf4
stimulates Snfl via direct protein-protein interactions or that Snf4
inhibits a negative effector of Snfl.
Mutations in 55/r/6 and TIIPI can suppress the derepressed
phenotype of SIr/FI mutant strains. It has been suggested, from
studying the epistatic relationships between different classes of
mutants, that sst'/6 and ruPl have direct negative regulatory effects
on carbon cataboìite repression. ssn6 mutations lead to high level,
gìucose insensitive expressjon of SIIC2 and other glucose repressìble
genes (schultz and carìson 1987). SSw6 encodes a nuclear
phosphoprotein which contains ìong tracts of poly(glutamine/atanìne)

25
residues and by ana'logy to other proteins containing these tracts,
Ssn6 is thought to be involved in transcriptiona'l repressìon. The
protein encoded by TUPI also contains polygìutamine tracts. No DNA
binding activity has been identified for Ssn6 or Tupl and therefore
it is possib'le that these proteins associate with or affect the
activity of DNA bi ndi ng protei ns, leading to transcriptional
repress i on .
A further gene, MIGI, is involved in glucose repression of SUC2
(Nehl in and Ronne 1990). Qverexpression of MIGI inhibits SUCZ
expression whereas a MIGI gene disruption leads to altered gìucose
repression of SI\C2. MIGI encodes a protein containing a CrH, zinc-
finger motif and has been found to bind to specific target sites
upstream of SUCT. Migl is therefore believed to be a DNA binding
trans-repressor of SI\C\, however the molecuìar ana'lysis of MIGI
suggests that it may also have an activator function which has not
yet been identified (Nehìin and Ronne 1990).
It has been proposed that other genes such as CIDI, REGI and
HXKZ are probably involved in the sensory processes of carbon
cataboìite repression and lead to an appropriate response by the cell
to the carbon source available for utilization. Mutations in RGRI
lead, to the expression of SUC2 resistant to glucose repression' and
to increased expression under glucose derepressing conditions (Sakai
et ol. 1988). Mo'lecuìar anaìysis of RGR! has shown that it is an
essential gene and affects SUCT expression possibly via an
interaction with the pathway which regulates intercellular levels of
cAMP (Sakai et o1.1990).
The regulation of the genes encoding enzymes necessary for the

26
utilization of various carbon sources in S. cerevisioe is via a
complex series of reguìatory genes which affect the induction and/or
their regulation by carbon cataboìite repression. Although nany genes
involved in carbon catabolite repression have been identified by
nutation, little is known of the proteins they code for and how these
molecules interact and function at the molecular ìevel.
1.4 GLOBAL GENE REGULATI0N IN A. nidulonsz
The most widely studied and best characterized mechanism of
g'lobal gene reguì ati on i n /. nidulons and /V. crossø i s that of
nitrogen metabolite repression. In these fungi the synthesis of a
large nunber of enzymes involved in the utilization of alternative
nitrogen sources are repressed by armonium and gìutamine (Arst and
Cove 1973, reviewed by Marzluf 1981, reviewed by }rliame et ø1. 1985).
The positively acting regulatory gene øreA mediates nitrogen
metabolite repression in A. nidulons. The oreA gene product is
thought to act at the level of transcription and is required for the
expression of structura'l genes subject to nitrogen metabol ite
repression. In wiìdtype A. nídulons gìutamine and amnonium prevent
the expression and/or activity of the oreA gene product (reviewed by
Arst and Scazzocchio 1985, reviewed by Wiame et ol. 1985). A large
number of oreA mutant alle'les, with diverse phenotypic effects, have
been identified and characterized. Loss of function mutations are the
most cornmon alleles and lead to an inability to utilize nitrogen
sources other than an¡monium or glutamine and to repressed levels of
enzymes subject to controt by øreA (reviewed by Arst and Cove 1973'
reviewed by Wiame eú ø2. 1985). Sone of these mutants have been found

27.
to be the result of translocations which interrupt the reading frame
while another was found to be a chain termination mutant (Kudla et
o1.1990). gther rarer alleles, such as the oreA-102 mutation, result
in elevated levels of some enzymes subject to nitrogen metabolite
repression (Hynes and Pateman 1970, Hynes 1975a, Arst 1977) in
addition to repressed levels for other enzyme activities regardless
of the presence of arnmonium or glutamine (Arst and Cove 1973' Arst
and Scazzocchio 1975, Hynes 1975a). This phenotype suggests that
receptor sites for the øreA gene product may differ in structure and
therefore affinity for the oreA protein, allowing it to activate øreA
regulated genes with different efficiencies (reviewed by lr{iane et ol.
1985) . ore| has been cloned and sequenced (Caddick et ol. 1986, Kudla
et ol. 1990) and the derived amino acid sequence contains a putative
DNA binding zinc-finger of the Cys-Xr-Cys-Xrr-Cys-Xr-Cys type' The
sequence is rich in the S(T)PXX motif which is characteristic for
DNA binding proteins (Suzuki 1989) . øreA also has a highly acidic
region which may be similar in structure to regions of regulatory
prote'ins having a transcriptional activation function (Giniger and
Ptashne L987, Hope eú ø1. 1988, Kudta et oI. 1990). Sequence ana'lysis
of a number of sre| mutant atleles has identified the regions of the
putative protein that are essential for oreA function and
specificity. The ore|-102 mutation was found to be a single amino
acid change in a conserved residue of the ore[ zinc finger,
demonstrating that particular residues within the loop structure are
probably invoìved in the specific recognition of ore[ target sites
and its affinity to these sites (Kudl a et ol. 1990).
The corresponding gene to qre| in fl. crosso is nit-Z (Reinert
and Marzluf 1975, Coddington 1976, Dunn-Coleman eú ø2. 1979). Like

28
orel, nit-Z is one of the major pos'itively acting regulatory genes in
,V. crosso which regulates the expression of sructural genes in
response to the nitrogen source available for utilization (Marzluf
1981, Fu and Marzluf 1987) . nit-2 was characterized by 'loss of
function mutations and although derepressed aìleles have not been
isoìated (Marzluf 1981), there seem to be functional similarities
from the study of mutants between nit-2 and oreA. The nit-Z gene
complements orel- mutants of A. nidulons and turn on the expression
of nirate reductase, acetamidase and other enzymes in A. nidulons
(Davis and Hynes 1987). This demonstrates that the activation
function and DNA specificity of the nít-Z and oreA gene products are
quite similar and that the upstream recognition elements of the genes
involved in nitrogen assirnilation must be similar in these two fungi.
'Íhe nit-2 gene has been sequenced and encodes a protein predicted to
contain a single zinc finger which is similar to that predicted for
the oreA protein (Fu and Marzluf 1990a). Furthennore' gel
retardation experiments and DNA footprinting studies have
demonstrated that the Nit2 protein binds in a sequence specific
manner to at least three sites in the 5' region of the N. crosss
nitrate reductase gene and to reìated sites in the upstrean promoter
regions of the nitrate and nitrite reductase genes of A. nidulons (Fu
and Marzluf 1990b). However, the mechanisn of nitrogen metabolite
repression in these fungi may not be conpleteìy alike.
nnr has been identified to be another major gene involved in
nitrogen metabolite repression in /t/. crssso which has a negative
function and is expressed constitutively. ln nnr mutants a number of
enzymes involved in nitrogen assimilation are expressed
constitutively (Premakumat et sl. 1980, Dunn-Coìeman eú oI. 1981)'

29.
nnr has been cloned (Fu et ol. 1988) and sequence ananlysis of nmr
has not revealed any DNA binding motifs in the putative protein'
However, the predicted Nmr protein shows sequence simiìarity to the
negatively acting reguìatory protein ArgrII involved in arginine
synthesis repression ìn yeast (Young et ol. 1990). It has been
suggested that while Á. nidulons does not appear to have a gene
equivalent to nnr, øreA may fulfill the functions of both the 'V'
crosso genes nit-Z and nnr. This is supported by the fact that some
orel alleles have constitutive expression of nitrogen assimilation
enzymes while no nit-Z mutants lead to derepression and nnr mutant
alleles show the derepressed phenotype (Fu et øl' 1988)'
0ther wide domain regulatory genes have been identified in A'
nidulsns. These include suAneth and potc| which appear to be
negative]y acting regulatory genes involved in sulphur repression,
and the regulation of enzymes involved in the catabolism of phosphate
sources respectively. Qther genes, such as pøcc, psl|, polB, polC'
polE and pølF have been shown to be involved in the regulation of
cel lular activities in response to changes in pH (Arst and
Scazzocchio 1985, Caddick et ø1. 1986a, 1986b) . These global
mechanisms of gene regulation in A. nidulons are less well
characterized and the roles that the products of these genes play
remain obscure. Although more is known about nitrogen metabolite and
carbon catabolite repression (discussed in the following section) in
A. nídulsns there is limited understanding of how the regulatory
genes or their products themselves are activated and inactivated in
response to the carbon and nitrogen sources avai lable for
utìlization. Furthermore, the expression of some genes involved in
the utilization of compounds as both nitrogen and carbon sources such

30.
as acetamide are subject to both nitrogen metabolite repression and
carbon catabolite repression and reìief of either of these two
repression mechanisms leads to derepressed expression of ømdS (Hynes
1970). The mechanisms by which these two forms of repression interact
wìth each other at the molecular level are yet to be understood'
1.5 CARBON CATABO LITE REPRESSI0N IN Á. nidulonsz
When ,4 . nidulons is grown in the presence of a source of carbon
catabolite repression such as glucose or sucrose' the synthesis of a
range of enzymes involved in the utilization of alternative carbon
sources is decreased. Reduced activity has been demonstrated for a
'large nunber of enzynes under repressing conditions includìng:
alcohol dehydrogenase (Page and cove 1972), acetamidase (Hynes 1970'
Hynes and Pateman 1970a and 1970b), pro'line permease and proline
oxidase (Arst and MacDonald 1975, Bailey and Arst 1975), NAD-Iinked
glutamate dehydrogenase (Kinghorn and Pateman 1973, Bailey and Arst
Ig75, Hynes lg74), intracelluar and extracellular proteases (Cohen
1973), D-quinate dehydrogenase, ß -galactosidase, a -glucosidase,
acetyl-CoA synthetase and isocitrate ìyase (Hynes and Kelly 1977'
Kelly and Hynes Ig77). Further to this, it has been shown in some
systems that carbon catabolite repression in A. niduløns acts at the
level of mRNA probably by controlling transcription, in for example,
pelA (Dean and Timberlake 1989) , focT (Katz and Hynes 1989a) , olc{
and oZdA (Feìenbok et ol. 1989), ølcR (Lockington et ol. 1987) and
lomA and ZømB (Katz and Hynes 1989b).
Catabolite inactivation is the process by which there is rapid
inactivation of enzyne activity as opposed to repression of

31.
synthesis, after the addition of D-glucose or sucrose to the growth
medium. Carbon catabol ite repression and catabol ite inactivation
occur independentìy and by different mechanjsms in yeast cells
(reviewed by Holzer 1976). This independence was demonstrated by the
characterization of yeast strains in which catabolite inactivation is
defective while the response to carbon catabolite repression remained
unaltered (Entian Ig77). The presence of these two independent
processes complicates the interpretation of the mutant phenotypes
showìng altered responses to carbon metabolism and assinilation in
yeast. It has been shown that the activities of phosphoenolpyruvate
carboxy'lase, fructose-1,6-diphosphatase, isocitrate lyase, acetyl CoA
synthetase, malate synthase, NADP-mal ic enzyme, isocitrate
dehydrogenase, mal ate dehydrogenase, alcohol dehydrogenase and
acetamidase are not altered by D-glucose or sucrose in A' nidulons
(Ke'lly 1980). The absence of catabolite inactivation (at ìeast for
these enzymes) has therefore allowed for the easier interpretation of
mutants affected i n the regul ati on of carbon catabol i sm i n A '
nidulons.
As previously outlined, the role of cAMP in carbon catabolite
repression in yeast renains unclear. This is aìso the case with A'
nidulons. The exogenous addition of adenylate cyclase activators
such as gìucagon and isoproterenol or inhibitors of cAMP
phosphodiesterases such theophylline and aminophenline do not alter
the levels at which enzymes subject to carbon catabolite repression
are synthesized (Arst and Bailey 1977).

32.
1.5.1 Seìection of mutations affectinq carbon catabolite reoress i on
in A. nídulonsz
A number of methods have been used to isoìate strains of A.
nídulqns showing derepressed ìevels of various enzymes in the
presence of D-glucose and are therefore no longer subject to carbon
catabol ite repression.
Loss of function mutations in the ore|, gene which codes for a
positively acting regu'latory protein involved in ammonium repression
(Arst and Cove 1973), lead to an inabi'lity to utilize sole nitrogen
sources other than amnonium (discussed in section 1.4). These mutants
fail to derepress enzynes required for the utilization of other
nitrogen sources even in the absence of a source of nitrogen
repression. Enzymes for the utilization of compounds which can be
used as both nitrogen and carbon sources, such as acetamide and L-
proline are subject to both carbon catabolite repression and nitrogen
rnetabolite repression. The relief of either of these two nechanisms
of repression enables enzyme synthesis to occur (Hynes 1970, Arst and
MacDonaìd 1975). Therefore, oreA- mutant strains are able to utilize
acetamide and L-proìine as sole carbon and nitrogen sources (Arst and
Cove 1973) but not in the presence of D-gtucose or sucrose' due to
carbon catabolite repression of the synthesis of the enzynes required
to metabolize these compounds. Selection for growth of oreA- mutant
strains on media containing D-glucose with acetamide or L-proline as
the nitrogen sources has been used to isolate A. nidulons strains
having mutations resuìting in the synthesis of acetamidase and
enzymes for L-proline metabolism which are no longer sensìtive to
carbon catabolite repression by gìucose (Arst and Cove L973, Bailey

33
and Arst 1975, Hynes and Kelly 1977)-
Another seìection method utìlizes strains which are mutant for
pyruvate dehydrogenase activ'ity. Pyruvate dehydrogenase mutants are
unable to produce acetyì Co.A from pyruvate and therefore require an
alternative source of acetyl CoA (Romano and Kornberg 1968, 1969) '
This requirement can be supplemented by the addition and metaboìism
of acetamide or ethanol as the sole carbon source in the absence of
D-g'lucose or in the presence of derepressing carbon sources, such as
glycerol, lactose, meì ibiose or L-arabinose. The metabol ism of
acetamide or ethanol can only provide a source of acety'l CoA in the
absence of D-g1ucose, since acetamidase and alcohol dehydrogenase
synthesis are reguìated by carbon catabolite repression (gailey and
Arst 1975). Therefore, medium containing D-glucose together with
acetamide or ethanol has been used to select for pdtrA phenotypicalìy
revertant strains in which ethanol or acetamide could be utilized in
the presence of D-g1ucose, thereby identifying strains in which
a'lcohol dehydrogenase and acetamidase synthesis are no longer
sensitive to repression by D-glucose (Bailey and Arst 1975, Arst and
Bailey 1977).
0ther mutations have been identified in strains already affected
in carbon catabolite repression by selecting for the ability of
further mutations to suppress the phenotype of the parent strain' The
presence of derepressed levels of the enzymes alcohol dehydrogenase,
acetyl CoA synthetase and acetamidase in creA nutants can be scored
by their sensitivity to ally'l alcohol, fluroacetate and
fl uroacetami de i n the presence of D-gl ucose or sucrose' The
phenotypes shown by cre| alleles on these media abolishes the need to
score derepression of these enzymes in oreA:creA and pdhAzcreA double

34.
mutants, or assaying for enzyme activity. A'llyl alcohol js toxic to
A. ní.dulons when converted, via alcohol dehydrogenase to its toxic
product acrolein. cre| mutations lead to complete sensitivìty to
a'lìy'l alcohoì in the presence of D-glucose or sucrose, while wi'ldtype
strains are resistant due to the carbon cataboìite repression of
alcohoì dehydrogenase gene expression (Hynes and Keìly 1977). l'lhile
wildtype strains are sensitive, cre\ mutations lead to
hypersensitivity to fluroacetate and fluroacetamide in D-glucose or
sucrose medium. This is due to derepressed expression of the genes
coding for acetyl CoA synthetase and acetamidase which convert these
compounds to the toxic product flurocitrate (Hynes and Kelly L977).
Selection procedures have used media containing D-glucose or sucrose
together with the toxic anaìogues of acetamide, acetate and ethanol
(f'luroacetamide, fluroacetate and allyì alcohol respectively). This
enabled the identification of mutations in which the synthesis of the
enzymes involved in converting these compounds to toxic metabolites
are no ìonger derepressed in D-gìucose or sucrose media and possibly
even more sensitive to regulation by carbon catabolite repression
than wildtype strains (Keìly and Hynes 1977).
Finalìy, one other selection method resulted in the isolation of
a mutant affected in carbon catabolite repression. frP. mutants cannot
utilize D-fructose, D-sorbitol, D-mannitol or D-mannose as carbon
sources, leading to inhibited growth on these compounds in the
presence of D-glucose (reviewed by M'Cullough et ol. 1977). The
reasons for /rA mutants leading to this toxicity remain undefined.
creAd-30 was isolated as a spontaneous mutation in a /rA-1 strain
which conferred resistance to 1% D-mannitol in minimal mediun with 1%
L-arabinose as the carbon source, ôlthough the basis for this

35.
phenotype is not understood (Arst et ol. 1990).
These seìection methods have so far identified four loci
involved in carbon catabolite repression in ,4. nídulons which have
been designated cre|, cre}, creC and cre-34. cre[ has been mapped
to linkage group I (Baiìey and Arst 1975) while creB, creC and cre-34
are located on linkage group II with close linkage between creC and
cre-34 (Hynes and Keì \y 1977, Kel'ly and Hynes 1977) .
I.5.2 The phenotvpe of creA mutants:
Mutations in the creA gene have been the most cotrrnon mutations
identified as leading to an altered response to carbon catabolite
repression. Aì ì known cre| mutant al leles have a partial ly
derepressed phenotype regardless of the presence of D-glucose. That
is, they result in a failure to respond normal'ly to a source of
carbon catabolite repression. The reported creA mutant alleles are
alì recessive to the wildtype aìlele in diploid strains. So far no
crel alleles leading to the permanent repression of synthesis of
enzymes involved in the utilization of alternatjve carbon sources
have been identified even though efforts have been made to do so
using a variety of potential seìection procedures (Arst and Baiìey
t977).
cre$d-! and cre[204 have been shown to have similar phenotypic
affects and were isolated as suppressors of the øreA mutant phenotype
by their inabitity to utilize L-proline and acetanide respectively
in the presence of D-glucose (Arst and Cove 1973, Hynes and Kelly
lg77). creAd-1 leads to partial derepression of alcohol
dehydrogenase, proline oxidase and pennease' sorbitol dehyrogenase'

36
manni tol dehydrogenase, NAD-I i nked gl utamate dehydrogenase 'gal actoki nase and/or gaì actose permease and acetami dase, whi I e
remaining unaffected for the utilization of either repressing or
derepressing carbon sources (Arst and MacDonald 1975, Bailey and Arst
L975, Arst and Bailey 1977). The phenotypic effects of crePrd-l in
combination wjth a number of other mutations have been tested by Arst
and Bailey (1977). The pyc\-3 mutation leads to the loss of pyruvate
carboxy'lase activity (Skinner and Armitt 1972) and therefore strains
containing this mutation have a requirement for a source of
tricarboxyl ic acid cyc'le intermediates. In a pycA-3:creAd-1 double
mutant, cre[d-l allows pycA-3 to be at least partially supplemented
by (+)-tartrate, acetamide, ethanol, L-ornithine and L-proline in the
presence of D-glucose. This supplementation is prevented by D-gìucose
in pycA-3 singìe mutants, whi le in the double mutant carbon
catabolite repression of the enzymes converting these compounds to
TCA cycle intermediates is relieved (Bailey and Arst 1975).
Some mutants defective in sugar uptake have been shown to be
resistant to the toxic effects of L-sorbose and 2-deoxy-D-glucose
(Elorza and Arst 1971). However, cre{d-l strains remain sensitive to
these compounds and are unaffected in the uptake of D-glucose-laC
(Bailey and Arst 1975).
Phenotypic variation exists among the creA mutant alleles in
their abilities to suppress both oreA and pdhA mutant phenotypes for
growth on various compounds (Arst and Bailey 1977). cre\d-2 leads to
derepressed leveìs of proline oxidase and proline pennease while
carbon catabolite repression of acetamidase is unaffected in the
presence of a strong source of carbon catabolite repression. However,
creld-Zï results in the derepression of acetamidase but not of the

37.
enzymes for L-proline metabolism. In creAd-1 strains on the other
hand, both acetamidase and enzymes for L-proline metaboìism are
derepressed. Therefore, cre|d-I can Suppress the oreA mutant
phenotype on glucose medium containing either acetamide or L-proìine
as the sole nitrogen source. Variation is also demonstrated by the
ability of creA mutants to suppress the pdhA mutant phenotype. cre[d-
1 and cre|d-2 allow considerable utilization of ethanol by pdhA
strains in the presence of D-glucose while cre{d-ZïzpdhA double
mutants show poor suppìementatjon by ethanol in the same medium.
Therefore, creP.d-l and creAd-2 lead to greater reìief of carbon
catabolite repression of alcohol dehydrogenase than does creAd-25.
These studies demonstrate that creA mutations do not necessari'ly
affect the derepression of synthesis of the same enzymes subiect to
carbon catabolite repression, implying a non-hierachical
heterogeneity within this group of mutants (Arst and Bailey t977).
Such mutants cannot simpìy be null mutations, Since all the mutants
would be expected to have the same phenotype.
The extent to which various carbon sources are repressing or
derepressing have been scored by the abi'lity of øreA and pdhA mutants
to be suppìemented by nitrogen sources and sources of acetyl CoA
respectivety in the presence of a range of carbon sources. This has
shown that D-glucose, sucrose and D-xy'lose are strong sources of
carbon catabolite repression, while L-arabinose, lactose, melibiose,
meso-inositol and g'lycerol act as derepressing carbon sources in A.
nidulons. Intennediate between carbon sources that are repressing and
those that are derepressing, are D-mannose, D-ga'lactose, D-sorbitol,
D-fructose, D-rnannitol and ethanol (Arst and Bailey 1977).
creA mutant alleles lead to a very compact colony morphology. As

38
with the variation found for other phenotypes of creA alleles, the
affect on coìony morphology is also variable between alleles ' cre{ó-
30 has the most extreme affect and leads to a severe'ly compact
morphology (Arst et ol. 1990). creAd-2 also has an extreme affect on
colony morphology whiìe creAd-1 and crel204 strains have less affect.
creü225 on the other hand, shows littìe affect on colony morphoìogy
(Arst and Bailey 1977, Hynes and Keìly L977). Altered morpho'logy has
been reported in some yeast strains having defective carbon
catabolite repression (Denis and Maìvar 1990, Gosh eú ol. 1973,
Montencourt et ol. 1973, Sakai et ol. 1990) and also in other
organisms such as the crisp and.,¡Frost mutants of fl. crøsso whjch are
associated with altered cAMP levels within cells (Terenzi et ol'
1974, Scott 1976). Arst and Bailey (1977) have suggested that
altered regulation by carbon catabolite repression in Á. nidulons nay
lead to altered morphology due to the defective synthesis of cell
wall and membrane components such as carbohydrates, glucoproteins or
g'lycoì i pi ds.
Extracellular protease activity is subject to carbon cataboljte
repress'ion in wi ldtype ,4. nídulons and this activity can be assessed
by the clearing of casein in powdered milk in soìid medium (Cohen
1972, 1973). Therefore, cre mutants clear casein in the presence of
D-glucose or sucrose whereas wildtype strains do not (Hynes and Kelly
re77).
1.5.3 The phenotvpe of creB and creC mutants:
creBls and creC\7 were isolated and characterized by Hynes and
Keì]y (tg77) as affecting carbon catabolite repression by their

39.
üI
I
abiìity to suppress the phenotype of oreA2IT (Hynes 1975a) on Leo
sucrose medium using acetamide as the sole nitrogen source and
subsequently this suppression was shown not to be allele specific.
Mutations in creB and creC are recessjve and lead to the poor
utilization of a large number of carbon sources such as L-proìine, L-
alanine, gìuconate, D-glucuronate, L-arabinose, D-quinate, lactose,
succinate, L-tyrosine, L-glutamate, L-glutamine, L-asparagine, L-
ornithine, L-arginine, L-aspartate, D-fructose, L-rhamnose and D-
mannose. Hhile creBl5 and creCZ7 strains both show reduced growth on
these carbon sources, the creBl| phenotype being more extreme, creB
and creC mutants differ in their ability to utilize other carbon
sources. For exampìe, creBl$ leads to reduced utilization of D-
mannitol and pyruvate as sole carbon sources, while creCZ7 strains
are indistinguishable from wildtype A. nídulons for growth on these
compounds. The utilization of other carbon sources such as sucrose,
glucose, glycerol, acetate and ethanol is not affected in strains
containing creBl5 and creC27. ln creB and creC mutant strains the
enzymes found at altered leveìs in D-gìucose/sucrose grown myceìia
are closely related. Namely they are all enzymes of 2-carbon
metaboìism and include acetyl coA synthetase, acetamidase, alcohol
dedydrogenase, fructose 1-6-diphosphatase, isocitrate lyase, malate
synthase, NADP-1-isocitrate dehydrogenase and extacellular protease.
In contrast to the creA alleles, the phenotypes for the creB and crec
mutants are not restricted to derepression phenotypes. The synthesis
of some enzynes such as alcohol dehydrogenase are partial ly
derepressed in creB and creC mutants while the levels of other
enzymes are found to be reduced without their sensitivity to carbon
catabolite repression being altered as is the case with D-quinate
dehydrogenase. Extracelìular protease levels are found to be elevated
r

40
I
regardless of the presence or absence of glucose in creB and creC
strains as they are in creA mutant strains, while still other enzymes
such as o-glucosidase show an increase in the derepressed leveìs of
synthesis. creB and creC mutant alleles, like creA alleles, do not
affect the uptake or metabolism of D-gìucose itseìf but there is
evidence that they do lead to a reduced uptake of L-glutamate and L-
proline. creBl5 and creC27 also show hypersensitivjty to fluroacetate
and fluroacetamide and sensitìvity to a1 lyl a'lcohol in glucose
medium. This is also simiìar to the phenotype of creA mutant alleles
on these media (Hynes and Kel'ly L977, Kelly and Hynes 1977, Kelly
1e80).
One other locus has been identified by the cre-34 mutation as
beìng involved in carbon catabolite repression in A. níduløns. cre-34
was isolated by Kelly and Hynes (L977) as a spontaneous mutation jn a
creC¿7 strain, reversing the sensitìvity of creC27 to fluroacetamide
in glucose medium. cre-34 was mapped to within 3-6 map units of
creCL7 and is recessivetowildtype. Further to this, it also
reìieves sensitivity to fluroacetamide jn the presence of a strong
source of carbon catabol ite repression and decreases growth on
acetamide, L-pro'line and L-glutamate as nitrogen sources in creC27
and creA204 strains. cre-34 results in tighter carbon catabolite
repression by sucrose of acetyl CoA synthetase and isocitrate ìyase
as compared to wildtype strains and lower levels of these enzymes in
the presence but not absence of sucrose or glucose. cre-34 leads to
the reversal of the alìyì alcohol sensitivity in gìucose medium of
creC27 and affects utilization of L-proline, L-glutamate and milk
powder clearing in the presence but not in the absence of glucose'
Kelty and Hynes (1977') poìnt out that these properties suggest that
ft:,&
I
I{
r
I

41.
r¡ID
I
some aspect of the utilization of these compounds is subject to
greater carbon catabolite repression in cre-34 strains.
1.5.4 Propo sed roles for crel, creB, creC and cre-34 gene products:
0n the basis of the observed effects of mutations in cre[, creB
and creC and cre-34 tentative roìes for the products of these genes
have been proposed.
Bailey and Arst (1975) suggested that creA probably codes for a
negatively acting wide domain reguìatory molecule involved in carbon
catabolite repression, where a nonnal functional creA product is
required for the repression of genes involved in the utiìization of
alternative carbon sources. That creL is a wide domain regu'latory
gene is suggested by virtue of the number and range of enzymes that
are no longer subject to carbon catabolite repressjon in creA mutant
strains, and that these aìleles show phenotypic heterogeneity. The
creA gene product is proposed to be negatively acting since aìl known
mutant alleles are recessive and have the phenotype of derepression.
If these alleles are loss of function alleles then mutants leading to
permanent repression would be expected to be rare in a negatively
acting regulatory system. Aìthough dominant or semi-dominant
mutations leading to failure to derepress the synthesis of a variety
of enzymes in the absence of a source of carbon catabolite repression
wouìd be expected to occur rarely, extensive searches have not
identified any showing this phenotype (Arst and Bailey 1977).
tI
j
The creB and creC gene products have been shown to
specific to the control of enzymes for the utilization of
carbon sources. The lack of phenotypic heterogeneity among
be more
2-carbon
alleles
r

42
isolated (felly, pers. cofnm.) impìies that creB and creC have a more
specìfic role in carbon cataboìite repression than does the creA gene
product. 0n the tentative assumption that the creB and creC mutant
al'leles are loss of function alleles, then these gene products may
also act negativeìy to control the synthesis of some enzymes.
However, their role is unclear since the phenotype of creB and creC
mutants is only partial derepression together with altered leveìs of
activity found for other enzymes. Positive regulation by these gene
products is compatible with the recessive nature of their poor
utilization of some carbon sources (Kel'ly and Hynes 1977 ' Kelìy
1e80) .
Kelly (1980) proposed that the phenotype and recessive nature of
the cre-34 mutation suggests a positive role for the cre-34 gene
product in carbon cataboìite repression, probably in the presence
but not absence of a source of carbon catabolite repression. This is
supported by the fact that enzyme synthesis is not affected in the
cre-34 mutant in the absence of glucose or sucrose.
1.5.5 Molecular studies of reoulation bv carbon catabol ite rePression
I
I
l
i
L
I
ri
i n ,4. nídulons z
only a limited amount of information is available to indicate
how creA acts at the molecular level to regulate the large nunber of
genes that are subject to carbon catabolite repression. Although the
cloning and pre'liminary characterization of creL are outlined in this
thesis, detailed molecular studies of the genes controlled by cre[
are also required in order to fully understand the molecular basis
for regulation by creA. Molecular studies have begun to determine the

43
mechanism by which creA acts to control the regulation of other genes
such as those invoìved in the utilization of acetamide, ethanol and
L-pro'line as carbon sources.
The utilization of ethanol by A. nidulons is controìled by
induction via the positiveìy acting regulatory gene olct and by
glucose repression. The two structuraì genes required for ethanoì
metabolism, olc| encoding alcohol dehydrogenase and oldA encoding
aldehyde dehydrogenase are subject to positive control by the oLcR
gene (Pateman et ol. 1983, Sealy-Lewis and Lockington 1984). These
three genes have been cloned and sequenced (Lockington et ol. 1985'
Gwyne et ol. 1987, Pickett et ol. 1987, Feìenbok et ol. 1988). The
expression of ølcR is subject to carbon catabolite repression and was
found to be derepressed in the cre|d-I strain (Lockington et ol.
1937). It has been demonstrated that carbon catabolite repressìon
via cre| acts independently on øZcA and oldA. Evidence for this
comes from the finding that these genes remain subject to carbon
catabolite repression in transformants containing a construct of the
øZcR coding region under the control of the promoter of the
glyceraìdehyde-3-phosphate dehydrogenase gene which is not subject to
carbon catabol i te repress i on , and hence I eads to derepressed
expression of the øZcR protein (Felenbok et ol. 1989). Therefore, the
cre| protein independently controls the expression of both the
structural genes and the positively acting regulatory gene oZcR
invoìved in the utilization of ethanol as a carbon source. Feìenbok
(1989) points out that since these three genes are subject to control
by the creA prote'in then conmon sequences in their promoter regions
may be expected to occur which identify a creA specific target site.
No putative sites from sequence comparisons have been identified

44
(Fetenbok et oI. 1988). However, other possible sequences which may
represent crel target sites have been suggested in the promoter
regions of olc|, prnB, qutB and ømdS which are all subject to
regulation by creA. Deìetion of a 13bp sequence at position -136 in
the upsteam region of øZcR has been carried out and this construct
used to transform wildtype A. nidulqns. Initial reports of
transformants possib'ly showing derepressed levels of øZcR expression
exist (Felenbok et ø1. 1989).
The regulation of the ømdS gene encoding acetamidase in A.
nidulons has been extensive'ly studied by both classical and molecular
techniques. A large number of 5' mutations that affect øndS
expression have been sequenced and this approach has been used to
identify target sites within the omdS control regìon for sone of the
proteins whìch regulate the expression of the gene (Hynes et ol.
1988, reviewed by Davis and Hynes 1989). However, if cre[ acts
directìy on the omdS promoter region to mediate regulation by carbon
catabolite repression, its target site remains to be identified.
0'Connell (1990) has investigated the upstream sequences of
genes regu'lated by carbon catabolite repression from a number of
fiìamentous fungi for putative creA-like target sites. It was found
that a core sequence of 7bp has sequence sinilarity in the upstream
controì regions of a number of genes including oldA from A. niger,
ølcR, olcl, qcu}, ocu|, ondS, foc|, prn} and qutE from A. nidullns
and qo-Z from /V. crosso which are all reguìated by carbon catabolite
repression. The significance of this sequence remains to be confirmed
since it was also foUnd (although with generally less overall
homo'logy) to be present in the upstream regions of other genes not
subject to regulation by carbon catabolite repression. Furthennore'

45
deletion anaìysis of this sequence from the upstream region of øldA
from A. niger was shown not to affect the expression of the gene
(0'Connel I 1990) .
The mechanisms by wh'ich the products of the cre[, creB and creC
genes act at the moìecular level to reguìate the ìarge number of
genes subject to carbon catabolite repression remain to be studied
and understood. Biochemical and genetical studies of nutants with an
altered repsonse to carbon catabol ite repress'ion have suggested
functions for these genes. This information must be coup'led with the
investigation of these genes and their products at the molecular
level in order to begin to expìain the mechanism of carbon
catabolite repression in Á. nídulons.
1.6 ATMS OF THIS STUDY:
This thesis outlines the characterization of the creA gene with
a view to determining how it may be involved in carbon catabolite
repression. Previous work reported on creA suggested that it is one
of the major genes involved in carbon catabolite repression in A.
nidulqns and for this reason the study of this gene was chosen as a
starting point to an understanding of carbon catabolite repression in
A. nidulons. This study set out to isolate and clone the creA gene
from A. nídulons and to determine as far as possible, from the
characterization of the gene, how creA may act at the moìecular
level. It was of interest to determine whether the putative product
of the creA gene showed sequence similarities to negative regulators
in support of creAs negative role as suggested by geneticaì studies.
This is in contrast to most regulators of transcription in A.

46.
niduløns and other eukaryotes, which are generally positiveìy acting.
At this stage it cannot be ruled out that creA acts positively in
that it may activate other molecules that derepress carbon catabolite
repressi on, or general transcripti on factors that acti vate
expression. However, analysis of the putative creL protein outlined
in this thesis shows that it contains a domain similar to that
found in other proteins known to be required for transcriptional
repression function, further suggesting that the creA protein acts
negatively in carbon catabolite repression.

