Embed Size (px)
Citation preview

Thanks to: Broad Inst., DARPA-BioComp, DOE-GTL, EU-MolTools,
NGHRI-CEGS, NHLBI-PGA, NIGMS-CECBSR, PhRMA, Lipper Foundation
Agencourt, Ambergen, Atactic, BeyondGenomics, Caliper, Genomatica, Genovoxx, Helicos, MJR, NEN, Nimblegen, ThermoFinnigan, Xeotron/Invitrogen
For more info see: arep.med.harvard.edu
BU BME retreat 23-Jun-2004 9:45-10:30 Seacrest, N. Falmouth, MA
Optimal Combinatorial Biology & Genome Engineering
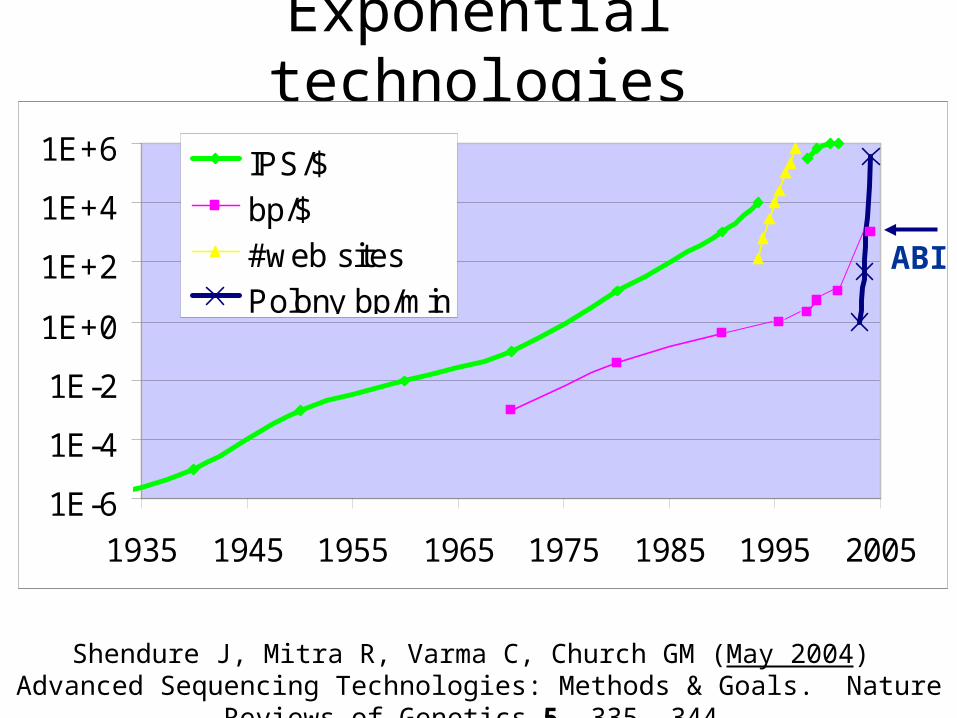
Exponential technologies
1E-6
1E-4
1E-2
1E+0
1E+2
1E+4
1E+6
1935 1945 1955 1965 1975 1985 1995 2005
IPS/$
bp/$
#web sites
Polony bp/min
Shendure J, Mitra R, Varma C, Church GM (May 2004) Advanced Sequencing Technologies: Methods & Goals. Nature Reviews of Genetics 5, 335 -344.
ABI

010101 0101001010001101010101001011001011001010001101010010010 111010
0101010101001010001101010101001011001011001010001101010010010111010
010101010010101101010
101001000101100100011010100100111010
01010100101101010101000010110010000101001001010
Programming cells with DNA
vs.
Digital computers simulating cellsCells simulating digital computers
Drugs & devices simulating human systems
01010100101101010101000010110010000101001001010
01010100101101010101000010110010000101001001010
01010100101101010101000010110010000101001001010
01010100101101010101000010110010000101001001010

Engineering complex systems (comparative genomics)
Stedman et al. (2004) [Masticatory] Myosin gene mutation correlates with anatomical changes in the human
lineage Nature 428, 415 - 418

