Embed Size (px)
Citation preview

The Cellular Protein SPT6 Is Required for Efficient Replication ofHuman Cytomegalovirus
Daniel Cygnar,a Stacy Hagemeier,a* Daniel Kronemann,a and Wade A. Bresnahana,b
Department of Microbiologya and Institute for Molecular Virology,b University of Minnesota, Minneapolis, Minnesota, USA
The human cytomegalovirus tegument protein UL69 has been shown to be required for efficient viral replication at low multi-plicities of infection. Several functions have been associated with UL69, including its ability to regulate cell cycle progression,translation, and the export of viral transcripts from the nucleus to the cytoplasm. However, it remains unclear which, if any, ofthese activities contribute to the phenotype observed with the UL69 deletion mutant. UL69 has been shown to interact with thecellular protein SPT6. The functional significance of this interaction has never been examined in the context of an infection. Toaddress this, we generated UL69 mutant viruses that were unable to interact with SPT6 and determined what effect these muta-tions had on virus replication. Abolishing UL69’s ability to interact with the SPT6 protein inhibited virus replication to levelsindistinguishable from those observed following infection with the UL69 deletion mutant. Surprisingly, abolishing UL69’s inter-action with SPT6 also resulted in the impairment of UL69 shuttling activity. Finally, we demonstrate that inhibition of SPT6 ex-pression by short hairpin RNA (shRNA) knockdown inhibits wild-type virus replication. Taken together, our results demon-strate that UL69’s ability to interact with SPT6 plays a critical role in viral replication.
Human cytomegalovirus (HCMV) belongs to the betaherpes-virus family and is a ubiquitous human pathogen. HCMV
infection is generally asymptomatic in healthy individuals. How-ever, considerable complications can arise in newborns or indi-viduals that are immunocompromised, such as transplant recipi-ents and HIV/AIDS patients (18). Like all herpesviruses, theHCMV virion contains a tegument layer that is composed of anumber of virally encoded proteins that are packaged in the virionand delivered to the host cell upon infection. A number of thesetegument proteins have been shown to play important roles inviral entry, gene regulation, immune evasion, DNA replication,and viral assembly (10, 11). The UL69-encoded tegument proteinhas previously been shown to be required for efficient viral repli-cation (9). Infection with a UL69 deletion mutant results in asevere growth defect that is multiplicity dependent. Even thoughthe growth phenotype of the UL69 deletion mutant has beenknown for years, the mechanism whereby UL69 contributes toviral replication has remained elusive. Several activities have beenassociated with UL69, including its ability to regulate viral geneexpression (9, 26), regulate translation (2), shuttle between thenucleus and cytoplasm (14, 16), interact with RNA (24), and reg-ulate cell cycle progression (9, 17). It is thought that many, if notall, of these activities are regulated by UL69’s interaction with hostcell proteins (2, 16, 20, 23, 25).
One of the proteins that has been shown to interact with UL69is the human homolog of the suppressor of Ty6 (SPT6) (25). SPT6is a highly conserved multifunctional protein that has been shownto interact with the C-terminal domain (CTD) of RNA polymer-ase (Pol) II and be involved in chromatin remodeling, transcrip-tional elongation, and mRNA export (3, 5, 7, 8, 12, 27). SPT6regulates chromatin structure by functioning as a putative histonechaperone that interacts with histone H3 and promotes the reas-sembly of nucleosomes in the wake of RNA Pol II. In addition,SPT6 has been identified as a classical transcription elongationfactor (7, 12) that can either individually or in conjunction withSPT4 and SPT5 (DRB sensitivity-inducing factor [DSIF]) stimu-late the rate of RNA Pol II elongation both in vitro and in vivo.
Interestingly, SPT6’s ability to function as a transcriptional elon-gation factor is independent from its chromatin remodeling activ-ity since SPT6-enhanced transcriptional elongation occurs on na-ked DNA (7). Finally, SPT6 can regulate mRNA export through itsinteraction with a cellular protein termed Iws1 (interacts withSPT6-1). Iws1 directly interacts with the nuclear export factorAly/REF, and depletion of Iws1 has been shown to lead to splicingdefects and nuclear retention of bulk poly(A) mRNAs (27).
Given that UL69 has been implicated in regulating the exportof viral mRNAs and other aspects of viral gene expression, weasked if UL69’s interaction with SPT6 is required for efficientHCMV replication. We demonstrate that viral mutants that areunable to interact with SPT6 display a growth phenotype identicalto that of the UL69 deletion virus. In addition, UL69 mutants thatare unable to bind SPT6 also display a defect in UL69’s nucleocy-toplasmic shuttling activity. Finally, we show that short hairpinRNA (shRNA)-mediated knockdown of SPT6 inhibits the repli-cation of wild-type (WT) HCMV. Taken together, our resultsdemonstrate that UL69’s interaction with SPT6 is important forefficient viral replication and also provide further insight into themechanism by which UL69 functions.
MATERIALS AND METHODSGeneration of allelic exchange shuttle vectors and mutant BACs. Prim-ers UL69FR1 (5=-CGCCAAGCTCGATTCGAACC-3=) and UL69FR2 (5=-CGTGCAGGTGGTCATCGACC-3=) were used to amplify the genomicregion corresponding to nucleotides 97887 to 100698 of the AD169 ge-
Received 9 November 2011 Accepted 2 December 2011
Published ahead of print 14 December 2011
Address correspondence to Wade A. Bresnahan, [email protected].
* Present address: University of Wisconsin, McArdle Laboratory for CancerResearch, Madison, Wisconsin, USA.
Copyright © 2012, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JVI.06776-11
0022-538X/12/$12.00 Journal of Virology p. 2011–2020 jvi.asm.org 2011
on July 27, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

nome which contains the UL69 open reading frame (ORF) and �300 bpof flanking sequence on both the 5= and 3= ends of the UL69 ORF. ThePCR product was TA cloned into the pGEM-T Easy vector (Promega) togenerate the pGEMT-UL69FR plasmid. To create the UL69-C496A,PP602/603AA, and E617A alanine substitution mutants, site-directedmutagenesis (Stratagene QuikChange kit) was performed according tothe manufacturer’s instructions with the C496A-S (5=-CCTGCAGTGCCACGAGGCTCAGAACGAGATGTGC-3=) and C496A-AS (5=-GCACATCTCGTTCTGAGCCTCGTGGCACTGCAGG-3=), PP602/603AA-S (5=-CCTCCCGCCCAGGCAGCGTCGCAACC-3=) and PP602/603AA-AS(5=-GGTTGCGACGCTGCCTGGGCGGGAGG-3=), and E617A-S (5=-GCGAGCTGGAAGCGGACGAAGACAGTG-3=) and E617A-AS (5=-CACTGTCTTCGTCCGCTTCCAGCTCGC-3=) primer sets, respectively, us-ing pGEMT-UL69FR as a template. The resulting constructs, termedpGEMT-UL69FR-C496A, pGEMT-UL69FR-PP602/03AA, and pGEMT-UL69FR-E617A, were subsequently sequenced to verify the incorporationof the proper mutations. The individual pGEMT-UL69FR constructswere then digested with NotI and ligated into the pGS284 shuttling vector(21) that was also digested with NotI to generate the shuttle vectorspGS284-C496A, pGS284-PP602/603AA, and pGS284-E617A. Bacterialartificial chromosomes (BACs) containing the UL69-C496A, PP602/603AA, and E617A mutations were generated by standard allelic exchangeprotocols (21) using the pADCREGFP�UL69 BAC (4, 13) and shuttlevectors pGS284-C496A, PP602/603AA, and E617A, respectively. All BACswere screened by restriction enzyme digestion, and the UL69 ORFs weresequenced to verify proper incorporation of the specific mutations.