47.
CHAPTER ThJO
MATERIALS AND METHODS.
2.1 MATERIALS.
General reagents: General chemi caì s, medi a requi renents and
supplements were of 'laboratory grade and were purchased fron Aldrich
Chemical Company Inc., BDH Ltd., S'igma Chemical Company, Oxojd Ltd.
and United States Biochemical Corp. . Antibiotics 'i sopropyl th'i ogaì actos i de ( I PTG) , S-bromo-4-ch I oro-3- i ndol yl -ß-D-
galactos'ide (X-qal) and nuclease free bovine serum albumjn were
purchased from Boehringer-Mannheim GmbH. Bleomycin was a gift from M'
J. Hynes.
Nucleotides and i : Unl abel I ed deoxyri bonucl eoti des and
dideoxyribonucìeotides urere purchased from Boehringer-Mannheim GmbH.
a-¡2P-dCTP and a-szP-dATP (3000 ci/nrmole) and Y-32P-dATP (4000
Ci/nrmole) were purchased from BRESATEC Pty. Ltd.. 0ììgonucleotides
for DNA sequencing and primer extension ana'lysis were purchased from
the Department of Microbiology, University of Adelaide.
Enzymes: Restriction endonucleases, calf intestinaì phosphatase,
polynucleotide kinase, sequencing grade kìenow fragment of E. coli
DNA polymerase I and proteinase K were purchased from Boehringer-
Mannheim GmbH. Bacteriophage T4 DNA ligase and f. coZi DNA polymerase

48
I were purchased from BRESATEC Pty. Ltd.. M-MLV reverse transcriptase
was purchased from Bethesda Research Laboratorjes Ljfe Technolog'ies
Inc. Lysozyme, ß-gìucuronidase, ribonuclease A and pronase were
purchased from Sigma Chemical Company. Novozyme was purchased from
Novo Industries.
2.2 BUFFERS AND STOCK SOLUTIONS:
2.2.I 1 X Reaction buffers:
Restrictjon endonucleases: Reaction buffers for restriction enzyme
digests were used as supplied by Boehringer-Mannheim GmbH.
Ligation buffer: 10mM MgClr, 10mM DTT, lmM spermid'ine, lmM ATP, 50mM
Tris.HCl (pH 7.5).
Ca'lf intestinal phosphatase buffer: 50mM Tris.HCl (pH 9.0), lmM
MgC'lr, 0.lmM ZnC'lr, lil spermidine.
Poìynucleotide kinase buffer: 50mM Tris.HCl (pH 7.5) ' 10mM MgCl, '
5mM DTT.
Nick translation buffer: 50rnM Tris.HCl (pH 7.5), 7.SmM Mg(CH.C00)r,
4mM DTT, 100u9/ml BSA.
HIN buffer (C-tests): 10mM Tris.HCl (pH 7.4), 10mM MgClr, 50mM NaCl.
TM buffer (DNA sequencing): 10mM Tris.HCl (pH 8.0), 10mM MgCìr.

49.
Stock solutions:
1 X Denhardtsz 0.02% (w/v) ficoll, 0.02% (w/v) polyvinlyrolidone,
0.02% (w/v) BSA (Pentax Fraction V).
AspergiZlus salt solution: (per ìitre) 269 KCI , 269 MgS0o.7Hr0, 769
trace el ement so'lut'ion, Znl CHCI3 asKH2P04, 50ml Aspergillus
preservati ve.
1 X Loading buffer: 0 .042v" (w/v) bromophenol blue, 0.042% (w/u)
xylene cyanoì , 6.67% sucrose.
2 X 149 Salts: (per litre) 12g NarHP04, 69 KHrPOo, 19 NaCl , 29 NHoC'l .
SM buffer: 100mM NaCì, 10mM MgS0o .7H20, 50mM Tri s. HCI (pH 7 .5) , 0. 1%
(w/v) ge'l ati n .
1 X SSC: 0.15M NaCl, 0.15M Na.CuHuOr.2H20, pH 7.2.
1 X SSPE: 0.18M NaCl, 10mM NaH,POo, LmM EDTA, pH 7.4.
I X STE: 10mM Tris.HCl (pH 7.5), 100mM NaCl, lmM EDTA.
1 X TAE: 40mM Tris, 20mM NaCH.COO, 2mM EDTA, pH 7.8.
1 X TBE: BgmM Tris, BgmM H.BO', 2mM EDTA, pH 8.4.
lXTE 10mM Tris.HCl (pH 8.0), lmM EDTA.

50
AspergílZus trace element solution: (per litre) 40mg NarBo0r, 400m9
CuSQo, 19 FePQo, 600mg MnSQo.HrQ, 800mg NarMoQo.2H20, 89 ZnSQ4'7H20,
2n1 CHCI3 as preservative.
AspergilZus vitamin solution: (per ìitre) 40mg p-aminobenzoic acid,
50mg aneurin.HCl, lmg D-biotin, 400mg inositoì, 100m9 nicotinic acid,
200mg calcium D-pantothenate, 100mg riboflavin, 50mg pyridoxine, 2ml
CHCI3 as preservative.
2.3 MEDIA:
2.3.1 Bacterial and veast growth medìa:
L-broth: I% (*/v) NaCì, 0.5% (w/v) yeast extract, I% (w/v) tryptone,
pH 7.5. Plates were solidified with 1.5% (w/v) agarose or 0xoid Class
I agar.
M9 qlucose minimal medium: 50% 2 X M9 Salts, 10mM MgSQo, lmM caC1r,
10mM thiamine.HCì,
Class I agar).
9.2," (wf v) D-glt¡cose, 50% (vlv) {lz (w/v) 0xoid
SOC: L-broth p]us: leo (w/v) Qxoid tryptone, I-6% (w/v) yeast
extract, pH 7.5.
YEPD: 1% yeast extract (w/v) , 2e" (wfv) peptone, 2% D-glucose. Plates
were solidified with l% Oxoid Cìass III agar.
2 X YT: 0.5% (w/v) NaCl , 1% tryptone, 1.6% (wlv) yeast extract, pH
7 .5.

51.
2.3 .2 Aspergi Z Zus grguv!!_ng4ig,
Acetate medium: 2% (v/v) AspergíZlus salt soìution, 50mM NaAc, pH
6.5. Pìates were solidified with L% (w/v) 0xoid Class I agar.
Carbon f ree medi um: 2% (v lv) AspergíZ Zus sal t sol ut'ion,
Plates were solidified with I% or 2.2% (wlv) 0xoid Class
Unless other'wise indicated, carbon sources were added to
concentration of 1%.
pH 6.5.
I agar.
a final
Complete medium: I% (w/u) D-glucose, 0.2% (w/v) peptone, 0-I5% (w/u)
caesein hydrolysate, 0. I% (wlv) yeast extract, 10mM ar¡nonium (+)-
tartrate, 2% (u/v) AspergiZlus salt solution, I% (v/v) Aspergíllus
vitamin solution, Zlugfnl riboflavin, pH 6.5. Plates were solidified
with 1% or 2.2% (w/v) 0xoid Class III agar.
Nitroqen free medium (q'lucose medium): 2% (v/v) Aspergillus salt
solution, I% (w/v) D-gìucose, pH 6.5. Plates were solidified with I%
or 2.2% (w/v) Qxoid Class I agar. Unless other'wise indicated,
nitrogen sources were added to 10mM.
Protoplast medium: lM sucrose , l% (w/v) D-gìucose, 2% (v/v)
Aspergíllus salt solutjon, pH 7.0 and solidified with 0.25% or leo
(w/v) Oxoid Class I agar for overlayer and underlayer of plates
respecti ve'ly.
Supplements: Growth supplements were added as required to the
fol lowi ng concentrations :
L-arginine 0. l2mglnl

52.
D-bi oti n
nicotinic acid
p-aminobenzoic acid
pyridoxal hydrochìoride
ri bofl avi n
sodium thiosuìphate
0.01ug/ml
1.0u9/m1
50.0u9/m'l
0.5u9/ml
2.5u9/n\
0.1% (w/v)
Antibiotics: Antibiotics were added to media to the foì lowing
concentrati ons :
bl enoxane
acri fl avi n
hygromyci n
1.0u9/ml
0.001% (w/v)
600u9/ml
2.4 E. coli STRAINS AND BACTERIOPHAGE:
E. col¿ strain DHI (Hanahan 1983) was used for general plasmid
maintenance. E. coli strain JM101 was used to screen recombinant
pl asmids using pUC19 as a vector and for propagating ÀM13
bacteriophage (Yannisch-Perron et ol. 1985). Recombinant trZAP and
ÀZAPII (Short et ql. 1988) bacteriophage were propagated on E. coli
strain NM538 (Frischauff et ø1. 1983) and recombinant À9t10
bacteriophage were propagated on E. coZ¿ strain C600 (Huynh et oI.
1985) . Aspergiltus nídulons cDNA ljbrarjes in IZAP, ÀZAPII and Àgt10
were obtained from lr{.E. Timberlake (Dean and Timberlake 1989). The
titers for the libraries were determined to be 5.0 X 108, 1.8 X 108
and 2.5 X 10? respectively.

53
2.5 FUNGAL STRAINS:
A list of AspergiZlus strains used in this study and thejr
genotypes are given in Table 2.5.1. For meanings of gene symbols see
Clutterbuck (1984).
Qther funga'l species used were Penícilliun chrysogenun obtained
from S. Andrews (S.A College of Advanced Education), Neurosporo
crøssø strain T391 obtained from D. Catcheside (Fì inders
University, South Austraìia), .Socchoromyces cerevísíoe genotype a/c
leuT; uro3-s|furo3; odel obtained from P. Langridge (l,laite
Institute, South Austral ia) and strai n 228Û 9û of Melonpsoro liní
obtained from J. Tinnnis (University of Adelaide, South Austral'ia).
2.6 PLASMIDS:
A list of plasmids used in this study is shown in Table 2.6.1'.
2.7 METHODS:
Unless othen¡ise stated, methods were carried out as described
in the references cited.
2.7 .I Genetic manipu lation and qrowth testinq of Aspergíllusz
Growth testing and meiotic analyses of A. nídulons were
carried out as in Cove (1966). Diploids were hap'loidized on l%
complete medium (Hastie 1970) containing lu'l/ml of 0.0752" benlate to
select for haploids, unless othen¡ise specìfied. A. niduløns strain

54.
Table 2.5.L: Genotypes of Aspergillus strains.
STRA I N GENOTYPE REFERENCE
A. nidulonsz
wi I dtype
c26. 1 .10
MSF
J7
1070
664
sA20
sA25
creAd -30
c43
c61
c62
A. nigerz
wi ì dtype
A. oryzøe?
wi I dtype
bíAI; niiA4 Cove and Pateman (1963)
yA2 pøbøAL; orgB2 UPshall (1986)
yAL ødE20 suAødEZ}l; ocrAli
golEl; pyroA4; foc\303; sB3;
n¿cB8; riboB} Kafer (1961)
pobolI; prn L353 Sharma and Arst (1985)
bíAI cre[d-I Bailey and Arst (1975)
biAI cre[204; niíA4 Hynes and Kelly (1977)
yAI creA220; ríboB2; oreA2IT Hynes (unpublished)
yll cre[225; ribo}2; sreLl!7 Hynes (unpub'lished)
bíAl creAd-3g Arst et ol. (1990)
biAI pobøAl creA204; orgB2 see text
yAI; nicï$ pyroA4; niíA4 riboBZ see text
Acrlli gotEI; føc\303; sB3; b¿41; riboBT see text
no mutant markers known Kelly and Hynes (1985)
no mutant marker known S. Andrews (pers conrm')

55
Table 2.6.I: Plasmids used in this study.
PLASMI D INSERT VECTOR REFERENCE
pM006
p3SR2
pPL3
pAmPh366-1
pANT-1
pANCI
pANC2
pANC3
pANC4
pANC5
pANC6
pANCT
pANCS
ørgB pUCl9 UPshal I (1986)
snds PBR322 HYnes et ol' (1983)
riboB PUC19 OakleY et ol' (1987a)
Pnr pUCB Austi n et ol. (1990)
Hyg, pUCl9 Punt et ol. (1987)
ørgB, creL PUC19 see text
orgB PUC19 rr
crel (2.6kb) PUC19 rr
crel (2.3kb) pUC19 '|r
crel (2.3kb) PACYC184 rr
crel (7.5kb) PUC19 rr
crel (7.5kb) PUCBX rr
riboB + cre[
f I anki ng regi ons PUCBX rr
pUCl9 with the Bomïl and XbøI
sites deleted from the polylinker.
pUCBX

56
MSF was used as the multiply marked strain for haploidization
analyses (Kafer 1961). Mutagenesis of specific strains l{as achieved
by expos.ing conidial suspensions touv (254.$nm) l'ight, ât a
distance of 7cm for ten minutes.
2.7.2 Transformation of Aspergíllusz
The isolation, preparation and transformation of A. nídulons
protoplasts with p'lasmid DNA was by the method of Tilburn et ol.
(1983). ArgB+ transformants were selected as in Berse et ol. (1983).
Selection of AmdS+ transformants was as in Tilburn et ol. (1983)'
RiboB+ transformants as described by Oakìey et ol. (1987a),
hygromycin B resistant transformants as in Punt et ol. (1987) and
bìeornycin resistant transformants as in Austin eú ol. (1990). The
selection of CreA+ transformants is outlined in the text (Chapter 3,
section 3.1.3).
2.7.3 Isolation of nucleic acids:
Aspergillus, Neurosporø and Peniciltíun DNA was ìsolated by the
method of Hynes et ol. (1983) from freeze dried myceìia which had
been harvested from overnight ìiquid cultures grown at 37oC. Specific
size fractions of genomic DNA for the construction of libraries were
recovered by spinning partìal digests through sucrose step gradients
(Griffith, 1979) or by e]ectrophoresing digests in agarose ge'ls of
the appropriate concentration for separat'ion, followed by sqeeze-
freezing the desired region of the ge] to recover the DNA by the
method of Thuring et ø1. (1975).

57.
DNA from Socchoronyces was jsolated from cultures grown at 30oC
by the same method as that for AspergiZZøs DNA preparations.
For the isolatjon of Aspergíllus RNA, cultures were grown
overnjght in liquid glucose media at 37oC, harvested and washed with
warm carbon free mediun and then transferred to appropriate prewarmed
medium for four hours. Cultures were then harvested and washed with
cold sterile distilled water and freeze dried overnight. Total RNA
was isolated from the freeze dried myceìia as in Rienert et ol.
(1e81).
For the large scale preparation of plasmid DNA, E. coli
harbouring p'lasmids were cultured in L-broth plus S0ug/ml ampicilìin
and amplified using chìoramphenicol to a final concentration of
170ug/ml. Ceììs were harvested by centrifugation at 35009 for 10
mi nutes , washed wi th 100rnì s 1 X STE and repel I eted. Pl asmi d
preparations were then made using either of the two following
methods:
(A). Cells were resuspended in 10mls of 25% sucrose; 50mM Tris.HCl pH
8.3; 50mM NaCl; 10mM EDTA plus an extra 2.5mls of 0.SmM EDTA.
Lysozyme was added to 2ng/nl and the cells incubated at room
temperature for 30 minutes and a further 20 minutes on ice. 15mìs of
2% Triton-X; 50mM Tris.HCl pH 8.3; 10nM EDTA was added and the
precipitate peì leted for 90 minutes at 20,000 r.p.m. To the
supernatant was added 40% PEG to a final concentration of lM, lM NaCl
and lmg of heat treated ribonuclease A and incubated overnight at
4oC. DNA was pelleted at 8,5009 for 10 minutes, resuspended in 4mls 1
X TE and extracted once with chloroform:isoamyl aìchol Qazl).
Plasmid DNA was purified on CsCl equilibrium gradients by the method
of Maniatis et ø1. (1982).

58
(B). Ceìls were resuspended in 4mls 15% sucrose; 25mM Tris.HCl pH
8.0; lQmM EDTA and incubated on ice for 40 minutes after the addition
of Smgs of ìysozyme. Smls of 0.2N NaQHi lvo SDS was added and the
solution held on ice for 10 minutes followed by the addition of 5mls
of 3M NaAc pH 4.6 and incubated on ice for a further 40 minutes. The
precipitate was pelleted at 5,0009 for 15 minutes and the supernatant
incubated with 100ug heat treated ribonuclease A at 37oC for 40
minutes. This was followed by a phenol:chloroform (1:1) and one
chìoroform extraction. DNA was precipitated by the addition of 1/10
volume 3M NaAc pH 5.5 and 2.5 volumes of ethanol. The pellet u,as
resuspended in 1.6mìs of water and the pìasmid DNA was precipitated
by the addition of 0.4m1 13% PEG , incubated on ice for t hour and
peìleted at 8,5009 for 10 minutes. Plasnid DNA was resuspended in I X
TE.
Smaìì scale preparation of pìasmid DNA was performed by the
alkaline lysis method of Ish-Horowicz and Burke (1981).
Bacterial genomic DNA was isolated fron overnight cultures.
Cells were pelìeted at 35009 for 10 minutes and incubated in lOmls
10mM Tris.HCl (pH 8.0), 10mM NaCl, 10mM EDTA, 0.5% SDS, 50ug/mì
proteinase K per gram of cells for 30 minutes at 37oC. The lysed
cells were then extracted three times with buffer saturated phenol.
Two volunes of ethanoì were added to the final aqueous phase and the
precipitated DNA spooled out. The DNA was washed with 70% ethanol,
resuspended in 1 X TE, RNAase treated , extracted with phenoì and
reprecipitated with ethanol.
DNA was prepared from lambda bacteriophage by the plate lysate
method of Maniatis eú ø1. (1982).

59.
For the preparation of singìe stranded DNA for sequencing,
singìe pìaques of M13 bacteriophage u,ere propagated in 2ml cultures
of E. colí in 2 x YT medium as described by Ausubel et ol. (1987).
Cultures were centrifuged at 3,5009 for 10 minutes and 1.5mìs of the
supernatant was collected in a microcentrifuge tube. This was spun at
12,000 r.p.m for 10 minutes and lml of the supernatant was decanted
into a fresh microcentrifuge tube. 275u1 of 20% PEG 8,000; 2.5M NaCl
was added and the tubes incubated at room temperature for 15 ninutes.
Bacteriophage particles were peì leted by microcentrifugat'ion at
12,000 r.p.m for 10 minutes. The supernatant was decanted and
discarded. The tubes were spun for a further 2 ninutes and the
residuaì supernatant was removed with a drawn-out pasteur pipette.
The bacteriophage were resuspended in 200u1 of I X TE, extracted with
100u1 of tris-buffered phenol and 150u1 of the aqueous layer being
collected. This was extracted with 75ul of chloroform and 100u1 of
the aqueous ìayer was collected. M13 DNA was precipitated by the
addition of lluì of 4M LiCl and 275u1 of ethanol, incubated at -70"C
for 15 minutes and collected by microcentrifugation at 12,000 r.p.m.
Pellets were resuspended in 12ul of I X TE. For double stranded
sequenc'ing, 15ug of purified plasmid in 1 X TE was incubated for 15
minutes at 37oC with 5ul of lN Na0H, lmM EDTA. The sanple was spin
dialysed through sepharose CL-68 equilibrated in 1 X TE. 7ul of spin
dia'lysate was used per sequencing reaction.
2.7.4 General recombinant DNA methods:
DNA manipulation and modification: General methods were used as
I aboratoryindicated by the suppìiers of enzymes or as outlined in
manuals (Maniatis et ø2. 1982; Ausubeì et ø1. 1987).

60
Transformatjon of E. coli: Transformation of E. coZ¿ was performed
either by electroporation using a Biorad Gene Pulser Version 1.0 and
the methods of the supplier or by heat shocking as outlined in
Maniatis et ol. (1982). Ceìls were made competent for heat shock
transformation by the following method: 50ml cultures in late log
phase were pe'lleted f or 2 minutes at 10,000 r.p.m., resuspended jn
25mls 0.lM MgCl, and re-peìleted. The cells were resuspended in 25mls
of 50mM CaCì, and jncubated on ice lor 20 minutes. This was foìlowed
by a final sp'in down and the cells resuspended for storage in a
total of Smls of 50mM CaClr, 15% glyceroì. Cells were made competent
for transformation by eìectroporation by the foìlowing procedure:
500m1 cultures were grown to an 0.D.uoo of 0.4, pel'leted for 10
minutes at 5,0009 and resuspended on ice in 250mls of cold I0%
g1ycerol. Cells were repe'lleted and resuspended in lOmls of cold L0%
gìyceroì twice and 5mls of cold IOv" g'lyceroì once. The final pe'llet
was resuspended in 2.5mls coìd 10% glyceroì and the cells were stored
at -80"C in 100u1 aliquots. These cells were thawed on ice and
incubated with DNA for 1 minute on ice prior to eìectroporation. lml
of SOC medium was added to eìectroporated cells and incubated for 1
hour prior to plating out.
2.7 .5 Gel electrophoresis:
Agarose geì electrophoreseis of DNA was carried out in gels made
from 1 X TAE buffer and an appropriate concentration of agarose. DNA
was recovered from agarose gel s by the squeeze-freeze method of
Thuring et oI. (1975). Bacteriophage lambda DNA digested with HdndlII
was used as the moìecular weight marker.
..¡
LI
',ü
1
þ

61.
.liil
If¡
RNA was electrophoresed in L.5% (w/v) agarose, 8.0%
formaldehyde, 10mM sodium orthophosphate (pH 7.0) geìs with 10mM
sodium orthophosphate (pH 7.0) as the running buffer. 10ug or 20ug
sampìes of total RNA were prepared for electrophoresis by the
addition of two voìunes of {50% (v/v) formamide; 12% (vlv)
formaldehyde; 10mM sodìum orthophosphate (pH 7.0)) and heated for 10
minutes at 65oC.
DNA sequencing reactions were eìectrophoresed in gels containing
4-8% (w/v) po'lyacrylamide, sM urea, 1X TBE. Some sequences were also
resolved by the additjon of 25% (vlv) deionized formamide to the gel
mix. The dimensions of the sequencìng gels was 450 X 170 X 0.2rrn and
these were run at 1500 - 2000 volts.
2.7.6 32P Labellin of DNA:
DNA probes were prepared for hybridizations by nick
translation. Specifically, reactions contained 250n9 of DNA to be
labelled, 0.lrnM each of 3 dNTP's and lug/ml DNAase jn 1 X nick
translation buffer. This was jncubated at room temperature for 30
minutes, heated to 65"C for 5 minutes and placed on ice. 20uCi 0-
32PdNTP and 10 units of E. coZi DNA polymerase I were added and the
reaction carried out for 30 minutes at 16"C. Unincorporated
nucleotides were removed using Bio-Rad Bio-Gel P60 columns.
Strand specifìc probes were created by annea'ling recombinant M13
clones to a reverse sequencing primer (25mer) obtained from BRESATEC
pty. Ltd.. dATP, dGTP and dTTP were added to a final concentration of
0.5mM, together with 40uCi of cr-sz-P-dCTP and one unit of Klenow. The
reaction was extended for 30 minutes at 37oC and chased with 0.5mM
rI
;
?

62.
dNTP's for a further 15 minutes. Unincorporated nucleotides
removed us'ing Bio-Rad Bio-Gel P60 columns. These probes were
denatured prior to hybridization to RNA.
were
not
ì
ürüI
I
3
I
Molecuìar weight markers were end labelled by combining up to
5ug of DNA with 0.08mM each of three dNTP's, lQuCi cr-32PdNTP and I
unit of Klenow in 1 X low salt restriction enzyme buffer. The
reaction was incubated at room temperature for 30 minutes and
unincorporated nucleotides removed using Bio-Rad Bio-Gel P60 columns.
2.7.7 Nucleic acid blottins and hvbridization conditions:
Southern blotting, Northern blotting and slot blotting of RNA
and DNA using a Schlejcher and Schuell minifold II apparatus was to
Zeta-Probe menbrane (Bio-Rad) by the al kal i transfer methods
reconrnended by the supp'lier. Plaque lifting to nitrocellulose was
performed by the method of Benton and Davis (1977). Biodyne membrane
(Patl) was used for colony blots and carried out by the SDS/NaOH
method of Maniatis eú ø1. (1982).
Hybridization of nick translated probes to Zeta-Probe membrane
and washing of filters after hybridization was performed by the
methods recorrnended by the supplier. The sane conditions were used
for coìony blots on Biodyne except that l$ug/rnl E. coli DNA was
i ncl uded i n the prehybri di zati on sol uti on . For ni trocel I ul ose,
filters were prehybridized for 4 hours at 42"C using 50% formamide, 4
X SSPE, 10 X Denhardts, 0. l% (w/v) SDS and 3Oug/ml sonicated
denatured salnon . spenn DNA. Hybridization of denatured probes was
under the same conditions except for the addition of L0% (w/v)
dextran sulphate. Filters were washed to a final stringency of 0'1 X

63.
SSC; l.O% SDS at 65"C unless other'wise stated. Autoradiography was
carried out using intensifying screens except for the autoradiography
of quantitative slot blot analyses.
2.7.8 Quantification of DNA coov number and nRNA levels:
creA mRNA levels and DNA copy number were determined by
hybridization of slot blots of serial dilutions of DNA and total RNA
using the insert of pANC4. The quantity of DNA or RNA in each series
was standardized using the orgB gene of pM006 or the riboB gene of
pPL3 in the case of transformants. The signal on autoradiograms was
quantified using a LKB Ultroscan XL enhanced laser densitometer. The
absorbance units detected were plotted against the amount of RNA
blotted to obtain data points within a linear range for the X-ray
fi lm. The arbitrary value of absorbance units/ug for the crel.
specific probe was corrected using the other probes and values were
then normalized against samples indicated.
2.7.9 DNA sequencing:
The creA clone was subcloned into the bacteriophage vectors
M13mp18 and M13mp19 for single stranded DNA sequencing using standard
cloning techniques. Compìementarity tests (c-tests) were carried out
to determine the orientation of M13 clones by combining 5ul of two
sìngìe stranded preparations and 8ul of 10 X HIN buffer, boiling for
three minutes and leaving at roon temperature for 30 rninutes prior
to electrophoresi s. DNA sequencing of both pl asmi d and sing'l e
stranded DNA was performed by the dideoxynucleotide chain termination
method (Sanger et oL 1976) using Sequenase Version 2 kits (United
tI
J
!

64
I
States Biochemical Corp.) or by the following procedure using Klenow:
Z.5ng of Bio-Labs -40 sequencing primer or a specifjcally synthesized
oìigonucìeotide was annealed to 8ul of tempìate DNA in I X TM buffer
by heating the mix to 100"C and then cooled sìowly to room
temperature. The annealed DNA was added to a tube containing 20uCi of
o-32P-dCTP and 1 unit of Klenow. 2.5u1 of this was a'liquoted into
four tubes containing one No (Tabìe 2.7.1) solution (2.Ou.l) and 2.0u1
of the corresponding ddNTP. The dideoxynucleotides ddATP, ddCTP,
ddGTP and ddTTP were aliquoted from stock solutions of 0.3mM'
0.05mM, 0.15mM and 0.5mM respect'ive'ly. The reactions were incubated
at 37oC for 15 minutes and then 2.5u1 of chase (0.5tnM dCTP, 0.5mM
dGTP, 0.5mM dTTP, 0.5mM dATP and 0.1 units/ul K'lenow) was added to
each tube and the reactions incubated for a further 15 minutes. 4uì
of 'loading buffer (lQmg/ml bromophenol blue, lQmg/ml xylene cyanol,
lmM EDTA in deionized fonnamide) was added to stop the reactions.
Samp'les were denatured at 100"C for 10 minutes and 2ul was loaded
onto the sequencing gel. After eìectrophoresis, the ge'l was fixed in
{20*" (v/v) methanol, IO% (v/v) acetic acìd} for 20 minutes, dried and
exposed to X-Ray film overnight. Compressions and pauses in the
banding pattern were resotved by substituting dITP for dGTP in the
reactions or by incubating reactions at 50"C or by the use of
formamide in the gel mixture (see section 2.7.5).
Computer anaìysis of sequence data was carried out using Staden,
NIH programs and the Genetics Computer Group Sequence Analysis
Software Package Version 6.1 (Devereux 1984). Database comparisons
were to the Genbank, EMBL, NBRF-Nucleotide and NBRF-Protein
databases.

65.
Table 2.7.L: dNTP solutions for DNA sequencing.
Ao To Go co
0.SmM dATP
0.5m4 dTTP
0.snM dGTP
0.5rrü.1 dCTP
lXTE
4.3u1
43ul
43ul
lul
8.7u1
43ul
4.3uì
43uì
1ul
8.7u1
43ul
43ul
4.3uì
1uì
8.7u1
29ul
29ul
29ul
lul
I2u1

66.
2.7.L0 Primer extension analysis:
Primer extension anaìysis was carried out by procedure 1, for
RNA sequencing, outlined in Geliebter et ø1. (1986) except that the
ddNTP's were omitted fron the transcription buffer. A specific
oligonucleotide was end labelled with Y-32-P-dATP using
polynucìeotide kinase and annealed to RNA at a temperature determined
by: anneal ing temp. in oC = tt(Ç+Ç)+2(A+T)-5.

67.
CHAPTER THREE
THE CLONING OF creA FROM
Asperg¿ Z ltts n ídu Zorts -
This chapter descrjbes the cìoning and the prel iminary
characterization of the creA gene from Aspergillus nidulons. A
specific clon'ing strategy was chosen, previously shown to be useful
for the isolation of regulatory genes, which have low levels of
expression and are of unknown sequence. This strategy 'involved the
transformation of a strain containing a creA mutant alleìe wjth a
genonic library, se'lection for complementation of the mutation and
the physical rescuing of tranforming sequences from the compìemented
transformant. Simiìar techniques have been employed by a number of
workers to isolate reg'ions containing both structural and regulatory
genes of interest from ,{. nídulons including the regulatory genes
omdï (Andrianopou'lous and Hynes 1988) , føcB (Katz and Hynes 1989a)
and brZA (Boy'l an et ol. 1987) and the structural genes yA (Yel ton et
ol. 1985) , ocuD (Balìance and Turner 1986) , pyr| (Qakley et ot.
1987a) and riboB (Oakley et o1.1987b). Evidence ìs presented in this
section which indicates that the region which has been cloned from A.
nidulons contains the creA 1ocus.

68
3.1 THE CLONING 0F creA:
3.1.1 The construction of a wildtvpe qenomic library from A
nídulsns z
DNA from wìldtype ,4. nídulons was partially digested with Mbol
in reactions to optimize the number of fragments in the 6-15kb size
range. Fragments of this size range were purified and ligated jnto
the EomHI sjte of pM006. Ligation reactions were transformed into E.
coli strain DHI. Plasmid harboring co'lonies were selected on medium
containing ampiciìlin and pooìed into four batches. Large scale
preparations of plasmid l'ibrary were made from cultures incubated for
only three doub'l ings without ampì ification. In total , these
libraries represented 12,000 clones with an average jnsert size of
9.0kb and therefore there was a greater than 95% chance that a
sequence of interest was represented among these clones.
The number of clones required =
ln (l-probability sequence being present)
average insert size
ln 1-size of genome
3.I.2 The identification of a non-revertabìe creA- allele:
Al lyl al cohol i s toxi c to A. nídulons when converted, v'ia
aìcohol dehyrogenase, to its toxic product acrolein. l.liìdtype strains
are resistant to atìyl alcohol in the presence of D-gìucose or
sucrose due to carbon catabolite repression of alcohol dehydrogenase
enzyme activity. However, creA mutatjons Iead to compìete sensitivity
((((

69.
to 2.5mM allyì alcohol in sucrose or D-glucose media because alcohol
dehydrogenase act'ivity is no longer subiect to carbon catabolite
repression (Hynes and Ke'lìy t977). Therefore, comp'lementing
transformants or revertants of a creA mutant strain may be identified
by selection for resistance to aìlyì alcohol on D-glucose or sucrose
medium. However, mutations in the structural gene for alcohol
dehydrogenase, olc|, can also lead to a resistant phenotype. These
mutants can be di sti ngui shed from wi I dtype strai ns by thei r
resistance to aìlyì alcohol in glycerol medium (Ciriacy, 1975) and
their poor utiìization of ethanol as a carbon Source. The extreme
toxicìty of acroìein in A. nídulons provides a powerful means of
selecting for strains which do not have a'lcohol dehydogenase enzyme
activity when grown in the presence of D-gìucose or sucrose. This
provided a means by which it was possible to select for
complementation of creA mutant alleles with the wildtype ìibrary on
D-gìucose or sucrose medium containing 2.SmM alìyl aìcohol. For this
selectìon system to be successful in identifying true complementation
of cre| mutant alleles, it was important to determine the frequency
with which the creA mutant aììeles could phenotypicalìy revert to
wiìdtype on sucrose pìus allyì aìcohoì medium.
Conidial suspensions of strains containing the mutant alleles
creld-L or crel204 were either spread directly, of after UV
mutagenesis, onto sucrose medium containing 2.5mM aìlyl alcohol.
Colonies that grew (Figure 3.1.1) were screened for growth on ethanol
minimal medium and those that could not utilize ethanol as a sole
carbon Source were cl assi fi ed as mutati ons affecting al cohol
dehydrogenase activity. Colonies which could utilize ethanol as a
carbon source, showed sensitivity to a'l ìyl alcohol in glycerol

70.
Figure 3.1.1: Spontaneous and induced reversion of creAd-30 and
crelt204. A. nídulons strains 1070 and 664 are plated on sucrose
media containing 2.5mM aìlyl alcohol. Conidial suspensions of each
strain were pìated out either directly (spontaneous) or after UV
mutagenesis (induced) and grown for two days at 37'C in order to
determine if these strains couìd phenotypically "revert" to CreA+'
Strain 1070 showed high background growth and had a high "reversion"
frequency on this medium.

664
1070
Spontaneous I n duced
/\

7r.
medium, and were resistant to allyl alcohol on sucrose medium were
classjfied as CreA+ "revertants" (Table 3.1.1). Strain 664,
containing the cre[204 allele did not show any spontaneous or induced
phenotypic reversion to CreA+. This strain uras crossed into an orgB2
background (stra'in C26.1.10) to g'ive strain C43 (b¿41 pobo{I cre\204;
ørg}2) and was used as the recipient strain for transformations with
the pìasmid tibraries. The strain containing the creAd-l allele
(1070) produced many mutants affecting alcohol dehydrogenase activìty
and also potential CreA+ "revertants" when plated out in the Same
manner, on sucrose pìus aìlyì alcohol medjum. The relatively high
"reversion" rates seen for cre|d-L was probably strain dependent,
aì though thi s al I eì e r/,,as not tested i n a di fferent geneti c
background. Since there was an increased frequency of both AlcA+ and
CreA+ phenotypic classes, this did not necessarily indicate a highly
revertable allele. Such high mutat'ion rates could possibly have been
due to a defective DNA repair system in strain 1070. Ten of these
CreA+ "revertants" were chosen for further characterization. These
phenotypic revertants segregated creAd-1, progeny when outcrossed to
strain J7 which indicated that these strains contained phenotypic
suppressors of the creAd-1 allele. Diploids were constructed between
these ten strains and MSF, in an attempt to map some of these
suppressors to a particular linkage group by haploidization analysis.
Initiaììy, some of these potentiaì suppressors appeared to map to
linkage group V or VII, however these strains seemed to be highly
unstable and were found to spontaneously aquire mutations which could
be characterized as Alc- or others that were shown to have poor
utilization of ethanol as the sole carbon source. The latter group
were not classified as Alc- since, unlike Alc- strajns, they
remained sensitive to alìyl alcohol in the presence of glycerol' This

72.
Table 3.1.1: Spotaneous and induced "reversion" of crePró-L and
creA204.
creA204 creAd-t
Spont. Induced*
1 . 1x10- s"reversion" freq. 1.3x10-6
no. tested 40
no. Alc- 40
no. CreA+ 0
46
Spont.
1.4x10-5
37
33
4
I nduced*
0.038
48
40
I46
0
Conidial suspensions were plated onto sucrose minimal
containing 2.5ilr1 al lyl alcohol and 10rN anmonium tartrate.
were screened on l% ethanot plus 10mM anmoniun chloride.
* Conidia were treated with UV, with a survival rate of 10%.
medi un
Col oni es

73.
suggested that these strains had ìevels of alcohol dehydrogenase
activity which were capable of converting a sufficient amount of
allyl alcohol to acrolein to cause toxicity while remaining subiect
to carbon catabol ite repression. The difficulties inherent in
maintaining the stabi'l'ity of these strains did not allow for the full
characterization of their genotypes as part of this work. This
analysis made it clear that strain 664 containing the cre\204 allele
was more appropriate for this work.
3.1.3 Complementation of the cre\204 mutation:
Strain C43 carrying the crel204 allele was transfonned with
preparati ons of the wi I dtype genomi c I i brari es i n pltl006. Transformed
protoplasts were either (a) plated onto minimal protoplast medium to
select for arginine prototrophy and then screened for complementation
of the creA204 mutation by repìica plating to sucrose minimal medium
p'lus 2.5mM alìyì alcohol or, (b) plated onto minimal medium se'lect'ing
for arginine prototrophy for ten hours and then overlayered with
sucrose minimal medium p'lus 2.5mM allyl alcohoì directly selecting
for complementation of both orgB2 and cre\204.
In a total of eight experinents these selection techniques
produced approximately one hundred and fifty colonies that grew on
sucrose and ally'l alcohol nedium. Three of these colonies (which
arose on the same transformation plate and subsequently were deened
to have originated from a single transformant) were able to utilize
ethanoì as a sole carbon source, and were therefore not ølc- mutants.
The remaining colonies showed.poor utilization of ethanoì as a carbon
source and were resistant to altyl alcohol in gìycerol medium, and

74.
$rere classified as øZc- mutants.
These complemented transformants (T41a, T41b and T41c) were
tested for growth on a range of media to rule out the possibiìity
that the observed phenotype was alcohol dehydrogenase specific (see
Figure 3.1.2b for compìementation for growth on sucrose plus allyl
alcohoì med'ium) by determining whether they also showed
comp'lementation for some of the other phenotypes due to creA mutant
alleles. T41a, b and c showed wildtype morphology on comp'lete medium
(Figure 3.1.2a), unlike the compact morphology of creA mutant
strains. l'lhile wildtype Á. nidulons strains are sensitive, creA
mutations lead to hypersensitivity to fluroacetate and fìuroacetamide
in glucose medium due to the derepressed expression of genes coding
for the enzymes converting these compounds to flurocitrate (Hynes and
Kelty lg77). T41a, b and c grew slightly better than wiìdtype on
glucose medium containing these compounds (figure 3.I.2c and d),
indicating repressed levels of acetyl-Co-A synthetase and acetamidase
in the presence of a source of carbon catabol ite repression.
Extracelluìar protease activity is subject to carbon catabolite
repression in wildtype A. nídulons and this activity can be assessed
by the cìearing of casein in powdered milk in solid media (Cohen 1972
and Cohen 1973). The cre[204 mutation showed distinct clearing of
milk in the presence of glucose whereas T41a, b and c showed ìower
levels of clearing (Figure 3.1.2e). Therefore prelininary growth
testing showed that T41 was conplemented for the creL204 allele of
strain C43.