DNA RNA Proteins
Metabolites
Replication rate
Environment
Biosystems Engineering Integrating Measures & Models
Microbes Cancer & stem cells Darwinian optimaIn vitro replicationSmall multicellular organisms
RNAiInsertionsSNPs
interactions
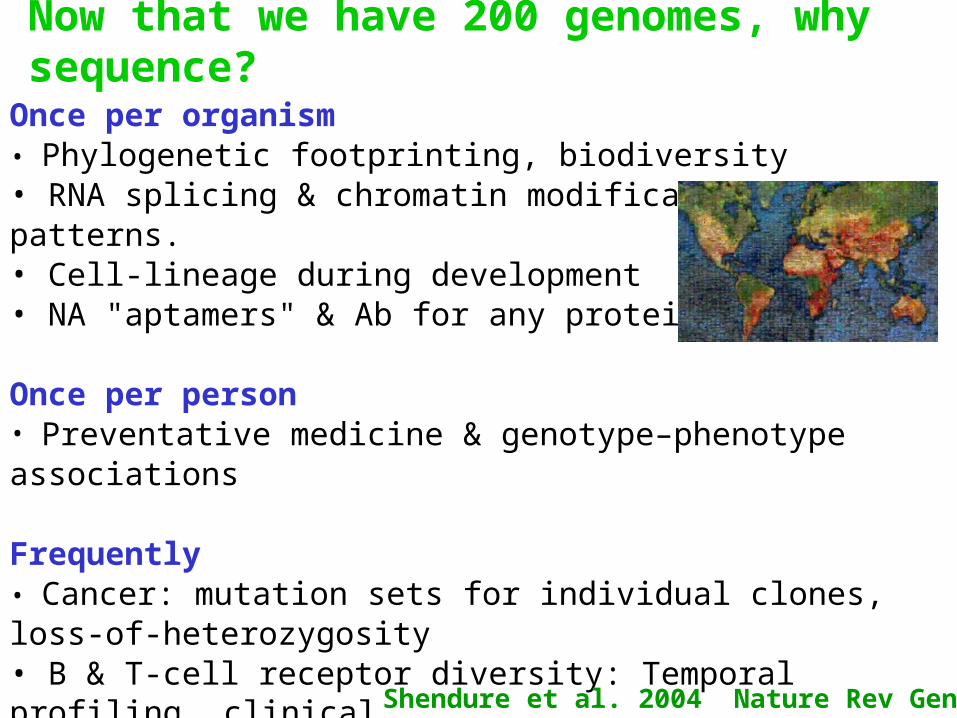
Now that we have 200 genomes, why sequence?
Once per organism• Phylogenetic footprinting, biodiversity• RNA splicing & chromatin modification patterns.• Cell-lineage during development• NA "aptamers" & Ab for any protein
Once per person• Preventative medicine & genotype–phenotype associations
Frequently• Cancer: mutation sets for individual clones, loss-of-heterozygosity• B & T-cell receptor diversity: Temporal profiling, clinical • New & old pathogen "weather map", biowarfare sensors• DNA computing & lab selections
Shendure et al. 2004 Nature Rev Gen 5, 335.

Why 'single molecule' sequencing?
(1) Single-cell analyses , e.g. Preimplantation (PGD)
(2) Co-occurrence on a molecule, complex, cell e.g. RNA splice-forms(3) Cost: $1K-100K "personal genomes"http://grants.nih.gov/grants/guide/rfa-files/RFA-HG-04-003.html
(4) Precision: Counting 109 RNA tags (to reduce variance)
(~5e5 RNAs per human cell)Fixed 5e3 5e4 5e6 5e9 (goal) Costs EST SAGE MPSS Polony-FISSeq (polymerase colony)

Polony Fluorescent In Situ Sequencing Libraries
Greg PorrecaAbraham Rosenbaum
1 to 100kb Genomic1 to 100kb Genomic
L R
M
L R
PCRbead
Sequencingprimers
Selectorbead
2x20bp after MmeI2x20bp after MmeI
Dressman et al PNAS 2003 emulsion

Cleavable dNTP-Fluorophore (& terminators)
Mitra,RD, Shendure,J, Olejnik,J, Olejnik,EK, and Church,GM (2003) Fluorescent in situ Sequencing on Polymerase Colonies. Analyt. Biochem. 320:55-65
Reduce
or
photo-cleave
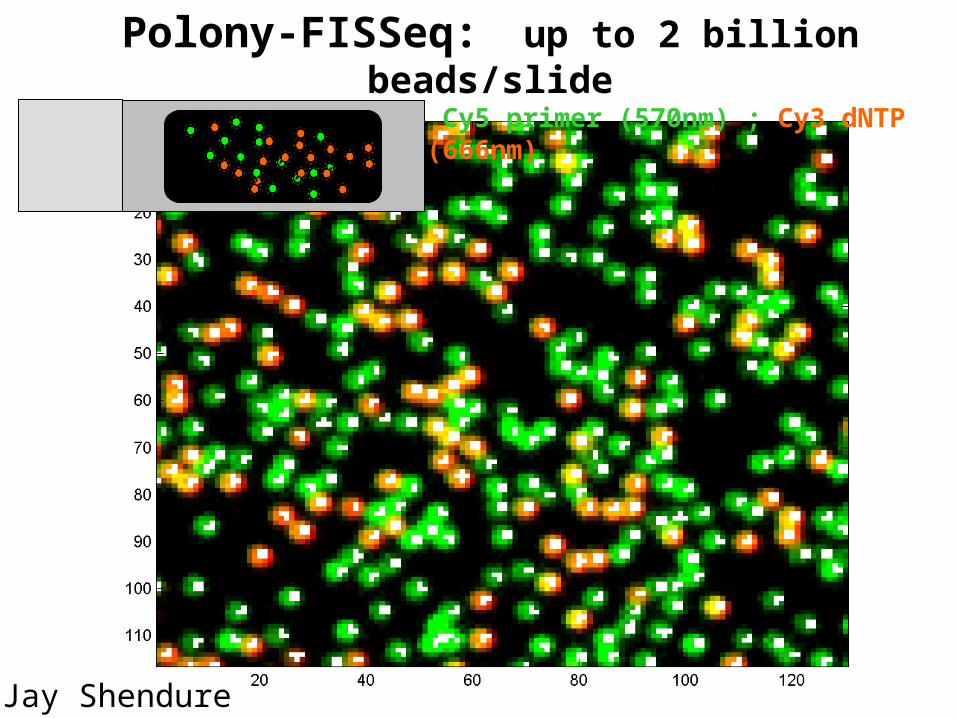
Polony-FISSeq: up to 2 billion beads/slideWhite= Fe-core pixels, Cy5 primer (570nm) ; Cy3 dNTP (666nm)
Jay Shendure

