Embed Size (px)
Citation preview

at SciVerse ScienceDirect
Biochimie 95 (2013) 1023e1032
Contents lists available
Biochimie
journal homepage: www.elsevier .com/locate/biochi
Research paper
The C-terminal domain of CblD interacts with CblC and influencesintracellular cobalamin partitioningq
Carmen Gherasim a,1, Luciana Hannibal b,1, Deepa Rajagopalan a,Donald W. Jacobsen b,c,d,**, Ruma Banerjee a,*
aDepartment of Biological Chemistry, University of Michigan Medical Center, Ann Arbor, MI 48109-0600, USAbDepartment of Cellular and Molecular Medicine, Lerner Research Institute, Cleveland Clinic, Cleveland, OH 44195, USAc School of Biomedical Sciences, Kent State University, Kent, OH 44242, USAdDepartment of Molecular Medicine, Cleveland Clinic Lerner College of Medicine, Case Western Reserve University, Cleveland, OH 44106, USA
a r t i c l e i n f o
Article history:Received 3 September 2012Accepted 1 February 2013Available online 14 February 2013
Keywords:CobalaminTrafficHomocystinuriaB12
q This work was supported by grants from the N(DK45776 to RB and HL71907 to DWJ) REFER MS Set* Corresponding author. University of Michigan, B
Medical Center Dr., Ann Arbor, MI 48109-5606, USA.** Corresponding author. Department of Cellular andLerner Research Institute, Cleveland Clinic, 9500 EuclUSA. Tel.: þ1 216 444 8340.
E-mail addresses: [email protected] (D.W. Jaco(R. Banerjee).
1 Equal contributors.
0300-9084/$ e see front matter � 2013 Elsevier Mashttp://dx.doi.org/10.1016/j.biochi.2013.02.003
a b s t r a c t
Mutations in cobalamin or B12 trafficking genes needed for cofactor assimilation and targeting lead toinborn errors of cobalamin metabolism. The gene corresponding to one of these loci, cblD, affects boththe mitochondrial and cytoplasmic pathways for B12 processing. We have demonstrated that fibroblastcell lines from patients with mutations in CblD, can dealkylate exogenously supplied methylcobalamin(MeCbl), an activity catalyzed by the CblC protein, but show imbalanced intracellular partitioning of thecofactor into the MeCbl and 50-deoxyadenosylcobalamin (AdoCbl) pools. These results confirm that CblDfunctions downstream of CblC in the cofactor assimilation pathway and that it plays an important role incontrolling the traffic of the cofactor between the competing cytoplasmic and mitochondrial routes forMeCbl and AdoCbl synthesis, respectively. In this study, we report the interaction of CblC with four CblDprotein variants with variable N-terminal start sites. We demonstrate that a complex between CblC andCblD can be isolated particularly under conditions that permit dealkylation of alkylcobalamin by CblC orin the presence of the corresponding dealkylated and oxidized product, hydroxocobalamin (HOCbl). Aweak CblC$CblD complex is also seen in the presence of cyanocobalamin. Formation of the CblC$CblDcomplex is observed with all four CblD variants tested suggesting that the N-terminal 115 residuesmissing in the shortest variant are not essential for this interaction. Furthermore, limited proteolysis ofthe CblD variants indicates the presence of a stable C-terminal domain spanning residues w116e296.Our results are consistent with an adapter function for CblD, which in complex with CblC$HOCbl, orpossibly the less oxidized CblC$cob(II)alamin, partitions the cofactor between AdoCbl and MeCblassimilation pathways.
� 2013 Elsevier Masson SAS. All rights reserved.
Although only two human enzymes use cobalamin (or de-rivatives of B12) as a cofactor, its intracellular handlers are many[1,2]. Clinical genetics studies have led to the recognition of at leastseven loci (cblAeF and cblJ) that support B12 functions in addition tothe two encoding the B12-dependent enzymes, methionine
ational Institutes of HealthAs per style.iological Chemistry, 1150 W.Tel.: þ1 734 615 5238.Molecular Medicine, NC-10,
id Ave. Cleveland, OH 44195,
bsen), [email protected]
son SAS. All rights reserved.
synthase (cblG) and methylmalonyl-CoA mutase (mut) [3e5]. Withthe recent identification of all seven genes encoding proteinsdedicated to intracellular B12 assimilation and trafficking [6e13],elucidation of their biochemical functions has just begun, providingearly insights into a complex intracellular pathway for B12 pro-cessing. Given the relative rarity of the B12 cofactor (30e700 nM inhuman tissues [14]) and its reactivity in all three biologically rele-vant cobalt oxidation states, we have proposed that navigation ofcobalamin from its point of exit from the lysosome to its destina-tions in the cytoplasm (methionine synthase) and mitochondrion(methylmalonyl-CoA mutase) respectively, relies on protein escorts[1,2]. B12 trafficking can be classified into (i) the early or commonpathway, (ii) the methylcobalamin (MeCbl) branch and (iii) the 50-deoxyadenosylcobalamin (AdoCbl) branch (Fig. 1). Functions in theearly pathway are shared for assimilation of MeCbl and AdoCblneeded by methionine synthase and methylmalonyl-CoA mutase,

Fig. 1. Schematic representation of a model for the cobalamin trafficking pathway.Proteins corresponding to the nine genetic complementation groups are designated asCblA-J and Mut (for methylmalonyl-CoA mutase). CblC is proposed to function as thecobalamin donor to cytoplasmic methionine synthase (CblG), which associates withmethionine synthase reductase (CblE). We propose that CblD functions as an adapter,facilitating complex formation and/or cofactor transfer from CblC. The mechanism bywhich B12 enters the mitochondrion is not known and a putative transporter is shownin blue for purposes of illustration. The question marks denote the uncertainty in themechanism for B12 import into the mitochondrion and its loading onto CblB. The formof cobalamin bound to CblC after dealkylation is shown as cob(II)alamin for simplicity.We note however that the dealkylation product is cob(I)alamin, which is rapidlyoxidized to cob(II)alamin and then HOCbl in vitro. Cob(II)alamin is presumed to bestabilized under the reducing intracellular conditions and is also the product of CblC-catalyzed decyanation of CNCbl.
C. Gherasim et al. / Biochimie 95 (2013) 1023e10321024
respectively and include the cblF, cblJ, cblC and cblD loci. Defects inthese loci typically lead to combined methylmalonic aciduria andhomocystinuria, as expected for impaired activity of both B12 en-zymes. Defects in the MeCbl branch (cblG and cblE) lead to isolatedhomocystinuria whereas mutations in the AdoCbl branch (cblA,cblB and mut) lead to isolated methylmalonic aciduria. The excep-tion to this tidy classification is the cblD locus, which showscomplexity, in that depending on the location of the pathogenicmutation, patients exhibit either combined or isolated methyl-malonic aciduria and homocystinuria [15]. The CblD protein ispredicted to have an eleven-residue mitochondrial leader sequenceat the N-terminus [9,16]. Mutation analyses reveal that defects atthe N- and C-termini of the encoded CblD2 protein are associatedwith methylmalonic aciduria and homocystinuria respectively,suggesting that either different isoforms or distinct protein do-mains support the MeCbl versus the AdoCbl branches of thepathway [9,16]. Initiation codons at positions 1, 62 and 116 in goodKozak contexts are located on themRNA encoding CblD, supportingthe possibility that internal initiation sites can be used to produceCblD variants [16]. However, western blot analysis reveals thepresence of only full-length CblD protein in wild-type human fi-broblasts [16]. In contrast, the shorter variants corresponding toinitiation at the 62 and 116 start sites were seen in fibroblasts froma patient with homozygous nonsense mutation that results intermination of CblD at Ser20. Hence, internal initiation at Met62and Met116 is likely to be important in a subset of cblD patients
2 CblD is also referred to as MMADHC (for methylmalonic aciduria type D withhomocystinuria) [9]. Since this nomenclature does not aptly describe the hetero-geneity in this patient group, i.e. not all individuals exhibit combined methyl-malonic aciduria and homocystinuria, we use CblD to refer to the gene product ofthe cblD locus.
with mutations that result in premature termination of the proteinimpacting AdoCbl synthesis but allowing MeCbl synthesis [16].