75.
Figure 3.I.2: Growth testing of T41a, T41b and T41c.
Plates contain:
(A). Compìete medium.
(g) . sucrose mediun contain.ing 2.5mM al]yì alcohol .
(c). D-glucose medium containing lomg/ml fluroacetate.
(D) . D-glucose medium containing lOmg/ml fluroacetamide.
(E). D-glucose medium containing 1% mi'lk powder'
(A), (B), (c) and (D):
Top left : wiìdtYPe A- nidulons.
Top right z cre\204.
Bottom left to right : T41a, T41b and T41c'
(E) :
Top 'left z creÃ204.
Top right : wi ldtYPe ,4. nídulons '
Bottom left to right : T41c, T41b and T41a'


76.
3.1.4 Rescue of transforminq sequences:
Genomic DNA prepared from T41 was parially digested with BonHI,
PstL and XhoI separately with the aim of generating fragments
containing both pM006 vector sequences and the complementing sequence
of interest. These partiaì digests were recircularized by ligation
and transformed into E. coli strain H8101. Colonies harbouring pM006
vector sequences were selected on medium containing ampicillin.
Twenty ampicillin resistant colonies resulted from this experiment,
of which nine, nine and two of these originated from partial digests
using BanHI, PstI and XhoI respectively. Preliminary restriction
enzyme digest analysis of plasmids isolated from these colonies
showed that six different pìasnids were represented. Those which
appeared to be the same were pooled for large scale plasmid
preparations and used to re-transform ,4. nidulons strain C43. Each
plasmid preparation was tested for its ability, on transformation of
strain C43, to either (A) cornplement both orgB2 and cre\204 by
se'lecting for transformants on minimal medium for 10 hours and then
overlayering with sucrose medium containing 2.5mM aìlyl alcohol or
(B) compìement creA aìone by plating transformed protoplasts onto
media containing arginine and overlayering with sucrose medium plus
alìyt alcohol after 16 hours. One of the plasmid pools complemented
both the orgB2 and creA204 mutations (Fìgure 3.1.3). Eight of the
twenty rescued plasmids were represented in this plasmid pool which
complemented C43 for both mutations, of which all originated from
religated Bomïl partial digests of T41 DNA. Further restriction
digest analysis demonstrated that these 8 plasmids were identical. A
restriction map of this plasmid, pANC1, is shown in Figure 3.1.4.

77.
Fi gure 3.1 .3: Comp'lementati on of C43 wi th pANCI '
Plates are:
Right: control of untreated protop] asts pl ated onto
protoplastmediumwithalìsupplementsaddedexcept
argenìneandover.layedwithprotoplastmedium
containing 2.5mM alìYl alcohol'
Left top: pANCI treated protopìasts plated onto protopìast
medium with all supplements added except argenìne.
Manyofthesetransformantsappearedtoturnyellow
whengrownformorethanfìvedaysat3ToC.However'
onlygreencolonieswereobservedwhentreated
protopìasts were plated at low density and only
greentransformantscou]dbeis'olatedwhenye.llow
regions were subcu'ltured' The reason for the
initiat yelìow appearance of some of these
transfonnants remains unclear'
Left botton: pANCI treated protopìasts plated onto protopìast
mediumwithallsupplementsaddedexceptargenine
andoverlayedwithprotoplastmediumcontaining
2.srnM aì lYl al cohol .
Therefore, PANCl
mutati ons .
complemented C43 for both the creA204 and orgB2

la'a
tÛII
,.G-t

!,
,I'1
r
I
78.
Figure 3.1.4: Partial restriction map of pANC|. The regions of pANCI
that were subcloned into pUCl9 to give pANC2, pANC3 and pANC4 are
i ndi cated.
Restriction site abbreviations:
B - BønHl
Bg - Bglll
E - EcoRI
S - SølI
X - Xbol

E x
pAll02pAtGS
pAllG4
r lkbl
Ì::i,-: -æg"tr
---

79.
riff
Southern hybridization analysjs of Xbol cut wildtype DNA using
pANCI as a probe showed two bands of hybridization, one of 3.3kb
correspondjng to the native orgB sequence and another of
approximateìy 10.skb corresponding to the cloned sequence. XboI
digested T41 DNA had two extra bands of approximately 3.7kb and 9.7kb
indicating that transforming sequences had not integrated into either
the org\ native ìocus or the native site for the cloned sequence
(Figure 3.1.5) .
3.1.5 The localization of cre[ within pANCI:
Restriction fragments of pANCI were subcloned into pUCl9 and
transformed into strain C43 in order to ìocalize a smaller fragment
capable of complementing the creL204 mutation alone. Transformation
experiments using these plasmids were p]ated out, selecting
separately for orgB+ and creA+ transformants in each case. Southern
analysis of pANCI digests probed with pUCl9 confirmed that the 2.9kb
Xbøl - fcoRl fragnent of pANCI represented pUC19 sequences. pANC2
contained the 3.0kb XboI fragment of pANCI (see Figure 3.1.4) and
complemented the orgB2 mutation when transformed into strain C43.
pANC3 contained the 2.8kb Xbol fragment and pANC4 the 2.3kb BønHI -
XboI fragment of pANCI (see Figure 3.1.4). Transformants of strain
C43 obtained with both pANC3 and pANC4 showed complenentation of
cre[204 but not orgB2. The insert of pANC4 was used for further
ana'lyses since the smaìl 0.5kb BonHI - Xbol fragment in pANC3
represented a sequence which would not have been continuous with the
2.3kb Bomïl - Xbol fragment in the genome, having resulted from
circularization at the BømHI site when the plasmid was generated.
Thi s was confi rmed by compari ng Southern bl ots of wi I dtype ,4.
IlrI
r

80
Figure 3.1.5: Southern blot analysis of wildtype A. nidulons and
transfonnant T41. Each track contains 2ug of DNA digested with XboI'
The probe used was PANC1.
Tracks: 1. T41
2. wt A. nídulons
3. C43
Bacteriophage lambda DNA digested with HíndIII was used as a marker
of molecular weight.
ii
¡
:
II
lt,
I'I
II
r
I
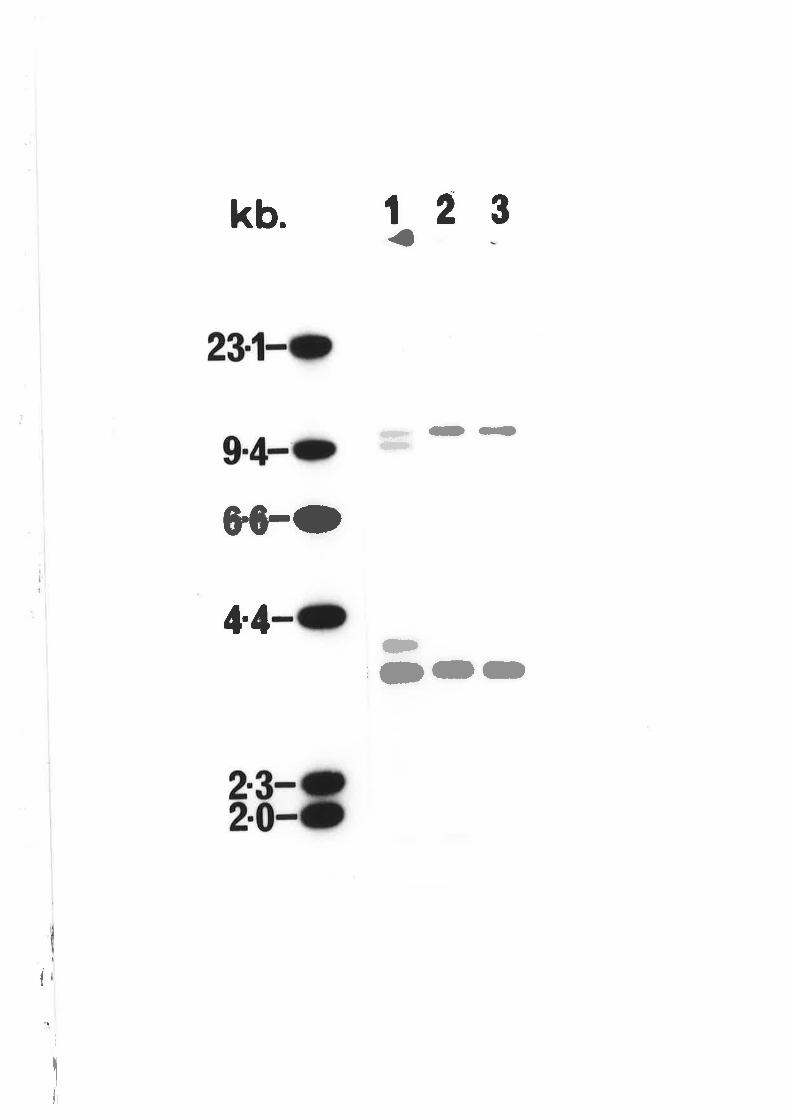
ll
i
I
(tte ,
-
(ÞrD
v.v
(D-00
'ql
*.f-
?] 7 I

81.
nidulons DNA digested with various restriction enzymes and probed
wjth pANC3 and pANC4. Faint bands of hybridization to wildtype DNA
are seen when probed with pANC3 which are not present when probed
with pANC4 (Figures 3.I.7 and 4.1.1). A partìal restriction map of
the pANC4 insert is shown in Figure 3.1.6. These analyses also
demonstrate that all bands can be accounted for by the restriction
map of pANC4 and therefore it was unlike'ly that the insert had
rearranged during the rescue procedures.
pANC4 was also found to comp'lement the cre\220, creA225,
cre$d-! and creAd-30 alleles. This ruled out the poss'ibility that
it contained an alleìe spec'ific suppressor o1 cre|. Hybridization of
pANC4 to genomi c Xbol dìgests of DNA from strains carrying the
alleì es cretlzz}, cre225 and creAd-l (Figure 3.1.8) showed that these
mutants urere not the result of any gross deletions, insertions or
rearrangements in the genome since hybridization was seen to the same
10.5kb band as seen for the wildtype and the cre\204 strains'
3.2 ANALYSIS 0F CreA+ TRAI{SFORMANTS:
DNA was isolated from 16 phenotypical ty creA+ pANC3
transformants of strain C43, designated TC43A - TC43P' Slot blot
analysis of serial dilutions of the DNA samples using the insert of
pANC4 as a probe showed that all transfonnants contained transforming
sequences and the approximate number of copies of cre[ in these
tranfonnants was determined. DNA loadings were standardized using the
singìe copy riboB gene of A. nidulons from pPL3. Copy numbers nanged
from I for those that were the result of a gene repìacement event to
about 15 in the case of TC43E (fa¡le 3.2.1).

82
Figure 3.1.6:
direction of
arrow.
Partial restriction map of the insert in pANC4'
transcription of the creA gene is indicated by
The
the
Restriction site abbreviations :
B - Bonïl
E - fcoRV
Ps - PstI
Pv - Pvutl
R - RsoI
S - SølI
X - Xbol
The Xbol site is shown in brackets since it is not a genonic site
and resulted from the subcloning of pANCI-

Ps Pv RE S
.200bo ,r

83
Figure 3.1.7: Southern blot analysis of wildtype A. nidulons DNA cut
with various restriction enzymes and probed with pANC3. Each track
contains 2ug of DNA cut with:
1. Bon}J.l
2. Bglll
3. EcoRI
4. Híndlll
5. Pstl
6. Pvull
7. SøcI
8. SmøI
9. SøZI
10. Xbol
tI. Xhol
12. Clol
Bacteriophage lambda DNA digested with HíndIII was used as a marker
of moìecular weight.
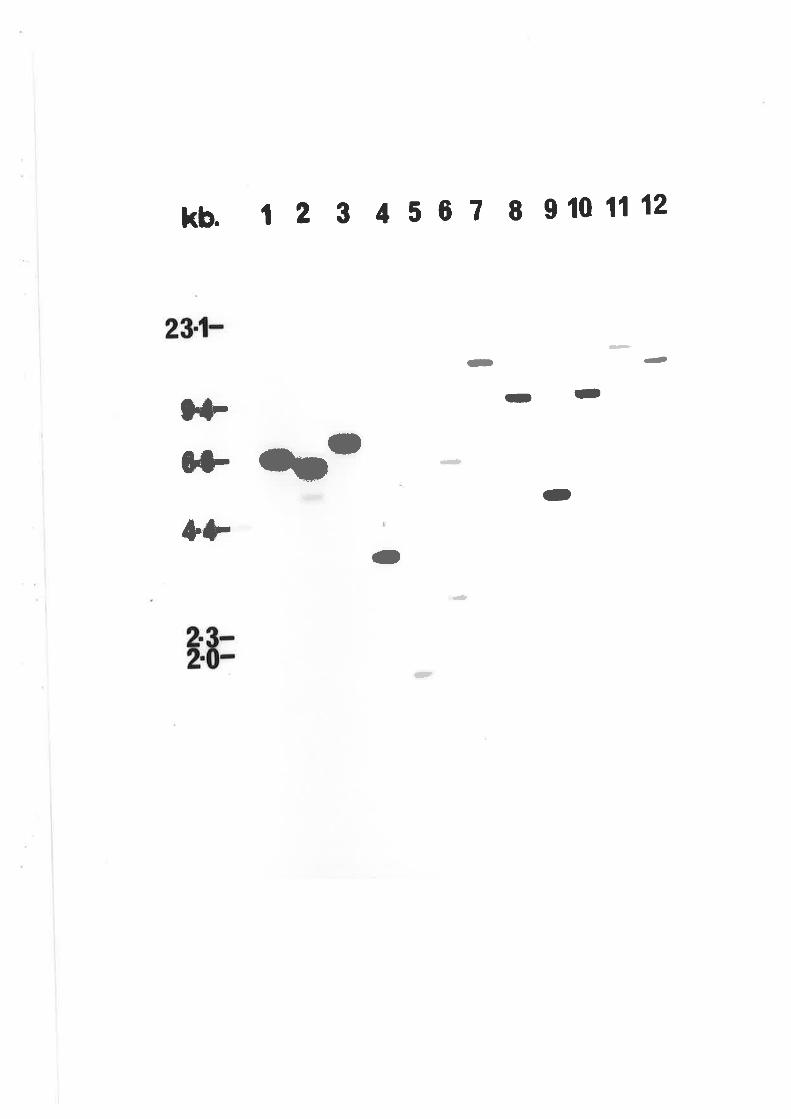
kb. 12 3 4 567 8910 11 12
2ü'l-
- -
+={F -lt
(-tilF
4"*-
to -,#
ta.-
ffi e

84
Figure 3.1.8: Southern blot analysis of XöøI digested DNA from
wildtype A. nidutons and strains containing creA mutant alleles' The
amounts of DNA in each track are not equal.
Tracks are: 1-3. wildtype A. nídulons'
4. 664 (crel'204).
5. 1070 (creAd-l).
6. C43 (cre{204).
7. cre\204-
8. creA220-
9. creA225-
Bacteriophage lambda DNA digested with HindIII was used as a marker
of molecular weight.

I g, IlttT ü9fD--n¡¡
kb.
2&Þ-(DI- r-)
-$4-rÞ
->
-D
&þ-
&F

I
I
I
85
Table 3.2.L: cre[ copy number in CreA+ transformants.
COPY NUMBER TRANSFORMANTS
1
2
3-6
15
TC43A, TC43J, TC43M.
TC43B, TC43F, TC43K.
TC43C, TC43D, TC43G, TC43H, TC43I, TC43L, TC43N'
TC430, TC43P.
TC43E.

86
Southern hybridization of Xbol digested DNA using pANC4 as a
probe showed all transformants contained pANC4 sequences and retained
the 10.5kb native creA band except TC43H, in which the native band
was disrupted by transforming DNA (figure 3.2.1). TC43H was
outcrossed to straìn J7 and among 485 progeny scored, none were of
cre{ mutant phenotype. This indicated that this transformant arose
from a honologous integration at the creA locus, and that the clone
was of creA and not a suppressor of the creA mutant phenotype. TC43H
was meiotically stable, as no progeny of creL mutant phenotype were
found among 465 progeny of a selfed cleistothecia. These resuìts,
where integration has occurred at the homologous genomic sequence,
and the comp'lementing sequence is inseparable from the creA mutation
through meiosis is further evidence that the clone contains creA.
75 pANC3 transformants of the cre\204 strain were tested for
their ability to utilize a range of compounds as sole carbon sources
at a concentration of I% in appropriately supplemented minimal media
with lOmt'l anmonium tartrate as the nitrogen source. Compounds tested
were D-gìucose, glycerol, D-sorbitol, sucrose, ethanoì, maltose,
acetate, lactose (at 0.5%) , D-mannitoì , D-galactose, D-mel ibiose,
raffinose, L-arabinose, L-sorbose and quinic acid. Utilization of L-
threonine, acetanide, 2-pyrrolidinone, ß-alanine, p-aminobenzoic
acid, L-proìine and L-gìutamic acid were tested as both carbon and
carbon and nitrogen sources at a finaì concentration of 50mM in the
media. There uras variation between transformants on synthetic
complete medium, and only these same differences were evident for
al I the carbon sources tested. Therefore, none of the cre[+
transformants could be identified as showing appreciably different
uti I i zati on of these carbon sources as compared to wi ì dtype Á.

87
Figure 3.2.1: Southern blot analysis of A' nídulons
transformants. Each track contains 2ug of DNA digested with
pANC4
Xbø[.
The probe used was PANC4.
Tracks are: 1. wildtype A.
2. C43
3. T41
4. TC43A
5. TC43B
6. TC43C
7. TC43D
8. TC43E
9. TC43F
10. TC43G
11. TC43H
nídulsns
Bacteriophage lambda DNA digested wilh HíndlII was used as a marker
of molecular weight.

kb. 1 2 E 4 qg7 ôe19 11
23.
-
-
9
6.
Ort, -f -
-
_--
#Ò'
-
0.
.*

88
nidulons. An increase in the number of copies of cre[ may not
necessarily be expected to ìead to detectable differences in the
utilization of various carbon sources since the creA mutant alleles
themselves do not show any such differences in growth (Arst and Cove
1973, Bai'ley and Arst 1975, Hynes and Kelly 1977). This homogeneous
phenotype u,as also to be expected if the creA gene product has a
regulatory role as opposed to being directly involved in the
metabolism of carbon sources. Growth differences in plate testing may
therefore onìy be detected by other approaches such as titration
anaìyses in multicopy transformants or by specific plate testing
which may show variation in the extent to which metabolic functions
are subject to carbon catabolite repression by creA. Transformants
TC43A - TC43H were tested for their ability to grow on sucrose media
containing alìyl aìcohol at concentrations ranging from 2.5rûl to
100mM. Those transformants with more than two extra copies of cre{
grew consistently better than wildtype on these media, suggesting
tighter repression of alcohol dehydrogenase synthesis.
3.3 EXPRESSION 0F creA IN A. nídulons:
Strand specific probes were generated from the insert of pANC4
which had been cloned into M13 mp18 and mp19. Each of these was used
separately as probes to slot blots of total RNA isoìated from
wiìdtype A. nídulons grown with gìucose as the sole carbon source.
The differentiat hybridization between these probes to RNA showed
that the creA gene is transcribed in the direction fron the BonHI
site to the XbøI site, with respect to the restriction nrap of pANC4
(see Figure 3.1.6) .

89
Total RNA was isolated from wiìdtype and creA mutant strains
grown in the presence of various carbon sources with 10nùll an¡nonium
tartrate or 10mM ammonium orthophosphate for acetate medium acting as
the nitrogen sources. Northern blot ana'lysis using the insert of
pANC4 as a probe showed that wildtype Á. nidulons and the mutant
strain cre\204 produce a major mRNA species of approximately 2.Okb in
ìength (Figure 3.3.1). Under the growth conditions used, creL mRNA
levels in wildtype A. nidulons in the presence of glucose were of the
same order of rnagnitude as that for the orgB transcript, in that
sinilar signals were given by hybridization to both transcripts when
probed with pANCI (Figure 3.3.2). Relative levels of creA message
were determined by sìot blot anaìysis of RNA sampìes probed with the
insert of pANC4. Levels were calculated relative to wildtype A.
nidulons RNA from 1% D-gìucose grown mycelia (Table 3.3.1). Two fold
higher levels were found in Lv" L-arabinose and l% glycerol grown
cultures, but not in 50mM D-quinate or 50mM L-proline grown cultures.
Similar levels of creA expression were found for the mutant strain
creA204 as for wiìdtype A. niduløns in D-gìucose grown mycelia. Inglycero'|, L-arabinose and acetate grown cultures of creA204, cre|
expression was increased 1.5 - 2.5 fold as in the wiìdtype strain.
The aìtered levels of the creA message present under these different
growth conditions as determined by Northern anaìyses are insufficient
to account for the regulatory actions of cre[ and its phenotypic role
in carbon catabolite repression. Although transcriptionaì regulation
is still possible, the activation/inactivation of the CreA protein in
reponse to some external stimulus is 'like'ly to be involved in the
regulation of carbon catabolite repression by creA
Slot blot and Northern analyses u,ere aìso performed using the

90.
Figure 3.3.1:
A. nidulons
approximateìY
pAl{C4.
Tracks are:
Northern blot analysjs of RNA isolated from wildtype
and creA204 mutant strain. Each track contains
20ug totaì RNA. The probe used was the insert of
1. wiìdtype Á. nídulqns from D-glucose grown myceìia.
2. wildtype ,4. nídulons from g'lycero'l grown myceì'ia'
3.wildtypeA.nídutqnsfromL-arabinosegrownmycelia.
4. cre[204 fron D-gìucose grown mycelia'
5. cre[204 from glycerol grown mycelia'
6. cre[204 frorn L-arabinose grown myce'lia'

| 2 3 4 5 6
zkb+> rO

91.
Figure 3.3.2: Northern blot analysis of RNA jsolated from wiìdtype
A. nidulons D-g'lucose grown mycelia. The probe used was pANCI' The
creA and ørgB transcripts are indicated
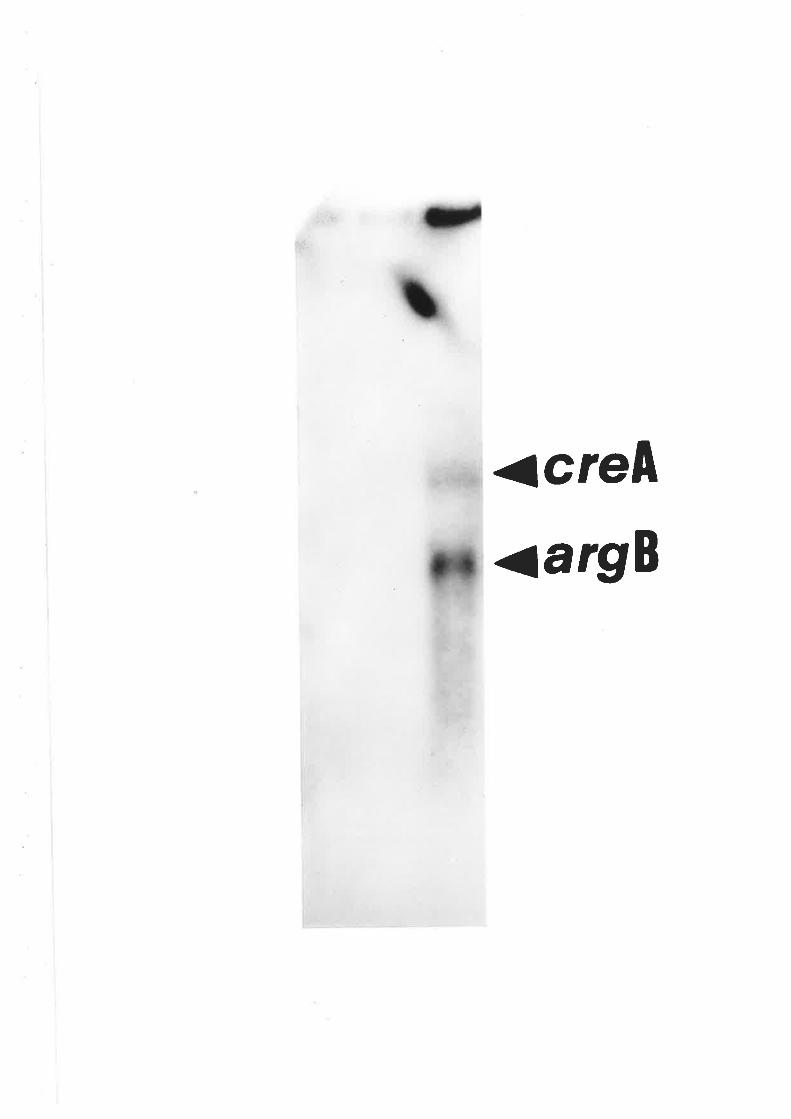
.ì..ir*
#

92.
Table 3.3.1: Relative levels of expression of cre{ jn wildtype A
nidulons and strain 664.
CARBON SOURCE t,'lï 664
D-gl ucose
L-arabi nose
glycerol
acetate
L-prol i ne
quinic acid
1.0
2.5
2.0
2.5
1.0
1.0
1.0
1.5
2.0
1.5
1.0
1.0
Relative values are rounded to the nearest 0.5 of a unit.
Table 3.3.2: Relative leveìs of expression of creA in cre[ mutant
strains and CreA+ transformants.
CARB0I{ SOURCE 1070 creA220 creA225 TC43D TC43E
D-gl ucose
L-arabi nose
1.0
1.0 4.0
3.0
1.5
20.0 6.0
30.0
Relative values are rounded to the nearest 0.5 of a unit.

93
insert of pANC4 as a probe to RNA isolated from the strains carrying
the mutant alleles creAd-1, creA220 and creA225 (Table 3.3.2 and
Figure 3.3.3). Strain 1070, carrying the creAd-l allele, had levels
of creA mRNA comparable with D-g'lucose grown wildtype A. nidulons
when both D-glucose and L-arabinose were present as carbon sources.
However, in the presence of D-glucose, but not L-arabinose, 1070
expresses another transcript which is ìarger than the native cre[
message. A ìarger message was also present in the strains carrying
the creA220 and creA225 alleles which is expressed regardless of
whether D-glucose or L-arabinose acts as the carbon source. creA225,
in contrast to strains carrying the other mutant alleles, showed
higher levels of creA expression in the presence of D-gìucose than
when L-arabinose was the sole carbon source. These analyses showed
that, of the mutant al I el es avai I abl e, al ì expressed creA
transcript(s) in some form. It is therefore possible that these
alleles may not represent complete ìoss of function alleles and that
the derepressed phenotypes of the creA mutants may not be the result
of a true null allele.
Total RNA was also prepared from the creA multicopy
transformants TC43E and TC43D to determine whether or not increased
copy number ìed to increased levels of crel. messenger RNA. TC43E and
TC43D grown with D-gìucose as the carbon source had approximateìy six
times, and twenty times respectively, the level of crel. mRNA
expression as that found in sirnilarly grown wildtype A. nídulans.
This was increased to thirty times that found in wildtype D-glucose
grown mycelia when TC43E was grown with L-arabinose as the carbon
source (Table 3.3.2). Thus despite showing increased levels of cre{
expression TC43E still showed regu'lated creA expression. The level of

94
Figure 3.3.3: Northern blot analysis of RNA isolated from wildtype
A, nídulons and creA mutant strains probed with the insert of pANC4'
Each track contains approximately 20ug of total RNA'
Tracks are:
1. t{ildtype A. nidulons from D-gìucose grown mycelia'
2. cre[220 from D-glucose grown mycelia'
3. creA220 from L-arabinose grown mycelia'
4. cre[225 from D-gìucose grown mycelia'
5. cre[225 from L-arabinose grown rnycelia'
6. cre[d-l fron D-glucose grown mycelia'
7. creAd-l from L-arabinose grown mycelia'

t2 3 4 567
O *¡ *&IÈ
zkb+; tot

95.
cre| expression found in TC43D, with two copies compared to 15 in
TC43E, showed that higher message level s did not necessari ly
correlate with higher creA copy number or with the observation of
apparentìy "tighter" repression on sucrose pìus ally'l alcohol media.
This, along with the observation that TC43D and TC43E produce
multiple transcripts are probab'ly both effects of the positions of
integration of transforming sequences (Figure 3.3.4).
3.4 SUMMARY AND CONCLUSIONS:
The cre| gene from, spergíllus nidulons has been cloned by
complementation of a non-revertable mutant allele using a genomic
ìibrary and p'lasmid rescue techniques. The rescued sequence was
subcloned and a 2.3kb fragment was identified which compìements
severaì cre\ mutant alleles. Northern anaìyses showed that cre[
encodes a transcript of approximateìy 2.Okb in length and that the
levels of this transcript varied by up to two foìd, depending on the
carbon source. All the mutant atleles that were tested showed
expression of a creA transcript. Transformants containing one or
more copies of crel greu, as wiìdtype on a large range of carbon
sources, but for transformants with two or more extra copies of creA
there was evidence for tighter carbon catabolite repression

96
4!
t,
I
r
{
Figure 3.3.4: Northern blot analysis of RNA isolated from wildtype
A. nídulons and pANC4 transfonnants of strain C43. Tracks contain
20ug of total RNA. The insert of pANC4 was used as a probe.
Tracks are:
1. Wildtype A. nidutons from D-glucose grown myceìia'
2. TC43D fron L-arabinose grown mycelia'
3. TC43D frorn D-glucose grown mycelia'
4. TC43E from D-glucose grown mycelia.
5. TC43E from L-arabinose grown nycelia'
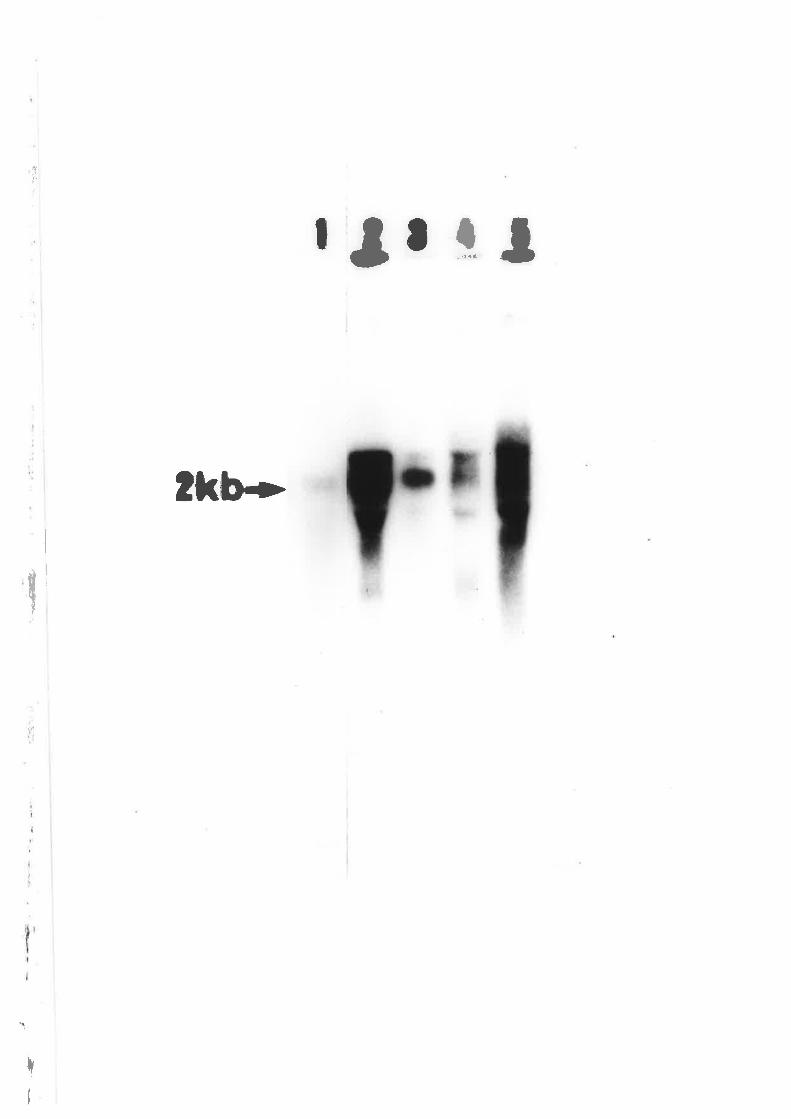
r!.
I
t t¡
T T
1
i
2frbt+
Ì
I

97.
dìtf
J
CHAPTER FOUR
ANALYSIS OF creAsl-3o AND THE
PHENOTYPE OF A creA NULL ALLELE -
Experiments using the creAd-30 allele are presented jn this
chapter and they provide additional evidence that the clone isolated
and described in Chapter 3 is of the creA region. The molecular
analysis of the mutant allele creAd-30 along with the fjndings'in the
previous chapter that all mutant alleles express creA transcripts,
demonstrates the importance of constructing a true loss of function
creA aìlele. Work presented in this section, where a loss of function
creA allele was constructed by reverse genetics, shows that creA is
an essential gene, and that homologous genes are probably present in
a number of other fung'i, especiaìly those more cìosely related to A.
nidulons.
4.1 MOLECULAR LOCALIZATION OF THE INVERSION BREAKPOINT IN CTEAd-30:
The creAd-30 mutation is the most phenotypica'lly extreme cre[
allele known and results in a very compact coìony morphoìogy. cre[d-
30 was identified by Arst and co-workers (Arst et ol. 1990) as a
spontaneous mutation in a strain containing the fr[-l mutation. /rA-1
leads to the jnhjbition of growth by D-fructose, D-sorbitoì, sucrose
and D-mannitoì (Roberts 1963). The creAd-30 mutation conferred
resistance to the toxicity of 1% D-mannitol in the presence of L% L-
I
r
I

98
d'{F
I
arabinose with SmM L-glutamate as the nitrogen source in a /rA-lstrain. The basis for this resistance is unknown. Linkage analysis
showed that the creAd-30 mutation is a result of a pericentric
inversion, with one breakpoint occurring in creA on the left arm of
chromosome I and the other occurring between the markers binÈ and yA
on the right arm of chromosome I. Since creAd-30 was the result of an
inversion with one breakpoint occurring within the cre[ locus it has
been presumed that the phenotype, of derepression, is that of a loss
of function allele. The identification and genetic characterization
of creAd-30 in Arst's laboratory provided a means to show
unequivocally that the cloned fragment was a clone of the cre{
region. Using the cìoned gene, the inversion breakpoint of creAd-30
could be mapped to a particular position within the creA gene itself.
The clone could then be used to determine whether the creAd-30 allele
was a true loss of function alìele at the level of mRNA, an important
point underpinning the genetical hypothesis of negative control.
Genomic DNAs fron wi ldtype A. nídulons and creAd-30 were
singularly and doubly digested with a number of restriction enzymes,
sites for which were present within the insert of pANC4, and
Southerns of these digests were probed with the insert of pANC4
(Figure 4.1.1). For each of the singìe and double digests, the
patterns of hybridization to the insert of pANC4 differed between the
two strains, indicating that the cloned fragment represented a region
which was disrupted in creAd-30. The expected patterns of
hybridization to digests of wi ìdtype DNA, with respect to the
restriction map of pANC4 are shown in Table 4.1.1, and all bands were
present in the wildtype strain. The altered bands found in the mutant
strain creAd-30 are also shown. These altered bands represent fusion
'i"
I
!