• # of bases sequenced (total) 23,703,953
• # bases sequenced (unique) 73
• Avg fold coverage 324,711 X
• Pixels used per bead (analysis) ~3.6
• Read Length per primer 14-15 bp
• Insertions 0.5%
• Deletions 0.7%
• Substitutions (raw) 4e-5
• Throughput: 360,000 bp/min
Polony FISSeq Stats
Current capillary sequencing 1400 bp/min (600X speed/cost ratio, ~$5K/1X)
(This may omit: PCR , homopolymer, context errors)Shendure

CD44 Exon Combinatorics (Zhu & Shendure)
• Alternatively Spliced Cell Adhesion Molecule• Specific variable exons are up-or-down-regulated in
various cancers (>2000 papers)• v6 & v7 enable direct binding to chondroitin sulfate,
heparin…
Zhu,J, et al. Science. 301:836-8.

V1
V2
V3
V4
V5
V6
V7
V8
V9
V1
0
RNA exon examples
auto-regridded
& quan-titated
Zhu,J, Shendure,J, Mitra, RD, Church, GM (2003) Science. 301:836-8.Single Molecule Profiling of Alternative Pre-mRNA Splicing.

Zhu J, Shendure J, Mitra RD, Church GM. Science 301:836-8. Single molecule profiling of alternative pre-mRNA splicing.
EXON PATTERN Eph4 Eph4bDD TOTALEph4 FRATIO LSTP-PV------------7-8-9-10 609 764 1373 1.17 1E-4--------------8-9-10 320 390 710 1.13 3E-2----------6-7-8-9-10 431 251 682 -1.85 4E-18------4-5-6-7-8-9-10 218 216 434 -1.08 2E-1----------------9-10 68 143 211 1.96 7E-7--------5-6-7-8-9-10 86 39 125 -2.37 2E-6----3-4-5-6-7-8-9-10 40 56 96 1.30 9E-2------4-5---7-8-9-10 16 74 90 4.30 2E-9--2-3-4-5-6-7-8-9-10 44 28 72 -1.69 1E-21-2-3-4-5-6-7-8-9-10 22 5 27 -4.73 3E-4--------5---7-8-9-10 5 19 24 3.53 3E-3----3-4-5---7-8-9-10 1 15 16 13.95 4E-4--2-3-4-5---7-8-9-10 1 10 11 9.30 5E-3
Eph4 = murine mammary epithelial cell line
Eph4bDD = stable transfection of Eph4 with MEK-1 (tumorigenic)
CD44 RNA isoforms