CblD belongs to an uncharacterized protein family with limitedsequence similarity to the ATPase component of a putative ABCtransporter [9]. However, CblD is missing theWalker A, Walker B andtheABCsignaturemotifs characteristic of all classicalABC transporters.In principle, CblD might be a B12-binding protein which accepts thecofactor from CblC and delivers it to the client enzymes, methioninesynthase andmethylmalonyl-CoAmutase. In support of this proposalis the presence of a signature DxHxxG-39/40–gxSxLxxxx–17/18-xxxxGGx B12-binding sequence [17], albeit incomplete. Alternatively,CblD might influence the activity of CblC either at the level of B12processing (i.e. its dealkylation and decyanation activities) or at thelevel of its interactions with downstream client proteins facilitatingtransfer of B12 from the CblC donor to the cytoplasmic and mito-chondrial acceptors. Finally, CblD could be involved further down-stream i.e., in docking of the cofactor to the target enzymes,methionine synthase and methylmalonyl-CoA mutase, respectively.
In this study, we have purified four forms of recombinant hu-man CblD with varying N-terminal start sites at residues 1, 12, 62and 116, respectively. We demonstrate that CblD does not bind B12
nor exhibits ATPase activity. All four CblD variants form complexeswith and exhibit selectivity for the B12 cofactor bound form of CblCcompared to apo-CblC. We propose that CblD might function as anadapter for complexation of CblC$cob(II)alamin to acceptor pro-teins: methionine synthase in the cytoplasm and either adenosyl-transferase or a transporter in the mitochondrion (Fig. 1). Insupport of this model, we demonstrate that fibroblasts from cblDpatients dealkylate [57Co]-MeCbl but that the subsequent parti-tioning of the cobalamin product toMeCbl versus AdoCbl is affectedby the location of the mutation in CblD. Mutations near the N- andC-termini of the protein diminish AdoCbl and MeCbl synthesis,respectively, while mutations in the middle affect both pathways.Hence, while CblD is not required for the activity of CblC per se, itaffects partitioning of the processed cobalamin product of CblC intothe cytoplasmic and mitochondrial pathways.
1. Materials and methods
1.1. Materials
All chemicals were purchased from Sigma unless otherwisestated. [57Co]-MeCblwas synthesized by the reaction of [57Co]-cob(I)alaminwithmethyl iodide (Sigma)underdimred light andanaerobicconditions as described previously [18]. Pfu polymerase, dNTPs andrestriction enzymes were from NEB. Advanced DMEM and Dulbec-co’s phosphate buffered saline (PBS) were purchased from Gibco.
1.2. Cloning of CbD variants
The cDNA encoding CblD in the pcDNA3.2/V5/GW/D-TOPOvector was generously provided by Dr. Brian Fowler (UniversityChildren’s Hospital, Basel). Four start sites (at positions 1, 12, 62 and116) were used to clone the gene into the pET-28b(þ) vector(Novagen) using the NcoI/XhoI and NdeI/XhoI restriction enzymesites to generate the C- and N-terminal 6�His-tagged constructs,respectively. Oligonucleotides containing the first and the last 24bases of cblD gene with the restriction sites at their 50-end, wereused as primers as described below. The underlined bases corre-spond to the NdeI and XhoI restriction sites that were designed intothe forward and reverse primer sequences, respectively.
Forward M1: 50-CATATGGCCAATGTGCTTTGTAACAGAGCC-30,Forward S12: 50-CATATGGTTTCCTATCTCCCAGGATTTTGC-30,Forward M62: 50-CATATGGGACCCTTTGGACCTCAAGATCAG-30,

C. Gherasim et al. / Biochimie 95 (2013) 1023e1032 1025
Forward M116: 50-CATATGGCACAATATGTGAATGAATTTCAG-30,Reverse: 50-CTCGAGTTAATTTCCACTTAATTTCTTCAT-30.
1.3. Expression and purification of CblD proteins
The Escherichia coli strain BL21(DE3) was freshly transformedwith the desired plasmid constructs and grown in LuriaeBertani(LB) medium containing kanamycin (50 mg/ml) at 37 �C, inducedwith 0.25 mM isopropyl b-D-1-thiogalactopyranoside (IPTG) (atA600 nm ¼ 0.5e0.7), and then grown overnight (w12 h) at 20 �C. Thecells were harvested by centrifugation and the cell pellet was storedat �80 �C until further use.
Recombinant CblD was prepared by resuspending the cell pel-lets in lysis/binding buffer (50 mM Tris, pH 8.0, 300 mM KCl, 15 mMimidazole (Buffer A)) supplemented with 10 mM b-mercaptoe-thanol, lysozyme (0.2 mg/ml) and one tablet of Complete-EDTAprotease inhibitor (Roche). Cells were disrupted by sonication andcentrifuged at 20,000 � g for 1 h and the supernatant was appliedto a 15 ml nickelenitrilotriacetic acid column (Qiagen). The columnwas washed with 50 mM Tris, pH 8.0, 300 mM KCl, 50 mM imid-azole until the flow-through was protein-free and recombinantCblD was eluted with a 50e400 mM imidazole gradient. The frac-tions of interest identified by SDS-PAGE were pooled, concentratedand the buffer was exchanged with 100 mMHepes, pH 7.0, 150 mMKCl, 10% glycerol (Buffer B).
1.4. Size exclusion chromatography
The oligomeric state of the CblD variants was determined byloading 1e2.5 mg protein on a Superdex 200 column (1.6 � 80 cm)in Buffer B at a flow rate of 0.5 ml/min. The column was calibratedusing gel filtration standards from Bio-Rad.
1.5. Binding of B12 derivatives and ATP to CblD
Binding of MeCbl, AdoCbl and HOCbl to CblD was examined byisothermal titration calorimetry (ITC) and fluorescence or UVevisible spectroscopy. Fluorescence measurements were made onan RF-5301 PC Shimadzu spectrofluorimeter at 280 nm excitationwavelength (slit width, 3 mm) and the emission was observed be-tween 300 and 380 nm (slit width, 3 mm). The experiments wereperformed at 20 �C in a fluorescence quartz cuvette and successivealiquots (1e2 ml) of B12 stock solution (5e15 mM) were added to a0.5 mM CblD solution in Buffer B. Similarly, binding of ATP to CblDwas determined using 10e100 mM stock solutions. In the ITC ex-periments, CblD (10e40 mm) was titrated with twenty four 12-mlaliquots of a 200e800 mm solution of B12 or ATP in Buffer B at10.0� 0.1 �C. The calorimetric signals were integrated, and the datawere analyzed with Microcal ORIGIN software.
1.6. Effect of CblD on B12 binding to CblC
The binding of CNCbl and MeCbl to CblC in the presence orabsence of CblD was monitored by ITC as previously described [19].CblC and CblD (5e40 mM) were added at a 1:1 ratio and titratedwith 100e800 mM of B12.
1.7. ATPase activity of CblD
The ATPase activity of CblD forms was carried out using acontinuous assay coupled to pyruvate kinase/lactate dehydrogenase[20]. The reactions were carried out at 22 �C in 50 mM TriseCl, pH8.0, 5 mM MgCl2. The reaction mixtures were supplemented with200 mM NADH, 3 mM phosphoenolpyruvate, 2e20 U pyruvate
kinase, 2e20 U lactate dehydrogenase and 1mMATP and incubatedfor5minat22 �Cprior to additionof theCblDprotein.ATPhydrolysiswas followed by measuring the decrease in absorbance at 340 nm.
1.8. Effect of CblD on the catalytic activities of CblC
Anaerobic decyanation and aerobic dealkylation reactionscatalyzed by CblC were monitored as described previously [19,21]with the exception that CblD was added to the reaction mixtureat a 1:1 ratio with CblC.
1.9. Analysis of CblC:CblD complexes
Complex formation between CblD and CblC was tested byincubating 10e15 mg of CblC with an equal amount of CblD in thepresence or absence of 100 mm MeCbl, CNCbl or HOCbl and 1 mMGSH at 20 �C in 100 mM Hepes, pH 8, 150 mM KCl, 10% glycerol.After 30 min, the mixtures were analyzed by polyacrylamide gelelectrophoresis under nondenaturing conditions on a 4e20%gradient gel (Bio-Rad) over w2 h at 4 �C.