Cell culture and virus propagation. Human foreskin fibroblast (HFF)cells and human embryonic kidney 293T cells were cultured in Dulbecco’smodified Eagle’s medium (DMEM) supplemented with 10% (vol/vol)fetal bovine serum (HyClone), 100 units/ml penicillin, and 100 �g/mlstreptomycin in an atmosphere containing 5% CO2 at 37°C. Recombinantviruses were generated by cotransfecting purified BAC DNA and pCGN-pp71 into UL69-complementing HFF cells as previously described (13)and harvesting the virus when the cells displayed a 100% cytopathic effect(CPE). Viral stocks were then generated by infecting noncomplementingcells at a high multiplicity, as described previously (9). The UL69 openreading frame was subsequently sequenced from the corresponding virusstocks to confirm that the proper mutations were incorporated and thatno other mutations were present within the open reading frame.
Expression plasmids. The URH49-HA expression vector was previ-ously described (13). Hemagglutinin (HA)- and Flag-tagged UL69 andFlag-tagged eIF4a1 expression plasmids were generated using the pENTR/D-TOPO kit (Invitrogen) to construct appropriate entry vectors. EachORF was PCR amplified with gene-specific primers using Advantage HDpolymerase (Clontech) with either wild-type or mutant pGEMT-UL69FRplasmids or the pOTB7-eIF4a1 cDNA plasmid (Open Biosystems) as thetemplate. The PCR product was purified and cloned into the pENTR entryvector according to the manufacturer’s instructions. ORFs were thentransferred to HA or Flag pCI-Neo Gateway-compatible destination vec-tors using LR Clonase II (Invitrogen) as previously described (19). Thevectors were then sequenced to confirm the fidelity of each open readingframe.
Immunoprecipitation and Western blotting. Immunoprecipitationsand Western blotting were performed as previously described (13, 19).Briefly, 293T cells were transfected by the calcium phosphate method(Promega) or HFF cells were infected with virus as indicated below. Cellswere lysed in NP-40 lysis buffer (150 mM NaCl, 50 mM Tris-HCl, 10 mMEDTA [pH 8.0], 0.75% NP-40), and cellular debris was removed by cen-trifugation. Immunoprecipitations were performed by incubating 300 �gof total protein with the indicated antibodies for 3 h at 4°C. Protein com-plexes were collected on protein A/G agarose beads, washed three timeswith NP-40 lysis buffer, and separated by SDS-PAGE. Proteins were trans-ferred to a nitrocellulose membrane (Optitran; Schleicher & Schuell) andprobed with primary and secondary antibodies. Immunoreactive proteins
were detected by an enhanced chemiluminescence (ECL) system(Thermo Scientific).
Heterokaryon shuttling assay. Heterokaryon assays were performedas previously described (13). Briefly, HFF cells were infected at a multi-plicity of 0.5 PFU/cell with the indicated viruses. Murine 3T3 cells wereoverlaid on the infected HFF cells at 72 h postinfection and allowed toattach for 4 h. Cells were then incubated with DMEM containing cyclo-heximide (50 �g/ml) for 30 min and then fused with a 50% (wt/vol)solution of PEG-8000 in Hanks balanced salt solution (lacking Mg2� andCa2�) for 2 min. Cells were washed several times with phosphate-bufferedsaline (PBS) and incubated for 3 h with complete DMEM containing 50�g/ml cycloheximide. Cells were then fixed in 4% paraformaldehyde,permeabilized, and stained for UL69 (T. Shenk, Princeton University) andUL44 (1202S; Rumbaugh Goodwin Institute).
Immunofluorescence. HFF cells or heterokaryons on glass coverslipswere washed in PBS and fixed with 4% paraformaldehyde at room tem-perature for 20 min. Cells were washed once with PBS and permeabilizedwith PBS-T (PBS, 0.1% Triton X-100, 0.05% Tween 20). Coverslips wereincubated with blocking solution (PBS, 0.05% Tween 20, 0.5% bovineserum albumin [BSA], 1% goat serum) for 30 min at room temperature.Primary and secondary antibody incubations were performed in a humid-ified chamber at 37°C for 1 h each. Cells were stained with Hoechst, andcoverslips were mounted onto slides with 90% glycerol. Images were cap-tured on a Zeiss Axiovert 40 CFL microscope with a Jenoptik ProgRes C10camera running ProgRes CapturePro v. 2.8.0 software.
Lentivirus generation, shRNA knockdown, and cell viability assays.An SPT6 shRNA lentivirus vector was obtained from Open Biosystems(clone ID TRCN0000019732; full hairpin sequence, CCGGCGCCTTGTACTGTGAATTTATCTCGAGATAAATTCACAGTACAAGGCGTTTTT). Lentivirus was generated by cotransfecting lentivirus plasmid intoHEK293T cells using Arrest-In transfection reagent and a proprietarypackaging mixture (Open Biosytems) according to the manufacturer’sinstructions. At 48 and 72 h posttransfection, lentivirus particles werecollected from the cell supernatant, cleared of cell debris by centrifuga-tion, filtered through a 0.45-�m syringe filter, and stored at �80°C. HFFcells were then transduced two times with lentivirus supplemented with 8�g/ml Polybrene at 37°C for 3 h each time. Cells were washed with PBS,and the complete culture medium was placed back on the cells. Mediumcontaining 1.5 �g/ml puromycin was added to the cells 24 h after trans-duction. Following 72 h of selection, the medium was replaced and thecells were used for the indicated experiments. To quantify SPT6 mRNAand protein levels, cell lysates were collected at 7 days posttransductionand assayed. Total RNA was isolated using TRIzol reagent. RNA was re-verse transcribed using an oligo(dT) primer and Superscript II ReverseTranscriptase (RT) (Invitrogen) according to the manufacturer’s instruc-tions. Transcript levels were quantitated by a QuantiFast Sybr green (Qia-gen) quantitative PCR (qPCR) with SPT6-specific primers SPT6 RT-Fwd(TAGACAATGGTGTCACCGGCTTCA) and SPT6 RT-Rev (CATGATGCGGCAGTGAACACTCAT) on a Bio-Rad IQ5 real-time thermal cyclerand analyzed using the IQ5 software. Protein lysates were collected andanalyzed by Western blotting as described above. Cell viability of knock-down cells was examined by incubating knockdown cells (either 72 h or 7days posttransduction) with 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) reagent (Invitrogen) for 4 h at 37°C.Cells were then lysed with a 10% SDS and 0.01 M HCl solution andincubated for 2 h at 37°C. Absorbance values were read on a MolecularDevices plate reader set at 595 nm.
Antibodies. The following antibodies were obtained from commercialsources and used for immunoprecipitations: rabbit polyclonal anti-SPT6(803A; Bethyl Laboratories), mouse monoclonal anti-HA (16B12; Cova-nce), and rabbit polyclonal anti-Flag (F7425; Sigma). The following anti-bodies were used for Western blotting: mouse monoclonal anti-IE1/2(1203), mouse monoclonal anti-UL44 (1202S), and mouse monoclonalanti-UL99 (1207) (all from the Rumbaugh-Goodwin Institute), mousemonoclonal anti-HA (16B12; Covance), mouse monoclonal anti-Flag
Cygnar et al.