DNA RNA Proteins
Metabolites
Replication rate
Environment
Biosystems Engineering Integrating Measures & Models
Escherichia Darwinian optima Prochlorococcus mutant suboptimality
Homo
RNAiInsertionsSNPs
interactions
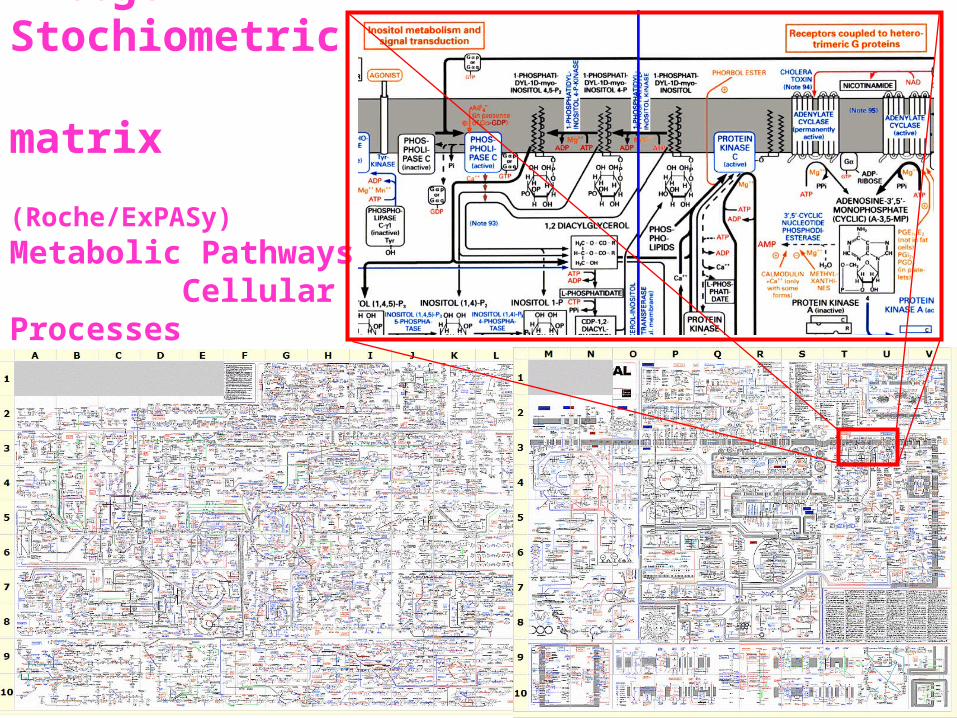
Integer Stochiometric matrix
(Roche/ExPASy)
Metabolic Pathways Cellular Processes
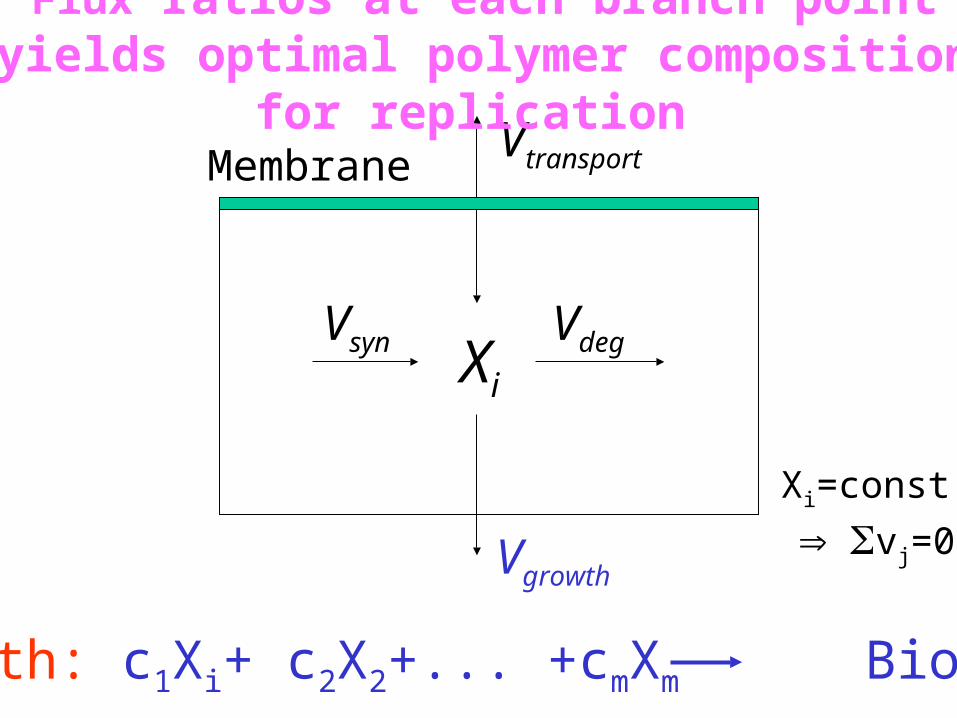
Xi
MembraneVtransport
Vsyn Vdeg
Vgrowth
Growth: c1Xi+ c2X2+... +cmXm Biomass
Flux ratios at each branch point yields optimal polymer composition for replication
Xi=const.
vj=0
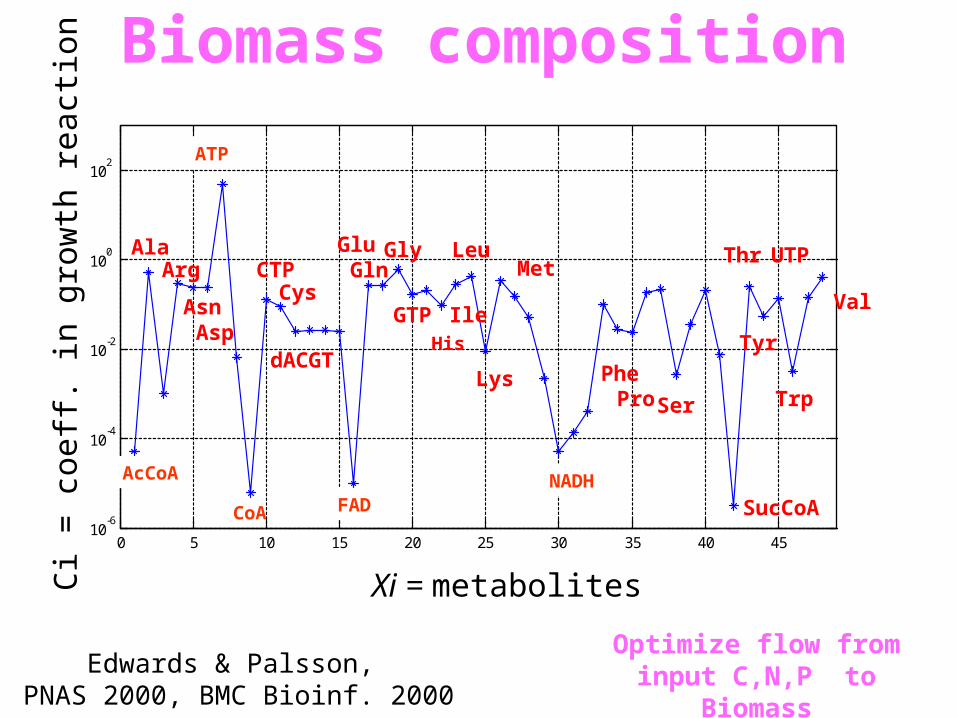
0 5 10 15 20 25 30 35 40 4510
-6
10-4
10-2
100
102
AcCoA
CoA
ATP
FADNADH
Xi = metabolites
Ci =
coe
ff. i
n gr
owth
rea
ctio
nBiomass composition
Edwards & Palsson, PNAS 2000, BMC Bioinf. 2000
Optimize flow from input C,N,P to Biomass
GTP
Trp
LeuAlaArg
Gly
Cys
Ser
Asn Asp His
CTPUTP
SucCoA
Val
Glu Gln
PhePro
Ile
Lys
Met
Tyr
Thr
dACGT