I
99.
Figure 4.1.1: southern bìots of wildtype ,4. nidulons and creAd-30
DilA probed with pANC . For each pair of tracks DNA from the wìldtype
crel strain is on the left and from the creAd-30 strain on the
right. Digests are:
First panel: 1. Bon|l 2. EcoRY 3. PsúI 4. PvuII 5- Xbol 6. xhol
7. BonHllEcoRV 8. PstI (longer exposure) '
Second panel: 1. Bom1l/PstI 2. Bon1l/Pvull 3. Bom1llXboI
4. EcoRYlXbøl 5- PvulIlPstI 6 ' PstllXbøl
7. PvuIIlXbol 8. PvuII/PstI (longer exposure)
9. PstIlXbol (longer exposure).
In the longer exposure tracks of PstI singìe and double dìgests'
faint bands of hybridization to a 150bp fragment in the creAd-3O
strain are visible, while in the wildtype a doublet (150bp and
250bp) is present). In interpreting these blots several other points
should be noted: 1. Tracks 83 and B8 wildtype: the faint band at
4.4kb results from partial digestion and is not present in other
blots . 2. Tracks 83, 88, C5, C8 and c9 cre[d-3Q: the faint band
below the strong lkb band is a fusion band. 3. Track 87 creAd-3O:
two fusion bands are present - a faint one at approximately lkb and
a strong one foming a doublet with the largest band. 4. Track 84
creAd-30: the "band" at 4.4kb is not present in other blots and is
believed to be an artifact whilst the strong band is probably a
doublet containing a fusion band. Qnly one fusion band is apparent
in PstI and Pvul\ digests of creAd-30 DNA, because the breakpoint
is probably too close to the Pvull/PstllPvulI site cluster to allow
hybridization to the other fusion band'
r
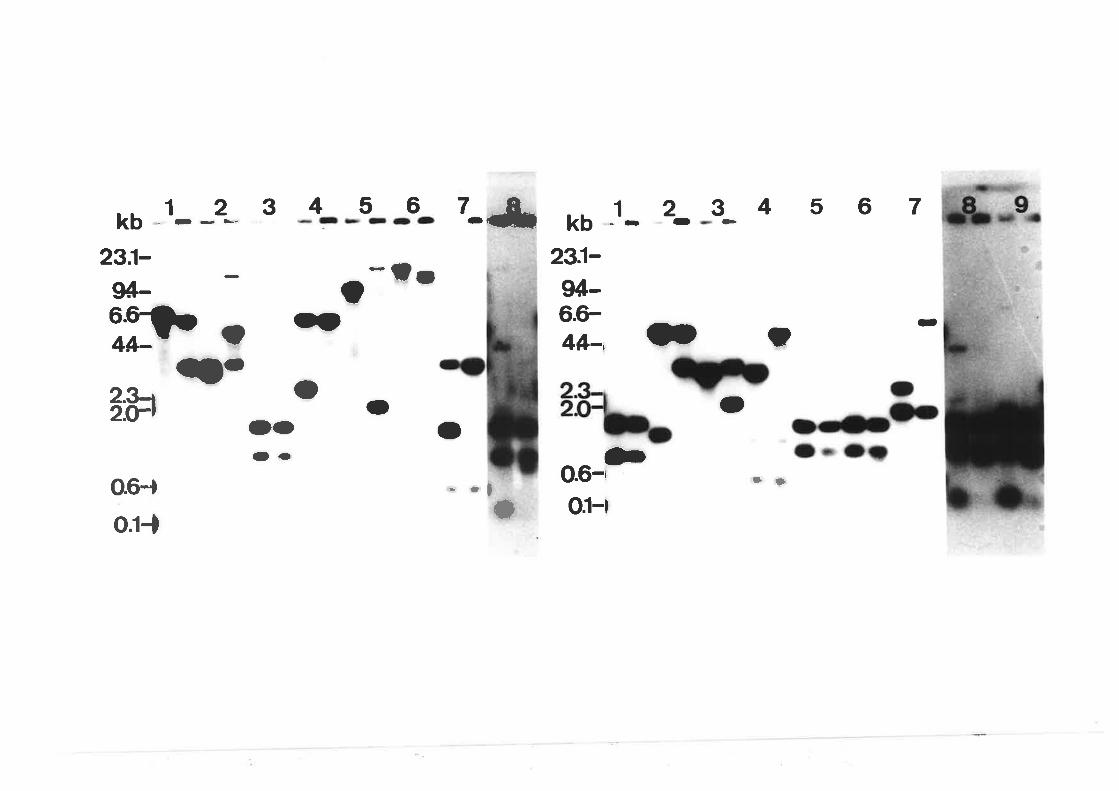
kb
231-94-
0.6.|
o.1-l
I- .fr kb231-ç),f[-6.6-44-,
1 723456
O
1 2-
34567
O ?
È-
o
.-- -
-l¡r .1
?iFo
e.) rt
l|oO2.3-r
2.V)o
OO(t-
O
-O.6-'
Ol-leO
ia ¡t

100
Table 4.1.1 Expected bands of hybidization of pANC4 (insert) to
wildtype A. nidulons DNA. For each restriction enzyme the sizes of
the expected bands of hybridization to ,4. nidulons genomìc DNA are
listed. These sizes were generated from the restriction map of pANC4.
*indicates the bands altered in creAd-30 digests.
RESTRTCTION ENZYME(S) BANDS OF HYBRIDIZATION (KB)
BonHI
EcoRV
PsT I
Pvull
Xbol
XhoI
BonïllEcoRV
BonHI I Pstl
Bonïl f PvuII
BonH.l I Xbol
EcoRY I Xbol
Pvull I Pstl
PstI/XboI
Pvu|l I XbaI
>2.3*
>1 .5x
>0.8
>1 .1
>2.3*
>2.3*
1 .5*
0.8
r.2*
>2.3*
>1 .5*
>0.8
>0.8
>1.2
>0.425 0.475
0. 15 0.25* >1 . 1
>I.2 (band in mutant is a doubìet)
0.475
0. 15
>1.1
0.475
0.15
0. 15
>1.1
>0.425
0.25*
>0.425
0.25*
0.25*
>1.1
>1 .1
>1.1

01.
fragments between the cre[ cloned region and stretches of DNA
flanking the creAd-30 inversion breakpoints. It was evident that in
the PsúI single and double digests the creAd-3O strain onìy showed
hybridizatjon to the l50bp fragment of the two smaller bands, while
the wildtype strain showed hybridization to the same 150bp fragment
pìus a 250bp fragment. Bands of hybridization present in aìl other
digests could be accounted for by a breakpoint occurring in the 250bp
PstI fragment of the cloned gene. Since only one fusion band is
apparent in the Pvull and PstI digests of creAd-30 DNA, the
breakpoint within the 250bp PsúI fragment is probably located very
close to the Pvull/Pstl/Pvull (overlapping sites identified by
sequencing) cluster and as such hybridization to one of the fusion
bands could not be detected.
Total RNA was prepared from wildtype,4. nídulans and the cre[d-
30 strain grown with 1% D-glucose and 1% L-arabinose serving as the
carbon sources in minimal medium. Northern anaìysis using the pANC4
insert as a probe (Figure 4.1.2) showed that creA message was present
in this strain, but the creAd-30 mutation gives rise to an altered
cre[ transcript. When cultures were grown with 1% L-arabinose as a
carbon source the creAd-30 strain expressed a transcript which is
ìarger than the wiìdtype creA message. When D-g'lucose is used as the
carbon source thi s message i s aì so present together wi th an
additional transcript which is smaller than the wildtype message. The
presence of these two transcripts in D-glucose grown cultures may
indicate that the 3'end of creA has been fused to a gene which is
expressed when D-g'lucose but not L-arabinose is present as the carbon
source. The larger message, which is expressed in both growth
conditions, probably represents the 5' end of the creA transcript

t02.
Figure 4.1.2: Northern blot analysis of wildtype Á. nidulons and
. creAd-30 RNA probed with the insert of pANC4'
Tracks are:
1. t{ildtype A. nidulons from D-glucose grown mycelia'
2. cre[d-3O from D-glucose grown myceìia'
3.wildtypeA.nidulonsfromL-arabinosegrownrnycelia.
4. cre$d-30 from L-arabinose grown mycelia'
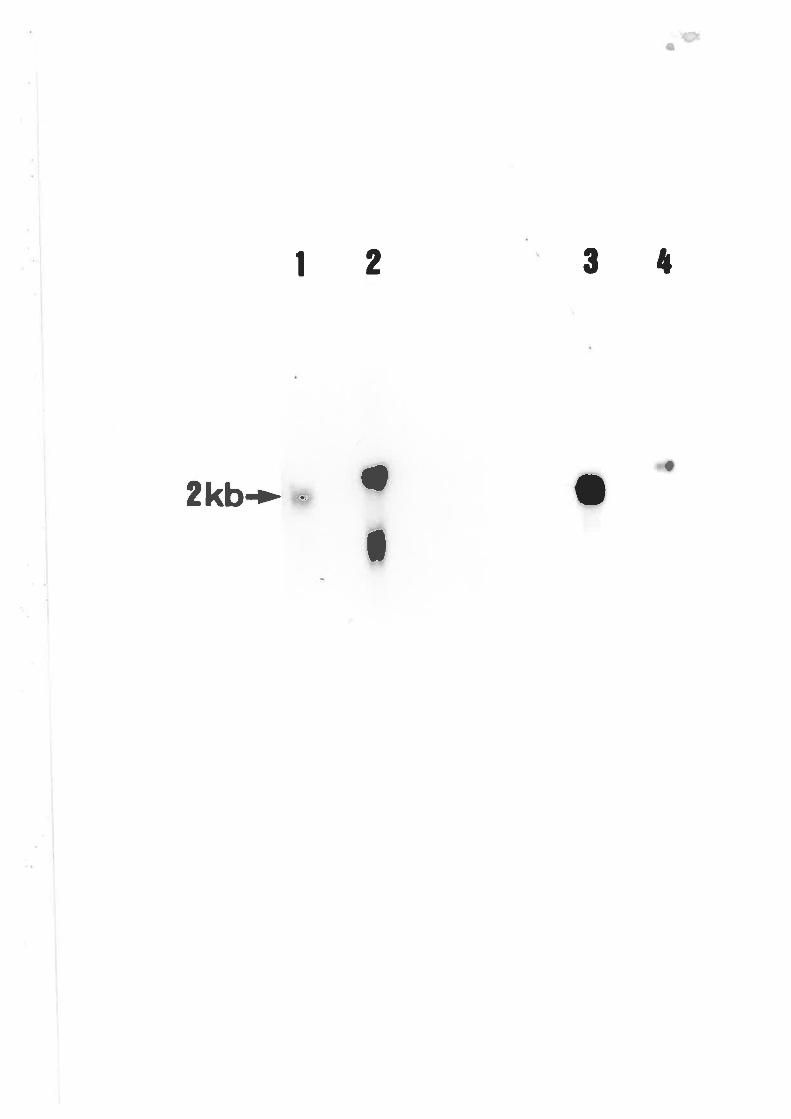
r2 34
¡a
ta
Zkb+ +:

103.
which has extended through the inversion breakpoint in creAd-30.
Therefore, the creAd-30 mutation does not represent a formal lack of
functjon alìele making it necessary to contruct a gene repìacement
strain to determine the phenotype of a lack of function mutation.
Chromosomal rearrangements' leading to the expression of truncated
protein products do not necessari ìy represent loss of function
alleles and this has been demonstrated for a number of mutant genes.
For exampìe, the ore|'-I8 mutation results fron a translocation
breakpoint within the oreL locus and leads to an inability to utiìize
nitrogen sources other than anmonium and glutamine. oreA'-18 can
revert intracistronicalìy via further reamangements invoìving the
3'end of the gene such that the gene is transcribed and a functional
protein expressed (Arst et ol. 1989) which lacks the non-essential
336 N-terminal amino acids (Kudla et ol. 1990). Studies have also
shown that a ìarge deletion of the 3' end of the ølcR gene resulted
in a gene stitl capabìe of weak partial complementation of the
otcül2| aìlele (Lockington 1984). Furthe*o"", nany oncogenes are
cellular genes that have been activated by translocation (reviewed by
Haluska et ol. 1987).
4.2 CONSTRUCTION OF A cTeA DELETION STRAIN:
4.2.1 The isolation of a larqer creA qenomic cìone:
The creA clone isolated by marker rescue was only 2.3kb in size.
Although this clone contained the required sequences to complement
the creA mutant alìeìes, Northern analyses showed that this genomic
clone was not much ìarger than the corresponding rnessage it detected.
It was therefore necessary for the construction of a cre[ deìetion

104.
strain to isolate a ìarger genomic clone which contained a sufficient
amount of cre[ flanking sequence.
The creA clone in pANC4 was shown to hybridize to a 7.5kb EcoRI
fragrnent by Southern analysis. DNA from wildtype Á. nidulons was
digested with EcoRI to completion and fragments in the size range 6 -
8kb were purified. These fragments were cloned into the EcoRI site of
pUC19 and transformed into E. coZi strain JM101 to create a partial
plasmid library. 900 colonies harbouring recombinant plasmids were
screened using the insert of pANC4 as a probe. 4 colonies were
identified by this method as harbouring sequences homologous to the
pANC4 insert. Southern anaìysis of mini plasmid preparations from
these colonies showed that two of these contained EcoRI inserts of
the correct size which were homo'logous to the origina'l clone in
pANC4. This plasmid was designated pANC6 and a partial restriction
map is shown in Figure 4.2.I.
4.2.2 The construction of a creA qene replacement vector:
The ríboï gene in pPL3 was chosen as the transformation
selection marker to be used for the construction of a plasmid to
repìace the wildtype A. nidulons cre[ gene. The 7.5kb EcoRI insert of
pANC6 was purified and ligated into the EcoRI site of pUCBX (pUC19 in
which the EomHI and Xbol sites had been deleted from the polyìinker).
This plasmid was designated pANCT and was digested with BonH.l and
Xbol to cut out the 3.5kb EomHl-XbøI fragment containing the cre[
gene. The larger fragment generated, containing pUCBX along with
sequences flanking the creA gene, was purified and end filled. This
was then blunt end ligated with the 2.3kb KpnI fragment of pPL3 and

105
Figure 4.2.L: Partial restriction map of the insert in pANC6.
Triangles indicate the extent of the insert in pANC4'
Restriction site abbreviations:
B - BomHI
E - EcoRI
Ev - fcoRV
H - Híndlll
Ps - Pstl
Pv - PvulI
S - SolI
Sc - SøcI
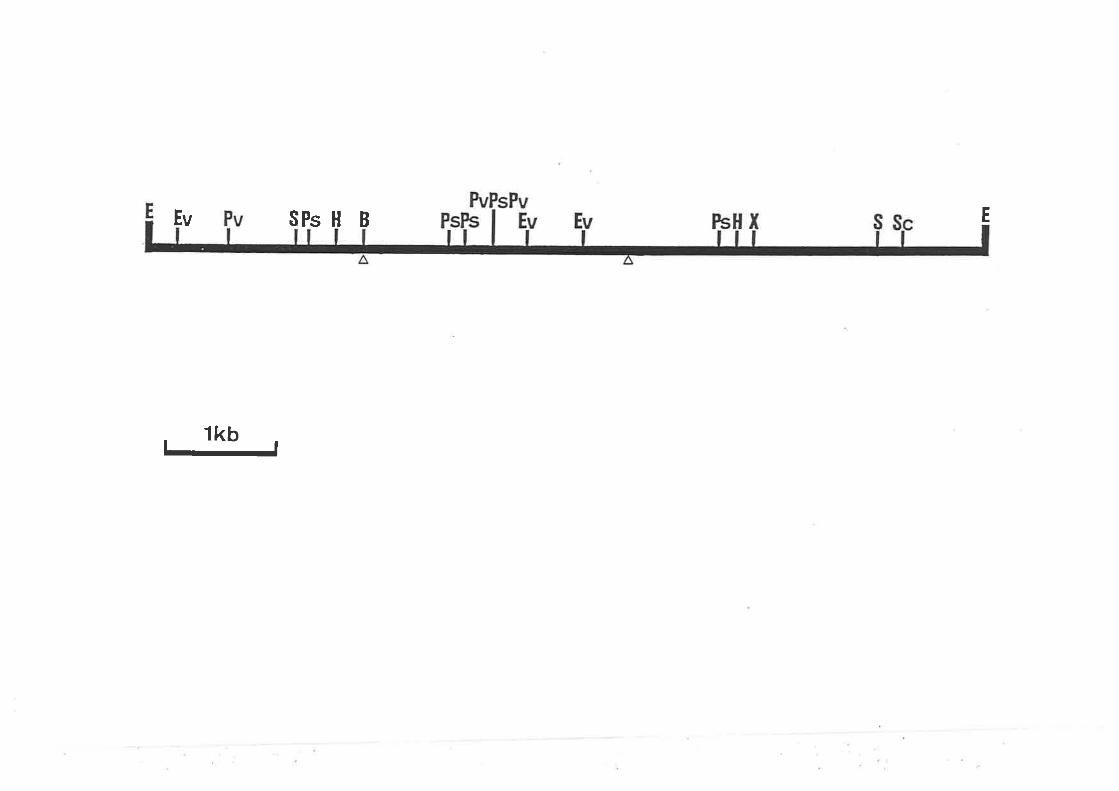
Ev
| 1kb I
SPs H B E

106.
containing the ríboB gene which had also been end filled- The
ligation mixture was transformed into E. col¿ strain DHl and colonies
harbouring the desired plasmid construct were identified using
dupìicate colony blots probed separately with the 2.3kb Kpnl insert
of pPL3 and the insert of pANC6. A restriction map of this 9.2kb
plasmid, pANC8, is shown in Figure 4.2.2. It contains the riboB gene
inserted between 1.95kb and 2.lkb of sequence normally flanking the
creA gene in the genome. pANCS was aìso used as a probe to Southerns
of pUC19, pANC6, pANCT and pPL3 digested with the appropriate
restriction enzymes in order to confirm that the desired construct
v,,as correct. pANCS used as a probe to Northerns of wildtype A.
nidulons total RNA showed that only one message was detected which
corresponded to the rióoB transcript (Figure 4.2.3). Therefore, pANCS
did not contain any sequences which, if transcribed, could produce
message homologous to creA.
4.2.3 The creation of a creA qene reolacement strain of Á. nidulons:
The ,4. nidulons strains c61 (yAl; pyro\4; nícB}; nii[4 riboB})
and C62 (b¿41; AcrALi gotELT foc\303; sB31' ríboB?) were both derived
from a cross between J7 and MSF and were transformed with the gene
replacenent construct pANC8. RiboB+ transformants were selected on
appropriateìy suppìemented protoplast media. Thirty RiboB+
transfonnants of strain C61 (TC611 - TC6130) and twenty eight of
strain C62 (TC621 - TC6228) were purified for further analysis. 0n
minimal medium at I of these transformants except TC622 showed
wi'ldtype morphology. The colony morphology of TC622 was extremely
compact, not unlike that expected for a creA mutant Strain.
Similarly, this transformant showed very weak growth on sucrose

107.
Figure 4.2.2: Partial restriction map of the insert in pANC8. The
open region contains the riboB gene of A. nídulons and the coloured
regions are sequences flanking the creA gene in pANC6.
Restriction site abbreviations :
B - BonHI
E - EcoRI
Ev - EcoRV
H - Híndlll
Ps - Pstl
Pv - Pvull
S - SOII
Sc - SocI
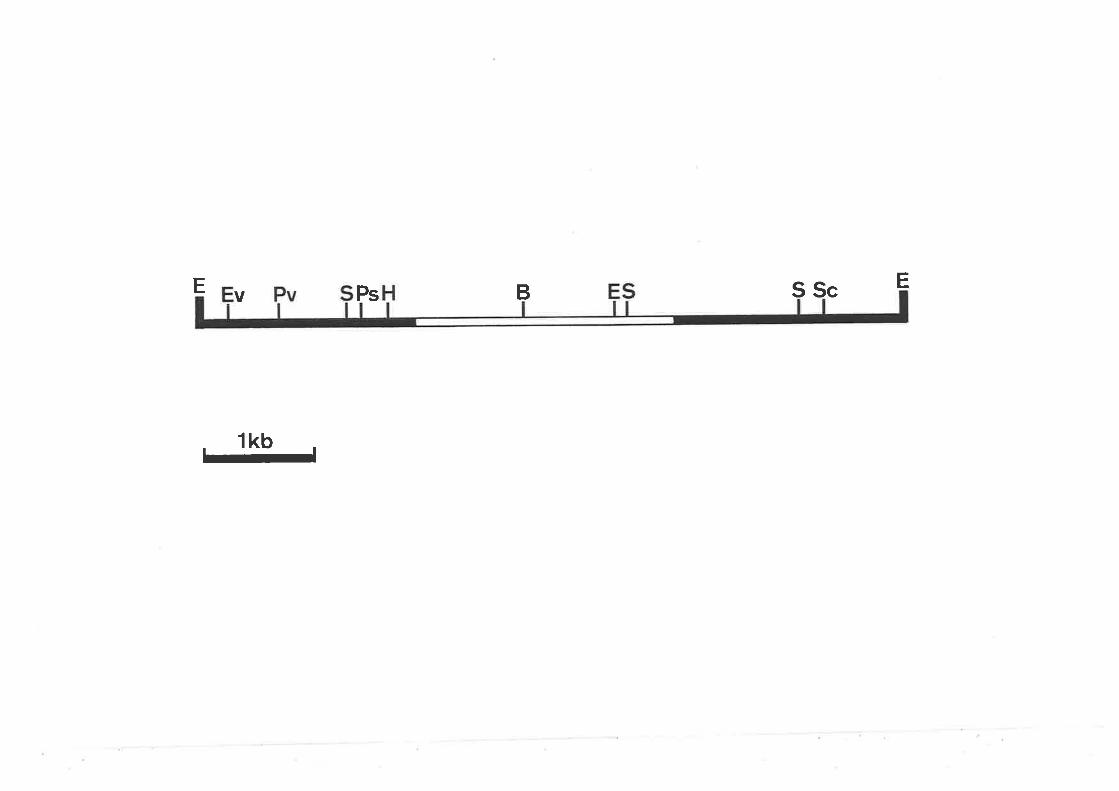
EBvE Ps SSc
hlL

108.
Figure 4.2.3: Northern bìots of wi ldtype ,4. nidulons RNA isolated
from D-glucose grown mycelia. Each pair of tracks contains
approximateìy 20ug of tota'l RNA.
Panels were probed with:
A.KpnlfragnentfrompPL3containingtheriboBgene.
B. pANC8.
c. Bonïl-xbol fragment from pANC6 containing the creA gene'

-{plt ,.tb t
ora)a
--rv g

109.
medium containing 2.5mM al ìy'l alcohol , whi le the rest of the
transformants had wiìdtype leveìs of growth on this medium. DNA was
isolated from each of these 58 RiboB+ transformants. Southerns of DNA
sampìes digested with EcoRI were probed with the 3.5kb BonHI - Xbql
fragment from pANCT in order to determine whether or not any of these
transformants were the result of a gene repìacement event at the creA
locus. Alì the transformants, including TC622, showed hybridization
to the native cre| EcoRI fragment at about 7.5kb and therefore no
transformant had the native creA gene repìaced with ríboB (for
example see Figure 4.2.4'). Transforrnants resuìting from a gene
replacement event with pANCS would not be expected to show
hybridization to this EomHI-Xbøl fragment from pANCT (Figure 4-2.5) -
At the same time as these experiments were being undertaken, pANCS
t{as also used to transform a dip'loid strain since the possibi I ity
existed that a true creA null allele may be lethal, iî which case a
gene replacement event could not be obtained in a hapìoid strain.
A dip'loid strain, C63, was constructed between strains C61 and
C62 and used in transformation experiments wìth pANC8. RiboB+
transformants of C63 were seìected on protoplast minimaì mediun with
10m,il sodium nitrate as the nitrogen Source, thus maìntaining
selection for the diploid state. Fifty of these transfonnants
(TC631 - TC6350) were purified and picked onto 1% cornpìete medium
containing various concentrations of benlate for the formation of
haploid sectors. The rationale for this was that if a cre[ gene
replacenent event had occurred in one of the chromosomes(I) in the
diploid, and this event was lethal in hap'loid cel ìs, then
transfonnants which were the result of a gene replacement could be
identified as segregating either only green or only yellow hap'loid

110.
!,
ü
r
Figure 4.2.4: Southern blot of EcoRI digested DNA from pANCS
transformants of strai n C62 probed with the BonHl-XboI fragnent of
pANC6 containing the creA gene. The amounts of DNA in each track are
not equal.
Tracks are: 1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
Bacteriophage lanbda DNA digested with HíndIII was used as a marker
of molecular weight.
c62
TC62l2
TC6213
TC62r4
TC6215
TC6216
TC62r7
TC6218
TC6219
TC6220
rc622l
TC6222
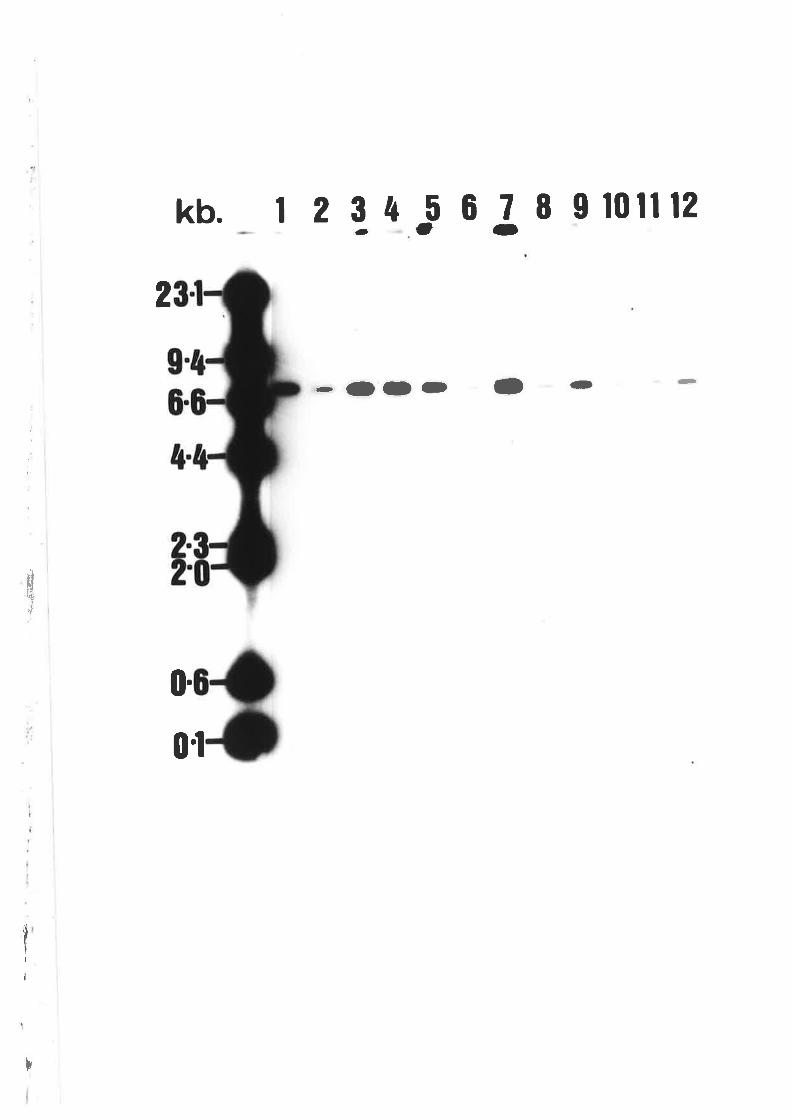
¿l
9 l0ll l22 3 4 5 6 7I.DeaDIkb.
0'l
2
.-1-,- tDO¡ O
$,l
0.
tI
!

t,
111 .
Figure 4.2.5: StrategY
nidulons.
used for deleting the creA gene from A
(a). The crel gene present within a 7.5kb fcoRl fragrnent in the
genome.
(b). Insert of pANCS constructed to repìace the cre\ gene. It
contains the ríbo} gene inserted between sequences that flank
cre[ in the genome.
(c). The 2.6kb and 3.3kb EcoRI fragments expected in the genorne of
a creA deletion strain created by recombination events between
fìanking sequences in (a) and (b).
I
'i
I
I
r
{

++++++riboB
** 9EA *********+++
+++++
+++
++
++++
xBE E
(a) 7.5kb
E E
E
(D
(c)
EE
6.3kb
6.3 kb
tlkbl
,*** ** ** *****l riboB r ++++++++++++

I
TI2.
I
sectors on complete benlate medium. Since conidial coìour
distinguishes haploids on the basis of which chromosome I is present,
transformants could be screened by eye.
0f the 50 RiboB+ transformants screened, two failed to produce
hapìoids of both colours. TC6330 fails to sector green while TC6332
fails to sector ye'l'low haploids. Both these transformants segregated
all other chromosomes as determined from the genotypes of hap'loid
progeny. Alì haploids derived fron both transformants were
phenotypicaì'ly Ribo- and therefore the transforming sequences were
located on the chromosome I not represented in haploids.
Similarly, TC6330 and TC6332 also failed to produce hapìoids of
both colours when hapìoids were selected on benlate medium containing
glyceroì or L-arabinose or fructose as the sole carbon source'
el ì iminating the possibi'l ity that haploids unabìe to uti ì ize
particular carbon sources were segregating. The failure to segregate
haploids of both colours was detennined not to be temperature
specific by the selection of hapìoids at 25"C, 30oC, 37oC and 42oC on
media containing the various carbon sources.
DNA was isolated from strains c61, c62 and C63 and the
transformants TC6330 and TC6332. Duplicate Southerns of EcoRI digests
of these samples were probed with (I) pÞ006 plus the 3.5kb BonHI
Xbol fragment of pANCT and (II) pUCl9 pìus the 3.5kb Bonïl - XbøI
fragment of pANCT (figure 4.2.6). The autoradiogram of fiìter (I)
showed that approximateìy equal anounts of each DNA sample were
present as shown by the intensities of the larger band corresponding
to the single copy of the ørgB gene. Hybridization to the creA native
band on this filter showed that, ôt least in the case of TC6330'
!1

113.
Figure 4.2.6: Southern ana'lyses of gene disrupted diploids TC6330
and TC6332. Each track contains 2ug of DNA digested with fcoRl from:
1. C63
2. TC6330
3. TC6332
4. C61
5. C62
From right to ìeft the panels were probed with:
1. pM006 and the 3.skb Bonïl-xbøI fragment from pANCT containing
the creA gene.
2. pUC19 and the 3.skb Bont.l-xbol fragnent from pANCT containing
the creA gene.
3. The 6.9kb Bon},l-xbol fragment of pANCT containing pucl9 and
sequences flanking creA in the genome'
4. pUC19.
N.B. þlhile the band of hybridization corresponding to the native
cre| band in c61 is not as intense as that for c62 in panels 1 and
2, equal intensities were consistently seen on repeat fjlters'
Autoradiograms were exposed without the use of intensifying screens'
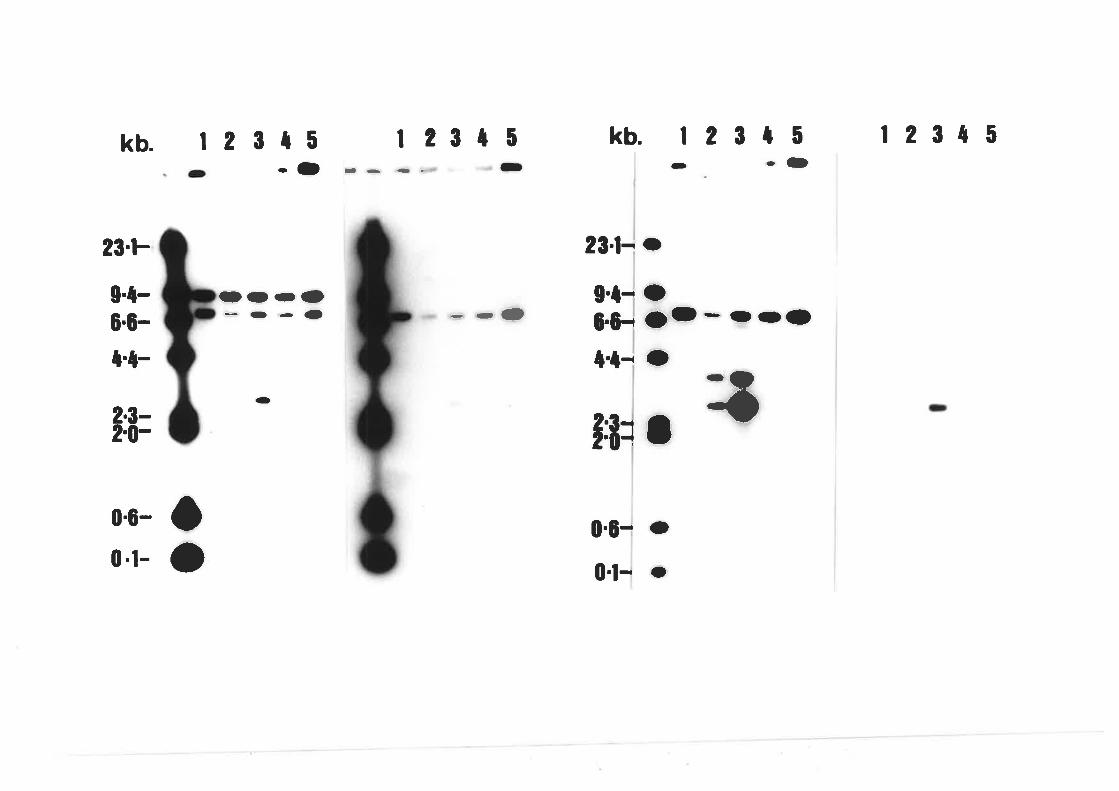
kb. l2 3¡5-(D
23.t-
t 2 3 ¡ 5
-
kb. l2 3t5--
23.1- O
9'l- (D
6.0-r ¡O - (DotD
l'lr O
îFT
0'0- l'
0'l- o
12345r-It---
9'[-6'0-
4.1-
2.!-2'0-
O(DorD(--(D
(-
- -lO
oÇ
{
0.6- o0.1- o

114.
there appeared to be onìy half the amount of hybrization when
compared to the diploid parent strain. The smaller band hybridizing
in TC6332 represents pUCl9 sequences as shown when the same samp'les
were probed with pUCl9 alone. TC6330 did not contain any pUCl9
sequences and therefore appeared to be the result of a direct gene
replacement event of one of the native creA genes in the dipìoid.
Further to this, when the same samples were probed with the larger
Bonïl - XbqI fragment of pANCT (containing pUCBX and the cre[
fl anki ng regions) TC6330 showed about hal f the amount of
hybridization to the native band at 7.5kb as compared to the parent
strain and to two smaller bands of 2.6kb and 3.3kb. It can be
determined from the restriction rnap of the insert of pANCS that this
is the expected pattern of hybridization for a gene replacement
diploid strain (Figure 4.2.5). TC6332 probab'ly has nore than one copy
of pANC8. l.lhile TC6330 showed patterns of hybridization expected for
a gene replacement event, it may also have a small amount of cre[
flanking sequences integrated into another site ìn the genome.
Autoradiograms exposed for more than a week showed a very faint band
of hybrìdization at approximately 500bp, to TC6330 DNA probed with
cre¡ flanking sequences. DNA from six haploids derived from TC6330,
each having a unique chromosome compìement were screened by Southern
analysis using the creA flanking sequences as a probe to determine to
which ìinkage group these extra sequences could be assigned. None of
the haploids contained extra transfonning sequences and therefore
these must have been integrated into the sane chromosome I as the
gene replacement event in the diploid. As far as can be determined
this does not interfere with the gene repìacement event. Therefore,
analysis of TC6330 at the molecular level demonstrated that it is a
transformant resuìting from a gene repìacement event of one of the

115 .
creA loci.
The gene disruption plasmid was used to transform two hapìoid
strains. 0f the many transformants obtained, none had a cre[ mutant
phenotype or a true disruption of the CreA locus. This result
suggests the hypothesis that a disruption of the CreA locus tp give a
null aìlele reults in a lethal phenotype. This hypothesis would
predict that a gene disruption could be obtained in a dipìo'id where
the undisrupted allele couìd complement the nulì allele. Such a
dipìoid would have the phenotype that it could not be broken down to
give a haploid sector bearing the chromosome carrying the disrupted
aììele. This experiment was carried out and two diploid transforrnants
with this phenotype were obtained. As predicted by the hypothesis,
both of these transformants were shown by Southern analysis to have a
gene replacement of one of the two creA alleles. Thus it is likely
that the creA product is essential for cell viability'
The creA mutant alìeles isoìated by various selection procedures
would therefore represent a class of alleles with altered function
rather than total loss of creA function for which mutants could not
have been identified in the haploid state. This raises the question
as to whether cre| acts in a posìtive or negative manner in
regulating the expression of genes subject to carbon catabolite
repression. Previous anaìyses have assumed creA alleles to result
from loss of function.
4.2.4 Veqetative versus conidial lethality of the creL nul l
phenotype:
Although it was quite evident that a creA null aììele could not