2012 jvi.asm.org Journal of Virology
on July 27, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

(F1804; Sigma), rabbit polyclonal anti-SPT6 (801A; Bethyl Laboratories),and mouse anti-tubulin (TU-02) and mouse monoclonal anti-green flu-orescent protein (GFP) (sc-9996) (both from Santa Cruz Biotechnology).The mouse monoclonal anti-UL69 antibody was provided by T. Shenk(Princeton University). Alexa Fluor 546-conjugated Fab2 goat anti-mouse or Alexa Fluor 488-conjugated Fab2 goat anti-rabbit antibodies(Molecular Probes) were used for immunofluorescence.
RESULTS
Previous reports have suggested that the interaction betweenUL69 and the cellular protein SPT6 is important for UL69 func-tion (25). However, to date, there have been no studies examiningwhat role UL69’s interaction with SPT6 plays during a productiveHCMV infection. Therefore, we tested several UL69 mutants fortheir ability to bind SPT6 and determined if these mutants wouldsupport virus replication. To begin, we screened a panel of sevendifferent UL69 mutants for their ability to interact with SPT6. Thefirst mutant, termed C496A, contains a cysteine-to-alanine sub-stitution at position 496 and has previously been described as anSPT6-binding mutant (25). We also tested two mutants termedPP602/603AA and E617A, which are alanine substitution mutantsthat have previously been described as nucleocytoplasmic shut-tling mutants (14). In addition, we tested a mutant termed mUAPwhich contains 4 alanine substitutions at positions 22, 23, 25, and26 and has previously been shown to abolish UL69’s ability to bindthe cellular UAP56 and URH49 proteins (16). Finally, we testedthree control mutants termed L503A, PP598/599AA, and ED619/620AA, all of which represent alanine substitution mutants at sitesadjacent to the other mutations described (14). To assay for SPT6binding, cells were transfected with expression constructs that ex-press either HA-tagged wild-type UL69 or one of the UL69 mu-tants. Forty-eight hours posttransfection, cell lysates were har-vested and immunoprecipitations were performed with an SPT6antibody. Immune complexes were separated by SDS gel electro-phoresis, transferred to nitrocellulose membranes, and analyzedby Western blotting using an anti-HA antibody to detect UL69. Asshown in Fig. 1A, we identified two UL69 mutants that were un-able to interact with SPT6. As expected, the C496A mutant wasunable to bind SPT6 (lane 3). In addition, the previously describedPP602/603AA shuttling mutant was also unable to bind SPT6(lane 6). All other UL69 mutants tested retained their ability tointeract with SPT6, including the E617A mutant. Like the PP602/603AA mutant, the E617A mutant was previously described as ashuttling mutant, suggesting that residues 602 and 603 may beinvolved in both SPT6 binding and shuttling activity. To deter-mine if either of these functions are required for virus replication,we incorporated the C496A, PP602/603AA, and E617A mutationsinto the viral genome to generate individual UL69 viral mutants.Figure 1B depicts the UL69 ORF and the location of each specificmutation. These UL69 mutants were then assayed for viral repli-cation, SPT6 binding activity, and shuttling activity.
SPT6 binding correlates with viral replication. To generatethe specific UL69 mutant viruses, we utilized a previously de-scribed system in which the UL69 ORF was replaced with a selec-tion cassette containing both kanamycin resistance and LacZgenes by using allelic exchange protocols (13). This pAD�UL69BAC was then used as the parental BAC to generate the specificUL69 point mutant BACs. The mutant BACs were then screenedby restriction enzyme digestion, Southern blotting, and direct se-quencing to verify that the mutations were properly inserted and
that no other subsequent mutations were incorporated into theUL69 open reading frame (data not shown). The viruses werereconstituted by transfecting mutant UL69 BAC DNA into com-plementing HFF cells and harvesting the viruses when a 100%cytopathic effect was observed (4, 13). Viral stocks were generatedby infecting noncomplementing cells at a high multiplicity of in-fection, and the titers of the stocks were determined by plaqueassay on complementing cells. Growth curves were then per-formed by infecting noncomplementing cells at a multiplicity of0.01 PFU/cell with the wild-type, C496A, PP602/603AA, E617A,or �UL69 virus or the appropriate revertant virus. Virus was har-vested at the indicated times postinfection, and viral titers weredetermined by plaque assay on complementing cells. As shown inFig. 2A and B, both the C496A and PP602/603AA mutants dis-played a growth phenotype similar to that of the �UL69 mutant.Replication of both the C496A and PP602/603AA mutants wasreduced by approximately 2 logs compared to that of the wild-typevirus. However, when the mutations were repaired, the C496Aand PP602/603AA revertant viruses replicated to levels similar tothat observed for the wild-type virus, demonstrating that the phe-notype is not the result of a secondary mutation elsewhere in thegenome. We observed no replication defect with the E617A mu-tant virus (Fig. 2C).
We next examined the expression of several viral genes follow-ing infection with each virus. HFF cells were infected at a multi-plicity of 0.01 PFU/cell and protein lysates were harvested at 24-hintervals for 5 days and assayed for UL69, IE1, IE2, UL44, andUL99 expression by Western blotting. There was a delay and de-crease in UL69, UL44, and UL99 expression following infectionwith the �UL69, C496A, and PP602/603AA mutants (Fig. 3A,middle panel) compared to that observed following infection withthe wild-type virus. This delay and decrease in protein expressionwas not observed following infection with the �UL69, C496A, andPP602/603AA revertant viruses (Fig. 3A, bottom panel). Therewas no significant difference in IE1 and IE2 expression levels at
FIG 1 Identification of UL69 mutants unable to bind SPT6. (A) 293T cellswere transfected with HA-tagged wild-type or mutant UL69 expression vec-tors. At 48 h posttransfection, protein lysates were collected and an equalamount of protein was immunoprecipitated (IP) with anti-SPT6 antibody.Immune complexes and cell lysates were separated by SDS-PAGE and ana-lyzed by Western blotting. (B) Schematic representation of the UL69 ORF andlocation of the mUAP, C496A, PP602/603A, and E617A point mutations.