Minimization of Metabolic Adjustment (MoMA)
Linear Programming (LP) to find optima, Quadratic (QP) to find closest points
x,y are two of the 100s of flux dimensions
Wild-typeoptimum
Mutantoptimum
Mutantinitially
(closest point)
Mutant Wild type(feasible flux polyhedra)
Objective function = growth flux hyperplanes
Segre, Vitkup, & Church PNAS 99: 15112-7

12C 13C
MS/NMR FluxRatio Data

0 50 100 150 2000
20
40
60
80
100
120
140
160
180
200
1
2
3
456
78
9
10
11121314
15
16
17 18
-50 0 50 100 150 200 250-50
0
50
100
150
200
250
1
2
3456
78
910
11121314
1516
17
18
Experimental Fluxes
Pre
dic
ted
Flu
xes
-50 0 50 100 150 200 250-50
0
50
100
150
200
250
1
2
3
456
78
910
111213
14
15
16
1718
pyk (LP)
WT (LP)
Experimental Fluxes
Pre
dic
ted
Flu
xes
Experimental Fluxes
Pre
dic
ted
Flu
xes
pyk (QP)
=0.91p=8e-8
=-0.06p=6e-1
=0.56p=7e-3
Flux Data C009-limited

Reproducibility of mass competition
Correlation between two selection experiments
Badarinarayana, et al. Nature Biotech.19: 1060

Essential 142 80 62Reduced growth 46 24 22
Non essential 299 119 180 p = 4∙10-3
Essential 162 96 66Reduced growth 44 19 25
Non essential 281 108 173 p = 10-5
MOMA
FBA
Competitive growth data
2 p-values
4x10-3
1x10-5
Position effects Novel redundancies
On minimal media
negative small selection effect
Hypothesis: next optima are achieved by regulation of activities.
LP
QP

Motif Co-occurrence, comparative genomics, RNA clusters, and/or ChIP2-location data
P= 10-6 to 10-11
Genome Res. 14:201–208Bulyk, McGuire,Masuda,Church

Synthetic testing of DNA motif combinations
1.3 2.4 (1.3 in argR)
1.1 1.3
0.7 2.5
0.2 1.4
1.4 3.5
RNA Ratio (motif- to wild type) for each flanking gene
Bulyk, McGuire,Masuda,Church Genome Res. 14:201–208

Systems Biology Loop
Synthesis /Perturbation
Model
Experimental design
(Systematic)
Data
Proteasome targetingGenome Engineering

Engineering BioSystems Perturbations
Action Specificity %KO "Design"
Small molecules (drugs) Fast Varies Varies Hard
Antibodies Fast Varies Varies Hard
RNAi Slow Varies Medium OK
Insertion "traps" Slow Yes Varies Random
Proteasome targeting Fast Excellent Medium Easy
Homologous recombination Slow Perfect Complete Easy

Programming proteasome
targeting
Janse, DM, Crosas,B Finley,D & Church, GM (2004) Localization to the Proteasome is Sufficient
for Degradation.