1.10. Limited proteolysis of CblD proteins
CblD protein variants (15 mg) were incubated with trypsin (2%w/w) at room temperature for 10 min. The limited proteolysis re-actions were stopped by addition of 1 ml TLCK (10% w/v). Thedigested proteins were then separated by electrophoresis on a 12%SDS-PAGE gel.
1.11. Cell culture and [57Co]-MeCbl metabolic labeling
Normal and cblD mutant fibroblasts were grown in AdvancedDMEM (Gibco) supplemented with 10% fetal bovine serum (FBS).Normal human skin fibroblast (HFF) was kindly provided by theCell Culture Core of the Lerner Research Institute, Cleveland Clinic.The two normal cell lines used in the present study were generatedfrom discarded tissue from circumcised donors of 1 and 3 monthsof age, respectively as described previously [18]. Human foreskintissue was obtained for establishment of control skin fibroblastcultures after informed consent was received. The InstitutionalReview Board of the Cleveland Clinic approved the protocol. DavidRosenblatt (McGill University) kindly provided human cblDmutantskin fibroblasts from patients with either isolated or combinedhomocystinuria and methylmalonic aciduria (WG2024, WG3280,WG3583 andWG3745) obtained as described previously [9,22]. TheRoyal Victoria Hospital Research Ethics Board (Montréal, Canada)approved the research protocol for procurement of patient skinbiopsies.
For metabolic labeling experiments, cells were split at a ratio of1:2 and [57Co]-MeCbl was added to achieve a final concentration of0.125 nM (specific activity: 379 mCi/mg MeCbl). After 48 h, cellswere harvested, total cobalamins extracted with 80% aqueousethanol and the intracellular cobalamin profile determined asdescribed previously [18]. The cell cultures were protected fromlight at all times to prevent photolysis of the alkylcobalamins.
1.12. Determination of homocysteine and methylmalonic acid inculture medium
Cells were cultured as described and at the end point of theexperiment, the culture medium was collected, centrifuged at1000 rpm for 10 min (to remove dead cells), filtered using a0.22 mm filter (Millipore) and stored at 4 �C until further use. Totalhomocysteine in culture medium was determined using mono-bromobimane and HPLC with fluorescence detection [23]. The

Fig. 2. Purification of recombinant CblD forms. (A) Boundaries of the CblD constructsused in this study. The predicted mitochondrial leader sequence (MLS) is indicated. (B)SDS-PAGE analysis of protein standards (lane 1) full-length CblD (FL, lane 2), CblD-ND11 (lane 3), CblD-ND61 (lane 4) and CblD ND115 (lane 5). An equal amount ofprotein (7 mg) was loaded in each lane and visualized by Coomassie blue staining.
C. Gherasim et al. / Biochimie 95 (2013) 1023e10321026
concentration of methylmalonic acid in conditioned culture me-dium was determined by gas chromatography and mass spec-trometry in the Department of Clinical Pathology, Cleveland Clinicby a method modified from Hoffmann et al. [24]. Briefly, methyl-malonic acid was extracted from conditioned culture mediumalong with tri-deuterated methylmalonic acid (internal standard,MSD Isotopes) using Bond-Elute SAX solid phase extraction col-umns (Varian). The extracted acid was then derivatized withcyclohexanol to form a dicyclohexyl ester. The derivatized sampleswere resolved by gas chromatography and detected by massspectrometry using selective ion monitoring. Values werenormalized to intracellular protein concentration determined bythe bicinchoninic acid assay (Thermo Scientific) using bovineserum albumin as a standard.
2. Results
2.1. Purification of recombinant CblD variants
Based on sequence analysis using MitoProt II and PSORT II, theCblD protein sequence is predicted to include a putative mito-chondrial targeting sequence that spans residues 1e12 at the N-terminus to give the CblD DN11 variant [9]. Furthermore, two in-ternal Met62 and Met116 codons have been shown to function asalternative translation initiation sites giving rise to DN61 andDN115-CblD variants [16]. We purified the recombinant full-length,DN11-, DN61- and DN115-CblD proteins to >95% homogeneity(Fig. 2). Since we found that the C-terminal His-tagged constructswere considerably less stable than the corresponding N-terminal
tagged ones, only the latter were further characterized in this study.CblD as isolated, is devoid of a visible absorption spectrum sug-gesting the absence of a bound cofactor. These wild-type CblDvariants were also devoid of a metal cofactor as assessed by plasmaemission spectroscopy [25] at the Chemical Analysis Laboratory,University of Georgia, Athens (not shown). Size exclusion chro-matography revealed that CblD is a monomer with estimatedmolecular masses of 35 kDa, 33.5 kDa, 28.5 kDa and 22.4 kDa,respectively for the full-length and the three N-terminal truncatedvariants. All CblD variants displayed a tendency to form high mo-lecular weight aggregates.
2.2. Binding of B12 by CblD
Despite the presence of a weakly conserved B12-binding motif inCblD, we found no evidence for binding of MeCbl, AdoCbl or HOCblto this protein as determined by fluorescence, ITC and UVevisiblespectroscopic studies. To test whether the 6�His-tag on CblDcould be interfering with B12 binding, we cleaved it from the pu-rified recombinant protein using thrombin. However, the absenceof a His-tag did not promote B12 binding to CblD under the condi-tions employed in our assay.
2.3. CblD-catalyzed ATP hydrolysis
The putative ATPase activity of CblD was assessed using a pre-viously described continuous assay [20]. Despite the sequencesimilarity with the ATPase component of the Salmonella entericaABC transporter, CblD does not hydrolyze ATP.
2.4. Effect of CblD on B12 binding and processing by CblC
The presence of either full-length or DN11 CblD had essentiallyno effect on the affinity of CblC for CNCbl (Kd ¼ 11.4� 1.6 mM versus14.2� 2.1 mM)orMeCbl (Kd¼0.22�0.02 mMversus 0.19�0.02 mM).Similarly, the CblC-catalyzed rates for the reductive decyanation ofCNCbl and the dealkylation of MeCbl by glutathione reported pre-viously [19,21] were comparable in the presence or absence of full-length, DN11 or DN115 CblD (not shown).