UL69/SPT6 Interaction Required for HCMV Replication
February 2012 Volume 86 Number 4 jvi.asm.org 2013
on July 27, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

early times after infection with any of the UL69 mutants comparedto infection with the wild-type virus. However, as with the otherproteins assayed, we observed an increase in IE1 and IE2 levels atlate times postinfection in cells infected with the wild-type viruscompared to infection with the �UL69, C496A, and PP602/603AA mutants (Fig. 3A, top and middle panels). This delay anddecrease in gene expression observed following infection with the�UL69, C496A, and PP602/603AA mutants is likely due to thereplication defect observed at 4 days postinfection (Fig. 2A and B)
and the viruses’ inability to efficiently spread to uninfected cells atlate times after infection. To test this, we examined viral geneexpression after infecting cells at a multiplicity of 3 PFU/cell wherethe UL69 growth defect is limited (9) and all cells are infected in asynchronous fashion. UL69 expression was again delayed andslightly lower at 36 and 72 h postinfection following infection withthe C496A and PP602/603AA mutants (Fig. 3B, middle panel)compared to that observed following infection with the wild-typevirus. However, by 108 h postinfection, UL69 protein levels weresimilar to those observed following wild-type infection. Levels ofIE1, IE2, UL44, and UL99 expression observed following infectionwith the �UL69, C496A, or PP602/603AA mutant were similar tothose observed following infection with the wild-type virus (Fig.3B, middle panel). These results suggest that the decreased proteinlevels observed at a multiplicity of 0.01 PFU/cell were largely dueto the growth defect observed with these mutants and their inabil-ity to efficiently produce infectious virus and spread, rather than adefect in viral gene expression. Since we observed a delay anddecrease in UL69 expression at both multiplicities, it raised thepossibility that UL69 may be regulating its own transcription. Totest this, we assayed for UL69 RNA abundance by qPCR followinginfection with either the wild-type virus, the C496A virus, or thePP602/603AA virus. We observed no significant difference in thelevels of UL69 RNA abundance when comparing the C496A andPP602/603AA mutants to the wild-type virus (data not shown),suggesting that the delay in UL69 expression is not due to a defectin UL69 transcription.
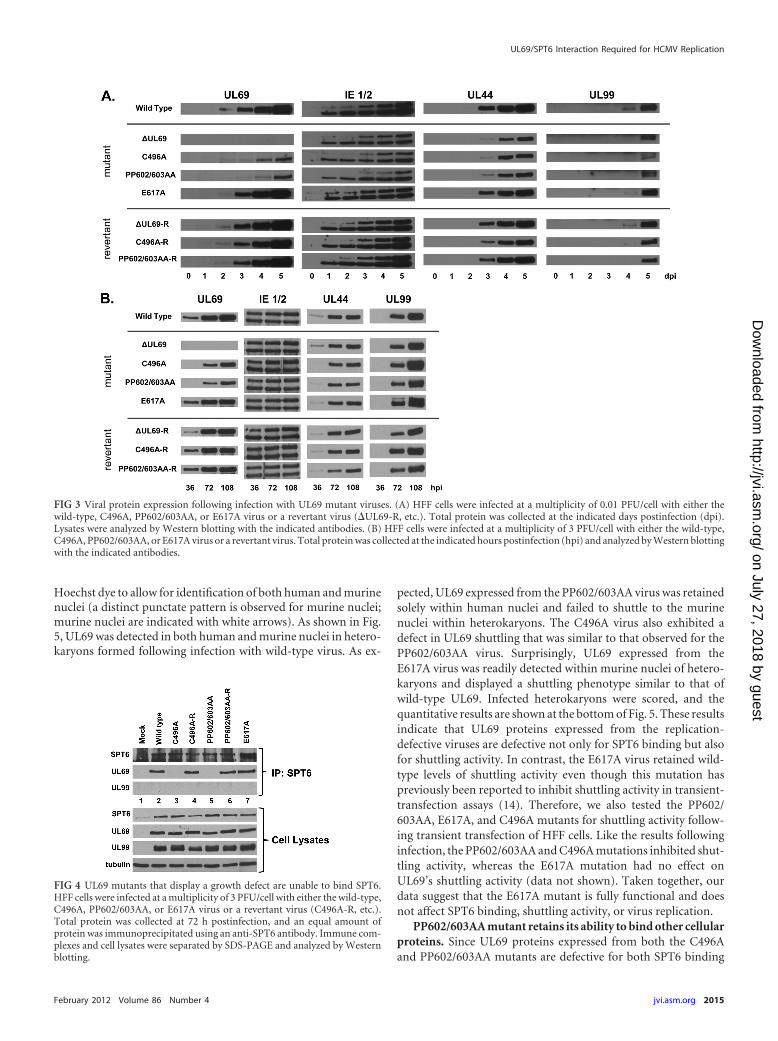
C496A and PP602/603AA mutants fail to interact with SPT6during infection. We next assayed for UL69’s ability to bind SPT6during infection. HFF cells were infected with wild-type, mutant,or revertant virus, and lysates were harvested 72 h postinfectionand subjected to immunoprecipitation with an antibody againstSPT6. As shown in Fig. 4, we could readily detect an interactionbetween UL69 and SPT6 in cells that had been infected with eitherthe wild-type virus (lane 2), the E617A virus (lane 7), or the rever-tant viruses (lanes 4 and 6). However, we were unable to detect aninteraction between SPT6 and UL69 in cells infected with theC496A (lane 3) or PP602/603AA (lane 5) mutants. We were alsounable to detect an interaction between SPT6 and the UL99 viralprotein following infection with any of the viruses, confirming thespecificity of the SPT6/UL69 interaction. These data confirm theresults obtained from the transient-transfection experiments (Fig.1A) and suggest that the interaction between UL69 and SPT6 isimportant for efficient viral replication.
UL69 mutants defective for SPT6 binding are defective fornucleocytoplasmic shuttling. The two mutants that inhibitUL69’s ability to interact with SPT6 also displayed a dramaticdefect in viral replication. One of these mutants, PP602/603AA,has also been shown to be defective for shuttling activity intransient-transfection heterokaryon experiments (14). However,it is not known if the C496A mutant has a similar shuttling defect.This raised the possibility that the SPT6 interaction may be in-volved in supporting UL69 shuttling activity. We assayed theC496A, PP602/603AA, and E617A mutants for UL69 shuttlingactivity during virus infection. HFF cells were infected with eitherwild-type or mutant virus, and heterokaryon assays were done aspreviously described (13). At 72 h postinfection, HFF cells werefused with murine 3T3 fibroblasts to form heterokaryons. Thecells were then fixed and stained with antibody against UL69 orthe nonshuttling UL44 protein. Cells were also stained with
FIG 2 Growth curve analysis of UL69 mutant viruses. (A) HFF cells wereinfected at a multiplicity of 0.01 PFU/cell with either the wild-type (�), �UL69(Œ), C496A (�), or C496A revertant (C496A-R) (p) virus. (B) HFF cells wereinfected at a multiplicity of 0.01 PFU/cell with either the wild-type (�), �UL69(Œ), PP602/603AA (�), or PP602/603AA revertant (p) virus. (C) HFF cellswere infected at a multiplicity of 0.01 PFU/ml with either the wild-type (�),�UL69 (Œ), or E617A (�) mutant virus. Total virus was collected at the indi-cated days postinfection, and viral titers were determined by plaque assay oncomplementing cells. Results are the averages of three independent experi-ments.
Cygnar et al.