Synthetic Genomes & Proteomes. Why?
• Test or engineer cis-DNA/RNA-elements •Access to any protein (complex) including post-transcriptional modifications• Affinity agents for the above.• Mass spectrometry standards, protein design• Utility of molecular biology DNA-RNA-Protein
in vitro "kits" (e.g. PCR, SP6, Roche)
Toward these goals design a chassis:• 115 kbp genome. 150 genes.• Nearly all 3D structures known.• Comprehensive functional data.

PURE translation utility (yet room for improvement)
Removing tRNA-synthetases, RNases & proteases makes feasible:
Optimal mRNA structure & codon usage
Lee et al. 2004 J Immunol Methods. 284:147-57. Selection of scFvs specific for HBV DNA polymerase using ribosome display.
Forster et al. 2003Programming peptidomimetic syntheses by translating genetic codes designed de novo. PNAS 100:6353-7.
Klammt et al. 2004 Eur J Biochem. 271:568-80. High level cell-free expression & specific labeling of integral membrane proteins.
Shimizu et al. 2001 Nat Biotechnol. 19:751-5. Cell-free translation reconstituted with purified components.

in vitro genetic codes
5'
mS yU eU
UGGUUG CAG
AAC... GUU A 3'GAAACCAUG
fM TN V E
| | | | | || | |
5' Second base 3'
U
A
C
C U
mSyU
eU
A C U
G
A
0
500
1000
1500
2000
2500
3000
3500
30 40 50 60 70 80
3H-E dpm
time (min.)
fM yU mS eU E |
Forster, et al. (2003) PNAS 100:6353-7
80% average yieldper unnatural coupling.
bK = biotinyllysine , mS = Omethylserine eU=2-amino-4-pentenoic acid yU = 2-amino-4-pentynoic acid

Mirror world : resistant to enzymes, parasites, predators
L-amino acids & D-ribose (rNTPs, dNTPs)
Transition: EF-Tu, peptidyl transferase, DNA-ligase
D-amino acids & L-ribose (rNTPs, dNTPs)
Dedkova, et al. (2003) Enhanced D-amino acid incorporation into protein by modified ribosomes. J Am Chem Soc 125, 6616-7

Escherichia coli Mycoplasma 3D structureColiphage 29 DNA polymerase + +Coliphage P1 Cre recombinase - + >Coliphage Lox/Cre recombinase site - +Coliphage T7 RNA polymerase + + >Coliphage T7 RNA polymerase initiation site + + >Coliphage T7 RNA polymerase termination site + +RNase P RNA + -RNase P protein + + >RNase P site/RNA primer for DNA polymerase + +Small subunit 16S ribosomal RNA + +All 21 small subunit ribosomal proteins (1-21) + except 1,21 +Large subunit 5S ribosomal RNA + +Large subunit 23S ribosomal RNA + +Large subunit 23S rRNA G2445>m2G methylase: unknown ? -Large subunit 23S rRNA U2449>dihydroU synthetase: unknown ? -Large subunit 23S rRNA U2457>pseudoU synthetase ? -Large subunit 23S rRNA C2498>Cm methylase: unknown ? -Large subunit 23S rRNA A2503>m2A methylase: unknown ? -Large subunit 23S rRNA U2504>pseudoU synthetase ? -All 33 large subunit ribosomal proteins (1-7,9-11,13-25,27-36) + except 25, 30 +Translational initiation factor 1 + +Translational initiation factor 2 + +Translational initiation factor 3 + +Translational elongation factor Tu + +Translational elongation factor Ts + +Translational elongation factor G + +Translational release factor 1 + +Translational release factor 2 - +Translational release factor Gln methylase + +Translational release factor 3 - +Ribosome recycling factor + +33/45 Transfer RNAs (see Fig. 2) 29/33 +tRNA(I) C34>lysidine synthetase ? +tRNA(R) A34>I deaminase ? +tRNA(ASV) U34>cmo5U (=V) synthetase: unknown - -tRNA(R) U34>2sU Cys desulfurase - +tRNA(R) nm5U34 methylase ? +tRNA(R) U34>cmnm5U GTPase ? +tRNA(R) U34>cmnm5U synthetase ? +tRNA(R) cmnm5U34>nm5U,mnm5U synthetase ? -tRNA(R) G37 N1-methylase + +tRNA(RNIKM) A37>t6A N6-threonylcarbamoyl-A synthetase: unknown + -tRNA(CLFSWY) A37>i6A synthetase - +tRNA(CLFSWY) i6A37>s2i6A(ms2i6A) synthetase - +All 22 aminoacyl-tRNA synthetase subunits (20 enzymes) + except G subunit, Q + except G subunitMet-tRNA formyltransferase + +Chaperonin DnaK + +Chaperonin GroEL + +Chaperonin GroES + +
Total genes = 150Forster & Church
Oligos for 150 & 776
synthetic genes(for E.coli minigenome & M.mobile whole genome
respectively)