116.
be segregated out in hapìoidization analysis, the question remained
as to whether the phenotype was lethality or failure to fonn
conidia. It frôy, in some instances, be difficult to distinguish
between haploid aconidial mutants and aneuploid sectors during
haploidization. However, when non-conidiating sectors were picked
onto 1% cornplete medium they failed to show the vegetative myce'liaì
growth expected for aconidial mutants and were probably aneup'loid. In
addition to this, a 'large number of non-conidiating sectors from
haploidization pìates were scraped into a suspension and plated out
at low density onto 1% compìete medium with no evidence of colonies
which grew but failed to conidiate.
A further experiment to show that the creA null allele is lethal
in vegetative ceìls prior to the development of conidia was carried
out using mycelial cultures of TC6332. TC6332 was used in this series
of experiments in preference to TC6330 so that haploids containing
the deletion aìlele could be selected for by the absence of both
riboflavin and biotin from the growth media. Hapìoids from this
diploid containing the wi ldtype al lele, which is on the same
chromosome as the óiAl narker, are auxotophic for biotin and are
selected against under these conditions. TC6332 was grou,n overnight
in liquid cultures supplenented with sodium thiosulphate, pyridoxal
hydrochloride, nicotinic acid and ammonium tatrate as the nitrogen
source and various concentrations of benlate (or without benlate),
thereby allowing hapìoid celìs with the genotype bi{+ riboB+ to grow
simultaneously selecting against brAl haploids having the wi'ldtype
creA allele. Mycelial cultures wene harvested, ground coarsely in a
coffee grinder and spread onto 1% simi'larly supp'lemented minimal
medium containing various concentrations of benlate. Mycelia that

tI7.
continued to grow on such media must therefore be biA+ and ribo9+.
All of the resulting myceìial growth on these plates eventually
conidiated and were found to be diploid. No biA+ rib9+ areas of
growth were haploid or failed to conidiate. This was consistent with
the hypothesis that a creA nulì allele is lethal in vegetative cells
as opposed to failing to form conidial progenito¡ ceìls or conidia
thenselves. If the latter was the case then areas of haploid
vegetative growth should be identifiable. These experiments suggest
therefore that the creA null allele is recessive lethal in vegetative
ceì I s.
4.2.5 Rescuinq the abilitv of TC6330 to form haploids containing
chromosome I having the gene replacement:
The failure of TC6330 to form haploids containing one member of
the chromosone I pair was due to the inabiìity to form haploids
containing a creA gene repìacenent. Such a siiuation should be able
to be rescued by inserting a wildtype copy of creA into any
chromosome except the chronosome I carrying the wildtype aìlele inTC6330.
In an attempt to demonstrate this, TC6330 was co-transformed
with the wildtype cre[ gene in pANCT and pAmPh366-1 conferring
resistence to blenoxane as a doninant selectable marker. Diploid
transfonnants were selected on minimal protoplast nediun contain'ing
sodium nitrate as the nitogen source and blenoxane. Twenty bleomycin
resistant transfonnants were picked onto I% complete medium
containing benlate. Four of these diploid transformants appeared to
segregate both green and yellow haploid sectors unlike the parent

118.
strain which on'ly segregates yellow hapìoids. The genotypes of six
green haploids derived fron each of these transformants were
determined. All were found to be b¿41 aìong with riboB2, while biAl
and ribo9+ are on the same chromosome in the untransformed diploid
parent. These haploids were also creA+ as determined by growth on
sucrose pìus aìlyl alcohoì mediun. Furthermore, DNA from one of these
green haploids was shown by Southern analysis to have the native creA
band intact when probed with the insert of pANC7. Therefore, these
haploids must have arisen by mitotic recombination events in the
dipìoid transfonnants rather than being CreA+ transfonnants. Such an
apparently high frequency of mitotic recombination, which is normaìly
a rare event, may be explained by the use of blenoxane as the
transformation selection system. The antibiotic bleomycin is
cytotoxic because it causes both single and doubìe stranded breaks in
DNA (Waring 1981) and it is 'likeìy that such an effect may increase
the frequency of mitotic recombination events in dip'l-oid cells.
Bleomycin may therefore be of use in experiments where mitotic
reconbination is required.
In further attempts to rescue the phenotype of the cre[ nul I
aììele, TC6330 and TC6332 were co-transfonned with either pANCT and
pAN7.1 carrying the dominant selectable narker for hygromycin
resistance, or pANCT and p3SR2 containing the øndS gene. Very few
Hyg' and AmdS+ transformants of TC6330 and TC6332 were obtained, the
reason for which remains unclear. None of these transformants
produced both yeìlow and green hapìoid sectors during haploidization
analysis and were probably not co-transformants. Further attempts are
in progress to obtain larger numbers of transfonnants of these
strains to increase the possibiìity of identifying stable co-

119 .
transfonnants.
4.3 SEQUENCES H0M0L0G0US T0 creA FR0M Á. nidulønsz
The cre{ cìone from A. nidulons was used as a probe to DNA
samples from various organisms. Southern hybridizations using this
probe were performed at low stringency (37"C). Sequences hybridizing
to creA were found in DNA from A. níger, A. oryzoe, /l/. crosso,
Penicílliun chrysogenum and S. cerevísioe. The genome sizes of these
fungi are given in the legend to Figure 4.4.1. As shown in F'igure
4.4.1, the strongest hybridization was seen to DNA from A. niger with
detectable, but lesser amounts of hybridization to the other fungi.
It was not unexpected that if creL is an essential wide domain
regulatory gene that similar sequences would be found in fungi
closely related to Aspergillus since such classes of genes are
expected to be highly conserved. This supports further the idea that
cre[ has a function which is central to the mechanism of carbon
catabolite repression rather than being specific to a particular
netabolic pathway. No hybridization was detected when the creA clone
was hybridized at low stringency to DNA from E. colí, ltlelonpsors lini(flax rust), Nicotiøno tobocun, Snínthopsis crøssícsudoto or Hono
sopiens. Fai lure to detect hybridization in these samples may
reflect their large genone sizes (listed in the 'legend to Figure
4.4.1) and/or their evolutionary distance from ,4spergíllus.
4.4 SUI'O,IARY AND COI{CLUSIOI{S:
The clone which has been isolated fron Aspergillus nídulqns by
narker rescue techniques has been shown to be the region which is

t20.
Fi gure
probed
gene.
4.4.1: Southern blot of DNA isolated from
with the insert of pANC6 containing the A'
various fungi
nidulons creA
Tracks are:
1. Sug A. niger DNA digested with EonHI'
2. 15ug rV. crossø DNA digested with EomHI'
3. 5ug S. cerevis¿øe DNA digested with XbøI'
4. 5ug ,4. niger DNA digested with bomHl'
5. 15ug Á. oryzoe DNA digested with BønHI'
6. 15ug P. chrysogenun DNA digested with 8ømHI'
Hybri di zati ons u,ere carri ed out at 37"C usjng the same condi ti ons
outlined in Chapter 2. Autoradiograms were exposed for one week at
-80oC using ìntensifying screens.
ilo hybridization was detected to E. colí, M. Lini, N. tobocun, s'
crossícoudsto or H. sopíens DNA samples.
Haploid genome sizes:
E.co-i-4.5X103kb(BrittenandKnohnelg63).S. cerevisíse - 1.8 X 104kb (Ogur et ol' 1952)'
lt/. crosso - 4.3 X 104kb (Horowitz and Mcleod 1960) '
Aspergilzus* - 3 X 104kb (Timberlake 1978, Brody and
Carbon 1989).
S. cross icoudøto - 4 X 106kb (Hayman et ol ' 1932) '
H. soPiens - 3 X 106kb (l.{hite 1973) '
,V. tobøcun - 2 X 104kb (Bennett et ol' 1976)'
M. tini - 6 X 104kb (f '¡rmi s et oI ' 1990) '
*A. nidulons, A. niger, A. oryzae and P. chrysogenum probably have
sirni I ar hapì oi d genome si zes .

kb. f2I kb.
2?;l-
2-þ?0-
ITIr|nÇ Çt
9'l-F0-
['[-
:-4;;r

r21.
disrupted in the creAd-3O alìele. Analysis of creAd-30 shows that
thjs allele is a result of a translocation breakpoint occurring at a
position corresponding to the 250bp PsúI fragment in the cloned
region. The pericentric inversion in creAd-30 leads to an altered
creA message being expressed, suggesting that creAd-30 is not a true
loss of function allele. In order to construct a creA gene disruption
strain, a ìarger clone of the region has been isolated. This was used
to create a diploid strain containing a creA null aììeìe. As far as
can be determined, analysis of this strain demonstrated that a creA
null alìele is lethal in haploid cells. Although creA appears to be
an essential gene and the mutant al leles confer altered cre[
function, the way in which creA acts at the molecular level remains
unclear. Molecular characterization of the creA gene may suggest the
role creA has in carbon catabolite repression and whether or not ithas features suggestive of a gene encoding a negative'ly acting
repressor or an activator molecule.

t22.
CHAPTER FIVETHE PHYSICAL CHARACTERTZATION OF
THE Asperg¿ZZus níd¿tlarts creA GENE-
The molecular characterization of the cloned creA gene from A.
nídulons and computer analyses of the putative CreA protein are
described in this chapter. The physical analysis has included the
determination of the nucleotide sequence of the creA gene and the
structure of its transcript. Computer analyses of the derived amino
acjd sequence of crel have shown that it contains a number of
features characteristic of reguìatory and DNA binding proteìns. Amjno
ac'id sequence comparisons between the putative CreA protein and other
proteins of known function ìead to suggest-ions as to how CreA
functions at the molecular leveì.
5.1 THE PHYSICAL ANALYSIS 0F creA:
5.1.1 Nucleotide sequence analvsis of creA and characterization of
the codinq reqion:
Subclones of a 2.7kb region starting at the internal EonHI site
in pANC6 and spannìng the creA gene were sequenced using the strategy
outlined in Figure 5.1.1. The direction of transcription of the creA
gene is from the internal BomHI site in pANC6 towards the XboI site
(as reported in Chapter 3, section 3.7). The complete nucìeotide
sequence of this 2.7kb region containing the creA gene is shown jn

L23.
FIGURE 5.1.1: Partial restriction map of the insert in pANC6 showing
the strategy used for the sequencing of the A. nidulqns creA gene
and its flanking regions. The extent and direction of sequence
obtained fron the insert of pANC6 is indicated by the arrows below
the restriction map. Sequence data obtained from cDNA clones ìs aìso
shown. The positions of the start (ATG) and end (TAA) of transìation
are indicated by the vertical arrours.
Restriction site abbreviations : B
E
Ev
H
Ps
Pv
S
Sc
X
BqmH.l
EcoRI ,
fcoRV
Híndlll
PsÚ I
Pvull
Sol I
.SocI
Xbol
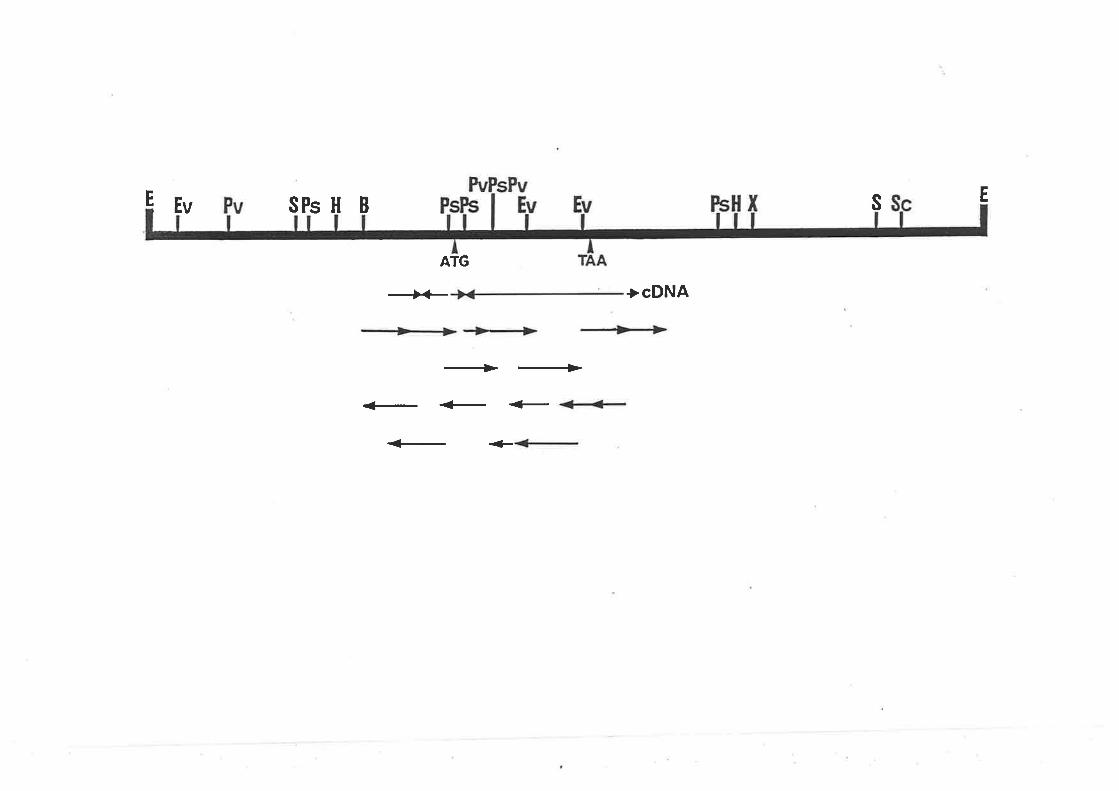
E Ev SPs H BEs
ATG
->+- +cDNA
-+#
# <_ #
<- +

r24.
Figure 5.I.2.
In order to identify the codìng region of the creA gene, twelve
cDNA cl ones were i sol ated from the cDNA 'l i brari es descri bed i n
Chapter Two. These cDNA clones were isolated either using the entire
BomHI-XbøI insert of pANC4 or the I.zkb BonHl-PstI fragment
containing the 5' region of the gene as probes to pìaque lifts of
these libraries. In total, seven cDNA clones of different 'lengths
were isolated. Two of these, designated pCDl and pCDS were subcloned
and sequenced and found to span the entire genomic sequence from
nucleotide position -599 through to 1602. Therefore, sequence
analys'is of these cDNA clones demonstrates that the creA gene does
not contain any ìntrons. The region covered by these cDNA clones is
indicated in Figure 5.I.2. Furthermore, a computer search failed to
reveal any putatjve intron splice sites or consensus lariat sequences
which would allow intron excision and the maintenance of the reading
frame. Aìthough the majority of genes anaìysed from Á. nídulons and
other filamentous fungi have been shown to contain short introns of
less than 100bp in length, a number contajn no introns (reviewed by
Gurr et ø2. 1987) including the gene involved in annnonium repression,
ore|, which has a single long ORF in the sequenced region of 2l57bp
in length (Kudì a et ol. 1990).
The entire sequence was compared to nucleic acid data bases to
determine if any sequence similarities couìd be found between the
creL sequence and other genes of known function. No significant
sequence similarities could be identified although overaì'l sequence
similarity was shown between the creA sequence and that of the brl[gene from Á. nidulons. However, more precise alìgnments of these two
sequences failed to show any extended stretches of homology and the

125.
FIGURE 5.L.2: Nucleotide sequence of the A. nídulons creA gene and
its flanking regions. Nurnbering of nucleotides begins with the first
nucleotide of the start codon as +1. Putative TATAA and CAAT boxes
are underlined. The major start point of transcription is indicated
by a solid wide arrow. The extent of cDNA clones pCDl and pCDS are
shown by smalì arrow heads and wide open arrows respectively' The 3'
end of pDCS aìso denotes the position of polyadenylation of the creA
transcript. Possible signaìs for po'lyadenylation are indicated with
asterisks. The putative amino acid sequence is shown below the
corresponding codons. The boxed regions contain the two putative
zinc fingers and an alanine rich region is indicated by stars under
each of the residues. The sequence is continued overleaf.
It1,[
i
I
*

-827
-760
-693
-626
-559
-492
-425
-358
-29r
-224
-t57
-90
-23
GGATCCCCAAGCAGCAGGCGATCTGGAATGAGCACGTTCTTTTTTTATTTTCTTTTCTTTTTGCCCT
ATTTGGTAATTTTTTGTCTTTTTTTTGG
TCTTTTTTTTTTTTTTTTTTTTTTCTTTTCTTTTTTGCCCCCçAAJATCTTCATTCATTCCCCACAC{}+AAAGTTCTCACILIA'JCTTTTTCTTTTCTCCCTCCTTGCTCCGATTCCGATAACCCTCCCCCTCTCC
GTAGGCTCAACCTGTCTTTTTGCCAACTCCCCTCCCCCGAGTTCCGTTTCATTCTTCTTCCCCCCAT
CCTCCATTCCGGCTCTCATTCTTATATCCTCCTTCCCCCAGATTG TTCTCCAGATTTTTTTTTTCTT
CCTCTCTCTTCCTTCTGATCCTTTCCTCGACTCCGTATCATTCCCTCGACTTCATACATCCCGACGC
CGCCACATACCAACACATCGTTTCCCAACTAGGACCACCTACCTATATCGGCAGCTCTTCAGCGTGG
CGTCTTACCTCCCTTCGATAAAGG AAAGTCGTCATTTTGGTAATTTCGCCTTCTTTTTTTACGACTG
GCTCTCAGTCAAGCACACAAACAACAACTGTTCTCATTCTG CTGATTCTCGAAATCCCATCTCG TCC
TGGAAAACCGACCTAGCACGG CTTAGTGTGGTTGGTGATCGTCTCG CG CCGAGCTAACCCCTGGCTG
TGAGAACATCCTTTTTCCTCGGCTTATCACMCCCTTTTTAGCTCCGTCCCACCCTGG TCTCCTCGG
AGCTGCAGAAGGACGAGCTTCAC ATG CCG CAA CCA GGA TCG TCA GTG GAT TTCMet Pro Gln Pro Gly Ser Ser Val Asp Phe
31 TCA AAT CTG CTG AAC CCT CAA AAC AAC ACG GCC ATC CCT GCC GAA GTC TCC
Ser Asn Leu Leu Asn Pro Gln Asn Asn Thr Ala Ile Pro Ala Glu Val Ser
82 AAC GCT ACA GCT AGT GCT ACC ATG GCT TCA GGA GCC AGT CTG TTG CCA CCT
Asn Aìa Thr Ala Ser Ala Thr Met Ala Ser Gly Ala Ser Leu Leu Pro Pro
133 ATG GTG AAG GGC GCT CGC CCG GCT GCA GAG GAA GCT CGT CAG GAC CTT CCT
Met Val Lys Gly Ala Arg Pro Ala Aìa Glu Glu Ala Arg Gln Asp Leu Pro
184 CGA CCA
Arg ProTAC AAG TGC CCC CTG TGC GAG CGC GCC TTC CAC CGC CTA GAA CAC
|d
ï
r s Pro Leu Glu A Ala Phe His A Leu Glu His
235 CAA ACA AGA CAC ATT CGC ACT CAC ACT GG
Gln Thr Arg His lle Arg Thr His Thr Gl
337 TCG CGA ATC CAT AACSer Arg Ile His Asn
TI GAA AAG CCCyl elu Lys Pro
lcnr ecc rcc cAc
luis nta Cys Gìn
286 C CCC GGC TGC TCG AAG CGT TTC AGT CGC TCA GAT GAG CTT ACC CGG
Phe Pro Gly Cys Ser Phe Ser Arg Ser Asp Glu Leu Thr Arg HiA
AACI CCC AAC TCA AGA CGT GGA AAC AAG GCT CAA CAC
nsnl lro Asn Ser Arg Arg Gìy Asn Lys Ala Gln His
388 CTG GCG GCA GCC GCC GCA GCT GCALeu Ala Ala Ala Ala Ala Ala Ala+++++++
GCT GCG AAC CAA GAT GGT AGC GCG ATG
AJ,a AJ-a Asn Gìn Asp Gly Ser Ala Met++tI
,
439
490
541
GCG
Ala
TCTSer
TTC
AAC
Asn
GCT
Ala
TCC
AACAsn
ccrPro
AAC
GCT
Ala
GTC
Vaì
TAT
GGA
Glv
TCTSer
GCC
TCASer
CAAGln
AAC
ATGMet
GTC
Val
cAc
ATG
Met
GGG
Glv
ATG
cccPro
TCCSer
CGC
ccrPro
ccGPro
TCG
cccPro
GACAsp
AAT
AGC
Ser
ATTIle
CTG
AAALys
TCG
Ser
AGC
cccPro
cccPro
ccc
ATCIle
ccGPro
TAC
ACT CGA
Thr Arg
CAC TCTHis Ser
TCT CGT
I

,li
HI
592
643
694
745
796
949
1000
1051
ITO2
1 153
T2T3
1280
t347
1414
1481
1548
1615
1682
t749
1816
Phe
ACC
Thr
GCG
Ala
AGTSer
TCASer
GAG
Gìu
TATTyr
TCTSer
TGG
Trp
Ser
AGTSer
TCTSer
cAcHis
GCC
Ala
GATAsp
TATTyr
cGcArg
CGG
Arg
Asn
GAAGlu
CAAGln
CATHis
TACTyr
TCTSer
Tyr
CGG
Arg
GTCVal
ATG
Met
GCC
Aìa
TATTyr
ccGPro
GAG
Gìu
AGG
Arg
Ala
GCG
Ala
GAG
Glu
TATTyr
ATCIle
GCG
Ala
AACAsn
GTG
Val
ATAIle
Asn
TCASer
CGTArg
GGT
Glv
TCCSer
TCASer
CAAGln
cAcHis
TGTcys
His
TCASer
GATAsp
cccPro
cAcHis
CATHis
ccTPro
AGC
Ser
TAAEnd
Met
GGC
Glv
GAAGlu
cGcArg
AGC
Ser
cGcArg
Arg
ATG
Met
AGT
Ser
CATHis
ATGMet
GTC
Val
Ser
GATAsp
TTTPhe
GGC
Glv
AGC
Ser
AAG
Lys
Asn
ATCIle
GGA
Glv
AGC
Ser
CGT
Arg
CGT
Arg
Leu
AACAsn
TTCPhe
AGG
Arg
TCCSer
TCASer
SerV
CTTLeu
cGcArg
GGA
Glv
CATHis
AGAArg
Pro
CTTLeu
TCTSer
CTTLeu
TCG
Ser
ccrPro
Tyr
GCT
Ala
GGT
Glv
ccTPro
CAC
His
AACAsn
Ser Arg
ACG GCC
Thr Ala
CAA CGT
Gìn Arg
TCT CTTSer Leu
GAG GATGlu Asp
TCA CCC
Ser Pro
847 AAC TCG ACT GCT CCT TCT TCG CCT ACC TTC TCC CAC GAC TCC TTAAsn Ser Thr Ala Pro Ser Ser Pro Thr Phe Ser His Asp Ser Leu
898 ACT CCT GAC CAC ACG CCA TTG GCT ACG CCC GCC CAT TCG CCA CGA CTG AAG
Thr Pro Asp His Thr Pro Leu Ala Thr Pro Ala His Ser Pro Arg Leu Lys
TCT CCC
Ser Pro
CCA TTG TCG CCG AGT GAG CTA CAT CTG CCC TCA ATC CGT CAC CTA TCG CTTPro Leu Ser Pro Ser Glu Leu His Leu Pro Ser Iìe Arg His Leu Ser Leu
CAC CAC ACT CCG CGT CTC CGT CCA ATG GAG CCC CAG GCC GAG GGA CCC AATHis His Thr Pro Arg Leu Arg Pro Met Glu Pro Gln Aìa Glu Gìy Pro Asn
AACAsn
ccrPro
TCCSer
CAT GTT GGC CCA AGC ATA AGC GAT ATC ATG
His Vaì Gìy Pro Ser Ile Ser Asp Ile Met
GAA AAC TTC CGA TAC CTC AGG TGC CCA AAG
Gl u Asn Phe Arg Tyr Leu Arg Cys Pro Lys
ATCCTAGCGGGTTTACTTCAGTCTCTTCATCAACCGCAA
'l'
ATTCCGTTGCTGGTGGTGACTTGGCTGAGAGGTTCTAATCCGGCCAAAAAACTTCGTTTTCTTGTTA
GGCGTACGAAAGATATAGACCTTTGCATTTCTGGTTGATTCATGGGCATCATTGGTGTCACGGAATT
AGGTTGTTTGACGATTCTTCACACTGTTTGAGTACACTATTTTGCGAGGCGTTGCCCCTATAGACGG
ATTATCCCTTGCCTTCACACACAAGTCTTGTTTTCATTCTATTCACTTCTCTTCTTTGACACCTATT
Y
ACACCAACGTTTTATCTCTTTCCTTTCGAGATCCTCTCCTATCGGACAAGCTCCTCAGCAGTTTACT
IATTTTCTTGAGGTTACACTTCITNNTCNÏNTNCNNNAGAACGAGTTTCCTTGAATGCCAACACTGTA
CCATTTATCTCTTTCCGTATTAAATGAATACTCCTCATGAGGACTACAACTGGACTGCATTTTTCTA
GCGACTATGGAGTACCCCGCATAGTCTCCACAGAGCATCCTCCATATCCATAGCTTATACAGCCACT
GCCGTCAGTAACTTGTTAGTACATCTATCTTATATCTTACTGATAAGAAGGCTTAGTTAATCTCCATtII
I
3
CTGGAGGTGTAGATACTGTAGTCTATAGACACTTGCTCAATATC

t26.
EIt,;l
I
simi larity probably results from the hìgh frequency of serine
residues in the two encoded proteìns.
Codon usage within the creA gene is shown in Table 5.1.1. Many
genes from fungi, especially those that are highly expressed, have
been shown to have a bias against A and for C in the third position
of codons (reviewed by Ballance 1986, reviewed by Gurr et ol. 1987).
The codon usage jn creA is not random and some degree of codon bias
is present. However, codon bias is not marked jn the creA coding
region which may be a reflection of the low levels of expression of
this gene in,4. nídulons. Low codon bias is also found in the coding
regions of the regulatory genes oreA from A. nidulons (Kudla eú ol.
1990) and nnr from /V. crosso (Young et ol. 1990) and therefore may be
more a feature of reguìatory rather than of structural genes from
filamentous fungi. Furthermore, reduced codon bjas has also been
correlated with those funga'l genes for which there is a low turnover
of both its transcrìpt and the encoded proteìn'within cells (revjewed
by Gu'r et ol. 1987). AGN codons for serjne and arginine have been
found to be used infrequentìy in genes from filamentous fungi. In the
cre| gene there are 22 AGN codons out of 391. This frequency is
unusuaìly high with the CreA protein having a high serine content in
which 16 out of the 59 serine residues are encoded by AGN codons.
Filamentous fungal genes show a preference for the termination codon
TAA and this is used for the termination of translation in the creA
transcrì pt.
5.1.2 The 5' region of the creA gene:
þ
The major start point of transcription was mapped by primer

127.
Table 5.1.1: Codon usage in the creA gene of A. nidulons.
Glv
GAT 8GAC 4
Val cTc 3GTA O
GTT 1
GTC 5
Ala cCG 7GCA 4GCT 15GCC 13
Ser AGT 7AGC 9TCG 11TCA 13TCT 11TCC I
Lys
Asn AAT 3AAC 19
Met
Ile
ATG T2
ATAATTATC
Thr ACG 4ACA 2ACT 6ACC 4
Cys TGTTGC
GGG
GGA
GGT
GGC
I745
1
5
54
31
8372
I8
39
9L7
79
1316
Glu GAG 10GAA 7
Asp
Arg AGG
AGACGG
CGA
CGT
cGc
Trp
End
TGG 1
Tyr TATTAC
Leu TTGTTACTG
CTACTTcrc
His CATcAc
Pro CCG
ccAccrccc
II
AAG
AAA
Phe TTTTTC
Gln CAG
CAA
227
3335
1110 TGA O
TAG O
TAA 1
T
{i
ti
i

r28.
extension analysis (Figure 5.1.3) and was positioned at nucleotide
-589. Although some fungaì genes have been shown to have a unique
start site for transcription jnitiation, many genes have multìp'le
initiation si tes. Evi dence of 5' heterogeneity for the cre{
transcript is shown by the difference between the major start point
as mapped by primer extension analysis and the 5' end of the cDNA
clone pCDS which begins at nucleotjde posìtion -604 in the genomjc
sequence. However, aìthough pCDS is probably a full length cDNA clone
it has an anomalous tract of more than thirty thym'idine residues
upstream of the 5' end of homology to the genomic sequence. This can
not be accounted for by the presence of an intron which brings
together the ìong stretches of oìigo (d)T present between nucleotides
-788 and -658 in the genomic sequence. Furthermore, it is not
possible that this represents a polyA tail at the 3'end of a gene
since ìong 0RF's are not present in the complement of the sequence of
pCD5. It was therefore assumed that this region was an artifact
created during the construction of this cDNA ìibrary. Presumably, the
start site determined from primer extension analysis represents the
major start point of transcription and other minor start sites,
'including the one represented by the 5' end of pCD5, may have been
indicated by ìonger exposures of the primer extension products- The
region between the putative TATAA-Iike sequence and the major start
point of transcription is pyrimidine rich where 20120 bases are
pyrimidines. This is aìso seen in the pronoters of other funga'l
genes, particularly in genes from S. cerevisioe which are highìy
expressed. Pyrimidine rich promoter sequences are also found to be
cofflnon in highìy expressed genes from ,4spergillus (l,{ard and Turner
1986, Clements and Roberts 1986, Punt et ol. 1988, Ward et ø1. 1988).
The major start point of transcription of the ,4. niduløns creA gene

L29.
FIGURE 5.1.3: Primer extension analysis for the mapping the 5' end
of the A. nidulons crel mRNA transcrìpt. A Y-32P-labelled
o'l ìgonucleotide primer of the sequence TCTAACAAGAGGTCTAAA'
compìementary to nucleotides -454 to -436 (Figure 5.1'2') was
annea'led at 43"C to total mRNA isolated from wildtype A' nídulons
mycelia from glucose grown cultures and extended with M-MLV reverse
transcriptase by the method of Geliebter et ol. (1986) as described
in Section 2.7.10. The products of the extension (tracks 1-3) and of
a dideoxynucleotide sequencing reaction (tracks T, C, G and A) were
electrophoresed on a 6% polyacry'lamide sequencing gel (Section
2.7.5). The clone sequenced for the molecular weight marker was
pANCT primed with the same oligonucleotide as above.
The tracks are: 1. primer extension reaction using 20ui total mRNA'
2. Primer extension reaction using 10ug total nRNA'
3. Primer extension reaction without mRNA'
The length of the extended product was 154 nucleotides which allows
the positioning of the major start point of transcription at -589 in
the nucleotide sequence (figure 5.1.2).
Exposure of the extension reactions to X-ray film was carried out at
-80oC using an intensifying screen.

CG AOaç
t 2 3 T
/t
û#'
ËÊ'
üt
ët''l4
#
*4"
ÒüI
'I
Ìj
¡.

130.
is located at the first purine after this pyrimidine rich region
which is the case for other AspergiZZus genes. Deletion of a small
pyrimidine rich region of the A. nidulons trpC promoter results in
transcription initiat'ing from heterogeneous positions (Hamer and
Timberlake 1987). Therefore, this sequence may play a role in
directing transcriptional start points rather than affecting high
levels of expression.
The 5' untranslated region of the creA transcript is 589bp in
'length which is relativeìy long for fungal genes. Although
considerable variation exists in the length of the 5' untranslated
mRNA of fungaì genes, most commonly this region is 30-70bp in length
(reviewed by Gurr eú oZ. 1987). However, other genes from A. nídulons
have been shown to have unusually long 5' untranslated regions, for
exampìe, ana'lysis of a cDNA clone for the gene biøG from A. nidulons
demonstrated that its nRNA has an untranslated 5' region that is 885
bases 'long (Doonan and Morris 1989). As more'genes from fungi are
characterized it is becoming evident that greater variation exists
in the length of mRNA untranslated regions.
A TATAA-Iike sequence is present at about 30bp upstream from the
major start point of transcription in many fungal genes (reviewed by
Gurr eú ø2. 1987). The promoter region of the creA gene contains a
TATAA-Iike sequence begining at -26 to the maior start point of
transcription. Furthermore, another A-T rich sequence is present at -
731 in the genomic sequence (142bp upstream fron the start point of
transcription) resembles a TATAA box. The CAAT motif is also present
in the promoters of many eukaryotic genes (reviewed by Gurr et ø1.
1987) . These are usual'ly at about -70 to -90 upstream from the start
point of transcription. Multiple CATT box sequences are present at

131.
62bp upstream from the major start point of transcription in the creA
sequence. However, the roìe of TATAA and CAAT boxes in the promoter
regions of fungaì genes remains unclear and whether or not they
affect expression levels of the genes jn which they occur can on'ly be
determined frorn mutational anaìyses of these promoters. Deletions of
TATAA boxes from yeast genes have demonstrated that the resulting
constructs have severe'ly reduced leveìs of transcription without
affecting regulation. This suggests that the TATAA box may represent
at least part of a promoter element in yeast genes (reviewed by
Guarente 1984) . Thi s i s probably not enti rely the case for al l
promoters from fi'lamentous fungi. Such sequences have not been found
in many genes from these fungi and may be deleted from some
fiìamentous fungal promoters without affecting expression (Hamer and
Timberlake 1987).
The preferred bases inrmediateìy preceeding the start codon in
most fungal genes are TCA'in positions -5 to -J (gallance 1986). The
sequence inrmediately upstream from the start codon in creA (TTCACATG)
resembles that of other fungal genes although the TCA is located at
nucleotide positions -4 to -2.
The 5' fegion of the creA gene contains large b'locks of C-T rich
sequence. This includes long stretches of T's, the longest being a
run of 22 I's. The significance of these 5' sequences is unclear
although C-T rich regions are found in many fungal promoters. In
eukaryotes, the upstream regions of genes often have altered
chromatin structures (Nussinov et ol. 1984, Nussinov 1985, reviewed
by Nussinov 1990). Physicaì chemical studies have suggested that
oligo (dA-dT) tracts in DNA sequence can fonn unique conformations
and cause DNA bendi ng. Extreme di ffj cuì ty was experienced in

1.32.
sequencing through the oliqo d(T) stretches in the cre[ upstream
region, characterized by large compressìons and pauses on sequencing
gels. This may have been due to secondary structures in the DNA
caused by the oligo d(T) runs. Similar regions containing oligo (dA-
dT) tracts have been ìocated upstream from promoter elements (Bossi
and Smith 1984, Plaskon and Wartelì 1987) and within promoters in
protein binding sites and origins of replication (Koepse'l and Khann
1986, Snyder et ol. 1986, Zahn and Blattner 1987, Inokuchi et ol.
1988). Studies on the interaction between T-antigen binding and SV40
have denonstrated that sequences fìanking oligo (dA-dT) tracts are
bound to this proteìn. The oligo (dA-dT) tract is itself not jnvoìved
in protein binding but instead 'is required for bringing the fìanking
regions into the proper position for T-antigen binding (Maroun and
Olson 19S8). 0'ligo (dA-dT) tracts have also been found to occur
frequently in intergenic regions in the genome of S. cerevísioe and
have been shown to function, in either orientation, as promoter
elements (Struhl 1985a, 1985b). These eìements may function by
association with the nuclear scaffolding in yeast cells (Gasser and
Laenrmli 1986) or by interfering with nucleosome formation (Kunkel and
Martinson 1981, Prunell 1982). Qther reports suggest that oìigo d(T)
stretches on the DNA coding strand may act as UAS's by excluding DNA
binding proteins (Russell et ol. 1983, Chen et ol. 1987) or by acting
as protein bindjng sites themselves (Lue et ø1. 1989, Thiry-Blaise
and Loppes 1990). Recently it has also been demonstrated that a
singte oìigo (dA-dT) sequence is the site for the initiation of
transcription of two divergent'ly transcribed yeast genes (Schlapp and
Roedel 1990)

133.
5.1.3 The 3' region of the creA gene:
Like the 5' untransìated region of the creA transcript, the 3'
untranslated region is aìso very long, extending 428bp from the 3'
end of cDNA clone pCDS. A]though extensive 3' untranslated
sequences are uncommon in genes fron A. nidulons, they are not
unknown. For examp'le, the 3' untranslated portion of the oreA
transcript is reported to be 539 bases 'long (Kudl a et ol. 1990). The
site of poìyadenylation of the creA transcript was detennined by the
presence of a small poly(A) taiì of six adenosine residues at the 3'
end of pCDS. The addition of poly(A) usuaììy occurs approximately
20bp downstream of the consensus sequence AATAAA or rel ated sequences
such as ATAA or AATA in fungaì genes (reviewed by Gurr et ol. 1987).
These smaller core sequences occur in the 3' region of the creA gene
25 and 32bp upstream from the site of polyadenylation and may be the
signals for poladenylation of the creA transcript.
5.2 ANALYSIS 0F THE PUTATM CreA PROTEIN:
The predicted CreA protein is 390 anino acids 'in length (Figure
5.1.1) and exhibits several features characteristic of regulatory
and DNA binding proteins. These structural motifs are discussed in
the following sections along with the generaì features of the protein
as predicted from computer analyses. Comparison of the entire CreA
protein sequence with the GenEMBL database showed that the top one
hundred best score protein sequences identified as having sequence
similarity to CreA were almost exclusiveìy sequences containing a
zinc finger DNA binding rnotif. The results of further more detailed
sequence comparisons are discussed below.