2.5. Complex formation between CblD and CblC
Since CblD does not appear to influence B12 binding or the B12processing activities of CblC, we next looked for evidence for in-teractions between these two proteins. In the absence of B12, CblCbehaves like a higher order oligomer that barely migrates into thegel (Fig. 3A, lane 11). Addition of CNCbl in the presence or absence ofglutathione induced an electrophoretic shift in CblC which ranwithan apparentmolecularmass ofw250 kDa (Fig. 3A, lanes 12e13). Thepresence of AdoCbl and/or glutathione had no effect on the CblDelectrophoretic pattern that migrated with a molecular mass ofw50 kDa (Fig. 3A, lanes 14e16), although the protein behaves as amonomer with a predicted molecular size of w35 kDa by sizeexclusion chromatography (not shown). When CblC and CblD werepre-incubated, a new band was visualized but only in the presenceof B12 (compare lane 6 with lanes 2e5). Addition of glutathioneversus dithiothreitol to the CblD:CblC$B12 mixture enhanced com-plex formation (compare lanes 2e5 versus 7e10), in particular thecomplexes formed in the presence of MeCbl and AdoCbl. Since theprotein samples were incubated for 30min prior to being loaded onthe native gel, CblCwould have catalyzed the dealkylation ofMeCbl,and to a lesser extent, of AdoCbl, by GSH leading toHOCbl formation(via air oxidationof the product, cob(I)alamin). These results suggestthat when an alkylcobalamin substrate is bound to CblC, it inhibitscomplex formationwith CblD. Instead, CblD preferentially interacts
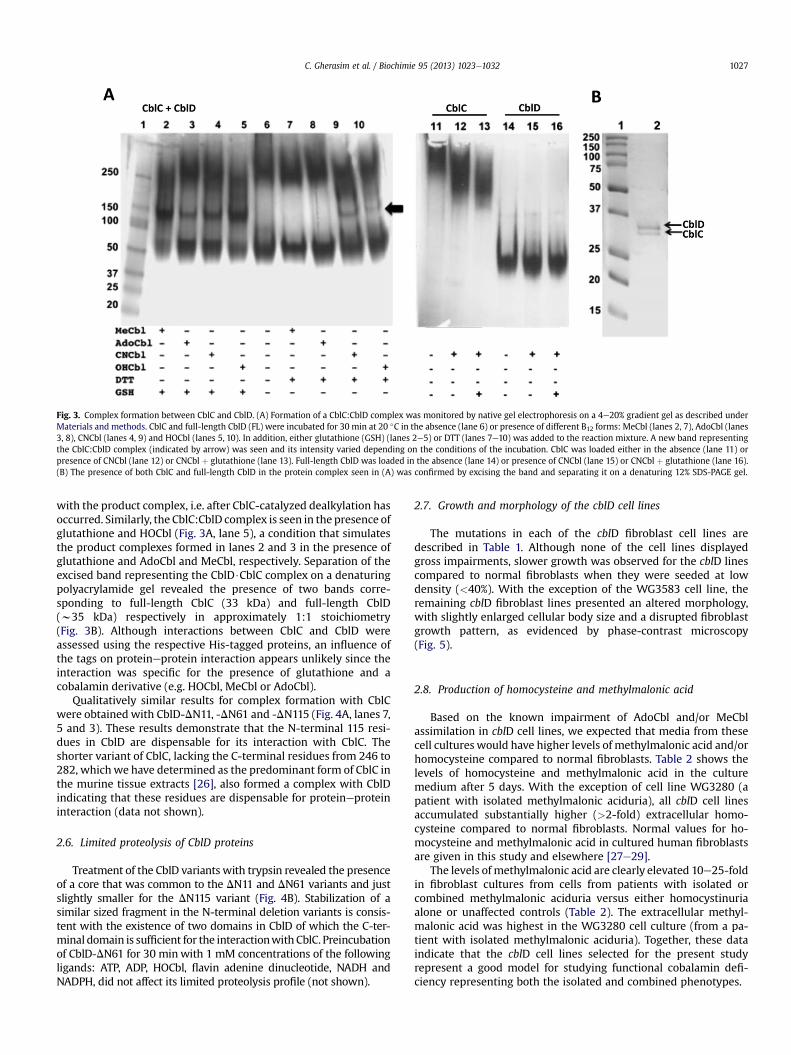
Fig. 3. Complex formation between CblC and CblD. (A) Formation of a CblC:CblD complex was monitored by native gel electrophoresis on a 4e20% gradient gel as described underMaterials and methods. CblC and full-length CblD (FL) were incubated for 30 min at 20 �C in the absence (lane 6) or presence of different B12 forms: MeCbl (lanes 2, 7), AdoCbl (lanes3, 8), CNCbl (lanes 4, 9) and HOCbl (lanes 5, 10). In addition, either glutathione (GSH) (lanes 2e5) or DTT (lanes 7e10) was added to the reaction mixture. A new band representingthe CblC:CblD complex (indicated by arrow) was seen and its intensity varied depending on the conditions of the incubation. CblC was loaded either in the absence (lane 11) orpresence of CNCbl (lane 12) or CNCbl þ glutathione (lane 13). Full-length CblD was loaded in the absence (lane 14) or presence of CNCbl (lane 15) or CNCbl þ glutathione (lane 16).(B) The presence of both CblC and full-length CblD in the protein complex seen in (A) was confirmed by excising the band and separating it on a denaturing 12% SDS-PAGE gel.
C. Gherasim et al. / Biochimie 95 (2013) 1023e1032 1027
with the product complex, i.e. after CblC-catalyzed dealkylation hasoccurred. Similarly, the CblC:CblD complex is seen in the presence ofglutathione and HOCbl (Fig. 3A, lane 5), a condition that simulatesthe product complexes formed in lanes 2 and 3 in the presence ofglutathione and AdoCbl and MeCbl, respectively. Separation of theexcised band representing the CblD$CblC complex on a denaturingpolyacrylamide gel revealed the presence of two bands corre-sponding to full-length CblC (33 kDa) and full-length CblD(w35 kDa) respectively in approximately 1:1 stoichiometry(Fig. 3B). Although interactions between CblC and CblD wereassessed using the respective His-tagged proteins, an influence ofthe tags on proteineprotein interaction appears unlikely since theinteraction was specific for the presence of glutathione and acobalamin derivative (e.g. HOCbl, MeCbl or AdoCbl).
Qualitatively similar results for complex formation with CblCwere obtained with CblD-DN11, -DN61 and -DN115 (Fig. 4A, lanes 7,5 and 3). These results demonstrate that the N-terminal 115 resi-dues in CblD are dispensable for its interaction with CblC. Theshorter variant of CblC, lacking the C-terminal residues from 246 to282, whichwe have determined as the predominant form of CblC inthe murine tissue extracts [26], also formed a complex with CblDindicating that these residues are dispensable for proteineproteininteraction (data not shown).
2.6. Limited proteolysis of CblD proteins
Treatment of the CblD variants with trypsin revealed the presenceof a core that was common to the DN11 and DN61 variants and justslightly smaller for the DN115 variant (Fig. 4B). Stabilization of asimilar sized fragment in the N-terminal deletion variants is consis-tent with the existence of two domains in CblD of which the C-ter-minal domain is sufficient for the interactionwith CblC. Preincubationof CblD-DN61 for 30 minwith 1 mM concentrations of the followingligands: ATP, ADP, HOCbl, flavin adenine dinucleotide, NADH andNADPH, did not affect its limited proteolysis profile (not shown).
2.7. Growth and morphology of the cblD cell lines
The mutations in each of the cblD fibroblast cell lines aredescribed in Table 1. Although none of the cell lines displayedgross impairments, slower growth was observed for the cblD linescompared to normal fibroblasts when they were seeded at lowdensity (<40%). With the exception of the WG3583 cell line, theremaining cblD fibroblast lines presented an altered morphology,with slightly enlarged cellular body size and a disrupted fibroblastgrowth pattern, as evidenced by phase-contrast microscopy(Fig. 5).
2.8. Production of homocysteine and methylmalonic acid
Based on the known impairment of AdoCbl and/or MeCblassimilation in cblD cell lines, we expected that media from thesecell cultures would have higher levels of methylmalonic acid and/orhomocysteine compared to normal fibroblasts. Table 2 shows thelevels of homocysteine and methylmalonic acid in the culturemedium after 5 days. With the exception of cell line WG3280 (apatient with isolated methylmalonic aciduria), all cblD cell linesaccumulated substantially higher (>2-fold) extracellular homo-cysteine compared to normal fibroblasts. Normal values for ho-mocysteine and methylmalonic acid in cultured human fibroblastsare given in this study and elsewhere [27e29].
The levels of methylmalonic acid are clearly elevated 10e25-foldin fibroblast cultures from cells from patients with isolated orcombined methylmalonic aciduria versus either homocystinuriaalone or unaffected controls (Table 2). The extracellular methyl-malonic acid was highest in the WG3280 cell culture (from a pa-tient with isolated methylmalonic aciduria). Together, these dataindicate that the cblD cell lines selected for the present studyrepresent a good model for studying functional cobalamin defi-ciency representing both the isolated and combined phenotypes.