Up to 760K Oligos/Chip18 Mbp for $700 raw (6-18K genes)
<1K Oxamer Electrolytic acid/base 8K Atactic/Xeotron/Invitrogen Photo-Generated Acid Sheng , Zhou, Gulari, Gao (U.Houston) 24K Agilent Ink-jet standard reagents 48K Febit 100K Metrigen 380K Nimblegen Photolabile 5'protection Nuwaysir, Smith, Albert
Tian, Gong, Church

Improve DNA Synthesis CostSynthesis on chips in pools is 5000X less expensive per
oligonucleotide, but amounts are low (1e6 molecules rather than usual 1e12) & bimolecular kinetics slow with square of concentration decrease!)
Solution: Amplify the oligos then release them.
10 50 10 => ss-70-mer (chip)
20-mer PCR primers with restriction sites at the 50mer junctions
Tian, Gong, Sheng , Zhou, Gulari, Gao, Church
=> ds-90-mer
=> ds-50-mer
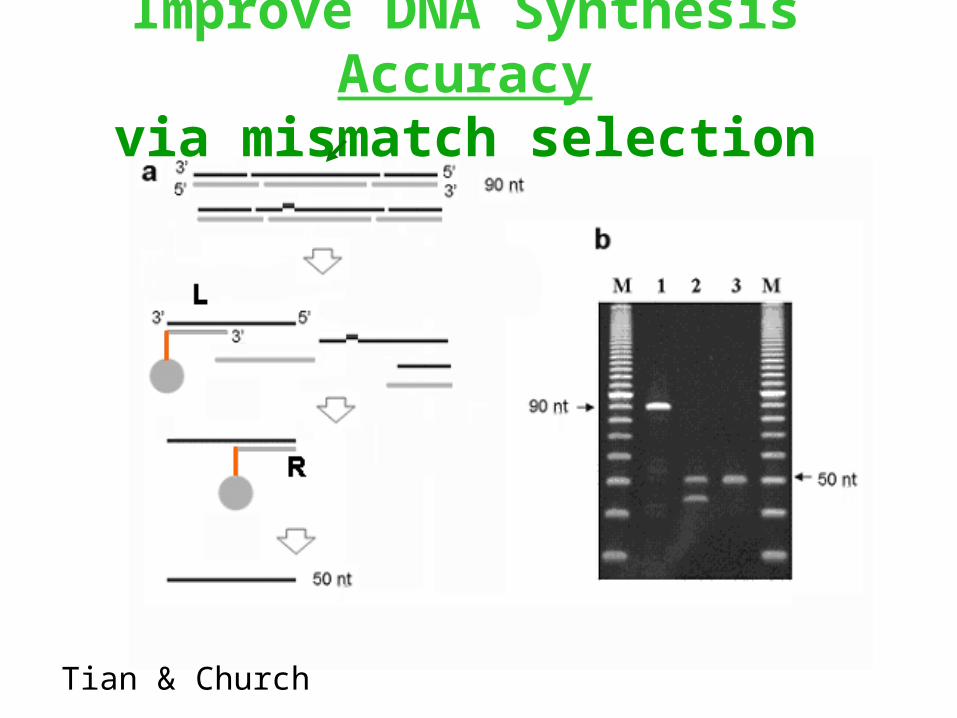
Improve DNA Synthesis Accuracyvia mismatch selection
Tian & Church

Genome assembly
Challenges: 1. Tandem, inverted and dispersed repeats (hierarchical assembly, size-selection and/or scaffolding)2. Reduce mutations (goal <1e-6 errors) to reduce # of intermediates 3. >30 kbp homologous recombination (Nick Reppas)
Stemmer et al. 1995. Gene 164:49-53. Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides.
50
75
125 225 425 825 … 100*2^(n-1)