2014 jvi.asm.org Journal of Virology
on July 27, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from
Hoechst dye to allow for identification of both human and murinenuclei (a distinct punctate pattern is observed for murine nuclei;murine nuclei are indicated with white arrows). As shown in Fig.5, UL69 was detected in both human and murine nuclei in hetero-karyons formed following infection with wild-type virus. As ex-
pected, UL69 expressed from the PP602/603AA virus was retainedsolely within human nuclei and failed to shuttle to the murinenuclei within heterokaryons. The C496A virus also exhibited adefect in UL69 shuttling that was similar to that observed for thePP602/603AA virus. Surprisingly, UL69 expressed from theE617A virus was readily detected within murine nuclei of hetero-karyons and displayed a shuttling phenotype similar to that ofwild-type UL69. Infected heterokaryons were scored, and thequantitative results are shown at the bottom of Fig. 5. These resultsindicate that UL69 proteins expressed from the replication-defective viruses are defective not only for SPT6 binding but alsofor shuttling activity. In contrast, the E617A virus retained wild-type levels of shuttling activity even though this mutation haspreviously been reported to inhibit shuttling activity in transient-transfection assays (14). Therefore, we also tested the PP602/603AA, E617A, and C496A mutants for shuttling activity follow-ing transient transfection of HFF cells. Like the results followinginfection, the PP602/603AA and C496A mutations inhibited shut-tling activity, whereas the E617A mutation had no effect onUL69’s shuttling activity (data not shown). Taken together, ourdata suggest that the E617A mutant is fully functional and doesnot affect SPT6 binding, shuttling activity, or virus replication.
PP602/603AA mutant retains its ability to bind other cellularproteins. Since UL69 proteins expressed from both the C496Aand PP602/603AA mutants are defective for both SPT6 binding
FIG 3 Viral protein expression following infection with UL69 mutant viruses. (A) HFF cells were infected at a multiplicity of 0.01 PFU/cell with either thewild-type, C496A, PP602/603AA, or E617A virus or a revertant virus (�UL69-R, etc.). Total protein was collected at the indicated days postinfection (dpi).Lysates were analyzed by Western blotting with the indicated antibodies. (B) HFF cells were infected at a multiplicity of 3 PFU/cell with either the wild-type,C496A, PP602/603AA, or E617A virus or a revertant virus. Total protein was collected at the indicated hours postinfection (hpi) and analyzed by Western blottingwith the indicated antibodies.
FIG 4 UL69 mutants that display a growth defect are unable to bind SPT6.HFF cells were infected at a multiplicity of 3 PFU/cell with either the wild-type,C496A, PP602/603AA, or E617A virus or a revertant virus (C496A-R, etc.).Total protein was collected at 72 h postinfection, and an equal amount ofprotein was immunoprecipitated using an anti-SPT6 antibody. Immune com-plexes and cell lysates were separated by SDS-PAGE and analyzed by Westernblotting.
UL69/SPT6 Interaction Required for HCMV Replication
February 2012 Volume 86 Number 4 jvi.asm.org 2015
on July 27, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

and shuttling, it raised the possibility that these mutations are notspecific for SPT6 binding and/or shuttling but rather are nonspe-cific mutations that may render the UL69 protein nonfunctional.To address this, we assayed these mutant proteins for their abilityto interact with other UL69-binding proteins. Previous reportshave demonstrated that UL69 interacts with the eukaryotic initi-ation factor 4a1 (eIF4a1) and the 49-kDa UAP56-related helicase(URH49) protein and is able to dimerize with itself. Unfortu-nately, we could not perform these assays in the context of aninfection due to the lack of a URH49 antibody and the require-ment that UL69 needs to be tagged with two different epitopes to
demonstrate dimerization. Therefore, we utilized cotransfectionexperiments to assay for UL69 binding to eIF4a1 and URH49 andits ability to dimerize. Cells were cotransfected with vectors ex-pressing Flag-tagged wild-type, C496A, PP602/603AA, or E617AUL69 along with a vector expressing eIF4a1, HA-tagged URH49,or HA-tagged wild-type UL69. At 48 h posttransfection, cell ly-sates were harvested and coimmunoprecipitations were per-formed using the indicated antibodies. As shown in Fig. 6, thewild-type, PP602/603AA, and E617A UL69 proteins were capableof binding eIF4a1 and URH49. In addition, these mutants were allcapable of dimerizing with wild-type UL69. However, we wereunable to detect an interaction between C496A UL69 and any ofthe binding partners tested (Fig. 6). Identical results were obtainedwhen the mutants were tested in a yeast two-hybrid assay for theirability to bind eIF4a1, URH49, and UL69 (data not shown). Cu-mulatively, our results suggest that the C496A mutation may re-sult in a global functional defect in UL69 since this mutant wasdefective in every assay in which it was tested. Therefore, it isdifficult to draw specific conclusions about the function of UL69using this mutant. However, unlike the C496A mutation, thePP602/603AA mutations specifically abolished UL69’s interactionwith SPT6 (Fig. 1A) while retaining its ability to interact witheIF4a1 and URH49 and dimerize with wild-type UL69 (Fig. 6).Furthermore, we conclude that residues 602 and 603 are requiredboth for binding SPT6 and for UL69 shuttling activity.
FIG 5 UL69 mutants that display a growth defect lack shuttling activity. HFFcells were infected at a multiplicity of 0.5 PFU/cell with the indicated viruses.HFF cells were fused with 3T3 fibroblasts to form heterokaryons 72 h postin-fection. Cells were fixed and stained for UL69 or UL44. Nuclei were visualizedwith Hoechst stain. Virally encoded GFP was used to aid in identifying infectedheterokaryons. Murine nuclei are indicated with white arrows. The bar graphrepresents the quantitation of shuttling-positive heterokaryons.
FIG 6 The PP602/603AA mutant retains its ability to bind other proteins.Cells were cotransfected with empty vector or Flag-tagged UL69 expressionvectors and HA-tagged URH49, eIF4a1, or wild-type UL69 expression vectors.Total protein was collected and quantified 48 h postinfection. Immunopre-cipitations were performed using the indicated antibodies. Immune com-plexes and cell lysates were separated by SDS-PAGE and analyzed by Westernblotting.
Cygnar et al.