134.
5.2.I The zinc finger motif:
The derived CreA protein sequence contains two putative zinc
finger DNA binding motifs (Klug and Rhodes 1987) that closely
resembìe the CrH, zinc fingers first recognized in the transcription
factor TFIIIA fromXenopusloevís (Milleret al. 1985). A ìarge
number of proteins from a variety of eukaryotic organisms have
subsequent'ly been found to contain regions that resemble the zinc
finger of TFIIIA. Some of these proteins are known to be
transcriptionaì activators or function as repressors (reviewed by
Levine and Manley 1989, Nehlin and Ronne 1990). The first zinc finger
in CreA begins at residue 64 in the amino acid sequence and is
cìoseìy followed by a second zinc finger begining at residue 92
(Figure 5.1.1). Figure 5.2.1 demonstrates the striking amino acid
similarity between the two zinc fingers in CreA and other zinc finger
domains from a variety of proteins and suggests that CreA is a DNA
binding protein. The concensus sequence for C"H, zinc fingers is Cys-
Xr-o-Cys-X.-Phe-Xu-Leu-Xr-Hi s-Xr-o-His, aìong with conserved fl anking
residues and is characterized by specificalìy spaced, conserved
amino acid residues that are arranged so that the cysteine and
histidine residues are tetrahedralìy co-ordinated to a single atom of
zinc. Both these zinc fingers are of the C"H" type and their
predicted structures based on the model for the conformation of these
sequences are shown in Figure 5.2.2. There is evidence suggesting
that this organization allows the conserved hydrophobic phenylalanine
and leucine residues to form hydrophobic interactions thereby giving
stability to the tip of the finger (reviewed by Bray and Thiesen
1990). The first zinc finger in CreA does not have this usuaì'ly
conserved leucine residue but instead has a glutamine at this

135.
FIGURE 5.2.1: Alignment of the putative CreA zinc finger donains
with the amino acid sequences of other proteins containing the C"H,
zinc finger motif. Numbers after the proteins specify the number of
the finger represented. Arrows ìndicate the cysteine and histidine
residues of the CrHr motifs.
The protein sequences are:
TFIIIA - TranscriPtion factor
et qI. 1985).
IIIA from X. loevís (Uiller
BrlA - Protein involved in developnentaì regulation fron A.
nídulons (Adams et ol. 1988).
- Proteìn encoded by a gene which is deleted in Wilms'
tumor (Cal ì et ol. 1990) .
- Reguìator of ADH2 from s. cerevisíoe, (Hartshorne
et ø1. 1986).
- Zinc finger protein from X' laevis (Rltaba et
oL. 1987) .
(Page et ø1. 1987).
(Rosenberg et ol.
cerevisioe (Nehl in
}'lT
ADRI
Xfi n
TFD
Kr
MIGl
- Human testis determining factor
- Kruppel from D. nelonogoster
1e86) .
- repressor of SIJC2 gene fron S'
and Ronne 1990).

CreA
TFTIIA
BrlÄ
I,il
ADRL
Xfi¡
sp1
TFD
Kr
l"IIGl-
YK
HA
YI
FP
FK
HV
rc-cQ
CF
CK
CK
C I^I
PL
FP
SA
EE
EP
VP
KT
GL
GL
PE
MT
PG
PI
FP
v-cGC
DC
GC
GC
GC
ER
SK
GA
EK
NG
HR
RL
RS
KN
SL
RQ
RS
RS
RR
EK
RS
SR
RD
RL
RS
L-
I-
K-
I-
S-
KT
THTA
HNN
HTG
HTG
HSK
HSK
HTG
HSG
HKN
HQN
HPE
HTG
HTG
HTN
T
2
a
2
t_
2
3
D G
R
R
R
K
K
K
R
K
A
R
A
G
R
A
K
C
S
R
K
R
A
R
F
F
Y
F
F
F
F
F
F
F
F
F
F
F
H
S
N
T
K
S
S
T
V
M
K
T
H
S
D
D
S
D
G
H
E
D
H
L
A
H
F
H
H
E
L
L
L
L
L
L
0
L
K
r
S
S
K
K
T
T
T
R
R
K
R
T
R
R
H
H
H
H
H
H
H
H
T
A
O
I
M
M
M
R
I
TEHQTRHIR
DELTRHSR
wKLQAHLC
HHLTRHSL
EHLKRHMK
DNLNAHYT
31
FQC
YPC
YKC
FAC
YPC
FEC
HÀC
HAC
cQ
CN
CE
CP
CG
CD
CH
CV
RT
QK
RV
KT
KN
KL
RI
RT
3
1_
2
1
2

136.
FIGURE 5.2.2: Amino acid sequence of zinc fingers I and II of CreA
drawn to illustrate the finger motifs as proposed by Miller et ol'
(1985). High'ly conserved residues are circled'

.rtR-S
tr
.zn:.
ï
H-B-[ \sD'EIE
IH
I
T
BI
K
I
s
AI
B
I
E
G s
]t
R
\
/
\ll
aIL
I
P
K
PR
\
T-G - E-K-p H.A
-Zn'
T/ -0t
FF
t

r37.
position. Hhether or not the structure of the first zinc finger in
CreA is affected by this difference is not clear. However, the firstzinc finger in the MIG 1 protein from yeast also has a glutamine in
this position. Furthermore, the alignment of the amino acid sequences
of CreA and MIG I show striking similarities between the entire zinc
finger regions of both proteins (Figure 5.2.3). MIG 1 is required for
glucose repression of the suc2 gene from S. cerevisíoe and also like
the cre[ gene, encodes a protein containing two consecutive zinc
fingers. The zinc fingers in this repressor protein were identified
as being very similar to the zinc fingers of the human early growth
response proteins (Egr 1 and 2) and the zinc finger domains in a
protein encoded by a gene which is deleted in Wilms tumor. MIG t has
been shown to bind to two target sites approximately 500bp upstream
from the suc2 gene (Nehlin and Ronne 1990). That CreA binds to
specific DNA sequences via its putative zinc fingers and whether
both domains are invoìved in binding remains to be confinned.
It seems clear that TFIIIA - like zinc fingers are responsible
for sequence specific DNA binding and that this activity requires the
presence of more than one finger domains in tanden (or perhaps
dimers) of singìe finger proteins. For exampìe, BrlA which is
involved in the transcriptionaì regulation of developmental genes in
A. níduløns, has two zinc fingers which are both required for
activity of the protein (Adams et ø1. 1990). Another conserved
region in zinc finger proteins is the sequence between two successive
zinc finger domains called the H-C tink which spaces repeated finger
domains along the DNA helix. The H-C link in the CreA protein shows
some sequence simiìarity to the consensus sequence TGEKPHA (reviewed
by Bray and Thiesen 1990) (Figure 5.2.4).

138.
FIGURE 5.2.3: Comparison of the amino acid sequences of the zinc
finger regions from creA (ttris thes'is) and MIG1 (Nehlin and Ronne
1990). Double dots indicate the same amino acids are present in
each sequence while sjngle dots show similar amino acids' Arrows
indicate the histidine and cystine residues of the CrH, motifs'
FIGURE 5.2.4: A comparison of the H-C ìink regions of zinc fingers
fron various proteins with the same region from CreA. Lower case
ìetters indicate dìfferences fron the consensus (Bray and Thiesen
1990). The Kruppel sequence is the consensus found for this protein
(Rosenberg et ol. 1986) where X denotes any amìno acid.
The protein sequences are:
BrlA - Protein ìnvolved in developmental regulation from
A. nídulons (Adams et ol. 1988).
Kruppel - Kruppel from D. melonogosúer (Rosenberg et ol.
1e86)
Mouse MKR - Mouse growth factor induced protein (christy et
ot. 1988) .
MIG1 - Repressot of sucl expression from s. cerevisioe
(Nehlin and Ronne 1990).
Spl - Human transcription factor Spl (Kadonaga et øl'
1987) .
HPFI - Human zjnc finger protein HPFI (Bellefroid et ol'
1e89) .
- Human zinc finger protein 7 (tani a et ol' 1990) '
protein 36 (Cunl iff and
ZFPT
zFP-36 - Human zinc finger
Trowsdale 1990).
- Mouse zinc finger
1e89) .
MFGl protein (Passananti et ol '

tt rr r t ttPRPYKCPLCERAFHRLEHQTRH I RTHTGEKPHACQFPGCSKRFSRSDELTRHSR I HNNCREA
MIGl
CREA
BRLA
KRUPPEL
MOUSE MKR
MIGl
Spl 1
2
HPFl
ZFPT
ZFP-36
MFGl
CONSENSUS HTGEKPYXC
PRPHACP I CHRAFHRLEHQTRHMRI HTGEKPHACDFPGCVKRFSRSDELTRHRRI HTN
HTG
HskHTG
HTG
HTG
HTG
HTG
HTG
HTG
HTG
HTG
hAc
hvcYXC
YEC
hAc
fMcfAcYKC
YPC
YEC
YKC
E
E
E
E
E
E
E
E
E
E
E
K
K
K
K
K
r
K
K
r
K
K
P
P
P
P
P
P
k
P
P
P
P

139.
There have been no reports of the successful crystallization of
an intact zinc finger, although singìe finger donains have been
ana'lysed. Predictions of molecular structure have arisen from two
dimensional and three dimensional NMR studies of finger fragments and
from computer modeììing of DNA-protein interactions (Berg 1988, Lee
et ol. 1989). These models indicate that most fingers fonn aß-turn -
o-helical structure where non-specific interactions occur between
positively charged amino acid side chains and phosphates of the DNA
backbone. Sequence specific interactions result from the recognition
of two nucleotide pairs by five amino acid residues situated on the
exposed helical surface and NH, terminus of the finger. The predicted
peptide structuraì features of the region spanning the two CreA zinc
fingers is shown in Figure 5.2.5. The predictions show that these
sequences may al so have structural simi ì arities as weì I as
differences to characterized zinc finger domains. Sone fingers such
as fingers 1,3 and 6 of TFIIA have significant structural differences
as predicted by protein modelling (reported by Bray and Thiesen
1990). However the functional contraints of these different
structures in these fingers and the corresponding dornains in CreA are
uncìear. The probability of the amino acids spanning the zinc
fingers in CreA being on the surface of the folded protein is shown
in Figure 5.2.5. The tips of the fingers (shown in Figure 5.2.2)
occur between residues 70-81 in zinc finger I and 100-111 in zinc
finger II. The amino acids in these regions are predicted to be on
the surface of the protein due to their hydrophobic nature and this
is consistent with the likeìihood that these regions do form the tips
of functional zinc fingers. The tips of zinc fingers are inportant
for DNA binding and the core amino acid residues in these regions
show variation between different zinc finger proteins and are

140.
FIGURE 5.2.5: Plotstructure of Peptidestructure (Devereux 1984) for
CreA amino acid sequence spanning the two zinc fingers from residue
50 to 150. Residues are numbered from I to 100 on the X axis'
Predictions are:
KD Hydrophi I icity - hydrophi t icity according to Kyte and
Dool ittle (1982) .
Surface Prob. - surface probabi'l ity according to Emini et ol '
(1e8s).
Flexibi t ity - flexibi I ity of peptide chain according to Karplus
and Schulz (Devereux 1984).
Jamson-t{olf Antigenic index - a rneasure of the probability that
the region of the protein is antigencic (Devereux 1984).
CF Turns - residue conformations most conmonly found in turns
according to Chou and Fasman (1978).
cF Alpha He]ices - chou and Fasman prediction of alpha-he'lix forming
regions that are not in conflict with other secondary structures
(Chou and Fasman 1978, Nishikawa 1983).
cF Beta Sheets - chou and Fasman prediction of beta-sheet stt'uctures
that are not in conflict with other secondary structures (Chou
and Fasman 1978, t{ishikawa 1983).
GOR Turns - predictions of turns in protein according to Garier-
Osguthorpe-Robson (Garnì er et ol. 1978) '
GOR Alpha Helices - Garnier-Qsguthorpe-Robson prediction of alpha-
heìix fonning regions (Garniet et ol. 1978)'
GOR Beta Sheets - Garnier-Qsguthorpe-Robson predìction of beta
sheet structures (Garni et et ol. 1978).
Glycosyl. Sites - predicted sites for gtycosyìation where the
residues have the composition NXT or NXS (Cohen et ø1. 1984) '
rt',!,
,f
fI
j
k

PL0TSTRUCTURE of: cFea .p2sPEPTIDESTFUCTURE of: cnea. pep
TFANSLATE of: cneag.seq check:50
rrrrrr!ll
?0, 1gg0 13:2550 to: 150
827 to: 10BB
NovembenCk: 1381.
4370 fnom:u 100
I5.0
KD Hydnophilicity_5.0
l0 '0
Sunface Fnob.
0.0
1.2
Flexibility0.8
L./
Jameson-l4o1f(Antigenic Inoex)
_1 .7
CF ïunns
CF Alpha Helices
GOR Tunns
GûH Alpha Helices
Glycosyl. SitesI
0
I
50 100
:=+>- -
ls.:- : -# Ì

t4r.
presumed to give zinc finger proteins their specificity to target
sites (Miller et o1.1985, Bellefrojd et ol. 1989). Comparison of the
amino acid residues within the tips of the zinc fingers from MIG 1
with other zinc fingers indicated that the central residues FSRSD in
zinc finger II are identical in a number of other zinc fingers
(Nehlin and Ronne 1990). An alignment of these zinc finger loops to
the same region in zinc finger II of CreA is shown'in Figure 5.2.6.
All (except Spl) contain the centraì residues FSRSD and a high degree
of conservation of flanking amino acids is also evident. The
sequences to which some of these proteins bind (Figure 5.2.6) have
been identified and their conservation has been noted (Chavrier 1990,
Nehlin and Ronne 1990, Nardelli et o1.1991). It will therefore be of
interest to determine whether CreA also binds to a similarly related
target sequence.
5.2.2 An alanine rich region:
The putative CreA protein sequence was compared to protein
sequences known to have transcriptional activation or -repression
functions as well as to many other regulatory proteins from various
organisms. This h,as done in an atternpt to define other domains
within the CreA protein which may indicate how CreA functions at the
molecular level.
Along with regions of amino acid similarity due to the presence
of zinc fingers, the aìanine rich region occurring after the second
zinc finger in CreA was identified as being simiìar to domains
present in a number of regulatory proteins. The alanine rich region
in CreA consists of nine consecutive alanine residues beginning at
$'r
rl
tI
j
3
I

il
'l
I
t42.
l{erve GF
Krox-20
MIGl
FIGURE 5.2.6: Comparison of zinc finger loop regions from CreA with
other similar zinc finger loops from various zinc finger proteins'
l{umbers refer to which zinc finger from the protein is given' Target
sequences to which sone of these proteins bind are also shown
(references appear in the text).
Sequences are from the following proteìns:
BrìA - Protein involved in developmental regulation from A'
nidulons (Rdams et oI. 1988).
Human EGR1 - Human early growth response protein 1 (suggs ef ol'
19eo) .
Hunan EGR2 - Human early growth response protein 2 (Joseph et ol'
1988).
Mouse EGR1 - Mouse early growth response protein 1 (Sukhatne et
øt. 1988) .
Mouse GFI - Mouse growth factor induced protein (christy et ol'
1e88).
Krox-24
- Human nerve growth factor (Changel ian et øL ' 1989) '
-Mouseseruminducjbletranscriptionaìactivator(Chavri er et ol. 1988) .
- Mouse early growth response protein (Janssen-Tinmen
et ol. 1989).
- Repressor of SuC2 expression from S' cerevisiøe
(Nehlin and Ronne 1990).
- Human transcriptìon factor Spl (Kadonaga et qL'
1e87).
1
lII
I
!
spl

PROTEIN
CREA 1
2
BRLA 2
HUMAN EGR1
HUMAN EGR2
MOUSE EGR1
MOUSE GFI
NERVE GF
Krox-20 1
Krox-24
MIG1 1
2
Sp1
ZINC FINGER LOOP
E
s
H
K
K
A H LEHQ
T
BINDING SEQUENCES
GCGGGGGCG
GCGGGGGCG
GCGCGGGCG
GCGGGGGCGG
CCGGGGGCG
GCGGGG
GGGGCGGGGC
GGGCGG
GGGGCGG
ll
K
a
A
R
FSRSD
FSRSD
FSRSD
FSRSD
FSRSD
FSRSD
FSRSD
FSRSD
NA
CDRRFSRSDELTRH
LEHQ
RFSRSDELTRH
N
H
RHRSDELRFc
HR
RF
R
E
E
R
c
c
c
RAF TRH
L
L
L
L
L
L
L
L
CDRR
CDRR
CDRR
CDRR
CDRR
CDRR
TRH
TRH
TRH
TRH
TRH
TRH
TRH
TRH
E
E
E
E
E
c
c

143.
position 131 in the amino acid sequence while a significant number of
aìanine residues are also present upstream of the zinc finger region
(Figure 5.2.L). Similar alanine rich regions occur in the repressor
proteins kruppel, engroíled and evenskípped from Drosophilo
nelonogoster (Figures 5.2.7-5.2.9). kruppeZ acts as a negative
regulator of transcription in D. melonogoster and represses the
expression of the pair-ruìe gene evenskipped (Goto et ol. 1989) and
the gap gene hunchbock (Jackle 1986) during the blastodenn stage of
D. nelonogoster development. It has been demonstrated that the DNA
binding function and transcriptional repression activities are
separate domains in the kruppel protein, as they are in most DNA
binding transcription factors. The repression function of kruppel has
been mapped to the NHr-terminal region of the protein showing the
simiìarity to the alanine rich region in CreA. Comparisons of the
repressor domain of kruppel to the amino acid sequences of
evenskipped and engroiled identified sirnilar alanine rich regions in
these proteins. It has aìso been reported tfrat tfre deletion of the
alanine rich region of evenskipped leads to the protein having a
reduced abitity to act as a repressor of transcription (Han and
Manley reported by Licht et o1.1990). It is therefore not surprising
that these proteins also showed homoìogy to the alanine rich region
in CreA. Significant similarity between CreA and engroíled (Figure
5.2.8) extends past the aìanine rich region and probably reflects the
high serine content of these regions. Alanine residues are relatively
hydrophobic and therefore probably promote the formation of c
helices. Alanine rich regions may therefore be important for protein-
protein interactions.
A number of eukaryotic transcription factors, including kruppel,

144.
FIGURE 5.2.7: ComPare progran output between putative CreA protein
and Kruppel protein from D. nelønogsster. Points are marked where
the two amino acid sequences are similar within the window size
indicated (Maizet and Lenk 1981, Devereux 1984). The .ìttled region
shov,,s the position of an alanine rich region in both sequences'

300I
200I
100nI
lrlll¡rrt
/,/ /.
//,/(oOñ
o¡
H(u
o)+
- 400
- 300
- 200
227.
2l./
tr///
co(oso+J
scncor,:l(Jo_0JqO
Jo_o_fc_
>¿
3iäs¿?
O:
o)O;
+2
3s:P
rJ
ãcnE
R
==
rE'.c
+;
OcJ uJcc
E+
.OãCJ
)o-f-OO
,/ //
,l/ ,,
/
/,
//i
a', '/
../ / '/'
,/,
z//
- 100
t,/t .
z'
-0
1 to 390-lrrrrlttt
Cnea . P
ep ck: 1, 38 1,

145.
FIGURE 5.2.82 Compare program output between putative CreA protein
and Engrailed protein fron D. nelonogoster. Points are marked where
the two amino acid sequences are similar within the window size
indicated (Maizel and Lenk 1981, Devereux 1984). The cìrcled region
shows the position of an alanine rich region in both sequences'

300I
,/
,/ ,' ,,/,,
/
200I
1000I
- 500
/ -400
/
/r//r,/
@ñ(o
^tr)H
ss)
cct
,27
/2,,,. .. ,'4.'rZ
',/ "
7%
r'/
/,I
//
cr)LOtr)o-]J
c.)cuC
U
ctt
)¿(JoqJ
tEOJ
.droLglcLrl
C-Ì Ð
(U--o.Ooo3C
D
q;c)
'.: o
uo)c
PÐã(nCqlrlucu
iïi€;C'd
+=
OcJ L!æ
.
Eg
.Oã(r.Jo_FO-
- 300
- 200
/,/,/.,/
//,,// /.2
/'r
.2 ,,'rz:/
/,/..,/ t .
;;l'/////
4
I,/
./'
- 100
t/
,1,
/,/
-l r
r r
r I
r r
r r
I r
r r
r I
r r
r ¡
-0
Cnea.P
ep ck. 1,381, 1 to 390

146.
FIGURE 5.2.g: Compare program output between putative CreA protein
and Evenskipped protein from D. melonogøster. Points are rnarked
where the two amino acid sequences are sinrilar within the window
size indicated (Maizel and Lenk 1981, Devereux 1984). The circìed
region shows the position of an aìanine rich region in both
sequences.

t-rOñ
È\
rf)H
roo)s
uJÐ(U
.4
3ñoiÌoU.O
O
@q;o2Eb
c
Ð1)
Â
(t)aoJ
,^oäi
IljCU
:trOC
J LLJE
:+,Oã(rJo_F
-Oo
0I
100200I
300I
y''//
,/'/ /,/ //
?
/2,
,//
- 300
- 200
- 100
//
///,,I//
,/
,i'2,/r--t-cr)o+
J
cticot¿C
J
aG)
o_EOJ
o_o_-l¿U)
C(Ð
L!
t%
,/,,/.
/..'/
/t//
,/tz
-l'
-0
Cnea.P
ep ck: 1,381, 1 to 390

t47 .
have been shown to have both activator and repressor functions.
Genetjcal data indicates that kruppel acts to positively reguìate
another gap gene, knírps, and further to this, the knirps promoter
has been found to contain a binding site for the kruppel protein
(Pankratz et ol. 1989). However, the actjvator domain of kruppel is
yet to be defined and mapped (Licht et ol. 1990). That activator and
repressor functions of the uZtrobíthorox protein from D. nelonogoster
can be separated (Krasnow et ol. 1989) suggests that this may be the
case for other eukaryotic transcription factors having both
activities (reviewed by Levine and Manley 1989). Therefore, although
these protei n domai n simi I ari ti es suggest that CreA coul d have
repressor function at the.level of transcription, it can not be ruled
out that it may also have positively acting regu'latory functions
invoìving the activation of transcription of some other (as yet
geneticaìly unidentified) genes controlled by CreA.
CycS (SSN6) acts as a negative regulator of gene expression
during glucose repression in .S. cerevisioe (Trumbly 1986) and like
the above mentioned D. nelonogoster genes it aìso has an alanine rich
region (Trumbly 1988) showing homology to the aìanine rich region of
CreA (Figure 5.2.10). However, deletion studies of the CycS protein
have indicated that this region is not important for at ìeast some of
the activities of Cyc8, although the functional significance of other
glutamine/alanine rich regions in this protein are yet to be ana'lysed
(Schultz et ol. 1990). The importance and/or s'ignificance of this
region for CreA function therefore remains to be determined through
the functional analysis of truncated CreA proteins.

148.
.
I
i
FIGURE 5.2.10: Compare program output between putative CreA protein
and CycS protein from S. cerevisise. Points are marked where the two
anino acid sequences are similar within the window size indicated
(Maizel and Lenk 1981, Devereux 1934). The circled region shows the
position of an alanine rich region in both sequences'

oON
I
oI
\\.\.
)ì...*.' \ ))ì\..
r t. \'
.\.'
. \.u..\
\ì ..
o- €
oo- (D
\
\
\'\
Þ.-(oO)
o-p
r@Lft
ol
ll(J
o_OJ
qco(J
CJ
\
\
\.* oov
oo- ru
o-T
06t o1 tr 'I BE 'I :13 dad ' eaJ3
çt8 'z
g0:60 166I
:slurod 0'0I :Ásua6uIJls 0¿
'6¿ ÁJenuel 0q'¿08 :ÁlISUeO
:M0purM luvdN0S
tud'BrÁrÁ :]o -L0tdl00

149.
5.2.3 The S/TPXX motifs:
The amino acid sequence motifs Ser-Pro-X-X and Thr-Pro-X-X
(S/TPXX) are found to occur more frequently in regulatory proteìns
such as the Drosophilo homeotic proteins and steroid hormone
receptors than in other DNA binding proteins such as the core
histones which are not directìy involved in gene regulation. It has
been suggested that these motifs assume a ß-turn structure and
contribute to the DNA binding capacity of proteins (Suzuki 1989). The
putative CreA protein contains 58 serine and 43 proìine residues and
e'ight SPXX and four TPXX motifs. The expected frequency (calculated
as in Suzuki 1989) for SPXX and TPXX motifs occurring at randon in
the CreA protein are 1.6 X 10-2 and 4.5 X 10-3 respectively, while
the observed frequencies are 2.0 X 10-2 and 1.0 X 10-2 respectiveìy.
Thus, these motifs occur more frequently in the CreA protein than
expected and more frequentìy than is found in general proteins (2.89
X 10-3 and 3.08 X 10-3 respectively) and may reflect the regulatory
nature of the protein. AreA is another zinc finger protein from A.
nidulons and like CreA, it also has a high frequency of these motifs
(Kudì a et ol. 1990).
5.2.4 PEST sequences:
It has been reported that many proteins with intracellular half
lives of ìess than two hours contain regions (PEST sequences) which
are rich in proline(P), glutamic acid(E), serine(S) and threonine(T)
and lack internal hydophobic residues which are flanked by several
positively charged amino acids. Since the expressjon of the creA gene
appears to be constitutive, it is possible that other mechanisms such

150 .
as those affectìng protein stabitity assist in reguìating the
activities of CreA within the cell. A computer prograrn generated by
Rogers et ol. (1986) was used to scan the CreA amino acjd sequence
for PEST sequences. Four PEST-Iike regions were identified using this
program of which two, located between positions 1-47 and 163-179, had
PEST-SC0RES of -5.5 and 0.97 respectively, which are similar scores
to those reported for proteins having short intracellular half lives.
However, the relative importance of these sequences, with respect to
other structural features of proteins, in affecting stability remains
to be cìearly understood. No. experimental evidence has been
reported to suggest the involvement of PEST sequences in protein
stabi I ity, however they rnay be useful for predict'ing the nature of
pnotei ns .
5.2.5 Other generaì features of the CreA protein:
There are four asparagine-X-serine/threonine motifs in the
protein that are potential sites for N-linked glycosylation (Marshall
1972, Modrow and Wolf 1986) ìocated at positions L8, 28, 191 and 283.
However, it has been reported that the presence of a proìine either
between the asparagine and the serine/threonine or C-tenninal to the
serine/threonine as is the case with the predicted site at 191, wilì
cornpleteìy suppress N-glycosylation (Bause 1983). Therefore, the
asparagine-X-serine motif at 191 is less likeìy to be a potential N-
glycosylation site.
The phosphoryìation/dephosphoryìation of proteins is a common
way in which the activities of proteins are regulated. Protein
ki nases alter the functions of thei r target proteins by

151.
phosphoryìating specific serine, threonine and tyrosine residues
(reviewed by Kemp and Pearson 1990). In the absence of regulation of
creA at the transcriptional level there remains the possibility that,
with the high serine content, the CreA protein i s
activated/inactivated by specific phosphoryìation. Numerous potential
sites for phosphorylation in the CreA protein are identified using
the PROSITE program (Bairoch 1990). These include consensus target
sites for cAMP-dependent protein kinase, protein kinase C and caesin
kinase II. The consensus sequence for cAMP-dependent protein kinase
occurs between positions 101-105 in the amino acid sequence, placing
it within the second zinc finger in the CreA protein. It would
therefore be of interest to determine if this site is in fact a
target for cAMP-dependent protein kinase and if so, what effect
phosphorylation/dephosphorylation would have on the binding capacity
of zinc finger II.
The Chou and Fasman (1978) and Garnier, '0sguthorpe and Robson
(1978) predictions as to the secondary structure of the putative CreA
protein are shown in Figures 5.2.II and 5.2.12.
5.3 SUI{'IARY:
2.7kb spanning the creA gene fron A. nídulons has been
sequenced. The derived amino acid sequence predicts that the creA
gene encodes a protein of 390 amino acids which contains two zinc
finger DNA binding motifs. The putative CreA protein is likely to be
phosphorylated as it has a high serine content and this may represent
a mechanism by which the activity of the protein is reguìated. A
repressor function is suggested for the CreA protein by the presence

152.
4,'il
II
I
FIGURE 5.2.I1: The predicted secondary structures in the putative
CreA protein according to Chou and Fasnan (1978). Helices are shown
with a sine wave, Beta sheets with a sharp saw-tooth wave' turns
with 180 degree turns and coils with a dull saw-tooth wave'
Hydrophilicity is superimposed over the wave and the size of these
syrnbols is proportional to the value of these measures' Possible
glycosylation sites are narked with o (Gribskov and Devereux 1986'
Devereux 1984).

PLOTSTBUCTURE of: crea.pep ck:TRÂNSL,ÀTE of: cneag.seq check 4370 froD: BZ7 to:
200
e50
Chou-Fasnan Pned lct10nNovenbeP 20. 1990 13:25
300
138 1
lOBB
0KO Hydnophilicity >-1.3
KO Hydnophobiclty >-1.3NH2
50
100
' =-¿:- - --æSê!-
c00H
-iF'

153.
r,
i
FIGURE 5.2.12: The predicted secondary structures in the putative
CreA protein according to Garnier et ol. (1978). Helices are shown
with a sine wave, Beta sheets with a sharp saw-tooth wave' turns
with 180 degree turns and coiìs with a dull saw-tooth wave'
Hydrophilicity is superimposed over the wave and the size of these
symbols is proportional to the value of these neasures. Possible
glycosylation sites are marked with o (Gribskov and Devereux 1986,
Devereux 1984).
I
I{r
r
I

PLOTSïBUCTURE of: cnea.pep ck: 1381IRANSLATE of: cneag.seq-check 4370 fpo!¡: 827 to: IOBB
NH2
6apnlep-osguthonpe{obson PFedlctlonNovemben 20. 1990 13:25
0
KD Hydnophlliclty >-1.3
EO K0 HydPophoblcity >-1.3
150
e00
¿50
' :ã+r- - -
H00c

t54.
of an alanine rich region which exists in other proteins where they
have been identified as being domains conferring repressor function.
That CreA may be a DNA binding repressor molecu'le is consistent with
the geneticaì interpretation of creA being a negative regu'lator
directly involved in carbon catabolite repression in A. nidulons.
Experiments are now unden¡lay in this laboratory to confirm whether or
not the CreA protein is able to bind to specific DNA sequences in the
upstream regions of genes known to be reguìated by the cre[ gene
product.
{

155.
CHAPTER S IX
CONCLUD I NG D I SCUSS TON .
The aims of the study outlined in this thesis were to isolate
and characterize the wide domain regulatory gene, creA, from
Aspergiltus nídulons. This study was undertaken in order to begin to
understand the molecuìar mechanisms of carbon catabolite repression
in /. nidulons and more specìfically to determine as far as possible
how the product of the creA gene may be involved in this type of gene
regu'l at i on .
A region contain'ing the creA gene was cloned by complementation
of the cre\204 mutation using a plasmid ììbrary followed by the
rescue of complementing sequences. This cìone was subsequentìy used
to isolate a larger genomic fragment containing the creA gene. The
clone was also shown to complement several mutant alleles, including
creAd-3Q which has a translocation breakpoint within the creA coding
sequence (Arst et oL. 1990). The clone was shown to be homologous to
a region which is disrupted in creAd-30 which allowed the inversion
breakpoint to be mapped to the 250bp PsúI fragment in the creA coding
regi on.
Northern analyses using the cloned sequence as a probe
demonstrated that creA is constitutively expressed at relativeìy low
levels, expression'levels only varying by about two fold depending on
the available carbon source. Increased expression levels were found

156.
in multicopy transformants and although these transformants grew as
w'iìdtype on a variety of carbon sources, there was evidence that
increased crel copy number may 'lead to "tighter" carbon catabolite
repression of at ìeast some enzyme activities subject to control by
the creA gene product. This could be investigated further by
determining the effects of multiple copies of the creA gene under
repressing and derepressing conditions on the mRNA levels for those
genes, including ølcR, øZcA and andS, which are subject to regulation
by creA.
Strains containing the cre\204, cre|d-I, creA220, creA225 and
creAd-30 mutations were all shown to produce cre| mRNA, therefore
suggesting that this group of alleìes were not total lack of
function creA mutants. A complete deletion of the creA coding region
was constucted in a diploid strain usìng in vitro techniques.
Analysis of this strain demonstrated that the phenotype for compìete
loss of function for cre| is lethality. The. creL mutant alleles
isolated by various workers to date must therefore represent a class
of mutants which have an altered function or lack some non-essential
domain while retaining the essential function of the protein.
The physicaì analysis of the cre| gene has included the
determination of the nucleotide sequence of a 2.7kb region spann'ing
the gene and the anaìysis of the structure of its transcript to
determine the extent of the coding region. Ana'lysis of the sequence
showed an open reading frame from the first ATG in the transcribed
region extending 1170bp to an infrane stop codon. The anaìysis of the
effects of ín vítro generated mutations or deletions is required to
determine the functional significance of C-T rich sequences and other
promoter-like elements in the upstream region of the creA gene. There

r57 .
are no introns in the creA gene and the derived amino acid sequence
predicts that the gene encodes a relatively small protein of 390
amino acids in size. The putative creA protein contains a number of
features characteristic of DNA binding reguìatory proteins including
numerous S/TPXX motifs, two consecutive zinc finger DNA binding
domains of the CrH, class and an alanine rich region. The alanine
rich region shows sequence similarity to domains in other proteins
known to be important for transcriptionaì repression.
The node of action of the creA gene product can be described by
at least two models. One predicts that the creA gene encodes a DNA
binding repressor protein involved in carbon catabolite repression,
and that complete derepression of alì the functions under creA
control is ìethaì to the cell. Alternatively, the creA gene could
encode a DNA binding repressor protein, but also has an essential
activator function which as yet has not been geneticalìy defined.
Many eukaryotic transcriptionaì repressors, including the homeobox
proteins and other zinc finger proteins, have been shown to contain
both repressor and activator domains (reviewed by Levine and Manley
1989). Some of these proteins can function as either depending on the
circunstances. For example, the relative contributions of these
opposing domains could result from the conformation of the protein
which can be influenced by severaì factors, including its affinity
for DNA binding sites and interactions with other proteins. The
comparison of activator dornains from various transcription factors
shows that although they are genera'lly rich in acidic amino acids,
there is a lack of a consensus sequence. It has been suggested that
this low specificity could be sufficient if interactions between
bound proteins are important for specific transcriptional activation

158.
(reviewed by Guarente 198S). The finding that a complete deletion of
the creA gene is lethal in a hap'loid strain supports the suggestion
that there could be an essentjal activator domain, as well as a
repressor domain, in the protein. It is interesting to specuìate that
the phenotypes of the creA mutant alìeles may resuìt from an impaired
repressor function, while the lethaìity of a null allele may result
fron the loss of an essential activator function. Information from
the sequences of creA mutants will determine which domain(s) in the
CreA protein are required for repressìon, while the analysís of
diploids transfonned with in vitro generated mutant alleles may
provide the necessary information required to determine whether an
activator as weìl as a repressor domain is present in the protein.
There are a number of possible mechanisms by which CreA may act
negativeìy to repress the transcription of regulatory and structural
genes subject to carbon catabol i te repressi on. These i ncl ude
repression mediated by the direct inhibition of binding of positively
acting transcription factors, by the blocking of the activation of
transcription or by inactivating the transcription initiation complex
itself. Competition between the binding of negative regulators and
particular positively acting transcription factors is cormonìy found
in prokaryotic gene regulation (reviewed by Renkawjtz 1990). This may
involve the competition between negative regulators and genera'l
transcription factors (such as RNA polymerase I) at or near
transcription start points or between more specific transcriptional
activators upstrean from the start point of transcri pti on. In
eukaryotes, it appears that repressors are conmonly found to prevent
the activation of transcription by positive factors which are bound
to their target sites (reviewed by Levine and Manley 1989, reviewed

159 .
by Renkawitz 1990). Repression in this case involves the binding of
activating and repressing proteins to adjacent non-overlapping DNA
sequences and proteìn-protein interactions between the repressor and
activator preventing the activator from making the correct contacts
with the transcription complex. Alternatively, it is aìso feasible
that CreA directìy represses transcription by binding to defined
sites, possibly even at a distance from the target promoters, and
interferes with the formation or activity of the basal transcription
compìex leading to a reduced rate of transcription initiation
(cornmonly caìled "silencing") (reviewed by Renkawitz 1990).
To further define how creA may act as a transcriptional
repressor it is important to determine the position of putative
binding sites in the upstream or promoter regions of its target
genes. Many of the genes subject to controì by carbon catabolite
repression and the product of the creA gene have been cloned. It is
therefore of interest to identify upstream seq[ence elements in these
genes whi ch are necessary and suffici ent for creA medi ated
reguìation. It is not known whether carbon repression acts via
regulatory genes or directly on the 5' regions of genes encoding
enzymes, or on both as appears to be the case for the genes involved
in ethanol metabolism in A. nídulons (Felenbok et ol. 1989). Features
of the derived amino acid sequence of the creA polypeptide indicate
that it is likely to be a DNA binding protein. Therefore, binding
of the protein to putative DNA target sites could bè investigated by
mobitity shift assays and footprinting studies. Although such
experiments are required to confirm that creA is a DNA binding
protein, they will aìso serve to ìocate the positions of such sites
and conditions under which binding occurs.