M 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21
DNA Templates
RNA Transcripts
All 30S-Ribosomal-protein DNAs & mRNAs synthesized in vitro
s190.5kb0.3kb
NimblegenXeotron/Atactic
Wild-type
DNA Templates
Tian, Gong, Sheng , Zhou, Gulari, Gao, Church
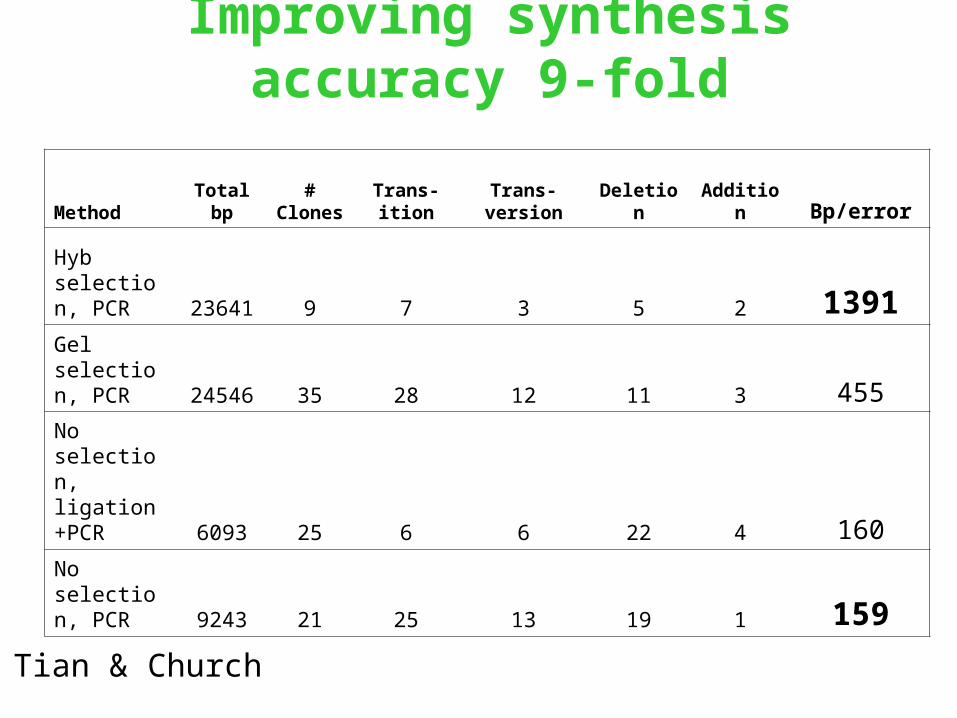
Improving synthesis accuracy 9-fold
MethodTotal
bp#
ClonesTrans-ition
Trans-version Deletion Addition Bp/error
Hyb selection, PCR 23641 9 7 3 5 2 1391Gel selection, PCR 24546 35 28 12 11 3 455
No selection, ligation+PCR 6093 25 6 6 22 4 160
No selection, PCR 9243 21 25 13 19 1 159
Tian & Church

Extreme mRNA makeover for protein expression in vitro
RS-2,4,5,6,9,10,12,13,15,16,17,and 21 detectable initially.
RS-1, 3, 7, 8, 11, 14, 18, 19, 20 initially weak or undetectable.
Solution: Iteratively resynthesize all mRNAs with less mRNA structure.
Tian & Church
20w 20m 17w 17m 16w 16m
10kd
W: wild-typeM: modified
Western blot based on His-tags

Enabling technologies
• Multi-Gene Assembly• Protein, peptidomimetic synthesis• CAD/CAM & Design for manufacturing
• Automated homologous recombination for E.coli & embryonic stem cells• Fidelity enhancements• Sequencing 107 bp/$ ($1K/human)

Thanks to: DOE-GTL, DARPA-BioComp, NIGMS-CECBSR,
NGHRI-CEGS, PhRMA, EU-MolTools, NHLBI-PGA,
Broad Inst., Lipper Foundation
Agencourt, Ambergen, Atactic, BeyondGenomics, Caliper, Genomatica, Genovoxx, MJR, NEN, Nimblegen, ThermoFinnigan, Xeotron/Invitrogen
For more info see: arep.med.harvard.edu
BU BME retreat 23-Jun-2004 9:45-10:30 Seacrest, N. Falmouth, MA
Optimal Combinatorial Biology & Genome Engineering

.

Improve DNA Synthesis accuracySynthesis on a chip pools of "construction" ~50-mers and two
complementary "selection" ~26-mers (Left & Right)
10 50 10 => ss-70-mer (chip)
Tian, Gong, Sheng , Zhou, Gulari, Gao, Church
=> ds/ss-50-mer (amplif/restrict)
10 26 10 => ss-56-mer (chip)
20-mer PCR primers (one biotinylated)
Biotin=> ss-76-mer (amplif/avidin)

Improve DNA Synthesis Accuracyvia D-HPLC or MutS
Smith & Modrich (1997) PNAS 94: 6847–50. Removal of polymerase-produced mutant sequences from PCR products. MutHLS Cleaves at GATC near mismatches. Lowers error rate from 6e-6 to 6e-7.
Bellanne-Chantelot et al. (1997) Mutat Res. 382:35-43. Search for DNA sequence variations using a MutS-based technology.
Mulligan & Tabone (2002) US Patent 6,664,112. Methods for improving the sequence fidelity of synthetic doublestranded-oligonucleotides.