2016 jvi.asm.org Journal of Virology
on July 27, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from
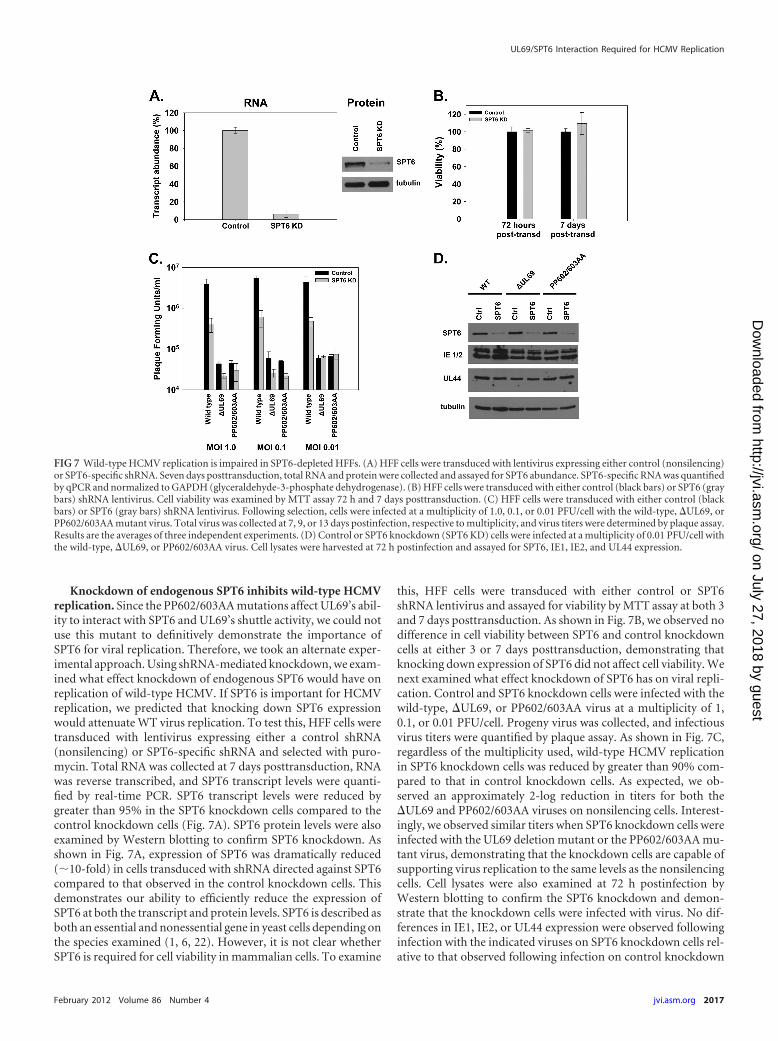
Knockdown of endogenous SPT6 inhibits wild-type HCMVreplication. Since the PP602/603AA mutations affect UL69’s abil-ity to interact with SPT6 and UL69’s shuttle activity, we could notuse this mutant to definitively demonstrate the importance ofSPT6 for viral replication. Therefore, we took an alternate exper-imental approach. Using shRNA-mediated knockdown, we exam-ined what effect knockdown of endogenous SPT6 would have onreplication of wild-type HCMV. If SPT6 is important for HCMVreplication, we predicted that knocking down SPT6 expressionwould attenuate WT virus replication. To test this, HFF cells weretransduced with lentivirus expressing either a control shRNA(nonsilencing) or SPT6-specific shRNA and selected with puro-mycin. Total RNA was collected at 7 days posttransduction, RNAwas reverse transcribed, and SPT6 transcript levels were quanti-fied by real-time PCR. SPT6 transcript levels were reduced bygreater than 95% in the SPT6 knockdown cells compared to thecontrol knockdown cells (Fig. 7A). SPT6 protein levels were alsoexamined by Western blotting to confirm SPT6 knockdown. Asshown in Fig. 7A, expression of SPT6 was dramatically reduced(�10-fold) in cells transduced with shRNA directed against SPT6compared to that observed in the control knockdown cells. Thisdemonstrates our ability to efficiently reduce the expression ofSPT6 at both the transcript and protein levels. SPT6 is described asboth an essential and nonessential gene in yeast cells depending onthe species examined (1, 6, 22). However, it is not clear whetherSPT6 is required for cell viability in mammalian cells. To examine
this, HFF cells were transduced with either control or SPT6shRNA lentivirus and assayed for viability by MTT assay at both 3and 7 days posttransduction. As shown in Fig. 7B, we observed nodifference in cell viability between SPT6 and control knockdowncells at either 3 or 7 days posttransduction, demonstrating thatknocking down expression of SPT6 did not affect cell viability. Wenext examined what effect knockdown of SPT6 has on viral repli-cation. Control and SPT6 knockdown cells were infected with thewild-type, �UL69, or PP602/603AA virus at a multiplicity of 1,0.1, or 0.01 PFU/cell. Progeny virus was collected, and infectiousvirus titers were quantified by plaque assay. As shown in Fig. 7C,regardless of the multiplicity used, wild-type HCMV replicationin SPT6 knockdown cells was reduced by greater than 90% com-pared to that in control knockdown cells. As expected, we ob-served an approximately 2-log reduction in titers for both the�UL69 and PP602/603AA viruses on nonsilencing cells. Interest-ingly, we observed similar titers when SPT6 knockdown cells wereinfected with the UL69 deletion mutant or the PP602/603AA mu-tant virus, demonstrating that the knockdown cells are capable ofsupporting virus replication to the same levels as the nonsilencingcells. Cell lysates were also examined at 72 h postinfection byWestern blotting to confirm the SPT6 knockdown and demon-strate that the knockdown cells were infected with virus. No dif-ferences in IE1, IE2, or UL44 expression were observed followinginfection with the indicated viruses on SPT6 knockdown cells rel-ative to that observed following infection on control knockdown
FIG 7 Wild-type HCMV replication is impaired in SPT6-depleted HFFs. (A) HFF cells were transduced with lentivirus expressing either control (nonsilencing)or SPT6-specific shRNA. Seven days posttransduction, total RNA and protein were collected and assayed for SPT6 abundance. SPT6-specific RNA was quantifiedby qPCR and normalized to GAPDH (glyceraldehyde-3-phosphate dehydrogenase). (B) HFF cells were transduced with either control (black bars) or SPT6 (graybars) shRNA lentivirus. Cell viability was examined by MTT assay 72 h and 7 days posttransduction. (C) HFF cells were transduced with either control (blackbars) or SPT6 (gray bars) shRNA lentivirus. Following selection, cells were infected at a multiplicity of 1.0, 0.1, or 0.01 PFU/cell with the wild-type, �UL69, orPP602/603AA mutant virus. Total virus was collected at 7, 9, or 13 days postinfection, respective to multiplicity, and virus titers were determined by plaque assay.Results are the averages of three independent experiments. (D) Control or SPT6 knockdown (SPT6 KD) cells were infected at a multiplicity of 0.01 PFU/cell withthe wild-type, �UL69, or PP602/603AA virus. Cell lysates were harvested at 72 h postinfection and assayed for SPT6, IE1, IE2, and UL44 expression.
UL69/SPT6 Interaction Required for HCMV Replication
February 2012 Volume 86 Number 4 jvi.asm.org 2017
on July 27, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

cells (Fig. 7D). Taken together, our data suggest that UL69’s inter-action with SPT6 plays an important role in regulating HCMVreplication.
Knockdown of endogenous SPT6 does not affect UL69 shut-tling activity. Since the PP602/603AA mutant affects both UL69’sability to shuttle and its ability to interact with SPT6, it is possiblethat UL69’s shuttling activity may be linked to SPT6 binding. Toaddress this, we assayed for UL69 shuttling activity following wild-type infection of SPT6 knockdown cells. First, we performed im-munofluorescent staining on both control and SPT6 knockdowncells to determine the level and homogeneity of the SPT6 knock-down. As shown in Fig. 8A, we could readily detect SPT6 withinthe nucleus of control knockdown cells. However, we were unableto detect significant levels of SPT6 within cells expressing the SPT6shRNA. Control and SPT6 knockdown cells were then infectedwith the wild-type virus and assayed for UL69 shuttling using theheterokaryon assay. As shown in Fig. 8B, UL69 was capable ofshuttling to murine nuclei within heterokaryons formed in bothcontrol knockdown cells and SPT6 knockdown cells, suggestingthat SPT6 may not be required for UL69 shuttling. Quantitativeresults are shown in Fig. 8C.
DISCUSSION
The tegument protein UL69 has previously been shown to be re-quired for efficient virus replication and to interact with multiplecellular and viral proteins (2, 16, 20, 23, 25). However, it is notclear which of these interactions contribute to virus replication. Inthis report, we have utilized a number of UL69 viral mutants andshRNA knockdown technology to examine the functional signif-icance of UL69’s interaction with SPT6 during HCMV infection.