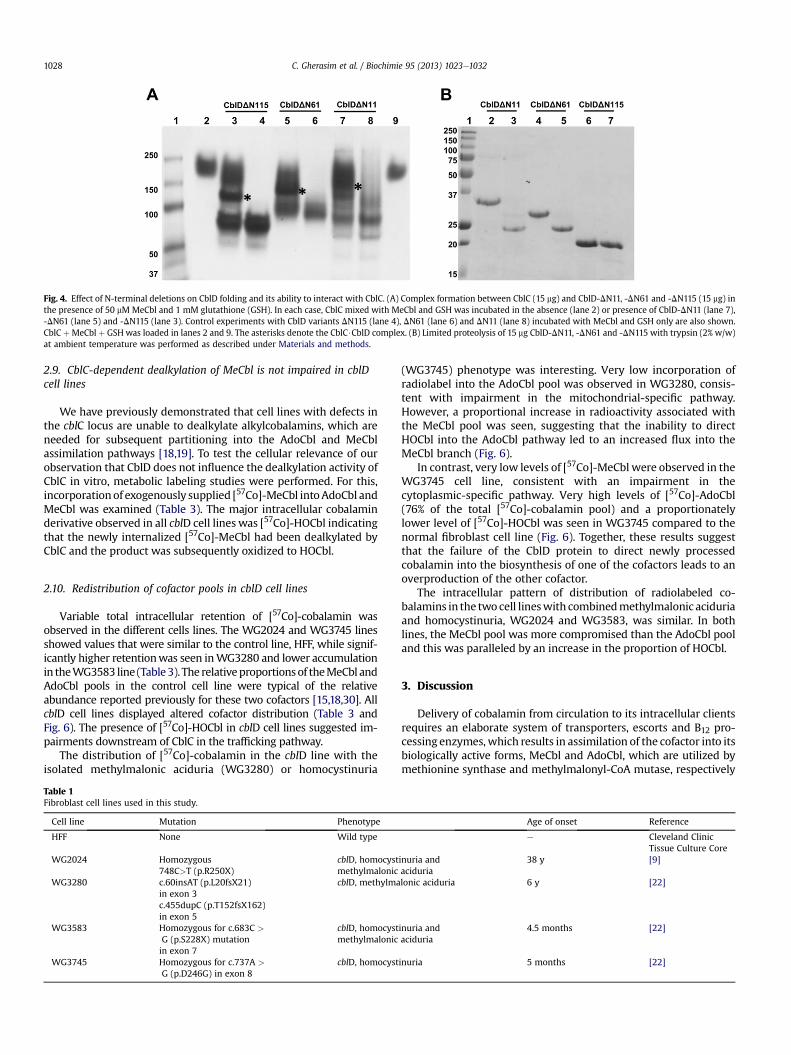
Fig. 4. Effect of N-terminal deletions on CblD folding and its ability to interact with CblC. (A) Complex formation between CblC (15 mg) and CblD-DN11, -DN61 and -DN115 (15 mg) inthe presence of 50 mM MeCbl and 1 mM glutathione (GSH). In each case, CblC mixed with MeCbl and GSH was incubated in the absence (lane 2) or presence of CblD-DN11 (lane 7),-DN61 (lane 5) and -DN115 (lane 3). Control experiments with CblD variants DN115 (lane 4), DN61 (lane 6) and DN11 (lane 8) incubated with MeCbl and GSH only are also shown.CblC þMeCbl þ GSH was loaded in lanes 2 and 9. The asterisks denote the CblC$CblD complex. (B) Limited proteolysis of 15 mg CblD-DN11, -DN61 and -DN115 with trypsin (2% w/w)at ambient temperature was performed as described under Materials and methods.
C. Gherasim et al. / Biochimie 95 (2013) 1023e10321028
2.9. CblC-dependent dealkylation of MeCbl is not impaired in cblDcell lines
We have previously demonstrated that cell lines with defects inthe cblC locus are unable to dealkylate alkylcobalamins, which areneeded for subsequent partitioning into the AdoCbl and MeCblassimilation pathways [18,19]. To test the cellular relevance of ourobservation that CblD does not influence the dealkylation activity ofCblC in vitro, metabolic labeling studies were performed. For this,incorporationof exogenously supplied [57Co]-MeCbl intoAdoCbl andMeCbl was examined (Table 3). The major intracellular cobalaminderivative observed in all cblD cell lines was [57Co]-HOCbl indicatingthat the newly internalized [57Co]-MeCbl had been dealkylated byCblC and the product was subsequently oxidized to HOCbl.
2.10. Redistribution of cofactor pools in cblD cell lines
Variable total intracellular retention of [57Co]-cobalamin wasobserved in the different cells lines. The WG2024 and WG3745 linesshowed values that were similar to the control line, HFF, while signif-icantly higher retentionwas seen inWG3280 and lower accumulationin theWG3583 line (Table3). The relativeproportionsof theMeCbl andAdoCbl pools in the control cell line were typical of the relativeabundance reported previously for these two cofactors [15,18,30]. AllcblD cell lines displayed altered cofactor distribution (Table 3 andFig. 6). The presence of [57Co]-HOCbl in cblD cell lines suggested im-pairments downstream of CblC in the trafficking pathway.
The distribution of [57Co]-cobalamin in the cblD line with theisolated methylmalonic aciduria (WG3280) or homocystinuria
Table 1Fibroblast cell lines used in this study.
Cell line Mutation Phenotype
HFF None Wild type
WG2024 Homozygous748C>T (p.R250X)
cblD, homocystmethylmalonic
WG3280 c.60insAT (p.L20fsX21)in exon 3c.455dupC (p.T152fsX162)in exon 5
cblD, methylma
WG3583 Homozygous for c.683C >
G (p.S228X) mutationin exon 7
cblD, homocystmethylmalonic
WG3745 Homozygous for c.737A >
G (p.D246G) in exon 8cblD, homocyst
(WG3745) phenotype was interesting. Very low incorporation ofradiolabel into the AdoCbl pool was observed in WG3280, consis-tent with impairment in the mitochondrial-specific pathway.However, a proportional increase in radioactivity associated withthe MeCbl pool was seen, suggesting that the inability to directHOCbl into the AdoCbl pathway led to an increased flux into theMeCbl branch (Fig. 6).
In contrast, very low levels of [57Co]-MeCbl were observed in theWG3745 cell line, consistent with an impairment in thecytoplasmic-specific pathway. Very high levels of [57Co]-AdoCbl(76% of the total [57Co]-cobalamin pool) and a proportionatelylower level of [57Co]-HOCbl was seen in WG3745 compared to thenormal fibroblast cell line (Fig. 6). Together, these results suggestthat the failure of the CblD protein to direct newly processedcobalamin into the biosynthesis of one of the cofactors leads to anoverproduction of the other cofactor.
The intracellular pattern of distribution of radiolabeled co-balamins in the two cell lineswith combinedmethylmalonic aciduriaand homocystinuria, WG2024 and WG3583, was similar. In bothlines, the MeCbl pool was more compromised than the AdoCbl pooland this was paralleled by an increase in the proportion of HOCbl.
3. Discussion
Delivery of cobalamin from circulation to its intracellular clientsrequires an elaborate system of transporters, escorts and B12 pro-cessing enzymes, which results in assimilation of the cofactor into itsbiologically active forms, MeCbl and AdoCbl, which are utilized bymethionine synthase and methylmalonyl-CoA mutase, respectively
Age of onset Reference
e Cleveland ClinicTissue Culture Core
inuria andaciduria
38 y [9]
lonic aciduria 6 y [22]
inuria andaciduria
4.5 months [22]
inuria 5 months [22]

Fig. 5. Morphology of cblD cell lines. With the exception of WG3583, all cblD cell lines presented morphological differences with respect to the normal cell line. In all cases, thegrowth was slower for cblD cell lines. However, all cell lines became confluent over time. An enlarged cellular body size and a disrupted fibroblast growth pattern were also evidentby phase contrast microscopy. Magnification: 100�.
C. Gherasim et al. / Biochimie 95 (2013) 1023e1032 1029
(Fig.1) [1,2].MeCbl is the predominant formof the cofactor in humanplasma [31], and following its entry into cells, it must be dealkylatedby the action of CblC to be subsequently partitioned to meet cellulardemands for the MeCbl and AdoCbl cofactor forms [19]. While therole of CblD is expected to be downstream of CblC, identification ofthe cblD gene did not provide obvious insights into the role of theencoded protein. The low sequence homology between residues 78
Table 2Cumulative levels of homocysteine and methylmalonic acid in culture medium after5 days.a
Cell lineID
Phenotype Homocysteine(mmol/mgprotein)
Methylmalonic acid(nmol/mgprotein)
HFF Control 0.016 � 0.004 0.90 � 0.12WG2024 Homocystinuria,
methylmalonic aciduria0.052 � 0.004 10.1 � 2.0
WG3280 Isolated methylmalonicaciduria
0.029 � 0.004 25.1 � 2.7
WG3583 Homocystinuria,methylmalonic aciduria
0.047 � 0.006 15.0 � 1.3
WG3745 Isolated homocystinuria 0.051 � 0.008 1.56 � 0.18
a The data represent the mean of three independent measurements � standarddeviation.
and 168 of CblD and the ATPase component of a bacterial ABCtransporter, suggests that it might hydrolyze ATP [9]. On the otherhand, based on the presence of a partially conserved canonical B12binding motif at the N-terminus of the protein, it was proposed thatCblD might be involved in binding the cofactor [9].