160.
Different proteins containing similar structural motifs such as
a zinc finger are able to recognize and bind to a variety of DNA
sequences in different target genes, ôlthough probabìy also with
different affinities. Investigations into the binding specifìcity and
affinities for different target sites of in vitro generated mutations
in the zinc finger regions of the CreA protein and their binding
sites could be carried out. This would allow the characterization of
important residues in the fingers required for their specificity, the
relative importance of both the fingers for CreA function and/or
binding and the critical nucleotides in their target binding sites.
Titration analysis is another valuable approach for studying
gene regulation. This involves the construction of strains carrying
muìtipìe copies of a target site for a trons-acting regulatory
protein which is present in cells in limiting amounts. It resultsìn
the titration of the regulatory molecule such that reduced levels are
insufficient for affecting complete regulatioñ of its target genes.
Titration studies in AspergiZZus have prov'ided valuabìe information
on the reìative affinities of different binding sites for trons-
acting regulatory proteins, the effects of mutations in regulatory
genes on the binding capacity of the protein and on the relative
amounts of regul atory proteins present in certain strai ns
(Andrianopoulos and Hynes 1988, Hynes and Kelly 1987, Hynes et ø1.
1988). Although this approach to studying gene regu'lation in
Aspergillus has only involved the titration of positively acting
regulatory proteins such as øndR and locB, sinilar experiments should
be possible to show the titration of transcriptional repressors, thus
defining target sites and reìative affinities to such sites. If CreA
is present in limiting amounts in cells and does in fact have a

161.
úrons-acting repressor function, transformants containing muìtipìe
copies of its target sites should lead to overexpression of the
regulatory and/or structural genes subject to direct regulation by
creL in the presence of glucose. Furthermore, anti-titration should
be possibìe by increasing creA copy number and expression in these
strai ns.
The cre[ clone from A. nidulons uras shown to hybridize to
sequences from a number of other fungi. It wilì be of interest to
isolate and characterize these sequences to determine if a corlmon
mechanìsm of carbon catabolite repression exists in these species.
This would also allow comparisons to the ,4. nidulons sequence to
identify conserved regions which may define further the important
regions for creA function. It would also be of interest to
investigate whether the deletion of such sequences from these other
species is lethal and to detennine if these genes are able to confer
creA-related functions in heterologous expression systems.
A'lthough this study has concentrated on the molecular analysis
of the creA gene, other loci have been genetical'ly identified as
being involved in carbon catabolite repression in A. nidulons (Hynes
and Keìly 1977, Kelly and Hynes 1977). Therefore, the isolation and
nolecular characterization of creB and creC, in particular, are
desirable in order to more futty understand the roìes of these genes
in carbon repression. Attempts to isoìate these genes using the same
techniques used to isolate creA are unden¡lay in this laboratory.
The control of gene expression by trons-acting regulatory genes
such as creA requires that the regulatory protein has a nunber of
functional properties. These include, a capacity to enter and

162.
accumulate in the nucleus, an ability to bind to specific target cís-
acting elements and/or to interact with other products that bind to
these sites leading to up or down regulation medjated by these other
molecuìes and following this, to activate or repress target gene
expression either directly or indirectly. Such transcription factors
must also respond accordingìy by some effect on one of these
activities, to specific environmental stimuli.
The identification of important domains using ín vítro generated
mutations and deletions and in vivo assays for the function of
regulatory gene products has been achieved for many regulatory
proteins including GCN4 (Hope and Struhl 1986), GAL4 (Johnson et ol.
1986), ADRI (Blumberg et ol. 1987) and NIT2 (Fu and Marzluf 1990).
It has become apparent fron some of these studies that a common
property of regulatory proteins is that relativeìy 'large regions of
these proteins may be deìeted without noticably altering their
function. For examp'le, mutational ana'lyses of the positive'ly acting
NIT2 reguìatory protein from /V. crosso have demonstrated that while
both the zinc finger domains and a downstream basic region are
critical for DNA binding and its transcriptionaì activation function,
deletion of 2I% of the C00H-terminus of the protein maintained fullactivity (Fu and Marzìuf 1990). Simi'larly, ôìthough anaìysis of the
putative CreA anino acid sequence has identified at least two domains
in the protein, one a DNA binding region and another possibly
required for repressor function, the molecular dissection of the CreA
protein shouìd allow the functional domains and the important regions
required for CreA activity to be identified.
studies reported in this thesis provide a solid
investigations into the precise nature and
The
further
basis for
mechani sms

163.
involved in creA mediated gene regulation as a model for gìobal gene
regulation in eukaryotes. Future analyses of the creA gene product
shouìd clarify the function of the protein at the molecular level.

164.
REFERENCES.
Abrams, E., Neigeborn, L. and Carlson, M. 1986. Molecular anaìysis of
SNF? and .S,VFS, genes required for expression of gìucose
repressibìe genes in Socchoronyces cerevisioe. Mol. Cell. Biol.
6: 3643-3651.
Adams, T.H., Boyìan, M.T. and Timberlake, l,l.E. 1988. brlA isnecessary and sufficient to direct conidiophore development in
Aspergíllus nidulons. Cell 54: 353-362.
Aiba, H., Fujimoto, S. and Ozaki, N. 1982. Molecular cloning and
nucleotide sequencing of the gene for E. colí cAMP receptor
protein. Nucleíc Acids Res. 10: 1345-1361.
Aiba, H. and Krakow, J.S. 1981. Isolation and characterization of the
amino and carboxyì proximal fragments of the adenosine cyclic
3',5'-phosphate receptor protein of Escåerichio colí. Bíochen.
202 4774-4780.
Altaba, 4.R., Perry-O'Keefe, H. and Me'lton, D.A. 1987. Xfinz an
embryonic gene encoding a multifingered protein in Xenopus. EMB0
J. 6: 3065-3070.
Anderson, W.8., Schneider, A.8., Emmer, M., Perlman, R.L. and Pastan,
I. 1971. Purification of and properties of the cyclic adenosine
3',5'-monophosphate receptor protein which mediates cycìic
adenosine 3',5'-monophosphate dependent gene transcriptìon in
Escherichio coli. J. Bíol. Chen. 246: 5929-5937.

165.
Anderson, l{.F., 0hlendorf, 0.H., Takeda, Y. and Matthews, B.hl. 1981
Structure of the cro repressor from bacteriophage ìambda and its
interaction with DNA. Noture 2902 754-758.
Andrianopouìos, A. and Hynes, M.J. 1988. Cloning and ana'lysis of the
positive'ly acting reguìatory gene øídR fron Aspergillus nidulsns.
llol. Cell. Biol . 8: 3532-3541 .
Arst, H.N. t977. Some genetical aspects of ornithine metabolism in
Aspergillus nidulons. Molec. Gen. Genet. 151: 105-110.
Arst, H.N. and Bai'ley, C.R. 1977. The reguìation of carbon metaboljsm
in Aspergillus nídulons. pp. 131-146. In J.E. Smith and J.A.
Pateman (eds.) Genetics and physiology of Aspergíllus. Academic
Press, Inc. N.Y.
Arst, H.N. and Cove, 0.J., 1973. Nitrogen netabolite repression in
Aspergillus nidulons. Molec. Gen. Genet. 126: 111-141.
Arst, H.N. and MacDonald, D.l.l. L975. A gene cluster in Aspergillus
nídulons with an internally ìocated cis-acting regulatory region.
Noture 254: 26-3I.
Arst, H.N. and Scazzocchio, C. I975. Initiator constitutive mutation
with an "up"-promoter effect in Aspergillus nidulons. Noture 2542
31-34.
Arst, H.N. and Scazzocchio, C. 1985. Formal genetics and molecular
biology of the control of gene expression in Aspergillus
nídulons. pp309-343. In J.W. Bennett and L.L. Lasure (eds.), Gene
manipulations in fungi. Academic Press, Inc., N.Y.
Arst, H.N., Tollervey, D. and Caddick, M.X. 1989. A translocation
associated loss-of-function mutation in the nitrogen metabolite
repression reguìatory gene of Aspergíllus nídulons can revert
intracistronical'ly . Molec. Gen. Genet. 2I5z 364-367 .

166.
Arst, H.N., Tol'lervey, D., Dowzer, C.E.A. and Kelìy. J.M. 1990. An
jnversion truncating the cre[ gene of Aspergillus nidulons
results in carbon cataboì ite derepression. l,lolec. Microbíol. 4:
851-854.
Austin, 8., Hall, R.M. and Tyìer, B.M. 1990. 0ptimized vectors and
selection for transformation of Neurosporo crossø and Aspergillus
nidulons to bìeomycin and phleomycin resistance, Gene 932 I57-
t62.
Ausubel, F.M., Brent, R., Kingston, R.E., Moore, D.D., Smith, J.4.,
Seidman, J.G. and Struhl, K. (eds.) 1987. Current protocols in
mol ecul ar bi o'logy 1987-1988. John lr{i 'ley and Sons , N. Y
Bailey, C. and Arst, H.N. 1975. Carbon catabolite repression in
Aspergillus nidulons. Eur. J. Biochen. 51: 573-577.
Bailey, R.B. and Woodward, A. 1984. Isolation and characterization of
a pleiotopic glucose repression resistant mutant of S.
cerevisioe. Molec. Gen. Genet. 193: 507-512.
Bairoch, A. 1990. Prosite, a dictionary åt protein sites and
patterns. Department de Biochimie Medicale, Universite de Geneva.
Baìlance, D.J. 1986. Sequences important for gene expression in
filamentous fungi. Yeost 2z 229-236.
Ballance, D.J. and Turner, G. 1986. Gene cìoning in Aspergillus
nidulons: isolation of the isocitrate lyase gene (øcuD). Molec.
Gen. Genet. 2022 27I-275.
Baum, J.4., Geever, R. and Giles, N.H. 1987. Expression of the qø:lF
activator protein. Identification of upstream binding sites in
the gø gene cluster and localization of the DNA-binding domain.
Mol. Cell. Biol. 7 ¡ 1,256-1266.

167.
Bause, E. 1983. Structural requi rements of N-gìycosyl ation of
proteins. Studies with proline peptides as conformational probes.
Biochenicol J. 209: 331-336.
Beato, M. 1989. Gene regulation by steroid hormones. CelZ 56: 335-
344.
Bellefroid, E.J., Lecoq, P.J., Bentida, A., Poncelet, D.4., Be'layew,
A. and Martial, J.A. 1989. The hurnan genome contains hundreds of
genes coding for finger proteins of the kruppeì type. DNA 8z 377-
387.
Bennett, M.D. and Smith, J.B. 1976. Nuclear DNA in angiosperms. Phí\.
Trons. R. Soc. London 82 742 227-224.
Benton, t{.D. and Davis, R.W. 1977. Screening À-gt recombinant clones
by hybridization to singìe pìaques ín situ. Science 196z 180-185.
Berg, J.M. 1988. Proposed structure for the zinc-binding doma'ins from
transcription factor IIIA and related proteins. Proc. Nøtl. Acod.
Sci. U.S.A. 85: 91-102.
Berg, J.M. 1990. Zinc fingers and other metal-binding domains.
Elements for interactions between macromolecules. J. Bíol. Chen.
2652 6513-6516.
Beri, R.J., l{hittington, H., Roberts, C.F. and Hawkins, A.R. 1987.
Isolation and characterization of the positive'ly acting gene QUIA
from ,4spergíllus niduløns. Nucleíc Acíds Res. 15: 7991-8001.
Berse, 8., Dmochowskô, 4., Skrzypek, M., l.leglenski, P., Bates, M.A.
and l.leiss, R.L. 1983. Cloning and characterization of the
ornithine carbamoyìtransferase gene from Aspergillus nidulons.
Gene 25: 109-117.

168.
d'{s
I
Beulìens, M., Mbonyi, K., Geerts, L., Ga'idines, D., Detremerie, K.,
Jans, A.l.l.H. and Therelein, J.M. 1988. Studies on the mechanism
of the gìucose-induced cAMP signal ìn g'lycolysis and glucose
repression mutants of the yeast Søcchoromyces cerevisioe. Eur. J.
Bíochen. I72z 227-23I.
Biggin, M.D. and Tjian, R. 1989. A purified Drosophilo homeodomain
protein represses transcription dn vitro. Cell 58: 433-440.
Blunberg, H., Eisen, 4., Sledziewski, 4., Bader, D. and Young, E.T.
1987. Two zinc fingers of a yeast reguìatory protein shown by
genetic evidence to be essential for its function. Noture 3282
443-445.
Bossi, L. and Smith, D.M. 1984. Conformational change in the DNA
associated with an unusual promoter mutation in a tRNA operon of
Salmonella. Cell 39: 643-652.
Boyìan, M.T., Mirabito, P.M., Wi I lett, C.E., Zinmerman, C.R. and
Timberlake, }{.E. 1987. Isoìation and physical characterization of
three essential conidiation genes fron Aspergillus nidulons.. Mol.
Cell. Bíol. 7: 3113-3118.
Bram, R.J. and Kornberg, R.D. 1985. Specific protein binding to far
upstream activating sequences in poìymerase II promoters. Proc.
Nøtl. Acod. Sci. U.S.A. 82; 43-47.
Bram, R.J., Lue, N. and Kornberg, R.D. 1986. A GAL famiìy of upstream
activating sequences in yeast: roles in both induction and
repression of transcription. EMB0 J. 5: 603-608.
Bray, P. and Thiesen, H-J. 1990. Putting the finger on DI{A: Meeting
review. The New Bíol. 2: 325-327.
Breunig, K.D. 1989. Glucose repression of LAC gene expression in
yeast is mediated by the transcriptional activator Lac9. Molec.
Gen. Genet. 216z 422-427.
IiiI
r
I

169.
Britten, R.J. and Kohne, D.E. 1968. Repeated sequences in DNA.
Science 16Iz 529-540.
Brody, H. and Carbon , J . 1989 . Eì ectrophoreti c karyotype of
AspergíIlus nidulons. Proc. Notl. Acod. Sci. U.S.A. 86: 6260-
6263.
Busby, S.J.l,l. 1986. Positive regulatjon in gene expression. In
I.R. Booth and C.F. Higgins (eds.), Regulation of gene expression
25 years on. Cambridge University Press. London, U.K.
Caddick, M.X., Arst, H.N. , Tayìor, 1.H., Johnson, R. I. and
Brownìee, 4.G., 1986c. Cloning of the regu'latory gene oreA
mediating nitrogen metabolite repression in Aspergillus nidulons.
EMB0 J. 5: 1087-1090.
Caddick, M.X., Brownìee, A.G. and Arst, H.N. 1986b. Phosphatase
reguìation in Aspergillus nidulonsz responses to nutritional
starvation. Genet. Res. 472 93-102.
Caddick, M.X., Brownìee, A.G. and Arst, H.N. 1986a. Regulation of
gene expression by pH of the growth medium in AspergíItus
nidulqns. Molec, Gen. Genet. 203: 346-353.
Caì1, K.M., Glasser, T., Ho, C.Y., Buckler,4.J., Pelletier, P.,
Haber, D.4., Rose, E.4., Kral,4., Yeger, H., Lewis, W.H., Jones,
C. and Housman, D.E. 1990. Isolation and characterization of a
zinc finger polypept'ide gene at the human chromosome 11 Wilms'
tumor locus. CelZ 60: 509-520.
Carlson, M. 1986. Genes at telomeres: fermentation gene families.
UCLA symp. 1,lol. Cell. Biol. new ser. 332 24L-250.
Carlson, M. 1987. Reguìation of sugar utilization in Socchqronyces
species. J. Bocteriol. 1692 4873-4877.
i
þ
i'j'\iJ
I

170.
rl
Carlson, M. and Botstein, D. L982. Two differently regulated mRNA's
with different 5' ends encode secreted and intrace'lluìar forms of
yeast invertase. Cell 28: 145-154.
Carlson, M., Qsmond, B.C. and Botstein, D. 1981. Mutants of yeast
defective in sucrose utilization. Genetics 98: 25-40-
Case, M.E. and Giles, N.H. 1975. Genetic evidence on the organization
and action of the qo-I gene product: a protein regulating the
induction of three enzymes in quinate catabolism in Neurosporo
crosso. Proc. Notl. Acod. Sci. U.S.A. 72: 553-557.
Celenza, J.L. and Carlson, M., 1986. A yeast gene that is essential
for release from glucose repression encodes a protein kinase.
Scíence 233: 1175-1180.
Celenza, J.L., Eng, F.J. and Carlson, M. 1989. Molecular analysis of
the SilF4 gene of Søcchoronyces cerevísioe: evidence for physica'l
association of the Snf4 protein with the Snfl protein kinase.
Mol. Cell. Biol. 9: 5045-5054.
Celenza, J.L., Marshall-Carìson, L. and Carlson, M. 1988. The yeast
SÍrlF3 gene encodes a glucose transporter homologous to the
manmalian protein. Proc. Nott. Acod. Sci. U.S.A. 85: 2130-2134.
Changelian, P.S., Feng, P., King, T.C. and Milbrandt, J. 1989.
Structure of the NGFI-A gene and deletion of upstream seguences
responsible for its transcriptional induction by nerve growth
factor. Proc. Nøtl. Acsd. Sc¿. U.S.A. 862 377-38t-
Chavrier, P., Lemaire, P., Relelant, 0., Bravo, R. and Charnay, P'
1988. Characterization of a mouse multigene family that encodes
zinc finger structures. Mol. Cell. Biol. 8z 1319-1326.
Chen, l{., Tabor,. S. and Struhl, K. 1987. Distinguishing between
mechanisms of eukaryotic transcriptional activation with
bacteriophage T7 RNA polymerase. CeLZ 50: 1047-1055.
¡
ìI

t7L.
Chou, P.Y. and Fasman, G.D. 1978. Prediction of the secondary
structure of proteins from their amino acid sequence. Adv. Enz-
47 z 45-147 .
Christy, 8.A., Lau, L.F. and Nathans, D. 1988. A gene activated in
mouse 3T3 cells by serum growth factors encodes a protein with
zinc finger sequences. Proc. Notl. Acod. Sci. U.S.A. 85: 7857-
786r.
Ciriacy, M. 1975. Genetics of alcohol dehydrogenase in Socchoronyces
cerevisíoe. Mutotion Res. 292 315-326.
Clenents, J.M. and Roberts, C.F. 1986. Transcription and processing
signal s in the 3-phosphogìycerate kinase (PGK) gene from
Aspergillus niduløns. Gene 44: 97-105.
Cì utterbuck, A. J . 1984. Loc i and I i nkage map of the fi I amentous
fungus Aspergillus nidulons. pp. 265-273. In S.J. 0'Brien
(ed.), Genetjc maps, vol 3. Cold Spring Harbor Laboratory, Cold
Spring Harbor, N.Y.
Coddington, A. 1976. Biochemical studies ån the n¿t mutants of
Neurosporo crssso. Molec. Gen. Genet. 145: 195-206.
Cohen, B.L. 1972. Amrnonium repression of extracellular protease in
Aspergillus nídulons. J. Gen. Micro. 7Iz 293-299.
Cohen, B.L. 1973. Reguìation of intracellular and extracellular
neutral and aìkaline proteases in Aspergillus nidulons. J. Gen-
Micro. 79: 3 I 1-320.
Cohen, G.H., Dietzschold, 8., Ponce, M., de Leon, M., Golube, E.,
Varrichio, 4., Pereira, L. and Eisenberg, R.J. 1984. Localization
and synthesis of an antigenic determinant of herpes sínplex virus
glycoprotein D that stinulates the production of neutral'izing
antibody. J. Virol. 49: 102-108.{r

r72.
{
Cossart, P. and Gicquel-Sanzey, B. L982. Cloning and sequence of the
crp gene of Escherichiø coli K L2. Nucleic Acids Res. 10: 1363-
1378
Cove, D.J. 1966. The induction and repression of nitrate reductase in
the fungus Aspergillus nidulons. Biochín. Biophs. Acto 113: 51-
56.
Cove, D.J. and Pateman, J.A. 1963. Independentìy segregating genetic
loci concerned with nitrate reductase activity in Aspergillus
niduløns. Noture l98z 262-263.
Cunliff, V. and Trowsda'le, J. 1990. Unpublished. genBank data base
accession X51760.
Darnelì, J.E. L982. Variety in the level of gene control in
eukaryotic cel1s. Noture 297: 365-371.
Davis, M.A. and Hynes, M.J. L987. Complementation of oreA-
regul atory gene mutati ons of Aspergillus níduløns by the
heterologous regulatory gene nit-2 of Neurosporo crosso. Proc.
Notl. Acsd. Sci. U.S.A. 84: 3753-3757.
Davis, M.A. and Hynes, M,J. 1989. Reguìatory genes in Aspergillus
níduløns. Trends Genet. 5: 14-19.
Dean, R.A. and Timberlake, W.E. 1989. Regulation of the Aspergillus
nídulons pectate ìyase gene (pe|A). Plont Cell Iz 275-284-
de Crombrugghe, 8., Busby, S. and Buc, H. 1984. Cyclic AMP receptor
protein: role in transcription activation. .Sc¿ence 224: 831-838.
Denis, C.L. and Malvar, T. 1990. The CCR4 gene from Socchøronyces
cerevisíoe is required for both non-fermentative and SPf-mediated
gene expression. Genetics l24z 283-29I.
Devereux, J. 1984. Genetjcs computer group sequence ana'lysis software
package version 6.1.. Nucleic Acids Res. 12:387-395.

r73.
Dickson, R.C., Geradot, C.J. and Martin, A.K. 1990. Genetic evidence
for similar negative regulatory domains in the yeast
transcriptional activators Gal4 and Lac9. Nucleíc Acíds Res. 18:
5213-5217.
Dickson, R.C. and Markin, J.S. 1980. Physiologica'l studies of ß -
galactosidase induction in Kluyveronyces løctis. J. Bocteriol.
1422 777-785.
Dodd, I.B. and Egan. J.B. 1987. Systematic method for the detection
of potential lambda Cro-like DNA-binding regions in proteins. J.
Mol. Biol. 194: 557-564.
Doonan, J.H. and Morris, N.R. 1989. The öimG gene of Aspergillus
nídulons required for compìetion of anaphase, encodes a homolog
of manrnalian phosphoprotein phosphatase 1. Cell 572 987-996.
Dowzer, C.E.A. and Kelìy, J.M. 1989. Cloning of the creA gene from
Aspergillus nidulons z a gene i nvol ved i n carbon catabol i te
repression. Curr. Genet. 15: 457-459.
Dunn-Coleman, N.S., Tomsett, A.B. and Garrett, H. L979. Nitrogen
metabolite repression of nitrate reductase in Neurosporo crøssøz
effect of the gln-1a locus. J. Bocteríol. 1392 697-700.
Dunn-Coìeman, N.S., Tomsett, A.B. and Garrett, R.H. 1981. The
reguìation of nitrate assimiìation in Neurosporo crossq2
biochemical analysis of nmr-l mutants. Molec. Gen. Genet. L82z
234-239.
Dynan, W.S. and Tjian, R. 1985. Control of eukaryotic rnRNA synthesis
by sequence specific DNA binding prote'ins. Noture 3162 774-778.
Ebright, R.H., Cossart, P., Gicquel-Sanzey, B. and Beckwith, J. 1984.
Mutations that alter the DNA sequence specificity of the
catabolite gene activator protein of E. coli. Noture 3llz 232-
235.

L7 4.
Ei len, E. , Pampeno, C. and Krakow, J.S. 1978. Production and
properties of the q-core derived from the cycl'ic adenosine
monophosphate receptor protein of Escheríchio coli. Biochen. 17z
2469-2473.
Elliot, C.G. 1960. The cytology of Aspergíllus nidulons. Genet. Res.
Iz 462-476.
Elorza, M.V. and Arst, H.N. t971. Sorbose resistant mutants of
Aspergillus nidulons. Molec. Gen. Genet. 111: 185-193.
Emini, E.4., Hughes, J.V., Per'low, D.S. and Boger, J. 1985. Induction
of hepatitis A virus-neutralizing antibody by a virus specìfic
synthetic peptide. J. Virol. 55: 836-839.
Entian, K-0.1977. Lack of carbon catabolite inactivation in a mutant
of Socchoromyces cerevisíoe with reduced hexokinase activity.
Molec. Gen. Genet. l58z 20L-210.
Entian, K-D. 1980. Genetic and biochemical evidence for hexokinase
PII as a key enzyme involved in carbon catabolite repression in
yeast . Molec. Gen. Genet. 178: 633-637
Entian, K-D. 1986. Gìucose repression: a comp'lex reguìatory system in
yeast. Mícro. Sci. 3: 366-371.
Entian, K-D., Frohlich, K.V. 1984. Socchøronyces cerevísiae mutants
provide evidence of hexokinase PII as a bifunctional enzyme with
catalytic and regulatory domains for triggering carbon catabolite
repression. J. Bøcteríol. 1582 29-35.
Eraso, P. and Gancedo, J.M. 1984. Catabolite repression in yeasts is
not associated with low levels of cAMP. Eur. J. Bíochen. L4lz
195-198.
Evans, R.M. 1988. The steroi d and thyroid hormone receptor
superfamily. Science 240: 889-895.

t75.
Felenbok, 8., Sequeva'l , D., Mathieu, M., Sibely, S., Gwynne, D.I. and
Davies, R.W. 1988. The ethanol regu'lon in Aspergillus nidulønsz
characterizatjon and sequence of the positive regu'latory gene
øZcR. Gene 73: 385-396.
Felenbok, 8., Sophianopouìou, V., Mathieu, M., Sequeval, 0.,
Kulmberg, P., Dialìinas, G. and Scazzocchio, C. 1989. Regulation
of genes involved in the utilization of carbon sources in
Aspergíllus nidulons. In H. Nevalainen and M. Penttila (eds.),
Proc. of the EMBQ-A]ko workshop on moì. biol. of filamentous
fungi, Helsinki . Foundotion for biotechnicøl ond índustriol
fermentøtion reseorch 6: 73-83.
Fincham, J.R.S., 1989. Transformation in fungi. Micro. Rev. 53: 148-
r70.
Fink, G.R. 1986. Translational control of transcription in
eukaryotes. Cell 45: 155-156.
Flick, J.S. and Johnston, M. 1990. Two systems of gìucose repression
of the â4t1 promoter in Socchoronyces cerevísioe. Mol. Cell.
Biol. I0z 4757-4769.
Fried, M. and Crothers, D.M. 1981. Equilibria and kinetics of ìac
repressor-operator interactions by polyacrylamide gel
electrophoresis. Nucleic Acids Res. 9: 6505-6525.
Frischauff , 4.M., Lehrach, H., Poustka, A. and Mumay, N. 1983.
Lambda repìacement vectors carrying poìylinker sequences. J. Mol.
Bíol. l70z 827-842.
Fu, Y.H. and Marzluf, G.A. 1987. Characterization of nít-Z, the maior
nitrogen regulatory gene of Neurosporo crøsso. Mol. Cell. Bíol.
7: 1691-1696.

176.
Fu, Y.H. and Marzluf, G.A. 1990a. nit-Z, the maior regulatory gene of
Neurosporo crosso, encodes a protein with a putative Zn finger
DNA-binding domain. Mol. Cell. Biol. 10: 1056-1065.
Fu, Y.H. and Marzluf, G.A. 1990b. Site-directed mutagenesis of the
"zinc-finger" DNA-binding domain of the nitrogen regu'latory
protein Nit2 of Neurosporo. Mol. Micro. 4z 1847-1852.
Fu, Y.H., Young, J.L. and Marzluf, G.A. 1988. Molecular cloning and
characterization of a negative-acting nitrogen regulatory gene of
Neurosporo crosso. Molec. Gen. Genet. 2I4z 74-79.
Furst, P., Hu, S., Hackett, R. and Hamer, D. 1988. Copper activates
metallothionein gene transcription by altering conformation of a
specific DNA binding protein. CelZ 55: 705-7L7.
Gancedo, J.M., Gancedo, C. 1986. Catabolite repression mutants of
yeast. FEIíS Mícro. Rev. 32¿ I79-I87.
Garnier, J., Qsugthorpe, D.J. and Robson, B. 1978. Analysis of the
accuracy and imp]ications of simp'le methods for predicting
the secondary structure of globuìar proteins. J. Mol Bíol. I20z
97-120.
Gasser, S.M. and Laenmìi, U.K. 1986. Cohabitation of scaffold binding
regions with upstream enhancer elements of three developmentaììy
regulated genes of D. nelonogoster. Cell 462 521-530.
Geever, R.F., Huiet, L., Baum, J.4., Tyler, 8.M., Patel, V.8.,
Rutledge, 8.J., Case, M.E. and Giles, N.H. 1989. DNA sequence'
organization and regu'lation of the go gene cluster of Neurosporo
crøssø. J. Mol. Biol. 207: 15-34.

r77 .
Geliebter, J. , Zeff, R.4., Melvold, R.l{. and Nathenson, S.G. 1986.
Mitotic recombination in germ cel ls generated two major
histocompatibility complex mutant genes shown to be identical by
RNA sequence analysis: Kbms and Kbm6. Proc. Nøtl. Acod. Scí.
U.S.A. 83: 3371-3375.
Ghosh, B.K., Montencourt, B. and Lampen, J.0. 1973. Abnonnal celI
envelope structure of a Socchoronyces mutant with invertase
formation resistant to hexoses. J. Bocteríol. 1162 I4I2-I420.
Gilbert, W.M., Majors, J. and Maxam, A. 1976. In Dahlem l{orkshop on
chromosomes . Abakon Verl agsgesel ì schaft, Berl i n . pp 167-178.
Giles, N.H., Case, M.E., Baum, J., Geever, R.F., Huiet, L., Patel, V.
and Tyler, B. 1985. .Gene organization and reguìation in the go
(quinic acid) cluster of Neurosporo crosso. Micro. Rev. 492
338-358.
Giniger, E. and Ptashne, M. 1987. Transcription in yeast activated by
a putative amphipathic aìpha helix linked to a DNA binding unit.
Nsture 3302 670-672.
Giniger, E., Varnuum, S.M. and Ptashne, M., 1985. Specific DNA
binding of GAL4, a positive regulatory protein of yeast. Cetl
40:767-774.
Goto, T., Macdonald, P. and Maniatis, T. 1989. Early and late
periodic patterns of evenskipped expression are controlled by
distinct regulatory elements that respond to different spatial
cues. Cell 572 413-422.
Grant, S., Roberts, C.F., Lamb, H., Stout, M. and Hawkins,4.R.1988.
Genetic regulation of the quinic acid utilization (QUT) gene
cluster in Aspergillus nidulons. J. Gen. Micro. 134: 347-358.

r78.
Gribskov, M. and Devereux, J. 1986. PEPLQT, a protein secondary
structure analysi s program for the UI.|GCG sequence analysi s
software package. Nucleíc Acids Res. 14:. 327-334.
Griffith, 0.M. 1979. Techniques of preparative, zonal, and continuous
flow centrifugation. Beckman Instruments Inc. U.S.A.
Guarente, L. 1984. Yeast promoters: positive and negative elements.
Cell 36: 799-800.
Guarente, L. 1988. UAS's and enhancers: cornmon mechani sm of
transcriptional activation in yeast and mammals. Cell 52: 303-
305.
Gurr, S.J., Unkles S.E. and Kinghorn J.R. 1987. In Kinnghorn, J.R-,
(ed). Gene structure in eukaryotic microbes. IRL Press, Oxford,
pp. 93-139.
Gwynne, D.I., Buxton, F.P., Sibely, S., Davies, R.W., Lockington,
R.4., Scazzocchio, C. and Sea'ly-Lewis, H.M. L987. Comparison of
the cis-acting control regions of two coordinateìy controlled
genes involved in ethanol utilization in Aspergillus nídulons.
Gene 5I¿ 205-2L6.
Haluska, F.G., Tsujimoto, Y. and Croce, C.M. 1987. Oncogene
activation by chromosome translocation in human malignancy. Ann.
Rev. Genet. 2I¿ 321-345.
Hamer, J.E. and Timberlake. l.l.E. 1987. Functional organization of the
Aspergíllus nídulons trpC promoter. Mol. CeIl. Biol. 7: 2352-
2359.
Han, K., Levine, M.S. and Manìey, J.L. 1989. Synergistic activation
and repression of transcription by DrosophiZø homeobox proteins.
Cell 56: 573-583.
Hanahan, D. 1983. Studies on transformation of Escheríchio coZ¿ with
plasmids. J. Mol. Biol. 166: 557-580.

179.
Hartshorne, T.4., Blumberg, H. and Young, E.T. 1986. Sequence
homoìogy of the yeast regu'latory protein Adrl with Xenopus
transcription factor IIIA. Noture 3202 283-287.
Hastie, A.C. 1970. Benlate induced instabi ì ity of Aspergillus
d'ipì oi ds. Noture 2262 77L.
Hawkins, 4.R., Lamb, H.K., Smith, M., Keyte, J.W. and Roberts, C.F.
1988. Molecular organ'ization of the quinic acid utilization (QUf)
gene cluster in Aspergíllus nidulons. Molec. Gen. Genet. 2L4:
224-23r.
Hawley, D.K., Maìan, T.P., Mu'lì igan, M. and McClure, l,f . 1982. In
R. Rodnguez and M. Chamberlin (eds.), Promoters: structure and
function. Praeger, N.Y. pp 54-68.
Hayashi, S. and Scott, M.P. 1990. What determines the specificity of
action of DrosopåiZø homeodomain proteins? CeIZ 63: 883-894.
Hayman, D.1., Ashworth, L.K. and Carrano, A.V. L982. The relative DNA
contents of the eutherian and marsupial X chromosomes. Cyto.
Cell. Genet. 342 265-270.
Ho'llenberg, S.M. and Evans, R.M. 1988. Multiple and cooperative
trans-activation domains of the human glucocorticoid receptor.
Cell 55: 899-906.
Hollenberg, S.M., Giguere, V. Sergui, P. and Evans, R.M. 1987.
Colocal ization of DNA-binding and transcriptional activation
functions in the hunan glucocorticoid receptor. Cell 49: 39-46.
Holzer, H. 1976. Catabolite inactivation in yeasts. Trend. Biochen.
Sci. 8: 178-181.
Hope, I.4., Mahadevan, S. and Struhì , K. 1988. Structural and
functional characterization of the short acidic transcriptional
activation region of yeast Gcn protein. Nøture 333: 635-640.

180.
Hope, I.A. and Struhl, K. 1986. Functional dissection of a eukaryotic
transcriptiona'l activator protein Gcn4 of yeast. Cetl 462 885-
894.
Horikoshi, M., Carey, M.F., Kakidani, H. and Roeder, R.G. 1988.
Mechanism of action of a yeast activator: direct effect of GAL4
derivatives on manmalian TFIID-promoter interactions. Cell 542
665-669.
Horowitz, N.H. and Mcleod, H. 1960. The DNA content of Neurosporo
nuclei . ||icro. Genet. Bull. L7: 196-198.
Huiet, L. and Gi'les, N.H. 1986. The qo repressor gene of Neurosporo
crosso: wi'ldtype and mutant nucleotide sequences. Proc. Notl.
Acod. Scí. U.S.A. 83: 3381-3385.
Huynh, T.V., Young, R.A. and Davis, R.l,l. 1985. Constructing and
screening cDNA libraries in lgtlO and À9t11. In D.M. Glover
(ed.), DNA c'loning: a practical appraoch Vol. IRL Press. 0xford,
U. K.
Hynes, M.J. 1970. Repressìon of amidase enzymes in Aspergillus
nidulons. J. Bscteríol. 1032 482-487.
Hynes, M.J. L974. The effects of the carbon source on glutamate
dehydrogenase activities in Aspergillus níduløns. J. Gen.
Mícro. 81: 165-170.
Hynes, M.J. t975. Studies on the roìe of the øreA gene in the
regulation of nitrogen catabolism in Aspergillus nidulons. Aust.
J. Biot. Sc¿. 282 301-313.
Hynes, M.J., Corrick, C.M., Ke]ìy, J.M. and Littìejohn, T.G. 1988.
Identification of the sites of action for regulatory genes
contro'lling the øndS gene of Aspergíllus nidulons. Mol- Cell.
Biol. 8: 2589-2596.