A number of point mutants have been described that nega-tively affect various UL69 functions, including its ability to shuttlebetween the nucleus and cytoplasm and to interact with variouscellular proteins (14, 16, 25). However, many of these mutantshave not been tested for their ability to interact with SPT6 orassayed for their effect on viral replication. Therefore, we exam-ined a panel of UL69 mutants to identify mutations that inhibitUL69’s ability to bind SPT6 (Fig. 1A). Using this approach, weidentified two UL69 mutants that abolished binding to SPT6. Asexpected, the C496A mutant was unable to bind SPT6. This mu-tation has previously been shown to block UL69’s ability to inter-act with SPT6 in yeast two-hybrid studies (25). The second mu-tant unable to bind SPT6 was the PP602/603AA mutant, whichhas also been described as a UL69 shuttling mutant (14). All othermutants tested retained their ability to interact with SPT6, includ-ing the E617A mutant, which has also been described as a shuttlingmutant (14). These results suggested that UL69’s shuttling activitymay be linked to its ability to bind SPT6 and that we may be able toseparate these two activities.
Therefore, we focused on the C496A, PP602/603AA, andE617A mutants for further characterization. We incorporatedthese three different UL69 mutants into the viral genome andexamined the replication phenotype of each recombinant virus.Replication of the C496A and PP602/603AA mutants was severelyimpaired and paralleled that of the UL69 deletion mutant (Fig. 2Aand B), suggesting that UL69’s interaction with SPT6 is importantfor viral replication. In support of this was the fact that the E617Amutant, which retained its ability to bind SPT6, replicated to wild-type levels (Fig. 2C). Unexpectedly, the two predicted shuttlingmutants (PP602/603AA and E617A) displayed opposite growth
phenotypes. Given the differences in the growth phenotypes be-tween the PP602/603AA and E617A mutants, we examined theability of all three mutants to shuttle between the nucleus andcytoplasm of infected cells. As expected, the PP602/603AA mutantwas defective for shuttling activity, but surprisingly, the C496Amutant exhibited a similar defect (Fig. 5). However, UL69 ex-pressed from the E617A mutant was fully capable of shuttlingbetween the nucleus and cytoplasm during infection (Fig. 5). Wealso tested the three mutants for their ability to shuttle followingtransient transfection of HFF cells. Like the results following in-
FIG 8 Knockdown of SPT6 does not inhibit UL69 shuttling activity. (A) HFFcells were transduced with lentivirus expressing either control or SPT6-specificshRNA. At 72 h posttransduction, cells were fixed and stained for SPT6. Nucleiwere visualized by Hoechst staining. (B) HFF cells were transduced with eithercontrol or SPT6 shRNA lentivirus. Knockdown cells were infected with wild-type HCMV at a multiplicity of 0.5 PFU/cell. At 72 h postinfection, hetero-karyon analysis was performed as described. Virally encoded GFP was used toaid identification of infected heterokaryons, and murine nuclei are indicatedwith white arrows. (C) The bar graph shows quantitation of shuttling-positiveheterokaryons on control or SPT6 knockdown cells.
Cygnar et al.
2018 jvi.asm.org Journal of Virology
on July 27, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

fection, the PP602/603AA and C496A mutations inhibited shut-tling, whereas the E617A mutation had no effect on UL69’s shut-tling activity (data not shown). The reason for the discrepancybetween our results and those previously published for the shut-tling activity of the E617A mutation is currently unknown butcould be due to the use of different cell types or expression vectorsin the heterokaryon assay. However, the entire UL69 open readingframe from the E617A mutant virus and expression plasmid wassequenced to confirm the specific point mutation and rule out thepossibility of a secondary mutation elsewhere in the ORF. Ourfindings clearly demonstrate that the E617A mutation has no ef-fect on viral replication, UL69 shuttling activity, SPT6 binding, orUL69’s ability to interact with eIF4a1, URH49, or wild-type UL69.
Since both the C496A and PP602/603AA mutant viruses dis-played growth phenotypes similar to the UL69 deletion mutantand both were inhibited for SPT6 binding and shuttling activity, itraised the possibility that these mutations may render the UL69protein nonfunctional. We tested this possibility by assaying eachof the UL69 mutants for their ability to interact with other cellularproteins that have previously been shown to bind UL69. Not onlywas the C496A mutant unable to interact with SPT6 (Fig. 1A), butit was also unable to interact with URH49 and eIF4a1 or dimerizewith wild-type UL69 (Fig. 6). These results suggest that mutationof amino acid 496 within UL69 may render the protein nonfunc-tional since this mutant was defective in every assay tested. How-ever, we cannot rule out the possibility that this protein remainsfunctional in other assays. The reason for this defect is unclear. Wedid observe a decrease in expression levels of the C496A UL69protein following either transient transfection or infection (Fig.1A and Fig. 3), suggesting that this mutation may affect the stabil-ity of the UL69 protein. In addition, the C496A mutation lieswithin the region that has been predicted to be required for UL69dimerization and/or multimerization (15). Therefore, it is possi-ble that UL69’s ability to dimerize may be required for its functionand/or its ability to form protein interactions. Unlike the C496Amutant, the PP602/603AA mutant retained its ability to interactwith URH49 and eIF4a1 and its ability to dimerize with wild-typeUL69, demonstrating that these mutations do not render theUL69 protein nonfunctional. It also demonstrates that the PP602/603AA mutations represent specific mutations that block SPT6binding. It also suggests that the SPT6-binding domain withinUL69 encompasses a region that includes amino acids 602 and603. Interestingly, we also observed lower protein levels at earlytimes postinfection with the PP602/603AA mutant virus. How-ever, unlike with the C496A mutant, there was no defect in expres-sion levels following transient transfection (Fig. 1A). The de-creased levels are not due to a defect in UL69 transcription sincewe observed similar levels of UL69 transcripts when comparingwild-type infection to PP602/603AA infection (data not shown).One possibility for the decreased protein levels is that the interac-tion between UL69 and SPT6 helps to stabilize the UL69 protein.Therefore, in the absence of SPT6 binding following infectionwith the PP602/603AA mutant, the stability of UL69 may be al-tered. Finally, the protein interaction results with the PP602/603AA mutant support our previous findings that demonstratedthat UL69’s interaction with URH49 is dispensable for virus rep-lication (13). They also suggest that UL69’s interaction witheIF4a1 may not be required for efficient virus replication, since thePP602/603AA mutant still retains its ability to bind eIF4a1 but
demonstrates a growth phenotype identical to that of the UL69deletion mutant.
Since abolishing UL69’s interaction with SPT6 correlated witha loss of shuttling activity, it is tempting to speculate that UL69’sinteraction with SPT6 is required not only for viral replication butalso for UL69 shuttling activity. To test this, we assayed for viralreplication and shuttling activity following infection of SPT6knockdown cells with wild-type HCMV. Knockdown of SPT6 didnot affect UL69’s ability to shuttle during infection, suggestingthat the interaction is not required for shuttling activity (Fig. 8B).However, caution should be taken when interpreting this result.SPT6 knockdown resulted in a significant reduction in availableSPT6 protein but did not result in a complete loss of SPT6 (Fig.7A). Therefore, it is possible that the residual amount of SPT6protein remaining after knockdown is below the limit of detectionby immunofluorescent staining but sufficient to allow for UL69shuttling activity. Alternatively, murine SPT6 may compensate forthe loss of human SPT6 upon heterokaryon formation, therebyallowing UL69 to shuttle in this assay. While these data suggestthese functions may not be linked, further studies will need to beperformed to confirm that UL69 shuttling is independent of theSPT6 interaction.