Full-length CblD was shown to interact with CblC in a bacterialtwo hybrid study and by surface plasmon resonance (SPR) [32],although the stoichiometry and ligand dependence of complexformation was not assessed since the CblD was only partially pu-rified. While our study was in progress, the stoichiometric inter-action between purified CblD-DN12 and CblD-DN61 and full-lengthCblC by SPR was reported [33]. In this study, we have used acombination of biochemical and cell biological approaches toelucidate complexation of four CblD variants with a full-length anda truncated variant of CblC and have evaluated the consequences ofCblD deficiency on MeCbl and AdoCbl processing.
We have expressed and purified four recombinant CblD variants(Fig. 2) and demonstrate that these monomeric proteins as isolated,are devoid ofmetal and organic cofactorswith detectable absorptionspectra. Furthermore, CblD does not appear to bind B12 or to hy-drolyze ATP. Based on these results, we propose that CblD functionsinstead as an adapter protein. A traffic junctionwhere CblDmight beinvolved is the transfer of the B12 cargo from CblC to acceptors in the

Table 3Total uptake and intracellular distribution of cobalamin derivatives.a
Cell line Total [57Co] retention(fmol Cbl/mg protein)
Intracellular Cbl (fmol/mg protein)
HOCbl AdoCbl MeCbl MeCbl:AdoCbl
HFF 0.65 � 0.06 0.23 � 0.05 (35%) 0.15 � 0.02 (23%) 0.27 � 0.01 (42%) 1.8WG2024 0.74 � 0.08 0.48 � 0.11 (65%) 0.14 � 0.02 (19%) 0.12 � 0.03 (16%) 0.86WG3280 3.58 � 0.32 1.37 � 0.22 (38%) 0.30 � 0.06 (8%) 1.91 � 0.52 (53%) 6.4WG3583 0.37 � 0.03 0.21 � 0.04 (57%) 0.10 � 0.02 (27%) 0.06 � 0.02 16%) 0.6WG3745 1.07 � 0.02 0.19 � 0.03 (18%) 0.81 � 0.17 (76%) 0.07 � 0.01 (6%) 0.09
a Values represent mean � standard deviation. Uptake was measured after 48 h of incubation with [57Co]-MeCbl.
C. Gherasim et al. / Biochimie 95 (2013) 1023e10321030
MeCbl and AdoCbl branches as shown in Fig. 1. This model is sup-ported by the enhanced interaction betweenCblC andCblD observedin thepresenceof glutathione andHOCbl (Fig. 3A), theprocessed andoxidized product of the CblC-catalyzed dealkylation of alkylcobala-mins. A weak complex is also observed in the presence of CNCbl.
While the shorter DN61 and DN115 variants are predicted to bephysiologically relevant only when mutations in cblD lead to iso-lated methylmalonic aciduria, their ability to retain complex for-mation with CblC indicates that the first 115 residues aredispensable for proteineprotein interaction. This is consistent withresults from a phage display analysis that identified five shortpeptides between residues 142 and 290 in CblD as being potentiallyimportant for binding to CblC [32]. Similarly, the last 37 residues inCblC present in the full-length form of the protein shown to beexpressed in human cell lines [33] but not in murine tissue [26] isnot needed for complexation with CblD. It is not known how B12 isdelivered to themitochondrion to support the AdoCbl branch of thepathway. A possible role for the N-terminal domain of CblD in thetransfer of the B12 from CblC to a mitochondrial transporter asdepicted in Fig. 1 can be considered.
The presence of a weak leader sequence at the N-terminuspredicts that CblD also resides in the mitochondrion. While local-ization in the mitochondrial compartment has not been shown,transfection of a CblD construct containing the strong aldehydedehydrogenase-2 mitochondrial leader sequence has been shownto improve the levels of AdoCbl in a cblD cell line with a combinedMMA/homocystinuria defect [16]. Based on mutational analysis ofCblD, the N-terminal domain is expected to be involved in targetingB12 bound to CblC to the mitochondrial pathway while the C-ter-minal domain could deliver it to the cytoplasmic acceptor, pre-sumably methionine synthase [9]. Interestingly, the presence ofCblC in the mitochondrion has been reported in a mitochondrialproteomic study [34]. Furthermore, cblC cell lines reportedlyexhibit elevated reactive oxygen species levels and a higher pro-pensity for undergoing apoptosis [35], suggestive of a mitochon-drial connection. However, the presence of CblC in themitochondrion has not been independently verified by localization
Fig. 6. Intracellular profile and distribution of [57Co]-cobalamins in normal and cblDmutant cell lines after 48 h in culture with [57Co]-MeCbl substrate.
or other studies and needs to be addressed before its involvementin mitochondrial delivery of the cofactor to the next protein in thetrafficking pathway, adenosyltransferase, can be assessed.
The cblD group is among the rarest of cobalamin disorders andalso themost complex in the heterogeneity of clinical presentationsthat range from isolated to combined deficiency in the MeCbl andAdoCbl branches of the trafficking pathway [15]. Cellular analysis ofthe fate of [57Co]-MeCbl in fibroblasts from cblD patients with iso-lated or combined B12 deficiency provide compelling evidence forthe role of CblD being downstream of CblC. Our study shows thatwhile MeCbl is dealkylated (a function of CblC) in cblD cell lines, thepartitioning of the resulting cofactor product between the cytosolicand mitochondrial compartments is significantly impaired. A pre-vious report on three of the four cblD cell lines used in this study(WG3280, WG3583 and WG3745) also revealed changes in theproportion of AdoCbl versus MeCbl when cells were fed [57Co]-CNCbl [22]. These results further support our conclusion that thefunction of CblD is downstream of CblC in the cobalamin traffickingpathway since we have shown that CblC decyanates CNCbl [21].
Perturbed intracellular cobalamin distribution in cblD cell linespoints to a role for CblD in guiding cofactor traffic betweencompeting pathways. In the WG3280 cell line, one allele has ap.L20fsX21mutation, i.e. a frame shift at L20 leads to termination atcodon 21 (Table 1). Hence, the translation product of this alleleinitiating at codon 1would lead to a truncated CblD product. On theother hand, internal initiation at codon 62 would give rise to ashorter CblD-ND61 product. The other allele has a frame shiftmutation leading to truncation at codon 162 (pT152fsX162), whichis expected to impair both the mitochondrial and cytoplasmicforms of CblD. In this cell line, there is both a large redirection of theintracellular cobalamin pool to MeCbl synthesis at the expense ofAdoCbl synthesis, and surprisingly, a net increase in the intracel-lular B12 pool size (Table 3). Curiously, the total radioactivity inAdoCbl is greater than in the control cell line, but theMeCbl:AdoCblratio is 6.4 in WG3280 versus 1.8 in the control.
One mechanism for the increase in total B12 could be viatranslational up-regulation of methionine synthase, which occursvia a B12-responsive IRES element that is sensitive to MeCbl but notAdoCbl [36]. Increased synthesis of methionine synthase wouldlead to increased production of MeCbl, which occurs in situ on theenzyme and to which it remains tightly bound. Methionine syn-thase, in turn, is stabilized by binding of B12 [37].
Isolated deficiency of methionine synthase occurs in theWG3745 line, which is homozygous for the D246G mutation inCblD. In this cell line, the total B12 levels are approximately in thesame range as in control, but a very high proportion (76%) is tied upas AdoCbl while MeCbl represents only 6% of the total pool and theMeCbl:AdoCbl ratio is 0.09. In contrast, in the control cell line,AdoCbl and MeCbl represent 23% and 42% of the total pool size,respectively (Table 3). The cofactor distribution is indicative ofgreater cobalamin availability for the mitochondrial assimilationpathwaywhen a mutation selectively impairs the cytoplasmic CblDfunction. It also suggests that under normal conditions, partitioningof cobalamin to methionine synthase and to MeCbl synthesis may

C. Gherasim et al. / Biochimie 95 (2013) 1023e1032 1031
be a priority. This is supported by the observation that a significantproportion of methylmalonyl-CoA mutase exists in the apo-form[38] whereas methionine synthase is largely present in the holo-enzyme form [39].