181.
Hynes, M.J., Corrick, C.M. and King, J.A. 1983. Isolation of genomic
clones containing the ørnds gene of Aspergillus níduløns and their
use in the analysis of structural and regulatory mutations. /tto¿.
Cell. Bíol. 3: 1430-1439.
Hynes, M.J. and Kelly J. 1977. Pleiotropic mutants of ,4. nídulons
altered in carbon metabol ism. Molec. Gen. Genet. 150: 193-204.
Hynes, M.J. and Pateman, J.A.J. 1970a. The genetic analysis of
reguìation of amidase synthesis in Aspergíllus nídulons I.
Mutants able to utiìize acrylamide. Molec. Gen. Genet. 108: 95-106.
Hynes, M.J. and Pateman, J.A.J. 1970b. The genetic analysis of
regulation of amidase synthesis in Aspergillus nidulons. II
Mutants resistant to fluroacetamide. Molec. Gen. Genet. 108: 107-
116.
Inokuchi, K., Nakayomô, A. and Hishinuma, F. 1988. Sequence-directed
bends of DNA heìix at the upstream activation sites of a-cell
specific genes in yeast. Nucleic Acids Res., 16: 6693-67LI.
Ish-Horowicz, D. and Burke, J.F. 1981. Rapid and efficient cosnid
cloning. Nucleíc Acids Res. 9: 2989-2998.
Jackle, H., Tautz, 0., Schuh, R., Seifert, E. and Lehmann, R. 1986.
Cross-regulatory interactions among the gop genes of Drosophílo.
Noture 324: 668-670.
Jackson, S.P. and Tjian, R. 1988. 0-glycosylation of eukaryotic
transcription factors: impl ications for mechanisms of
transcriptional regulation . Cell 55: 125-133.
Janssen-Tinmen, U., Lemaire, P., Mattei, M-G., Revelant, 0. and
Charnay, P. 1989. Structure, chromosome mapping and regulation of
the mouse zinc-finger gene krox-Z4, evidence for a corllnon
regulatory pathway for in¡mediate early serum-response genes. Gene
80:325-336.

182
Jarai, G. and Marzluf, G.A. 1990. Analysis of conventional and in
vitro generated mutants of nnr, the negatively acting nitrogen
reguìatory gene of Neurosporo crosso. Molec. Gen. Genet. 2222
233-240.
Jayaraman, P-S., Cole, J. and Busby, S. 1989. Mutational anaìysis of
the nucleotide sequence at the FNR-dependent nírB promoter in
Escherichía coli. Nucleic Acids Res. 17: 135-146.
Johnson, P.F., Landschulz, W.H., Graves, B.J. and McKnight, S.L.
1987. Identification of a rat liver nuclear protein that binds to
the enhancer core element of three animal viruses. Genes Devel.
1: 133-146.
Johnston, M. 1987. A model fungaì gene regulating mechanism: the GAL
genes of Socchoronyces cerevísíoe. Micro. Rev. 51: 458-476.
Johnston, S.4., Salmeron, J.M. and Dincher, S.E. 1987. Interactions
of positive and negative regulatory proteins in the gaìactose
regu'lon of yeast. Cell 50: 143-146
Joseph, L.J., Le Beau, M.M., Jamieson, G.4., Acharya, S., Shows,
T.8., Rowley, J.D. and Sukhatme, V.P. 1988. Molecular cloning,
sequencing and mapping of Egr-2, a human early growth response
gene encoding a protein with a zinc-binding finger structure.
Proc. Notl. Acad. Scí. U.S.A.85:7I64-7L68.
Kadonaga, J.T., Carner, K.R., Masiarz, F.R. and Tjian, R. L987.
Isolation of cDNA encoding transcrjption factor Spl and
functionaì anaìysis of the DNA binding domain. CelZ 1079-1090.
Kafer, E. 1961. The process of spontaneous recombination in
vegetative nucìei of Aspergillus nidulqns. Genetícs 45: 1581-
1609.

183.
Katz, M.E. and Hynes, M.J. 1989a. Isolation and anaìysis of the
acetate regulatory gene /ocB from Aspergillus nidulons. Mol.
Cell. Biol. 9: 5696-5701.
Katz, M.E. and Hynes, M.J. 1989b. Characterization of the ond[-
control 1ed lom| and lon} genes of Aspergíllus nídulons. Genetícs
I22:33I-339.
Kelìy, J.M. 1980. Plejotropic mutants of Aspergíllus nidulons
affected in carbon metabolism. Ph.D thesis, Dept. Genet. Human
Var. La Trobe University, Melbourne, Australia.
Kelly, J.M. and Hynes, M.J. L977. Increased and decreased sensitivity
to carbon catabolite repression of enzymes of acetate metabolism
in mutants of A. nidulons. Molec. Gen. Genet. 1562 87-92-
Kelly, J.M. and Hynes, M.J. 1985. Transformation of Aspergillus niger
by the omdS gene of Aspergillus nidulons. EMBÌ J. 4:475-479.
Kemp, B.E. and Pearson, R.B. 1990. Protein kinase recognition
sequence motifs. Trend. Bíochem. Scí. I5z 342-346.
Kinghorn, J.R. and Pateman, J.A. Lg73. NAò and NADP L-glutamate
dehydrogenase activity and anmonium reguìatjon in Aspergillus
nídulons. J. Gen. Micro. 78: 39-46.
Klevit, R.E., Herriott, J.R. and Horvath, S.J. 1990. Solution
structure of a zinc finger donain of yeast ADRl. Proteins 7z 2I5-
226.
Kìug, A. and Rhodes, D. L987. "Zinc fingers": a novel protein notif
for nucleic acid recognition. Trends Biochen. Sci. t2z 464-469.
Koepsel, R.R. and Khan, S.A. 1986. Static and initiator protein-
enhanced bending of DNA at a replication origin. Scíence 233:.
13 16- 13 18 .

184.
Krakow, J.S. and Pastan, I. 1973. Cyclic adenosine monophosphate
receptor: loss of cAMP-dependent DNA binding activity after
proteolysis in the presence of cyclic adenosine monophosphate.
Proc. Notl. Acod. Scf. U.S.A. 702 2529-2533.
Krasnow, M.4., Saffman, E.E., Kornfeld, K. and Hogness, D.S. 1989.
Transcriptionaì activation and repression by Ultrabithorax
proteins in cultured DrosophiZø ceìls. Cell 572 1031-1043.
Kudla, 8., Caddick, M.X., Langdon, T., Martinez-Rossi, N.M., Bennett,
F., Sibley, S., Davies, R.W. and Arst, H.N. 1990. The regulatory
gene oreA mediating nitrogen netabolite repression in Aspergillus
nidulons. Mutations affecting specificity of gene activation
alter a loop residue of a putative zinc finger. EMB0 J. 9: 1355-
1364.
Kuger, P., Godecke, A. and Breunig, K.D. 1990. A mutation in the Zn-
finger of the Gal4 homo'log Lacg results in glucose repression of
its target genes. Nucleic Acids Res. 18: 745-751.
Kunkel, G.R. and Mortinson, H.G. 1981. Nucleosomes will not fonn on
double stranded RNA or over po'ly dA- poly dT tracts in
recombinant DNA. Nucleíc Acíds Res. 9: 6869-6888.
Kyte, J. and Doolittle, R.F. 1982. A simple method for displaying the
hydropathic character of a protein. J. Mol. Bíol. L57: 105-132.
Landschulz, W.H., Johnson, P.F., Adashi, E.Y., Graves, B.J. and
McKnight, S.L. 1988b. Isolation of a recombinant copy of the gene
encoding C/EBP. Genes Devel. 2z 786-800
Landschulz, H.H., Johnson, P.F. and McKnight, S.L. 1988a. The leucine
zipper: a hypothetical structure corilnon to a new class of DNA
binding proteins. .Sc¿ence 240: 1759-1764.

185.
Lania, L., Donti, E., Pannuti, 4., Pascucci , 4., Pengue, G.,
Feìiciello, I., La Manti.t, G., Lanfrancone, L. and Peìicci, P-G.
1990. cDNA isolation, express'ion anaìysis and chromosomaì
location of two human zinc finger genes. Genonícs 6: 333-340.
Latchman, D.S. 1990a. Eukaryotic transcription factors. Biochen. J.
2702 281-289.
Latchman, D.S. 1990b. Gene reguìation. A eukaryotic perspective,
Unwin Hyman, London, U.K.
Laurent, 8.C., Treitel, M.A. and Carlson, M. 1990. The Snf5 protein
of Søcchoronyces cerevísiqe i s a gl utami ne and prol i ne ri ch
transcriptional activator that affects expression of a broad
spectrun of genes. Mol. Cell. Biol. 10: 5616-5625.
Lee, M.S., Gippert, G.P., Soman, K.V., Case, D.A. and l,{right, P.E.
1989. Three-dimensional soìution structure of a singìe zinc
finger DNA-binding domain. Science 245: 635-637.
Levine, M. and Manley, J.L. 1989. Transcriptional repression of
eukaryotic pronote rs. Cell 59: 405-408.
Licht, J.D., Grossel, M.J. Figge, J. and Hansen, U.M. 1990.
Drosophilø kruppel protein is a transcriptional repressor. Noture
346: 76-79.
Lockington, R., Scazzochio, C., Sequeval, D., Matthieu, M. and
Felenbok, B. 1987. Reguìation of ølcR, the positive regulatory
gene of the ethanol utilization regulon of Aspergíllus nídulons.
Mol. Mícro. Lz 275-28I.
Lockington, R.4., Sealy-Lewis, H.M., Scazzocchio, C. and Davies, R.H.
1985. Cloning and characterization of the ethanol utilization
regulon in Aspergíllus nídulons. Gene 33: 137-149.

186.
Lohr, D. and Hopper, J.E. 1985. The relationship of regulatory
proteins and DNase I hypersensitive sites in the yeast GALL
genes. Nucleic Acíds Res. 13: 8409-8423.
Lue, N.F., Buchman, A.R. and Kornberg, R.D. 1989. Activation of yeast
RNA poìymerase II transcription by a thymidine-rich upstream
element in vitro. Proc. Notl. Acød. Sc¿. U.S.A. 86: 486-490.
Mâ, H. and Botstein, D. 1986. Effects of null mutations in the
hexokinase genes of Socchoronyces cerevísioe on catabol ite
repression. l,lol Cell. Bíol.6z 4046-4052.
Magasanik, B. and Neidhardt, F. 1987. In F. Neidhardt (ed.),
Escherichio col¿ and Solnonellø typhinuríun. Vol I. American
Soc. Microbioì., Washington, D.C. pp 1315-1325.
Maizel, J.V. and Lenk, R.P. 1981. Enhanced graphic matrix analysis of
nucleic acid and protein sequences. Proc. Notl. Acod. Scf. U-5.A.
782 7665-7669.
Maniatis, T., Fritsch, E.F. and Sambrook, J. L9.82. Molecular cloning:
a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring
Harbor, N.Y.
Maroun, R.C. and Qlson, t{.K. 1988. Base sequence effects in double
hel i cal DNA I I . Conformati onal stati sti cs of rodl i ke chai ns .
Bíopolymers 27: 561-584.
Marshall, R.D. 1972. Glycoproteins. Annu. Rev. Bíochen. 4Lz 673-702.
Marzluf, G.A. 1981. Reguìation of nitrogen metabol ism and gene
expression in fungi . Micro. Rev. 45: 437-461.
Matern, H. and Holzer, H. 1977. Cataboìite inactivation of the
galactose uptake system in yeast. J. Bíol. Chen. 2522 6399-6402.

187.
Matsumoto, K., Toh, E.A. and Oshima, Y. 1981. Isolation and
characterization of dominant mutations resistant to carbon
catabolite repression of gaìactokinase synthesis in Socchoronyces
cerevisíoe. Mol. Cell. Biol. 1: 83-93.
Matsumoto, K., Uno, I., Ishikawa, T. and Oshimô, Y. 1983. Cyclic AMP
may not be involved in catabolite repression in Soccharonyces
cervísioez evidence fron mutants unable to synthesize it. J.
Bocteriol. 156: 898-900.
Matsumoto, K., Yoshimatsu, T. and Oshima, Y. 1983. Recessive
mutations conferring resistance to carbon catabolite repression
of gaìactokinase synthesis in Socchoronyces cerevísioe. J.
Bocteriol. 153: 1405-1414.
McCullough, W., Payton, M.A. and Roberts, C.F. L977. Carbon
metabolism in Aspergíllus nídulons. In J.E. Smith and J.A.
Pateman (eds.), Genetics and physiology of Aspergillus. Academic
Press. London, U.K.
McKay, D. and Steitz, T. 1981. Structure of cataboìite gene activator
protein at 2.94 resolution suggests binding to left-handed B-DNA.
Nøture 2902 744-753.
Mihara, H. and Kaiser, E.T.1988. A chemicaìly synthesized
Antennapedia homeodonain binds to a specific DNA sequence.
Scíence 2422 925-927.
Milìer, J., Mclachìan, A.D. and Kìug, A. 1985. Repetitive zinc-
binding domains in the protein transcription factor IIIA from
Xenopus oocytes. EMB0 J. 4z 1609-1614.
Mitchell, P.J. and Tjian, R. 1989. Transcriptional regulation in
mar¡nal ian cel ls by sequence-specific DNA binding proteins-
Scíence 245: 371-378.

188
Modrow, S. and l{olf, H. 1986. Characterization of two related
Epstein-Barr vi rus-encoded membrane protei ns that are
differentiaì ly expressed in Burkitt lymphoma and in vitro-
transformed cell lines. Proc. NotI. Acod. Sc¿. U.S.A. 83: 5703-
5707.
Moehle, C.M. and Jones, E.W. 1990. Consequences of growth media, gene
copy number, and reguìatory mutations on the expression of the
Pß81 gene of Socchoronyces cerevisíoe. Genetícs I24z 39-55.
Montencourt, 8.S., Kuo, S-C. and Lampen, J.0. 1973. Socchoronyces
mutants with invertase formation resistant to repression by
hexoses. J. Bacteríol. 1142 233-238.
Mulìer, M., Affoìter, M., Leupir, W., Qttin9, G., Wuthrich, K. and
Gehring, l{.J. 1988. Isolation and sequence-specific DNA binding
of the Antennapedia homeodomain. EMBÌ J. 7z 4299-4304.
Nardelli, J., Gibson, T.J., Vesque, C. and Charnay, P. 1991. Base
sequence di scrirnination by zinc-fi nger DNA-bi nding donai ns.
Noture 3492 175-178.
Nehtir, J. and Ronne, H. 1990. Yeast MIG1 repressor is related to the
mamnaìian early growth response and Wilms' tumour finger
proteins. EMB0 J. 9: 2891-2898.
Neigeborn, L. and Carlson, M. 1984. Genes affecting the regulation
of SUCZ gene expression by glucose repression in Socchoromyces
cerevisioe. Genetícs 108: 845-858.
Neigeborn, L. and Carlson, M. 1987. Mutations causing constitutive
invertase synthesis in yeast: genetic interactions with snl
mutations. Geneúics 1LSz 247-253.
Neigeborn, L. Rubin, K. and Carlson, M. 1986a. Suppressors of sf,Jf2
mutations restore invertase derepression and cause temperature-
sensitive lethality in yeast. Genetics II2= 74I-753-
üirt
I
I
r

189
!
Neìgeborn, L., Schwartzberg, P., Reid, R. and Carlson, M. 1986b.
Null mutation in the S/VF3 gene of Socchoromyces cerevísíoe cause
a di fferent phenotype than do previ ously i sol ated mi ssense
mutations. lloZ. Cell. Bíol. 6z 3569-3574.
Nishikawa, K. 1983. Assessment of secondary structure prediction of
proteins comparison of computerized Chou-Fasman method with
others. Bíochin. Bíophys. Acto 7482 285-299.
Nussinov, R. 1985. Large helical conformationaì deviation from ideal
B-DNA and prokaryotic reguìatory sites. J. Theor. Biol. 115: 179-
189.
Nussinov, R. 1990. Sequence signaìs in eukaryotic upstream regions.
Crit. Rev. Bíochen. Mol. Bíol. 25¿ L85-224.
Nussinov, R., Shapiro, 8., Lipkin, L.E. and Maize] , J.V. 1984. DNAase
I hypersensitive sites may be correlated with genomic regions of'large structural variation. J. Mol. Bíol. L77: 591-607.
0akley, C.E., t{ei'l , C.F., Kretz, P.L. and Oakley, B.R. 1987a. Cloning
of the riboB locus of Aspergíllus nidulons. Gene 53¿ 293-298.
Oakley, 8.R., Rinehart, J.E., Mitcheì1, 8.L., Oakley, C.E., Cannona,
C., Gray, G.L. and May, G.S. 1987b. Cloning, mapping and
molecular analysis of the pyrí (orotidine-S phosphate
decarboxylase) gene of Aspergillus níduløns. Gene 6l: 385-399.
0'Connel I , M.J. 1990. The structure and reguì ation of aldehyde
dehydrogenase encoding genes in Áspergillus níger and Aspergíllus
nidulons. Ph.D thesis. Dept. Genetics. University of Adeìaide,
Adelaide, Austraìia.
Oehler, S., Eismann, E.R., Kramer, H. and Muller-Hilì, B. 1990. The
three operators of the lac operon cooperate in repression. EMB0
J. 9z 973-979.
H
r
I
I
3
II

190.
f¡rlù
I
Ogur, M., Minckìer, S. and Lindegren, C.C. 1952. The nucìeic acids in
a poìyp'loid series of Socchoromyces. Arch. Biochen. Biophys. 40:
30-196.
0hkuma, Y., Horikoshi, M., Roeder, R.G. and Desplan, C. 1990.
Engrai led, a homeodomain protein can repress in vítro
transcription by competition with the TATA box-binding
transcription factor llD. Proc. Notl. Acod. Scí. U.S.A. 872 2289-
2293.
Omichjnski, J.G., Clore, G.M., Appella, E., Sakaguchi, K. and
Gronenborn, A.M. 1990. High-resoìution three-dimensional
structure of a single zinc finger from a human enhancer binding
protein in soìution. Bíochen. 292 9324-9334.
Oshima, Y. 1982. Regulatory circuits for gene expression: the
metaboìism of galactose and phosphate. In J.N. Strathern, E.W.
Jones and J.R Broach (eds.), The molecular biology of the yeast
Sacchøronyces. Metabol ism and gene expression. Cold Spring
Harbour Laboratory, Cold Spring Harbour, N.Y.
Page, D.C., Mosher, R., Simpson, E.M., Fisher, E.M.C., Mardon, G.,
Pollack, J., McGillivray, 8., de la Chapelle, A. and Brown, L.G.
1987. The sex-detennining region of the human Y chromosome
encodes a finger protein. CelZ 51: 1091-1104.
Page, M.M. and Cove, D.J. 1972. Alcohol and amine catabolisn in the
fungus Aspergillus niduløns. Bíochen. J. I27z 17.
Pan, T. and Coleman, J.E. 1990. Gal4 transcription factor is not a
"zinc finger" but forms a Zn(II)rCysu binucìear cluster- Proc-
Nstl. Acod. Scí. 872 2077-2081.
Pankratz, M.J., Hoch, M., Seifert, E. and Jackle, H. 1989. kruppel
requirement for knírps enhancement reflects overlapping gop gene
activities in the Drosophílø embryo. Noture 341: 337-339.
tI
;
Ì

191.
r
Parraga, G., Horvath, S.J., Eisen,4., Tayìor, l{.E., Hood, L., Young,
E.T. and Klevit, R.E. 1988. Z'inc-dependent structure of a singìe-
finger domain of yeast Adrl. Science 24lz 1489-L492.
Passananti, C., Felsani, 4., Caruso, M. and Amanti, P. 1989. Mouse
genes coding for zinc-finger containing proteins:
characterization and expression in di fferentiated cel I s . Proc.
Nøtl. Acod. Sci. U.S.A. 86: 94L7-942L.
Patel, V.8., Schweizer, M., Dykstra, C.C., Kushner, S.R. and Giles,
N.H. 1981. Genetic organizational regulation in the qo gene
cluster of Neurosporo crosso. Proc. Notl. Acod. Scí. U.S.A. 782
5783-5787.
Pateman, J.A., Doy, C.H., 0lsen, J.E., Norris, U., Creaser, E.H. and
Hynes, M.J. 1983. Regulation of alcohol dehydrogenase (ADH) and
aìdehyde (AìdDH) in Aspergillus nidulons. Proc. R. .Soc. London
B2I7z 243-264.
Pickett, M., Gwynne, D.I., Buxton, F.P., Elìiot, R., Davies, R.H.,
Lockington, R.4., Scazzocchio, C. and ieaìy-Lewis, H.M. Lg87.
Cloning and characterization of the øZdA gene of Aspergillus
nídulons. Gene 5L: 217-226.
Pìaskon, R.R. and Warteì ì , R.M. 1987. Sequence distributions
associated with DNA curvature are found upstream of strong E.
coZi promoters. Nucleic Acid Res. 15: 785-796.
Premakumar, R., Sorger, J.G. and Gooden, D. 1980. Physiological
characterization of a Neurosporo crosso mutant with impaired
regulation of nitrate reductase. J. Bocteriol. L44: 542-55I.
Prendergast, G.C. and Ziff, E.B. 1989. DNA binding motif. Noture 34Lz
392.
Prunnell, A. 1982. Nucleosome reconstitution on plasmid inserted poìy
(dA) - poly (dT). EMBî J. 1: 173-179.
rl
I
r

192.
Ptashne, M. 1986. A genetic switch: gene control and phage (ìambda).
Cambridge, Palo Alto and Calif; Cell Press, Blackwell Sci. Pub.
Ptashne, M. and Gann, A.A.F. 1990. Activators and targets. Noture
346:329-331.
Punt, P.J., Dingemanse, M.4., Jacobs-Meijsing, B.J.M., Pouwels, P.H.
and van den Hondeì, C.A.M.J.J. 1988. Isolation and
characterization of the g'lycera'ldehyde-3-phosphate dehydrogenase
gene of Aspergillus nidulons. Gene 692 49-57.
Punt, P.J.,0liver, R.P., Dingemanse, M.4., Pouwels, P.H. and van den
Hondel, C.A.M.J.J. 1987. Transfonnation of Aspergillus based on
the hygromycin B resistance marker from Escåeríchío colí. Gene
56: lL7-124
Redeman, N., Gauì, U. and Jackle, H. 1988. Disruption of a putative
Cys-zinc interaction eliminates the biological activity of the
Kruppel finger protein. Noture 3322 90-92.
Reinert, l.|.R. and Marzluf, G.A. L975. Genetic and metabolic control
of the purine catabolic enzymes of Neurosporo crosso. Molec. Gen.
Genet. 139: 39-55.
Reinert, W.R., Patel, V.B. and Giles, N.H. 1981. Genetic regulation
of the gø gene cìuster of Neurosporo crossø2 induction of qo
messenger ribonucleic acid and dependency on qo-L function. l4ol.
Cell. Biol. 1: 829-835.
Renkawitz, R. 1990. Transcriptional repression in eukaryotes. Trend.
Genet. 6: L92-L96.
Reznikoff, l'|.S., Maquat, L.E., Munson, L.M., Johnson, R.C. and
Mandecki, }{. 1982. In R. Rodnguez and M. Chamberlin (eds.),
Promoters: structure and function. Praeger, N.Y. pp 80-95.

193.
Reznikoff, W.S., t{inter, R.B. and Hurley, C.K. 1974. Repressor
binding sites, location in the loc operon. Proc. Notl. Acod.
Sci. U.S.A. 7Iz 2314-2318.
Riley, M.I., Hopper, J.E., Johnston, S.A. and Dickson, R.C. 1987.
Gal4 of Socchoronyces cerevisíoe activates the lactose-galactose
reguìon of Kluyveronyces løctís and creates a neu, phenotype:
glucose repression of the regulon. Mol. Cell. Bíol.7z 780-786.
Roberts, C.F. 1963. The genetic analysis of carbohydrate utilization
in Aspergillus niduløns. J. Gen. Mícro. 31:45-58.
Rogers, S., }{eììs, R. and Rechsteiner, M. 1986. Amino acid sequences
corrrnon to rapidty degraded proteins: the pest hypothesis. Scíence
234:364-368.
Romano, A.H. and Kornberg, H.L. 1968. Regulation of sugar utilization
by Aspergillus nidulsns. Biochin. Biophs. Acto 158: 491-493.
Romano, A.H. and Kornberg, H.L. 1969. Regulation of sugar uptake by
Aspergíllus nidulons. Proc. R. Soc. London.seríes B l73z 475-490-
Roseman, S. and Meadow, N.D. 1990. Signal transduction by the
bacterial phosphotransferase system. J. Biol. Chen. 265¿ 2993-
2996.
Rosenberg, U.8., Schroder, C., Preiss, 4., Kienlin, 4., Cote, S.,
Riede, I. and Jackìê, H. 1986. Structural homoìogy of the product
of the Drosophilo kruppel gene with Xenopus transcription factor
IIIA. Nsture 319: 336-339.
Russeì1, D.l{., Smith, M., Cox, D., l'lilliamson, V.M. and Young, E.T.
1983. DNA sequences of two yeast promoter-up mutants. Noture 3042
652-654.
Sakai, 4., Shimizu, Y. and Hishinuma, F. 1988. Isoìation and
characterization of mutants which show an oversecretion phenotype
in Socchoronyces cerevisioe. Geneú¿cs 119: 499-506.

194.
Sakai, 4., Shimizv, Y., Kandou, S., Chibazakura, T. and Hishinuma, F.
1990. Structure and moìecular ana'lysis of RGI1' a gene required
for glucose repression of Socchøromyces cerevisioe. Mol. Cell.
Bíol . 10 : 4130-4138.
Salmeron, J.M., Langdon, S.D. and Johnston, S.A. 1989. Interaction
between transcriptional activator protein Lac9 and negative
regulatory protein Gal80. Mol. Cell. Biol. 9z 2950-2956.
Sanger, R., Nicklen, S. and Coulson, A.R. 1977. DNA sequencing with
chain terminating inhibitors. Proc. Nøtl. Acod. Sci. U.S.A. 74:
5463-5467.
Sarokin, L. and Carlson, M., 1984. Upstream region required for
regulated expression of the gìucose repressible SUCZ gene of
Socchoîonyces cerevísise. Mol. Cell. Biol. 4z 2750-2757.
Sawadogo, M. and Roeder, R.G. 1985. Interaction of a gene-specific
transcription factor with the adenovirus major late promoter
upstream of the TATA box region. Cell 43: 165-175.
Schlapp, T. and Rodel, G. 1990. Transcription of two divergently
transcribed yeast genes initiates at a comnon oligo (dA-dT)
tract. Molec. Gen. Genet. 2232 438-442.
Schultz, J. and Carlson, M. 1987. Molecular analysis of S5/t6, a gene
functionally related to the Snfl protein kinase of Socchoronyces
cerevísiøe. líol. Cell. Biol. 7z 3637-3645.
Schul tz, J . , Marshal I -Car1 son , L. and Carì son, M. 1990. The N-
terminal TPR region is the functional dornain of Ssn6, a nuclear
phosphoprotein of Sscchoronyces cerevisíoe. Mol. Cell. Bíol. 10:
4744-4756.
Scott, Ï1.4. 1976. Adenosine 3':5'-cyclic monophosphate deficiency in
Neurosporo crqssø. Proc. Notl. Acsd. Sc¿. U.S.A. 732 2995-2999.

195.
Sealy-Lewis, H.M. and LockingtÒn, R.A. 1984. Reguìation of two
alcohol dehydrogenases in Aspergíllus níduløns. Curr. Genet.
8:253-259.
Selleck, S.B. and Majors, J.M. L987. In vivo DNA-binding properties
of a yeast transcription activator protein. l4ol. Cell. Bíol. 7z
3260-3267.
Sharma, K.K. and Arst, H.N. 1985. The product of the reguìatory gene
of the proline catabolism gene cluster in Aspergíllus nidulons is
a positive acting protein. Curr. Genet. 9: 299-304.
Skinner, V.M. and Armitt, S. 1972. Mutants of Aspergíllus nidulons
lacking pyruvate carboxylase. FEBS Lett. 202 16-18.
Short, J.M., Fernandez, J.M., Sorge, J.A. and Huse, }l.D. 1988. A
bacteriophage expression vector with in vivo excision
properties. Nucleic Acids Res. 16: 7583-7600.
Simpson, R. 1980. Interaction of the cAMP receptor protein with the
løc pronoter. /VucZeic Acids Res. 8: 759-766.
Snyder, M., Buchman, A.R. and Davis, R.W. 1986. Bent DNA at a yeast
autononously replicating sequence. Noture 3242 87-89.
Sorger, P.K. and Pelham, H.R.B. 1988. Yeast heat shock factor is an
essential DNA-binding protein that exhibits temperature-dependent
phosphorylation . Cell 542 855-864.
Struhl, K. 1985a. Nucleotide sequence and transcriptional mapping of
yeast pet56-hís3-ded| gene region. Nucleíc Acíds Res. 13: 8587-
8601.
Struhl, K. 1985b. Naturaìly occurring poly (dA-dT) sequences are
upstream promoter elements for constitutive transcription in
yeast. Proc. Nstl. Acod. Sci. U.S.A. 822 84L9-8423.

196.
Suggs, S.V., Katzowitz, J.1., Tsai-Morris, C. and Sukhatme, V.P.
1990. cDNA sequence of the human cellular early growth response
gene fgr-I. Nucleic Acíds Res. 18: 4283-4287.
Sukhatme, V.P., Cao, X., Chang, L.C., Tsai-Morris, C-H.,
Stamenkovich, D., Ferreira, P.C.P., Cohen, 0.R., Edwards, S.4.,
Shows, T.8., Currau, T., Le Beau, M.M. and Adamson, E.D. 1988. A
zinc finger encoding gene coreguìated with c-fos during growth
and differentiation and after cellular depolarization. Cell 53:
37-43.
Suzuki, M., 1989. SPXX, a frequent sequence motif in gene regulatory
proteins. J. Mol. Biol. 207: 61-84.
Tajima, M., Nogi, Y. and Fukasawa, T. 1986. Dupl icate upstream
activating sequences in the promoter region of the Soccharonyces
cerevisioe GALT gene. Mol. Cell. Bíol. 6: 246-256.
Terenzi, H.F., Flawia, M.M. and Torres, H.N. L974. A Neurosporo
crosso morphologicaì mutant showing reduced adenyìate cyclase
activity. Biochín. Biophys. Res. Connun. 58: 990-996.
Thiry-Bìaise, L.M. and Loppes, R. 1990. Deletion analysis of the
,4R64 promoter of Socchoromyces cerevísíoe: a poly (dA-dT) stretch
involved in gene transcription. Molec. Gen. Genet. 223: 474-480.
Thuring, R.t{.J., Sanders, J.P.M. and Borst, P. L975. A freeze-squeeze
method for the recovery of long DNA from agarose geìs. Anol.
Biochen. 662 213-220.
Tilburn J., Scazzocchio, C., Taylor, G., Zabicky-Zissman, J.
Lockington, R. and Davies, R.H. 1983. Transfonnation by
integration in Aspergíllus nídulons. Gene 262 205-22I.
Timberìake, }{.E. 1978. Low repetitive DNA content in Aspergillus
nidulons. Scíence 2022 973-975.

197.
Timberìake, l'l.E. and Marshaì1, M.A. 1988. Genetic regulation of
development in Aspergíllus nidulans. Trend. Genet. 4z L62-L69.
Tinmis, J.N., l'lhisson, D.1., Binns, A.M., Mayo, G. and Mayo J. 1990.
Deletion as a means of isoìating avirulence genes in flax rust.
Theoret. App. Genet. 79: 411-416.
Trumbly, R.J. 1986. Isolation of Socchoronyces cerevis¿øe mutants
constitutive for invertase synthesis. J. Bocteríol. 1662 LL23-
T127.
Trumbly, R.J. 1988. Cloning and characterization of the CÍCB gene
mediating glucose repression in yeast. Gene 73: 97-lLI.
Ullmann, A. and Danchin, A. 1983. Role of cyclic AMP in bacteria.
Adv. Cyclic Nucleotíde Res. 15: 1-53.
Unger, 8., Clore, G.M., Gronenborn, A.M. and Hillen, l{. 1983.
Specific DNA binding of the cAMP receptor protein within the Zoc
operon stabilizes double-stranded DNA in the presence of cAMP.
EI,\BO J. 2z 289-293.
Upshaì1, A. 1986. Genetic and molecular characterization of orgB+
transformants of Aspergillus nidulqns. Curr. Genet. 10: 593-599.
Vannus, H.E., Perlman, R.L. and Pastan, I. 1970a. Regulation of the
løc mRltA synthesis by cyclic AMP and glucose. J. Biol. Chen.245z
2259-2267.
Varmus, H.E., Perlman, R.L. and Pastan, I. 1970b. Regulation of loc
transcription in Escherichío coli by cyclic AMP. Studies with
DNA-RNA hybridization and hybridization competition. J. Bíol.
Chen. 2452 6366-6372.
Ward, M. and Turner, G. 1986. The ATP synthase subunit 9 gene of
Aspergíllus nídulons: sequence and transcription. Molec. Gen.
Genet. 205: 331-338.

198.
Hard, M., l{ilson, L.J., Carmona, C.L. and Turner, G. 1988. The oZíC3
gene of Aspergíllus nigerz isolation, sequence and use as a
selectable marker for transformation. Curr. Genet. 14: 37-42.
Weber, I., Takio, K., Titani, K. and Steitz, T. 1982. The cAMP-
binding domains of the regu'latory subunit of cAMP-dependent
protein kinase and the catabolite gene activator protein are
homologous. Proc. Notl. Acød. Sci. U.S.A. 792 7679-7683.
}{hite, M.J.D. 1973. Animal cyto'logy and evolution. Cambridge
University Press, London, U.K. p39-41.
}liame, J., Grenson, M. and Arst, H.N. 1985. Nitrogen catabolite
repression in yeasts and fi lamentous fungi . Advon. l|icro.
Physiol. 26t 1-88.
l,lills, C. 1990. Regulation of sugar and ethanol metabolism in
Socchoronyces cerevísíoe. Crít. Rev. Biochem. llol. Biol. 252 245-
280.
Wray, L.V., Wittle, M.M., Dickson, R.C. and Riley, M.I. 1987.
Characterization of a positive regulatory gene, LAC9; that
control s induction of the 'l actose-gal actose regulon of
Kluyveronyces loctis¡ structural and functional relationships to
GAL4 of Socchoronyces cerevísíoe. Mol. Cell. Bíol. 7z 1111-1121.
l{u, H-M. and Crothers, D. 1984. The locus of sequence-directed and
protein-induced DNA bending. Noture 308: 509-513.
Yannisch-Perron, C., Vieira, J. and Messing, J. 1985. Inproved M13
phage cloning vectors and host strains: nucleotide sequences of
the M13np18 and pUCl9 vectors. Gene 33: 103-119.
Yelton, M.M., Timberlake, W.E. and van den Hondel, C.4.M.J.J.1985. A
cosmid for seìecting genes by comp'lementation in Aspergillus
niduløns: selection of the developmentaìly regulated yA locus.
Proc. NotI. Acod. Sc¿. 82: 834-838.

199.
Young, J.L., Jarai , G., Fu, Y-H. and Marzluf , G.A. 1990. l{ucleotide
seguence and analysis of /VllR, a negative-acting regulatory gene
in the nitrogen circuit of Neurosporo crosso. Molec. Gen. Genet.
2222 120-128.
Zahn, K. and Blattner, F.R. 1987. Direct evidence for DNA bending at
the lanbda replication origin. Science 236: 416-422.
Zinunennann F.K. and Scheal, I. L977. Mutants of Socchoronyces
cerevísíoe resistant to carbon catabolite repression. lilolec. Gen.
Genet. 154: 75-82.





