However, knockdown of SPT6 inhibited wild-type replicationby greater than 90%, underscoring the importance of SPT6 inHCMV replication. The mechanism by which SPT6’s interactionwith UL69 regulates viral replication is still uncertain. SPT6 func-tions as both a chromatin remodeling protein and a transcrip-tional elongation factor. Therefore, it is likely that the interactionbetween UL69 and SPT6 is critical for regulating the rate of tran-scription that occurs on individual viral or cellular genes or thatthe interaction is required for the proper chromatin modificationsneeded to allow for efficient gene expression. Further studies willbe required to address these possibilities. Regardless of the mech-anism, our results demonstrate that the interaction between UL69and SPT6 plays an important role in controlling HCMV replica-tion.
ACKNOWLEDGMENTS
We are grateful to Tom Shenk (Princeton) for the monoclonal antibodyrecognizing UL69. We thank Steve Rice and Stacia Phillips for helpfuldiscussions and for critically reading the manuscript.
This work was supported by NIH grant AI059340 (to W.A.B).
REFERENCES1. Al-Rawi N, Laforce-Nesbitt SS, Bliss JM. 2010. Deletion of Candida
albicans SPT6 is not lethal but results in defective hyphal growth. FungalGenet. Biol. 47:288 –296.
2. Aoyagi M, Gaspar M, Shenk TE. 2010. Human cytomegalovirus UL69protein facilitates translation by associating with the mRNA cap-bindingcomplex and excluding 4EBP1. Proc. Natl. Acad. Sci. U. S. A. 107:2640 –2645.
3. Bortvin A, Winston F. 1996. Evidence that Spt6p controls chromatinstructure by a direct interaction with histones. Science 272:1473–1476.
4. Cantrell SR, Bresnahan WA. 2006. Human cytomegalovirus (HCMV)UL82 gene product (pp71) relieves hDaxx-mediated repression of HCMVreplication. J. Virol. 80:6188 – 6191.
5. Chiang PW, et al. 1996. Identification and analysis of the human andmurine putative chromatin structure regulator SUPT6H and Supt6h.Genomics 34:328 –333.
6. Clark-Adams CD, Winston F. 1987. The SPT6 gene is essential for growthand is required for delta-mediated transcription in Saccharomyces cerevi-siae. Mol. Cell. Biol. 7:679 – 686.
7. Endoh M, et al. 2004. Human Spt6 stimulates transcription elongation byRNA polymerase II in vitro. Mol. Cell. Biol. 24:3324 –3336.
UL69/SPT6 Interaction Required for HCMV Replication
February 2012 Volume 86 Number 4 jvi.asm.org 2019
on July 27, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

8. Hartzog GA, Wada T, Handa H, Winston F. 1998. Evidence that Spt4,Spt5, and Spt6 control transcription elongation by RNA polymerase II inSaccharomyces cerevisiae. Genes Dev. 12:357–369.
9. Hayashi ML, Blankenship C, Shenk T. 2000. Human cytomegalovirusUL69 protein is required for efficient accumulation of infected cells in theG1 phase of the cell cycle. Proc. Natl. Acad. Sci. U. S. A. 97:2692–2696.
10. Kalejta RF. 2008. Functions of human cytomegalovirus tegument pro-teins prior to immediate early gene expression. Curr. Top. Microbiol.Immunol. 325:101–115.
11. Kalejta RF. 2008. Tegument proteins of human cytomegalovirus. Micro-biol. Mol. Biol. Rev. 72:249 –265.
12. Kaplan CD, Morris JR, Wu C, Winston F. 2000. Spt5 and Spt6 areassociated with active transcription and have characteristics of generalelongation factors in D. melanogaster. Genes Dev. 14:2623–2634.
13. Kronemann D, Hagemeier SR, Cygnar D, Phillips S, Bresnahan WA.2010. Binding of the human cytomegalovirus (HCMV) tegument proteinUL69 to UAP56/URH49 is not required for efficient replication of HCMV.J. Virol. 84:9649 –9654.
14. Lischka P, Rosorius O, Trommer E, Stamminger T. 2001. A noveltransferable nuclear export signal mediates CRM1-independent nucleo-cytoplasmic shuttling of the human cytomegalovirus transactivator pro-tein pUL69. EMBO J. 20:7271–7283.
15. Lischka P, Thomas M, Toth Z, Mueller R, Stamminger T. 2007. Mul-timerization of human cytomegalovirus regulatory protein UL69 via adomain that is conserved within its herpesvirus homologues. J. Gen. Virol.88:405– 410.
16. Lischka P, Toth Z, Thomas M, Mueller R, Stamminger T. 2006. TheUL69 transactivator protein of human cytomegalovirus interacts withDEXD/H-Box RNA helicase UAP56 to promote cytoplasmic accumula-tion of unspliced RNA. Mol. Cell. Biol. 26:1631–1643.
17. Lu M, Shenk T. 1999. Human cytomegalovirus UL69 protein inducescells to accumulate in G1 phase of the cell cycle. J. Virol. 73:676 – 683.
18. Pass R. 2001. Cytomegalovirus, p 2675–2706. In Knipe DM, et al (ed),Fields virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
19. Phillips SL, Bresnahan WA. 2011. Identification of binary interactionsbetween human cytomegalovirus virion proteins. J. Virol. 85:440 – 447.
20. Rechter S, et al. 2009. Cyclin-dependent kinases phosphorylate the cyto-megalovirus RNA export protein pUL69 and modulate its nuclear local-ization and activity. J. Biol. Chem. 284:8605– 8613.
21. Smith GA, Enquist LW. 1999. Construction and transposon mutagenesisin Escherichia coli of a full-length infectious clone of pseudorabies virus, analphaherpesvirus. J. Virol. 73:6405– 6414.
22. Swanson MS, Winston F. 1992. SPT4, SPT5 and SPT6 interactions: ef-fects on transcription and viability in Saccharomyces cerevisiae. Genetics132:325–336.
23. Thomas M, et al. 2009. Cytomegaloviral protein kinase pUL97 interactswith the nuclear mRNA export factor pUL69 to modulate its intranuclearlocalization and activity. J. Gen. Virol. 90:567–578.
24. Toth Z, Lischka P, Stamminger T. 2006. RNA-binding of the humancytomegalovirus transactivator protein UL69, mediated by arginine-richmotifs, is not required for nuclear export of unspliced RNA. Nucleic AcidsRes. 34:1237–1249.
25. Winkler M, aus dem Siepen T, Stamminger T. 2000. Functional inter-action between pleiotropic transactivator pUL69 of human cytomegalo-virus and the human homolog of yeast chromatin regulatory proteinSPT6. J. Virol. 74:8053– 8064.
26. Winkler M, Rice SA, Stamminger T. 1994. UL69 of human cytomeg-alovirus, an open reading frame with homology to ICP27 of herpessimplex virus, encodes a transactivator of gene expression. J. Virol.68:3943–3954.
27. Yoh SM, Cho H, Pickle L, Evans RM, Jones KA. 2007. The Spt6 SH2domain binds Ser2-P RNAPII to direct Iws1-dependent mRNA splicingand export. Genes Dev. 21:160 –174.
Cygnar et al.
2020 jvi.asm.org Journal of Virology
on July 27, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from





