The combined phenotype of the cblD disease leads to a milderdefect in the biosynthesis of AdoCbl and MeCbl compared to theoutcome in the isolated cases (Table 3). The mechanistic basis ofthis finding is presently unknown. While all the cblD fibroblastsshow perturbations in cofactor partitioning toMeCbl versus AdoCblrelative to the control cell line, net diminution in the cofactor poolis consistently observed only for MeCbl (i.e. in cell lines from pa-tients with isolated or combined homocystinuria). This is consis-tent with our model that CblD plays a role in cofactor loading on tomethionine synthase, the site of MeCbl synthesis (Fig. 1). Incontrast, the AdoCbl pool size is not a predictor of associatedmethylmalonic aciduria. Indeed, in two cell lines frompatients withcblD-associated methylmalonic aciduria, WG2024 and WG3280,AdoCbl levels are equal to or higher than in the control cell line(Table 3). This suggests that loading of methylmalonyl-CoA mutasewith AdoCbl is impacted by mutations in the mitochondrial CblDbut AdoCbl synthesis per se is not. Based on this observation, wetentatively propose that CblD functions in the transfer complexbetween adenosyltransferase (CblB) and methylmalonyl-CoAmutase (Mut), which presumably exists in complex with its G-protein chaperone (CblA) in the cell (Fig. 1). In the bacterialorthologs of these mitochondrial proteins, adenosyltransferasesynthesizes and then directly transfer AdoCbl to the mutase$CblAcomplex [40e42]. In bacteria, orthologs of CblD and CblC are notfound; hence the details of the cofactor docking mechanisms arelikely to be different from the mammalian pathway.
In summary, our study maps the involvement of CblD down-stream of CblC in the cobalamin trafficking pathway and revealsthat the C-terminal domain is sufficient for interaction with CblC.Since CblD does not appear to bind B12 directly, at least under theconditions tested in this study, we suggest that CblD might play arole in delivery of the cofactor from CblC to methionine synthase inthe cytoplasm and possibly, to a mitochondrial acceptor. Themitochondrially-directed CblD might be similarly involved incofactor loading onto adenosyltransferase (CblB), which synthe-sizes AdoCbl and/or further downstream, i.e. cofactor docking tomethylmalonyl-CoA mutase.
Funding
This work was supported by grants from the National Institutesof Health (DK45776 to RB and HL71907 to DWJ).
Acknowledgments
We acknowledge Austin Stevens for his assistance with theinitial characterization of the CblD-DN115 variant. This work uti-lized the Morphology and Image Analysis Cores of the MichiganDiabetes Research and Training Center funded by NIH5P60DK20572 from the National Institute of Diabetes & Digestive &Kidney Diseases.
References
[1] R. Banerjee, B12 trafficking in mammals: a case for coenzyme escort service,ACS Chem. Biol. 1 (2006) 149e159.
[2] R. Banerjee, C. Gherasim, D. Padovani, The tinker, tailor, soldier in intracellularB12 trafficking, Curr. Opin. Chem. Biol. 13 (2009) 477e484.
[3] R.A. Gravel, M.J. Mahoney, F.H. Ruddle, L.E. Rosenberg, Genetic complemen-tation in heterokaryons of human fibroblasts defective in cobalamin meta-bolism, Proc. Natl. Acad. Sci. U.S.A. 72 (1975) 3181e3185.
[4] H.F. Willard, I.S. Mellman, L.E. Rosenberg, Genetic complementation amonginherited deficiencies of methylmalonyl-CoA mutase activity: evidence for anew class of human cobalamin mutant, Am. J. Hum. Genet. 30 (1978) 1e13.
[5] M.I. Shevell, D.S. Rosenblatt, The neurology of cobalamin, Can. J. Neurol. Sci. 19(1992) 472e486.
[6] D. Coelho, J.C. Kim, I.R. Miousse, S. Fung, M. du Moulin, I. Buers, T. Suormala,P. Burda, M. Frapolli, M. Stucki, P. Nurnberg, H. Thiele, H. Robenek, W. Hohne,N. Longo, M. Pasquali, E. Mengel, D. Watkins, E.A. Shoubridge, J. Majewski,D.S. Rosenblatt, B. Fowler, F. Rutsch, M.R. Baumgartner, Mutations in ABCD4cause a new inborn error of vitamin B12 metabolism, Nat. Genet. 44 (2012)1152e1155.
[7] F. Rutsch, S. Gailus, I.R. Miousse, T. Suormala, C. Sagne, M.R. Toliat,G. Nurnberg, T. Wittkampf, I. Buers, A. Sharifi, M. Stucki, C. Becker,M. Baumgartner, H. Robenek, T. Marquardt, W. Hohne, B. Gasnier,D.S. Rosenblatt, B. Fowler, P. Nurnberg, Identification of a putative lysosomalcobalamin exporter altered in the cblF defect of vitamin B12 metabolism, Nat.Genet. 41 (2009) 234e239.
[8] J.P. Lerner-Ellis, J.C. Tirone, P.D. Pawelek, C. Dore, J.L. Atkinson, D. Watkins,C.F. Morel, T.M. Fujiwara, E. Moras, A.R. Hosack, G.V. Dunbar, H. Antonicka,V. Forgetta, C.M. Dobson, D. Leclerc, R.A. Gravel, E.A. Shoubridge, J.W. Coulton,P. Lepage, J.M. Rommens, K. Morgan, D.S. Rosenblatt, Identification of the generesponsible for methylmalonic aciduria and homocystinuria, cblC type, Nat.Genet. 38 (2006) 93e100.
[9] D. Coelho, T. Suormala, M. Stucki, J.P. Lerner-Ellis, D.S. Rosenblatt,R.F. Newbold, M.R. Baumgartner, B. Fowler, Gene identification for the cblDdefect of vitamin B12 metabolism, N. Engl. J. Med. 358 (2008) 1454e1464.
[10] C.M. Dobson, T. Wai, D. Leclerc, H. Kadir, M. Narang, J.P. Lerner-Ellis,T.J. Hudson, D.S. Rosenblatt, R.A. Gravel, Identification of the gene responsiblefor the cblB complementation group of vitamin B12-dependent methylmalonicaciduria, Hum. Mol. Genet. 11 (2002) 3361e3369.
[11] C.M. Dobson, T. Wai, D. Leclerc, A. Wilson, X. Wu, C. Dore, T. Hudson,D.S. Rosenblatt, R.A. Gravel, Identification of the gene responsible for the cblAcomplementation group of vitamin B12-responsive methylmalonic acidemiabased on analysis of prokaryotic gene arrangements, Proc. Natl. Acad. Sci.U.S.A. 99 (2002) 15554e15559.
[12] N.A. Leal, S.D. Park, P.E. Kima, T.A. Bobik, Identification of the human andbovine ATP: cob(I)alamin adenosyltransferase cDNAs based on complemen-tation of a bacterial mutant, J. Biol. Chem. 278 (2003) 9227e9234.
[13] D. Leclerc, A. Wilson, R. Dumas, C. Gafuik, D. Song, D. Watkins, H.H.Q. Heng,J.M. Rommens, S.W. Scherer, D.S. Rosenblatt, R.A. Gravel, Cloning and mappingof a cDNA for methionine synthase reductase, a flavoprotein defective in pa-tients with homocystinuria, Proc. Natl. Acad. Sci. U.S.A. 95 (1998) 3059e3064.
[14] J.M. Hsu, B. Kawin, P. Minor, J.A. Mitchell, Vitamin B12 concentrations in hu-man tissue, Nature 210 (1966) 1264e1265.
[15] T. Suormala, M.R. Baumgartner, D. Coelho, P. Zavadakova, V. Koich, H.G. Koch,M. Berghauser, J.E. Wraith, A. Burlina, A. Sewell, J. Herwig, B. Fowler, The cblDdefect causes either isolated or combined deficiency of methylcobalamin andadenosylcobalamin synthesis, J. Biol. Chem. (2004) 42742e42749.
[16] M. Stucki, D. Coelho, T. Suormala, P. Burda, B. Fowler, M.R. Baumgartner,Molecular mechanisms leading to three different phenotypes in the cblDdefect of intracellular cobalamin metabolism, Hum. Mol. Genet. 21 (2012)1410e1418.
[17] C.L. Drennan, S. Huang, J.T. Drummond, R.G. Matthews, M.L. Ludwig, How aprotein binds B12: a 3.0 A X-ray structure of B12-binding domains ofmethionine synthase, Science 266 (1994) 1669e1674.
[18] L. Hannibal, J. Kim, N.E. Brasch, S. Wang, D. Rosenblatt, R. Banerjee,D.W. Jacobsen, Processing of alkylcobalamins in mammalian cells: a role forthe MMACHC (cblC) gene product, Mol. Genet. Metab. 97 (2009) 260e266.
[19] J. Kim, L. Hannibal, C. Gherasim, D.W. Jacobsen, R. Banerjee, A human vitaminB12 trafficking protein uses glutathione transferase activity for processingalkylcobalamins, J. Biol. Chem. 284 (2009) 33418e33424.
[20] J.G. Nørby, Coupled assay of Naþ, Kþ-ATPase activity, Methods Enzymol. 156(1988) 116e119.
[21] J. Kim, C. Gherasim, R. Banerjee, Decyanation of vitamin B12 by a traffickingchaperone, Proc. Natl. Acad. Sci. U.S.A. 105 (2008) 14551e14554.
[22] I.R. Miousse, D. Watkins, D. Coelho, T. Rupar, E.A. Crombez, E. Vilain,J.A. Bernstein, T. Cowan, C. Lee-Messer, G.M. Enns, B. Fowler, D.S. Rosenblatt,Clinical and molecular heterogeneity in patients with the cblD inborn error ofcobalamin metabolism, J. Pediatr. 154 (2009) 551e556.
[23] D.W. Jacobsen, V.J. Gatautis, R. Green, K. Robinson, S.R. Savon, M. Secic, J. Ji,J.M. Otto, L.M. Taylor Jr., Rapid HPLC determination of total homocysteine andother thiols in serum and plasma: sex differences and correlation withcobalamin and folate concentrations in healthy subjects, Clin. Chem. 40 (1994)873e881.
[24] G. Hoffmann, S. Aramaki, E. Blum-Hoffmann, W.L. Nyhan, L. Sweetman,Quantitative analysis for organic acids in biological samples: batch isolationfollowed by gas chromatographic-mass spectrometric analysis, Clin. Chem. 35(1989) 587e595.
[25] J.B.J. Jones, Elemental analysis of soil extracts and plant tissue by plasmaemission spectroscopy, Commun. Soil Sci. Plant Anal. 8 (1977) 349e365.
[26] M. Koutmos, C. Gherasim, J.L. Smith, R. Banerjee, Structural basis of multi-functionality in a vitamin B12-processing enzyme, J. Biol. Chem. 286 (2011)29780e29787.
[27] J.F. Kolhouse, S.P. Stabler, R.H. Allen, Identification and perturbation ofmutant human fibroblasts based on measurements of methylmalonic acid

C. Gherasim et al. / Biochimie 95 (2013) 1023e10321032
and total homocysteine in the culture media, Arch. Biochem. Biophys. 303(1993) 355e360.
[28] E.V. Quadros, S.C. Lai, Y. Nakayama, J.M. Sequeira, L. Hannibal, S. Wang,D.W. Jacobsen, S. Fedosov, E. Wright, R.C. Gallagher, N. Anastasio, D. Watkins,D.S. Rosenblatt, Positive newborn screen for methylmalonic aciduria identifiesthe first mutation in TCblR/CD320, the gene for cellular uptake oftranscobalamin-bound vitamin B(12), Hum. Mutat. 31 (2010) 924e929.
[29] L. Hannibal, P.M. DiBello, M. Yu, A. Miller, S. Wang, B. Willard, D.S. Rosenblatt,D.W. Jacobsen, The MMACHC proteome: hallmarks of functional cobalamindeficiency in humans, Mol. Genet. Metab. 103 (2011) 226e239.
[30] L. Hannibal, A. Axhemi, A.V. Glushchenko, E.S. Moreira, N.E. Brasch,D.W. Jacobsen, Accurate assessment and identification of naturally occurringcellular cobalamins, Clin. Chem. Lab Med. 46 (2008) 1739e1746.
[31] R.C. Chu, J.A. Begley, P.D. Colligan, C.A. Hall, The methylcobalamin metabolismof cultured human fibroblasts, Metabolism 42 (1993) 315e319.
[32] M. Plesa, J. Kim, S.G. Paquettea, H. Gagnona, C. Ng-Thow-Hinga, B.F. Gibbsb,M.A. Hancockb, D.S. Rosenblatt, J.W. Coultona, Interaction between MMACHCand MMADHC, two human proteins participating in intracellular vitamin B12metabolism, Mol Genet Metab 102 (2011) 139e148.
[33] J.C. Deme, I.R. Miousse, M. Plesa, J.C. Kim, M.A. Hancock, W. Mah,D.S. Rosenblatt, J.W. Coulton, Structural features of recombinant MMADHCisoforms and their interactions with MMACHC, proteins of mammalianvitamin B12 metabolism, Mol Genet Metab 107 (2012) 352e362.
[34] D.J. Pagliarini, S.E. Calvo, B. Chang, S.A. Sheth, S.B. Vafai, S.E. Ong, G.A. Walford,C. Sugiana, A. Boneh, W.K. Chen, D.E. Hill, M. Vidal, J.G. Evans, D.R. Thorburn,S.A. Carr, V.K. Mootha, A mitochondrial protein compendium elucidatescomplex I disease biology, Cell 134 (2008) 112e123.
[35] E. Richard, A. Jorge-Finnigan, J. Garcia-Villoria, B. Merinero, L.R. Desviat,L. Gort, P. Briones, F. Leal, C. Perez-Cerda, A. Ribes, M. Ugarte, B. Perez, Geneticand cellular studies of oxidative stress in methylmalonic aciduria (MMA)cobalamin deficiency type C (cblC) with homocystinuria (MMACHC), Hum.Mutat. 30 (2009) 1558e1566.
[36] S. Oltean, R. Banerjee, A B12-responsive IRES element in human methioninesynthase, J. Biol. Chem. 280 (2005) 32662e32668.
[37] K. Yamada, T. Kawata, M. Wada, T. Isshiki, J. Onoda, T. Kawanishi, A. Kunou,T. Tadokoro, T. Tobimatsu, A. Maekawa, T. Toraya, Extremely low activity ofmethionine synthase in vitamin B12-deficient rats may be related to effects oncoenzyme stabilization rather than to changes in coenzyme induction, J. Nutr.130 (2000) 1894e1900.
[38] W.A. Fenton, A.M. Hack, H.F. Willard, A. Gertler, L.E. Rosenberg, Purificationand properties of methylmalonyl coenzyme A mutase from human Liver,Arch. Biochem. Biophys. 214 (1982) 815e823.
[39] Z. Chen, S. Chakraborty, R. Banerjee, Demonstration that the mammalianmethionine synthases are predominantly cobalamin-loaded, J. Biol. Chem. 270(1995) 19246e19249.
[40] D. Padovani, T. Labunska, B.A. Palfey, D.P. Ballou, R. Banerjee, Adenosyl-transferase tailors and delivers coenzyme B12, Nat. Chem. Biol. 4 (2008)194e196.
[41] D. Padovani, R. Banerjee, A G-protein editor gates coenzyme B12 loading and iscorrupted in methylmalonic aciduria, Proc. Natl. Acad. Sci. U.S.A. 106 (2009)21567e21572.
[42] M. Lofgren, R. Banerjee, Loss of allostery and coenzyme B12 delivery by apathogenic mutation in adenosyltransferase, Biochemistry 50 (2011)5790e5798.





























